
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of the carbide microstructure by N- and S-functionalization of the support in MoxC/CNT catalysts†
Benjamin
Frank‡
,
Zai-Lai
Xie§
,
Klaus
Friedel Ortega¶
,
Michael
Scherzer
,
Robert
Schlögl
and
Annette
Trunschke
*
Department of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany. E-mail: trunschke@fhi-berlin.mpg.de; Tel: (+49)3084134457
First published on 15th December 2015
Abstract
A series of catalysts based on molybdenum carbide nanoparticles supported on carbon were prepared by carburization of an oxidic Mo precursor impregnated on differently treated multi-walled carbon nanotubes (CNTs) and reference carbons, respectively. The effects of surface defects and decoration of the support with heteroatoms (O, N, and S), as analyzed by IR and Raman spectroscopy as well as by TPD, were investigated. The catalysts were characterized by XRD, N2 physisorption, and electron microscopy. The catalytic performance in steam reforming of methanol was used as a probe to indicate changes in the catalyst surface during catalytic action. The surface chemistry of the carbon supports influences the process of carburization and the nature of resulting supported MoxC (nano) particles. This includes crystal phase composition (α- and β-MoxC) and crystallite as well as particle diameter. However, if the surface decoration of the support is limited to oxygen groups, these differences are not reflected in the catalytic action, which is almost identical for oxygen functionalized carriers. A significant modification of the catalytic performance can only be achieved by surface modification of a CNT support with S- or N-containing functionalities, which causes changes in the lattice constant of the resulting carbide compared to reference systems. These changes are sensitively reflected in activity and CO2/CH4 product ratio in steam reforming of methanol.
Introduction
The catalytic performance of group VI metal carbides in hydrogen transfer reactions, such as hydrogenation, dehydrogenation, isomerization of alkanes, ammonia decomposition, Fischer–Tropsch synthesis, or electrocatalysis makes them highly attractive candidates for the substitution of expensive noble metals that are nowadays required for these processes.1,2 The pure high surface area carbides for use in heterogeneous catalysis are accessible by controlled carburization of oxidic precursors,3 whereas dispersed carbide nanoparticles can also be stabilized on conventional high surface area support materials, such as carbon or alumina.4–6The synthesis and application of carbon-supported molybdenum carbide catalysts has been frequently reported.6–10 The dispersion of MoxC nanoparticles strongly depends on the carburization conditions such as heating rate and gas composition, but also on Mo loading and the degree of surface functionalization,1b,6–8 which can be achieved, for example, by nitric acid treatment.6 It is concluded that surface oxygen groups are needed to anchor the impregnated Mo precursor, although a systematic study has not been performed to investigate this effect in detail. A quantitative analysis indicates that the maximum loading of highly dispersed Mo species on the carbon surface is approximately 20 wt%,8,11,12 which is supported by experimental data.6,8
Besides the structural stabilization of the carbide nanoparticles, the intrinsic structure and defect chemistry of the carbon support will also affect both the carburization process and product properties. From the reports of Solymosi et al. one can compare the catalytic activities of Mo2C/C composites based on activated carbon13 and on multi-walled CNTs,5 respectively, in steam reforming of ethanol. An effect on both the activity and selectivity is observed showing that the CNT support lowers the activity but increases the selectivity to the target-product H2. The characterization of the catalysts, however, is limited to X-ray photoelectron spectroscopy (XPS).
Our recent report8 highlighted the impact of synthesis conditions, such as heating rate of the carburization in CH4/H2, composition of gas atmosphere, and Mo loading, on the physical and chemical properties and catalytic performance of CNT-supported MoxC catalysts in steam reforming of methanol (SRM). We showed that MoC/CNT is a stable catalyst under mild reaction conditions applied, whereas several previous studies pointed at the (partial) oxidation of the carbide phase in the presence of steam at higher temperatures and H2O concentrations, respectively, or including metal dopants.25a–d Electronic and structural features of the CNT support might also play an important role here.1b In the present study, we focus on the impact of carbon surface properties, such as surface functionalization, in particular, incorporation of heteroatoms, defect density, and specific surface area on the nature of MoxC nanoparticles as well as their reactivity in steam reforming of methanol applied as probe reaction.
Experimental
Synthesis
Commercial multi-walled CNTs (Baytubes C 150 HP®) as well as reference multi-walled CNTs obtained from Shandong Dazhan were pre-treated by refluxing in 65% HNO3 (500 ml per 10 g) for 2 h. The product was washed with deionized water until neutral pH and dried in air at 110 °C for 1 day (oCNT and oCNT(ref), respectively). 1 g of oCNT was further treated in a stream of 10% O2/He at 673 K for 5 h14 (O2–oCNT) or stirred in 50 ml of 30% H2O2 at 65 °C for 24 h15 (H2O2–oCNT), respectively. The pristine Baytubes were also treated in HNO3 vapor (HNO3–CNT).16 Sidewall functionalization of Baytubes C150 HP® (Bayer) based on the diazotization of sulfanilic acid with isoamyl nitrite in water at 60 °C (molar ratio of CNT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sulfanilic acid = 1000:50) followed by removal of unreacted component through sonication and washing in 2,5-dimethylfuran resulted in S–oCNT. N-doped MWCNTs (Bayer)17 were stirred in 3 M HNO3 (100 ml per 1 g) at room temperature for 24 h, washed with deionized water until neutral pH and dried in air at 110 °C for 3 days (oN-CNT). This mild acid treatment was also performed for high surface area nanographite (Timrex HSAG 300, oNG) and activated carbon (Chemviron Carbon 114A, oAC).
