
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd/C catalysts based on synthetic carbons with bi- and tri-modal pore-size distribution: applications in flow chemistry
X.
Fan
*a,
V.
Sans
b,
S. K.
Sharma
c,
P. K.
Plucinski
c,
V. A.
Zaikovskii
d,
K.
Wilson
e,
S. R.
Tennison
f,
A.
Kozynchenko
f and
A. A.
Lapkin
*g
aSchool of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, UK. E-mail: xiaolei.fan@manchester.ac.uk
bDepartment of Chemical and Environmental Engineering, University of Nottingham, Nottingham, UK
cDepartment of Chemical Engineering, University of Bath, Bath, UK
dBoreskov Institute of Catalysis, Novosibirsk, Russia
eEuropean Bioenergy Research Institute, School of Engineering and Applied Science, Aston University, Birmingham, UK
fMAST Carbon International Ltd, Basingstoke, UK
gDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, UK. E-mail: aal35@cam.ac.uk
First published on 19th November 2015
Abstract
Two new types of phenolic resin-derived synthetic carbons with bi-modal and tri-modal pore-size distributions were used as supports for Pd catalysts. The catalysts were tested in chemoselective hydrogenation and hydrodehalogenation reactions in a compact multichannel flow reactor. Bi-modal and tri-modal micro-mesoporous structures of the synthetic carbons were characterised by N2 adsorption. HR-TEM, PXRD and XPS analyses were performed for characterising the synthesised catalysts. N2 adsorption revealed that tri-modal synthetic carbon possesses a well-developed hierarchical mesoporous structure (with 6.5 nm and 42 nm pores), contributing to a larger mesopore volume than the bi-modal carbon (1.57 cm3 g−1versus 1.23 cm3 g−1). It was found that the tri-modal carbon promotes a better size distribution of Pd nanoparticles than the bi-modal carbon due to presence of hierarchical mesopore limitting the growth of Pd nanoparticles. For all the model reactions investigated, the Pd catalyst based on tri-modal synthetic carbon (Pd/triC) show high activity as well as high stability and reproducibility. The trend in reactivities of different functional groups over the Pd/triC catalyst follows a general order alkyne ≫ nitro > bromo ≫ aldehyde.
Introduction
Driven by the demand for cleaner, intensified, chemical processes structured reactors e.g. micro-, compact1–4 and monolith reactors5 offer a potential to replace the conventional reactor technologies, such as stirred tanks and trickle bed reactors. Structured catalytic reactors are particularly suited to smaller scale manufacturing of active pharmaceutical ingredients, speciality chemicals and intermediates. In comparison with conventional reactors one of the most important advantages of the structured reactors is the potential to integrate several functionalities in a single unit, i.e. heat transfer, mixing, sensing and even specific responsive/‘smart’ functionalities, for example a chemical valve membrane for controllable delivery of reactants.6 This may result in improvements in process safety, higher throughput, lower operating costs and most significantly, shorter development time of the new technologies.Integrating heat exchangers into the structured reactors is a common method of improving thermal efficiency of chemical processes, to avoid hot spots formation, thermal runaways and to minimise heat losses, which helps to improve the overall process efficiency.2,4,7–9 One example demonstrated in the early investigations is the continuous oxidation with molecular oxygen in a compact reactor-micro heat exchanger.2 Structured chemical reactor–heat exchangers also promote the development of new, otherwise impossible, chemical processes. For instance, coupling the endothermic steam reforming reaction with an exothermic catalytic combustion in a mm-scale structured monolith reactor.7,8
Most of the studied structured reactors use reaction channels with hydraulic diameters in the range of 0.05 to 5 mm,1,2,7–9 which promote the intensification of heat and mass transfer and safe plant operation. Intensified heat and mass transfer are important for practical heterogeneous catalytic processes. The absence of transport limitations is critical for obtaining reliable quantitative kinetics10 and is essential for increasing yield and selectivity. In conventional reactors, however, especially in large-scale batch reactors, reaction conditions free from heat and mass transfer limitations are hard to achieve.
Small intensive reactors offer the possibility of scaling by numbering up to attain high overall process throughput.1,11 However, this approach is frequently met with the main drawback of compact and micro-reactors, i.e. clogging. Channel-clogging induced by e.g. precipitate formation, impure feeds or in processing of solids is a well-reported problem of small-channel reactors.11
For heterogeneous catalytic microreaction systems another potential limitation is the immobilisation of catalysts in the reaction channels. The most popular method is coating catalysts onto reactor walls.12–14 This method, however, produces single-use catalytic reactors, due to difficult replacement of catalytic coating in the case of catalyst deactivation. Furthermore, catalytic wall coating would have to be optimised for each particular catalyst/reaction, offering little operational flexibility. Recently, microreactors with exchangeable catalytic plates were designed and tested for heterogeneous reactions (e.g. selective hydrogenation of 3-methyl-1-pentyn-3-ol over palladium).15,16 Such design offers more flexibility than conventional designs with fixed catalytic bed. However, special treatment to the exchangeable catalytic plate is still needed, i.e. growing carbon nanofibres on the plate, to increase the available surface area.
An alternative approach is to use catalytic packing in micro-reaction channels,17 which features an easier catalyst handling (unloading and repacking) and less chance of channel clogging. Catalysts used in the mm-scale packed-beds can be developed by well-understood conventional methods. In the early investigations, the use of a mm-scale packed-bed reactor for several important reaction classes has been demonstrated, i.e. oxidation,2,3,18 hydrogenation,19 C–C coupling and multi-step synthesis.20 Synthetic mesoporous carbons were used as supports and were found to be particularly suitable for applications in the mm-scale packed-bed reactors.
There are different requirements for heterogeneous catalysts for microreactors though, due to a markedly different hydrodynamics on the macro and micro scale, the need to operate in the kinetic reaction regime, and the need for a simple catalyst loading procedure. The particle sizes in micro-packed-beds would necessarily be in the order of tens to hundreds of micrometres, thus potentially resulting in significant pressure drops across reactors, especially in the liquid- and multi-phase processes of interest in this study. Therefore, there is a requirement towards catalyst particle shape that would minimise pressure drop. It was shown earlier, that microspherical catalytic particles produce lower pressure drop in a three-phase reaction and result in a faster establishment of a steady-state regime following initial catalyst bed re-packing.18 In addition to the lower pressure drop associated with spherical catalyst, spherical catalyst is also very easy to handle, i.e. easy filling and emptying of the catalytic channels for a pilot plant reactor.
Due to intensified mass and heat transfer the reaction rates in microreactors are expected to be higher than in conventional batch reactors. Therefore, catalyst structure that would lead to the kinetic reaction regime, without internal mass transfer limitations is even more critical. This requires optimisation of a catalyst internal structure in terms of site activity, stability, number and accessibility.
Hierarchical porous materials with well-developed mesoporous properties received much attention in recent years, especially in the liquid-phase systems, due to the reduced mass transfer limitation in comparison with microporous solid hosts.21 Apart from the enhanced diffusion of reactants and products to and from the catalytic sites, mesoporous hosts also allow the reaction with larger molecules21–23 and promote preparation of heterogeneous catalysts with controlled particle sizes.
