
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The selective oxidation of n-butanol to butyraldehyde by oxygen using stable Pt-based nanoparticulate catalysts: an efficient route for upgrading aqueous biobutanol†
Inaki
Gandarias
*a,
Ewa
Nowicka
b,
Blake J.
May
b,
Shaimaa
Alghareed
b,
Robert D.
Armstrong
b,
Peter J.
Miedziak
b and
Stuart H.
Taylor
*b
aDepartment of Chemical and Environmental Engineering, University of the Basque Country (UPV/EHU), Alameda Urquijo s/n 48013, Bilbao, Spain. E-mail: inaki_gandarias@ehu.eus
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: taylorsh@cardiff.ac.uk
First published on 19th January 2016
Abstract
Supported Pt nanoparticles are shown to be active and selective towards butyraldehyde in the base-free oxidation of n-butanol by O2 in an aqueous phase. The formation of butyric acid as a by-product promoted the leaching of Pt and consequently the activity of the catalysts decreased upon reuse. Characterisation showed that the degree to which Pt leached from the catalysts was related to both the metal–support interaction and metal particle size. A catalyst active and stable (<1% metal leaching) in the aqueous reaction medium was obtained when Pt nanoparticles were supported on activated carbon and prepared by a chemical vapour impregnation method. The presence of n-butanol in the aqueous medium is required to inhibit the over oxidation of butyraldehyde to butyric acid. Consequently, high selectivities towards butyraldehyde can only be obtained at intermediate n-butanol conversion.
Introduction
Future sustainable societies will require the production of fuels and commodity chemicals from biomass through technically and economically feasible processes. For instance, butyraldehyde is a key intermediate in the chemical industry, as it is used in the production of 2-ethylhexanol, 2-ethylhexanoic acid, polyvinyl butyral and trimethylolpropane.1 Currently, the commercial method to obtain butyraldehyde is the hydroformylation of propylene, a petroleum derivative.2 A significantly greener approach would be the production of butyraldehyde through the selective oxidation of biobutanol,3 which can be obtained by the acetone–butanol–ethanol (ABE) fermentation of biomass. Hence, biobutanol could become a key building block in future biorefineries if its production cost is reduced by improving the industrial microbial strains, the fermentation technologies, and the separation processes.4Relatively high butyraldehyde yields have been reported from the gas phase selective oxidation of n-butanol.5 However, after the ABE fermentation process biobutanol is obtained in a highly dilute aqueous solution,6 therefore, from an economic and environmental point of view aqueous phase oxidation processes using molecular oxygen as the oxidant, and a highly active, easily separable and reusable heterogeneous catalyst would appear to be the best option.7
The catalytic oxidation of an alcohol is reported to be a two-step process. In the first step the substrate adsorbs onto the catalytic surface forming a hydrogen adatom and an alkoxide. The second step is the abstraction of the β-hydrogen by the metal site.8 As the metal sites and the sites of the support are involved in the catalytic cycle, smaller metal nanoparticles enlarge the interfacial periphery regions between the nanoparticles and the support and, thus, they increase the oxidation activity of the catalysts. Recently, we studied the aqueous phase base-free oxidation of n-butanol to butyric acid using Au–Pd nanoparticles supported on TiO2.9 The catalysts prepared by the sol-immobilisation method displayed the narrowest particle size distribution (average size 2.2 nm) and the best performance in n-butanol oxidation (92% butyric acid selectivity at 90% n-butanol conversion). Remarkably these catalysts were also stable under the operating conditions used: no metal leaching was observed and the catalysts maintained their activity during three consecutive reaction cycles. Very high activities have also been reported for Au–Pd/TiO2 catalysts prepared by sol-immobilisation in the oxidation of other alcohols.10
The selective production of butyraldehyde in the aqueous phase oxidation of n-butanol presents some challenges. It has been reported that non-aqueous solvents are required for the synthesis of aldehydes from the selective oxidation of the corresponding alcohols.11 In fact, butyraldehyde oxidises to butyric acid in the aqueous phase at 100 °C even in the absence of a catalyst.9 This has been related to the aldehyde reacting with water to generate a geminal diol which rapidly reacts to form the acid12 (see Scheme 1). Hence, to obtain high butyraldehyde yields, a catalyst that is active in the oxidation of n-butanol to butyraldehyde but does not catalyse its transformation into butyric acid has to be developed. Furthermore this catalyst has to be stable under the operating conditions used.
 | ||
| Scheme 1 Selective oxidation of n-butanol to butyraldehyde and butyric acid. | ||
Experimental
Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80 wt% hydrolysed) was freshly prepared and used as the stabiliser. NaBH4 (Sigma Aldrich, 0.1 M aqueous solution) was also freshly prepared and used as the reducing agent. To a mixed aqueous solution of metals, the required amount of a PVA solution was added (PVA/metals (wt/wt) = 1.2) with vigorous stirring for 15 minutes. NaBH4 was then added rapidly, such that the NaBH4:total metal ratio (mol/mol) was 5. After 30 minutes of sol generation, the colloid was immobilised by adding the support (TiO2: Degussa P25, C: Cabot Vulcan XC-72R and CeO2: Sigma Aldrich) and one drop of concentrated H2SO4. The amount of support material and metal precursor was calculated so as to have a total metal loading of 1 wt%. The metal molar ratio for the supported bimetallic or trimetallic catalysts was 1:1 and 1:1:1 (unless otherwise stated). After 2 h, the slurry was filtered, the catalyst washed thoroughly with distilled water and dried at 120 °C for 16 h. These catalysts were designated as metal(s)/supportSOL_PVA. Some catalysts were reduced under a flow of 5 vol% H2/Ar at a temperature of 400 °C. These catalysts were designated as metal(s)/supportSOL_PVA_Red400.
000, 80 wt% hydrolysed) was freshly prepared and used as the stabiliser. NaBH4 (Sigma Aldrich, 0.1 M aqueous solution) was also freshly prepared and used as the reducing agent. To a mixed aqueous solution of metals, the required amount of a PVA solution was added (PVA/metals (wt/wt) = 1.2) with vigorous stirring for 15 minutes. NaBH4 was then added rapidly, such that the NaBH4:total metal ratio (mol/mol) was 5. After 30 minutes of sol generation, the colloid was immobilised by adding the support (TiO2: Degussa P25, C: Cabot Vulcan XC-72R and CeO2: Sigma Aldrich) and one drop of concentrated H2SO4. The amount of support material and metal precursor was calculated so as to have a total metal loading of 1 wt%. The metal molar ratio for the supported bimetallic or trimetallic catalysts was 1:1 and 1:1:1 (unless otherwise stated). After 2 h, the slurry was filtered, the catalyst washed thoroughly with distilled water and dried at 120 °C for 16 h. These catalysts were designated as metal(s)/supportSOL_PVA. Some catalysts were reduced under a flow of 5 vol% H2/Ar at a temperature of 400 °C. These catalysts were designated as metal(s)/supportSOL_PVA_Red400.
