
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible cyclometalation at RhI as a motif for metal–ligand bifunctional bond activation and base-free formic acid dehydrogenation†
L. S.
Jongbloed
a,
B.
de Bruin
a,
J. N. H.
Reek
a,
M.
Lutz
b and
J. I.
van der Vlugt
*a
aHomogeneous, Bioinspired & Supramolecular Catalysis, van 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.i.vandervlugt@uva.nl
bCrystal & Structural Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH, The Netherlands
First published on 11th November 2015
Abstract
Reversible cyclometalation is demonstrated as a strategy for the activation of small protic molecules, with a proof-of-principle catalytic application in the dehydrogenation of formic acid in the absence of an exogenous base. The well-defined RhI complex Rh(CO)(L) 1, bearing the reactive cyclometalated PN(C) ligand L (LH = PNCH = 2-di(tert-butylphosphinomethyl)-6-phenylpyridine), undergoes protonolysis of the Rh–CPh bond with weak protic reagents, such as thiols and trifluoromethanesulfonamide. This system also displays bifunctional metal–ligand protonolysis reactivity with formic acid and subsequent decarboxylation of the formate complex. Density functional theory (DFT) calculations show that H2 evolution from putative Rh(CO)(H)(LH) complex A is very facile, proposedly encompassing formal C–H oxidative addition at Rh to give Cvia agostic intermediate B and subsequent reductive elimination of H2. Complex 1 is a catalytically competent species for base-free formic acid dehydrogenation, with the intermediacy of formate complex 4. DFT calculations reveal accessible barriers for involvement of a flanking phenyl group for both initial activation of formic acid and release of H2, supporting a cooperative pathway. Reversible C–H activation is thus a viable mechanism for metal–ligand bifunctional catalysis.
Introduction
The application of reactive ligands for metal–ligand bifunctional bond activation and subsequent cooperative catalysis receives much attention.1 Among the different reactive ligand designs, systems bearing a proton-responsive group (showing reversible deprotonation activity) are particularly attractive and versatile. Generally, two strategies to incorporate such a fragment (an ‘internal base’) within the ligand structure that can easily activate substrates co-exist: i) a site in the coordination sphere of a metal center and ii) a site at a location not directly connected to the metal center (2nd coordination sphere). Well-known designs implementing the latter strategy operate via reversible dearomatization by deprotonation of functionalized picoline,2 aminopyridine,3 or pyridone fragments.4 Regarding the strategy involving proton-responsive groups in the coordination sphere of a transition metal, reversible deprotonation of metal-bound functionalized amines5 has been successfully applied in a variety of catalytic transformations.Metal–carbon bonds are typically rather strong, but their bond energy can be influenced by e.g. strain or non-ideal orbital overlap, as present in cyclometalated species. Reversible cyclometalation at late transition metals using strong acids has been well-documented for stoichiometric scenarios,6–8 but examples with low-valent metal ions such as RhI and applications of this type of reactivity in catalytic turnover are rare, to the best of our knowledge. Metal–ligand bifunctional catalysis by reversible cyclometalation has been postulated as a possible mechanism with a few systems (Fig. 1). Mashima et al. discussed this strategy for the dehydrogenative silylation of phenylpyridines catalyzed by a cyclometalated iridium complex.9 A similar ‘roll-over’ mechanism was suggested for base-free transfer hydrogenation with a ruthenium catalyst.10 The cooperativity of a cyclometalated fragment in the ligand structure has also been proposed, on the basis of computational studies, to be suitable for the dehydrogenation of ammonia-borane.11–13 However, it was experimentally proven that this mechanism occurs most likely only in the early stage of catalysis13 or as a way to generate an active species.12
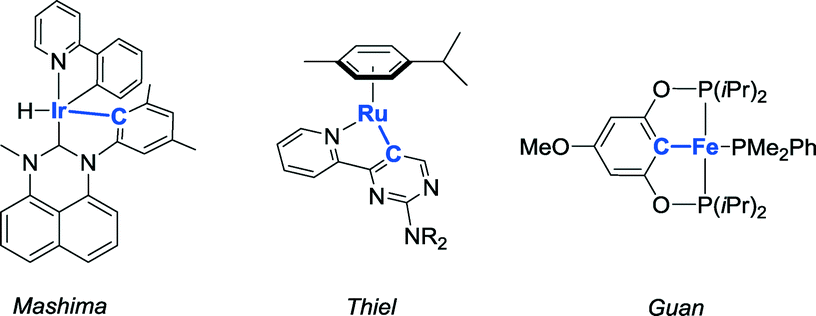 | ||
| Fig. 1 Complexes that have been proposed to act as cooperative catalysts for different types of transformations via reversible cyclometalation. | ||
Computational studies by Vanka et al. indicate that reversible cyclometalation can not only be useful for NH3BH3 dehydrogenation but can also be a suitable mechanism for formic acid dehydrogenation to CO2 and H2.11 Dihydrogen is considered a key component of many future renewable energy solutions, but efficient and reversible storage and release of H2, e.g. in organic liquids such as formic acid (FA), is essential for a hydrogen-based economy. Most catalytic systems for the dehydrogenation of HCOOH to H2 and CO2 require the presence of an exogenous base,14 which not only decreases the overall hydrogen content from 4.4 wt% (for pure HCOOH) to 2.3 wt% (for a typical 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 HCOOH/NEt3 mixture) but also necessitates post-catalysis processing for fuel cell applications (removal of volatile amines).15 Hence, catalytic formic acid dehydrogenation should ideally be performed in the absence of such an exogenous base, but to date, only a limited number of systems capable of base-free formic acid dehydrogenation have been reported.16
:2 HCOOH/NEt3 mixture) but also necessitates post-catalysis processing for fuel cell applications (removal of volatile amines).15 Hence, catalytic formic acid dehydrogenation should ideally be performed in the absence of such an exogenous base, but to date, only a limited number of systems capable of base-free formic acid dehydrogenation have been reported.16
Given our interest in the design of reactive ligand systems for cooperative bond activation reactions and catalytic processes,17 we wondered whether reversible C–H activation in the coordination sphere of a metal could serve as a new methodology to facilitate e.g. formic acid dehydrogenation. In such a strategy, a metal–carbon fragment should function as an internal base for the activation of a suitable protic substrate. A hypothetical cooperative mechanism based on reversible cyclometalation as a bond-activation concept involves i) M–C bond-assisted E–H bond activation and ii) ligand-assisted Y–H bond reductive elimination after productive conversion of the activated M–E moiety into a product-like M–Y fragment (Scheme 1). Reversible cyclometalation by protonation of the M–C bond might result in a weakly coordinating agostic C–H bond.18 This fragment could be viewed as masking a vacant site at the metal center, without significant perturbations (structural or electronic) of the global ligand framework, unlike what is often encountered for other reactive ligands. An agostic C–H interaction might also assist in stabilizing catalytically relevant intermediates, with beneficial implications for the overall energy profile of a potential reaction path.