:sulfanilic acid = 1000:50) followed by removal of unreacted component through sonication and washing in 2,5-dimethylfuran resulted in S–oCNT. N-doped MWCNTs (Bayer)17 were stirred in 3 M HNO3 (100 ml per 1 g) at room temperature for 24 h, washed with deionized water until neutral pH and dried in air at 110 °C for 3 days (oN-CNT). This mild acid treatment was also performed for high surface area nanographite (Timrex HSAG 300, oNG) and activated carbon (Chemviron Carbon 114A, oAC).
Aliquots of 1 g of each precursor were impregnated with 3 ml of a 117 mmol L−1 solutions of (NH4)6Mo7O24·4H2O, respectively, to achieve a final MoxC loading of 20 wt%. Accordingly, samples are denoted as MoC/y-(o)CNT, where y identifies the treatment of the carbon support. The resulting pastes were thoroughly kneaded in a mortar followed by drying in air at 110 °C for 1 day.
In a typical carburization procedure, a catalyst mass nominally containing 0.5 mmol Mo (e.g., 250 mg MoC/oCNT) was placed in a quartz tubular reactor (7 mm inner diameter) in a stream of 37 ml min−1 of a 20 vol% CH4/H2 mixture. After elution of H2O and O2 the reactor temperature was linearly increased by 5 K min−1 up to 700 °C and kept for 2 h, followed by cooling to ambient temperature in the reducing atmosphere.
Characterization
Temperature-programmed desorption (TPD) of CO was performed immediately after carburization without passivation or air contact of the catalyst. The reactor was flushed with 5 vol% CO/Ar for 10 min at ambient temperature and subsequently flushed with He until no CO was detectable by on-line mass spectrometry (MS, Pfeiffer GAM 200) and gas chromatography (Varian CP-4900 Micro GC). In a He stream of 30 ml min−1 the temperature was linearly increased by 10 K min−1 up to 500 °C. Further characterization of the catalysts was performed after catalytic testing. Passivated samples were analysed by SEM and (HR)TEM, energy-dispersive analysis of X-rays (EDX), X-ray diffraction (XRD), and N2 physisorption. SEM images were obtained with a Hitachi S-4800 FEG microscope (1.5 kV) equipped with an EDAX Genesis EDX detector (15 kV). TEM bright field images were taken with an Philips CM200 microscope equipped with an LaB6 cathode at an acceleration voltage of 200 kV. TEM data presented in ESI† were obtained with an image corrected Titan 80–300 Cs operated at 300 keV. For TEM analysis the samples were dry deposited onto holey carbon coated Cu grids. The XRD measurements were performed on a Bruker AXS D8 ADVANCE DAVINCI diffractometer equipped with a nickel filter and a LYNXEYE position sensitive detector (Cu Kα1+2 radiation) in Bragg–Brentano geometry (fixed divergence slit). N2 physisorption was performed at 77 K and p/p0 = 0.05–0.3 after drying the sample in vacuum at 200 °C for 2 h. ATR-FTIR spectra were collected using a Varian 670 spectrometer fitted with a MCT detector and a germanium crystal (512 scans, 4 cm−1 resolution). The finely powdered specimen was deposited without diluent on the ATR crystal.Catalytic testing
After CO TPD the catalysts were tested for their catalytic performance in the steam reforming of methanol (SRM) at 250 °C in 100 ml min−1 of a 1 vol% CH3OH/1 vol% H2O/He mixture. After 2 h on stream, the flow rate was varied between 100 and 10 ml min−1, followed by decreasing the temperature in 10 K steps to 200 °C at 100 ml min−1. Reaction products were quantified by GC analysis. Contact of the catalyst with H2O (reactant) and CO2 (product) ensured the mild passivation of the Mo2C surface, which is required prior to final exposure of the catalyst to ambient for its characterization.Results and discussion
Characterization of support materials
To investigate the impact of carbon surface properties on resulting MoxC/C catalysts a variety of support materials have been investigated. Based on the HNO3-treated commercial multi-walled CNTs (oCNT, Baytubes® C150HP)8 different methods of surface treatment have been applied, which include oxidation processes as well as a specific decoration with sulfonic acid groups. Also the impact of N-doping of CNTs has been tested. Nano-graphite, activated carbon as well as a different type of CNTs serve as reference materials.Nanostructured carbon supports used for Mo impregnation provide specific surface areas in the range of 200–350 m2 g−1 (Table 1) that justify a MoxC loading of 20 wt%.