One catalyst was prepared using the same procedure but with polyvinylpyrrolidone (PVP, 1 wt% aqueous solution, Sigma Aldrich, MW = 10000) instead of PVA as the stabilising agent.
Catalyst testing
Catalytic reactions were carried out using a 50 mL Colaver glass reactor. The reactor was charged with 10 mL of an aqueous solution of n-butanol (0.54 mol L−1 or 9.1 mol L−1) and 72 mg of catalyst. The glass reactor was sealed, purged three times with oxygen and then pressurised with oxygen to 3 bar. This pressure was maintained throughout the experiment; hence as the oxygen was consumed in the reaction it was continuously replenished. The reaction mixture was heated to the desired temperature (100 °C) and stirred with a magnetic stirrer at 750 rpm for 6 hours. The reactor vessel was then cooled in an iced bath for 10 minutes. Following this, the reactor was slowly opened and 50 mL of an ethanol solution containing a known amount of 2-butanol (external standard) was added. The catalyst was separated by filtration and centrifugation and the liquid was analysed by GC-FID (Varian Star 3400 cx with a 30 m CP-Wax 52 CB column). The C-balance obtained in all the experiments was in the range 95–103%. Besides butyraldehyde, butyric acid and butyl-butyrate no other product formation was detected. When the reaction was carried out only with the support, C and TiO2, no n-butanol conversion was observed.Catalyst characterisation
Metal leaching analyses were also performed using an Agilent 7900 ICP-MS instrument. Reaction mixtures were diluted with H2O to give a maximum 5 vol% of organic media. The percentage of total metal leached was quantified through comparing the metal content of fresh catalysts with the metal content of reaction solutions.
Results and discussion
Activity test results
A series of Au, Pd, and Pt bimetallic (metal molar ratio 1:1) and trimetallic (metal molar ratio 1:1:1) catalysts supported on TiO2 with a total metal nominal content of 1 wt% were prepared by the sol-immobilisation (SOL) method using polyvinyl alcohol (PVA) as the stabilizing agent. As previously mentioned, this method allows for the synthesis of very active catalysts with a small and narrow nanoparticle size distribution.13 These catalysts were tested in the aqueous phase oxidation of n-butanol and results are shown in Fig. 1. A blank reaction without adding any catalyst showed no conversion of n-butanol.
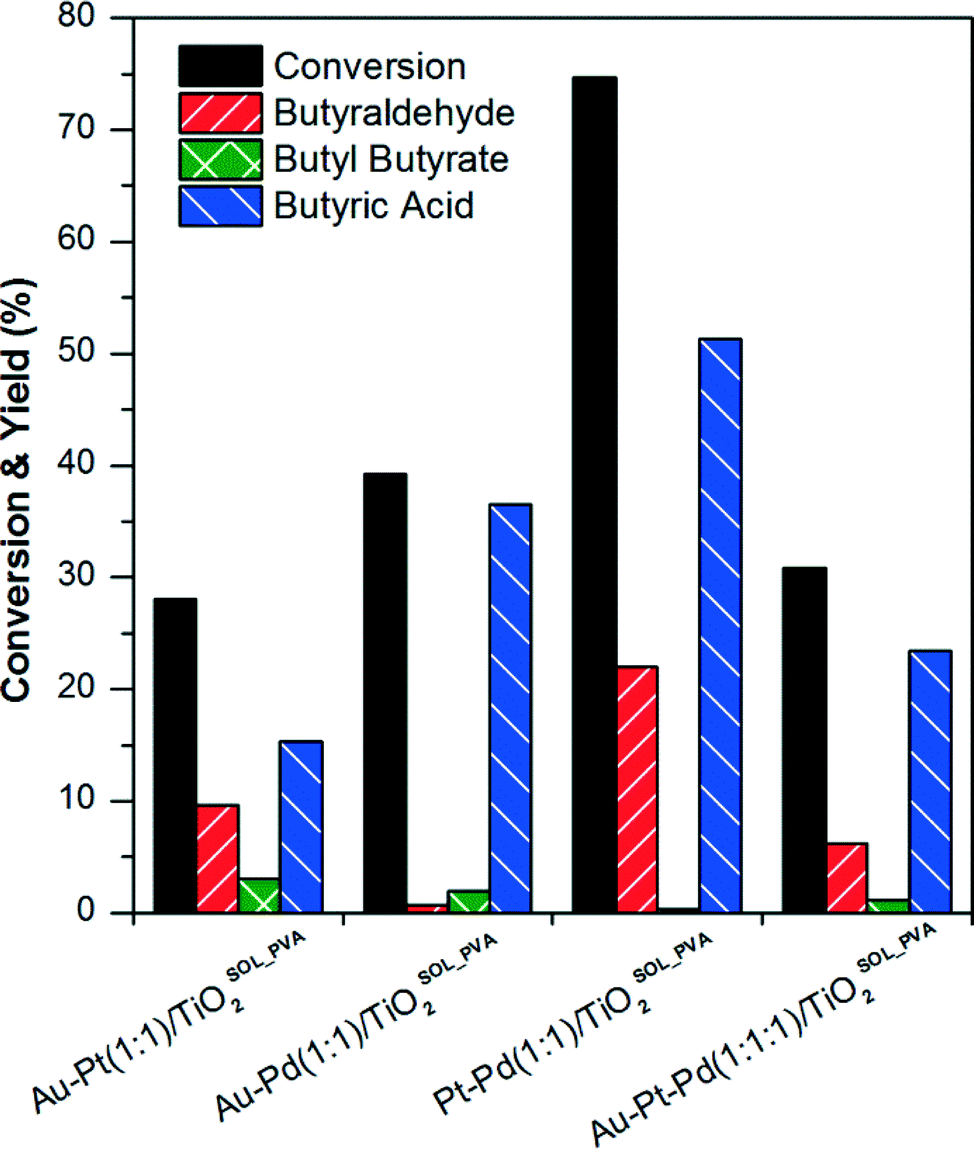 | ||
| Fig. 1 n-Butanol conversion and yield of butyraldehyde, butyl butyrate and butyric acid obtained with different bimetallic and trimetallic catalysts prepared by sol-immobilisation. Reaction conditions: 100 °C, 3 bar O2, 6 h, 10 mL of n-butanol solution (0.54 mol L−1), 0.072 g of catalyst. | ||
Each noble metal promoted the formation of different products. Butyraldehyde yields increased with the catalyst containing Pt, while the presence of Pd led to the formation of butyric acid. In the reaction with catalysts containing Au, higher selectivity to butyl butyrate was observed. The highest n-butanol conversion and yield of butyraldehyde was obtained with the bimetallic Pt–Pd(1:1)/TiO2SOL_PVA catalyst. Compared to the most active catalyst, bimetallic Pt–Pd, the n-butanol conversion significantly decreased when the three metals (Au–Pt–Pd) were present on the catalyst.