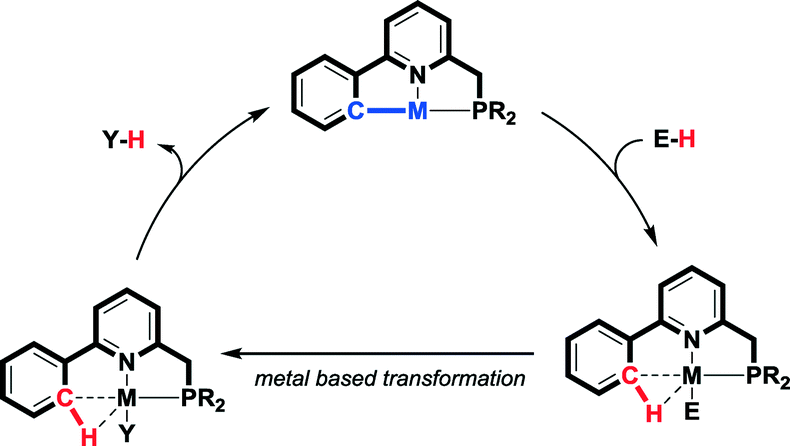 | ||
| Scheme 1 Proposed pathway involving reversible cyclometalation for metal–ligand bifunctional bond activation and catalysis. | ||
Recently, we synthesized cyclometalated RhI complex 1 bearing the deprotonated derivative of ligand LH that can act both as a neutral bidentate PN-ligand and as anionic tridentate PNC-system, depending on the reaction conditions.19 Based on these initial results, we speculated that the ligand-based reactive carbon center in the coordination sphere of RhI could be employed as an internal base for the activation and conversion of functionalized protic substrates and that the flexidentate character of the ligand could be beneficial in catalysis. We previously studied the activation of alkynes,20 activated amines21 and thiols22,23 using proton-responsive PN and PNP ligands coordinated to late transition metals using dearomatization/aromatization cooperativity. In this article, we describe the reactivity of the Rh–C bond toward related substrates and we report on the base-free dehydrogenation of formic acid as a proof-of-principle for the use of reversible cyclometalation in metal–ligand bifunctional catalysis.
Results and discussion
Reactivity of 1 toward weak protic donors – thiols
Cyclometalated complex 1 was shown to be susceptible to Rh–C cleavage by ethereal HBF4 as a strong acid. This generates a RhI complex with an agostic Rh–(CPh–H) bond in the solid state, possibly via protonation of the metal to create a RhIII(hydride) intermediate, with subsequent C–H reductive elimination. Furthermore, facile methylation at the cyclometalated carbon results from the reaction of 1 with MeI. Based on these initial results, the activation of less reactive substrates was investigated. Initial attempts to activate alcohols or phenylacetylene at r.t. did not result in Rh–C cleavage, based on NMR spectroscopy. This may point toward either a pKa mismatch between these protic substrates and the metal–carbon bond as an ‘internal base’ or to unfavorable steric interference that prevents formal oxidative addition at the metal center.Aliphatic thiols did react smoothly with 1, judging from the rapid color change of the solution from red to light-yellow (Scheme 2). 31P NMR spectroscopy was found to be very useful in monitoring the chemistry at the Rh–C bond trans to the phosphine donor. Hence, while 1 appears as a doublet at δ 76.31 ppm (1JRh–P = 101 Hz), the reaction with 1,3-propanedithiol led to a doublet at δ 69.75 ppm (1JRh–P = 151.7 Hz) for complex 2. A strong IR-band for the carbonyl was present at ν 1938 cm−1 (Δν of 5 cm−1vs.1), while the pyridine signals were significantly shifted downfield in the 1H NMR spectrum. These data suggest the decoordination of the pyridine donor and thus the monodentate P-coordination of the PNCH framework, induced by the tendency of the thiolate fragments to bridge metal centers. This hypothesis was corroborated by X-ray crystal structure determination of the single crystals of 2 grown from a concentrated acetone-d6 solution (Fig. 2). The geometry around each RhI-center is square planar and the overall structural features with a gem-dithiolate core resemble those reported in the literature.24
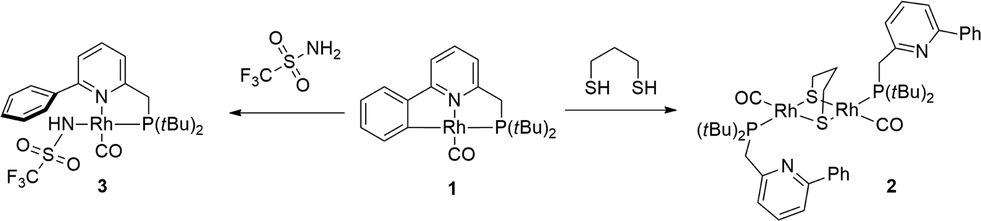 | ||
| Scheme 2 Reactivity of RhI complex 1 toward 1,3-propanedithiol and trifluoromethanesulfonamide. | ||
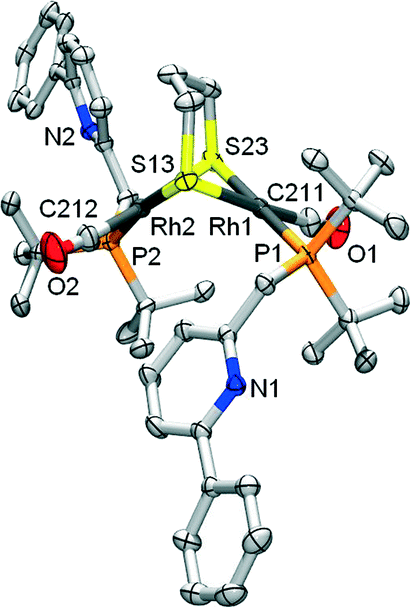 | ||
| Fig. 2 ORTEP plot (50% displacement ellipsoids) for complex 2. Selected bond lengths (Å) and angles (°): Rh1–P1 2.3163(5); Rh1–C211 1.827(2); Rh1–S13 2.3940(5); Rh1–S23 2.3784(5); Rh2–P2 2.3154(6); Rh1–C212 1.808(2); Rh2–S13 2.3833(5); Rh2–S23 2.3948(5); Rh1⋯Rh2 3.0845(2); P1–Rh1–S13 94.682(18); S13–Rh1–S23 82.762(18); P2–Rh2–S23 93.690(19); S13–Rh2–S23 82.642(18). Angle sums Rh1: 359.99(10); Rh2: 360.44(12)°. Dihedral angle between S–Rh–S planes: 61.27(3)°. | ||
Similar spectroscopic observations were made when 1 was allowed to react with benzyl mercaptan.25 This behaviour is strikingly different from the chemistry observed for Ni-complexes with dearomatized tridentate PNP ligands,22 although for CuI, this PNP scaffold was shown to adopt a dinucleating coordination mode.23 Because of the decoordination of the pyridine from the metal, we did not pursue catalytic (hydro-addition) transformations involving thiols, as the proposed cooperative nature of reversible cyclometalation requires the proximity of the C–H bond to the metal, with the pyridine acting as directing group. Also, trifluoromethylsulfonamide reacts rapidly with the Rh–C bond (Scheme 2), which resulted in a 31P shift for the resulting amide complex 3 at δ 103.7 ppm (1JRh–P = 152 Hz). The combined spectroscopic data are similar to the previously reported Pd(CH3)(RPN)(triflamide) species,21 so coordination of the triflamide trans to phosphorus is proposed, although this compound could not be obtained as single crystalline material.
