
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methanol steam reforming catalysts derived by reduction of perovskite-type oxides LaCo1−x−yPdxZnyO3±δ†
Jagoda
Kuc
a,
Matthias
Neumann
b,
Marc
Armbrüster
c,
Songhak
Yoon
a,
Yucheng
Zhang
d,
Rolf
Erni
d,
Anke
Weidenkaff
e and
Santhosh Kumar
Matam
*a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland. E-mail: Santhosh.matam@empa.ch; matamsanthosh@yahoo.com
bMax-Planck-Institut für Chemische Physik fester Stoffe, D-01187 Dresden, Germany
cMaterials for Innovative Energy Concepts, Institute of Chemistry, Faculty of Natural Sciences, Technische Universität Chemnitz, D-09107 Chemnitz, Germany
dElectron Microscopy Center, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
eMaterials Chemistry, Institute for Materials Science, University of Stuttgart, D-70569 Stuttgart, Germany
First published on 25th September 2015
Abstract
Methanol steam reforming (MSR) catalysts are derived from perovskite-type oxides LaCo1−x−yPdxZnyO3±δ by reductive pretreatment. The unsubstituted LaCoO3±δ (LCO) and LaCo1−x−yPdxZnyO3±δ (Co substituted with Pd and/or Zn) are synthesized by a citrate method and characterized by different techniques. The perovskite-type oxides exhibit a rhombohedral crystal structure and a comparable surface area (≈8.5 (±2) m2 g−1). The temperature-programmed reduction (TPR) shows low (100 °C < T < 450 °C) and high (T > 450 °C) temperature reduction events that correspond to partial and complete reduction of the non-rare-earth metal ions, respectively. At high temperatures, Pd–Zn alloy nanoparticles are formed exclusively on Pd- and Zn-containing LaCo1−x−yPdxZnyO3±δ, as evident from high angular annular dark-field scanning transmission electron microscopy (HAADF-STEM). The CO2-selective MSR performance of the catalysts strongly depends on the reductive pretreatment temperature, catalyst composition (i.e., the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn molar ratio and the degree of Co substitution) and reaction temperature. Only LaCo1−x−yPdxZnyO3±δ catalysts show a low-temperature CO2 selectivity maximum between 225 and 250 °C, while all catalysts present similar high-temperature selectivity maxima at T > 400 °C. The former is missing on LCO, LaCo1−xPdxO3±δ or LaCo1−yZnyO3±δ. Pd–Zn nanoparticles facilitate Zn(OH)2 and Co(OH)2 formation exclusively on LaCo1−x−yPdxZnyO3±δ, as evident from in situ XRD under steam atmosphere. This indicates the important role of Pd–Zn nanoparticles in the low-temperature CO2 selectivity, which is improved from 0 to 76% at 225 °C on LCO and LaCo0.75Pd0.125Zn0.125O3±δ, respectively. The high-temperature CO2 selectivity is governed by the bulk catalyst composition and the occurrence of reverse water gas shift reaction.
:Zn molar ratio and the degree of Co substitution) and reaction temperature. Only LaCo1−x−yPdxZnyO3±δ catalysts show a low-temperature CO2 selectivity maximum between 225 and 250 °C, while all catalysts present similar high-temperature selectivity maxima at T > 400 °C. The former is missing on LCO, LaCo1−xPdxO3±δ or LaCo1−yZnyO3±δ. Pd–Zn nanoparticles facilitate Zn(OH)2 and Co(OH)2 formation exclusively on LaCo1−x−yPdxZnyO3±δ, as evident from in situ XRD under steam atmosphere. This indicates the important role of Pd–Zn nanoparticles in the low-temperature CO2 selectivity, which is improved from 0 to 76% at 225 °C on LCO and LaCo0.75Pd0.125Zn0.125O3±δ, respectively. The high-temperature CO2 selectivity is governed by the bulk catalyst composition and the occurrence of reverse water gas shift reaction.
1. Introduction
Catalytic methanol steam reforming (MSR) has great potential to supply H2 for fuel cells, especially for mobile applications such as proton exchange membrane fuel cells (PEMFCs).1–3 Methanol (CH3OH) as a fuel has several advantages: (i) it can be a carbon-neutral renewable feedstock,4 (ii) its gravimetric H2 density is higher than those of either compressed H2 gas or liquid H2 (ref. 5) and (iii) it is easier to distribute with existing infrastructure and safer to handle than compressed H2.1,6 The MSR reaction proceeds according to eqn (1).| CH3OH + H2O ⇌ CO2 + 3H2 ΔH° = 49 kJ mol−1 | (1) |
| CH3OH ⇌ CO + 2H2 ΔH° = 91 kJ mol−1 | (2) |
| CO2 + H2 ⇌ CO + H2O ΔH° = 41 kJ mol−1 | (3) |
:Zn ratio is lower than one.10 It is further demonstrated that ZnPd in combination with ZnO is responsible for the high CO2 selectivity.14 The synthesis of supported ZnPd nanoparticles by reductive decomposition of ternary hydrotalcite-like compounds (HTlcs) is reported.5 The structural flexibility of HTlcs allows a large number of elemental (reducible and non-reducible) combinations in which all metal cations are in close interaction making HTlcs potential precursors for the formation of intermetallic nanoparticles. This method provides a basis for deriving well-dispersed ZnPd nanoparticles that show a CO2 selectivity of 61% (at 250 °C, 14% CH3OH conversion).5
In this study, we report perovskite-type oxides as novel precursors for deriving MSR catalysts. To this end, well-defined single-phase perovskite-type oxides with the general formula ABO3 are employed. A and B represent a rare earth metal cation coordinated to 12 oxygen atoms and a transition metal cation surrounded by 6 oxygen atoms in octahedral coordination, respectively.15 The physical and chemical properties of the materials can be tailored for specific applications by partial substitution of A and/or B site cations with suitable ones, whilst preserving the perovskite crystal structure.16,17 These metal cations are in close interaction due to the covalent nature of the bonds in the compounds that exhibit remarkable catalytic properties, for example, LaFe0.57Co0.38Pd0.05O3 as a three-way catalyst.18 This study concludes that Pd located in the crystal structure emerges from the crystal to the surface under reducing environment, and reintegrates into the crystal upon oxidation. Based on the former concept, the novel MSR catalysts are derived for the first time by reductive pretreatment of perovskite-type LaCo1−x−yPdxZnyO3±δ. The CO2 selectivity of the catalysts is sensitive to the LaCo1−x−yPdxZnyO3±δ composition and the degree of Pd–Zn alloy formation.
2. Experimental
2.1 Catalyst preparation
A series of single-phase perovskite-type oxides with the general formula LaCo1−x−yPdxZnyO3±δ were synthesized by the amorphous citrate method.17,19 Metal salt precursors La(NO3)3·6H2O (Alfa Aesar, 99.9%), Co(NO3)3·6H2O (Alfa Aesar, 98%), Zn(NO3)3·6H2O (Sigma Aldrich, 98%) and Pd(NO3)3 (Alfa Aesar, 4.42% w/w Pd cont.) were used. As a chelating agent, citric acid monohydrate (Alfa Aesar, 99%) was utilized. Required amounts of metal precursors were dissolved in 150 mL of deionized water and subsequently, citric acid was added in a metal-to-acid molar ratio of 1:1.1. The resulting aqueous solution was carefully evaporated in a rotary evaporator at 70 °C and 100 mbar to obtain a viscous solution, which was then dried overnight at 80 °C in a vacuum oven operated between 50 and 70 mbar. The resulting solid foam-like material was ground in a mortar and subjected to calcination in synthetic air at 800 °C for 2 h. With this method, perovskite-type oxide catalysts with varying Pd and Zn molar ratios (0.09, 0.38, 1.0 and 1.15) were prepared. Also, LaCo1−x−yPdxZnyO3±δ catalysts having different degrees (x and y = 0.05, 0.15 and 0.25) of Co substitution with Pd and Zn (at a 1:1 Pd:Zn molar ratio) were synthesized. For comparative purposes, reference perovskite-type oxides of unsubstituted LaCoO3±δ (LCO), palladium-substituted LaCo0.873Pd0.127O3±δ (LCPO) and zinc-substituted LaCo0.89Zn0.11O3±δ (LCZO) were also prepared. For the sake of clarity, the catalyst compositions are abbreviated. For example, LCPZO-1.15 indicates the presence of La, Co, Pd and Zn, while the number defines the Pd:Zn molar ratio in the catalyst. Similarly, the second number in a catalyst like LCPZO-1-0.15 indicates a Pd and Zn (at a 1:1 ratio) substitution level of 15 mol% in LaCo0.85Pd0.075Zn0.075O3±δ. The catalyst composition, Pd and/or Zn content and BET surface area are reported in Table 1.