Based on these results a series of Pt–Pd/TiO2SOL_PVA catalysts was prepared in order to determine the effect that the Pt:Pd ratio has on both n-butanol conversion and yield of butyraldehyde. As can be seen in Fig. 2A, the maximum n-butanol conversion was obtained with the Pd–Pt(1:1)/TiO2SOL_PVA catalyst. This indicates a synergistic effect between Pt and Pd, which promotes the oxidation of n-butanol. However, butyraldehyde yield increased with increasing Pt:Pd ratio and the maximum was achieved for the monometallic Pt/TiO2SOL_PVA catalyst. Similar behaviour was reported for the selective oxidation of benzyl alcohol using a combination of Au, Pt and Pd metals supported on TiO2.14 When Pt based catalysts were used, selectivity towards benzaldehyde was significantly higher when compared to the results obtained with Au–Pd based catalysts. These results suggest that Pt is not a good catalyst to oxidise aldehydes to acids. In bimetallic catalysts, Pt seems to act as either an electronic or strain modifier to Au and Pd and decrease activity in the oxidation of aldehydes.
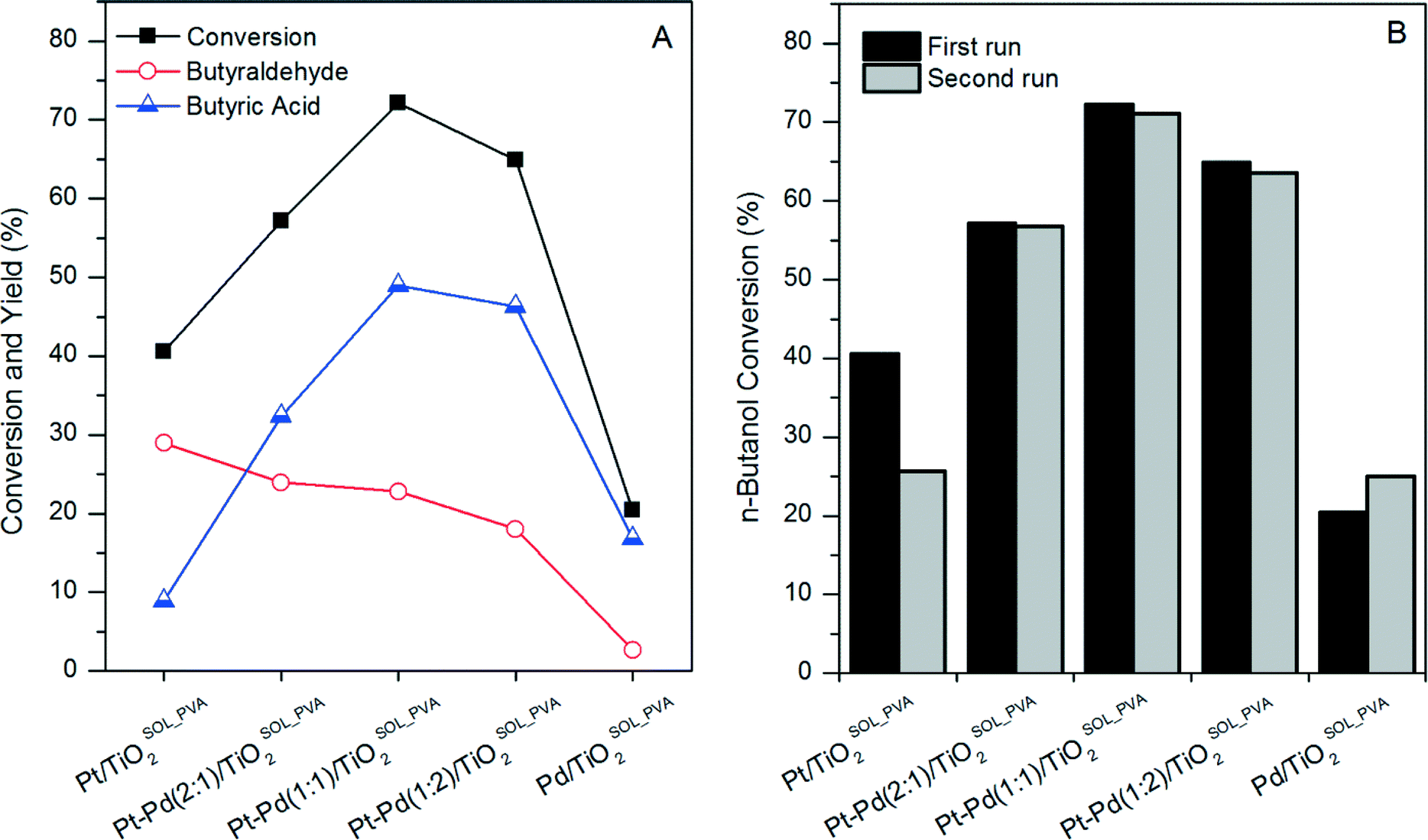 | ||
| Fig. 2 (A) n-Butanol conversion and yield of butyraldehyde and butyric acid for a series of Pt–Pd/TiO2Sol_PVA catalysts. (B) n-Butanol conversion of fresh and used Pt–Pd/TiO2Sol_PVA catalysts. 100 °C, 3 bar O2, 6 h, 10 mL of n-butanol aqueous solution (0.54 mol L−1), 0.072 g of catalyst. | ||
The stability of the catalysts was investigated by performing a second reaction with each used catalyst of the Pt–Pd/TiO2SOL_PVA series. In the case of the Pd/TiO2SOL_PVA catalyst, an increase in the n-butanol conversion was observed between the first and the second run. We have previously reported a similar effect for Au–Pd/TiO2SOL_PVA catalysts in the aqueous phase selective oxidation of n-butanol to butyric acid.9 It was observed that residual PVA was removed from the surface of the catalysts during the first activity test in aqueous solution at 100 °C. Residual PVA on the surface of SOL catalysts is known to have a negative effect on catalytic activity, through limiting the accessibility of reactants to the active sites.15 Therefore, the removal of PVA was responsible for the increase in n-butanol conversion in the second use. An increment in n-butanol conversion was also expected upon reuse of the other Pt–Pd/TiO2SOL_PVA catalysts. However, the activity decreased for all the bimetallic catalysts and for the monometallic Pt/TiO2SOL_PVA. In order to explain the decrease in activity, the metal content of fresh and used catalysts was measured by MP-AES. As can be observed in Table S1,† the Pt content of used samples was significantly lower than that of fresh samples, while the Pd content remained virtually unchanged. Consequently, Pt leaching was determined to be the reason for the decrease of activity for Pt containing catalysts.
