
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic production of hydrogen peroxide from water and dioxygen using cyano-bridged polynuclear transition metal complexes as water oxidation catalysts†
Yusuke
Isaka
a,
Kohei
Oyama
a,
Yusuke
Yamada
*b,
Tomoyoshi
Suenobu
a and
Shunichi
Fukuzumi
*acd
aDepartment of Materials and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp
bDepartment of Applied Chemistry and Bioengineering, Graduate School of Engineering, Osaka City University, Osaka, 558-0022, Japan
cDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea
dFaculty of Science and Technology, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-8502, Japan
First published on 28th December 2015
Abstract
Hydrogen peroxide was produced efficiently from water and dioxygen using [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst and cyano-bridged polynuclear transition metal complexes composed of Fe and Co as water oxidation catalysts in the presence of Sc3+ in water under visible light irradiation.
Hydrogen peroxide (H2O2) has merited increasing attention as an ideal energy carrier alternative to hydrogen, because an aqueous solution of H2O2 instead of gaseous hydrogen can be used as a fuel in a one-compartment fuel cell to generate electricity.1–14 The maximum output potential of an H2O2 fuel cell theoretically achievable is 1.09 V which is comparable to that of a hydrogen fuel cell (1.23 V).1–14 Thus, H2O2 production from water (H2O) and dioxygen (O2) using solar energy provides an ideally sustainable solar fuel in combination with power generation using an H2O2 fuel cell.15–17 It is highly desired to improve the catalytic activity for the photocatalytic production of H2O2 from H2O and O2 (ΔG° = 210 kJ mol−1, eqn (1)) using earth-abundant metal catalysts.15–17
| 2H2O + O2 → 2H2O2 ΔG° = 210 kJ mol−1 | (1) |
We report herein the photocatalytic production of H2O2 from H2O and O2 using [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst and structurally-definable and molecularly-ordered metal complexes, i.e., cyano-bridged polynuclear transition metal complexes composed of Fe and Co, as water oxidation catalysts (WOCs) in the presence of Sc3+ in water under visible light irradiation. Among various metal complex-based WOCs, metal complexes were found to play in some cases the role of precursors of actual WOCs.18,19 In contrast, cyano-bridged polynuclear transition metal complexes as they are have been proven to maintain absolutely high catalytic reactivity with high yield and quantum efficiency for water oxidation.20
The photocatalytic cycle is shown in Scheme 1, where the excited state of [RuII(Me2phen)3]2+ is oxidatively quenched by electron transfer to O2 to produce [RuIII(Me2phen)3]3+ and the O2•−–Sc3+ complex, which undergoes disproportionation in the presence of H+ to yield H2O2.15,17 Water is oxidised by [RuIII(Me2phen)3]3+ in the presence of a heteropolynuclear cyanide metal complex as a WOC to produce O2.
 | ||
| Scheme 1 Catalytic cycle of the photocatalytic production of H2O2 from H2O and O2 using [RuII(Me2phen)3]2+ as a photocatalyst and heteropolynuclear cyanide complexes (M3[M′(CN)6]2; M, M′ = different metals) as water oxidation catalysts. | ||
Heteropolynuclear cyanide complexes take a cubic structure provided that the contained metal ions allow octahedral coordination.21,22 Both the C and N atoms of cyanide interact with metal ions. When the number of N-bound metal ions is larger than that of C-bound metal ions, the N-bound metal ions need external ligands such as an aqua ligand to fulfil octahedral coordination.23,24 The number of external ligands can be controlled by considering charge compensation in a heteropolynuclear complex.23,24 Thus, heteropolynuclear cyanide complexes composed of different metal ions can be designable heterogeneous catalysts for water oxidation.