| Material | Chemical composition | Metal content (wt%) | BET (m2 g−1) | MSR performance at 225 °C | |||
|---|---|---|---|---|---|---|---|
| Pd | Zn | CH3OH con. (%) | CO2 selec. (%) | H2 concentration (ppm) | |||
| a Determined by ICP-OES analysis; other values are nominal values. | |||||||
| LCO | LaCo0.987(±0.024)O3±δa | — | — | 8.9 | 0.4 | 0 | 1500 |
| LCPO | LaCo0.862(±0.017)Pd0.123(±0.003)O3±δa | 5.10 (±0.05)a | — | 10.5 | 12.0 | 7.6 | 4200 |
| LCZO | LaCo0.874(±0.015)Zn0.106(±0.002)O3±δa | — | 2.75 (±0.01)a | 10.5 | 2.0 | 2.5 | — |
| LCPZO-0.09 | LaCo0.823Pd0.014Zn0.163O3±δ | 0.60 | 4.31 | 8.4 | 1.5 | 26.0 | — |
| LCPZO-0.38 | LaCo0.8Pd0.055Zn0.145O3±δ | 2.35 | 3.80 | 8.6 | 4.3 | 35.0 | — |
| LCPZO-1-0.05 | LaCo0.939(±0.036)Pd0.024(±0.001)Zn0.024(±0.001)O3±δa | 1.02 (±0.02)a | 0.61 (±0.02)a | 6.5 | 2.0 | 35.3 | 2800 |
| LCPZO-1-0.15 | LaCo0.830(±0.005)Pd0.073(±0.001)Zn0.072(±0.001)O3±δa | 3.11 (±0.02)a | 1.88 (±0.02)a | 7.5 | 8.3 | 53.3 | 4600 |
| LCPZO-1-0.25 | LaCo0.735(±0.030)Pd0.124(±0.005)Zn0.123(±0.004)O3±δa | 5.07 (±0.06)a | 3.09 (±0.01)a | 7.0 | 10.6 | 76.0 | 7200 |
| LCPZO-1.15 | LaCo0.763Pd0.127Zn0.11O3±δ | 5.35 | 2.85 | 7.0 | 11.0 | 24.0 | — |
2.2 Characterization
Similarly, in situ XRD during temperature-programmed reaction with steam (4 vol% H2O in N2, total flow: 100 ml min−1) was conducted between 100 and 550 °C to monitor the phase evolution as a function of temperature. The diffraction patterns were collected every 15 K. Prior to the experiments, the catalysts were reductively pretreated in 5 vol% H2 in N2 at 600 °C (5 K min−1) for 1 h and cooled to 100 °C. It should be mentioned that CH3OH was not included in the feed due to technical limitations of the in situ reaction chamber.
2.3 Methanol steam reforming (MSR)
Catalytic MSR activity experiments were carried out at ambient pressure in a plug flow quartz glass reactor (di = 6 mm, l = 400 mm) equipped with flow-bus operated gas manifold (Bronkhorst) and gas analyzing systems (GC and MS). The reactor was loaded with 100 mg (sieve fraction of 150–200 μm) of catalyst that was diluted with quartz glass particles of the same size in a 1:1 volume ratio to improve mass and heat transport, and firmly packed between two quartz wool plugs. The reactor was positioned vertically in a programmable tube furnace (controlled with Controltherm software, Nabertherm), and a K-type thermocouple was coaxially inserted into the catalyst bed to measure the temperature. Prior to the MSR reaction, the catalysts were reduced at either 300 or 600 °C in 25 vol% H2 in He (total flow: 50 ml min−1 He) for 1 h. After reductive pre-treatment, the catalysts were cooled to 100 °C. At this temperature, the desired MSR reaction mixture was introduced. The effect of the H2O-to-CH3OH volume ratio on the CO2 selectivity was assessed by varying the ratio between 1 and 1.3. The H2O and CH3OH streams were produced by passing He through two saturators containing deionized H2O and 99.997% CH3OH (SeccosSolv®, Merck), respectively. The resulting gas stream (100 ml min−1) with 4 vol% H2O and 3 vol% CH3OH was fed to the reactor. The reaction was carried out between 100 and 550 °C (2 K min−1), and the CH3OH conversion (%), H2O conversion (%), CO2 selectivity (%) and H2 concentration (ppm) were studied as a function of temperature. Additionally, an isothermal MSR test was performed for 80 h at 245 °C over LCPZO-1-0.15. The reactor outlet was connected to a gas chromatograph (3000A microGC, Agilent Technologies, equipped with PoraPLOT Q and Molecular Sieve 5A columns in combination with a thermal conductivity detector) to analyze the unconverted reactants (i.e., CH3OH and H2O) and reaction products (CO2 and CO). H2 was detected and quantified with a mass spectrometer (Pfeiffer Omni).
Under all reaction conditions tested in the study, the observed reaction products were H2, CO2 and CO. Other by-products such as CH4 or H2CO were not detected. Thus, the CH3OH conversion (XCH3OH (%)) is defined as:
 | (4) |
The CO2 selectivity (SCO2 (%)) is defined as:
 | (5) |
3. Results and discussion
3.1 Characterization
The composition and the surface area (SBET) of the catalysts are shown in Table 1. The ICP-OES data confirm the nominal Pd and Zn contents in the catalysts, indicating the efficiency of the synthesis method. The SBET of the unsubstituted LCO is 8.9 m2 g−1 which increases slightly (10.5 m2 g−1) upon substitution of Co with either Pd or Zn. This is in line with the literature25 which indicates that partial substitution of Co with Pd in LaCo0.95Pd0.05O3 decreases the perovskite crystallite size and hence increases the specific surface area. Accordingly, the crystallite size of 80 nm is determined for LCO. This size is decreased to 30 nm for LCPO. Contrarily, it appears that the substitution of Co with both Pd and Zn slightly decreases the SBET to around 7.5 (±1) m2 g−1 (Table 1).The XRD patterns of the catalysts are depicted in Fig. 1 and S1.† According to the PDF reference code 01-084-0848 (ref. 26), both unsubstituted LCO and substituted LaCo1−x−yPdxZnyO3±δ exhibit a single-phase rhombohedral perovskite crystal structure with the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c.27,28 These results indicate that the perovskite structure of LaCoO3±δ is preserved upon partial substitution of Co cations with Pd and/or Zn ions in LaCo1−x−yPdxZnyO3±δ, in line with our previous observations.28 The XRD reflections of the substituted LaCo1−x−yPdxZnyO3±δ are located at slightly lower 2θ angles compared to those of LCO. The slight shift in the 2θ angles indicates the unit cell expansion of the perovskite crystal due to the substitution of Co cations with Pd and/or Zn ions that have larger ionic radii than the Co ions.29 The phase purity of the rhombohedral perovskite crystal structure of the catalysts strongly depends on the degree of Co substitution. The phase purity is retained up to an x and y substitution level (in LaCo1−x−yPdxZnyO3±δ) of around 0.25. Above this, additional reflections assignable to PdO and ZnO are observed (not shown); hence such catalysts are not considered further.
c.27,28 These results indicate that the perovskite structure of LaCoO3±δ is preserved upon partial substitution of Co cations with Pd and/or Zn ions in LaCo1−x−yPdxZnyO3±δ, in line with our previous observations.28 The XRD reflections of the substituted LaCo1−x−yPdxZnyO3±δ are located at slightly lower 2θ angles compared to those of LCO. The slight shift in the 2θ angles indicates the unit cell expansion of the perovskite crystal due to the substitution of Co cations with Pd and/or Zn ions that have larger ionic radii than the Co ions.29 The phase purity of the rhombohedral perovskite crystal structure of the catalysts strongly depends on the degree of Co substitution. The phase purity is retained up to an x and y substitution level (in LaCo1−x−yPdxZnyO3±δ) of around 0.25. Above this, additional reflections assignable to PdO and ZnO are observed (not shown); hence such catalysts are not considered further.
