
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MeOx/Al2O3 and MeOx/CeO2 (Me = Fe, Co, Ni) catalysts for high temperature N2O decomposition and NH3 oxidation†
L. G.
Pinaeva
*a,
I. P.
Prosvirin
ab,
L. S.
Dovlitova
a,
I. G.
Danilova
a,
E. M.
Sadovskaya
ab and
L. A.
Isupova
a
aBoreskov Institute of Catalysis, Novosibirsk, Russia. E-mail: pinaeva@catalysis.ru
bNovosibirsk State University, Novosibirsk, Russia
First published on 30th October 2015
Abstract
A series of MeOx/Al2O3 and MeOx/CeO2 (Me = Fe, Co, Ni) compounds was prepared by incipient wetness impregnation and characterized by XRD, XPS, UV-vis DRS, differential dissolution phase analysis (DDPA). Their activity towards N2O decomposition and ammonia oxidation at 750–900 °C was shown to depend on oxygen mobility in the sample as characterized by steady state isotopic transient kinetic analysis (18O SSITKA) and dispersion of MeOx (Me = Fe) species. Obvious reverse “activity-FeOx dispersion” dependence was related to the change of contribution from oxygen transfer from the support to active sites through the Me–ceria or not improbably, the Me–alumina interface. It was shown that the high efficiency of CeO2 based samples in deN2O can be additionally increased by CeO2 supporting onto high surface area alumina.
1. Introduction
Secondary abatement processes of N2O formed as an undesirable by-product during the catalytic oxidation of ammonia into nitric oxide implicates a catalyst set up in the ammonia burner operating at 800–900 °C directly after the Pt–Rh gauzes.1 Bulk or supported mixed metal oxides were developed for catalytic N2O decomposition and put into practice in nitric acid plants before 2000 (Co2AlO4/CeO2, CuO/Al2O3, La0.8Ce0.2CoO3 and Fe/ZrO2).1 Moreover, alternative catalytic formulations were evaluated such as yttrium-doped zirconia,2 ceria–zirconia,3 A1−x A′xB1−yB′yO3 perovskites with A = La or Ca, B = Mn, Co or Fe, A' = Sr, B' = Cu or Ni,4–6 metal-substituted hexa-aluminates,7 Fe2O3–MeOx (Me = Al, Zr, Ce, La, Cu, Cr) obtained by co-precipitation,8,9 Co3O4 (ref. 10) and CoOx–CeO2 (ref. 11) obtained by calcinations of mixtures of individual oxides. The pilot plant reactor and real plant studies8 confirmed high activity and very good mechanical stability of the Fe2O3/Al2O3 catalysts prepared by co-precipitation as well as no decomposition of nitric oxide. Several aspects with respect to the reaction mechanisms, the structure–activity correlations, the role of various gas inhibitors as well as the strategies followed to adjust the local surface structure of the abovementioned noble metal-free metal oxides were analyzed in the review of Konsolakis.12Ageing of Pt–Rh gauzes is accompanied by decrease of activity and yield of NO + NO2 (NOx), whereas N2O production increases. Taking account of the tendency in the last decade to reduce platinum metal loading in the reactor (including the number of gauzes and wire thicknesses), at gauzes deactivation noticeable slip of ammonia becomes possible followed by its reaction with NOx. Therefore, development of a bi-functional catalyst that is capable of efficient N2O abatement and NH3 oxidation to NOx becomes urgent for further optimization and the reduction of harmful emissions from the process of HNO3 production.
An idea for development of an oxide-based catalyst that is capable of converting slipping ammonia with high selectivity to NOx was successfully realized using a dual bed system, in which a Fe2O3–Al2O3-based extruded monolith followed fewer numbers of platinum gauzes.13 Later studies showed the efficiency of these compositions in both slipping NH3 oxidation to NOx and N2O abatement.14 At the same time, Al2O3-based honeycomb catalysts with the same geometry and FeOx-based active components supported by impregnation were substantially more active in both reactions and retained high activity after the run in a dual-bed system for 3 months.14
Earlier, we have shown that catalytic activity of Sr substituted La manganites in high-temperature N2O decomposition correlated with the coefficient of lattice oxygen self-diffusion.5 In the materials with high bulk oxygen mobility, the rates of oxygen transfer in the two-phase system “O2gas–surface–catalyst bulk” characterized by 16O/18O exchange and N2O decomposition additionally increased either by their decoration by structures characterized by a higher rate of heteroexchange (composites LaSrFeO4 (surface)–La0.4Sr0.6FeO3 (bulk)6) or by modification of near surface layers by hetero-cations (LaFeO3–CeO2 composites with Fe3+ ions inserted into a fluorite lattice15). In the former case,6 “activity – oxygen mobility” correlation was observed for reaction of NH3 oxidation as well. Pérez-Ramírez et al.16 showed that the reaction of NH3 oxidation to NOx over oxide catalysts (Fe2O3, CeO2, Cr2O3) follows a Mars–van Krevelen-type scheme with participation of lattice oxygen and reoxidation of the so-formed vacancies by both gas-phase O2 and bulk lattice oxygen. Due to this, the efficiency of NH3 oxidation to NOx, especially in oxygen deficient conditions, depended on the rate of bulk oxygen diffusion to surface vacancies.
In accordance with these findings, the idea to combine high activity towards N2O decomposition and NH3 oxidation (at high selectivity to NOx) in the same sample was checked. In addition to Fe/Al2O3(CeO2) samples, Co- and Ni- supported on Al2O3 and CeO2 were tried as well since a vast number of patent applications have claimed a relatively high NO selectivity (90–95%) using Co and Ni single metal oxides. Numerous attempts were made to commercialize Co3O4-based catalysts but substantially lower activity and reversible deactivation of Co-systems, due to reduction of Co3O4 by NH3 to the inactive CoO under reaction conditions in the upper parts of the bed, excluded the replacement of Pt gauzes by oxide catalysts. Nevertheless, these systems could be tried as the second catalyst in the dual bed system, especially since the stabilizing effect of CeO2 in Co3O4–CeO2 mixed oxides with low Co content is well-known.11 Since copper catalyzes ammonium nitrate decomposition, an extremely important safety problem may arise if copper leaches from the catalyst and accumulates in the fertilizer product.1 This completely excludes any Cu-based system for use in the conditions of the ammonia burner, although Cu/CeO2 revealed high efficiency in deN2O at lower temperatures17,18 and more probably can be located after the absorption column. In addition, it is well known that under these conditions, Cu-containing systems oxidize NH3 to N2 with high selectivity.
In the first stage, we showed that intrinsic catalytic activity of different Me/Al2O3 (Me = Fe, Co, Ni) samples obtained by impregnation towards N2O decomposition and NH3 oxidation was substantially lower compared to corresponding Me/CeO2 samples with similar surface Me concentration and correlated with the rate of oxygen exchange in the samples. Variation of Fe content in the Fe/Al2O3(CeO2) samples revealed noticeable reverse dependence of deN2O and NH3 oxidation activity due to the size effect. In accordance with this, we supported FeOx onto specially developed Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 characterized by high specific surface area, and formation of highly dispersed FeOx at increased Fe content in the sample finally resulted in N2O and NH3 conversion increase. For such samples, we focused on Fe oxide as the active component because in the case of Me = Co, Ni low active MeAl2O4 or NiOx phases were formed on the alumina surface. Using of the effect of high oxygen mobility in CeO2 was restricted by its low surface area at the temperature of reaction, independently on preparation method.3,11,20 Therefore, an attempt was made to disperse CeO2 using precipitation onto high surface area Al2O3.
19 characterized by high specific surface area, and formation of highly dispersed FeOx at increased Fe content in the sample finally resulted in N2O and NH3 conversion increase. For such samples, we focused on Fe oxide as the active component because in the case of Me = Co, Ni low active MeAl2O4 or NiOx phases were formed on the alumina surface. Using of the effect of high oxygen mobility in CeO2 was restricted by its low surface area at the temperature of reaction, independently on preparation method.3,11,20 Therefore, an attempt was made to disperse CeO2 using precipitation onto high surface area Al2O3.