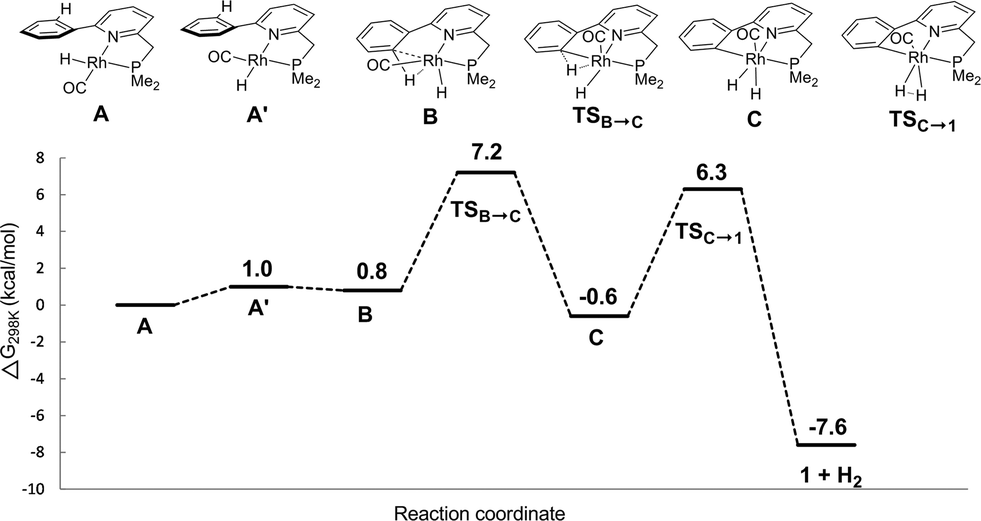 | ||
| Fig. 3 DFT (BP86, def2-TZVP, disp3) calculated free energy profile (ΔG0298K in kcal mol−1) of dihydrogen formation from hydride intermediate AMe, with methyl instead of tert-butyl groups. | ||
Catalytic dehydrogenation of formic acid
To capitalize on the apparent facile loss of H2 from putative species A in combination with the potential reactivity of the Rh–C bond in dehydrogenative catalysis and to illustrate the concept of reversible metalation for metal–ligand bifunctional substrate activation, we studied the dehydrogenation of formic acid as a proof-of-principle reaction. Addition of 20 molar equiv. of HCOOH to 1 in MeCN instantaneously resulted in a yellow complex that was characterized as formate derivative 4 (Scheme 3). Complex 4 (31P: δ 105 ppm, 1JRh–P 167 Hz) is the only species present at r.t., but upon warming to 55 °C in a closed NMR tube, deep-red species 1 is regenerated within 45 minutes. No trace of the remaining HCOOH was observed, and the formation of H2 was detected (Fig. 4).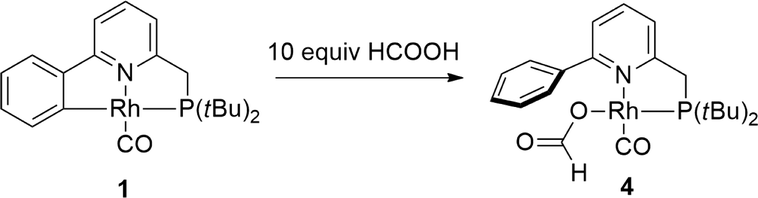 | ||
| Scheme 3 Reactivity of RhI complex 1 toward 10 molar equiv. of HCOOH. | ||
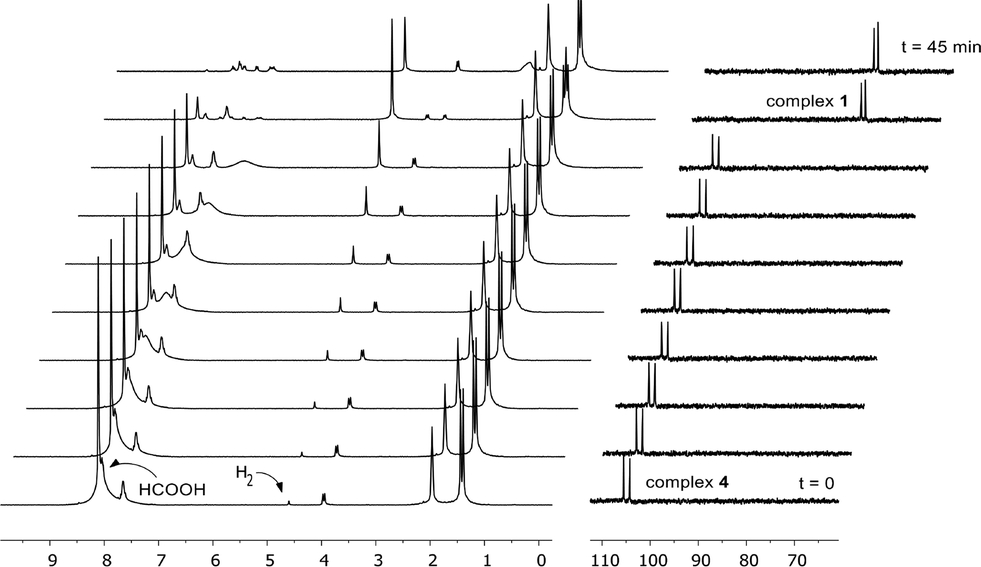 | ||
| Fig. 4 Catalytic experiment (0.02 mmol of cat. 1, 0.4 mmol of HCOOH, 2 mL of MeCN, 55 °C → 60 °C) in a 10 mm HP-NMR tube, monitored by 1H NMR (left) and 31P NMR spectroscopy (right) over a time-span of 45 min. The NMR spectra are stacked under an angle of 15°. | ||
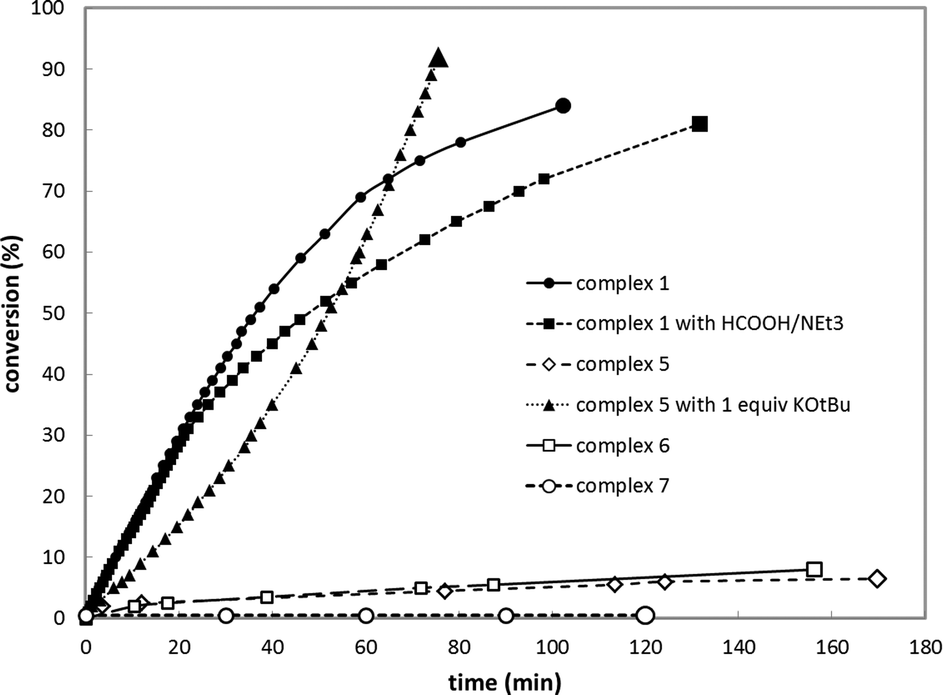
The use of HCOOD resulted in selective deuteration of both ortho-C–H groups on the phenyl ring in 1, which is in line with the cooperative activation of FA over the Rh–C bond. No deuteration of the methylene spacer was observed under these conditions, as confirmed by 2H NMR studies, excluding a role for this potentially reactive site during turnover. Smooth catalytic dehydrogenation of HCOOH was established using 0.5 mol% species 1 in dioxane at 75 °C, with a turnover frequency (TOF) of 169 mol mol−1 h−1 (see Fig. 5 and the ESI†). Addition of an external base (NEt3) did not affect the catalytic activity.26 Catalyst 1 showed reproducible performance during eight consecutive runs (a total TON of 1024). The gaseous fraction produced during reaction was analyzed by GC and no CO was found within the detection limit (δ = 10 ppm). Although the TOF achieved is still moderate under these (unoptimized) conditions, this represents the first example of base-free formic acid dehydrogenation using a RhI complex.27
 | ||
| Fig. 5 Catalytic dehydrogenation curves. | ||
Control experiments using complex 5 ([Rh(Cl)(CO)(PNH)]) bearing a bidentate PNH ligand, that lacks the flanking phenyl arm (Fig. 6)20,21 showed very low conversion in the absence of a base, likely due to blocking of the fourth coordination site by the chloride ligand. Upon addition of one equivalent of strong base to deprotonate the PNH ligand, the system showed a similar TOF but a different reaction profile including significant substrate inhibition, suggesting a different catalytic pathway for this catalyst compared to complex 1 (Fig. 4). This species likely follows a pathway involving ligand ‘dearomatization’. The known RhI-pincer complexes [Rh(CO)(PNN*)] (6) and [Rh(CO)(PCP)] (7) (PNN* = 6-di(tert-butyl)phosphinomethine-2,2′-bipyridine; see Fig. 5)28,29 barely exhibited activity, suggesting that low-coordinate geometries and the presence of a ligand with adaptable denticity are important.
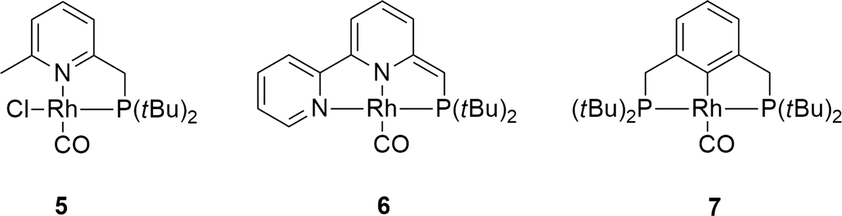 | ||