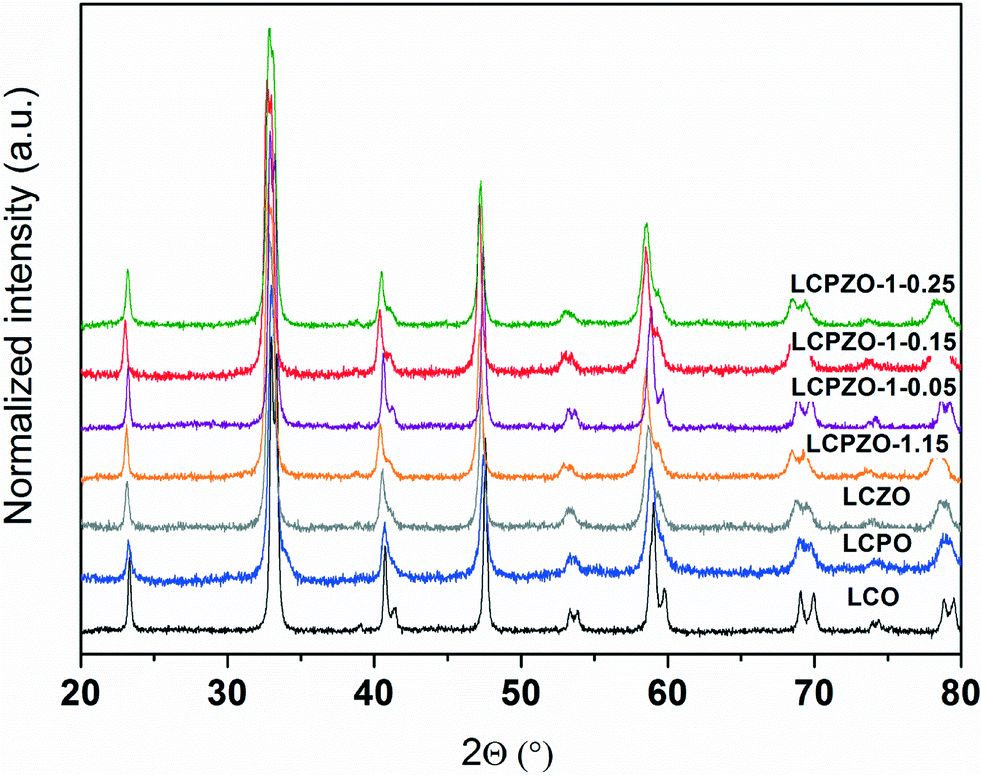 | ||
| Fig. 1 XRD patterns of the catalysts recorded at ambient atmosphere. | ||
The effect of substitution of Co with Pd and/or Zn on the reduction behavior of LaCo1−x−yPdxZnyO3±δ is investigated by temperature-programmed reduction (TPR) with hydrogen. The TPR profiles of the catalysts are shown in Fig. 2 and S2.† The unsubstituted LCO exhibits two main reduction events centered around 400 and 585 °C. The low-temperature reduction event consists of two signals indicating the reduction of two distinct Co oxide species with different reduction behaviors. Based on the literature,30 the low-temperature events are assigned to the partial reduction of Co3+ to Con+ (0 < n+ < 3), whereas the high-temperature peak can be assigned to the reduction of Con+ to Co0 which is in line with thermogravimetric analysis.16 These results suggest that the reduction of Co oxide in LaCoO3±δ is a two-step process. Interestingly, Pd-containing LCPO exhibits a low-temperature reduction peak between 100 and 300 °C with a maximum centered at 150 °C that contains a shoulder at 200 °C. The peak at 150 °C is attributed to the reduction of Pd2+,28,31 while the shoulder can be assigned to the partial reduction of Co3+. The high-temperature reduction peak is located at 520 °C and can be ascribed to the reduction of bulk cobalt oxide to elemental Co. These reduction events indicate that Pd decreases the reduction temperature of Co3+ in the perovskite. This can be anticipated from the hydrogen spillover effect.31 In contrast, Zn-containing LCZO shows two reduction events at relatively higher temperatures as compared to LCO and LCPO (Fig. 2). The low-temperature reduction peak is centered at 425 °C. The high-temperature peak exhibits two unresolved peaks at 620 and 650 °C. The latter peak could be due to the reduction of ZnO species. These reduction events suggest that Zn increases the reduction temperature of Co3+ in the perovskite.32 Based on these results, it can readily be seen in Fig. 2 that Pd and Zn-containing LCPZO-1.15, LCPZO-1-0.05, LCPZO-1-0.15 and LCPZO-1-0.25 catalysts also show the low- and high-temperature reduction events. The low-temperature signal is located between 170 and 240 °C, which is closer to the reduction event observed for Pd-containing LCPO at 150 °C than that for LCO. The high-temperature reduction event of the catalysts also matches the event observed for LCPO at 520 °C. The observed slight differences in the reduction events of the catalysts can be tentatively attributed to the variation in the substituent distribution.
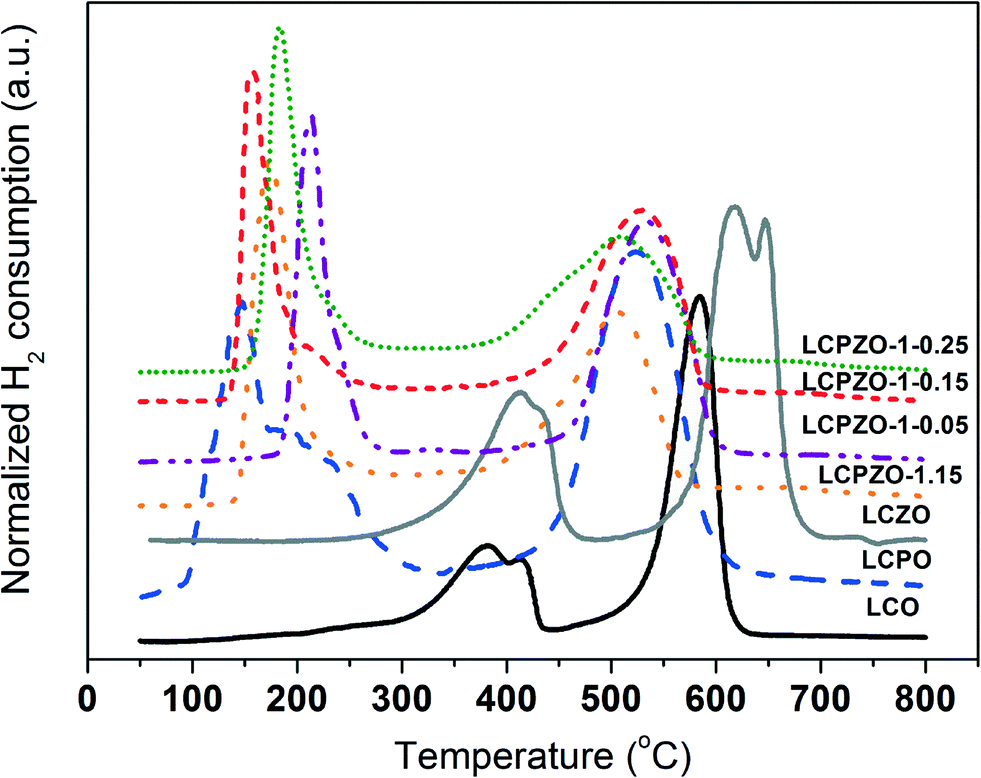 | ||
| Fig. 2 H2-TPR profiles of the catalysts. | ||
For better understanding of the reduction behavior, the materials are monitored during TPR by XRD to follow the evolution of phases as a function of temperature. The XRD patterns of the unsubstituted LCO during TPR are shown in Fig. 3. The rhombohedral crystal structure of LaCoO3±δ remains detectable up to 275 °C, indicating that the reduction of LCO does not take place. This is in good agreement with the corresponding H2-TPR data which show that the reduction begins only above 275 °C (Fig. 2). Above this temperature, noticeable changes in the XRD pattern of LCO can be observed. The changes include peak broadening and a shift in the peak positions to lower 2θ angles (i.e., larger d-spacing) due to lattice expansion caused by thermal effects. The former effect can be caused by the partial reduction of LCO, which in turn creates oxygen vacancies in the lattice. In order to maintain the overall charge neutrality in the crystal, reduction of Co3+ to a lower valence state (<3+) occurs. As a result, the average ionic radius of cobalt cations increases leading to CoO6 octahedral expansion.33 Further increase in oxygen vacancies leads to the formation of vacancy-ordered perovskite-related structures such as La3Co3O8 (PDF reference code: 01-089-1319)34 observed between 325 and 475 °C.35 Besides, the formation of other structures like La2Co2O5 could not be categorically determined due to the overlap of multiple XRD reflections at the same 2θ angles. Therefore, the formation of such phases cannot be ruled out. These results confirm that the low-temperature reduction event (at around 400 °C) observed on LCO during H2-TPR (Fig. 2) is due to the partial reduction of cobalt cations. Above 500 °C, the formation of cubic (PDF reference code: 03-065-3185)36 and hexagonal La2O3 (PDF reference code: 01-074-2430)37 as well as elemental Co (PDF reference code: 00-015-0806)38 is observed. Cubic La2O3 disappears between 575 and 600 °C, while hexagonal La2O3 and Co are sustained up to 800 °C (Fig. 3). These results are again in line with the H2-TPR profile of LCO indicating that complete reduction of the non-rare-earth metal ions is attained between 550 and 625 °C.