2. Experimental
2.1. Sample preparation
CeO2 and Al2O3 samples used as the support were obtained by calcination of the corresponding nitrates of chemical purity (99.5%) at 900 °C for 36 h. Al2O3-based supports were prepared by calcination of: 1) Al(NO3)3·9H2O of chemical purity (99.5%) at 1000 °C or 1200 °C for 6 h (denoted below as Al2O3-1000 and Al2O3-1200, respectively), or 2) the product of centrifugal thermal activation of commercial-grade alumina hydrate (hydrargillite)19 at 1000 °C for 19 h (Al2O3-C). To obtain the Ce/Al2O3 support, the product of thermochemical activation of hydrargillite was first calcined at 500 °C (SBET = 210 m2 g−1); then, CeO2 was precipitated onto this product from 0.75 M Ce(NO3)3·6H2O water solution using 1 M (NH4)2CO3 as a precipitation agent, so that the Ce:Al ratio in the final product was 1:1 (mole). The resultant precipitate was washed until the pH of the filtrate was 7, dried at 60 °C overnight, followed by calcination at 900 °C for 5 h.
All Me/support samples (Me = Fe, Co, Ni) were prepared by incipient wetness impregnation of the corresponding support by water solution of Me nitrate with added citric acid in 10 wt% excess to the stoichiometric amount to form the corresponding citrates and ethyleneglycol, dried in air at 150 °C for 3 h and then calcined at 900 °C for 4 h. In the commonly-used abbreviation nMe/support, “n” corresponded to Me weight content in the sample in terms of the corresponding metal, although oxide compounds obviously formed. For CeO2 and Al2O3-1200, the samples with Me concentration 6.7 × 1019 at m−2 have been prepared, which corresponded to Me content of around 2.5–2.7 wt% (Al2O3-1200) and 0.86–0.91 wt% (CeO2). For all supports, we also varied Fe content in the samples from 0.86 to 19.8 wt%. Bulk MeOx were obtained by calcination of the corresponding chemical purity nitrates at 900 °C for 6 h.
2.2. Sample characterization
X-ray powder diffraction (XRD) patterns were recorded using a Bruker D8 diffractometer with Cu Kα monochromatic radiation. Each sample was scanned in the range of 2θ from 10° to 70° with a step size of 0.05° in 2θ. Surface composition of the samples was investigated by X-ray photoelectron spectroscopy (XPS) using a SPECS spectrometer with Al-Kα irradiation (hν = 1486.6 eV). The binding energy (BE) scale was preliminarily calibrated by the position of the peaks of Au 4f7/2 (84.0 eV) and Cu 2p3/2 (932.67 eV) core levels. UV-vis diffuse reflectance spectra (UV-vis DRS) were obtained using a UV-2501 PC Shimadzu spectrometer with an IRS-250A diffusion reflection attachment in the 11 000–54 000 cm−1 range using BaSO4 as a reference. The edge energy (or band gap, Eg) for allowed transitions was determined by finding the intercept of the straight line for the low-energy rise of a plot of [F(R∞) hν]2versus hν,21 where hν is the incident photon energy and F(R∞) is the Kubelka–Munk function.The method of differential dissolution phase analysis (DDPA) was used to reveal the composition of the phases (including poorly crystallized, but without information about the oxygen content therein) formed in the supported samples, including possible decoration of one phase by another.22 For this, about 10 mg of the sample was loaded in a microreactor and dissolved in the water based solvent with the composition changing from the lower towards higher acidity in the following order: 1) HCl (pH = 2), 2) (1 ÷ 3) M HCl, while the temperature increased continuously from 20 °C to 90 °C, 3) (3.6 M) HF and running the reactor with a flow rate of 3.6 ml min−1. Change of the outlet mixture composition with time was analyzed by atomic emission spectroscopy (BAIRD spectrometer) using the following spectral lines (nm) of the elements: Fe–238.2, Al–308.2, Ce–413.8, Co–238.8 with 5% accuracy of measurements at sensitivity level 10−3 μg ml−1.
Kinetics of oxygen exchange was characterized by steady state isotopic transient kinetic analysis (SSITKA). For SSITKA experiments, the sample (g = 0.025 g) was loaded into a reactor (quartz tube, i.d. = 3 mm) and heated to 800 °C in an 0.5% vol. 16O2 + He flow and kept at this temperature for 30 minutes, whereupon the gas mixture was replaced stepwise by the same one containing 18O2 and Ar (1 vol%) as an inert tracer. The gas flow rate for all mixtures amounted to 16.7 cm3 s−1. All responses were analyzed as time variation of the 18O atomic fraction in the gas phase αg(t) = (16O18O + 218O2)/(2ΣOiOj), where ΣOiOj = 16O2 + 16O18O + 18O2.23
The catalytic activity for samples with particles of 250–500 μm in size was measured in a fixed-bed U-shaped reactor (3 mm i.d. quartz tube) at ambient pressure in the temperature range 750–900 °C and contact time ~10−3 s. For NH3 oxidation, a mixture of 1% NH3 in air or 1% NH3 + 2% O2 in N2 was fed to the reactor charged by 0.015 or 0.043 g of the sample (see details in the text) with a flow rate 25 l h−1. Outlet mixture composition was measured by IR spectroscopy. For N2O decomposition, a gas mixture consisting of 0.15 vol% N2O (+3% O2 + 3% H2O) in He flowed through the reactor with a flow rate of 60 l h−1. For samples characterized by high specific surface area (Al2O3-C, Ce/Al2O3 based), we failed to reach reasonably low values of N2O conversion at 800 °C. Therefore, the data obtained at the same sample loading (0.038 g) and flow rate (60 l h−1) were considered to analyze the effect of Fe concentration. Outlet mixture composition was analyzed by an on-line gas chromatograph with Porapack T (i.d. = 3 mm, l = 3 m) and NaX (i.d. = 3 mm, l = 2 m) columns.