| Sample | S BET/m2 g−1 | I D/IG | O/mmol g−1 | S or N/mmol g−1 |
|---|---|---|---|---|
| a Heteroatom concentration determined from CO, CO2, SO2, NO, and N2 release during TPD up to 850 °C. b n N/(nN + nC) as determined by EDX. | ||||
| oCNT | 209 | 2.05 | 1.9 | — |
| O2–oCNT | 323 | 2.48 | 1.4 | — |
| H2O2–oCNT | 270 | 2.47 | 1.9 | — |
| HNO3–CNT | 342 | 2.75 | 2.9 | — |
| oCNT(ref) | 284 | 1.98 | 2.5 | — |
| S–oCNT | 226 | 2.27 | 1.1 | 0.39 |
| oN–CNT | 206 | 6.30 | 1.3 | 0.03 (6.8%b) |
| oNG | 303 | 1.67 | 1.6 | — |
| oAC | 741 | 15.01 | 3.5 | — |
The abundance of micropores in the activated carbon reference oAC results in a higher value of 741 m2 g−1. For nanographite (oNG), Raman spectroscopy (Fig. 1) reveals dominant features at around 1350, 1580, and 1620 cm−1, referred to as D1, G, and D2 bands, respectively, indicating defective/amorphous (D1, D2) as well as crystalline/graphitic (G) carbon domains.18 Notably, the rise of the G+ band in CNT-based materials at 1590 cm−1, which is associated with C-atom vibrations along the c-axis of the tube19 (see the corresponding range in IR spectra, Fig. 1), leads to a weak blue-shift of the superimposed G-feature. It is seen that oxidative post-treatment of oCNT further increases the specific surface area, which is accompanied by an increasing ID/IG ratio observed by Raman. The increase in surface area can thus be referred to surface roughness induced by oxidative etching of the CNT basal plane. Here, harsh vapor HNO3 treatment (HNO3–CNT) leads to substantial degradation documented by the highest ID/IG value of 2.75. oCNT(ref) shows higher crystallinity at a higher specific surface area (ID/IG = 1.98), pointing at a qualitative difference between both CNT precursor materials. On the other hand, acid-treated N-doped oN–CNTs are rich in bulk and surface structural defects, which is supported by TEM analysis showing a low degree of crystallinity and a significant fraction of fishbone carbon nanofibers.17 The two reference materials nanographite oNG and activated carbon oAC were chosen due to very high and very low degree of structural order, respectively, with ID/IG ratios ranging from 1.67 to 15.01. The pronounced structural disorder in oAC and oN–CNT is supported by the appearance of a broad D4 band located at around 1200 cm−1 (Fig. 1).
 | ||
| Fig. 1 Normalized Raman spectra (left) and ATR spectra (right) of functionalized carbon support materials before impregnation with Mo precursor. | ||
The rich surface chemistry of functionalized carbon supports before Mo loading is documented in the content of O, N, and S atoms as determined by TPD (Table 1) as well as in infrared spectra (Fig. 1). It is supposed and intended that a significant fraction of N and S released during carburization (only up to 700 °C), is implemented in MoxC particles formed on these supports. Bands in the range of 1000–1320 cm−1 indicate C–O stretching vibrations attributed to alcohols, ethers, and carboxylic acids. These are dominating in oAC and absent in oNG, however, present in all other carbon supports. The high intensity of the corresponding bands in oAC is not due to a high surface density of the oxygen-containing functional groups, but related to the high specific surface area of oAC. A strong OH bending feature of carboxylic and sulfonic acid groups at 1423 cm−1 is visible for oCNT, oCNT(ref), and S–oCNT, whereas shoulders of this feature are weakly indicated in the spectra of the other materials.
The range of 1475–1600 cm−1 is dominated by aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the graphitic lattice of the (nano)carbons. CO stretching vibrations at 1670–1820 cm−1 are pronounced in the spectra of oCNT derived materials, oNG, and oAC. The oN–CNT material has no feature in this range, however, shows bands (shoulders) at 1279 and 1612 cm−1, which can be interpreted as Caryl–N stretching and N–H bending vibrations in amines, and CN stretching vibrations, respectively. Instead, S–oCNT shows an additional broad band located at 1120 cm−1, which is located in the typical range of asymmetric (1186 cm−1) and symmetric (1062–41 cm−1) S–O stretching vibrations of benzene sulfonic acid.20 In Fig. 1 the frequency range up to 4000 cm−1 is not shown due to the absence of significant features according to C–H or O–H stretching vibrations.
C stretching vibrations of the graphitic lattice of the (nano)carbons. CO stretching vibrations at 1670–1820 cm−1 are pronounced in the spectra of oCNT derived materials, oNG, and oAC. The oN–CNT material has no feature in this range, however, shows bands (shoulders) at 1279 and 1612 cm−1, which can be interpreted as Caryl–N stretching and N–H bending vibrations in amines, and CN stretching vibrations, respectively. Instead, S–oCNT shows an additional broad band located at 1120 cm−1, which is located in the typical range of asymmetric (1186 cm−1) and symmetric (1062–41 cm−1) S–O stretching vibrations of benzene sulfonic acid.20 In Fig. 1 the frequency range up to 4000 cm−1 is not shown due to the absence of significant features according to C–H or O–H stretching vibrations.