A series of heteropolynuclear cyanide metal complexes containing different metal ions, Co3[Fe(CN)6]2, Co3[Co(CN)6]2, Cu3[Co(CN)6]2, Co[Ni(CN)4], Fe3[Cr(CN)6]2, Mn3[Fe(CN)6]2, Co3[Mn(CN)6]2, Co3[Fe(CN)6]2, Co[Pd(CN)4] and Co[Pt(CN)4], were prepared according to the literature.20Fig. 1 shows the time profiles of the production of H2O2 from H2O and O2 in an aqueous solution containing [RuII(Me2phen)3]2+, Sc(NO3)3 and a heteropolynuclear cyanide metal complex under visible light irradiation with a xenon lamp using a UV light cut filter (λ > 420 nm). The amount of produced H2O2 was determined by spectroscopic titration with an acidic solution of [TiO(tpypH4)]4+ complex (Ti-TPyP reagent).25 Among the various heteropolynuclear cyanide complexes, Fe3[Co(CN)6]2 exhibited the highest catalytic reactivity.
 | ||
| Fig. 1 Time courses of production of H2O2 from H2O and O2 in an O2-saturated aqueous solution (2.0 mL) of [Ru(Me2phen)3]2+ (100 μM), Sc(NO3)3 (100 mM) and a heteropolynuclear cyanide metal complex (1.0 mg) under photoirradiation of visible light (λ > 420 nm) with a xenon lamp using a UV light cut filter at room temperature. The employed heteropolynuclear cyanide complexes are Fe3[Co(CN)6]2 (blue square), Co3[Co(CN)6]2 (red right triangle), Cu3[Co(CN)6]2 (green diamond), Co[Ni(CN)4] (orange regular triangle), Fe3[Cr(CN)6]2 (grey inverse triangle), Mn3[Fe(CN)6]2 (dark orange right triangle), Co3[Mn(CN)6]2 (pink square), Co3[Fe(CN)6]2 (light green circle), Co[Pd(CN)4] (blue circle) and Co[Pt(CN)4] (purple diamond). | ||
A series of heteropolynuclear cyanide complexes (FexCo1−x)3[Co(CN)6]2 (x = 0, 0.10, 0.50, 0.75, 0.90 and 1) were prepared by mixing an aqueous solution of K3[CoIII(CN)6], CoII(NO3)2 and FeII(ClO4)2 with various Fe/Co ratios of the (FexCo1−x) moiety ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1. All of the synthesised complexes were isostructural with Prussian blue as confirmed by the powder X-ray diffraction patterns (Fig. S1†). A schematic drawing of the complex is shown in Fig. 2. The Co and Fe ion contents of each compound were determined by X-ray fluorescence measurements (Table S1†). The size of (FexCo1−x)3[Co(CN)6]2 particles remains about the same (260–300 nm) irrespective of the Co-to-Fe ratio as indicated by DLS measurements (Fig. S2†).
:0 to 0:1. All of the synthesised complexes were isostructural with Prussian blue as confirmed by the powder X-ray diffraction patterns (Fig. S1†). A schematic drawing of the complex is shown in Fig. 2. The Co and Fe ion contents of each compound were determined by X-ray fluorescence measurements (Table S1†). The size of (FexCo1−x)3[Co(CN)6]2 particles remains about the same (260–300 nm) irrespective of the Co-to-Fe ratio as indicated by DLS measurements (Fig. S2†).
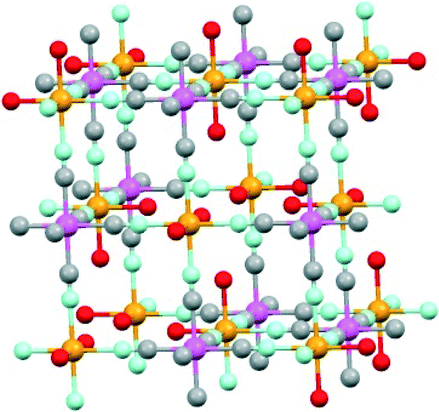 | ||
| Fig. 2 A schematic drawing of (FexCo1−x)3[Co(CN)6]2 where x = 0, 0.10, 0.50, 0.75, 0.90 and 1. Ions are colour-coded: N-bound CoII and FeII (orange), C-bound CoIII (pink), C (grey), N (blue) and O (red). | ||
The catalytic reactivity of (FexCo1−x)3[Co(CN)6]2 with various x values was examined for the production of H2O2 from H2O and O2 in an O2-saturated aqueous solution of [Ru(Me2phen)3]2+ (100 μM), Sc(NO3)3 (100 mM) and (FexCo1−x)3[Co(CN)6]2 (1.0 mg) under photoirradiation of visible light with a xenon lamp using a UV-light cut filter (λ > 420 nm) at room temperature as shown in Fig. S3.† The initial rate of production of H2O2 increased with increasing Fe-to-Co ratio in the (FexCo1−x) moiety (FrFe), reaching a maximum at FrFe = 0.75, and then decreased as shown in Fig. 3. The catalytic reactivity of (Fe0.75Co0.25)3[Co(CN)6]2 (1) was 4.5 and 1.5 times enhanced as compared to those of Co3[Co(CN)6]2 and Fe3[Co(CN)6]2.