3. Results and discussion
3.1. Phase composition
 | ||
| Fig. 1 XRD patterns of: (A) Al2O3-1200 (1) and (2.5–2.7) Me/Al2O3-1200 (Me = Co(4), Fe (2), Ni(3)) samples, (B) Fe/Al2O3-C samples with different Fe content, and (C) Ce/Al2O3 (1) and 9.9Fe/Ce/Al2O3 (2) samples. | ||
| Sample | S BET, m2 g−1 | Phase composition | d XRD, Å |
|---|---|---|---|
| Al2O3-1200 | 4 | α-Al2O3 (a = b = 4.756 Å, c = 12.986 Å) | 370 |
| Al2O3-1000 | 7 | α-Al2O3 (a = b = 4.760 Å, c = 12.997 Å) | 300 |
| Al2O3-C | 71 | α-Al2O3 (a = b = 4.771 Å, c = 13.020 Å) | 480 |
| θ-Al2O3, κ-Al2O3, γ-Al2O3 | 100 (γ-Al2O3), 140 (θ-Al2O3) | ||
| 2.5Fe/Al2O3-1200 | 4.0 | α-Fe2O3 (a = b = 5.035(0) Å, c = 13.741 Å) | 260 |
| α-Al2O3 (a = b = 4.761 Å, c = 12.997 Å) | 500 | ||
| 2.7Co/Al2O3-1200 | 4.0 | CoAl2O4 or Co3O4 (a = b = c = 8.104 Å) | 260 |
| α-Al2O3 (a = b = 4.761 Å, c = 12.997 Å) | 520 | ||
| 2.7Ni/Al2O3-1200 | 4.0 | NiO (a = b = c = 4.178 Å) | 240 |
| NiAl2O4 | ≤120 | ||
| α-Al2O3 (a = b = 4.760 Å, c = 12.996 Å) | 580 | ||
| 2.5Fe/Al2O3-C | 60 | α-Al2O3 (a = b = 4.778 Å, c = 13.039 Å) | 510 |
| θ-Al2O3, κ-Al2O3, γ-Al2O3 | Highly dispersed | ||
| 6.6Fe–Al2O3-C | 46 | α-Al2O3: 31% – a = b = 4.770 Å, c = 13.030 Å | 510 |
| 69% - a = b = 4.795 Å, c = 13.057 Å | 470 | ||
| θ-Al2O3, κ-Al2O3, γ-Al2O3 | Highly dispersed | ||
| 13.2Fe–Al2O3-C | 33 | α-Al2O3: 18% – a = b = 4.760 Å, c = 13.000 Å, 82% – a = b = 4.795 Å, c = 13.057 Å, | 510 |
| 520 | |||
| Traces of θ-Al2O3, κ-Al2O3, γ-Al2O3 | Highly dispersed | ||
| Fe2O3 (a = b = 5.032 Å, c = 13.702 Å) | 250 | ||
| CeO2 | 1.4 | CeO2 (a = b = c = 5.412 Å) | 680 |
| 0.91Co/CeO2 | 1.4 | CeO2 (a = b = c = 5.412 Å) | 630 |
| Co3O4 (a = b = c = 8.084 Å) | 460 | ||
| 0.86Fe/CeO2 | 1.4 | CeO2 (a = b = c = 5.411 Å) | 550 |
| 0.91Ni/CeO2 | 1.4 | CeO2 (a = b = c = 5.411 Å) | 600 |
| 2.5Fe/CeO2 | 1.4 | CeO2 (a = b = c = 5.412 Å) | 650 |
| traces α-Fe2O3 | — | ||
| CeO2/Al2O3 | 63 | CeO2 (a = b = c = 5.411 Å) | 400 |
| θ-Al2O3 and traces of γ-Al2O3 | Highly dispersed | ||
| 9.9Fe/Ce/Al2O3 | 35 | CeO2 (a = b = c = 5.411 Å) | 460 |
| θ-Al2O3 and traces of γ-Al2O3 | Highly dispersed | ||
| α-Fe2O3 (a = b = 5.035 Å, c = 13.741 Å) | 320 |
Although Co3O4 and Co–Al–O spinels are characterized by the same symmetry group, increased lattice parameters of Co compounds in 2.7Co/Al2O3-1200 sample (Fig. 1A) (a = b = c = 8.104 (1) Å) compared with those in the 0.91Co/CeO2 one or in bulk Co3O4 (a = b = c = 8.084 (0) Å) favor formation of CoAl2O4 in the former,24 probably in the mixture with Co3O4. In the 2.7Ni/Al2O3-1200 and 2.5Fe/Al2O3-1200 samples, additional NiO with admixture of highly dispersed Ni–Al–O compounds of spinel structure and α-Fe2O3, respectively, were detected. The lattice parameters of NiO and Fe2O3 in the supported samples were exactly the same as for bulk MeOx (not presented in Table 1 for clarity). However, slight increase of α-Al2O3 lattice parameters in all Me/Al2O3 samples points to the probability of non-substantial Men+ penetration into the alumina lattice. Taking account of the slightly larger radii of Me3+ ions compared with that of Al3+ (69–74 pm and 67.5 pm, respectively), such an assumption looks quite reasonable.
FeOx supported on Al2O3-C resulted in continuous decrease of the peaks corresponding to θ-, κ-, γ-Al2O3 with an increase of Fe content from 2.5 to 13.2 wt% (Fig. 1b). Instead of these, a second α-Al2O3 phase with an increased lattice parameter (a = b = 4.795 Å, c = 13.057 Å) becomes evident and increases in intensity (Table 1), which more obviously points to efficient incorporation of Fe3+ ions into the lattices of low temperature aluminas thus promoting their transition to α-Al2O3 at a lower temperature.25 This fact agrees well with decrease of SBET value from 70 m2 g−1 for Al2O3-C to 22 m2 g−1 after supporting 19.8% of Fe (pattern of the last sample not presented for brevity).
In the patterns of (0.86–0.91)Me/CeO2 samples, traces of crystalline MeOx were observed only for the case of Me = Co, and some α-Fe2O3 was detected for substantially higher Fe content (sample 2.5Fe/CeO2). Nevertheless, unlike alumina-based samples, neither sintering of CeO2 nor change of CeO2 lattice parameters were noted after Me supporting (Table 1). Considering that the radius of Fe3+ ions (0.049–0.078 pm, depending on coordination and spin state) is smaller than that of Ce4+ ions in the typical cubic fluorite lattice (0.097–0.101 nm), the expected lattice shrinkage took place at substantial Fe content in the mixed Fex–Ce1−x oxides.26 However, at x < 0.05, even slight unit cell expansion was observed and related by authors to partial Ce4+ reduction to the larger (1.23 Å) Ce3+ ion27 during calcination in the presence of Fe3+. Therefore, penetration of supporting Me into the CeO2 lattice cannot be excluded, especially taking account of concreted nanocrystallites present in the initial support.
In the 9.9Fe/Ce/Al2O3 sample, α-Fe2O3 was detected with lattice parameters and dXRD value similar to that in 13.2Fe/Al2O3-C and 2.5Fe/Al2O3-1200 samples. Although Fe supporting onto Ce/Al2O3 resulted in noticeable drop of the surface area, like in Al2O3-C based samples, it did not facilitate formation of α-Al2O3 (Tables 1 and 2, Fig. 1C). For similar CeO2/Al2O3 samples, it was shown that it is Ce in the lower oxidation state that stabilizes alumina toward the formation of low surface area phases up to 1100 °C or 1200 °C under oxidizing and reducing conditions, respectively.28 Since CeAlO3 crystallites at high temperature and under reducing conditions were observed by XRD, stabilization of Ce3+ is more probably due to interaction with the OH groups of alumina. Absence of such alumina-bonded hydroxyl groups can prevent penetration of Fe3+ into the bulk alumina and the phase transition observed in the case of the Al2O3-C sample.