Carboxylic acids are known to be good chelating agents and to accelerate the leaching of supported platinum,16 therefore the extent of leaching in aqueous butyric acid solutions of varying concentration was investigated. Three samples of Pt/TiO2SOL_PVA catalysts were stirred for 16 hours in 10 mL of water, 10 mL of 0.07 M aqueous butyric acid solution, and in 10 mL of 0.5 M aqueous butyric acid solution. No significant Pt leaching was observed for the samples that were stirred in water (Table S2†), however around 40% leaching was detected for the two samples which were stirred in aqueous butyric acid solution. It is noteworthy that the degree of leaching was independent of the organic acid concentration, and even small concentrations of butyric acid in the reaction media can greatly affect the stability of the Pt/TiO2SOL_PVA catalyst.
One of the major challenges in liquid-phase oxidation with solid catalysts is to prevent leaching of the active species.17 Different strategies were applied in order to increase the stability of Pt supported nanoparticles for the reaction in an aqueous medium. Results from these investigations are summarised in Table 1. The percentage of metal leached was determined by measuring the amount of Pt in the post-reaction solution using MP-AES or ICP-MS techniques and comparing this with the metal content of fresh catalysts. No significant differences were noted between MP-AES and ICP-MS analyses. The first approach to increase catalyst stability was to apply a heat treatment at 400 °C and reduce the catalyst under a flow of hydrogen (Table 1, entry 1). Alternatively, polyvinylpyrrolidone (PVP) was used instead of PVA as the stabilising agent in the sol-immobilisation catalyst preparation method (Table 1, entry 2). Unfortunately, Pt leached in both cases.
| Entry | Catalyst | Conversion n-butanol (%) | Selectivity (%) | Pt leaching (%) | Pt particle sizea (nm) | ||
|---|---|---|---|---|---|---|---|
| Butyraldehyde | Butyric acid | Butyl-butyrate | |||||
| a Average Pt particle size measured from representative TEM images of fresh catalysts. b Pt content in the reaction medium measured by MP-AES. c Pt content in the reaction medium measured by ICP-MS. | |||||||
| 1 | Pt/TiO2SOL_PVA_Red400 | 32.1 | 73.1 | 26.9 | 0.0 | 51.4b | — |
| 2 | Pt/TiO2SOL_PVP | 31.4 | 78.8 | 21.2 | 0.0 | 31.4b | — |
| 3 | Pt/TiO2CVI_Red400 | 54.2 | 53.7 | 45.4 | 0.7 | 60.0b | — |
| 4 | Pt/ASACVI_Red400 | 4.3 | 100 | 0.0 | 0.0 | 3.1b | — |
| 5 | Pt/Al2O3CVI_Red400 | 16.0 | 77.0 | 23.2 | 0.0 | 58.5c | — |
| 6 | Pt/CSOL_PVA | 85.9 | 21.2 | 78.9 | 0.0 | 25.6c | 1.758 |
| 7 | Pt/CSOL_PVA_Red400 | 91.8 | 23.2 | 67.9 | 8.8 | 4.8c | 2.215 |
| 8 | Pt/CCVI_Red400 | 77.6 | 37.9 | 61.7 | 0.4 | 0.3c | 2.754 |
| 9 | Pt/CeO2SOL_PVA_Red400 | 30.5 | 21.1 | 78.2 | 0.0 | 1.51c | 1.251 |
| 10 | Pt/CeO2CVI_Red400 | 54.4 | 52.4 | 47.6 | 0.0 | 0.95c | 2.429 |
It is well known that both the catalyst preparation method and metal precursors used can affect the interaction between nanoparticles and the support, and hence affect catalyst stability.18 In our previous work on n-butanol oxidation, the Au–Pd/TiO2 catalysts prepared by wet impregnation, physical mixing, or deposition–precipitation showed significantly lower activity than the catalysts prepared by sol-immobilisation.9 This was ascribed to the larger Au–Pd nanoparticles obtained with these methods. Therefore, a suitable catalyst preparation method should yield small nanoparticles that are stable under reaction conditions.
Previous work on Pt/TiO2 catalysts prepared by chemical vapour impregnation (CVI) reported an average nanoparticle size of <3 nm. These catalysts were very active for the selective oxidation of toluene.19 Following this preparation method a Pt/TiO2CVI_Red400 catalyst was prepared and tested for the aqueous phase oxidation of n-butanol. As can be observed in Table 1, entry 3, this catalyst was relatively active and selective towards butyraldehyde, however, it was not stable. Based on the inefficiency of the different strategies with the TiO2 support, it was decided to synthesise catalysts with different supports: amorphous silica-alumina (ASA), γ-alumina, activated carbon and ceria. Application of Pt/ASACVI_Red400 and Pt/Al2O3CVI_Red400 in the oxidation reaction resulted in the lowest n-butanol conversion, while the highest activity was obtained with the catalysts supported on activated carbon. In terms of stability, less than 1% leaching was observed for the Pt/CCVI_Red400 and Pt/CeO2CVI_Red400 catalysts. Remarkably, it was possible to synthesise active and stable Pt nanoparticles using the CVI method and C or CeO2 as supports.
Catalyst characterisation
To better understand their catalytic performance, Pt/C and Pt/CeO2 catalysts prepared by both sol-immobilisation and CVI were characterised by XPS and TEM. XPS analysis (Table 2) of Pt/C samples reveal exclusively metallic Pt, with a Pt(4f7/2) binding energy of 70.9 eV (±0.2 eV), regardless of the preparation technique. In contrast to the Pt/C samples, analysis of CeO2 supported Pt catalysts prepared by sol-immobilisation and CVI methods exhibited Pt0 and Pt2+ states at 71.0 and 72.5 eV respectively. Exact determination of the Pt0/Pt2+ is difficult as it has been shown that Pt clusters, depending on their size, can not only have an effect on the binding energy, but also a significant effect on the full width at half maximum (FWHM) of the peak, which in turn will affect the quantification. Despite the samples being reduced, it is believed the presence of Pt2+ may be a result of incorporation of Pt at Ce+4 sites within the CeO2 lattice as shown by Bera et al.20 and may account for the presence of Ce3+ detected in the ceria support. These results suggest a relatively strong Pt–CeO2 interaction, which might be responsible for the relatively low Pt leaching observed with CeO2 supported catalysts (Table 1, entries 9 and 10). In the case of C-based catalysts, the lower Pt leaching observed as compared to TiO2 based catalysts can be ascribed to the well known high resistance of carbon supports to acidic and chelating media.21| Catalyst | Pt binding energy (eV) | Concentration (at%) | |||
|---|---|---|---|---|---|
| Pt | C | Ce | O | ||
| Pt/CSOL_PVA | 70.7 (100%) | 0.04 | 98.24 | — | 1.72 |
| Pt/CSOL_PVA_Red400 | 70.9 (100%) | 0.04 | 98.14 | — | 1.82 |
| Pt/CCVI_Red400 | 70.9 (100%) | 0.01 | 96.23 | — | 3.76 |
| Pt/CeO2SOL_PVA_Red400 | 70.8 (82%)/72.7 (18%) | 0.19 | 65.82 | 9.38 | 24.61 |
| Pt/CeO2CVI_Red400 | 71.4 (75%)/72.6 (25%) | 0.15 | 42.58 | 17.94 | 39.34 |
Representative TEM images and associated particle size distributions (PSDs) of the Pt/CSOL_PVA and Pt/CSOL_PVA_Red400 are shown in Fig. 3. Reduction of the catalysts at 400 °C led to a slight increase in the average particle size from 1.8 to 2.2 nm, which is likely to be due to a slight sintering effect at the elevated temperature. As shown in Table 1 (entries 6 and 7) there was significantly lower Pt leaching in the reduced sample. This could be ascribed to agglomeration of the smaller, more labile metal particles into larger particles which would be less susceptible to leaching. The activity of the catalyst before and after the reduction step is very similar, suggesting that the smaller particles are not involved in the reaction.