 | ||
| Fig. 3 Initial rates of H2O2 production plotted vs. fraction of Fe (FrFe). H2O2 was produced from H2O and O2 in an O2-saturated aqueous solution (2.0 mL) of [Ru(Me2phen)3]2+ (100 μM), Sc(NO3)3 (100 mM) and (FexCo1−x)3[Co(CN)6]2 (1.0 mg), where x = 1 (black square), 0.90 (inverse red triangle), 0.75 (orange circle), 0.50 (green diamond), 0.10 (purple triangle) and 0 (blue diamond) under photoirradiation of visible light (λ > 420 nm) with a xenon lamp using a UV light cut filter at room temperature. The time courses of H2O2 production are shown in Fig. S3.† | ||
The rate of H2O2 production was enhanced 2.9 times when N-bound Co ions in Co3[Co(CN)6]2 were thoroughly replaced with Fe ions as shown in Fig. 3. Therefore, the water oxidation reactivity of N-bound Fe ions was higher than that of N-bound Co ions. On the other hand, the peak attributed to CN ligand stretching observed in the IR spectra of Fe3[Co(CN)6]2 red-shifted as N-bound FeII ions were replaced with CoII ions (Fig. S4†). This is because an FeII ion can accept electrons from bonding orbitals of CN ligands rather easily than a CoII ion because of its low LUMO level. The electron-rich CN ligands can stabilise high-valence metal ions that form in the water oxidation process. Therefore, the volcano-type dependence of the rate of H2O2 production on FrFe is considered to be a result of those two contradictory effects of FrFe on the water oxidation reaction where a complex with a large FrFe would contain more active sites for water oxidation while a complex with a small FrFe would easily stabilise high-valence metal ions formed during water oxidation.
The catalytic activity of (FexCo1−x)3[Co(CN)6]2 was also examined in the photocatalytic oxidation of water with persulfate (Na2S2O8) using [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photocatalyst. The photocatalytic cycle is given in Scheme 2, where the excited state of [Ru(bpy)3]2+ was oxidatively quenched by Na2S2O8 to produce [Ru(bpy)3]3+, which oxidises water in the presence of (FexCo1−x)3[Co(CN)6]2 acting as a WOC to evolve O2. The time courses of O2 evolution in the photocatalytic water oxidation with Na2S2O8 in the presence of [Ru(bpy)3]2+ and (FexCo1−x)3[Co(CN)6]2 are shown in Fig. S5.† The O2 evolution rate was maximised at FrFe = 0.75 which also gave the most effective WOC for photocatalytic H2O2 production (Fig. 4). Catalytic O2 evolution by water oxidation was also confirmed when [RuIII(Me2phen)3]3+ was added to an aqueous suspension of 1 at pH 3.0, the same pH condition as that in the H2O2 production reaction (Fig. S6†).