| Sample | Sample dissolved, weight% | Me, weight% of the sample in dissolved portion | Element | Distribution of elements dissolved during different steps but O, mole% | |||
|---|---|---|---|---|---|---|---|
| Total | HCl (pH = 2) | 0.33 M HCl | 3.6 M HF | ||||
| 2.5Fe/Al2O3-1200 | 5 | 2.53 | Al | 38.1 | — | — | 38.1 |
| Fe | 61.9 | — | 18.4 | 43.5 | |||
| 2.7Co/Al2O3-1200 | 2 | 0.43 | Al | 82.1 | — | — | 82.1 |
| Co | 17.9 | 0.4 | — | 17.5 | |||
| CeO2 | 4 | — | Ce | 100 | — | — | 100 |
| 0.91Co/CeO2 | 95 | 0.93 | Ce | 98.2 | — | 98.2 | — |
| Co | 1.8 | Traces | 0.39 | 1.41 | |||
| 0.86Fe/CeO2 | 54 | 1.0 | Ce | 96.8 | 96.8 | — | |
| Fe | 3.2 | 0.60 | 1.86 | 0.74 | |||
| 0.91Ni/CeO2 | 11 | 0.81 | Ce | 91.3 | — | — | 91.3 |
| Ni | 8.7 | Traces | 7.8 | 0.9 | |||
 | ||
| Fig. 2 DDPA spectra of: (A) 2.5Fe(2.7Co)/Al2O3-1200, (B) CeO2 and 0.91Co/CeO2 samples. | ||
CeO2 did not dissolve in HCl, and very slow dissolution (about 4% of the total sample weight in 7 minutes) took place after HF feeding (Fig. 2B, Table 2). Me/CeO2 samples dissolved in substantially milder conditions, but the character and the rate of their dissolution depended on the nature of Me. Therefore, already 95 weight% of the 0.91Co/CeO2 sample dissolved in these conditions. Increase of solubility can be related first of all to formation of Co–Ce–O solid solution with Co/Ce = 0.004 and including all soluble Ce (dissolved in (1 ÷ 3) M HCl). This obviously means that Co3+ ions are able to insert into more dispersed (~10 nm) concreted microcrystallites and modify the lattice of larger (~100 nm) particles increasing their solubility. The rest of Co dissolved slowly as an oxide in the most harsh conditions (3.6 M HF) and more probably, can be related to the well-crystallized Co3O4 detected by XRD (Table 1). Absence of any Co in the same flow during dissolution of the 2.7Co/Al2O3-1200 sample points to the formation of well-crystallized Co–Al spinel therein. The quantity of 0.86Fe/CeO2 sample dissolved was somewhat lower (54 wt% of the total) and all Fe passed into solution as it was found for 2.5Fe/Al2O3-1200 sample (Fig. S1†). It was included into Fe oxide/hydroxide whose solubility in HCl (pH = 2) and at the substantially higher acidity (3.6 M HF) can be due to different degree of crystallinity or defect structures formed. The parallel dissolution of the remaining Fe and Ce in the flow of (1 ÷ 3) M HCl with the stoichiometry Fe/Ce = 0.02 points to formation of Fe–Ce–O solid solution, more probably, with participation of CeO2 composites characterized by smaller sizes of crystallites. Unmodified ceria is stable even in 3.6 M HF and can be related to composites with largest crystallites. We cannot exclude that both unmodified CeO2 and Ce–Fe–O solid solution are decorated by highly defective Fe oxide/hydroxide species dissolved in weak HCl. In the case of the 0.91Ni/CeO2 sample (Fig. S1†), most of the supported Ni dissolved individually in (1 ÷ 3) M HCl, while the parallel Ni and Ce dissolution took place in the more rigid conditions (3÷6 M HF) and included only 10 weight% of the sample.
Therefore, in the 2.7Co/Al2O3-1200 sample, Al3+ ions partially replace Co3+ in the spinel structure, forming both highly disordered and well-crystallized Co–Al–O species, while incorporation of Fe3+ ions into the boundaries of the micro-composites results in the anchoring of Fe2O3 crystallites to the alumina surface. Me–ceria interaction results in formation of Me–Ce–O solid solutions in Me/CeO2 samples; their content and degree of disorder in the sample change in the following order: 0.91Co/CeO2 > 0.86Fe/CeO2 > 0.91Ni/CeO2 ≥ CeO2.
3.2. Microstructure
UV-vis DR spectra of different Co- and Fe-based catalysts have been presented in Fig. 3a and b, respectively. CeO2 was characterized by strong absorbance bands at 36500 and 29000 cm−1 attributed to Ce4+ ← O2− charge transfer and the interband transitions, respectively,29 and a band gap energy (Eg) value of 3.35 eV, which is consistent with the reported value of 3.3 eV.29–31 The spectra of 0.91Co/CeO2 (Fig. 3a) and 0.86Fe/CeO2 (Fig. 3b) samples are in fact a superposition of those for corresponding bulk oxides and CeO2, excluding the red shift of absorption edge of CeO2 (Eg values of 3.18 and 3.22 eV for 0.91Co/CeO2 and 0.86Fe/CeO2, respectively) and additional absorption band at ~20000 cm−1 (Co case) and 23500 cm−1 (Fe case). A minimal red shift of the absorption edge compared to that in the support was found for the 0.91Ni/CeO2 sample (Eg value of 3.27 eV, spectrum not shown for brevity). We already observed these spectral characteristics for a Fe/CeO2 sample prepared in the same way15 and explained their appearance by O2 adsorption on the structural defect, more obviously, clusters of oxygen vacancies arose after the entry of the dopant ions into the bulk of CeO2. As proposed in,30 a change in the value of the absorption edge indicates that either (1) Me ions in Me/CeO2 samples substitute into the CeO2 lattice; or (2) the isolated Me ions form a mixed Me–Ce oxide and strongly interact with CeO2 leading to the electronic structure changes. In our case, the value of the red shift correlated well with content of Me–Ce mixed solution in the sample, as determined by DDPA.
 | ||
| Fig. 3 UV-vis DR spectra of: A – (1) 2.9Co/Al2O3-1200, (2) CoAl2O4 supplied by Aldrich, (3) 0.93Co/CeO2, (4) CeO2, (5) Co3O4; and B – (1) FeOx, (2) 0.86Fe/CeO2, (3) 2.7Fe/Al2O3-1200, (4) CeO2. | ||
In the spectrum of the 2.9Co/Al2O3-1200 sample, an intense triplet of bands at about 15900, 17000 and 18200 cm−1 with the shoulder at 13800 cm−1 was detected and related to the transitions of 4A2 → 4T1(4P) of the tetrahedral Co2+ ions in Co–Al–O spinel and cobalt oxide, respectively.32 The presence of the same triplet in the reference CoAl2O4 sample (Aldrich) supports formation of Co–Al–O spinel with admixture of Co3O4.
3.3. Surface composition
As follows from the X-ray photoelectron spectra of Ni 2p, Fe 2p, and Co 2p levels in Me/Al2O3-1200 and Me/CeO2 samples presented in Fig. 4, the nature of Me surface compounds depends on the nature of the support. Usually, no difference between various Co compounds can be reliably drawn from binding energies of either Co 2p1/2 or Co 2p3/2 signals10 without accounting for the position of their satellites arising due to the shake-up process of the Co2+ compound in the high spin state. Indeed, BE values characterizing the Co 2p3/2 peak position in CoO, CoOOH and Co3O4 are indistinguishable (~780.0 eV). Higher, but also very close, BE values have been reported for CoAl2O4 and Co(OH)2 (781.9 and 781.2 eV, respectively).33–38 However, for CoO, Co(OH)2 or CoAl2O4, the satellite – main signal distances fall in the range 5–6.5 eV,34,35,39,40 while the satellites in the spectrum of pure Co3O4 and CoOOH should be located approximately 9–10 eV away from the main signals.34,36,39 In this regard, the BE value of the Co 2p3/2 signal at 781.6 eV with satellite at 786.6 eV in the 2.7Co/Al2O3-1200 sample (Fig. 4a) excluded CoO, Co3O4 and CoOOH from our consideration, and either Co(OH)233 or CoAl2O434,37,38 could form on the surface. According to DDPA data, Co(OH)2 could be responsible for about 0.4% of Co compounds (Table 2, corresponds to Co dissolved in HCl at pH = 2); therefore, most of the surface Co included in the Co–Al spinel structures. To ensure the correctness of such assignment, the spectrum of CoAl2O4 supplied by Aldrich and additionally calcined at 900 °C for 5 h has been presented as well. In the 0.91Co/CeO2 sample, the position of the main Co 2p3/2 peak shifted to lower BE values (779.9 eV), while the shoulder at 781.9 eV was still present resulting in the ratio of their intensities of ~3:2. Definite assignment of the peak at 779.9 eV to individual CoO, Co3O4 or CoOOH was impossible since the presence of satellites at 786.7 and 790.8 eV agrees with formation of any of these compounds. In addition, in the XPS spectrum of the bulk Co3O4 sample characterized by the same lattice parameters as the 0.91Co/CeO2 sample (Table 1), the intensity of the satellite peak was substantially lower. Hence, different oxide/hydroxide-like Co compounds, including those in Co–Ce–O solid solution, have been formed on the surface of the 0.91Co/CeO2 sample. In particular, Co(OH)2 in this case could result from hydration of highly dispersed species.
 | ||
| Fig. 4 (A) Co2p, (B) Ni2p, (C) Fe2p spectra of Me/Al2O3-1200(CeO2) samples (Me = Co, Ni, Fe) with similar (~7 × 1019 at/m2) concentration of Me, and Fe 2p spectrum of 3.8Fe/Ce/Al2O3 sample (C). | ||
The Ni 2p3/2 peak at 856.3 eV in the 2.7Ni/Al2O3-1200 sample (Fig. 4b) can be due to both NiAl2O4 (BE ranging from 855.2 to 857.2 eV) and Ni(OH)2 (855.6–856.6 eV),41–45 the former being detected as well by XRD (Fig. 1, Table 1). NiO state, characterized by lower BE value (854.2 eV),45 was detected in the 0.91Ni/CeO2 sample together with Ni(OH)2 (NiO:Ni(OH)2) = 1:1) resulting in the widening of both the main Ni 2p peak and the energy shake-up satellite peak about 6 eV above the main one. As in the 0.91Co/CeO2 sample, Ni(OH)2 detection could indicate the presence of highly dispersed NiO species both in Al2O3 and CeO2-based samples. In this case, their relative content in the samples is higher compared with the Co case.