| Fig. 6 Reference complexes that have been included in this study on RhI mediated dehydrogenation of formic acid. | ||
Mechanistic considerations
Based on these catalytic results and supported by DFT calculations, two catalytic cycles are conceivable (Scheme 4). The first intramolecular path involves reversible cyclometalation as the key element. The cooperative activation of formic acid over the reactive Rh–C fragment to form formate species 4 proceeds with a moderate barrier of 17.4 kcal mol−1. The transition state for a concerted hydride–proton-transfer step30 could not be found, most likely because the hydride would be located in an unfavourable axial position (filled dz2 orbital) at Rh. Alternatively, HCOOH could also oxidatively add to form a RhIII intermediate that can undergo reductive elimination of the CPh–H bond. This option could not be ruled out by DFT calculations, as charged species cannot be compared to neutral species in gas phase calculations (see the ESI†). The resting state 4, which lies −1.9 kcal mol−1 lower in energy than 1, converts to monohydride Avia rate-limiting β-H elimination (18.2 kcal mol−1 relative to 4) concomitant with CO2 release. Subsequent C–H oxidative addition via the RhI(C–H) agostic species B (a close analogue of a previously isolated cationic derivative19) and facile release of H2 from RhIII intermediate C regenerate 1 as the active catalyst. The reversible C–H metalation pathway, providing a hemilabile aryl moiety, is also proposed to stabilize the Rh-species between turnovers.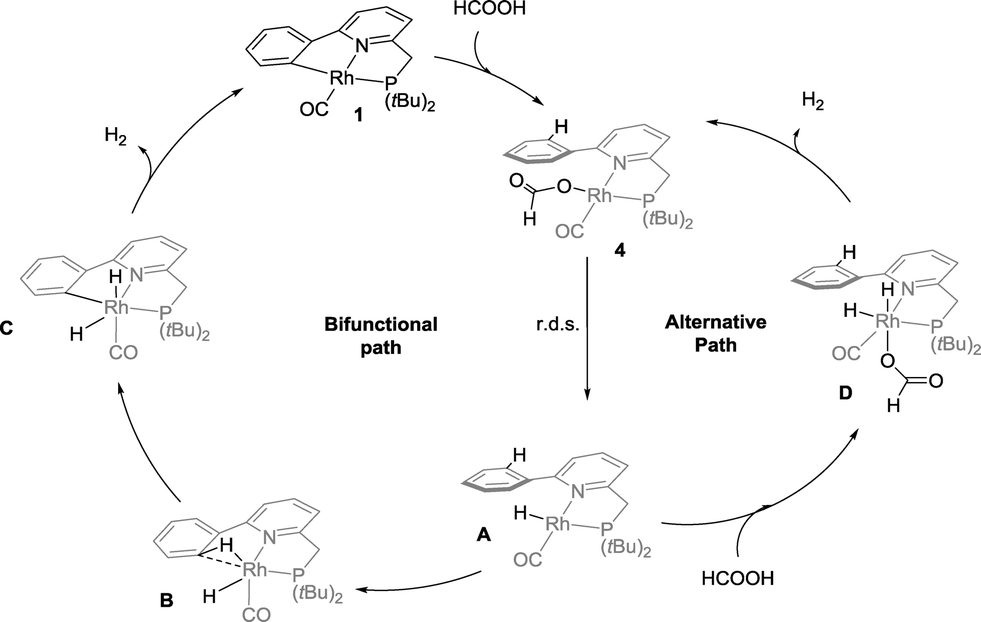 | ||
| Scheme 4 Proposed mechanism for the base-free cooperative dehydrogenation of formic acid using 1 as catalyst. The DFT calculated values for the relative transition state barriers are shown in kcal mol−1. | ||
A second, non-cooperative path has very similar reaction barriers and shares the same rate-limiting step (from 4 to A), followed by oxidative addition of a second molecule of HCOOH to form dihydride intermediate D, which lies 0.8 kcal mol−1 higher in energy than A. Dihydride D generates H2via reductive elimination with a TS barrier of 5.3 kcal mol−1. Given the near-identical overall reaction profiles (with a shared rate limiting step with a barrier of ∼18 kcal mol−1), both mechanisms are likely catalytically competent and thus co-exist under catalytic conditions, regenerating red species 1 during and/or after catalysis. The involvement of the cooperative path is supported by selective deuteration experiments, the isolation of an agostic C–H model complex as a relevant intermediate19 and the spectroscopic observation of 4 in the presence of 10 equivalents of formic acid, followed by the regeneration of 1 with the conversion of HCOOH and release of H2.
Conclusions
We have shown that reversible cyclometalation may be successfully employed as a motif for cooperative bond activation processes. Complex 1 readily reacts with thiols and activated amines, which leads to the protonation of the anionic carbon of the reactive flexidentate31 ligand L. DFT calculations show that the release of dihydrogen is facile from putative monohydride complex A. The reaction of cyclometalated complex 1 with a small excess of formic acid results in formate adduct 4. To demonstrate the potential of reversible cyclometalation in metal–ligand bifunctional catalysis, we have successfully employed RhI catalyst 1 in the base-free dehydrogenation of formic acid. Experimental observations in combination with DFT studies support the cooperative mode of action based on reversible cyclometalation as a feasible mechanism.Experimental