Recently, MAST Carbon Ltd developed novel phenolic-resin-derived activated carbons with different pore structures, i.e. microporous (mean pore diameter ≈ 0.65 nm)24 or hierarchical bi-modal micro-mesoporous carbon materials (mean pore diameter in a range of 1 nm to 60 nm).25 These carbons are based on phenolic resin technology and manufactured using a patented molecular templating process. The carbon formed in the process is known as glassy or vitreous carbon. The process also allows the production of complex shapes from beads to honeycomb monolith without any loss of performance.25 In this work, Pd catalysts were prepared by a conventional method based on two carbons possessing bi- and tri-modal pore size distributions. Performance of the flow catalytic system in terms of space-time yield conversion and selectivity for continuous chemoselective hydrogenation and hydrodehalogenation of various substrates was evaluated. This is the first systematic analysis of catalysts made from bi- and tri-modal pore size distribution supports specifically for micro-reactor and flow chemistry applications.
Experimental
Phenolic-resin-derived synthetic carbon and reagents
The phenolic-resin-derived synthetic carbons with tailored pore structures, i.e. bi-modal micro-mesoporous and tri-modal micro-mesoporous structures were produced in the form of beads and provided by MAST Carbon International (UK). The synthetic method of such synthetic carbons is described elsewhere.25 Besides micro-spherical carbon particles (with different particle diameters), debris and aggregates of very small particles are found altogether. Therefore, preliminary treatment of synthetic carbon, i.e. sonication in methanol to break up aggregates and sieving, was performed for producing carbon support samples with a uniform particle diameter. In this study, particle fraction with diameters 110–150 μm was used. All chemicals used in this study were obtained from Sigma-Aldrich and Fisher Chemicals, and used without further purification.Preparation of Pd/C catalysts
An anaerobic incipient wetness method was used to prepare approximately 3% wt Pd/C catalysts utilising standard Schlenk technique.26 Palladium(II) acetylacetonate, Pd(C5H7O2)2, dissolved in dried, degassed tetrahydrofuran (THF, 1.5 mL, corresponds to the pore volume possessed by 1 g synthetic carbon) was used as the precursor to produce Cl-free catalysts. The precursor solution was added into carbon support under N2 and the incipient wetting impregnation was carried out at room temperature for 24 h. The catalysts were evacuated overnight at 300 K, then at 323 K for 2 h to remove any THF, and transferred to a desiccator. The as-prepared catalyst was packed in reaction channels of the structured reactor and reduced in situ with hydrogen flow at 333 K. The denotations of Pd/biC and Pd/triC were used for Pd-supported on the bi-modal micro-mesoporous synthetic carbon and Pd-supported on the tri-modal micro-mesoporous synthetic carbon catalysts, respectively.Materials characterisation
High-resolution transmission electron microscopy (HR-TEM) images were obtained using a JEOL JEM-2010 system (200 kV accelerating voltage, with the resolution limit of 0.14 nm). Low temperature nitrogen adsorption experiments were performed using a Micromeritics ASAP-2010 volumetric system with N2 as probing gas. Powder X-ray diffraction (PXRD) analysis was performed by using a Philips 4 kW X-ray generator (PW1730) with a Cu Kα X-ray source (λ = 0.154 nm) and the diffractometre goniometre (PW1820) controlled via Philips (PW1877 PC-APD) software. All the samples were scanned over the 2θ range of 30–55° at a rate of 0.01° per step with a scan time of 1.25 s per step. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos Axis HSi instrument equipped with Mg Kα X-ray source and charge neutraliser. High-resolution spectra were recorded at normal emission using analyser pass energy of 40 eV and an X-ray power of 225 W. Energy referencing was employed using the valence band and adventitious carbon at 285 eV. Spectra were Shirley background-subtracted across the energy region and fitted using CasaXPS Version 2.3.15. Inductively coupled plasma optical emission spectroscopy (ICP-OES) was performed using a Thermo Scientific iCAP 6300 Duo ICP-OES instrument. Samples were accurately weighed and then acid digested in a mixture of nitric and perchloric acids. After digestion, hydrochloric acid was added and then it was reheated. The solution was then transferred to a volumetric flask and made up with deionised water for ICP analysis to measure the concentration of Pd present.Structured compact reactor and continuous catalytic reactions
The structured multichannel reactor was assembled by diffusion bonding of thin shims (316 stainless steel) incorporating etched through channels within them. Assembly of shims created imbedded static mixers, micro-heat exchangers, catalyst filters and reaction channels. Details of the design of the reactor and of the auxiliary continuous catalytic system used are given elsewhere.2,3,18,19,27 Gas chromatography (GC, Varian CP-3900, CP Sil 5 CB column) was used to analyse reaction products. Catalysts (ca. 0.5 g) were packed in 3 × 3 × 100 mm reaction channels and the loaded catalysts were repetitively used for different reactions under different conditions and sufficient solvent washing was performed after each reaction.Hydrogenation of various substrates in the compact reactor was carried out to evaluate the catalytic performance of the Pd-supported on synthetic carbon catalysts under continuous-flow conditions, the space-time yields (STY in kg L−1 h−1) are calculated based on eqn (1).