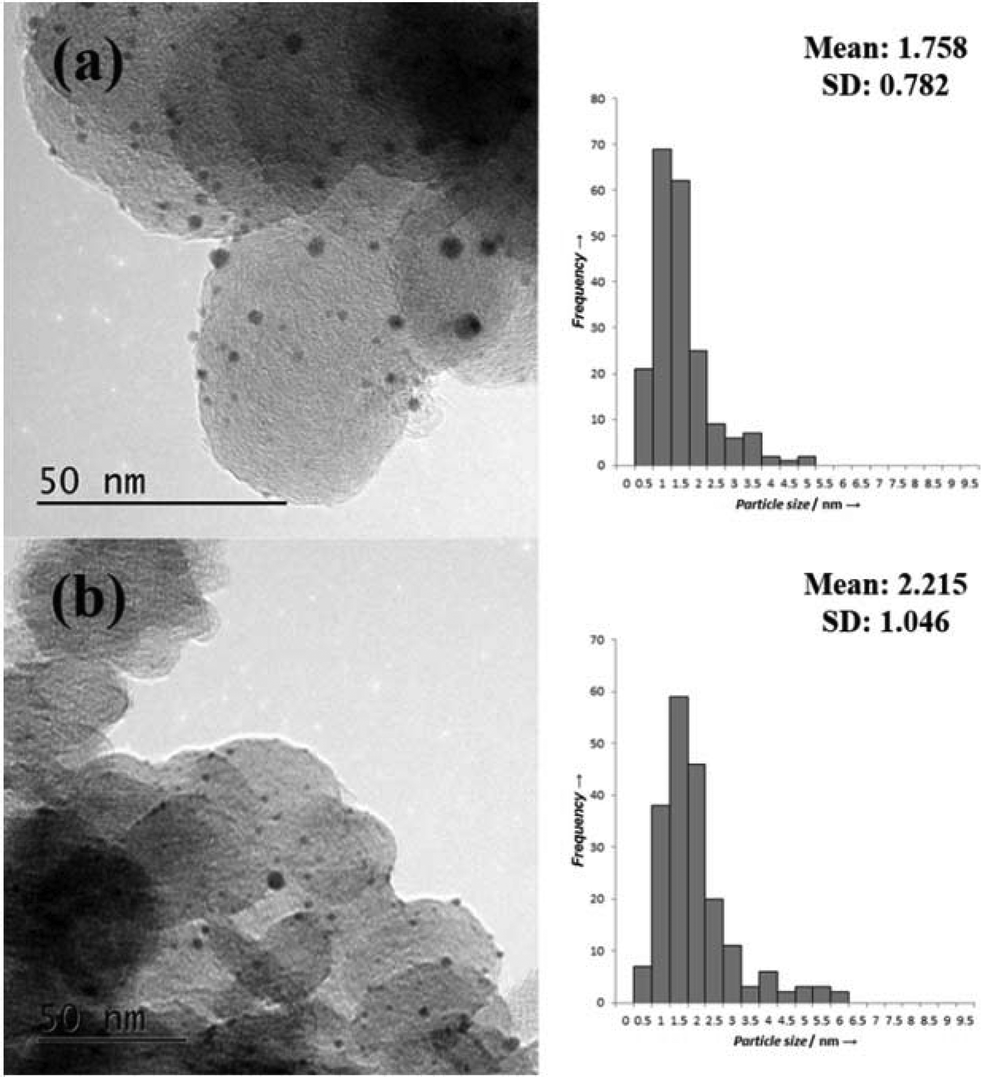 | ||
| Fig. 3 TEM images and associated PSDs of Pt/CSOL-PVA, Pt/CSOL-PVA_Red400. | ||
TEM characterisation of the Pt/CCVI_Red400 catalyst identified the presence of large particles (>30 nm), in both fresh and used samples. Although not shown in the particle size distribution of Fig. 4a and b respectively, these were included in calculations. The presence of these clusters in the catalyst, shown in Fig. S1a† along with an example of large metal nanoparticles (S1b†) indicates that the CVI process was not fully successful at distributing the metal salt. The mean particle size, 2.7 nm, is larger for the catalysts prepared by CVI compared to those prepared by sol-immobilisation. The presence of the large particles led to much broader standard distributions than were observed for sol immobilisation catalysts.