General considerations
All reactions were carried out under an atmosphere of nitrogen using standard Schlenk techniques. The reagents were purchased from commercial suppliers and used without further purification. THF, pentane, hexane and Et2O were distilled from sodium benzophenone ketyl. CH2Cl2 was distilled from CaH2, and toluene from sodium under nitrogen. The NMR spectra (1H, 1H{31P}, 31P, 31P{1H}, 31P-1H and 13C{1H}) were measured on a Bruker DRX 500, Bruker AV 400, Bruker DRX 300 or on a Bruker AV 300 spectrometer. The IR spectra (ATR mode) were recorded with a Bruker Alpha-p FT-IR spectrometer. The high-resolution mass spectra were recorded on a JMS-T100GCV mass spectrometer using field desorption (FD).Complex 2, Rh2(SCH2CH2CH2S)(CO)2(κ1-P-1H)2
To a solution of 1 (10 mg, 23 μmol) in CH2Cl2 (1 mL) was added 1,3-propanedithiol (1.1 μL, 23 μmol), resulting in an immediate color change from red to dark yellow. The solvent was evaporated to yield 2 in quantitative yield (11 mg). 1H NMR (300 MHz, 298 K, CD2Cl2, ppm): δ 8.44 (d, J = 6.3 Hz, 2H), 8.14–8.07 (m, 4H), 7.63–7.40 (m, 10H), 4.21–3.82 (m, 4H, CH2P), 2.95–2.69 (m, 4H), 2.42–2.30 (m, 2H), 1.53 (d, 3JPH = 12.7 Hz, 18H, PtBu2), 1.41 (d, 3JPH = 12.9 Hz, 18H, PtBu2). 31P NMR (121 MHz, 298 K, CD2Cl2, ppm): δ 69.75 (d, 1JRhP = 151.7 Hz). 13C NMR (75 MHz, 298 K, CD2Cl2, ppm): δ 190.58 (dd, JRhC = 73.4 Hz, JCP = 14.9 Hz, CO), 157.28 (s, Py-C), 155.81 (s, Py-C), 139.48 (s, Ph-C), 136.22 (s, Py-CH), 128.71 (s, Ph-CH), 128.58 (s, 2C, Ph-CH), 126.75 (s, 2C, Ph-CH), 124.71 (d, J = 2.8 Hz, Py-CH), 117.89 (s, Py-CH), 38.67 (s, SCH2CH2), 37.31 (d, J = 16.2 Hz, PC(CH3)3), 36.93 (dd, J = 15.7, 1.3 Hz, CH2P), 31.71 (d, J = 13.0 Hz, SCH2CH2), 30.16 (dd, J = 17.4, 3.8 Hz, PC(CH3)3). IR (ATR, cm−1): vCO 1938. HRMS (FD): m/z calcd for C44H62N2OP2Rh2S2: 966.18888 [M–CO]+; found: 966.18386.Complex 3, Rh(NHSO2CF3)(CO)(κ2-P,N-1H)
To a solution of 1 (12 mg, 27 μmol) in CH2Cl2 (1 mL) was added trifluoromethylsulfonamide (4 mg, 27 μmol), resulting in a color change from red to orange within 5 min at room temperature. The solvent was evaporated to yield 3 in quantitative yield (16 mg). 1H NMR (300 MHz, 298 K, CD2Cl2, ppm): δ 8.20–8.12 (m, 2H, Ph), 7.90 (t, J = 7.8 Hz, 1H, Py), 7.68–7.59 (m, 3H, Ph), 7.52 (t, J = 8.3 Hz, 2H, Py), 3.75 (d, 2JPH = 9.3 Hz, 2H, CH2P), 1.41 (d, 3JPH = 14.1 Hz, 18H, PtBu2), 1.14 (s, 1H, NH). 31P NMR (121 MHz, 298 K, CD2Cl2, ppm): δ 103.70 (d, 1JRhP = 152.0 Hz). 19F NMR (282 MHz, 298 K, CD2Cl2, ppm): δ −78.68. 13C NMR (75 MHz, 298 K, CD2Cl2, ppm): δ 189.59 (dd, JRhC = 75.5 Hz, JCP = 17.7 Hz, CO), 161.58 (s, Py-C), 161.50 (dd, J = 4.7, 1.8 Hz, Py-C), 139.03 (s, Py-CH and Ph-C), 130.57 (s, Ph-CH), 128.62 (s, Ph-CH), 128.52 (s, Ph-CH), 124.17 (s, Py-CH), 121.48 (d, J = 9.2 Hz, Py-CH), 120.88 (q, JCF = 325.5 Hz, CF3), 35.32 (dd, J = 20.8, 2.3 Hz, CH2P), 34.78 (d, J = 20.1 Hz, PC(CH3)3), 28.92 (d, J = 4.2 Hz, PC(CH3)3). IR (ATR, cm−1): vCO 1973. HRMS (FD): m/z calcd for C22H30F3N2O3PRhS: 593.07219 [M]+; found: 593.07219.Complex 4, Rh(OCH(O)(CO)(κ2-P,N-1H))
To a solution of 1 (4.4 mg, 10 μmol) in CDCl3 (0.6 mL) was added formic acid (9.2 mg, 200 μmol), resulting in an immediate color change from red to yellow at room temperature. Due to its unstable nature, this species was only characterized in situ using NMR spectroscopy. 1H NMR (400 MHz, 298 K, CDCl3, ppm): δ 8.01–7.94 (m, 2H, o-Ph), 7.82 (ddd, J = 7.8, 7.8, 1.0 Hz, 1H, Py), 7.57–7.39 (m, 5H, 2Py, m-Ph, p-Ph), 3.73 (d, 2JPH = 9.6 Hz, 2H, CH2P), 1.38 (d, 3JPH = 14.3 Hz, 18H, PtBu2). 31P NMR (162 MHz, 298 K, CDCl3, ppm): δ 105.29 (d, 1JRhP = 166.8 Hz).Complex 6, Rh(Cl)(CO)(κ2-P,N-2-methyl-6-((di-tert-butylphosphino)-methyl)pyridine))