| STY = (cinitial·Y·M)/τ | (1) |
| τ = V/(∑F) | (2) |
Results and discussion
Characterisation of synthetic carbons and Pd/C catalysts
The isotherms of low temperature N2 adsorption on the bi-modal and tri-modal carbons are shown in Fig. 1a. For both synthetic carbons, there is a clear two-step increase in the amount of nitrogen adsorbed over the whole relative pressure range (type II adsorption isotherms), which indicates the presence of micro and mesoporous structure. The sharp linear increase in the amount adsorbed of nitrogen and the round knee in the isotherm at low relative pressures (p/p0 < 0.05) represent the presence of micropores. The mesoporous structure of the bi-modal carbon was evidenced by the consecutive increase in the amount of adsorbed N2 at medium and high relative pressures (0.05 < p/p0 < 0.42), and the hysteresis loop in the relative pressure range of 0.40–0.90. However, the isotherm of the tri-modal carbon exhibits a steeper increase at a relative pressure up to p/p0 = 0.8, indicating the presence of another type of mesopores within the tri-modal samples. This hypothesis is confirmed by analysing the resulting pore size distributions, as shown in Fig. 1b. As one can see, in comparison with the bi-modal carbon, the pore size distribution of the tri-modal carbon sample clearly shows the presence of two types of mesopores with diameters of ca. 6.5 nm and 42 nm and micropores with diameters of ca. 0.8 nm. The pore size distribution of the bi-modal carbon centres at approximately 1 nm and 42 nm, accounting for micropores and mesopores. Such mesoporous structures are advantageous for applications that require rapid mass transport (diffusion) or pore accessibility to active sites. The calculated Brunauer–Emmett–Teller (BET) surface areas of the carbon were 1326 m2 g−1 for the bi-modal sample and 970 m2 g−1 for tri-modal. The hierarchical mesoporous structure of tri-modal carbon contributes to the bigger mesoporous volume (ca. 1.57 cm3 g−1) than the one of the bi-modal carbon (ca. 1.23 cm3 g−1). Mesopores were found to represent the majority part of the pore structures of both carbons. The mesoporous volume for both samples is one magnitude higher than the micropore volume (ca. 0.1 cm3 g−1) obtained by t-plot method using the slit-shaped pore model. Both the mesoporous and microporous volume of the phenolic-resin-derived synthetic carbons were found decreased after Pd impregnation, i.e. ca. 20% for mesopore volumes and ca. 40% for micropore, suggesting that Pd may be deposited (i) both in micro and in mesopores or (ii) in mesopores with the blockage of micropores.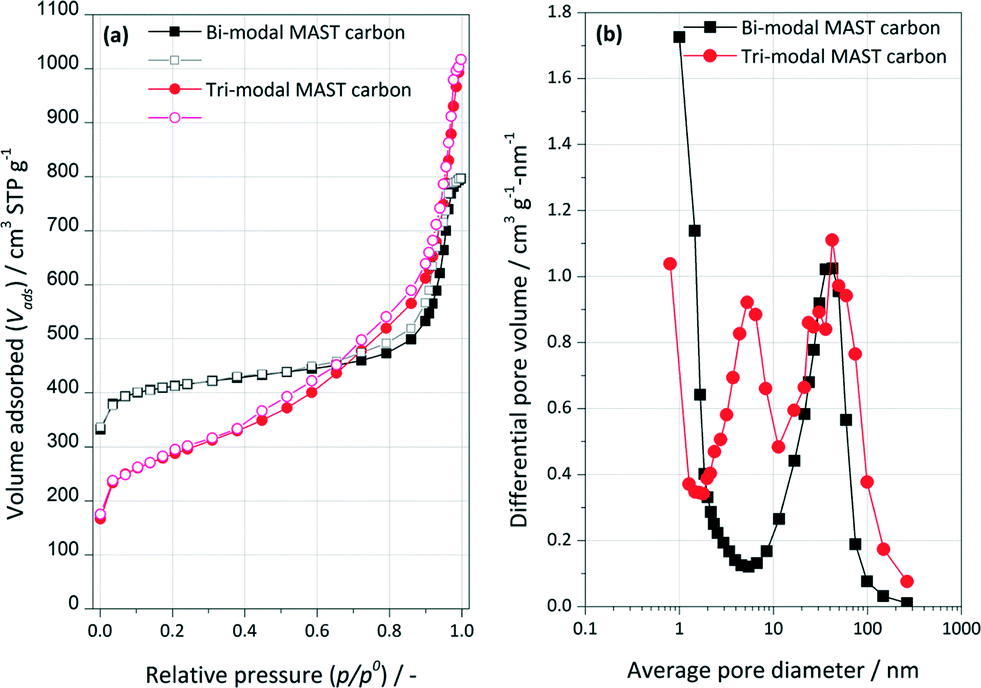 | ||
| Fig. 1 (a) Nitrogen adsorption (solid scatters) and desorption (open scatters) isotherms measured at 77 K and (b) pore size distributions curves for bi-modal and tri-modal synthetic carbons. | ||
The structure and distribution of Pd nanoparticles on the catalysts were analysed by HR-TEM microscopy. Carbon microspheres were mechanically ground, dispersed in ethanol and sonicated; a sample droplet was placed onto the TEM sample grid and evaporated. In general, it appears that the tri-modal carbon promoted a better size distribution of Pd nanoparticles. Fig. 2a and b show the HR-TEM microscopic images for the fresh catalyst of Pd/triC. The homogeneity of Pd in size (ca. 5 nm) and even Pd distribution within the pores of the carrier were observed. For the catalysts based on the bi-modal carbon, on the other hand, Pd species of three different sizes are found. A large number of dispersed particles of Pd with particle size of ca. 1 nm were found for the Pd/biC catalysts, indicating the possibility of Pd deposition within micropores of bi-modal carbons. HR-TEM analysis (Fig. 2c and d) also showed a size variation of Pd nanoparticles with large aggregated particles with ca. 30 nm diameters. Therefore, one can see that two-tiered mesopores in the tri-modal synthetic carbon has a beneficial effect on promoting uniform size distribution of metal nanoparticles i.e. possibly by allowing the diffusion of Pd species into the hierarchical mesoporous structure and restricting the growth of Pd nanoparticles by the presence of 6.5 nm mesopores. While for the bi-modal synthetic carbon, the aggregation of Pd species in its mono-sized mesopores forming large Pd clusters cannot be avoided.
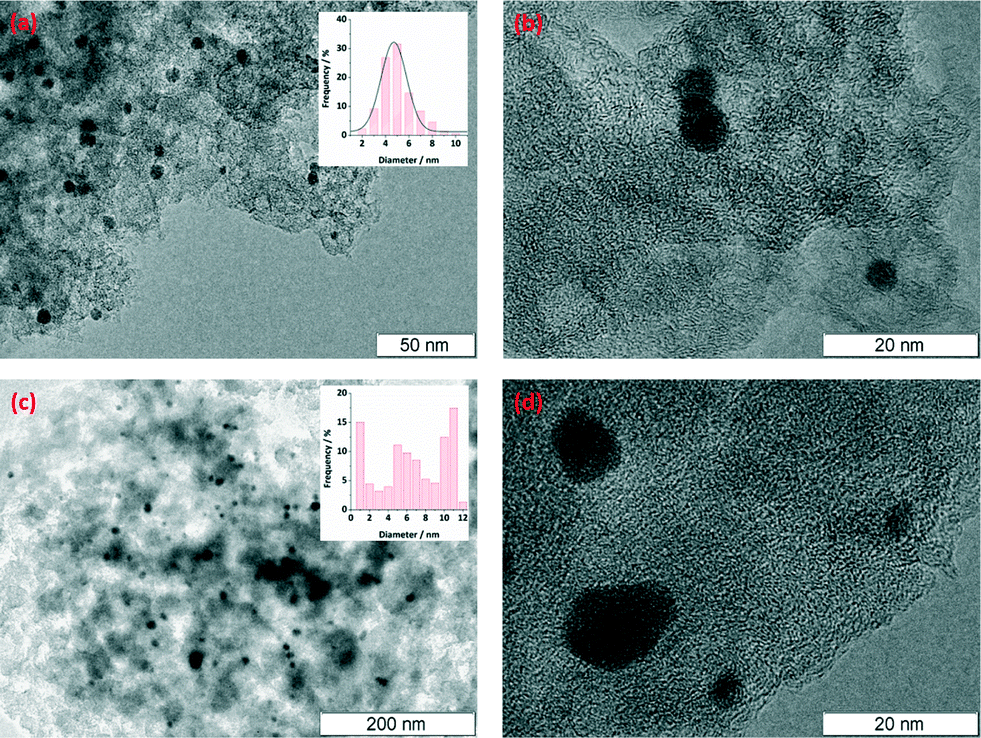 | ||
| Fig. 2 HR-TEM microscopic images of Pd nanoparticles on (a–b) Pd/triC fresh catalysts; (c–d) Pd/biC fresh catalysts. | ||
Fig. 3a shows the PXRD patterns of the reduced Pd catalysts and synthetic carbons. The synthetic carbon was revealed with a peak at the 2θ value of ca. 43° corresponding to the (100) reflection of the graphite layers. The peaks at 2θ = 40.1° and 46.7° are the supported Pd phases which are related to the (111) and (200) reflections of crystalline Pd(0). The average Pd particle diameters for each catalyst were estimated using integral breadth analysis and Scherrer formula28,29 on the Pd (111) diffraction peak. For the Pd/triC catalysts, the estimated average particle size by PXRD is 5.5 nm that corresponds well with the result based on HR-TEM analysis. PXRD analysis yielded an average Pd particle diameter of 9.8 nm for the Pd/biC catalyst.