 | ||
| Fig. 3 XRD patterns of LCO during TPR (above) and subsequent TPO (below). | ||
Subsequently, the material is cooled from 800 °C to room temperature in the same gas mixture, flushed with N2 for 10 minutes and subjected to temperature-programmed oxidation (TPO) between room temperature and 800 °C. The XRD patterns of LCO obtained during TPO are shown in Fig. 3. It is interesting to note that during re-oxidation, elemental Co is sustained up to 225 °C. At 275 °C, the formation of the Co3O4 phase (PDF reference code: 01-076-1802)39 is observed. Further heating leads to the disappearance of Co3O4 at around 600 °C; however, the hexagonal La2O3 phase is still sustained. Above this temperature, simultaneous disappearance of La2O3 and appearance of the rhombohedral LCO phase are observed. The XRD reflections attributable to the single-phase perovskite are completely restored above 700 °C, which is close to the calcination temperature of the catalysts (i.e., 800 °C). These results suggest the occurrence of bulk structure reversibility of LaCoO3±δ upon a redox cycle.18
The in situ XRD patterns of Pd and Zn-substituted LaCo1−x−yPdxZnyO3±δ catalysts during H2-TPR are also recorded. The behavior of the catalysts during TPR is similar as evident from the XRD patterns of the catalysts (data not presented). As an example, the in situ XRD patterns of LCPZO-1-0.15 are shown in Fig. 4. However, the XRD patterns of Pd and Zn-substituted LaCo1−x−yPdxZnyO3±δ are different from those of the unsubstituted LCO. The main differences observed are in the phase transition temperature and composition. In general, the reduction temperature of substituted LaCo1−x−yPdxZnyO3±δ decreases with increasing Pd content in the catalyst, in line with the H2-TPR data. The thermal expansion of the substituted perovskite lattice occurs at relatively lower temperatures, as compared to the unsubstituted LCO. Similarly, the onset temperature required for the successive phase transitions is significantly lower for the substituted materials compared to that for LCO. For example, the rhombohedral crystal structure of LCPZO-1-0.15 is sustained only up to 100 °C, while that of the unsubstituted LCO is maintained up to 275 °C. The low stability of the perovskite structure is attributed to the H2 spillover effect due to Pd (ref. 31) that enhances the reduction of Co3+. The XRD reflections of LCPZO-1-0.15 shift towards lower angles at around 125 °C, indicating the occurrence of detectable reduction. This is again in agreement with the corresponding H2-TPR data which show that the reduction starts at around 125 °C (Fig. 2). The formation of the La3Co3O8 phase is observed at 175 °C and is visible up to 500 °C. The formation temperature is much lower than that observed for unsubstituted LCO. Above 500 °C, elemental Co is formed. Between 475 and 550 °C, the formation of cubic and hexagonal La2O3 phases is observed. Above 575 °C, the cubic La2O3 phase disappears and the XRD pattern is dominated by hexagonal La2O3 and elemental Co. This is in line with the H2-TPR profile of LCPZO-1-0.15 that shows complete reduction of the non-rare-earth ions at around 600 °C.
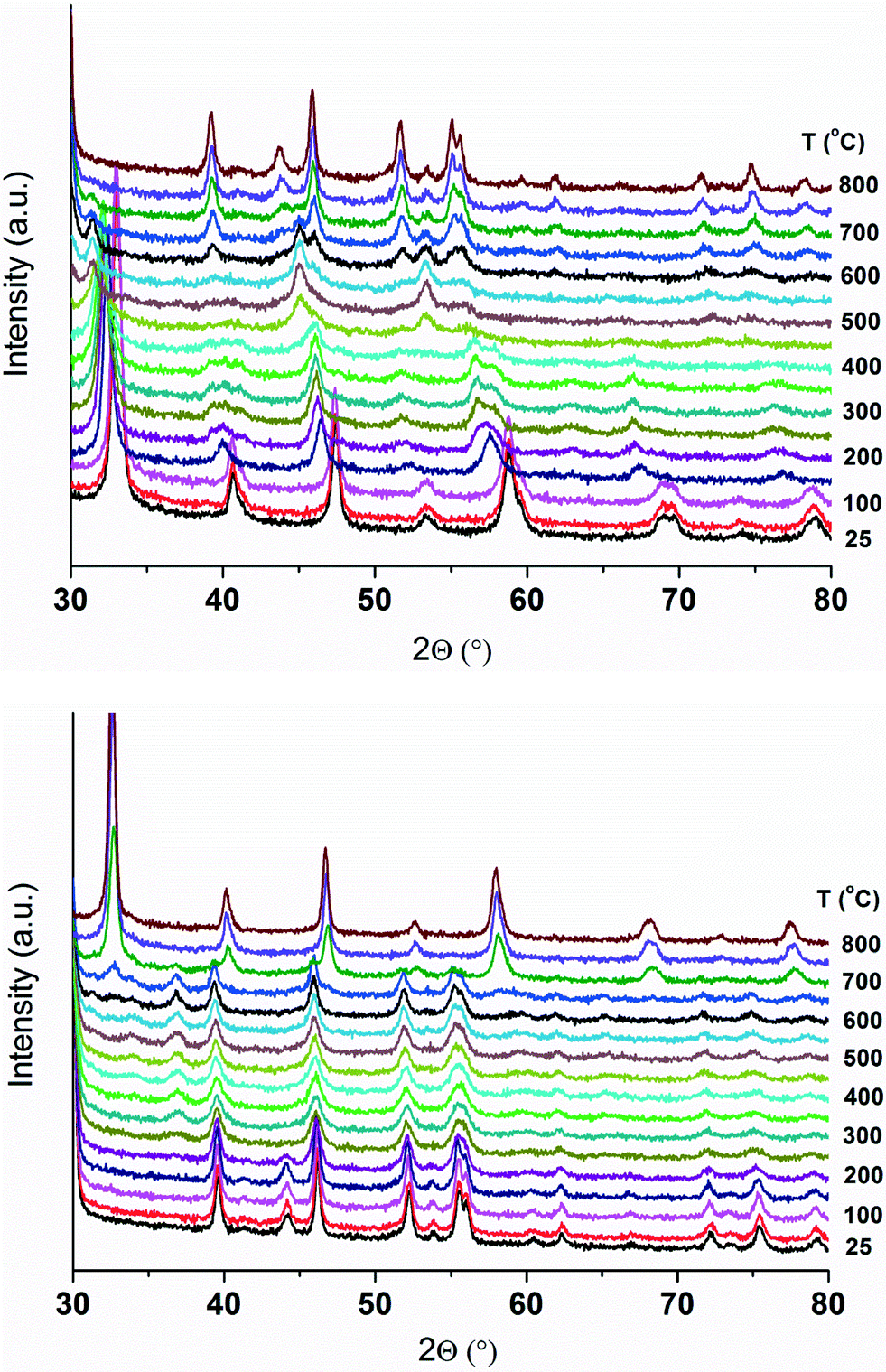 | ||
| Fig. 4 XRD patterns of LCPZO-1-0.15 during TPR (above) and subsequent TPO (below). | ||
During subsequent TPO, hexagonal La2O3 and elemental Co are sustained up to 200 °C as evident from Fig. 4. The partial oxidation of Co to Co3O4 is observed between 225 and 600 °C, and hexagonal La2O3 is visible up to 650 °C. Above this temperature, the rhombohedral crystal structure of the perovskite appears and thereafter dominates the XRD pattern. In general, the effect of Co substitution with Pd/Zn on the re-oxidation during TPO appears to be minimal as evident from the phase composition and its transition temperatures that are comparable for both unsubstituted LCO and LCPZO-1-0.15. In line with this, TPO completely restores the rhombohedral perovskite crystal structure of the unsubstituted LCO and LCPZO-1-0.15 (Fig. S3†) without altering the phase composition of the material, indicating the self-regeneration of the catalyst, at least in the bulk, during a redox cycle.
To summarize the TPR investigation, the reduction of the unsubstituted and substituted materials is a two-step process involving low- and high-temperature reduction events. In situ XRD reveals that the phase composition is temperature dependent. Oxygen-deficient perovskite, cubic/hexagonal La2O3 and elemental Co phases are formed during TPR. The oxygen-deficient perovskite phases are observed between 175 and 500 °C. The complete reduction of the catalysts occurs, except for La3+ as expected, at around 600 °C. The literature review indicates that upon reductive treatment of classical catalysts like Pd/ZnO, the intermetallic compound ZnPd is formed.3 The temperature required for the formation of such species strongly depends on the nature of the catalyst. The formation of ZnPd is typically identified by XRD reflections at 2θ of 41.2° and 44.2° in the case of Cu radiation.13,40 Thus, the formation of ZnPd (also elemental Pd and Zn as well as ZnO) in substituted LaCo1−x−yPdxZnyO3±δ catalysts during TPR is carefully analyzed. However, the identification of ZnPd reflections is not straightforward from the XRD patterns of the materials due to reflections arising from other phases derived from the perovskite-type oxides such as oxygen-deficient perovskite-type oxides, Co and La2O3 that overlap at the same 2θ angles. Therefore, the materials are further analyzed by HAADF-STEM to assess the state of Pd and/or Zn species on the catalyst surface.