To a solution of 2-methyl-6-((di-tert-butylphosphino)methyl)-pyridine (0.025 g, 0.010 mmol) in CH2Cl2 (0.5 mL) was added a solution of [Rh(CO)2(μ-Cl)]2 (0.019 g, 0.005 mmol) in CH2Cl2 (2 mL) and the reaction mixture was stirred overnight. After evaporation of the solvent, the product was washed with pentane (1 mL), yielding the desired complex as yellow powder (0.038 g, 0.092 mmol, 92%). 1H NMR (300 MHz, 298 K, acetone-d6, ppm): δ 7.77 (virtual t, J = 7.7 Hz, 1H, Py), 7.46 (d, J = 7.7 Hz, 1H, Py), 7.23 (d, J = 7.7 Hz, 1H, Py), 3.93 (d, 2JPH = 9.6 Hz, 2H, CH2P), 3.10 (s, 3H, Py–CH3), 1.32 (d, 3JPH = 13.9 Hz, 18H, PtBu2). 31P NMR (121 MHz, 298 K, CDCl3, ppm): δ 106.12 (d, 1JRhP = 165.0 Hz). 13C NMR (75 MHz, 298 K, acetone-d6, ppm): δ 191.85 (dd, 1JRhC = 73.4, 2JCP = 14.5 Hz, CO), 163.78 (s, Py–C), 162.52 (d, J = 3.9 Hz, Py–C), 139.74 (s, Py–CH), 124.56 (Py–CH), 121.46 (d, J = 9.0 Hz, Py–CH), 36.05 (dd, 1JCP = 20.3 Hz, 2JRhC = 2.3 Hz, CH2P), 35.42 (d, 1JCP = 20.7 Hz, PC(CH3)3), 29.48 (d, 2JCP = 4.5 Hz, PC(CH3)3), 28.19 (s, Py–CH3). IR (ATR, cm−1): νCO 1958. HRMS(FD): m/z calcd C16H26ClNOPRh: 417.04956 [M]+; found: 417.04984.Catalytic dehydrogenation experiments
In a typical experiment, compound 1 (10 μmol) was added to the solvent (1 mL) in a 5 mL Schlenk tube equipped with a condenser and connected to a water replacement set-up. The reaction mixture was heated to the desired temperature (e.g. 75 °C) and stirred for 10 minutes. Formic acid (75 μL, 2 mmol) or the azeotrope HCOOH/NEt3 (187 μL, 2 mmol HCOOH) was added to the reaction mixture and the evolved gas was collected. In the case of complex [RhCl(CO)(PNH)], one equivalent of potassium tert-butoxide in THF (1 M) was added at r.t. to abstract the chloride ligand. After stirring this mixture for 5 min, 75 μL HCOOH was added. The mixture was rapidly heated to 75 °C and the evolved gas was collected. The set-up was calibrated with a Brooks flow meter type 1054-3C and the evolved gases were analyzed with a G·A·S Compact GC (Rt-MSieve 5A 20 m × 0.32 mm + Rt-Q-bond 2 m × 0.32 mm).X-ray crystal structure determination of complex 2
C45H62N2O2P2RhS2, Fw = 994.85, yellow block, 0.25 × 0.19 × 0.09 mm3, monoclinic, P21/n (no. 14), a = 12.7487(4), b = 19.7725(6), c = 18.4361(5) Å, β = 103.046(1)°, V = 4527.3(2) Å3, Z = 4, Dx = 1.460 g cm−3, μ = 0.93 mm−1. 60826 reflections were measured on a Bruker Kappa ApexII diffractometer with a sealed tube and a Triumph monochromator (λ = 0.71073 Å) at a temperature of 150(2) K up to a resolution of (sinθ/λ)max = 0.65 Å−1. The X-ray intensities were measured on a Bruker Kappa ApexII diffractometer with a sealed tube and a Triumph monochromator (λ = 0.71073 Å) at a temperature of 150(2) K. The intensities were integrated with the Eval15 software.32 Multi-scan absorption correction and scaling was performed with SADABS33 (correction range 0.67–0.75). 10392 reflections were unique (Rint = 0.039), of which 8330 were observed [I > 2σ(I)]. The structure was solved with Patterson superposition methods using SHELXT.34 Least-squares refinement was performed with SHELXL-97 (ref. 35) against F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were located in difference Fourier maps and refined using a riding model. 508 parameters were refined with no restraints. R1/wR2 [I > 2σ(I)]: 0.0255/0.0542. R1/wR2 [all refl.]: 0.0396/0.0580. S = 1.023. Residual electron density between −0.32 and 0.32 e Å−3. CCDC 1422009 contains the supplementary crystallographic data for this paper.