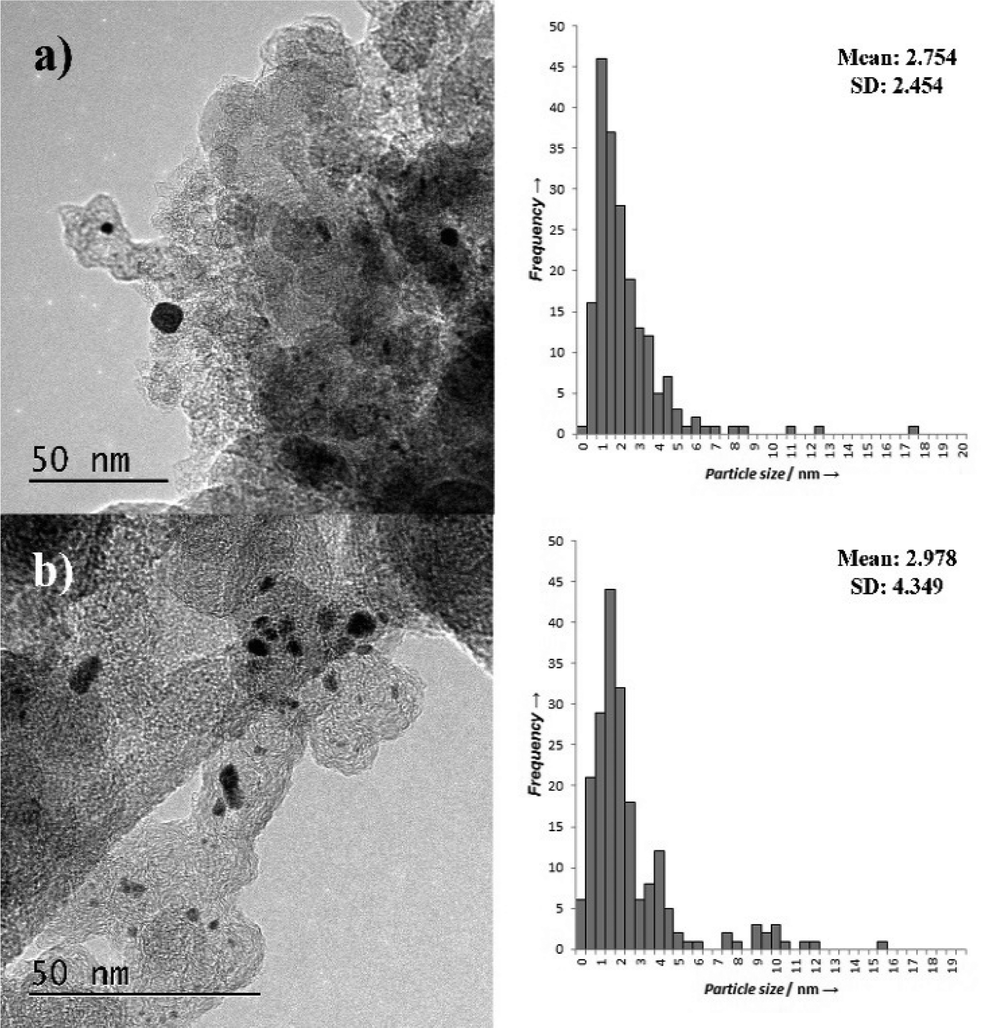 | ||
| Fig. 4 TEM images and associated PSDs of Pt/CCVI_Red400 a) fresh and b) used. | ||
The used Pt/CCVI_RED400 catalyst showed a slight increase in average particle size compared to the fresh catalyst (Fig. 4b), although this may be skewed by the larger particles that were present in the analysed region. The bi-modal nature of the particle size distribution makes it difficult to objectively analyse the average particle size. It is interesting to note that this is the catalyst which displayed the lowest degree of Pt leaching and that the PSD in the range of 1–7 nm is comparable for both the fresh and used catalysts.
Fig. S2† shows TEM micrographs for the Pt/CeO2SOL_PVA_Red400 and the Pt/CeO2CVI_Red400 catalysts. Both catalysts exhibit relatively low Pt leaching compared to the carbon supported catalysts. This suggests the degree of leaching is also related to the Pt-support interaction rather than solely the particle size, as observed in the XPS results. The average particle size of the Pt/CeO2SOL_PVA_Red400 catalyst (1.2 nm) is the smallest of all the catalysts analysed. Following the same pattern as the carbon supported catalyst the CVI method gave a slightly larger average particle size than the sol immobilisation method (2.4 nm). In this case however, no larger particles were observed by TEM. Comparing the activity of both CeO2 supported catalysts, higher n-butanol conversion and selectivity towards butyraldehyde was obtained with the catalyst showing the higher average particle size (see Table 1, entries 9–10).
In summary TEM data suggests that the degree to which Pt is leached from catalysts is related to both the metal–support interaction and metal particle size. The metal–support interaction is affected by the type of support and by the catalyst preparation method. For the catalysts with the same support, the smallest metal particles appear more susceptible to leaching and the activity of the catalysts suggests that it is not necessarily the smallest particles which are the most active for this oxidation reaction.
A process for the production of butyraldehyde and butyric acid
The Pt/CCVI_Red400 catalyst was selected for further analysis as it showed the highest Pt stability under the operating conditions, in addition to relatively high n-butanol conversion and selectivity towards butyraldehyde. Fig. 5 shows the temporal evolution of products. The selectivity towards butyraldehyde decreased and the selectivity towards butyric acid increased with increasing n-butanol conversion. As was mentioned in the introduction, butyraldehyde will undergo uncatalysed oxidation to butyric acid at 100 °C in water. However, relatively high butyraldehyde selectivities can be obtained at n-butanol conversions of lower than 40%, which suggests that n-butanol inhibits the conversion of butyraldehyde to butyric acid. In order to prove this hypothesis, three more experiments under standard reaction conditions (100 °C, 3 bar O2, t = 6 h) were carried out. With (i) an aqueous solution of 4 wt% of butyraldehyde and no catalyst, a 94% conversion of the aldehyde into butyric acid was observed. A similar conversion of butyraldehyde (97%) was measured when (ii) the same reaction was conducted but adding Pt/C as catalyst. However, with (iii) an aqueous solution of 4 wt% of n-butanol and 1 wt% of butyraldehyde and no catalyst, no conversion of n-butanol was observed and only 5% of the aldehyde was converted into butyric acid. These results demonstrate that in an aqueous phase the oxidation of butyraldehyde to butyric acid is mainly uncatalysed and that n-butanol is acting as the inhibitor for this uncatalysed oxidation. The same effect was studied through electron paramagnetic resonance (EPR) spin trapping experiments for the benzyl alcohol/benzaldehyde and octanol/octanal systems.22 It was determined that the alcohols inhibit the formation of the acids by intercepting the peroxy radicals, which otherwise play a key role in the aldehyde uncatalysed oxidation. Based on this mechanism, at high conversion less n-butanol is available to act as an inhibitor and therefore uncatalysed oxidation of butyraldehyde would become more favourable.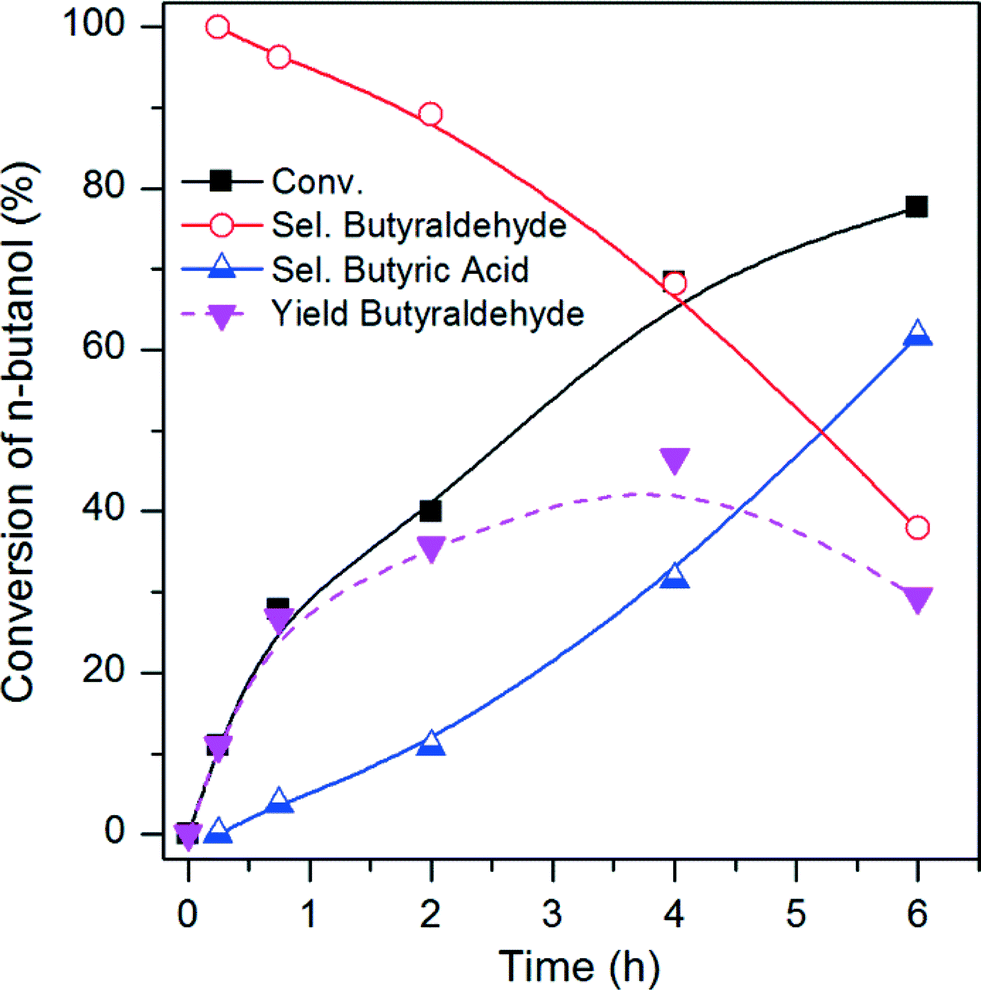 | ||