While the BE value of the Fe 2p3/2 spectra in the 0.86Fe/CeO2 sample at 710.4 eV (Fig. 4c) indicates a preferential Fe3+ state in the oxide, its high energy shift (BE = 711.8 eV) detected in both the 2.5Fe/Al2O3-1200 specimen and all Al2O3-1000, Al2O3-1200 and Al2O3-C supported samples can be related to formation of Fe3+ oxyhydroxide.46 In the 3.8Fe/Ce/Al2O3 sample, both states of Fe3+ have been detected also, but the fraction of Fe3+ state in the oxide is somewhat higher than in Fe/Al2O3-based samples. Therefore, FeOx species anchor both to the Al2O3 and CeO2 surfaces.
In the Ce 3d spectra of the 3.8Fe/Ce/Al2O3 sample, the normal complex form due to shake-down satellites from an O 1s to Ce 4f electron transfer observed for CeO2 and 0.86Fe/CeO2 was supplemented by the features marked as v' and u' due to anion defects and Ce3+ (Fig. S2†).47 This means that besides smaller crystallized CeO2 species, X-ray amorphous CeAlO3 and probably isolated Ce3+ species can reasonably form after CeO2 precipitation onto high surface area alumina,48,49 thus stabilizing alumina toward the formation of low surface area phases, as proposed in reference.27
Therefore, spinel-like surface structures including Al have been formed on the surface of the 2.7Co/Al2O3-1200 sample and probably their mixture with Ni(OH)2 in the 2.7Ni/Al2O3-1200 sample. It is quite reasonable to suppose that above 700 °C, surface Fe oxyhydroxide formed after Fe supporting onto Al2O3 and all Ni- and Co-containing hydroxylated species convert to corresponding oxides, and thus similar types of surface compounds participate in the reaction in both Al2O3-1200, CeO2 and CeO2/Al2O3 based samples.
Table 3 lists the data on the surface concentration of Me in the different Al2O3-1200 and CeO2 based samples. Similar values were measured in the (2.5–2.7)Me/Al2O3-1200 and 0.91Co(0.86Fe)/CeO2 samples with the same as related per surface unit (6.7 × 1019 at m−2) content of Me therein that means similar MeOx (Co–Al–O) species dispersion (DMe) therein. Its relative value can be estimated as follows: DMe = Mes/CMe, where Mes is the surface atomic concentration of Me in the sample as measured by XPS (Table 3) [%] and CMe is total content of Me in the samples [atoms/m2]. Noticeably higher content of Ni in the near surface layers of the CeO2 based sample can be explained either by lower degree of Ni penetration into CeO2, resulting in the formation of Ni–Ce–O solid solutions (Table 2) or by higher dispersion of NiOx species, which agrees well with high content of hydroxylated species.
| Sample | S BET, m2 g−1 | Mes, at% (by XPS) | Feexp, rel. units | DMe, rel. units |
|---|---|---|---|---|
| CeO2 based | ||||
| 0.86Fe/CeO2 | 1.3 | 1.0 | 1.3 | 1.5 |
| 0.91Co/CeO2 | 1.3 | 1.2 | — | 1.8 |
| 0.91Ni/CeO2 | 1.3 | 2.0 | — | 2.7 |
| 2.5Fe/CeO2 | 1.3 | 1.5 | 2.1 | 0.72 |
| Al2O3-1200 and Al2O3-1000 based | ||||
| 2.5Fe/Al2O3-1200 | 4 | 1.1 | 4.4 | 1.6 |
| 2.7Co/Al2O3-1200 | 4 | 1.0 | — | 1.5 |
| 2.7Ni/Al2O3-1200 | 4 | 1.4 | — | 2.1 |
| 0.86Fe/Al2O3-1200 | 4 | 1.1 | 4.4 | 4.8 |
| 2.5Fe/Al2O3-1000 | 7 | 1.4 | 9.8 | 3.7 |
| Al2O3-C based | ||||
| 2.5Fe/Al2O3-C | 61 | 0.46 | 28.1 | 10.5 |
| 6.6Fe/Al2O3-C | 46 | 1.3 | 59.8 | 8.5 |
| 13.2Fe/Al2O3-C | 33 | 2.2 | 72.6 | 5.1 |
| 19.8Fe/Al2O3-C | 22 | 1.6 | 35.2 | 1.7 |
| CeO2/Al2O3 based | ||||
| 3.8Fe/CeO2/Al2O3 | 50 | 1.3 | 65.0 | 16.0 |
3.4. 18O SSITKA
Fig. 5 shows the time dependencies of exchanged oxygen for different samples as related to unit of the surface (NO) calculated using the formulaewhere αginput-isotope fraction in the inlet mixture (0.95), CO2 − O2 concentration (mol mol−1), U–flow rate of the reaction mixture (mole/s), and NA–Avogadro number. It is seen that for (2.5–2.7)Fe(Co)/Al2O3-1200 samples, NO(t) dependencies (at higher rate of exchange on 2.5Fe/Al2O3-1200, as determined from the slope of No curves) reached the plateau after about 15 s after isotope admission corresponding to exchanged 4.9 × 1020 and 3.2 × 1020 O atoms per g for 2.5Fe/Al2O3-1200 and 2.7Co/Al2O3-1200 samples, respectively. Taking account of the quantity of supported Me (2.7 × 1020 atoms per g), one can suppose that within the accuracy of measurement, solely the oxygen of Fe2O3 and Co oxidic compounds (Co3O4 + Co3−xAlxO4) participated in the exchange. The integral quantity of exchanged oxygen in the 2.5Fe/Al2O3-C sample (7.9 × 1020 atoms per g) substantially exceeds that in supported Fe oxide. Provided substantially more dispersed FeOx species have been formed in this sample compared with those in 2.5Fe/Al2O3-1200 one (Table 3), it is more probably support oxygen that can transfer to O2 molecule through developed Fe-alumina interface.

 | ||
| Fig. 5 Time dependence of the quantity of exchanged oxygen (NO) in 18O SSITKA experiments for different Me/Al2O3 and Me/CeO2 samples. | ||
Substantially more profound and faster oxygen exchange took place on CeO2 based samples, obviously involving the bulk of the support starting from the very low time after 18O2 admission. The rate of exchange changed in the following order: 0.91Co/CeO2 ≥ 0.86Fe/CeO2 > 0.91Ni/CeO2 ≈ CeO2 and correlated with values of the red shift of the CeO2 band gap energy (Eg) in turn depending on the content of Me–Ce mixed solution in the sample characterized by increased degree of disorder. Therefore, oxygen transfer from fluorite lattice to the surface by extended vacancies arising after insertion of Men+ ions into the fluorite lattice can be responsible for enforced rate of isotope exchange on Me/CeO2 (Me = Co, Fe) samples compared with pure CeO2.