Carburization of catalyst precursors
The catalyst precursors obtained by impregnation of the different supports with an aqueous solution of ammonium heptamolybdate (AHM) and subsequent drying have been carburized in a methane–hydrogen mixture. The processes of carburization of bulk and carbon supported MoxC catalysts in CH4/H2, H2, and He atmospheres were discussed in detail in our previous studies.8,21 Briefly, the following transformations can be identified from the H2O traces recorded in the course of temperature-programmed carburization (Fig. 2): (i) decomposition of the AHM precursor into MoO3, (ii) reduction of MoVI to MoIV, (iii) partial carburization of MoIV to a non-stoichiometric oxy carbide MoOxCy, and (iv) complete carburization of MoOxCy into MoxC. The profiles of H2O evolution, the evolution of other gases (CO, CO2) (not shown), as well as consumption of H2 and CH4 (not shown), i.e., peak positions and relative intensities, do not significantly differ for oxygen-functionalized carbon supports indicating that the process of carburization is similar on carbon materials doped with oxygen. Notably, the intensity of the final carburization peak (iv) correlates with the presence of poorly dispersed β-Mo2C in the final catalysts as will be shown by XRD below. | ||
| Fig. 2 Area-normalized MS traces of H2O evolution (m/z 18) during carburization of carbon-supported MoxC catalysts. | ||
A clear difference, however, is seen for the N-doped carbon support, where the carburization begins at lower temperature. The profile of H2O evolution is broader and shows additional peaks. The incorporation of N into the MoxC lattice (nitridation) cannot be excluded. Features of initial AHM decomposition are seen on oN–CNT, whereas the final (bulk) carburization peak seems to exist only as a shoulder at the high-temperature edge of the main peak. The profiles suggest that N doping has a positive impact on the carburization as revealed by a peak shift to lower temperatures.
Reactivity in steam reforming of methanol
Steam reforming of methanol (SRM) was chosen as a probe reaction due to a complex selectivity pattern,8 which is expected to sensitively indicate changes in the catalyst structure. In the reaction network of SRM, MeOH can react to CO2 or CO,22 which in the presence of H2 can further react to CH4 and higher alkanes/alkenes via Fischer–Tropsch synthesis over Mo-based catalysts.23 At normal pressure, however, only small amounts of C2+ hydrocarbons (<5%) were detected. Online-MS analysis of the reaction products reveals that the catalysts typically approach a stable catalytic performance after 2 h time-on-stream at 250 °C. The deactivation after 24 h is negligible. Fig. 3 (top) displays the MeOH conversion as a function of contact time. For MoC/oCNT the rapid increase in conversion is followed by a slow-down of the reaction rate after reaching ∼50% conversion. This may be referred to inhibition by the main reaction products H2 and CO2 as observed over Cu-based catalysts.24 Similar to previous reports25,26 the reaction mainly yields CO2, CO and CH4. In the following, oxygen-functionalized carbons will be called heteroatom-free supports, since oxygen can be considered as a native surface modification. N-, and S-containing supports are referred to as heteroatom-doped materials. Surprisingly, all heteroatom-free carbon supported systems show almost identical activity in terms of MeOH conversion, which is a clear difference to a previous study comparing CNTs and activated carbon as support materials for MoxC catalysts.25 N-doping leads to higher activity, whereas S-doping reduces the activity.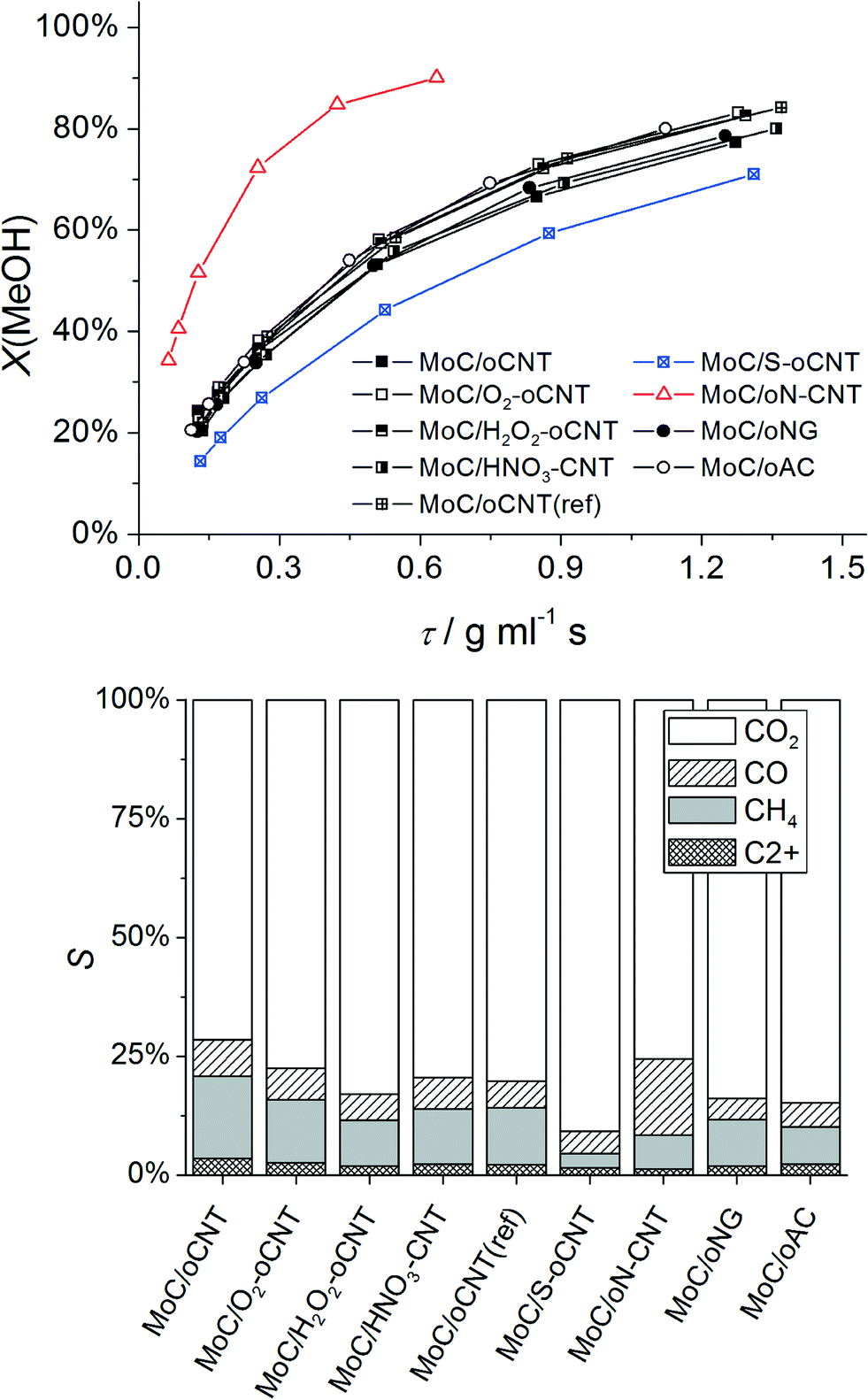 | ||
| Fig. 3 Conversion of MeOH in the SRM reaction over carbon-supported MoxC catalysts as a function of the contact time; reaction conditions: 250 mg catalyst, 10–100 ml min−1 of 1% MeOH/1% H2O/He, 250 °C (top); selectivity of the main reaction products at MeOH conversion of 50% (interpolated) (bottom). | ||