The HAADF-STEM images of fresh and reduced LCPZO-1-0.15 are shown in Fig. 5. The fresh catalyst shows only perovskite crystals that can be seen as bright large particles (Fig. 5A). These crystals are around 50 nm in size and are a solid solution of perovskite-type oxide LCPZO-1-0.15 that contains Pd and Zn within the crystal sites of Co. Any precipitation of Pd and/or Zn from the crystal as nanoparticles is not observed, which is in excellent agreement with the XRD data that reflect only the rhombohedral crystal phase (Fig. 1). The reductive pretreatment at 600 °C results in the formation of Pd–Zn nanoparticles as evident from the bright spots (Fig. 5B). The composition of the nanoparticles is confirmed by an EDX line profile (Fig. 5C). These Pd–Zn nanoparticles are homogeneously dispersed on the hexagonal La2O3 substrate and the size distribution of the particles is narrow as evident from the histogram (Fig. 5D). The average particle size is determined to be around 3.5 (±1) nm, which is indeed below the detection limit of XRD (Fig. 4). Such Pd–Zn nanoparticles are not observed after reductive pretreatment at 300 °C (data not shown). Interestingly, these nanoparticles disappear upon re-oxidation as evident from Fig. 5E indicating the dissolution of Zn and Pd into the perovskite crystal. These observations infer the occurrence of a self-regeneration process within the perovskite crystal under a redox cycle, in line with in situ XRD (Fig. S3†) and the literature.18 In comparison, reduction of LCPO at 600 °C also yields Pd nanoparticles as evident from Fig. 5F. However, these Pd nanoparticles are larger than the Pd–Zn nanoparticles observed on LCPZO-1-0.15 and their size distribution is broader (between 4 and 10 nm). These results may suggest that the presence of Zn restricts not only the particle size but also the size distribution, which is similar to the effect of Sn or P on Pt,41,42 indicating that base elements promote better dispersion of noble metals. Based on these observations, it can be concluded that the reductive pretreatment at 600 °C leads to the formation of highly dispersed Pd and Pd–Zn nanoparticles on LCPO and LCPZO-1-0.15, respectively.
 | ||
| Fig. 5 High angular annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of the catalysts: fresh LCPZO-1-0.15 – after calcination at 800 °C for 2 h (A); reduced LCPZO-1-0.15 – after reduction at 600 °C for 1 h (B); re-oxidized LCPZO-1-0.15 – after re-oxidation at 800 °C for 1 h (E); reduced LCPO – after reduction at 600 °C for 1 h (F). EDX data obtained by a line scan on a Pd–Zn nanoparticle (inset) on the reduced LCPZO-1-0.15 (C) and particle size distribution on the reduced LCPZO-1-0.15 (D). | ||
3.2 Methanol steam reforming (MSR)
The MSR performance of the perovskite-type oxide-derived materials are evaluated as a function of reductive pretreatment temperature, H2O:CH3OH ratio, Co substitution with Pd and/or Zn, Pd:Zn molar ratio, Pd and Zn concentration (at 1:1 Pd:Zn molar ratio) and time on stream (stability).
 | ||
Fig. 6 The effect of reductive pretreatment temperature on the MSR performance of LCO (■, □) and LCPZO-1-0.15 ( , , ). Reductive pretreatment at 300 °C (open symbols) and 600 °C (closed symbols) is studied with an H2O:CH3OH ratio of 1.3. ). Reductive pretreatment at 300 °C (open symbols) and 600 °C (closed symbols) is studied with an H2O:CH3OH ratio of 1.3. | ||
Differently, the substituted LCPZO-1-0.15 exhibits very interesting results after the two reductive pretreatments. Reduction at 300 °C results in slightly higher overall activity than that at 600 °C, while the overall CO2 selectivity of the catalyst is lower after pretreatment at 300 °C. At a comparable CH3OH conversion of around 25% at 250 °C, CO2 selectivities of around 25 and 65% are observed after pretreatment at 300 and 600 °C, respectively (Fig. 6). The activity and selectivity data corroborate two important aspects of the catalyst that are derived from the characterization results: (i) reductive pretreatment at 300 °C results in the formation of elemental Pd (as evident from H2-TPR, see Fig. 2) which is known to decompose methanol with high CH3OH conversion activity and (ii) pretreatment at 600 °C forms highly dispersed Pd–Zn nanoparticles and enhances the methanol steam reforming selectivity. The activity data reflect the literature, concluding that elemental Pd is active but not selective for the MSR reaction,43 while ZnPd is more selective.13,44 It is also suggested that complete alloy formation is not a prerequisite for achieving high CO2 selectivity, but elimination of small monometallic Pd particles (<2 nm) formed during reductive pretreatment is essential.45 In line with these observations, the CO2 selectivity profile of the LCPZO-1-0.15 catalyst consists of two distinct maxima. The low-temperature maximum centered at around 240 °C, which is not present in the profile of the unsubstituted LCO, indicates the important role of Pd–Zn in CO2 selectivity. The high-temperature CO2 selectivity maximum assigned to above 400 °C is identical over all the catalysts studied here and is tentatively attributed to the bulk catalyst composition. The contribution of rWGS (eqn (3)) cannot be excluded.43 Based on these observations, the reductive pretreatment temperature of 600 °C is implemented for further studies.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :CH3OH ratio.
An important reaction parameter that can influence CO2 selectivity is the H2O-to-CH3OH ratio in the MSR feed. According to eqn (1), H2O-to-CH3OH ratio of one should yield selective MSR performance giving rise to CO2 and H2, without CO formation. The formation of CO is a result of CH3OH decomposition which depends on the catalyst composition and its H2O activation capability. Thus, the effect of the H2O-to-CH3OH ratio on the MSR performance of the catalysts is studied by employing two different ratios, namely 1 and 1.3 (not shown). The composition with excess H2O (i.e., decreased CH3OH concentration) in the feed yields better CH3OH conversion and CO2 selectivity in the whole temperature range than the stoichiometric feed composition. At 240 °C, around 16% CH3OH conversion and 64% CO2 selectivity are obtained with the feed ratio of 1.3, while they become significantly lower at the stoichiometric feed ratio (6% conversion and 50% CO2 selectivity). The superior CO2 selectivity of the catalyst appears to be constant in the whole temperature range studied. These results suggest that the H2O-to-CH3OH ratio of 1.3 yields better MSR performance, especially CO2 selectivity, and hence is utilized for further studies.
:Zn molar ratio (in the catalyst precursor LaCo1−x−yPdxZnyO3±δ) on the CO2 selectivity is methodically studied.
:CH3OH ratio.
An important reaction parameter that can influence CO2 selectivity is the H2O-to-CH3OH ratio in the MSR feed. According to eqn (1), H2O-to-CH3OH ratio of one should yield selective MSR performance giving rise to CO2 and H2, without CO formation. The formation of CO is a result of CH3OH decomposition which depends on the catalyst composition and its H2O activation capability. Thus, the effect of the H2O-to-CH3OH ratio on the MSR performance of the catalysts is studied by employing two different ratios, namely 1 and 1.3 (not shown). The composition with excess H2O (i.e., decreased CH3OH concentration) in the feed yields better CH3OH conversion and CO2 selectivity in the whole temperature range than the stoichiometric feed composition. At 240 °C, around 16% CH3OH conversion and 64% CO2 selectivity are obtained with the feed ratio of 1.3, while they become significantly lower at the stoichiometric feed ratio (6% conversion and 50% CO2 selectivity). The superior CO2 selectivity of the catalyst appears to be constant in the whole temperature range studied. These results suggest that the H2O-to-CH3OH ratio of 1.3 yields better MSR performance, especially CO2 selectivity, and hence is utilized for further studies.
:Zn molar ratio (in the catalyst precursor LaCo1−x−yPdxZnyO3±δ) on the CO2 selectivity is methodically studied.
 | ||
Fig. 7 The effect of Co substitution with Pd and/or Zn on the MSR performance of LCO (■, □), LCPO ( , , ), LCZO ( ), LCZO ( , , ), and LCPZO-1.15 ( ), and LCPZO-1.15 ( , , ). Reductive pretreatment at 600 °C and an H2O:CH3OH ratio of 1.3 are employed. ). Reductive pretreatment at 600 °C and an H2O:CH3OH ratio of 1.3 are employed. | ||
:Zn molar ratio.
The Pd and Zn molar ratio in LaCo1−x−yPdxZnyO3±δ is varied between 0 and 1.15 (Table 1), which is based on the literature13,44 and Pd–Zn phase diagram.48 The MSR performance of the catalysts is compared with that of the unsubstituted LCO at 225 °C in Fig. 8. The CH3OH conversion increases from 0.5 to 10.5% with increasing Pd:Zn molar ratio from 0 to 1. Upon further increasing the ratio from 1 to 1.15, the conversion remains the same. The CO2 selectivity also increases significantly from 0 to 57% with increasing Pd:Zn ratio up to 1. Above this level, however, the CO2 selectivity significantly drops to 23% for LCPZO-1.15. These observations indicate the importance of the Pd:Zn molar ratio in the LaCo1−x−yPdxZnyO3±δ precursor for CO2-selective MSR performance, in line with previous reports on unsupported ZnPd (ref. 10, 49) and supported Pd/ZnO.13 Such studies conclude that catalysts with a Pd:Zn ratio close to one present CO2-selective MSR activity and excess Pd promotes the detrimental CH3OH decomposition reaction (eqn (2)), which is also evident from the LCPO performance as discussed above. Based on this finding, an attempt is made to further improve the CO2 selectivity of the catalysts by increasing the total Pd and Zn content in LaCo1−x−yPdxZnyO3±δ while keeping the molar ratio (Pd:Zn) of 1 and the phase purity (single-phase rhombohedral crystal structure) of the perovskite-type oxide catalyst precursor.
 | ||
Fig. 8 The effect of the Pd:Zn molar ratio on the MSR performance of the catalysts is compared at 225 °C: LCO (■, □), LCPZO-0.09 ( , , ), LCPZO-0.38 ( ), LCPZO-0.38 ( , , ), LCPZO-1-0.15 ( ), LCPZO-1-0.15 ( , , ) and LCPZO-1.15 ( ) and LCPZO-1.15 ( , , ). CH3OH conversion (closed symbols) and CO2 selectivity (open symbols). ). CH3OH conversion (closed symbols) and CO2 selectivity (open symbols). | ||
:1 Pd:Zn molar ratio).