3.5. Catalytic activity
Effect of the support nature. Among Al2O3-1200-based samples with similar surface concentration as supported Me (6.7 × 1019 at m−2), catalytic activity in N2O decomposition at T = 750–900 °C can be ordered as follows: 2.5Fe/Al2O3-1200 > 2.7Co/Al2O3-1200 > 2.7Ni/Al2O3-1200 > Al2O3-1200 (Fig. 6). The (0.86–0.91)Me/CeO2 samples with quite close, both calculated (6.7 × 1019 at m−2) and real (Table 3), surface concentration of Me as in (2.5–2.7)Me/Al2O3-1200 samples revealed substantially higher intrinsic activity. In the case of Co and Ni containing samples such increase of the activity could be partly related to the formation of principally different state of active component (oxides/hydroxides in CeO2 instead of corresponding Co(Ni)–Al–O spinels located on the surface of Al2O3). Nevertheless, a similar effect observed for Fe based samples characterized by similar type of active species on both supports obviously points to the importance of increased oxygen mobility of CeO2, which in turn was substantially more active than Al2O3. The general scheme of N2O decomposition obeys the Langmuir–Hinshelwood mechanism:
| N2O + S → N2O-S | (1) |
| N2O-S → N2 + O − S | (2) |
| 2O-S ↔ O2 + 2S | (3) |
| O-S ↔ Oss + S | (4) |
 | ||
| Fig. 6 Temperature dependence of the activity of different (2.5–2.7) Me/Al2O3-1200 (open symbols) and (0.86–0.91) Me/CeO2 (solid symbols) samples (Me = Co, Fe, Ni) in the reaction of N2O decomposition. | ||
Effect of Fe content and FeOx dispersion. With variation of Fe content in Al2O3-1200-based samples from 0.89 to 6.6 wt% (Fe was chosen as the most active among Me) we observed the inverse “activity (as related both to sample surface unit and to one Fe atom) – Fe content” dependence (Fig. 7A). The same Fes values for 0.86Fe/Al2O3-1200 and 2.5Fe/Al2O3-1200 samples (Table 3) indicate decrease of FeOx species dispersion (DFe, Table 3) at higher Fe content. Finally, similar rates of N2O decomposition were measured for bulk Fe2O3 and all Fe/Al2O3-1200 samples (Fig. S3†), although in the former case the Fes value should be obviously higher. Therefore, activity of surface Fe sites decreases substantially with enlargement of FeOx species.
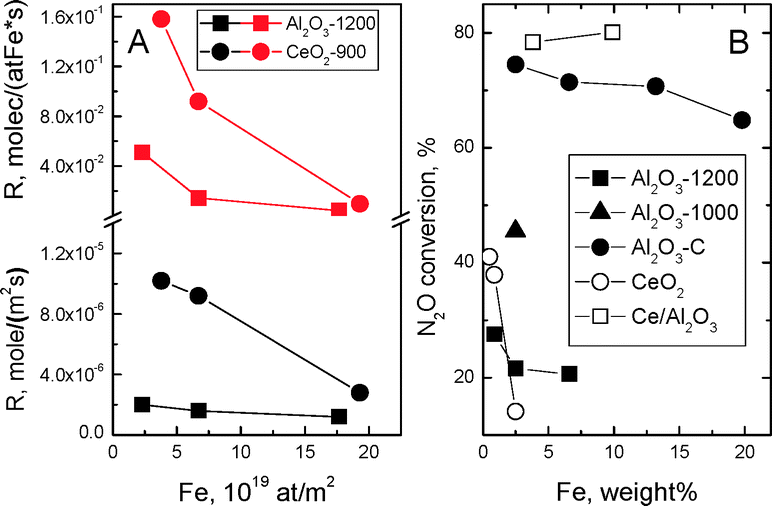 | ||
| Fig. 7 Reaction rate (A) and N2O conversion as measured at the same sample loading and flow rate (B) at 800 °C depending on Fe content in Al2O3-1200, Al2O3-1000, Al2O3-C, CeO2 and Ce/Al2O3 based samples. | ||
With variation of Fe content in CeO2-based samples from 0.5 to 2.5 wt% (i.e. at similar interval of supported Fe to that in the case of Al2O3-1200-based samples as related per surface unit and thus similar dispersion of FeOx species) their higher activity due to increased mobility of oxygen is more prominent at lower quantity of supported Fe. However, activity drop was substantially more prominent, although even higher Fes values were measured for the 2.5Fe/CeO2 sample than for the 0.86Fe/CeO2 one (Table 3). Supposing the rate of oxygen transfer from the bulk to active sites should depend on the length of MeOx–CeO2 interface, its decrease at enlargement of FeOx species (Table 3), revealed as start of oxygen exchange at higher temperature (Fig. S4†) should make an additional negative contribution to the activity.
Effect of the support surface area. In accordance with observed effect of FeOx dispersion on activity, use of the support characterized by higher specific surface area allows obtaining more dispersed FeOx species at substantially higher total Fe content (Table 3). This opens up additional possibilities for increase of conversion as measured at the same sample loading. Indeed, at a similar quantity of supported Fe, N2O conversion increased with SBET value of Al2O3 (Fig. 7B).
Unfortunately, for Fe/Al2O3-C samples we failed to perform catalytic tests at reasonably low conversion values for correct calculation of the reaction rate values. This fact and substantial negative effect of the quantity of supported Fe on SBET value (Tables 2 and 3) complicated analysis of parameters determining the activity. To overcome this problem, we calculated the concentration of exposable FeOx species (Feexp) per weight unit by the following formula: Feexp = Fes*SBET, where Fes is the surface concentration of Fe as measured by XPS (Table 3), and compared conversion values were measured for the same catalyst loading as dependent on both Fe content in the sample and Feexp. It turned out that N2O conversion decreased with increase of Fe content from 2.5% to 19.8% (Fig. 7B) or FeOx dispersion drop (Table 3), but this order cardinally differed from that for Feexp (13.2Fe/Al2O3-C > 6.6Fe/Al2O3-C > 19.8Fe/Al2O3-C > 2.5Fe/Al2O3-C). Therefore, similar to Fe/Al2O3-1200 samples, it is the size effect of FeOx species that seems to be the dominant factor determining descending character of “conversion-Fe content” dependence. Similar reverse dependence was already observed earlier for lower (500–650 °C) temperatures.51 Taking account of SSITKA data obtained for 2.5Fe/Al2O3-1200 and 2.5Fe/Al2O3-C samples, higher activity of smaller FeOx species can be due to growing ability of oxygen supply to reduced active sites S from the alumina regions adjacent to developed MeOx-alumina interface (step 4). Oxygen (3 vol%) and H2O (3 vol%) addition into the reaction mixture, in accordance with considered abovementioned scheme of the mechanism, decreased the observed N2O conversion, but did not change the order of activity (Fig. S5†).
Effect of CeO2 dispersion. Using the effect of high oxygen mobility of CeO2 for the increase of the efficiency of N2O decomposition (Fig. 7B) is restricted by its low surface area under the reaction conditions.3,20 Therefore, in addition to traditionally used increase of concentration of active sites by supporting more active component, we tried to disperse CeO2 by supporting onto high surface area Al2O3. Only Fe oxide was used in this case to avoid formation of low active Ni (Co)–Al spinels. The 3.8Fe/Ce/Al2O3 and 9.9Fe/Ce/Al2O3 samples revealed that the highest N2O conversion (Fig. 7B) was due to the part of FeOx that was anchored to the surface of higher dispersed ceria particles (Table 1, Fig. 4c). At the same time, the size effect, at least for the sample 3.8Fe/Ce/Al2O3, cannot be completely excluded (compare DFe values in the Table 3 for the samples 3.8Fe/Ce/Al2O3 and 6.6Fe/Al2O3-C with similar SBET).