| Fig. 5 Evolution of n-butanol conversion, selectivity towards main products and yield of butyraldehyde with reaction time. 100 °C, 3 bar O2, 6 h, 10 mL of n-butanol or butyraldehyde aqueous solution (0.54 mol L−1), 0.072 g of catalyst (Pt/CCVI_Red400), substrate:Pt molar ratio 1400:1. | ||
Therefore, at each operating condition there will be an optimum n-butanol conversion at which the maximum butyraldehyde yields would be obtained. Under the operating conditions shown in Fig. 5 the maximum butyraldehyde yield would correspond to a value of ca. 47%, at ca. 68% n-butanol conversion. At higher conversion, the rate of butyraldehyde oxidation exceeds the rate of its formation from n-butanol and therefore selectivity decreases.
After fermentation, biobutanol is normally purified via a two stage process. In the first step the water content is reduced through membrane pervaporation, solvent extraction, or adsorption.23 Following these processes, the concentration of n-butanol is normally lower than 80 wt%, and therefore, phase separation occurs with an n-butanol rich phase containing 79.9 wt% of n-butanol and a predominately aqueous phase containing 7.7 wt% n-butanol at 20 °C. In the second step purified n-butanol is obtained through distillation. For butyraldehyde and butyric acid production it would be more economic to use the n-butanol rich stream, rather than the purified n-butanol one, since water is produced during n-butanol oxidation. Regardless of the initial water concentration in the n-butanol stream, another separation step is required to purify the butyric acid formed. Therefore, an experiment was carried out with an initial concentration of 80 wt% n-butanol in water (9.1 mol L−1), 24 h and a metal to substrate ratio of 1:11863. Selectivity values obtained were 40% butyraldehyde, 51% butyric acid and 11% butyl butyrate at 18% n-butanol conversion. Even though the yield of butyraldehyde from butanol is yet to be optimised, this result indicates that it is possible to obtain butyraldehyde from concentrated n-butanol solutions. Moreover, the leaching of Pt was investigated and found to be lower than 1%, demonstrating high stability of Pt/CCVI_Red400 catalysts even at high initial concentrations of n-butanol.
Fig. 6 shows a simplified process flowsheet for the production of butyraldehyde and butyric acid from biobutanol. In the first reactor, a biobutanol rich aqueous stream (80 wt% butanol and 20 wt% water) would be selectively oxidised using a Pt/CCVI_RED400 catalyst. Besides butyraldehyde and unreacted n-butanol, which is required to inhibit butyraldehyde uncatalysed oxidation, butyric acid and butyl butyrate would also be part of the product stream leaving the first reactor. Butyraldehyde recovery from this stream through distillation is challenging due to non-idealities and the formation of azeotropes. Therefore the first step would involve a water removal process like water pervaporation, solvent extraction or adsorption. Following this the most volatile compound, butyraldehyde, would be separated through distillation. The heavier compounds, biobutanol, butyric acid and butyl butyrate, would be introduced into a second reactor loaded with a Au–Pd/TiO2 catalyst to produce butyric acid. Small amounts of water (around 20 wt%) would also be fed into this reactor to promote the hydrolysis of butyl butyrate. At 100 °C and with a highly active and stable Au–Pd/TiO2SOL_PVA catalyst this reaction shows high selectivity9 and the stream leaving the reactor would mainly consist of water and butyric acid. After a final distillation, purified butyric acid would be obtained.
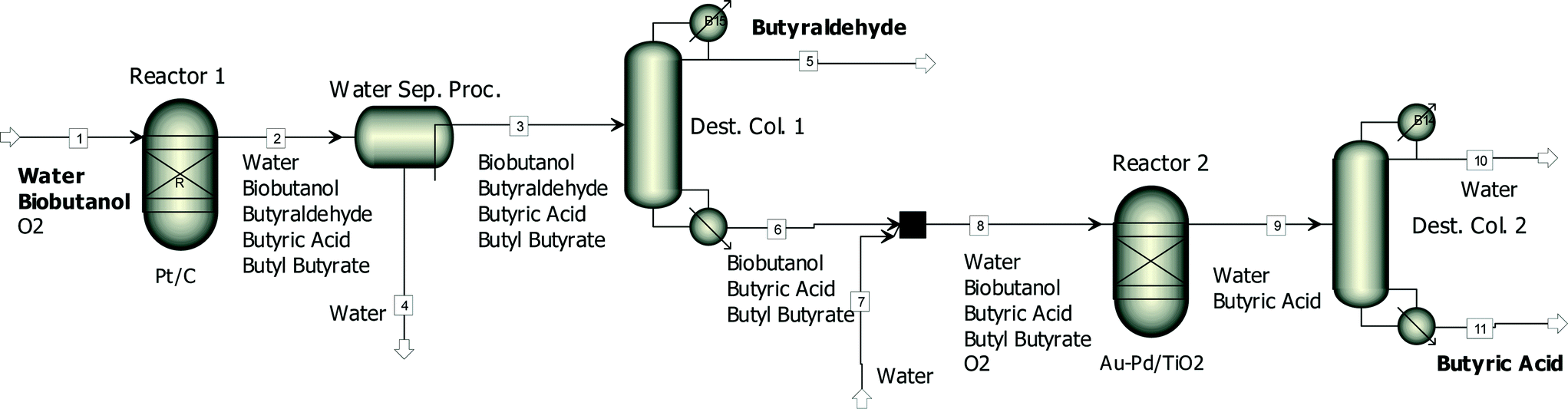 | ||
| Fig. 6 A simplified diagram of a possible process for the production of butyraldehyde and butyric acid from concentrated biobutanol. | ||
Although the economic viability of the process and effect of impurities within fermentation-derived biobutanol feeds should be assessed, this work shows that butyraldehyde and butyric acid can be obtained from an n-butanol rich aqueous solution using active and stable Pt/CCVI_Red400 and Au-Pd/TiO2SOL_PVA catalysts.