DFT calculations
Geometry optimizations were carried out with the Turbomole program package36 coupled to the PQS Baker optimizer37via the BOpt package,38 at the ri-DFT level using the BP86 (ref. 39) functional and the resolution-of-identity (ri) method.40 We optimized the geometries of all stationary points at the def2-TZVP basis set level,41 using Grimme's dispersion corrections (disp3 version)42 and a tight energy grid (m5). The identity of the transition states was confirmed by following the imaginary frequency in both directions (IRC). All minima (no imaginary frequencies) and transition states (one imaginary frequency) were characterized by calculating the Hessian matrix. ZPE and gas-phase thermal corrections (entropy and enthalpy, 298 K, 1 bar) from these analyses were calculated using standard thermodynamics.Acknowledgements
This work was funded by the European Research Council (ERC, Starting Grant 279097, EuReCat to J. I. v. d. V.). We thank Sander Oldenhof for useful discussions and practical tips on FA dehydrogenation chemistry, Ed Zuidinga for MS measurements and Christophe Rebreyend and Sandra de Boer for supplying compounds 7 and 8, respectively. The X-ray diffractometer at Utrecht University was funded by NWO.Notes and references
- Reviews: D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC; D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef CAS PubMed; O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363–375 CrossRef; V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef; T. Ikariya, Top. Organomet. Chem., 2011, 37, 31–54 CrossRef; D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef.
- Reviews: J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed; C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef PubMed; C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef PubMed; J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef PubMed.
- H. Li, B. Zheng and K.-W. Huang, Coord. Chem. Rev., 2015, 293–294, 116–138 CrossRef CAS; B. Bichler, C. Holzhacker, B. Stöger, M. Puchberger, L. F. Veiros and K. Kirchner, Organometallics, 2013, 32, 4114–4121 CrossRef PubMed; D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef PubMed.
- Overview: W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, New J. Chem., 2013, 37, 1860–1866 RSC See also: C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400–402 RSC; J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed; R. Kawahara, K. Fujita and R. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12790–12794 CrossRef PubMed.
- Reviews: B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed; S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef; S. Kuwata and T. Ikariya, Dalton Trans., 2010, 39, 2984–2992 RSC.
- RhIII: J. R. Krumper, M. Gerisch, A. Magistrato, U. Rothlisberger, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2004, 126, 12492–12502 CrossRef CAS PubMed.
- IrIII: Y. Gloaguen, L. M. Jongens, M. Lutz, J. N. H. Reek, B. de Bruin and J. I. van der Vlugt, Organometallics, 2013, 32, 4284–4291 CrossRef CAS; F. Morandini, B. Longato and S. Bresadola, J. Organomet. Chem., 1977, 132, 291–299 CrossRef; J. A. Raskatov, S. Spiess, C. Gnamm, K. Brödner, F. Rominger and G. Helmchen, Chem. – Eur. J., 2010, 16, 6601–6615 CrossRef PubMed PtII (and IrIII): S. Musa, R. Romm, C. Azerraf, S. Kozuch and D. Gelman, Dalton Trans., 2011, 40, 8760–8763 RSC AuIII: D. A. Smith, D.-A. Roşca and M. Bochmann, Organometallics, 2012, 31, 5998–6000 CrossRef PdII: A. D. Getty and K. I. Goldberg, Organometallics, 2001, 20, 2545–2551 CrossRef RuII: J. Becker, T. Modl and V. H. Gessner, Chem. – Eur. J., 2014, 20, 11295–11299 CrossRef PubMed NiII: D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef PubMed.
- Roll-over complexes: L. Maidich, G. Zuri, S. Stoccoro, M. A. Cinellu, M. Masia, A. Zucca and U. Sassari, Organometallics, 2013, 32, 438–448 CrossRef CAS; B. Butschke and H. Schwarz, Chem. Sci., 2012, 3, 308–326 RSC; J. Kwak, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778–17788 CrossRef PubMed; A. Zucca, G. L. Petretto, M. L. Cabras, S. Stoccoro, M. A. Cinellu, M. Manassero and G. Minghetti, J. Organomet. Chem., 2009, 694, 3753–3761 CrossRef.
- G. Choi, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2013, 135, 13149–13161 CrossRef CAS PubMed.
- L. Taghizadeh Ghoochany, C. Kerner, S. Farsadpour, F. Menges, Y. Sun, G. Niedner-Schatteburg and W. R. Thiel, Eur. J. Inorg. Chem., 2013, 4305–4317 CrossRef CAS.
- K. Ghatak, M. Mane and K. Vanka, ACS Catal., 2013, 3, 920–927 CrossRef CAS.
- C. Boulho and J.-P. Djukic, Dalton Trans., 2010, 39, 8883–8905 RSC.
- P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS PubMed.
- Selected recent examples: R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS; E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef PubMed; G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS PubMed; S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 Search PubMed; A. Boddien, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge, G. Laurenczy and M. Beller, Energy Environ. Sci., 2012, 5, 8907–8911 Search PubMed.