Effect of the support nature. Among (2.5–2.7) Me/Al2O3-1200 samples, the 2.5Fe/Al2O3-1200 one was the most active (Fig. 8A) and characterized by the highest selectivity to NO + NO2 (86–88%). As a result, the orders for the activity and the efficiency of NOx formation evaluated as NOxyield = Conversion × NOxselectivity were similar to that for the activity of N2O decomposition: 2.5Fe/Al2O3-1200 ≫ 2.7Co/Al2O3-1200 > 2.7Ni/Al2O3-1200–Al2O3-1200. In addition, the N2O yield value calculated in a similar manner as for NOxyield on the 2.5Fe/Al2O3-1200 sample in this temperature interval was lower than 2%, which is comparable to that for Pt–Rh gauzes,1 while continuous increase of N2O yield with temperature was noted for all other samples. At lower O2 concentration in the inlet mixture (2.5%), the yield of NOx decreased to 77–78%, mainly because of lower selectivity. For Co- and Ni-based samples and for pure Al2O3-1200, negative changes of conversion and NOx selectivity were substantially more prominent.
 | ||
| Fig. 8 Temperature dependence of NH3 conversion and N2O yield for (2.5–2.7) Me/Al2O3-1200 (A) and (0.86–0.91) Me/CeO2-900 (B) samples. Catalyst loading 0.015 g. | ||
We failed to calculate the rate of ammonia oxidation in all temperature intervals due to non-differential reactor operating conditions. Hence, to compare intrinsic activity of samples characterized by different SBET values, we tested catalytic properties of (0.86–0.91) Me/CeO2 samples at catalyst loading of 0.043 g corresponding to the same surface area as for Al2O3-1200-based samples. In these conditions, at 750 °C, complete NH3 conversion was measured on all (0.86–0.91)Me/CeO2 samples, and 96% for CeO2 that is obviously due to capability of CeO2 itself to donate lattice oxygen for ammonia oxidation.16 Even at the same catalyst loading as for Al2O3-1200-based samples (0.015 g), i.e. at about a 3 times lower number of Me sites therein (Table 3), higher values of NH3 conversion were observed for corresponding CeO2 based samples (Fig. 8B). This increase, at least in the case of Fe-based samples, is exclusively due to the ability of the support to donate oxygen to FeOx, since substantially lower conversion values were measured on CeO2 and bulk FeOx characterized by similar SBET values. N2O yield values were reasonably lower than on corresponding Al2O3-1200-based samples. In addition, at 800 °C, the order of NH3 conversion and NOx yield (not shown for brevity), 0.91Co/CeO2 ≥ 0.86Fe/CeO2 > 0.91Ni/CeO2 ≈ CeO2, was exactly the same as for the efficiency towards oxygen exchange therein (Fig. 4).
Strong decrease of both NH3 oxidation52 and N2O decomposition32 over Co oxide at high temperatures was shown to be due to Co3O4 reduction to CoO. We did not observe such deactivation of the 0.91Co/CeO2 sample with temperature both at N2O decomposition (Fig. 6), as in Co3O4–CeO2 mixed oxides with low Co content,11 and at ammonia oxidation (Fig. S7B†), although Co3O4 reduction under TPD conditions was observed (Fig. S6†). It obviously can be explained by higher resistance of the Co–Ce interface towards reduction due to some oxygen supply from CeO2 by reaction (4). Nevertheless, at lower temperature, the 0.91Co/CeO2 sample with Co state closer to Co3O4 was already substantially more active than 0.87Fe/CeO2 both in N2O decomposition (Fig. S7A†) and NH3 oxidation (Fig. S7B†).
Effect of Fe content and FeOx dispersion. Decreasing dependence of NH3 conversion vs. Fe content in Al2O3-1200 and CeO2-based samples (0.015 g sample loading for all these experiments) was observed (Fig. 9), similarly to the reaction of N2O decomposition, and thus can be related to size effect. Moreover, for the Fe/CeO2 case, it was more prominent due to attenuation of oxygen transfer processes through the Fe–CeOx interface (Fig. S4†). Nevertheless, unlike the reaction of N2O decomposition, FeOx dispersion is not a predominant factor for NH3 conversion any more. Therefore, at Fe content lower than 3 × 1019 atoms per m2 (the case with Fe/Al2O3-C and Fe/Ce/Al2O3 samples), the NH3 conversion value depended mainly on the quantity of exposable Fe (Feexp).
 | ||
| Fig. 9 Dependence of NH3 conversion (solid symbols) and NOx yield (open symbols) on Fe content in different Fe/(Al2O3,CeO2) samples. Catalyst loading 0.015 g. | ||
At the same time, these were larger FeOx species that converted NH3 to NOx more selectively in all Al2O3-based samples (Fig. S8†), finally resulting in quite similar (77–81%, in the case of Fe/Al2O3-1200 samples) or growing with Fe content (up to 79% for Fe/Al2O3-C) NOx yield values (Fig. 9). In accordance with findings of Pérez-Ramírez and Kondratenko16 on bulk Fe2O3, the desired reaction follows a Mars–van Krevelen-type scheme involving the participation of lattice oxygen in the NH3 conversion to NO. The degree of reduction of the oxide surface was shown to determine the product distribution. One can reasonably suppose that for small species, the FeOx surface should be in a more reduced state because of a smaller rate of dissociative activation of O2 molecules on the Fe–Al interface. In addition, a growing contribution from low selective NH3 oxidation on Al2O3 should be accounted as well.
We also believe that the more oxidized state of FeOx anchored to CeO2, resulting from more efficient O transfer through the Fe–Ce interface, causes higher selectivity to NOx in the 3.8Fe/Ce/Al2O3 and 9.9Fe/Ce/Al2O3 samples compared to the Fe/Al2O3-C ones with similar dispersion. In addition, substantially smaller values of N2O yield were measured on Fe/Ce/Al2O3 samples, which agree with their higher activity towards N2O decomposition.
Conclusions
We have shown that replacement of Al2O3 by CeO2 as the support for MeOx (Me = Fe, Co, Ni) characterized by increased lattice oxygen mobility resulted in substantial increase of the rates of 16O/18O exchange at 800 °C, N2O decomposition and NH3 oxidation on corresponding samples. Oxygen transfer from the fluorite lattice to the surface by extended oxygen vacancies arose after insertion of Men+ ions into the fluorite lattice was shown to be responsible for the enforced rate of isotope exchange.Formation of low active spinel-like Me–Al–O structures restricted the application of Co and Ni oxides in any Al2O3 containing catalysts. For CeO2- and Al2O3-based samples with supported FeOx, an obvious size effect was observed in both reactions. Therefore, highly dispersed FeOx was substantially more active due to change of contribution of oxygen supply from the support (both ceria and, probably, alumina) to reduced surface sites through Fe–CeOx interface thus increasing the rate of O2 desorption. However, smaller species oxidized NH3 to NOx less selectively.
Using high surface area Al2O3-C, the samples with high content of dispersed FeOx in the weight unit were synthesized to increase observable N2O conversion. To increase the surface area of CeO2 and thus efficiently use the promoting effect of oxygen mobility, CeO2 was dispersed onto alumina by precipitation. Fe/Ce/Al2O3 samples with optimal content of Fe revealed superior activity in N2O decomposition and NH3 oxidation to NOx compared with Fe/Al2O3 with similar SBET and dispersion of FeOx species values.
Acknowledgements
The authors thank Mrs. I. Kharina for preparation of the Al2O3-C sample.Notes and references
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117 CrossRef.
- P. Granger, P. Esteves, S. Kieger, L. Navascues and G. Leclercq, Appl. Catal., B, 2006, 62, 236 CrossRef CAS.
- P. Esteves, Y. Wu, C. Dujardin, M. K. Dongare and P. Granger, Catal. Today, 2011, 176, 453 CrossRef CAS.
- D. V. Ivanov, E. M. Sadovskaya, L. G. Pinaeva and L. A. Isupova, J. Catal., 2009, 267, 5 CrossRef CAS.
- Y. Wu, X. Ni, A. Beaurain, C. Dujardin and P. Granger, Appl. Catal., B, 2012, 125, 149 CrossRef CAS.
- D. V. Ivanov, L. G. Pinaeva, L. A. Isupova, E. M. Sadovskaya, I. P. Prosvirin, E. Yu Gerasimov and I. S. Yakovleva, Appl. Catal., A, 2013, 457, 42 CrossRef CAS.
- J. Pérez-Ramírez and M. Santiago, Chem. Commun., 2007, 619 RSC.
- G. Giecko, T. Borowiecki, W. Gac and J. Kruk, Catal. Today, 2008, 137, 403 CrossRef CAS.
- J. Kruk, K. Stołecki, K. Michalska, M. Konkol and P. Kowalik, Catal. Today, 2012, 191, 125 CrossRef CAS.
- E. Wilczkowska, K. Krawczyk, J. Petryk, J. W. Sobczak and Z. Kaszkur, Appl. Catal., A, 2010, 389, 165 CrossRef CAS.
- E. Iwanek, K. Krawczyk, J. Petryk, J. W. Sobczak and Z. Kaszkur, Appl. Catal., B, 2011, 106, 416 CrossRef CAS.
- M. Konsolakis, ACS Catal., 2015, 5, 6397 CrossRef CAS.
- V. A. Sadykov, L. A. Isupova, I. A. Zolotarskii, L. N. Bobrova, A. S. Noskov, V. N. Parmon, E. A. Brushtein, T. V. Telyatnikova, V. I. Chernyshev and V. V. Lunin, Appl. Catal., A, 2000, 204, 59 CrossRef CAS.
- L. Pinaeva, E. Sutormina, L. Isupova, N. Kulikovskaya and A. Marchuk, RU Pat., 2 430 782 C1, 2010 Search PubMed.
- L. Pinaeva, L. Isupova, I. Prosvirin, E. Sadovskaya, I. Danilova, D. Ivanov and E. Gerasimov, Catal. Lett., 2013, 143, 1294 CrossRef CAS.
- J. Pérez-Ramírez and E. Kondratenko, J. Catal., 2007, 250, 240 CrossRef.
- M. Zabilskiy, B. Eravec, P. Djinović and A. Pintar, Chem. Eng. J., 2014, 254, 153 CrossRef CAS.
- M. Konsolakis, S. A. C. Carabineiro, E. Papista, G. E. Marnellos, P. B. Tavares, J. Agostinho Moreira, Y. Romaguera-Barcelay and J. L. Figueiredo, Catal. Sci. Technol., 2015, 5, 3714 CAS.
- L. Isupova, Yu. Tanashev, I. Kharina, E. Moroz, G. Litvak, N. Boldyreva, E. Paukshtis, E. Burgina, A. Budneva, A. Shmakov, N. Rudina, V. Kruglyakov and V. Parmon, Chem. Eng. J., 2005, 107, 163 CrossRef CAS.
- I. Danilova, E. Slavinskaya, V. Zaikovskii, A. Ivanova, A. Boronin, R. Gulyaev and Yu. Amosov, Kinet. Catal., 2010, 51, 143 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627 CrossRef CAS.
- V. Malakhov, N. Boldyreva, A. Vlasov and L. Dovlitova, J. Anal. Chem., 2011, 66, 458 CrossRef CAS.
- L. G. Pinaeva, E. M. Sadovskaya, A. P. Suknev, V. B. Goncharov and B. S. Bal'zhinimaev, in Mass Spectrometry Handbook, ed. M. S. Lee, Wiley, Hoboken, 2012, pp. 1229–1256 Search PubMed.
- S. Kurajica, J. Popović, E. Tkalčec, B. Gržeta and V. Mandić, Mater. Chem. Phys., 2012, 135, 587 CrossRef CAS.
- O. Kirichenko, V. Ushakov, E. Moroz and M. Vorob'eva, Kinet. Catal., 1993, 34, 739 CAS.
- Z. Zhang, D. Han, S. Wei and Y. Zhang, J. Catal., 2010, 276, 16 CrossRef CAS.
- A. Gupta, A. Kumar, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2009, 21, 4880 CrossRef CAS.
- A. Piras, S. Colussi, A. Trovarelli, V. Sergo, J. Llorca, R. Psaro and L. Sordelli, J. Phys. Chem. B, 2005, 109, 11110 CrossRef CAS PubMed.
- A. Bensalem, J. C. Muller and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 153 RSC.
- R. Zhang, J. T. Miller and C. D. Baertsch, J. Catal., 2012, 294, 69 CrossRef CAS.
- R. C. Olegário, E. C. F. Souza, J. F. M. Borges, J. B. M. Cunha, A. V. C. Andrade, S. R. M. Antunes and A. C. Antunes, Dyes Pigm., 2013, 97, 113 CrossRef.
- C. He, M. Paulus, W. Chu, J. Find, J. A. Nickl and K. Kohler, Catal. Today, 2008, 131, 305 CrossRef CAS.
- I. G. Casella and M. R. Guascito, J. Electroanal. Chem., 1999, 476, 54 CrossRef CAS.
- H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2005, 231, 145 CrossRef CAS.
- M. H. Kim and K.-H. Choo, Catal. Commun., 2007, 8, 462 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem., 2010, C114, 111 Search PubMed.
- X. Duan, M. Pan, F. Yu and D. Yuan, J. Alloys Compd., 2011, 509, 1079 CrossRef CAS.
- N. Srisawad, W. Chaitree, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, J. Nanomater., 2012, 2012, 1 CrossRef.
- D. Barreca and C. Massignam, Chem. Mater., 2001, 13, 588 CrossRef CAS.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49 CrossRef CAS.
- D. Nikolova, R. Edreva-Kardjieva, G. Gouliev, T. Grozeva and P. Tzvetkov, Appl. Catal., A, 2006, 297, 135 CrossRef CAS.
- P. Dufresne, E. Grimblot and J. P. Bonnelle, J. Phys. Chem., 1981, 85, 2344 CrossRef CAS.
- E. Laurent and B. Delmon, J. Catal., 1994, 146, 281 CrossRef CAS.
- C. Jiménez-González, Z. Boukha, B. de Rivas, J. J. Delgado, M. A. Cauqui, J. R. González-Velasco, J. I. Gutiérrez-Ortiz and R. López-Fonseca, Appl. Catal., A, 2013, 466, 9 CrossRef.
- P. Prieto, V. Nistor, K. Nouneh, M. Oyama, M. Abd-Leftil and R. Diaz, Appl. Surf. Sci., 2012, 258, 8807 CrossRef CAS.
- S. Suzuki, K. Yanagihara and K. Hirokawa, Surf. Interface Anal., 2000, 30, 372 CrossRef CAS.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114, 12909 CAS.
- J. Z. Shyu, W. H. Weber and H. S. Gandhi, J. Phys. Chem., 1988, 92, 4964 CrossRef CAS.
- C. Ge, L. Liu, Z. Liu, X. Yao, Y. Cao, C. Tang, F. Gao and L. Dong, Catal. Commun., 2014, 51, 95 CrossRef CAS.
- M. Zabilskiy, P. Djinović, E. Tchernychova, O. Tkachenko, L. Kustov and A. Pintar, ACS Catal., 2015, 5, 5357 CrossRef CAS.
- P. Pomonis, D. Vattis, A. Lycourghiotis and C. Kordulis, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2043 RSC.
- M. C. Goromonzi, S. K. Raman, B. D. Padalia, A. Manohar and P. N. Koul, React. Kinet. Catal. Lett., 1980, 15, 131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01381j |
| This journal is © The Royal Society of Chemistry 2016 |