The product selectivity at MeOH iso-conversion (X = 50%) are shown in Fig. 3 (bottom). CO2 is the main product, which is formed via steam reforming of methanol. CO and CH4 are by-products formed via reverse water-gas shift and methanol decomposition or by methanol reduction and COx hydrogenation, respectively. Three parameters have been selected to quantify changes in the product selectivity of MoxC/C catalysts, namely the apparent activation energies of CO2 and CH4 formation, respectively, and the CO2/CH4 product ratio at X(MeOH) = 50% (Table 2). The CO2/CH4 ratio as a function of MeOH conversion (ESI,† Fig. S1) suggests that both CO2 and CH4 are primary products of the reaction; however, except for MoC/oN–CNT, secondary methanation of CO2 also occurs. It is observed that all heteroatom-free carbon supported systems behave very similar. A significantly higher selectivity to CO2 is obtained by using S–oCNT as the support, whereas the oN–CNT supported catalyst shows a CO2/CH4 ratio similar to heteroatom-free carbon supported systems, however, at a higher CO selectivity (Fig. 3 (bottom)). Fig. S1† furthermore reveals a different trend in product formation over MoC/oN-CNT, that is, increasing CO2 selectivity with increasing MeOH conversion, which points at a different reaction network of parallel and consecutive reactions. Water-gas shift could play a dominant role here, transforming excess CO formed by MeOH decomposition into CO2, whereas the methanation of CO and CO2 appears less relevant. The apparent activation energies Ea determined for CO2 and CH4 formation explain the catalytic performance pattern. The CO2/CH4 product ratio strictly follows the difference Ea(CH4) − Ea(CO2) (ESI,† Fig. S2). For heteroatom-doped S–oCNT and oN–CNT samples, the activities observed are in good agreement with observed Mo carbide dispersion as discussed later. N-doping of the carbon support increases both the Mo carbide dispersion and the resulting activity of the catalyst, whereas S-doping has the adverse effect. However, also electronic effects can contribute to the improved performance. For instance, it was found that small amounts of nitrogen (<5%) can strongly affect the rate of ammonia synthesis over both fcc and hcp MoxC catalysts.27 Accordingly, a bulk α-MoC1−x catalyst was reported to form a surface carbosulfide layer during hydrodesulfurization of thiophene, which lowers both CO adsorption capacity and catalytic activity.28
| Samplea | E a (CO2) | E a (CH4) | S(CO2)/S(CH4)c | S BET | CO ads.e |
|---|---|---|---|---|---|
| a Internal sample ID to clearly identify the catalyst batch are provided in ESI. b In kJ mol−1. c T = 250 °C, X(MeOH) = 50%. d In m2 g−1. e In μmol g−1. | |||||
| MoC/oCNT | 97 | 102 | 4.1 | 163 | 66.4 |
| MoC/O2–oCNT | 93 | 104 | 5.8 | 173 | 79.5 |
| MoC/H2O2–oCNT | 91 | 106 | 8.6 | 137 | 20.8 |
| MoC/HNO3–CNT | 93 | 107 | 6.9 | 136 | 47.0 |
| MoC/oCNT (ref) | 95 | 109 | 6.6 | 149 | 41.9 |
| MoC/S–oCNT | 85 | 109 | 30.9 | 145 | 6.5 |
| MoC/oN–CNT | 79 | 99 | 10.6 | 116 | 80.0 |
| MoC/oNG | 93 | 107 | 8.7 | 58 | 45.6 |
| MoC/oAC | 86 | 105 | 10.8 | 528 | 41.7 |
Post-catalytic characterization
After SRM tests the catalyst samples were characterized by XRD, N2-physisorption, and electron microscopy. Due to the contact with H2O and CO2 acting as mildly oxidizing agents29 during SRM no surface passivation by low-concentrated O2 was needed prior to exposition to ambient conditions. XRD confirms the pervasive transformation of AHM into fcc α-MoC and hcp β-Mo2C (Fig. 4). The full patterns are shown in the ESI† (Fig. S3). Nitride or sulfide phases are not observed, however, doping cannot be excluded. Although the catalysts have been used in a catalytic reaction involving potential oxidants such as H2O or CO2 and even after long-term exposition to ambient conditions the patterns give no rise to oxidic bulk phases. For comparison, the pattern of molybdenum oxy carbide MoOC is characterized by a shift of the fcc pattern to higher angles,30 which is indicative for a lattice contraction as a result of partial C–O-substitution. This is not the case here, where the detected peaks fall in line with the reference pattern for α-MoC.31 Peak analysis reveals a crystallite diameter of approx. 1–2 nm. | ||