Conclusions
The selective production of butyraldehyde from aqueous n-butanol is challenging as butyraldehyde oxidises to butyric acid in the presence of water at 100 °C reaction temperature. Nonetheless, for the green production of butyraldehyde, water is the preferred solvent as biobutanol is obtained in a highly dilute aqueous stream after the ABE fermentation process. Supported Pt/TiO2 catalysts prepared by the sol-immobilisation method showed high activity and selectivity towards butyraldehyde, however Pt was observed to leach, even in the presence of small concentrations of butyric acid.In searching for stable metal nanoparticles, Pt based catalysts were synthesised by different methods and using different supports. Catalyst characterisation by XPS and TEM suggests that the observed Pt leaching is related to both the metal–support interaction and the size of the metal nanoparticles. Using carbon (Cabot Vulcan XC-72R) as the support and CVI as the preparation method an active and selective catalyst (Pt/CCVI_Red400) was synthesised. By changing the catalyst support and preparation method we also reduced Pt leaching from 58% to <1% under the operating conditions used (100 °C, 3 bar of O2). The study of product evolution with reaction time revealed that butyraldehyde selectivity decreased with increasing n-butanol conversion. This was ascribed to n-butanol acting as a radical inhibitor in the uncatalysed oxidation of butyraldehyde to butyric acid. Hence, in an aqueous phase reaction high selectivity of butyraldehyde can only be obtained at medium/low n-butanol conversions. The Pt/CCVI_Red400 catalyst was also active, selective and stable when concentrated aqueous solutions of n-butanol (80 wt%) were utilised.
Acknowledgements
Financial support to I. Gandarias by the Department of Education of the Basque Government (“Programa Postdoctoral de Perfeccionamiento de Doctores”) is gratefully acknowledged. Dr. David J. Morgan is gratefully acknowledged for his work on XPS measurements.Notes and references
- M. Mascal, Biofuels, Bioprod. Biorefin., 2012, 6, 483 CrossRef CAS.
- C. W. Kohlpaintnera, R. W. Fischera and B. Cornils, Appl. Catal., A, 2001, 221, 219 CrossRef.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- S. B. Bankar, S. A. Survase, H. Ojamo and T. Granstrom, RSC Adv., 2013, 3, 24734 RSC.
- (a) J. Requies, M. B. Güemez, P. Maireles, A. Iriondo, V. L. Barrio, J. F. Cambra and P. L. Arias, Appl. Catal., A, 2012, 423–424, 185 CrossRef CAS; (b) J. Requies, M. B. Güemez, A. Iriondo, V. L. Barrio, J. F. Cambra and P. L. Arias, Catal. Lett., 2012, 142, 50 CrossRef CAS.
- M. Vane, Biofuels, Bioprod. Biorefin., 2008, 2, 553 CrossRef.
- T. Lu, Z. Du, J. Liu, H. Ma and J. Xu, Green Chem., 2013, 15, 2215 RSC.
- J. Feng, C. Ma, P. J. Miedziak, J. K. Edwards, G. L. Brett, D. Li, Y. Du, D. J. Morgan and G. J. Hutchings, Dalton Trans., 2013, 42, 14498 RSC.
- I. Gandarias, P. J. Miedziak, E. Nowicka, M. Douthwaite, D. J. Morgan, G. J. Hutchings and S. H. Taylor, ChemSusChem, 2015, 8, 473 CrossRef CAS PubMed.
- (a) A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036 RSC; (b) G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem. Int. Ed., 2011, 50, 10136 CrossRef CAS PubMed; (c) V. Peneau, Q. He, G. Shaw, S. A. Kondrat, T. E. Davies, P. Miedziak, M. Forde, N. Dimitratos, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2013, 15, 10636 RSC.
- (a) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (b) J. M. H. Dirkx and H. S. J. van der Baan, J. Catal., 1981, 67, 1 CrossRef CAS.
- R. Anderson, K. Griffin, P. Johnston and P. L. Alsters, Adv. Synth. Catal., 2003, 345, 517 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551 CrossRef CAS PubMed.
- Q. He, P. J. Miedziak, L. Kesavan, N. Dimitratos, M. Sankar, J. A. Lopez-Sanchez, M. M. Forde, J. K. Edwards, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2013, 162, 365–378 RSC.
- (a) S. Kunz and E. Iglesia, J. Phys. Chem. C, 2014, 118, 7468 CAS; (b) Y. Zhao, L. Jia, J. A. Medrano, J. R. H. Ross and L. Lefferts, ACS Catal., 2013, 3, 2341 CrossRef CAS.
- I. Bakos, T. Mallat and A. Baiker, Catal. Lett., 1997, 43, 201 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- (a) J. R. H. Ross, Heterogeneous Catalysis, Elsevier, Amsterdam, 2012 Search PubMed; (b) M. L. Toebes, J. A. van Dillen and K. P. de Jong, J. Mol. Catal. A: Chem., 2001, 173, 75 CrossRef CAS.
- M. M. Forde, L. Kesavan, M. I. Bin-Saiman, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, S. H. Taylor, C. J. Kiely and G. J. Hutchings, ACS nano, 2014, 8, 957 CrossRef CAS PubMed.
- (a) P. Bera, K. C. Patil, V. Jayaram, G. N. Subbanna and M. S. Hegde, J. Catal., 2000, 196, 293 CrossRef CAS; (b) P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Chem. Mater., 2003, 15, 2049 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2003, 81, 547 CrossRef CAS.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 Search PubMed.
- H. J. Huang, S. Ramaswamy and Y. Liu, Sep. Purif. Technol., 2014, 132, 513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01726b |
| This journal is © The Royal Society of Chemistry 2016 |