- Y. Gao, J. Kuncheria, R. J. Puddephatt and G. P. A. Yap, Chem. Commun., 1998, 2365–2366 RSC; A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed; J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef PubMed; J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC; R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed; S. Oldenhof, B. de Bruin, M. Lutz, M. A. Siegler, F. W. Patureau, J. I. van der Vlugt and J. N. H. Reek, Chem. – Eur. J., 2013, 19, 11507–11511 CrossRef PubMed; W.-H. Wang, S. Xu, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ChemSusChem, 2014, 7, 1976–1983 CrossRef PubMed; T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC; F. Bertini, I. Mellone, A. Ienco, M. Peruzzine and L. Gonsalvi, ACS Catal., 2015, 5, 1254–1265 CrossRef; J. Kethandaraman, M. Czaun, A. Goeppert, R. Haiges, J. P. Jones, R. B. May, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442–1451 CrossRef PubMed.
- J. I. van der Vlugt, M. Lutz, E. A. Pidko, D. Vogt and A. L. Spek, Dalton Trans., 2009, 1016–1023 RSC; J. I. van der Vlugt, M. A. Siegler, M. Janssen, D. Vogt and A. L. Spek, Organometallics, 2009, 28, 7025–7032 CrossRef CAS; R. C. Bauer, Y. Gloaguen, M. Lutz, J. N. H. Reek, B. de Bruin and J. I. van der Vlugt, Dalton Trans., 2011, 40, 8822–8829 RSC; Y. Gloaguen, W. Jacobs, B. de Bruin, M. Lutz and J. I. van der Vlugt, Inorg. Chem., 2013, 52, 1682–1684 CrossRef PubMed; F. G. Terrade, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Eur. J. Inorg. Chem., 2014, 1826–1835 CrossRef; D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574–11577 CrossRef PubMed; S. Oldenhof, F. G. Terrade, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Organometallics, 2015, 34, 3209–3215 CrossRef; D. L. J. Broere, L. L. Metz, B. de Bruin, J. N. H. Reek, M. A. Siegler and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 1516–1520 CrossRef PubMed; V. Vreeken, M. A. Siegler, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 7055–7059 CrossRef PubMed; Z. Tang, E. Otten, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Chem. – Eur. J., 2015, 21, 12683–12693 CrossRef PubMed; S. Oldenhof, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Chem. Commun., 2015, 51, 15200–15203 RSC.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395–408 CrossRef CAS; M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914.8 CrossRef PubMed.
- L. S. Jongbloed, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Chem. – Eur. J., 2015, 21, 7297–7305 CrossRef CAS PubMed.
- S. Y. de Boer, Y. Gloaguen, M. Lutz and J. I. van der Vlugt, Inorg. Chim. Acta, 2012, 380, 336–342 CrossRef CAS.
- S. Y. de Boer, Y. Gloaguen, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Dalton Trans., 2012, 41, 11276–11283 RSC.
- J. I. van der Vlugt, M. Lutz, E. A. Pidko, D. Vogt and A. L. Spek, Dalton Trans., 2009, 1016–1023 RSC.
- J. I. van der Vlugt, E. A. Pidko, R. C. Bauer, Y. Gloaguen, M. K. Rong and M. Lutz, Chem. – Eur. J., 2011, 17, 3850–3854 CrossRef CAS PubMed.
- M. A. F. Hernandez-Gruel, G. Gracia-Arruego, A. B. Rivas, I. T. Dobrinovitch, F. J. Lahoz, A. J. Pardey, L. A. Oro and J. J. Pérez-Torrente, Eur. J. Inorg. Chem., 2007, 5677–5683 CrossRef CAS; F. Monteil, R. Queau and P. Kalck, J. Organomet. Chem., 1994, 480, 177–184 CrossRef; W. D. Jones, J. Garcia and H. Torrens, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m2204–m2206 Search PubMed.
- Notably, the reaction of 1 with thiophenol gave different spectral features, with a doublet at 99 ppm (1JRh–P = 136.3 Hz) in MeCN-d3. When C6D6 was used as solvent, two species with similar shifts and coupling constants to complex 2 were observed, i.e. doublets at 66 ppm (1JRh–P = 152.1 Hz) and 72 ppm (1JRh–P = 152.9 Hz). The species interconvert when the solvent is changed in the same sample. These results indicate that monomeric complexes are formed when polar solvents are used in the reaction, but no conclusive structural evidence could be obtained..
- The catalytic dehydrogenation of formic acid is not faster when the azeotrope HCOOH/Et3N 5:2 is used, but the TOF can be increased as more equivalents of the azeotrope are added under the same conditions. Increasing the formic acid concentration without a base results in a loss of activity, presumably because dormant species 5 is formed at too high concentrations of acid..
- Formate decomposition: E. N. Yurchenko and N. P. Anikeenko, React. Kinet. Catal. Lett., 1975, 2, 65–72 CrossRef CAS CO2 hydrogenation: J.-C. Tsai and K. M. Nicholas, J. Am. Chem. Soc., 1992, 114, 5117–5124 CrossRef.
- Y. Gloaguen, C. Rebreyend, M. Lutz, P. Kumar, M. I. Huber, J. I. van der Vlugt, S. Schneider and B. de Bruin, Angew. Chem., Int. Ed., 2014, 53, 6814–6818 CrossRef CAS PubMed.
- K.-W. Huang, D. C. Grills, J. H. Han, D. J. Szalda and E. Fujita, Inorg. Chim. Acta, 2008, 361, 3327–3331 CrossRef CAS.
- S. Oldenhof, M. Lutz, B. de Bruin, J. I. van der Vlugt and J. N. H. Reek, Chem. Sci., 2015, 6, 1027–1034 RSC See also: S. Oldenhof, J. I. van der Vlugt and J. N. H. Reek, Catal. Sci. Technol., 2015 10.1039/C5CY01476J.
- R. Lindner, B. van den Bosch, M. Lutz, J. N. H. Reek and J. I. van der Vlugt, Organometallics, 2011, 30, 499–510 CrossRef CAS.
- A. M. M. Schreurs, X. Xian and L. M. J. Kroon-Batenburg, J. Appl. Crystallogr., 2010, 43, 70–82 CrossRef CAS.
- G. M. Sheldrick, SADABS, Universität Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 1–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
- R. Ahlrichs, Turbomole Version 6.5, Theoretical Chemistry Group, University of Karlsruhe, 2013 Search PubMed.
- Parallel Quantum Solutions, PQS version 2.4, Fayetteville, Arkansas (USA), 2001 CrossRefThe Baker optimizer is available separately from PQS upon request: I. Baker, J. Comput. Chem., 1986, 7, 385–395 CrossRef.
- P. H. M. Budzelaar, J. Comput. Chem., 2007, 28, 2226–2236 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic, crystallographic, catalytic and computational details. See DOI: 10.1039/c5cy01505g |
| This journal is © The Royal Society of Chemistry 2016 |