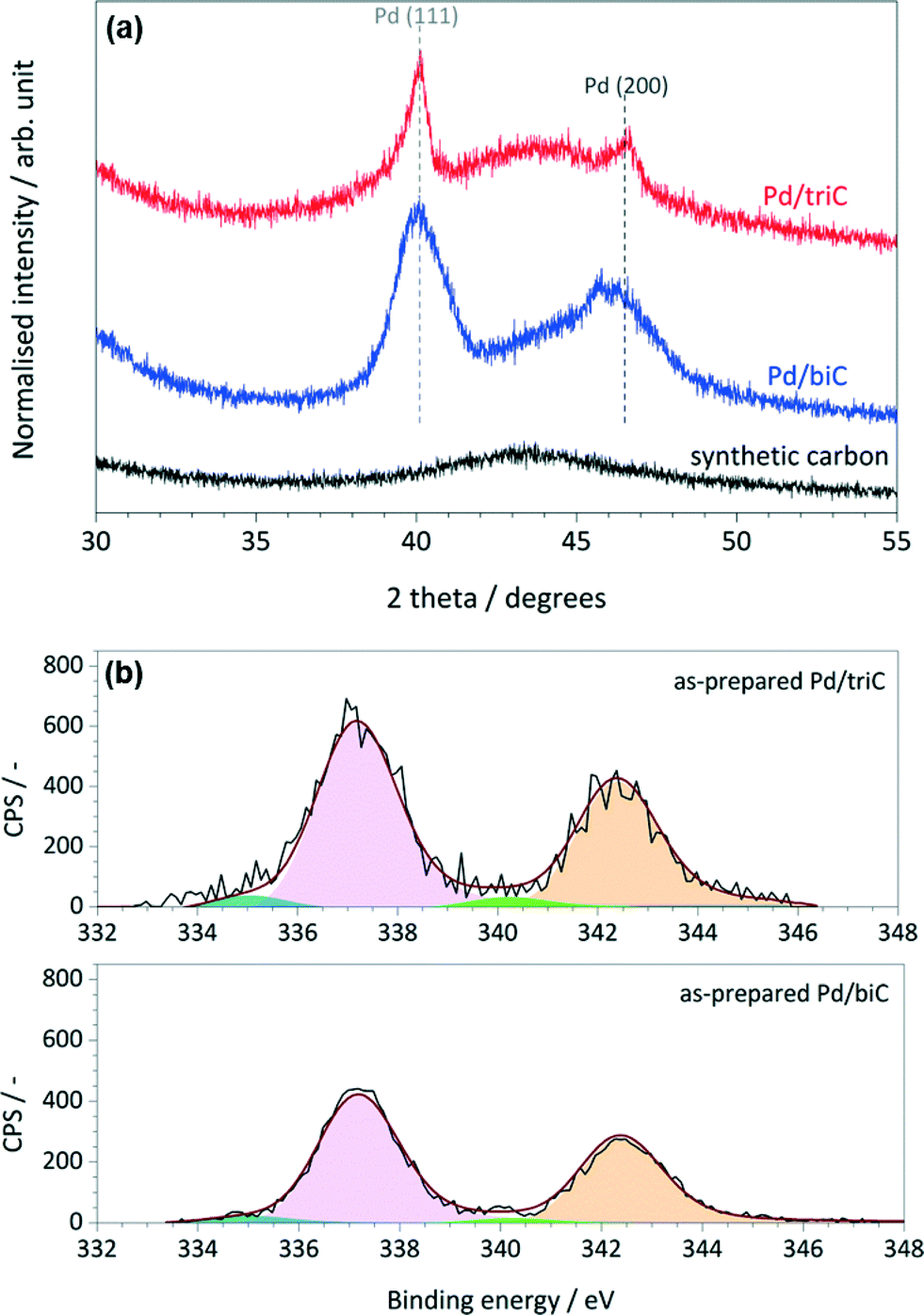 | ||
| Fig. 3 (a) XRD patterns of the synthetic carbon, Pd/triC and Pd/biC catalysts (reduced); (b) Pd 3d core level XPS spectra of the as-prepared Pd/triC and Pd/biC catalysts. | ||
Fig. 3b shows the Pd 3d XPS spectra along with the peak fittings (the normalised residual trace is about 10−2) for the as-prepared catalysts based on the synthetic carbons. The narrow feature of the peaks indicates that the accessible Pd species in the as-prepared samples possess mainly one oxidation state. For both catalysts, the dominating Pd 3d5/2 peaks with the binding energy (BE) of 337.1 eV were identified suggesting the presence of Pd species as their native oxidation state (Pd2+).30,31 For the as-prepared Pd/C catalysts, this is reasonable because the Pd catalyst (from palladium(II) acetylacetonate precursor) was only reduced in situ prior to the continuous-flow reactions. Pd 3d5/2 peaks corresponding to Pd0 species (BE = 335.4 eV) were also identified.
XPS quantitative analysis of the as-prepared Pd/C catalysts was performed to show the support effect on the dispersion of Pd species. The elemental composition for the surfaces of samples showed that the accessible Pd species in the Pd/triC catalyst was about 23% higher than the ones in the Pd/biC, i.e. 2.83 versus 2.17 wt%. This result suggests that the well-developed mesoporous structure of the tri-modal carbon promoted a better dispersion of Pd species in its hierarchical pore matrix than the bi-modal carbon conforming to the N2 adsorption analysis on the pore volumes of two synthetic carbons.