The effect of three different degrees of Co substitution (see Table 1) with Pd and Zn on the CO2 selectivity is studied. The overall MSR performance of the catalysts increases significantly with increasing concentration of Pd/Zn, especially the low-temperature CO2 selectivity (see Fig. S4†). Above 400 °C, the MSR performance of the catalysts is identical as noted above. Interestingly, the low-temperature CO2 selectivity maxima shift from 250 to 243 and 225 °C with increasing Pd and Zn concentration in LCPZO-1-0.05, LCPZO-1-0.15 and LCPZO-1-0.25, respectively. The MSR performance of the catalysts at 225 °C is compared in Fig. 9. At this temperature, the CH3OH conversion is much lower over LCO (0.5%) than that over LCPZO-1-0.05 (2.5%). Despite this, the CO2 selectivity of LCO is around 0% while that of LCPZO-1-0.05 is 35.3%. By increasing the Pd/Zn concentration from LCPZO-1-0.05 to LCPZO-1-0.15, not only CH3OH conversion (8.4%) but also CO2 selectivity (53.3%) improves. The CO2 selectivity further increases from 53.3% to 76.0% with the increase in the Pd/Zn content in LCPZO-1-0.25, while the conversion improves slightly to 10.7%. The MSR performance of the catalysts is in line with the H2O conversion and H2 concentration as shown in Fig. 9. At 225 °C, H2O is not converted on the unsubstituted LCO, hence CO2 formation does not also occur, indicating the absence of the MSR reaction (eqn (1)) as well as rWGS (eqn (3)). Thus, a H2 concentration of 1500 ppm at this temperature over LCO can be attributed to the CH3OH decomposition reaction (eqn (2)). This trend is altered for Pd and Zn-containing catalysts. By increasing the amount of Pd/Zn in the LCPZO-1-0.05, LCPZO-1-0.15 and LCPZO1-0.25 catalysts, H2O conversion increases linearly confirming the involvement of Pd/Zn in the H2O activation and hence in the selective MSR reaction. This is further supported by the H2 concentration at 225 °C (Fig. 9) which increases also linearly with the amount of Pd/Zn in the catalysts. The H2 formation rate is 1.9, 3.0 and 4.8 times higher on LCPZO-1-0.05, LCPZO-1-0.15 and LCPZO-1-0.25, respectively, than that on the unsubstituted LCO. These results confirm that the low-temperature MSR performance of the unsubstituted LCO is very poor, which can be greatly improved by substitution of Co with Pd/Zn. This can be attributed to the improved H2O activation ability of the catalysts. However, the high-temperature (>400 °C) MSR performance of the catalysts is comparable and can be ascribed to the bulk catalyst composition (i.e., La2O3 and Co). These phases may promote temperature-dependent rWGS (eqn (3)) and/or CH3OH decomposition (eqn (2)) reactions. An attempt is made to shed some light on the origin of CO2 selectivity of the catalysts by in situ XRD under steam atmosphere.
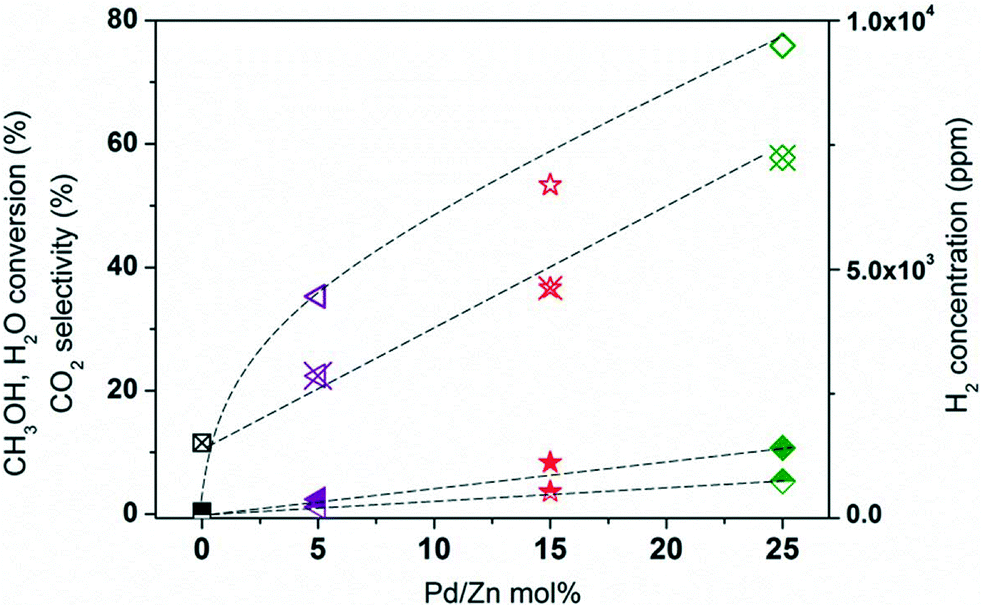 | ||
| Fig. 9 The effect of Pd/Zn concentration (at 1:1 Pd:Zn molar ratio) on the MSR performance of the catalysts is compared at 225 °C: LCO (squares), LCPZO-1-0.05 (triangles), LCPZO-1-0.15 (stars) and LCPZO-1-0.25 (diamonds). CH3OH conversion (closed symbols), H2O conversion (half-filled symbols), CO2 selectivity (open symbols), and H2 concentration (symbols with cross). | ||
3.3 In situ XRD under steam atmosphere
In situ XRD under steam atmosphere is studied over the unsubstituted LCO and substituted LCPZO-1-0.15 (Fig. 10). Prior to the experiments, the catalysts are reductively pretreated at 600 °C for 1 h followed by cooling down to 100 °C, which is identical to the pretreatment protocol utilized for MSR activity tests (section 3.2). At this temperature, hexagonal La2O3 (2θ = 26.3° and 30.1°) and Co (2θ = 44.5°) are identified on LCO. Upon introduction of H2O vapor (steam) at 100 °C, the hexagonal La2O3 phase gradually diminishes and disappears at around 150 °C. Subsequently, a new phase assignable to La(OH)3 (2θ = 27.4°, 28°, 39.6° and 48.7°) appears at 130 °C indicating the transformation of the hexagonal La2O3 phase to La(OH)3. Simultaneously, Co diminishes between 130 and 205 °C as evident from the XRD reflection at 2θ = 44.5° that disappears with temperature. This is accompanied by the emergence of the CoO phase at 2θ = 36.4° and 42.3°. The intensity of the reflections increases with temperature, suggesting the re-oxidation of elemental Co by steam. Besides these two phases, the LaOOH phase appears at around 350 °C and is visible up to 500 °C (not shown). Above this temperature, La2CoO4 (besides CoO) is observed, suggesting the formation of oxygen-deficient perovskite-type oxides under steam atmosphere. Based on these results, it can be suggested that La(OH)3, Co, CoO and LaOOH phases are present between 130 and 400 °C. | ||
| Fig. 10 In situ XRD patterns of LCO (above) and LCPZO-1-0.15 (below) during steam activation. For the sake of clarity, selected XRD reflections up to either 340 °C (LCO) or 430 °C (LCPZO-1-0.15) are shown. Refer to section 2.2 for experimental conditions. | ||
After reductive pretreatment at 600 °C, LCPZO-1-0.15 yields a mixture of cubic and hexagonal La2O3 and Co (see Fig. 10), in agreement with in situ XRD during TPR (Fig. 4). It is worth noting that during cooling from 600 to 100 °C, LCPZO-1-0.15 reacts below 250 °C with H2O that forms during the reductive pretreatment (see the bottom four patterns in Fig. 10). As evident from Fig. 10, the XRD reflections attributable to hexagonal La2O3 (2θ = 26.2° and 30°) diminish in intensity, while reflections assignable to cubic La2O3 (2θ = 26.5° and 43.8°) grow in intensity which is accompanied by a shift in the XRD reflections to slightly lower 2θ angles as compared to the reference La2O3.36 This is different from that observed for LCO which does not show any hydroxide phase formation while cooling from 600 to 100 °C after reductive pretreatment. Upon introduction of steam, LCO forms La(OH)3 at the expense of hexagonal La2O3 which is again different from that observed on LCPZO-1-0.15. Besides this, two new phases, Zn(OH)2 (2θ = 30.5°) and Co(OH)2 (2θ = 37.7°), are identified on LCPZO-1-0.15, which are not present (especially Co(OH)2) on LCO. The formation of hydroxide phases indicates the reactivity of the catalysts towards H2O molecules.
Upon introduction of steam at 100 °C, the hexagonal La2O3 phase starts to diminish (2θ of 30°) and disappears at around 160 °C, while the Zn(OH)2 phase grows in intensity along with Co(OH)2. However, the XRD reflection at 26.5° arising from cubic La2O3 is hardly affected. Unlike on LCO, the La(OH)3 phase is not observed on LCPZO-1-0.15. These results suggest different sensitivities/reactivities of the catalysts towards H2O. The observed phases are present up to 430 °C. Above this temperature (not shown), the following important phase transformations are observed: (i) the Zn(OH)2 phase disappears with simultaneous appearance of the ZnO phase that reflects at 2θ of 36.1°, 42.3° and 61.4° (PDF reference code: 03-065-0523),50 and (ii) Co(OH)2 diminishes and is perhaps transformed into CoO (PDF reference code: 01-071-1178)51 which overlaps at the same 2θ of ZnO. Above 500 °C, La2CoO4, cubic La2O3, ZnO and CoO are present.
By comparing LCO and LCPZO-1-0.15, it is evident that the evolution of phase composition under steam atmosphere is different and is a function of temperature. For example, at 225 °C, the La(OH)3 and CoO phases are mainly present on LCO, while Zn(OH)2, Co(OH)2 and cubic La2O3 are observed on LCPZO-1-0.15. However, the phase composition, such as oxygen-deficient perovskite-type oxides La2CoO4 and CoO, is similar over the two catalysts above 500 °C which can explain the identical high-temperature (>400 °C) CO2 selectivity pattern of the catalysts. The low-temperature (<400 °C) CO2 selectivity of the substituted LCPZO-1-0.15 can be attributed to Pd–Zn nanoparticles that promote hydroxide phases such as Zn(OH)2 which is not observed on the unsubstituted LCO. This is anticipated due to the fact that LCO does not contain Zn. Interestingly, both LCO and LCPZO-1-0.15 contain Co but the formation of Co(OH)2 is only observed on the latter catalyst.
4. Structure–activity relationships
The perovskite-type oxides LaCo1−x−yPdxZnyO3±δ exhibit a single-phase rhombohedral crystal structure after calcination as evident from XRD (Fig. 1 and S1†). The surface area of the catalysts varies between 6.5 and 10.5 m2 g−1, indicating similar physical and textural properties of the catalysts. The reduction behavior of the materials significantly depends on the nature of the substituent (i.e. Pd or Zn) of Co as evident from H2-TPR data (Fig. 2 and S2†). Compared with the TPR profile of LCO, the profiles of LCPO and LCZO evidently show that Pd decreases while Zn increases the reduction temperature, respectively. In the presence of both Pd and Zn, a moderate reduction behavior of the catalysts is observed which falls between the reduction temperatures of LCPO and LCZO. In any case, these catalysts are reducible in two steps as evident from the low- (<450 °C) and high-temperature (>450 °C) reduction events. In situ XRD during TPR shows that the low-temperature reduction step is due to the partial reduction of cobalt cations, while the high-temperature reduction event at 600 °C is attributable to the complete reduction of the perovskite-type oxide (except for LCZO) to its constituents like hexagonal La2O3 and elemental Co. The formation of neither elemental Pd nor Pd–Zn during TPR could be confirmed categorically from the dominant XRD reflections of La2O3 and elemental Co. However, reduction of Pd and Zn-containing catalysts at 600 °C results in the formation of homogeneously distributed Pd–Zn nanoparticles of around 3.5 (±1) nm as evident from the HAADF-STEM of LCPZO-1-0.15. Likewise, Pd nanoparticles are formed on LCPO. The size of Pd or Pd–Zn nanoparticles might have been below the XRD detection limit. Based on these results, the following scheme can be envisaged to explain the increased MSR selectivity of catalysts derived from their precursor perovskite-type oxides LaCo1−x−yPdxZnyO3±δ by reduction. According to Scheme 1, the rhombohedral crystal structure of the perovskite-type oxides transforms into its constituents, hexagonal La2O3 and Co, and highly dispersed Pd or Pd–Zn nanoparticles precipitate upon reductive treatment at 600 °C.52 These phases appear to be largely reversible upon TPO as evident from XRD (Fig. S3†) and HAADF-STEM (Fig. 5E). Such particles are not observed after reductive pretreatment at 300 °C. Based on this data, reductive pretreatment temperatures of 300 and 600 °C are investigated for the MSR reaction. The results indicate that the higher the reductive pretreatment temperature, the better the MSR performance of the catalysts, especially the CO2 selectivity which is temperature and composition (Pd:Zn ratio and Pd and/or Zn concentration) dependent. The selectivity profile comprises a low- (between 225 and 250 °C) and a high-temperature (>400 °C) maximum. The former is catalyst composition dependent, while the latter is not and is comparable over all the catalysts. The activity data indicate that the low-temperature CO2 selectivity is directly related to the H2O conversion activity of the catalysts, which is proportional to the Pd/Zn content of the catalyst. The Zn(OH)2 and Co(OH)2 phases exclusively form on Zn and Pd-containing LaCo1−x−yPdxZnyO3±δ catalysts as evident from in situ XRD under steam atmosphere. These two phases are not observed on either unsubstituted LCO or mono-metal Pd- or Zn-substituted LCPO and LCZO. The hydroxide phases are present between 100 and 400 °C, and above this temperature, they transform into the corresponding oxide and oxygen-deficient lanthanum cobaltite phases. The presence of hydroxide phases below 400 °C coincides well with the low-temperature CO2 selectivity maxima, which is dependent on the catalyst composition. Though the role of Co(OH)2 species in the CO2 selectivity is not clear, the role of ZnO is well documented. The dissociation of H2O and CH3OH is one of the initial steps in the MSR reaction and is promoted on ZnO, especially on the unsaturated surface. The remaining reaction might proceed over the Pd surface which is in close vicinity to the Zn oxide and/or hydroxide.53,54 These results are in line with a recent study which concludes that the low-temperature CO2 selectivity crucially depends on the presence of oxidized Zn species in contact with ZnPd during MSR.14,46 Therefore, the low-temperature CO2 selectivity of the catalysts in the present study is attributed to Zn-based species, including Pd–Zn nanoparticles.55 The temperature-dependent CO2 selectivity profiles below 400 °C over the catalysts are tentatively assigned to the dynamic behavior of the active/selective phase(s) as a function of temperature. The CO2 selectivity of the catalysts, regardless of the composition, is similar above 400 °C, suggesting the occurrence of rWGS reaction.43 Therefore, the stability of the LCPZO-1-0.15 catalyst during the MSR reaction is assessed at 245 °C over 80 h (Fig. 11). The reaction temperature corresponds to the low-temperature CO2 selectivity region of the catalyst. An initial CH3OH conversion of around 20% is observed which remains the same up to 30 h of time on stream. After that, the conversion slightly decreases and a conversion of around 13% is detected after 80 h of time on stream, which accounts to an overall loss of 35%. Interestingly, the CO2 selectivity continuously increases from 40 to 60% with time on stream and does not reach a plateau even after 80 h, indicating the continuous formation of the CO2-selective phase with time.10
 | ||
| Scheme 1 Schematic representation of the self-regeneration process upon a redox cycle: the rhombohedral crystal phase of a perovskite-type oxide LaCo1−x−yPdxZyO3±δ transforms into hexagonal La2O3, cubic Co and Pd–Zn alloy nanoparticles upon reduction at 600 °C in H2. These phases reassemble as rhombohedral LaCo1−x−yPdxZyO3±δ upon re-oxidation at 800 °C. | ||
 | ||
| Fig. 11 The stability of LCPZO-1-0.15 during MSR at 245 °C: CH3OH conversion (closed symbols) and CO2 selectivity (open symbols). GC was interrupted for a few minutes during 58.5 h of time on stream. Reductive pretreatment at 600 °C and an H2O:CH3OH ratio of 1.3 are employed. Refer to section 2.3 for experimental details. | ||
5. Conclusions
Homogeneously dispersed Pd–Zn nanoparticles with a narrow size distribution of 3.5 (±1) nm are derived by reduction of perovskite-type oxides LaCo1−x−yPdxZnyO3±δ at 600 °C for 1 h as evident from HAADF-STEM. The same reduction protocol also yields homogeneously dispersed Pd nanoparticles on LaCo0.87Pd0.13O3±δ containing Pd only, however with broad particle size distribution (4–10 nm). Evidently, Zn restricts the size and distribution of the nanoparticles. The Pd–Zn nanoparticles significantly improve the CO2-selective methanol steam reforming (MSR) performance of LaCo1−x−yPdxZnyO3±δ, while Pd nanoparticles mainly promote CO-selective methanol decomposition. Thus, the CO2-selective MSR performance of the catalysts strongly depends on the reductive pretreatment temperature and catalyst composition. The unsubstituted LaCoO3±δ (LCO) merely promotes the CH3OH decomposition reaction. Partial substitution of Co with either Pd (LCPO) or Zn (LCZO) does not improve the MSR performance. This is altered by Co substitution with both Pd and Zn. Furthermore, the MSR activity improves with increasing Pd/Zn molar ratio up to 1. The higher the degree of Co substitution with Pd:Zn (1:1), the better the MSR performance. Accordingly, the best MSR performance is achieved over LaCo0.75Pd0.125Zn0.125O3±δ with a CO2 selectivity of 76% at 225 °C. The CO2 selectivity profile of the substituted LaCo1−x−yPdxZnyO3±δ catalysts consists of two distinct maxima. The low-temperature (225–250 °C) CO2 selectivity is attributed to Pd–Zn nanoparticles which are responsible for the formation of Zn(OH)2 and Co(OH)2 under steam atmosphere, as evident from in situ XRD. Neither the low-temperature CO2 selectivity maximum nor these hydroxide phases are observed on un(or mono-metal)-substituted LCO (LCPO or LCZO). The high-temperature (>400 °C) CO2 selectivity maximum is identical on all catalysts studied and is attributed, in general, to the bulk catalyst composition (i.e., La2O3 and Co) and the occurrence of rWGS reaction. The substituted LaCo1−x−yPdxZnyO3±δ catalysts exhibit promising stability as evident from LaCo0.85Pd0.075Zn0.075O3±δ at 245 °C after 80 h of time on stream which shows a relative activity loss of 35% and increased CO2 selectivity from 40 to 60%.