| Fig. 4 XRD patterns of MoxC catalysts supported on differently functionalized carbon materials. Reference patterns: fcc α-MoC33 (blue) and hcp β-Mo2C (red).37 | ||
The nature of the carbide phase strongly differs among the samples: oCNT, HNO3–CNT, and oN–CNT supported carbide catalysts comprise mainly the cubic phase, whereas the reference supports oNG and oAC lead to the preferential formation of the hexagonal carbide structure. The oCNT derivatives O2–oCNT, H2O2–oCNT, and S–oCNT, as well as oCNT(ref) result in a mixture of both phases, typically with an excess of fcc MoC. The reason for the formation of the metastable32,33 α-MoC instead of β-Mo2C, which is predominantly reported to form under the synthesis conditions applied,3,4,6,29,34 is not fully understood. Han et al. suggest that the MoxC phase can be controlled over the Mo loading on an ordered mesoporous carbon (OMC) support.9 However, presented XRD diffractograms are difficult to interpret. It is more likely that the controlled reduction of the Mo precursor is the key factor for α-MoC synthesis. This has been successfully managed by MoO3 pre-reduction in a H2/n-butane mixture,33 or by adding 0.5% Pt to the Mo precursor to facilitate H2 activation.35 α-MoC proved a different performance than β-Mo2C in some catalytic reactions10,33,36 and is traditionally prepared via nitridation of MoO3 with NH3 to fcc γ-Mo2N followed by subsequent re-carburization with CH4.35
The specific surface areas of the CNT-based catalysts drop to 100–200 m2 g−1, which is caused by plugging of open CNT tips by agglomerates of MoC as well as by the much higher atomic mass of Mo as compared to C. The specific surface areas of MoC/oNG and MoC/oAC reference systems collapse to 58 and 528 m2 g−1, respectively. Having in mind similar activities and almost identical CO adsorption capacities for these samples (Table 2) we can exclude a significant impact of textural properties on the catalytic performances of MoC/C catalysts. For MoC/oCNT, the scanning (SEM) and transmission electron microscopy (TEM) analyses of the catalyst structure have been reported.8 The finely structured agglomerates of very small (<5 nm) α-MoC crystallites are located preferably at the outer CNT surface and at their tips, which were previously opened by harsh HNO3 treatment. They vary in size but rarely exceed a diameter of 10 nm. It was also shown that the CNT structure is intact after the carburization process,8 which indicates that CH4 rather than the support serves as the main carbon source for carbide formation. Similar results are obtained here: all nanotube-supported systems show the highly entangled structure without indication of significant MoC aggregation on the μm-scale, as exemplarily shown in the ESI† (Fig. S4a) for MoC/O2–oCNT. Contrarily, the oNG and oAC supported catalysts show signs of agglomeration as indicated by high contrast areas in the SEM images (backscatter mode, see arrows in Fig. S4b and c†). The appearance of such tens of nanometers sized agglomerates correlates with pronounced high-temperature peaks of H2O evolution during carburization (Fig. 2). Thus, we tentatively assign this feature to β-Mo2C bulk carburization as the presence of large agglomerates also correlates with a significant fraction of β-Mo2C (Fig. 4). Regarding the degree of surface functionalization (Table 1) an analogy exists between the interaction of the Mo precursor with the carbon surface and the nature of resulting supported MoC particles: well-anchored Mo species lead to finely dispersed nanoparticles of fcc α-MoC, whereas the poor interaction leads to large agglomerates of β-Mo2C, however, without any consequences for SRM catalysis.