Results of continuous-flow catalytic reactions
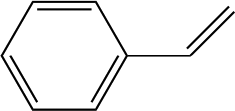
The space-time yields (STY in kg L−1 h−1) of hydrogenation reactions of various single-functional substrates in the compact reactor under continuous-flow conditions are summarised in Table 1. Hydrogen and substrate solutions were introduced using mass flow controller and HPLC pump and all results were collected under steady-state conditions. High efficiencies of hydrogenation of non-hindered triple bond were measured, namely quantitative yield of ethenylbenzene was achieved with ca. 65% conversion on all Pd-supported on synthetic carbon catalysts in a single pass with residence times (τ) of ca. 4 s for phenylacetylene hydrogenation. Such heterogeneous catalytic system based on a structured compact reactor can provide an intensive reacting environment with relatively high values of STY, i.e. 29.5 kg L−1 h−1 for Pd/triC, comparable to the state-of-the-art materials under continuous-flow configurations32–35 (e.g. about 50 kg L−1 h−1 reported by Vilé et al.)32 and outperforming the performance of conventional heterogeneous catalytic processes for fine chemical synthesis with the STY in a range of 1–10 kg L−1 h−1.36 Reactions were performed in an slight excess of hydrogen, e.g. mole flow-rate of substrate was 2.4 × 10−4 mol min−1 whereas mole flow-rate of hydrogen was ca. 4.4 × 10−4 mol min−1 under the conditions used (see note ‘a’ in Table 2). Due to consumption of hydrogen in the reactions very little gas was detected in the exit stream upon sampling. Ethylbenzene, as the product of consecutive hydrogenation of ethenylbenzene, was also detected in the hydrogenation of phenylacetylene (selectivity: 12% for Pd/biC, 8% for Pd/triC). Considering the high loading of palladium (3 wt%) and the excess of hydrogen, hydrides are expected to be formed accounting for the over-hydrogenation of the carbon–carbon triple bond.33| Entry | Substrate | Catalyst | Product | Space-time yielde [kg L−1 h−1] | Yieldf [%] |
|---|---|---|---|---|---|
| a Samples for GC analyses was taken after ten minute of starting the reaction. b Reaction conditions: Cinitial = 0.5 mol L−1; T = 333 K; p = 1 barg; Fliquid = 0.5 mL min−1; Fgas = 10 mL(STP) min−1; ethanol as the solvent. c Reaction conditions: Cinitial = 0.5 mol L−1; T = 318 K (for benzaldehyde hydrogenation); T = 328 K (for acetophenone hydrogenation); p = 9 barg; Fliquid = 0.5 mL min−1; Fgas = 8 mL(STP) min−1; ethanol as the solvent. d Reaction conditions: Cinitial = 0.5 mol L−1; T = 333 K; p = 9 barg; Fliquid = 0.5 mL min−1; Fgas = 20 mL (STP) min−1; ethanol as the solvent. e Conversion and selectivity were determined by GC analyses. For all GC analyses, mass balance error ≤1.5%. f SD for STY data is ±0.5 kg L−1 h−1. | |||||
| 1b |
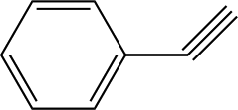
|
Pd/biC |
|
24.3 | 56 |
| Pd/triC | 29.5 | 60 | |||
| 2c |
|
Pd/biC |
|
22.3 | 55 |
| Pd/triC | 23.9 | 59 | |||
| 3c |
|
Pd/biC |
|
22.9 | 50 |
| Pd/triC | 25.7 | 56 | |||
| 4d |
|
Pd/biC |
|
81.3 | 97 |
| Pd/triC | 83.8 | 100 | |||
| Entry | Substrate | Conversion [%] | Product | Selectivity [%] |
|---|---|---|---|---|
| a Samples for off-line GC analyses was collected after ten minute of starting the reaction (steady-state operation). For all GC analyses, mass balance ≥98.5%. b Reaction conditions: Cinitial = 0.2 mol L−1; T = 333 K (for bromobenzene hydrodehalogenation); T = 383 K (for chlorobenzene hydrogenation); p = 1 barg; Fliquid = 0.5 mL min−1; Fgas = 6 mL(STP) min−1; triethylamine as additive; tetrahydrofuran as the solvent. c Reaction conditions: Cinitial = 0.5 mol L−1; T = 333 K; p = 1 barg; Fliquid = 0.5 mL min−1; Fgas = 14 mL(STP) min−1; tetrahydrofuran as the solvent. d Reaction conditions: Cinitial = 0.5 mol L−1; T = 333 K; p = 1 barg; Fliquid = 0.5 mL min−1; Fgas = 14 mL(STP) min−1; triethylamine as additive; tetrahydrofuran as the solvent. | ||||
| 1b |
|
43 |
|
100 |
| 2b |
|
26 | 100 | |
| 3c |
|
27 |
|
79 |
| 4d |
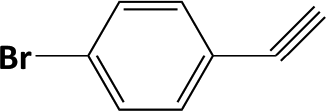
|
24 |
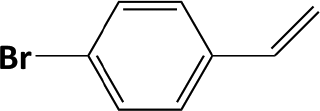
|
86 |
| 5d |
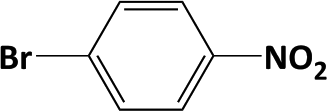
|
39 |
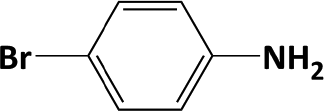
|
51 |
| 6d |
|
24 |
|
92 |
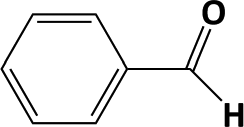
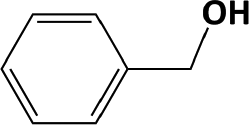
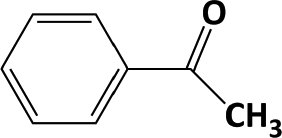
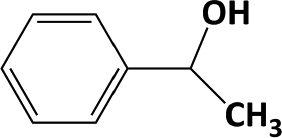
In Table 1, entries 2 and 3 list the results of hydrogenation of carbonyl groups (aldehyde and ketone) catalysed by the Pd/biC and Pd/triC catalysts. High selectivities to primary and secondary alcohols (>96%) with fairly good conversions (>55%) were achieved by using the combination of continuous-flow reactor and Pd on synthetic carbon catalysts. No over-hydrogenated products or hydrogenolysis products were detected (by GC) for the two model reactions under the current conditions. The possible reasons for the observed high selectivity can be attributed to the advantages of the multifunctional compact reactor: (i) good mixing, providing most likely plug flow regime, as was shown for a similar packed-bed system earlier,18 (ii) good control of isothermicity, and (iii) short residence time (ca. 5 s). From entry 2 and 3 in Table 1, one can see that, in terms of the yield of the target products, catalytic hydrogenation of aldehyde over all Pd/C candidates outperformed acetophenone hydrogenation by ca. 8%. This is because in general the reactivity towards nucleophiles of aldehydes is known to be higher than that of ketones, i.e. the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond energy of a carbonyl group: 170 kcal per mole for H2CO and 180 kcal per mole for R2CO. Furthermore, for acetophenone, the size of the phenyl group attached to the CO may also lower its reactivity (larger groups sterically hinder the approach of nucleophiles). However, for the performance of the compact reactor in terms of STY, the production of 1-phenylethanol is better than the one of benzyl alcohol due to the difference in molar masses.
O bond energy of a carbonyl group: 170 kcal per mole for H2CO and 180 kcal per mole for R2CO. Furthermore, for acetophenone, the size of the phenyl group attached to the CO may also lower its reactivity (larger groups sterically hinder the approach of nucleophiles). However, for the performance of the compact reactor in terms of STY, the production of 1-phenylethanol is better than the one of benzyl alcohol due to the difference in molar masses.