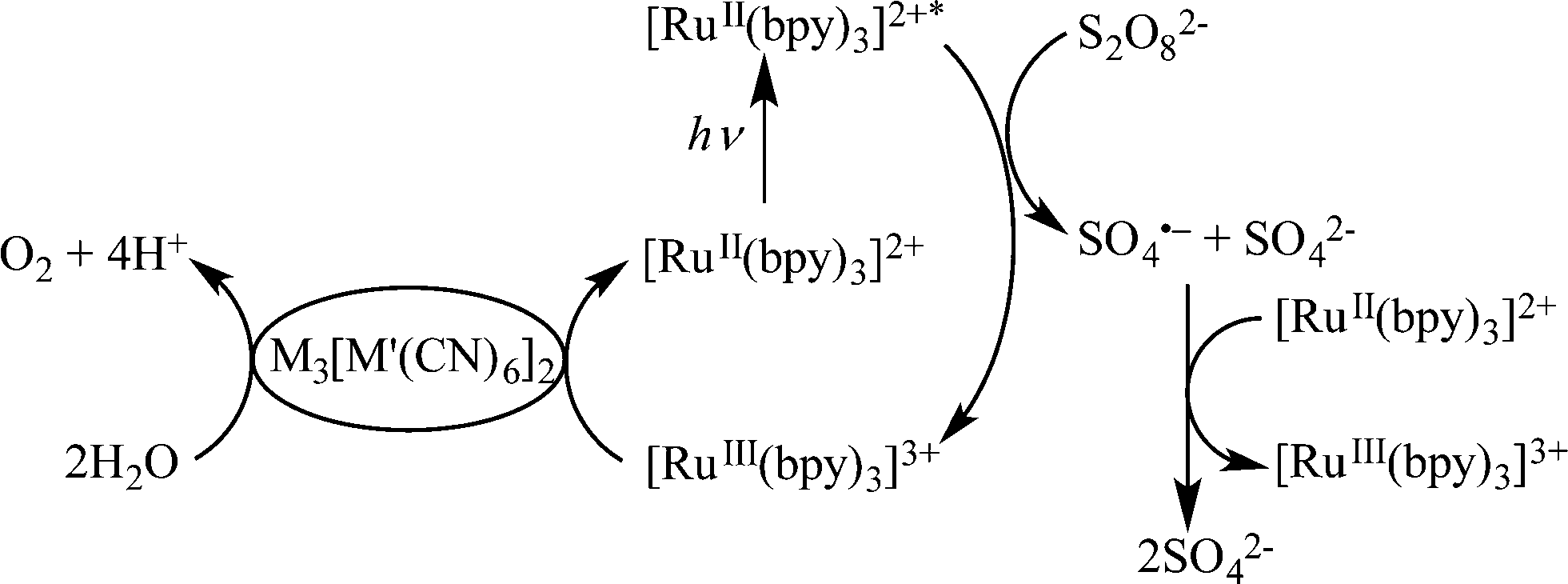 | ||
| Scheme 2 Photocatalytic cycle of water oxidation with Na2S2O8 using [Ru(bpy)3]2+ as a photocatalyst and (FexCo1−x)3[Co(CN)6]2 as a water oxidation catalyst. | ||
 | ||
| Fig. 4 Initial rates of O2 evolution plotted versus FrFe. O2 evolution was performed by photoirradiation (λ > 420 nm) of an aqueous phosphate buffer (2.0 mL) containing Na2S2O8 (5.0 mM), Ru[(bpy)3]2+ (100 μM) and (FexCo1−x)3[Co(CN)6]2 (1.0 mg), where x = 1 (black square), 0.90 (green diamond), 0.75 (red inverse triangle), 0.50 (orange circle) and 0 (blue diamond), at pH 8.0 and room temperature. The time courses of O2 evolution are shown in Fig. S5.† | ||
The dependence of the rate of production of H2O2 on the amount of 1 and [Ru(Me2phen)3]2+ was examined to obtain the optimised conditions where the amount of 1 is 1.0 mg and [[Ru(Me2phen)3]2+] = 100 μM (Fig. S7†). Under such optimised conditions, the quantum efficiency with λ = 450 nm and solar energy conversion efficiency with a solar simulator (HAL-320, Asahi Spectra Co., Ltd.) were determined to be 6.9% and 0.13%, respectively (Fig. S8†).26
1 was found to maintain its original catalytic activity for at least 5 repetitive cycles of photocatalytic production of H2O2 (Fig. S9†). There was no significant difference between the IR spectra as well as XRD patterns of 1 before the reaction and those of the precipitate obtained after centrifugation of the reaction solution, indicating the robustness of 1 under the reaction conditions (Fig. S10†). DLS data obtained after the reaction (Fig. S10c†) demonstrated no formation of significantly smaller nanoparticles such as metal oxides or hydroxides that could have been in situ formed with wide distribution of the particle size in many other cases of Co and other transition metal-based WOCs as reported previously.27–31 From the results mentioned above, we can conclude that the actual catalytically active species for water oxidation in H2O2 production is 1 as it is.32
In conclusion, cyano-bridged polynuclear complexes (FexCo1−x)3[Co(CN)6]2 act as effective water oxidation catalysts for the photocatalytic oxidation of H2O with O2 to produce H2O2 in an O2-saturated aqueous solution in the presence of [Ru(Me2phen)3]2+ and Sc(NO3)3 under visible light irradiation. The catalytic activity was maximised when the Fe-to-Co ratio in the (FexCo1−x) moiety of (FexCo1−x)3[Co(CN)6]2 was 0.75. This study provides a unique method to develop efficient catalysts for the photocatalytic water oxidation with O2 to produce H2O2 by changing the ratio of different metals contained in cyano-bridged polynuclear metal complexes.