The only strong differences in catalytic performance have been observed when the supporting CNT's were doped either with nitrogen or sulfur. To understand the reason, a high-resolution TEM analysis has been performed. In agreement with CO adsorption (Table 2), molybdenum carbide particles are highly dispersed on the inner and outer surface of oN-CNT (Fig. 5). The particle size distribution is, however, quite broad comprising also larger particles (ESI,† Fig. S5). A similar high dispersion has been determined by CO adsorption on O2–oCNT, while S–oCNT shows a particularly low dispersion (Table 2), which explains the lower activity of the latter catalyst.
 | ||
| Fig. 5 HRTEM image of MoC/oN–CNT. | ||
Based on electron diffraction, lattice expansion and/or contraction has been identified in oN–CNT compared to the MoC/O2–oCNT reference, which is less active in SRM in spite of similar dispersion according to CO adsorption (Table 2). The changes in lattice spacing might be an indicator for heteroatom doping of the carbide phase (see ESI,† Fig. S5 and Table S1). For the reference system MoC/O2–oCNT the statistical error of d-spacing in supported MoxC nanoparticles is within ±1% and defines the accuracy of this method. Clearly, the N- and S-containing systems show significantly larger deviations of up to 5–10% if referred to the un-doped system. Of course, these depend on the crystal phase considered (fcc or hcp), and it is far from a statistical analysis as only 5 particles per catalyst have been investigated. Nevertheless the results can be interpreted as a lattice distance modulation, which can be induced by heteroatom doping. This would be a reasonable explanation for the different catalytic behaviour observed in Fig. 3.
Conclusions
The preparation, characterization, and catalytic testing of CNT-supported MoxC catalysts are presented. The nature of the MoxC crystal phase is a sensitive function of the highly defective state of the carbon support structure, which kinetically controls the formation of favorable crystallization nuclei, leading to stabilized supported polycrystalline particles. The intimate interaction of the Mo precursor with the functionalized carbon surface leads to finely dispersed crystallites of α-MoC. For purely oxygen-decorated support materials, however, the nature of surface functionalization as well as the defect concentration or specific surface area is less relevant to the process of carburization. Instead, if the interaction is reduced by insufficient surface functionalization, larger agglomerates of β-Mo2C are formed. All materials synthesized are highly active and stable catalysts in the steam reforming of methanol. However, the control over MoxC phase and particle and crystallite size, respectively, cannot be transferred to the catalytic performance in this specific reaction, although the catalyst is apparently stable under the action of H2O and CO2 in the reactive gas phase. A different picture is obtained as soon as heteroatoms such as N or S are introduced to the support. The carburization as well as catalytic performance is influenced to enable control over activity and CO2/CH4 selectivity. N-doping increases dispersion and activity, while S-doping functions in the opposite way. Some indication for heteroatom doping has been found in MoC/oN–CNT, which might be the key to enhanced activity of this catalyst compared to the likewise highly dispersed carbide particles in the reference material MoC/O2–oCNT. However, further investigations are necessary to proof this hypothesis unambiguously.Steam reforming of methanol was used as a probe reaction rather than as the economically/industrially interesting pathway for H2 generation from liquid fuels. The experiments performed emphasise the impact of heteroatoms at the surface of nanostructured carbon supports on structure and catalytic performance of the supported phase. It is expected that trends and dependencies presented here to control the catalyst properties also provide important parameters to modify the activity and selectivity of CNT-supported MoxC catalysts in the synthesis of alcohols by hydrogenation of carbon monoxide.
Acknowledgements
The authors thank M. Hashagen, F. Rybicki, G. Weinberg, Dr. F. Girgsdies, and N. Pfänder for experimental assistance, and Dr. W. Xia (Ruhr-University Bochum) for providing HNO3 vapor treated CNTs. Financial support by the Federal Ministry of Education and Research (BMBF) within the CarboKat project (FKZ 03X0204C) of the Inno.CNT alliance is gratefully acknowledged.References
- (a) R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS; (b) W.-F. Chen, C.-H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943 RSC.
- S. T. Oyama, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley VCH, Weinheim, 2008, pp. 342–356 Search PubMed.
- J. S. Lee, S. T. Oyama and M. Boudart, J. Catal., 1987, 106, 125–133 CrossRef CAS.
- A. J. Brungs, A. P. E. York, J. B. Claridge, C. Márquez-Alvarez and M. L. H. Green, Catal. Lett., 2000, 70, 117–122 CrossRef CAS.
- R. Barthos, A. Széchenyi and F. Solymosi, Catal. Lett., 2008, 120, 161–165 CrossRef CAS.
- X. Li, D. Ma, L. Chen and X. Bao, Catal. Lett., 2007, 116, 63–69 CrossRef CAS.
- C. Sayag, M. Benkhaled, S. Suppan, J. Trawczynski and G. Djéga-Mariadassou, Appl. Catal., A, 2004, 275, 15–24 CrossRef CAS.
- B. Frank, K. Friedel, F. Girgsdies, X. Huang, R. Schlögl and A. Trunschke, ChemCatChem, 2013, 5, 2296–2305 CrossRef CAS.
- J. Han, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, ChemSusChem, 2012, 5, 727–733 CrossRef CAS PubMed.
- H. Wang, A. Wang, X. Wang and T. Zhang, Chem. Commun., 2008, 2565–2567 RSC.