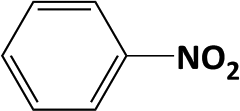
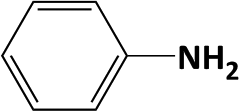
Hydrogenation of nitrobenzene to aniline is known as a highly exothermic reaction with a reaction enthalpy of 545 kJ mol−1.14 Good temperature control is essential to produce aniline and avoid generating intermediates which are formed in several parallel and consecutive reactions as suggested by Yeong et al.14 and Höller et al.37 All tested catalysts showed high selectivity towards the final amine product with excess supply of hydrogen at 2 s residence time (eqn (2)). The integrated micro heat exchangers showed high efficiency for removing the heat release by the reaction. The expected adiabatic temperature rise (ΔTad) for the conditions used (0.5 mol L−1 concentration, 100% conversion, 0.5 mL min−1 liquid flow-rate, 2 s residence time) was ca. 12 K (estimated by eqn (3)), whereas a temperature rise of <2 K was measured along the reaction channel, thus proving that isothermal condition was ensured during the reaction. In comparison with another study of this reaction using a microstructured falling film reactor (0.1 mol L−1 initial concentration, 333 K, 1 barg, 0.5 mL min−1 liquid flow-rate, 0.52 mL total reactor volume),14 a significant increase in aniline yield (max. ca. 70% vs. 98%) and space-time yield (0.4 kg h−1 L−1vs. <81 kg h−1 L−1) was achieved in the case of the packed-bed compact reactor (0.5 mol L−1 initial concentration, 333 K, 9 barg, 0.5 mL min−1 liquid flow-rate, 0.68 mL single channel volume). In the present study, the production capacity of a single channel (3 × 3 × 10 mm) in the compact reactor (for nitro benzene hydrogenation) was estimated as 1.62 kg per day and it is limited by the capacity of the mass flow controller used in current study, i.e. 20 mL(STP) min−1 maximum flow-rate (theoretical hydrogen consumption for reducing 0.5 mol L−1 nitrobenzene under conditions used is ca. 18 mL(STP) min−1).
| ΔTad = Q/(∑m·cP) = (ΔHR·cinitial·X·F·τ)/(∑m·cP | (3) |
For hydrogenation reactions, the catalytic results in terms of STY and yield achieved by Pd/triC are better than the ones of Pd/biC. This can be attributed to a better dispersion of Pd nanoparticles within the hierarchical mesoporous structures of the tri-modal synthetic carbon as evidenced by the bulk characterisation of catalysts. On the other hand, the poorer performance of the Pd/biC catalyst may be caused by the broad size distribution of Pd nanoparticles and observed formation of large Pd aggregates, leading to the leaching of Pd species during multi-phase catalytic reactions. The Pd contents in the spent samples (used for more than 10 hours under different reaction conditions) were determined with the ICP analysis. The amounts of Pd contained in the spent Pd/triC and Pd/biC were 2.7 and 2.2 wt%, respectively, whereas 3 wt% Pd in the fresh catalyst.
Pd nanoparticle morphology of the spent Pd/C catalysts was also examined by HR-TEM to check the effect of catalytic reactions on Pd nanoparticles within the porous structures of the synthetic carbons. As shown in Fig. 4a and b, for the Pd/triC catalyst, the catalytic reactions promoted broadening of Pd nanoparticles particle size distribution, resulting in a larger number of Pd particles ranging in size from 2 to 8 nm with majority of Pd nanoparticles still having the size of 5 nm. The same phenomenon was also noticed for Pd/biC catalysts (Fig. 4c and d), but with even broader size distribution, i.e. 1–15 nm. The post-reaction HR-TEM examination of Pd catalysts indicates that Pd species may not be stationary during liquid-phase catalytic hydrogenation reactions. By further comparing the spent Pd/biC with the spent Pd/triC, one can see that the packing of Pd nanoparticles in the spent Pd/biC is sparser and less uniform than that in the spent Pd/triC, confirming the structural effect of support on metal distribution.
 | ||
| Fig. 4 Post-reaction HR-TEM microscopic images of Pd nanoparticles on phenolic-resin-derived carbons. (a–b) Spent Pd/triC catalysts; (c–d) spent catalyst Pd/biC catalysts. | ||
The XPS survey spectra obtained from the spent catalysts are shown in Fig. 5. Comparing the XPS results of the as-prepared catalysts (Fig. 3b), the spectra of the spent samples exhibit an asymmetric broadening toward lower BE values. A distinct shift of Pd 3d5/2 peak towards lower BE (337.1 eV to 335.4 eV) was observed for both catalysts indicating the growth of the metallic Pd dispersed phase from the deposited Pd precursors on the synthetic carbons. Therefore, during the hydrogenation processes, part of the Pd species remained as reduced resulting Pd phases with various oxidation states during the reactions. The coexistence of metallic Pd and PdxOy/Pd0 interfaces are the possible active sites for the hydrogenation reactions.30 XPS analysis on the spent Pd/C catalysts also showed that the accessible Pd species on carbon surface were reduced, i.e. by ca. 8% for Pd/triC and ca. 11% for Pd/biC.
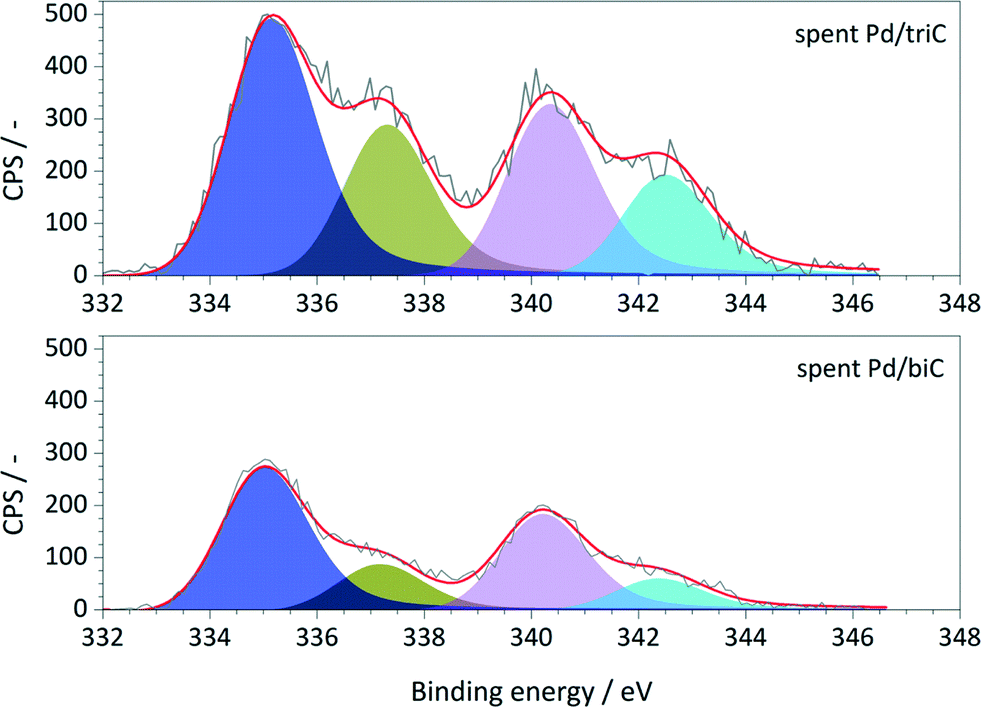 | ||
| Fig. 5 Pd 3d core level XPS spectra of the spent Pd/triC and Pd/biC catalysts. | ||
Since Pd species (e.g. leached Pd atoms from immobilised Pd nanoparticles) are believed to be transportable during catalytic reactions, an assumption can be made that the leaching of Pd species during reaction may be mainly attributed to the wash away of mobilised Pd species on carbon external surface and in large mesopores (42 nm). For the Pd nanoparticles within the well-developed secondary mesopores, despite the presence of migration most of Pd species can still be trapped within, and keep playing their active roles for catalytic reactions. Such solubilisation/redeposition of metal species from/on carbon materials were also noticed in our previous study of Heck/hydrogenation reactions within this compact reactor.20 Therefore, further investigation on the migration of metallic species within porous supports during catalytic reactions is necessary to fully understand the phenomena, hence for better design of metal nanoparticles supported catalysts.