Acknowledgements
The authors are thankful to Staatssekretariat für Bildung und Forschung (SBF project no. C11.0034) for financial support. The COST Action – CM0904 is also greatly appreciated. The authors acknowledge the Swiss National Science Foundation for funding the Micromeritics ASAP 2020c instrument through R'Equip (project no. 206021 128741/1).Notes and references
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608 CrossRef CAS PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881 CrossRef CAS PubMed.
- M. Armbrüster, M. Behrens, K. Föttinger, M. Friedrich, É. Gaudry, S. K. Matam and H. R. Sharma, Catal. Rev.: Sci. Eng., 2013, 55, 289 Search PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881 CrossRef CAS PubMed.
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. Navarro Yerga and M. Behrens, J. Catal., 2012, 293, 27 CrossRef CAS.
- Q. Zhang and R. J. Farrauto, Appl. Catal., A, 2011, 395, 64 CrossRef CAS.
- N. Iwasa, S. Masuda, N. Ogawa and N. Takezawa, Appl. Catal., A, 1995, 125, 145 CrossRef CAS.
- N. Iwasa, S. Kudo, H. Takahashi, S. Masuda and N. Takezawa, Catal. Lett., 1993, 19, 211 CrossRef CAS.
- H. F. Oetjen, V. M. Schmidt, U. Stimming and F. Trila, J. Electrochem. Soc., 1996, 143, 3838 CrossRef CAS.
- M. Friedrich, D. Teschner, A. Knop-Gericke and M. Armbrüster, J. Catal., 2012, 285, 41 CrossRef CAS.
- K. Faungnawakij, R. Kikuchi and K. Eguchi, J. Power Sources, 2006, 161, 87 CrossRef CAS.
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43 CrossRef.
- B. Halevi, E. J. Peterson, A. Roy, A. DeLariva, E. Jeroro, F. Gao, Y. Wang, J. M. Vohs, B. Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl and A. K. Datye, J. Catal., 2012, 291, 44 CrossRef CAS.
- M. Friedrich, S. Penner, M. Heggen and M. Armbrüster, Angew. Chem., Int. Ed., 2013, 52, 4389 CrossRef CAS PubMed.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981 CrossRef.
- A. Weidenkaff, Adv. Eng. Mater., 2004, 6, 709 CrossRef CAS.
- R. Robert, D. Logvinovich, M. H. Aguirre, S. G. Ebbinghaus, L. Bocher, P. Tomeš and A. Weidenkaff, Acta Mater., 2010, 58, 680 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164 CrossRef CAS PubMed.
- M. S. G. Baythoun and F. R. Sale, J. Mater. Sci., 1982, 17, 2757 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- A. Lebail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23, 447 CrossRef CAS.
- J. Rodriguezcarvajal, Phys. B, 1993, 192, 55 CrossRef CAS.
- P. Scherrer, Nachr. Ges. Wiss. Gottingen, Math.-Phys. Kl., 1918, 2, 98 Search PubMed.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79 CrossRef CAS.
- S. Sartipi, A. A. Khodadadi and Y. Mortazavi, Appl. Catal., B, 2008, 83, 214 CrossRef CAS.
- G. Thornton, B. C. Tofield and A. W. Hewat, J. Solid State Chem., 1986, 61, 301 CrossRef CAS.
- N. M. L. N. P. Closset, R. H. E. van Doorn, H. Kruidhof and J. Boeijsma, Powder Diffr., 1996, 11, 31 CrossRef CAS.
- A. Eyssler, A. Winkler, O. Safonova, M. Nachtegaal, S. K. Matam, P. Hug, A. Weidenkaff and D. Ferri, Chem. Mater., 2012, 24, 1864 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- L. Huang, M. Bassir and S. Kaliaguine, Appl. Surf. Sci., 2005, 243, 360 CrossRef CAS.
- M. Santhosh Kumar, A. Eyssler, P. Hug, N. van Vegten, A. Baiker, A. Weidenkaff and D. Ferri, Appl. Catal., B, 2010, 94, 77 CrossRef CAS.
- Z. Gong, X. Yin and L. Hong, J. Electrochem. Soc., 2010, 157, E129 CrossRef CAS.
- S. Miyoshi, J. Hong, K. Yashiro, A. Kaimai, Y. Nigara, K. Kawamura, T. Kawada and J. Mizusaki, Solid State Ionics, 2003, 161, 209 CrossRef CAS.
- O. H. Hansteen, H. Fjellvag and B. C. Hauback, J. Mater. Chem., 1998, 8, 2081 RSC.
- M. Radovic, S. A. Speakman, L. F. Allard, E. A. Payzant, E. Lara-Curzio, W. M. Kriven, J. Lloyd, L. Fegely and N. Orlovskaya, J. Power Sources, 2008, 184, 77 CrossRef CAS.
- H. Bommer, Z. Anorg. Allg. Chem., 1939, 241, 273 CrossRef CAS.
- H. Mueller-Buschbaum and H. G. von Schnering, Z. Anorg. Allg. Chem., 1965, 340, 232 CrossRef CAS.
- H. F. McMurdie, M. C. Morris, E. H. Evans, B. Paretzkin, J. H. de Groot, C. R. Hubbard and S. J. Carmel, NBS Monogr. (U. S.), 1966, 4, 10 Search PubMed.
- J. P. Picard, G. Baud, J. P. Besse and R. Chevalier, J. Less-Common Met., 1980, 75, 99 CrossRef CAS.
- S. Liu, K. Takahashi and M. Ayabe, Catal. Today, 2003, 87, 247 CrossRef CAS.
- M. Santhosh Kumar, D. Chen, A. Holmen and J. Walmsley, Catal. Today, 2009, 142, 17 CrossRef CAS.
- S. K. Matam, E. V. Kondratenko, M. H. Aguirre, P. Hug, D. Rentsch, A. Winkler, A. Weidenkaff and D. Ferari, Appl. Catal., B, 2013, 129, 214 CrossRef CAS.
- J. Kuc, S. K. Matam, M. Neumann, S. Yoon, P. Thiel, M. Armbrüster and A. Weidenkaff, Catal. Today, 2015 DOI:10.1016/j.cattod.2015.01.002G.
- G. Xia, J. D. Holladay, R. A. Dagle, E. O. Jones and Y. Wang, Chem. Eng. Technol., 2005, 28, 515 CrossRef CAS.
- A. Karim, T. Conant and A. Datye, J. Catal., 2006, 243, 420 CrossRef CAS.
- H. Lorenz, M. Friedrich, M. Armbrüster, B. Klötzer and S. Penner, J. Catal., 2013, 297, 151 CrossRef CAS PubMed.
- C. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, N. Memmel, M. Hävecker, R. Blume, D. Teschner, T. Rocha, D. Zemlyanov, A. Knop-Gericke, R. Schlögl and B. Klötzer, Angew. Chem., Int. Ed., 2012, 51, 3002 CrossRef CAS PubMed.
- B. Predel, SpringerMaterials – The Landolt–Börnstein Database, http://www.springermaterials.com/docs/pdf/10542753_2462.html?queryterms=%22pd-zn%22 Search PubMed.
- A. Pang Tsai, S. Kameoka and Y. Ishii, J. Phys. Soc. Jpn., 2004, 73, 3270 CrossRef.
- H. Karzel, W. Potzel, C. Schafer, M. Steiner, J. Moser, W. Schiessl, M. Peter, G. M. Kalvius, D. W. Mitchell, S. B. Sulaiman, N. Sahoo and T. P. Das, Hyperfine Interact., 1992, 70, 1067 CrossRef CAS.
- S. Sasaki, K. Fujino and Y. Takeuchi, Proc. Jpn. Acad., 1979, 55, 43 CrossRef CAS.
- J. Kuc, Y. Zhang, R. Erni, S. Yoon, L. Karvonen, A. Weidenkaff and S. K. Matam, Phys. Status Solidi RRL, 2015, 9(5), 282 CrossRef CAS.
- M. M. Natile, F. Poletto, A. Galenda, A. Glisenti, T. Montini, L. D. Rogatis and P. Fornasiero, Chem. Mater., 2008, 20, 2314 CrossRef CAS.
- A. Glisenti, A. Galenda and M. M. Natile, Appl. Catal., B, 2013, 453, 102 CrossRef CAS.
- J. Kuc, A. Weidenkaff and S. K. Matam, Top. Catal., 2015, 58(14), 905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01410g |
| This journal is © The Royal Society of Chemistry 2016 |