- I. F. Silva, M. Klimkiewicz and S. Eser, Energy Fuels, 1998, 12, 554–562 CrossRef CAS.
- Y.-C. Xie and Y.-Q. Tang, in Advances in Catalysis, Academic Press, 1990, pp. 1–43 Search PubMed.
- R. Barthos, A. Széchenyi, Á. Koós and F. Solymosi, Appl. Catal., A, 2007, 327, 95–105 CrossRef CAS.
- B. Frank, A. Rinaldi, R. Blume, R. Schlögl and D. S. Su, Chem. Mater., 2010, 22, 4462–4470 CrossRef CAS.
- Y. Peng and H. Liu, Ind. Eng. Chem. Res., 2006, 45, 6483–6488 CrossRef CAS.
- W. Xia, C. Jin, S. Kundu and M. Muhler, Carbon, 2009, 47, 919–922 CrossRef CAS.
- A. Wolf, V. Michele, L. Mleczko, J. Assmann and S. Buchholz, Method for Producing Nitrogen-Doped Carbon Nanotubes, Patent US20100276644, 2010 Search PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- G. Świderski, M. Kalinowska, R. Świsłocka, S. Wojtulewski and W. Lewandowski, Spectrochim. Acta, Part A, 2013, 100, 41–50 CrossRef PubMed.
- T. Cotter, B. Frank, W. Zhang, R. Schlögl and A. Trunschke, Chem. Mater., 2013, 25, 3124–3136 CrossRef CAS.
- Á. Mastalir, Á. Patzkó, B. Frank, R. Schomäcker, T. Ressler and R. Schlögl, Catal. Commun., 2007, 8, 1684–1690 CrossRef.
- M. Saito and R. B. Anderson, J. Catal., 1980, 63, 438–446 CrossRef CAS.
- B. Frank, F. C. Jentoft, H. Soerijanto, J. Kröhnert, R. Schlögl and R. Schomäcker, J. Catal., 2007, 246, 177–192 CrossRef CAS.
- (a) R. Barthos and F. Solymosi, J. Catal., 2007, 249, 289–299 CrossRef CAS; (b) Y. Ma, G. Guan, X. Hao, Z. Zuo, W. Huang, P. Phanthong, K. Kusakabe and A. Abudula, RSC Adv., 2014, 4, 44175 RSC; (c) Y. Ma, G. Guan, P. Phanthong, X. Hao, W. Huang, A. Tsutsumi, K. Kusakabe and A. Abudula, J. Phys. Chem. C, 2014, 118, 9485 CrossRef CAS; (d) Y. Ma, G. Guan, C. Shi, A. Zhu, X. Hao, Z. Wang, K. Kusakabe and A. Abudula, Int. J. Hydrogen Energy, 2014, 39, 258 CrossRef CAS.
- S. S.-Y. Lin, W. J. Thomson, T. J. Hagensen and S. Y. Ha, Appl. Catal., A, 2007, 318, 121–127 CrossRef CAS.
- R. Kojima and K. Aika, Appl. Catal., A, 2001, 219, 141–147 CrossRef CAS.
- J. S. Lee and M. Boudart, Appl. Catal., 1985, 19, 207–210 CrossRef CAS.
- W. Wu, Z. Wu, C. Liang, P. Ying, Z. Feng and C. Li, Phys. Chem. Chem. Phys., 2004, 6, 5603 RSC.
- I. F. Ferguson, J. B. Ainscough, D. Morse and A. W. Miller, Nature, 1964, 202, 1327–1328 CrossRef CAS.
- O. Matsumoto, Y. Yaguchi, Y. Shiota and Y. Kanzaki, High Temp. Sci., 1983, 16, 243–250 CAS.
- H. O. Pierson, in Handbook of Refractory Carbides & Nitrides, Noyes Publications, New Jersey, 1997, pp. 110–112 Search PubMed.
- C. Bouchy, I. Schmidt, J. R. Anderson, C. J. H. Jacobsen, E. G. Derouane and S. B. J. Derouane-Abd Hamid, J. Mol. Catal. A: Chem., 2000, 163, 283–296 CrossRef CAS.
- A. Hanif, T. Xiao, A. P. E. York, J. Sloan and M. L. H. Green, Chem. Mater., 2002, 14, 1009–1015 CrossRef CAS.
- J. S. Lee, L. Volpe, F. H. Ribeiro and M. Boudart, J. Catal., 1988, 112, 44–53 CrossRef CAS.
- G. S. Ranhotra, A. T. Bell and J. A. Reimer, J. Catal., 1987, 108, 40–49 CrossRef CAS.
- B. Lönnberg, J. Less-Common Met., 1986, 120, 135–146 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Analysis of catalytic performance data, full XRD patterns, SEM micrographs, and electron diffraction patterns. See DOI: 10.1039/c5cy01480h |
| ‡ Current address: UniCat BASF Joint Lab (BasCat), Hardenbergstraße 36, 10623 Berlin, Germany. |
| § Current address: State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 35002, PR China. |
| ¶ Current address: Faculty of Chemistry and CENIDE, Universität Duisburg-Essen, Universitätsstrasse 5–7, 45141 Essen, Germany. |
| This journal is © The Royal Society of Chemistry 2016 |