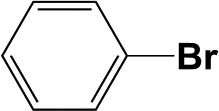
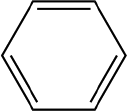
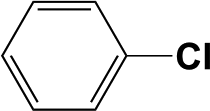
The Pd/triC catalyst was further tested with the hydrodehalogenation of aryl halides, see entries 1 and 2 in Table 2. For the liquid-phase hydrodehalogenation of bromobenzene, a 43% conversion was measured at 1 barg with 100% selectivity to benzene. For the less reactive chlorobenzene, a 26% yield of benzene was obtained for the Pd/triC catalyst at 333 K and 1 barg. Hydrodehalogenation reactions usually are carried out in a batch reactor at high temperatures (>500 K) and pressures (1 MPa)38 and may require hours of reaction time.39 Therefore, advantages of employing continuous mm-scale packed-beds are obvious from current study, i.e. simple operation, reasonable yield with short residence time (ca. 6 s) and high productivities with mild conditions.
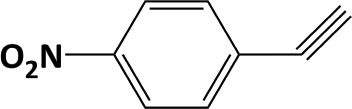
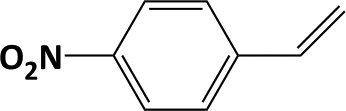
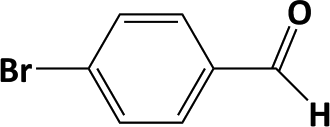
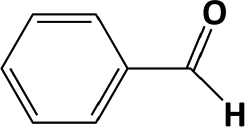
In developing practical hydrogenation processes for fine chemical and pharmaceutical industries one of the important questions is the chemoselectivity of a developed catalyst when dealing compound with multiple functional groups. Therefore, chemoselective hydrogenation of various multifunctional substrates over the Pd/triC catalyst was performed to assess its reactivity trend (entries 3–6 in Table 2) towards alkyne, halogen, nitro and aldehyde group. In the selective hydrogenation of a substrate containing terminal alkyne and nitro groups (i.e. 1-ethynyl-4-nitrobenzene, entry 3 of Table 2), the Pd/triC catalyst is more selective to the alkyne group than the nitro group giving 79% to 1-ethenyl-4-nitrobenzene as the hydrogenated product on a conversion of 27%. High selectivity to the alkyne group was also achieved by the Pd/triC catalyst in the selective hydrogenation of a halogenoarene with the terminal alkyne (i.e. 1-bromo-4-ethynylbenzene, entry 4 of Table 2). The catalytic activity of Pd/triC to nitro and bromo group was found comparable (entry 5 of Table 2). However, a slightly better selectivity to the nitro group than the one to bromo group was measured in the hydrogenation of 1-bromo-4-nitrobenzene, i.e. hydrogenated product of 4-bromoaniline 51% versus nitrobenzene 43%. When a substrate with bromo and aldehyde groups was used (e.g. 4-bromobenzaldehyde, entry 6 of Table 2), the Pd/triC catalyst has a tendency to cleave the bromo group from the benzene ring leading to a 92% selectivity to the hydrodehalogenated product of benzaldehyde.
In summary, the trend in the reactivity of different functional groups over the Pd/triC catalyst follows a general order alkyne ≫ nitro > bromo ≫ aldehyde. Currently, the nature of the metal on synthetic carbons and the type of the exposed crystal face area being carefully investigated in order to understand the intrinsic activity of the developed Pd-supported on synthetic carbon catalysts in chemoselective hydrogenation reactions.
Catalyst longevity and stability were studied in extended continuous runs for the Pd/triC catalyst. Results are shown in Fig. 6. For the Pd/triC catalysed hydrogenation reactions, the product yield stabilised at between 5–10 min after the start-up of the system and remained at that level for almost 5 h of continuous run. The Pd/triC catalyst used in the long experiments was not fresh catalyst and has been used several times (up to 50 h of cumulative reaction time) in previous experiments for reducing different substrates and only solvent washing was employed after each reaction to regenerate the catalyst bed. Furthermore, since the catalysts were used for over 50 h of reaction in previous tests, any activity loss of Pd/triC would have been apparent. Such catalyst longevity demonstrated in this work may have implications for industrial processes where catalyst stability is often of paramount importance.
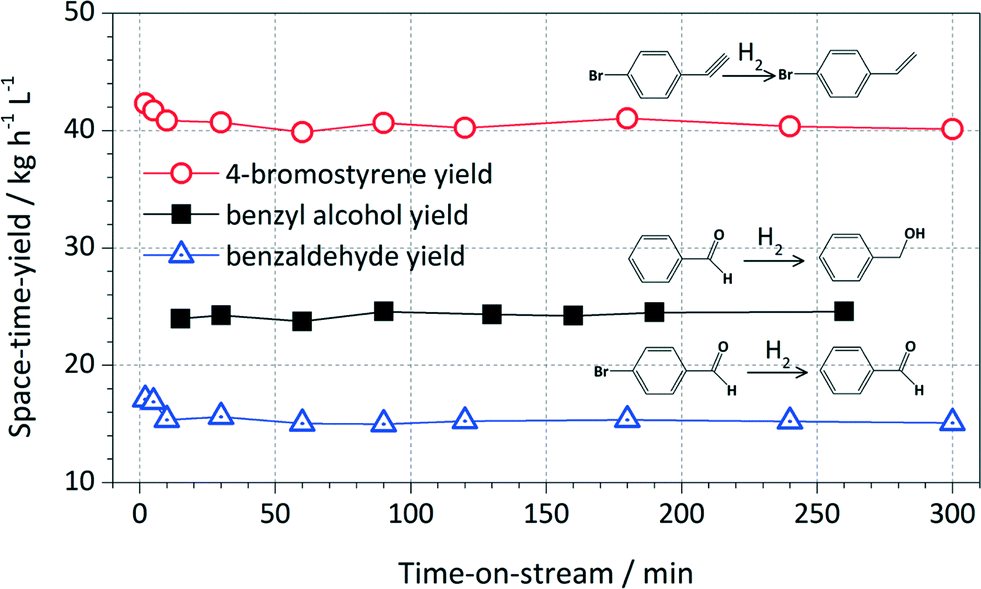 | ||
| Fig. 6 Yields of selective hydrogenation of various compounds over Pd/triC as a function of reaction time. Conditions: see notes (c) in Table 1 and (d) in Table 2. | ||
Conclusions
New synthetic carbons, with the pore structure specifically tailored for preparation of supported metal nanoparticles catalysts were used as supports for preparation of Pd catalysts by anaerobic incipient wetness method. N2 adsorption analysis showed that carbon supports possess micro-mesoporous bi-modal or tri-modal pore structures. HR-TEM, PXRD and XPS analyses of the catalysts showed that Pd nanoparticles were well dispersed within catalysts based on the tri-modal carbon (Pd/triC) with a monodispersed size of ca. 5 nm, whilst Pd catalysts based on the bi-modal carbon (Pd/biC) demonstrated a polydispersed distribution of Pd nanoparticle sizes.The developed Pd/C catalysts were evaluated in liquid-phase selective hydrogenation and hydrodehalogenation reactions in a structured compact reactor. The combination of integrated intensified reactor and specifically designed catalysts, especially for the Pd/triC catalyst, results in rapid and efficient reduction of various functional groups under mild conditions. The trend in the chemoselectivity of the Pd/triC catalys was evaluated as alkyne ≫ nitro > bromo ≫ aldehyde group. Due to the intensified heat transfer in the compact reactor, isothermal conditions were maintained for ensuring good selectivity and eliminating inherent hazards of exothermic reactions. The catalysts in the flow reactor system could be regenerated by solvent washing after the reaction. The developed Pd/triC catalyst demonstrated a good stability in long runs of model reactions and no deactivation of catalysts was detected in 5 hour continuous runs. The developed catalysts were reused up to 50 hours under flow conditions without loss of activity.