Acknowledgements
This work was supported by ALCA and SENTAN projects from JST (to S. F. and T. S.) and JSPS KAKENHI (Grant Nos. 24350069 and 15K14223 to Y. Y.).Notes and references
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- S. Fukuzumi and Y. Yamada, Aust. J. Chem., 2014, 67, 354–364 CrossRef CAS.
- L. An, T. Zhao, X. Yan, X. Zhou and P. Tan, Sci. Bull., 2015, 60, 55–64 CrossRef CAS.
- S. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS.
- Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334–7336 RSC.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 CAS.
- S. A. M. Shaegh, N. T. Nguyen, S. M. M. Ehteshami and S. H. A. Chan, Energy Environ. Sci., 2012, 5, 8225–8228 Search PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Chem. – Eur. J., 2013, 19, 11733–11741 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272–1274 CrossRef CAS PubMed.
- F. Yang, K. Cheng, T. Wu, Y. Zhang, J. Yin, G. Wang and D. Cao, RSC Adv., 2013, 3, 5483–5490 RSC.
- F. Yang, K. Cheng, X. Xiao, J. Yin, G. Wang and D. Cao, J. Power Sources, 2014, 245, 89–94 CrossRef CAS.
- X. Xiao, F. Yang, K. Cheng, X. Wang, J. Yin and K. Ye, J. Electroanal. Chem., 2014, 729, 103–108 CrossRef CAS.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 CAS.
- B. Reuillard, S. Gentil, M. Carrière, A. L. Goff and S. Cosnier, Chem. Sci., 2015, 6, 5139–5143 RSC.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa and S. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- Y. Isaka, S. Kato, D. Hong, T. Suenobu, Y. Yamada and S. Fukuzumi, J. Mater. Chem. A, 2015, 3, 12404–12412 CAS.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y. M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606–7616 CAS.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708–5716 CAS.
- Y. Yamada, K. Oyama, R. Gates and S. Fukuzumi, Angew. Chem., Int. Ed., 2015, 54, 5613–5617 CrossRef CAS PubMed.
- H. J. Buser, D. Schwarzenbach, W. Petter and A. Ludi, Inorg. Chem., 1977, 16, 2704–2710 CrossRef CAS.
- X.-P. Shen, Y.-Z. Li, Y. Song, Z. Xu and G.-C. Guo, Eur. J. Inorg. Chem., 2007, 1698–1702 CrossRef CAS.
- J. M. Herrera, A. Bachschmidt, F. Villain, A. Bleuzen, V. Marvaud, W. Wernsdorfer and M. Verdaguer, Philos. Trans. R. Soc., A, 2008, 366, 127–138 CrossRef CAS PubMed.
- M. Verdaguer and G. S. Girolami, in Magnetism: molecules to materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2005, vol. V Search PubMed.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst (Cambridge, U. K.), 1992, 117, 1781–1784 RSC.
- The light intensity was adjusted to 10 mJ cm−2 s−1 (Air Mass 1.5 (AM1.5)) at the sample position for the whole irradiation area (1.0 × 3.0 cm2) by using 1 SUN checker (CS-20, Asahi Spectra Co., Ltd.) at room temperature.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 79–89 CrossRef CAS.
- J. J. Stracke and R. G. Finke, ACS Catal., 2014, 4, 909–933 CrossRef CAS.
- J. D. Blackmore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef PubMed.
- The major reason for the decrease in catalytic activity of the system at prolonged reaction time (Fig. 1) may result from the gradual deactivation of [RuII(Me2phen)3]2+; however, no formation of RuO2 nanoparticles was confirmed by XRD as well as DLS data recorded after the reaction (Fig. S10†).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, X-ray diffraction patterns (Fig. S1 and S10b), X-ray fluorescence data (Table S1), DLS data (Fig. S2 and S10c), IR spectra (Fig. S3 and S10a), time courses of H2O2 production under various conditions (Fig. S4 and S7–S9), time courses of O2 evolution (Fig. S5 and S6) and estimation of the amount of evolved O2. See DOI: 10.1039/c5cy01845e |
| This journal is © The Royal Society of Chemistry 2016 |