Based on the results obtained in this study, it is clear that carbons with hierarchical mesoporous structures are good candidates for developing new catalysts even using conventional preparation methods for intensified continuous-flow catalysis in structured reactors. However, further investigation on catalysts–supports interaction within/on carbon porous structures during catalytic reactions will be essential for better designs of such transition metal supported on synthetic carbon catalysts.
Acknowledgements
Financial support from the Engineering and Physical Sciences Research Council (Engineering Functional Materials, EP/C519736/1), the Overseas Research Students Awards Scheme and the University of Bath Research Studentship is gratefully acknowledged. The assistance of Dr. Martin Jennings Dr. Chris Muryn (School of Chemistry, The University of Manchester) on ICP-OES and XPS analyses is acknowledged. KW acknowledges the Royal Society for the award of an Industry Fellowship.Notes and references
- W. Ehrfeld, V. Hessel and H. Löwe, Microreactors, WILEY-VCH, Weinheim, 2000 Search PubMed.
- D. V. Bavykin, A. A. Lapkin, S. T. Kolaczkowski and P. K. Plucinski, Appl. Catal., A, 2005, 288, 175–184 CrossRef CAS.
- P. K. Plucinski, D. V. Bavykin, S. T. Kolaczkowski and A. A. Lapkin, Catal. Today, 2005, 105, 479–483 CrossRef CAS.
- S. Ferrouillat, P. Tochon and H. Peerhossaini, Chem. Eng. Process., 2006, 45, 633–640 CrossRef CAS.
- M. T. Kreutzer, F. Kapteijn, J. A. Moulijn and J. J. Heiszolf, Chem. Eng. Sci., 2005, 60, 5895–5916 CrossRef CAS.
- A. Julbe, D. Farrusseng, D. Cot and C. Guizard, Catal. Today, 2001, 67, 139–149 CrossRef CAS.
- J. Frauhammer, G. Eigenberger, L. von Hippel and D. Arntz, Chem. Eng. Sci., 1999, 54, 3661–3670 CrossRef CAS.
- G. Kolios, J. Frauhammer and G. Eigenberger, Chem. Eng. Sci., 2002, 57, 1505–1510 CrossRef CAS.
- G. Kolb, J. Schurer, D. Tiemann, M. Wichert, R. Zapf, V. Hessel and H. Lowe, J. Power Sources, 2007, 171, 198–204 CrossRef CAS.
- U. K. Singh and M. A. Vannice, Appl. Catal., A, 2001, 213, 1–24 CrossRef CAS.
- S. L. Poe, M. A. Cummings, M. P. Haff and D. T. McQuade, Angew. Chem., Int. Ed., 2006, 45, 1544–1548 CrossRef CAS PubMed.
- J. Kobayashi, Y. Mori and S. Kobayashi, Adv. Synth. Catal., 2005, 347, 1889–1892 CrossRef CAS.
- J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, Science, 2004, 304, 1305–1308 CrossRef CAS PubMed.
- K. K. Yeong, A. Gavriilidis, R. Zapf and V. Hessel, Catal. Today, 2003, 81, 641–651 CrossRef CAS.
- S. R. A. de Loos, J. van der Schaaf, M. de Croon, T. A. Nijhuis and J. C. Schouten, Chem. Eng. J., 2011, 167, 671–680 CrossRef CAS.
- S. R. A. de Loos, J. van der Schaaf, M. de Croon, T. A. Nijhuis and J. C. Schouten, Chem. Eng. J., 2012, 179, 242–252 CrossRef CAS.
- M. W. Losey, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2001, 40, 2555–2562 CrossRef CAS.
- P. K. Plucinski, D. V. Bavykin, S. T. Kolaczkowski and A. A. Lapkin, Ind. Eng. Chem. Res., 2005, 44, 9683–9690 CrossRef CAS.
- X. Fan, A. A. Lapkin and P. K. Plucinski, Catal. Today, 2009, 147S, S313–S318 CrossRef.
- X. Fan, M. Manchon Gonzalez, W. Karen, S. R. Tennison, A. Kozynchenko, P. K. Plucinski and A. A. Lapkin, J. Catal., 2009, 267, 114–120 CrossRef CAS.
- A. Taguchi and F. Schuth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- H. Tamai, U. Nobuaki and H. Yasuda, Mater. Chem. Phys., 2009, 114, 10–13 CrossRef CAS.
- H. Tamai, J. Ogawa and H. Yasuda, J. Colloid Interface Sci., 2003, 260, 312–316 CrossRef CAS PubMed.
- A. Pigamo, M. Besson, B. Blanc, P. Gallezot, A. Blackburn, O. Kozynchenko, S. Tennison, E. Crezee and F. Kapteijn, Carbon, 2002, 40, 1267–1278 CrossRef CAS.
- S. R. Tennison, Appl. Catal., A, 1998, 173, 289–311 CrossRef CAS.
- N. Krishnanakutty and M. A. Vannice, J. Catal., 1995, 155, 312–326 CrossRef.
- X. Fan, H. S. Chen, Y. L. Ding, P. K. Plucinski and A. A. Lapkin, Green Chem., 2008, 10, 670–677 RSC.
- A. L. Patterson, Phys. Rev. Lett., 1939, 56, 978–982 CAS.
- J. L. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- R. Burch, E. Halpin, M. Hayes, K. Ruth and J. A. Sullivan, Appl. Catal., B, 1998, 19, 199–207 CrossRef CAS.
- A. L. Guimaraes, L. C. Dieguez and A. Schmal, J. Phys. Chem. B, 2003, 107, 4311–4319 CrossRef CAS.
- G. Vilé, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ACS Catal., 2015, 5, 3767–3778 CrossRef.
- G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J., 2014, 20, 5926–5937 CrossRef PubMed.
- A. J. McCue, F.-M. McKenna and J. A. Anderson, Catal. Sci. Technol., 2015, 5, 2449–2459 CAS.
- A. McCue and J. Anderson, Front. Chem. Sci. Eng., 2015, 9, 142–153 CrossRef CAS.
- E. Klemm, in Topical Workshop Catalysis, DFG Priority Program 1362, University of Stuttgart, Stuttgart, 2011 Search PubMed.
- V. Höller, D. Wegricht, I. Yuranov, L. Kiwi-Minsker and A. Renken, Chem. Eng. Technol., 2000, 23, 251–255 CrossRef.
- K. Konuma and N. Kameda, J. Mol. Catal. A: Chem., 2002, 178, 239–251 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4091 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |