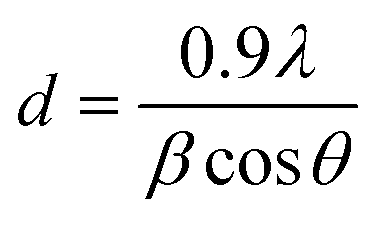Bifunctional hybrid catalysts derived from Cu/Zn-based nanoparticles for single-step dimethyl ether synthesis†
Received
9th July 2015
, Accepted 23rd August 2015
First published on 25th August 2015
Introduction
Fuel production and development of new routes to base chemicals from renewable resources are currently drawing much attention worldwide. The production of syngas (CO + H2), e.g. from biomass-derived feedstock and its conversion to methanol, dimethyl ether (DME), or gasoline, provide an attractive option.1 Recently, DME has attracted much interest not only as liquefied petroleum gas (LPG) for domestic applications and intermediate product for various base chemicals but also as a clean diesel substitute. Traditionally, DME is produced from syngas in a two-step process, where methanol is obtained over Cu-based catalysts in the first stage, followed by subsequent methanol dehydration over inorganic solid acids (e.g., γ-Al2O3 or zeolite) in the second stage. The overall conversion of syngas to DME (Scheme 1) described by eqn (4) involves the two consecutive reactions, methanol synthesis (1) and methanol dehydration (2), in parallel with the water–gas shift (WGS) reaction (3). The conversion of CO in (1) is limited by thermodynamics, typically leading to extensive recycling of non-converted syngas.2 If methanol synthesis is coupled in situ with its condensation to DME and immediately removed from the equilibrium in a tandem catalytic reaction ((1) + (2)), the thermodynamic equilibrium constraint may be circumvented. Hence, the single-step, syngas-to-DME (STD) process allows for higher CO conversions compared to the conventional two-step process and is receiving increased academic and industrial attention worldwide. The design of STD catalysts remains a crucial issue for enhancing catalytic efficiency.3
 |
| | Scheme 1 Reactions relevant in the conversion of syngas to DME. | |
In general, the preparation method significantly affects the performance of the catalyst in the STD process. Traditionally, the catalysts used in the STD process have been formulated by mechanically mixing the conventional methanol catalyst (e.g., Cu/ZnO/Al2O3) with a dehydration catalyst (e.g., γ-Al2O3 or zeolite such as H-ZSM-5).4 Besides the physical, wet or dry mixing of the two active components, a plethora of synthetic strategies for combining CO hydrogenation and methanol dehydration functionalities in bifunctional hybrid catalysts have been pursued, typically involving conventional mixed metal impregnation, co-precipitation impregnation, or co-precipitation sedimentation of the dehydration catalyst.5 Recently, some new synthetic approaches for bifunctional STD catalysts have been reported. Using a one-pot synthesis, small Cu nanoparticles, e.g., were obtained inside a mesoporous γ-Al2O3 matrix.6 This mesoporous Cu/γ-Al2O3 catalyst was highly active in the STD process, even though the catalyst didn't contain ZnO. Clearly, the balance between the methanol and dehydration functionalities is a critical issue in the design of the overall STD catalyst; their optimum ratio is further related to the operating conditions in the STD process. For the methanol synthesis, Cu/ZnO catalysts have been widely investigated over the last decades, including the doping with various promoters (e.g., Zr, Ga or Mn).7 Conventional γ-Al2O3 is recognized as a very efficient dehydration catalyst due to high DME selectivity, low cost, excellent lifetime and high mechanical resistance.3 The net acidic function for methanol dehydration can be further adjusted through the acidic strength and the number of acid sites. H-ZSM-5, e.g., was reported to show a high activity for the dehydration reaction in the STD process, combining strong acidity with a high density of acidic sites.8 Core–shell-like capsule catalysts were prepared by Tsubaki et al., where the methanol active component was enwrapped by a layer of the dehydration catalyst (i.e., a microsized zeolite shell).9 Although the CO conversion was only 5.59%9a and 9.48%9b for H-ZSM-5-encapsulated Cu/ZnO/Al2O3 and Pd catalysts, respectively, an increase in DME selectivity was observed as compared to the physically mixed system. An increase in CO conversion was further achieved for the silicoaluminophosphate-11 (SAPO-11) zeolite shell encapsulated Cu/ZnO/Al2O3 catalyst.9c Synergistic effects have been observed for bifunctional catalysts, if the two active components were finely dispersed and maintained in close contact.10 However, the interaction between the two functional components during catalyst preparation may also result in the deterioration of the textural and acidic properties of the dehydration catalyst and, thus, in a reduced efficiency for the STD process.11a Recently, the Cu/ZnO-based methanol active function was confined inside the pores of a mesoporous SBA-15 silica host prior to mixing with the dehydration component (i.e., H-ZSM-5) to minimize adverse interactions between the two types of active sites.11b By this, detrimental interactions and the sintering of Cu particles was avoided, resulting in an improved catalyst stability. Moreover, the coverage of active centers needs to be avoided. In general, texture, structure, and catalytic properties of the catalysts were not only affected by the type of applied synthetic methodology but also by the various preparation parameters, including concentrations, pH value, and aging time.12 Hence, these preparation routes face several challenges to balance the two functionalities, i.e. controlling dispersion and effective structure combination of the two active components as well as low reproducibility. In this context, well-defined nanoparticles as catalyst precursors may open up a path to model systems for contributing to the fundamental understanding of synthesis–structure–function relationships and to the future design of highly effective catalysts.13 Over the last decades, various techniques have been developed which enable the synthesis of highly uniform, nanoscopic metal particles with a predefined size and shape, e.g. by reduction of metal salts with divers reducing agents (such as polyols, borohydrides, amines, or organo aluminium compounds) in the presence of stabilizing ligands.14 These techniques for nanoparticle synthesis provide several advantages for catalyst design and may help to overcome the problems in the conventional preparation methods. Although the synthesis of Cu/Zn-based nanoparticles via colloidal approaches has been studied by several authors,15 their catalytic application has yet been limited to methanol synthesis. To the best of our knowledge, there have been no previous studies on the integration of predefined nanoparticles into bifunctional STD catalysts.
Herein, we report the design of new bifunctional, supported STD catalysts by taking advantage of highly uniform, nanoparticulate precursors. The well-defined Cu/Zn-based nanoparticles were synthesized in solution and subsequently supported on a conventional dehydration catalyst. This procedure ensured the intimate contact of the nanoparticulate methanol active component with the dehydration catalyst and enabled the individual optimization and balancing of the two active functionalities in the bifunctional catalyst. The characterization of the catalysts was carried out using electron microscopy (SEM-EDX, TEM), temperature programmed reduction (H2-TPR), N2-physisorption, powder X-ray diffraction (XRD), and X-ray absorption spectroscopy (XAS). The catalytic performance was examined in a continuously operating fixed bed reactor on a lab scale using a simulated biomass-derived, CO-rich synthesis gas (CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio 1:1).
:H2 ratio 1:1).
Experimental section
Materials
Copper(II) acetylacetonate (Cu(acac)2, Sigma Aldrich, 99.99%), diethylzinc (Zn(C2H5)2, Sigma Aldrich, ≥ 52 wt% Zn), toluene (Sigma Aldrich, 99.8%), tetrathydrofuran (THF, Sigma Aldrich), oleyl amine (OA, Sigma Aldrich, tech. grade), ethanol (absolute) were used without further purification. γ-Al2O3 (BET surface area 221 m2 g−1, pore size 7.7–9.2 nm) was purchased from Alfa Aesar. Unless specified otherwise, all steps of particle synthesis were carried out under inert conditions using dry solvents.
Synthesis of Cu- and Cu/Zn-based nanoparticles
Zn(C2H5)2 (12 mL, 1.2 M in toluene) was slowly added to Cu(acac)2 (10.5 g, 40 mmol) suspended in toluene (800 mL) at 40 °C and stirred for 24 h. The volatile components were eventually removed and the product was vacuum dried. 11.7 g Cu/Zn-based nanoparticles (CuZn-NP-1; mean particle diameter 6.2 ± 2.0 nm) were isolated as a black powder (elemental composition 16% Cu, 30% Zn) and further used for the preparation of bifunctional catalysts CZA-1 and CZA-2.
For catalysts CA-3, CZA-4 and CZA-5, the Cu nanoparticles were synthesized using a modified procedure previously reported by Son et al.16 Briefly, Cu(acac)2 (15.8 g, 60 mmol) was suspended in degassed OA (600 mL). The suspension was heated under stirring to 110 °C (heating rate: 20 °C min−1), which was maintained for 30 min. Next, the temperature was further increased to 250 °C (heating rate: 20 °C min−1), which was maintained for 60 min. The Cu nanoparticles were precipitated with ethanol (600 mL) and isolated by centrifugation. After redispersion in toluene (900 mL), the Cu nanoparticles (Cu-NP-3) were initially obtained as a burgundy solution, which gradually turned dark green on exposure to air. The as-obtained Cu nanoparticles were used as methanol active component in catalyst CA-3. The Cu/Zn-based nanoparticles, CuZn-NP-4 and CuZn-NP-5, were further synthesized from Cu-NP-3 and used as the methanol active component in catalysts CZA-4 and CZA-5, respectively. For this purpose, an amount of 5 mmol or 60 mmol Zn(C2H5)2 were additionally added to each 450 mL of Cu nanoparticles in toluene (Cu-NP-3) and hydrolyzed with a water loaded Ar stream for 24 h to yield the Cu/ZnO-based nanoparticles, CuZn-NP-4 and CuZn-NP-5, respectively.
Preparation of bifunctional STD catalysts
γ-Al2O3 was thoroughly purified with water, dried at 110 °C, and calcined at 550 °C for 5 h. The γ-Al2O3 pellets were crushed and sieved to the fraction of 80–160 μm. The bifunctional catalysts CZA-1, CA-3, CZA-4, and CZA-5 were prepared by dispersing the sieved fraction of γ-Al2O3 in the suspension of the nanoparticles CuZn-NP-1, Cu-NP-3, CuZn-NP-4, and CuZn-NP-5, respectively. The solvent was evaporated in vacuum to yield a black powder. The bifunctional catalyst was eventually treated in air at 350 °C for 4 h to remove the organic ligands of the nanoparticles. In the case of catalyst CZA-2 a different procedure was applied. Here, the nanoparticles (CuZn-NP-1) were directly calcined at 350 °C for 4 h, then pelletized, crushed, and sieved to the fraction of 80–160 μm. To yield the final catalyst CZA-2, this powder was physically mixed with γ-Al2O3 in a weight ratio of 1:1. In Table 1 the experimental parameters for the preparation of bifunctional catalysts are summarized.
Table 1 Experimental parameters of catalyst preparation
| Catalyst |
Nanoparticles |
γ-Al2O3 |
|
Impregnation of the nanoparticles on γ-Al2O3 as a dehydration catalyst.
Mechanical mixing of the calcined nanoparticles with the dehydration catalyst.
|
| CZA-1a |
CuZn-NP-1 (100 mL in THF, Cu 5.85 mg mL−L, Zn 10.96 mg mL−1) |
1.6 g |
| CZA-2b |
CuZn-NP-1 (1 g calcined powder) |
1.0 g |
| CA-3a |
Cu-NP-3 (450 mL in toluene, Cu 3.8 mg mL−1) |
1.4 g |
| CZA-4a |
CuZn-NP-4 (450 mL in toluene, Cu 3.8 mg mL−1, Zn 2.1 mg mL−1) |
2.5 g |
| CZA-5a |
CuZn-NP-5 (450 mL in toluene, Cu 3.8 mg mL−1, Zn 8.2 mg mL−1) |
5.0 g |
Characterization
The nanoparticles were analyzed with transmission electron microscopy (TEM) and elemental analysis (EDX) on a FEI Tecnai F20 ST TEM (operating voltage 200 kV), which was equipped with a field emission gun and EDAX EDS X-ray spectrometer (Si(Li) detecting unit, super ultra thin window, active area 30 mm2, resolution 135 eV (at 5.9 keV)). For TEM analysis, a small droplet of the dispersed sample was deposited on amorphous carbon-coated 400 mesh Ni grids and eventually air dried. For bifunctional catalysts, the solid powder was initially immersed in iso-propanol and the as-obtained suspension was deposited onto the TEM grid. Additionally, scanning electron microscopic (SEM) investigations were carried out on the bifunctional bulk catalysts with a DSM 982 Gemini SEM, equipped with a Schottky-type thermal field emission cathode (Co. Zeiss, Germany) and EDX. The elemental composition of the bifunctional catalysts was determined by SEM-EDX analysis of the bulk catalysts. In general, the particle sizes and size distributions were statistically evaluated from electron microscopy observations based on typically 100 particles.
X-ray absorption spectra (XANES and EXAFS) at Cu K and Zn K absorption edges were recorded at the XAS beamline of ANKA synchrotron in transmission mode.17 The nanoparticles and the calcined catalyst were measured as pellets pressed with cellulose, and the catalyst after reaction was sealed in a quartz capillary (diameter 0.5 mm, wall thickness 20 μm, Co. Hilgenberg) under inert conditions. The beam size was kept as 6 × 0.45 mm. A XAS spectrum of Cu foil was measured simultaneously and used for energy calibration. The spectra were analyzed using the ATHENA program from the IFFEFIT package.18 Fitting of the EXAFS spectra of Cu nanoparticles and the catalyst sample after catalysis was done using ARTEMIS software (IFFEFIT).18 After Fourier transformation of the k1-, k2-, and k3-weighted EXAFS function between 3 and 12 Å−1, data fitting was performed in R-space between 1 and 3 Å (corresponding to the first Cu–O and Cu–Cu shells). As model compounds for EXAFS fitting, Cu foil and Cu2O were chosen. From the fits of model compounds, S20 = 0.87 (Cu) and 0.72 (O) were obtained and used for fitting the catalyst spectra. To estimate the size of Cu clusters fitting of the Cu K edge EXAFS spectra was done (cf. Table S1, ESI†) offering the possibility to determine average coordination numbers which, in turn, depend on the particle size.19
Powder X-ray diffraction (XRD) studies were performed on a PANalytical X'Pert Pro X-ray diffractometer employing a Bragg–Brentano geometry with Cu Kα radiation and a Ni filter. The diffractograms were recorded from 5° to 120° over a period lasting 60 min. The reflections were compared to reference compounds reported in the Joint Committee of Powder Diffraction Standards (JCPDS) data base. The crystallite size d was calculated based on the reflections at 43° and 39° (2θ) for the Cu and the CuO phase, respectively, using the Scherrer equation,
| |  | (1) |
where 0.9 is a shape factor,
β the full width at half maximum of the diffraction peak and
θ the diffraction angle. LaB
6 (NIST) was measured in order to account for instrumental broadening.
The specific total surface areas of the catalysts (BET) were determined by N2-physisorption measurements employing a Quantachrome Nova 2000e instrument. Prior to physisorption measurements, the calcined catalyst (at least 20 mg) was degassed in vacuum at 230 °C for 12 h. The pore size distributions were calculated according to the Barret–Joyner–Halenda (BJH) method. For temperature programmed reduction (TPR) measurements, the calcined catalyst (minimum amount 120 mg) was initially dried in an Ar stream (30 mL min−1, heating rate 5 °C min−1, 200 °C, 60 min), and the reduction behavior (TPR) was subsequently analyzed from 20 °C to 300 °C in a reducing gas stream (6 vol% H2 in Ar; heating rate 2 °C min−1) with a Micromeritics AutoChem HP 2950. NH3 was used to investigate the acidity of the bifunctional catalysts. Temperature programmed desorption (TPD) measurements were performed with an Autochem HP 2950. The samples (at least 80 mg) were initially dried at 200 °C under He, and NH3 was subsequently loaded at 50 °C for 60 min. Desorption of NH3 was recorded with a TCD detector in the range of 50 to 600 °C (heating rate 4 °C min−1).
Catalytic tests
The catalytic properties of the catalysts in the STD reaction were investigated in a continuously operating laboratory scale plant equipped with a fixed bed reactor (volume 60.3 mL) and mass flow controllers (Co. Bronkhorst). The gas composition was analyzed by online gas chromatography (GC) using two columns (RESTEK RT®-U-Bond, RESTEK RT-Msieve) and a thermal conductivity detector (TCD) and a flame ionization detector (FID). The temperature of the exit line from the reactor to the GC was 120 °C. For GC analysis, Ar was used as a carrier gas (split ratio 20:1). The injector temperature and pressure were 200 °C and 3.5 bar, respectively; the total column flow was 2 mL min−1. The temperature in the GC oven was increased from 70 °C to 150 °C (10 °C min−1). The operating conditions used for catalyst testing have been selected according to previously established and published procedures.4a,c For all reactions, 2 g catalyst were loaded into the reactor and in situ reduced by heating to 240 °C in 2% H2 in Ar (total flow: 50 mL min−1; heating rate: 17 °C h−1). After keeping this temperature for another 2 h, the gas flow was switched to pure H2, while the temperature was further raised to 250 °C (heating rate: 10 °C h−1), and finally kept for 2 h at 250 °C. The reactor was then purged with argon and pressurized to 51 bar. The total gas flow was 100 mLNTP min−1 containing a (CO + H2):(N2 + Ar) mixture with a ratio 3:7, where the H2:CO ratio and Ar:N2 ratio were 1:1 and 6:1, respectively. CO conversion (XCO in %) was obtained by| |  | (1) |
with ṄCO,0 and ṄCO corresponding to CO flow (mol/s) at reactor inlet and at reactor outlet, respectively. For XCO,Cu, the CO conversion was normalized over the molar Cu content of the bifunctional catalyst (metal-based). The selectivity of component i (Si) was calculated based on the number of C atoms in the feed by| | 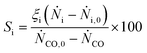 | (2) |
with ṄCO,0 and ṄCO corresponding to flow of component i (mol) at reactor inlet and at reactor outlet, respectively, and ξi to the number of carbon atoms in component i.
Results and discussion
Bifunctional STD catalysts were obtained in a two-step procedure from nanoparticle precursors. First, the Cu/Zn-based nanoparticles were synthesized via colloidal chemistry, then the nanoparticles were integrated in the conventional dehydration catalyst (i.e., γ-Al2O3).
A synthetic procedure for Cu/Zn-based nanoparticles was developed by reacting Cu(acac)2 with Zn(C2H5)2. The synthesis yielded particles in the lower nanometer size range and could easily be scaled-up to the required amount of catalyst. In contrast to other methods, no additional ligands, polymers, or surfactants were needed, which control both the particle nucleation and growth processes as well as the stabilization of the nanoparticles. After addition of Zn(C2H5)2 to the grey-blue suspension of Cu(acac)2, a dark red-brown, colloidal solution containing small Cu/Zn-based nanoparticles (CuZn-NP-1) was formed. TEM images confirmed the presence of 6.2 (±2.0) nm Cu nanoparticles (Fig. 1). The reaction of non-zero-valent transition metal precursors (group 8–10) with organometallic compounds, including AlR3, Grignard or organo Zn reagents, was previously studied by several groups.20 Organo Zn compounds were used for the synthesis of various coordination compounds with transition metal – Zn bonds and nanoparticles.21 Based on previous work by Wilke and co-workers, nanoparticle formation could proceed via replacement of anions in Cu(acac)2 by ethyl groups of Zn(C2H5)2.22 Cu-alkyl intermediates further decompose during reduction of the metal cation, yielding metallic Cu nanoparticles stabilized by Zn-containing species, probably Zn(acac)2 or Zn(acac)(C2H5). This was supported by GC analysis of the gas phase after nanoparticle synthesis, which confirmed the formation of various side products such as C2H4, C2H6, and C4H10. The formation of Zn(acac)2 was also observed by IR spectroscopy. The reduction of the copper species was confirmed by XAS spectra (Fig. 2). The X-ray absorption near edge structure (XANES) spectrum of the nanoparticles (CuZn-NP-1) showed the same edge features as metallic Cu, however it substantially differed from the spectrum of Cu metal after the absorption edge. The observed higher “white line” may result from interaction with O in a stabilizing Zn containing shell around the metallic Cu core of the nanoparticles. The flat structure without wiggles after the edge corresponded to very low average coordination numbers of Cu, indicating very small Cu nanoparticles. To obtain further information on the structure, Fourier-transformed (FT) k3-weighted extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3) were analyzed. At the Cu K edge, the spectrum showed backscattering peaks from O and from Cu, the latter at the same distance as in metallic Cu (0.254 nm, Table S1†). This further supported a model where the Cu surface was stabilized by O (or C) atoms from an organic Zn compound. Note that backscattering caused by neighboring atoms (C and O; Cu and Zn) cannot be distinguished by conventional EXAFS analysis due to their similar backscattering behavior. To exclude the formation of a Cu/Zn alloy, the Zn K edge spectra were analyzed in addition to the Cu K edge. In the case of the nanoparticles, only Zn–O backscattering from a first coordination shell and no Zn–Zn (or Zn–Cu) interaction was determined. Thus, Zn atoms may be a part of the organic shell without significant direct interaction with Cu atoms or with other Zn atoms. Note that the absence of Zn–Zn contributions at R = 3.1 Å further shows that no ZnO-particles are formed at this stage. Overall, the mechanistic details and intermediates of nanoparticle formation by this reaction remain unknown and may depend on the actual Cu2+/ZnEt2 ratio. As a reference, 11 nm Cu nanoparticles (Cu-NP-3) were synthesized by thermal decomposition of Cu(acac)2 in oleylamine (OA) after a modified procedure, which was initially described by Son et al.16 These particles were used to prepare a monometallic reference catalyst (CA-3). The addition of Zn(C2H5)2 and subsequent hydrolysis yielded CuZn-based nanoparticles (CuZn-NP-4, CuZn-NP-5) with different Cu/Zn ratios. The EDX analysis of these particles revealed that the Zn phase was formed in close contact around the Cu nanoparticles (Fig. 1b). The nanoparticles (CuZn-NP-1, Cu-NP-3, CuZn-NP-4, and CuZn-NP-5) were directly impregnated on the dehydration catalyst, γ-Al2O3, to yield the bifunctional catalysts CZA-1, CA-3, CZA-4, and CZA-5, respectively. The elemental composition of the catalysts after calcination is summarized in Table 2. Bifunctional catalysts derived from CuZn-NP-1 revealed C residues after the calcination step, most probably resulting from decomposition of an organic nanoparticle shell. The SEM images (Fig. S2†) of the bifunctional catalysts indicated that the Cu/ZnO particles were not always distributed completely homogeneous on the γ-Al2O3 support and formed some aggregated species. For the preparation of the hybrid catalyst CZA-2, the nanoparticles (CuZn-NP-1) were directly calcined and physically mixed with the dehydration catalyst. Fig. 4 shows TEM images of the two catalysts derived from CuZn-NP-1 nanoparticles by impregnation on γ-Al2O3 and physical mixing. High resolution TEM images (Fig. S3†) revealed the crystalline character of the particles.
 |
| | Fig. 1 (a) TEM image showing nanoparticles (CuZn-NP-1) obtained by reduction of Cu(acac)2 with Zn(C2H5)2. Inset: particle size distribution (mean particle diameter 6.2 ± 2.0 nm). (b) HAADF-STEM image (scale bar 100 nm) showing nanoparticles (CuZn-NP-5) obtained by hydrolysis of Zn(C2H5)2 in the presence of Cu nanoparticles (Cu-NP-3). Inset: EDX line profile along the red line. | |
 |
| | Fig. 2 XANES spectra at (a) Cu K and (b) Zn K edges of the nanoparticles NP (CuZn-NP-1) and the bifunctional catalyst (CZA-1) (calcined and after reaction) in comparison with reference spectra of bulk Cu, Cu2O, CuO, Zn, and ZnO. | |
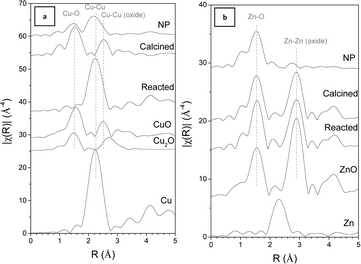 |
| | Fig. 3 k3-weighed Fourier-transformed EXAFS spectra (not corrected for the phase-shift) at (a) Cu K and (b) Zn K edges of the nanoparticles NP (CuZn-NP-1) and the bifunctional catalyst (CZA-1) (calcined and after reaction) in comparison with reference spectra of bulk Cu, Cu2O, CuO, Zn and ZnO. | |
Table 2 Physico-chemical characteristics of the bifunctional catalysts
| Catalyst |
Elemental composition (wt%) |
BET surface (m2/g) |
Pore size (nm) |
|
C 5%.
C 2%.
Properties of methanol active component only.
Bifunctional hybrid catalyst.
|
|
|
Al2O3 |
CuO |
ZnO |
|
|
| CZA-1a |
22.8 |
25.9 |
43.4 |
167d |
7.4d |
| CZA-2b |
50.0 |
12.0 |
36.0 |
31c |
3.8c |
| CA-3 |
51.3 |
48.6 |
— |
180d |
8.9d |
| CZA-4 |
48.3 |
31.0 |
20.7 |
159d |
9.2d |
| CZA-5 |
37.0 |
20.3 |
42.7 |
143d |
9.2d |
 |
| | Fig. 4 TEM images of the calcined CuZn-NP-1 nanoparticles on γ-Al2O3 in the bifunctional STD catalyst CZA-1 (a) and the calcined CuZn-NP-1 nanoparticles in CZA-2 (b). | |
The lattice spacings (indicated by arrows in Fig. S3†) were 0.246 nm, 0.281 nm, and 0.260 nm identifying ZnO particles. The 0.253 nm and 0.231 nm spacings were ascribed to CuO particles. The specific BET surface areas of the catalysts are summarized in Table 2. All bifunctional hybrid catalysts, CZA-1, CA-3, CZA-4, and CZA-5, revealed specific surface areas in the range of 143 to 180 m2 g−1. For these catalysts, the surface area of the mesoporous γ-Al2O3 dehydration catalyst (pore size 7.6–9.2 nm), initially 221 m2 g−1, was reduced after immobilizing the nanoparticles. Here, the blockage of pores by nanoparticles or agglomerates, which were formed during the impregnation and calcination procedures of catalyst preparation, could have contributed to the observed decrease in BET surface. A comparable trend for the decrease of surface and the plugging of the pore structure has also been observed for other bifunctional STD catalysts (e.g., obtained via coprecipiation).5d For the hybrid catalyst CZA-2, the nanoparticle precusor was directly calcined to yield the methanol active component, which revealed a BET surface area of 31 m2 g−1. The methanol active component was further physically mixed with the γ-Al2O3 dehydration catalyst (1:1 ratio), which averages to 126 m2 g−1 in the final hybrid catalyst. This procedure of catalyst preparation resulted in the smallest overall BET surface of the final hybrid catalyst. The results of XRD characterization of the bifunctional catalysts prepared from the Cu/Zn-based nanoparticle precursors (i.e., CZA-1, CZA-2, CZA-4, CZA-5) showed the reflections of a monoclinic CuO (JCPDS 01-089-5898) and a hexagonal ZnO phase (JCPDS 01-080-0074) (Fig. 5a). The Cu-based catalyst CA-3 revealed a small reflection at 36.4° (2θ) which was ascribed to the (111) reflection of a Cu2O (cuprite) phase (JCPDS 01-078-2076). In addition, a very diffuse reflection of low intensity was observed for CZA-1, CA-3, CZA-4, and CZA-5, which was consistent with the reflection of the commercial γ-Al2O3 dehydration catalyst at 45.8° (2θ) (JCPDS 00-010-0425). As the reflections were broad, some peaks of the CuO, Cu2O, ZnO, and γ-Al2O3 exhibited an overlap, e.g. the CuO and Cu2O reflections at 35.4° (2θ) and 36.4° (2θ), respectively, were covered by the ZnO reflection at 36.3° (2θ). No diffraction peaks assigned to mixed oxide species (e.g., CuAl2O4, JCPDS 01-071-0966) were detected. This also agreed with results of XAS analysis. The FT EXAFS spectra of the calcined catalyst CZA-1 revealed backscattering at Cu–O/Cu–Cu and Zn–O/Zn–Zn distances corresponding to CuO and ZnO oxide, respectively. In bulk CuO and ZnO, the Cu–Cu and Zn–Zn distances differ substantially, therefore the formation of mixed oxides was excluded. The XRD patterns displayed relatively broad reflections for all of the catalysts, indicating the small crystallite sizes. The size of the CuO particles, arising from initial Cu nanoparticle precursors after catalyst calcination, was estimated using the Scherrer equation (Table 3). With 7 nm, the catalyst CZA-1 revealed the smallest crystallite size. TEM analysis of the initial Cu nanoparticle precursor (CuZn-NP-1) showed a mean particle size of 6.2 (±2.0) nm, indicating that the initial size of the nanoparticles was preserved after the immobilization and calcination steps of catalyst preparation. The size of CuO crystallites in CA-3 and CZA-4 was 14 nm and 16 nm, respectively, which was also in good agreement with the size of the initial Cu nanoparticle precursor of 11 nm. All catalysts were isolated under inert conditions after catalytic testing and again characterized by XRD measurements (Table 3). Fig. 5b shows an exemplary XRD pattern for CA-3 after catalytic testing. For the CA-3, CZA-4, CZA-5 series of catalysts, all starting from the same size of Cu nanoparticles, the role of ZnO as a physical spacer and its effect on the sintering behavior was clearly demonstrated. With increasing amount of ZnO in the final hybrid catalysts, the sintering effects gradually reduced and the Cu crystallite sizes decreased after catalytic testing from 78 nm (CA-3, no ZnO) over 24 nm (CZA-4, 21 wt% ZnO) to 15 nm (CZA-5, 43 wt% ZnO). The role of ZnO with respect to the stabilization of a highly dispersed Cu phase has been previously mentioned by various authors.23 In agreement with results from XRD measurements, the XANES data (Fig. 2) and the FT EXAFS spectrum (Fig. 3) for catalyst CZA-1 after catalytic reaction revealed a metallic state of the Cu particles by Cu–Cu scattering at the Cu K edge with the same distance as in Cu metal (0.254 nm; corrected coordination number of 8.5). A minor backscattering signal derived from O suggested some oxidation of Cu at the particle surface, most probably during sample preparation, also leading to a smaller Cu–Cu coordination number than expected (cf. ESI†). At the Zn K edge, distances and intensities of Zn–O and Zn–Zn backscattering were consistent with those of the calcined catalyst, i.e. excluding both reduction of Zn or formation of alloyed CuZn species as well as the presence of a mixed phase. Despite the small increase in crystallite size, both the intimate ZnO and Cu contacts as well as the immobilization on γ-Al2O3 ensured an overall good stabilization of the methanol active component, whereas the non-supported particles in CZA-2 grew over calcination and catalyst testing steps to a final Cu crystallite size of 30 nm.
 |
| | Fig. 5 (a) XRD patterns of bifunctional catalysts prepared from different nanoparticulate precursors (CZA-1–CZA-5). (b) XRD patterns of the bifunctional catalyst CA-3 (1) initial, calcined catalyst and (2) reduced catalyst after catalytic testing. | |
Table 3 Crystallite size of the CuO and Cu particles in the bifunctional STD catalysts before and after catalysis, respectively, according to full width at half maximum of the XRD reflections at 38.8° (CuO) and 43.3° (Cu) (2θ)
| Catalyst |
CZA-1 |
CZA-2 |
CA-3 |
CZA-4 |
CZA-5 |
|
Very low intensity of the CuO reflections precluded particle size evaluation by XRD.
CuO crystallite sizes are slightly underestimated due to an overlapping of the (111) and (200) reflections.
|
| CuO crystallites (nm)b |
7 |
12 |
14 |
16 |
—a |
| Cu crystallites (nm) |
16 |
30 |
78 |
24 |
15 |
H2-TPR measurements were carried out to investigate the reduction behavior of the calcined catalysts. In Fig. 6, the H2-TPR profiles of the catalysts are given showing the different, rather broad and unsymmetric peak shapes. The catalysts CZA-1, CZA-2, and CZA-5 reveal a main reduction peak with a shoulder appearing at higher temperatures. The peaks and shoulders appearing at higher temperatures than the main reduction peaks have been attributed to the consecutive reduction of the different Cu species (Cu2+ → Cu+ → Cu0), indicating some strong interaction between CuO and ZnO.24 Although derived from the same nanoparticulate precursor (CuZn-NP-1), the two catalysts CZA-1 and CZA-2 differed in their reduction behavior. The TPR signals revealed a main reduction peak at 155 °C and 166 °C, respectively, and a shoulder at higher temperatures. Hence, the TPR signal observed for catalyst CZA-2 was significantly broader than for catalyst CZA-1. In general, the broader signals have been ascribed to a larger particle size distribution and the reduction of different copper species.25 A broader particle size distribution could be the consequence of nanoparticle calcination in absence of the support during the preparation of CZA-2. The main reduction temperature (Tm) of catalyst CZA-5 was shifted to lower temperatures (Tm 145 °C), whereas the corresponding catalyst CZA-4 with a lower ZnO content was shifted to higher temperatures (Tm 184 °C). Only for catalyst CZA-4, was a shoulder at temperatures below the main peak observed, which, in previous reports, has been attributed to the different reduction behavior of well dispersed CuO (shoulder) and bulk CuO species (Tm). Overall, various reduction profiles and temperatures have been reported for STD catalysts, ranging, for example, from Tm = 149–215 °C (i.e., catalysts prepared by coprecipitation/impregnation),5d to Tm = 210 °C with a shoulder at 176 °C (i.e., flame-made, Cu/ZnO/Al2O3-based catalyst),4c and Tm = 254 °C (i.e., CuZn nanoparticles sputtered on H-ZSM-5),29 to Tm = 330 °C (i.e., mesoporous Cu-γ-Al2O3 catalysts).6 A shift of the reduction profile to higher temperatures has been attributed to a strong metal support interaction or the formation of mixed oxide species.6,26 As compared to these results, the reduction temperatures observed for our nanoparticle-derived STD catalysts (145 °C to 184 °C) reside in the lower temperature range. NH3-TPD profiles measured for bifunctional catalysts CZA-1, CZA-4, and CZA-5 are summarized and compared to γ-Al2O3 in Fig. S4 ESI.† All of the catalysts showed desorption peaks in the range of 100–600 °C. The bifunctional catalysts displayed an additional peak around 280–295 °C in the TPD profiles. Similar peaks have been previously observed in NH3-TPD profiles of H-ZSM-5-based STD catalysts.5d The formation of new acid-sites was allocated to Al species in the co-precipitated CZA component of the catalyst, a process which can be excluded here. Overall, NH3-desorption at lower temperature is ascribed to weak acid sites, while peaks at higher temperatures correspond to stronger acid sites. In general, Lewis acid sites in γ-Al2O3 have been considered to be predominantly responsible for catalytic methanol dehydration to DME.8b,27
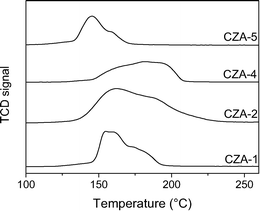 |
| | Fig. 6 TPR profiles of the bifunctional catalysts CZA-1, CZA-2, CZA-4 and CZA-5. | |
The performance of the catalyst systems in the STD reaction was screened with respect to CO conversion and DME selectivity in a continuously operating fixed-bed reactor (P = 51 bar, T = 250 °C). The operating conditions for catalyst screening were selected according to previously published results.4a,5d Simulated biomass-derived, CO-rich syngas (H2:CO ratio of 1:1) was employed and diluted with inert gas (70%) to avoid the formation of hotspots in the catalyst bed. Overall, the CO conversion depends on the actual operating conditions, e.g., reaction temperature, pressure, residence time, and H2:CO ratio.4a A decrease in the CO conversion was previously observed if syngas with a H2:CO ratio of 1:1 (CO-rich synthesis gas from sustainable, biomass-derived resources) was used instead of 2:1. It was further reported for STD catalysts with γ-Al2O3 as dehydration catalyst that the CO conversion increased with temperature, and a maximum was reached at 250 °C.4aTable 4 summarizes the results of the catalytic tests of the bifunctional catalysts. In these initial tests, all of the catalysts derived from the Cu/Zn-based nanoparticle precursors, i.e. CZA-1, CZA-2, CZA-4, and CZA-5, yielded DME from H2 and CO in a single reaction step with CO conversion ranging from 7 to 24%. The bifunctional reference catalyst CA-3, which was derived from pure Cu nanoparticles (Cu-NP-3), hardly revealed any catalytic activity at all, neither in the direct STD reaction nor in the methanol synthesis. Although many authors reported on the importance of the ZnO for methanol synthesis, active ZnO-free Cu catalysts for CO hydrogenation in the methanol and STD reaction have been described for H2:CO feeds.28 For mesoporous Cu-γ-Al2O3 catalysts, e.g., it was demonstrated that CO conversions of up to 72% and DME selectivities of 69% can be obtained (P = 50 bar, T = 310 °C, H2:CO ratio 2:1) for pure Cu-based catalysts.6 The large crystal size and, consequently, low Cu surface area may explain partly the observed, very low catalytic activity. Yet, the content in CuO was 48% for catalyst CA-3. Thus, the reduced Cu surface area probably did not account exclusively for the bad catalytic performance. The Cu surface was 3.35 m2 g−1 as estimated from mean particle diameters, and exceeded the Cu surface in catalyst CZA-2 (2.15 m2 g−1) with a CO conversion of 11% and DME selectivity of 64%. The crucial synergistic role of ZnO for methanol synthesis has been subject of many previous reports. Besides its role as a structural promotor supporting Cu dispersion, ZnO was further suggested to influence the reducibility of CuO, the particle shape of Cu, and the stabilization of defective sites on the Cu surface.23c,28 For a series of catalysts derived from the same type of 11 nm Cu nanoparticles (i.e., Cu-NP-3, CuZn-NP-4, CuZn-NP-5), the Cu nanoparticle size increased from 15 nm and 24 nm to 78 nm after 2.6 h time on stream according to the increasing molar Cu:Zn ratios (i.e., 1:2 (CZA-5), 3:2 (CZA-4), 1:0 (CA-3)). The CO conversion decreased in this direction from 20% over 7% to almost 0% and, accordingly, the reaction rate based on Cu weight decreased from 6.71 × 10−3 mmolCO s−1 gCu−1 over 1.56 × 10−3 mmolCO s−1 gCu−1 to almost no catalytic activity. In the reaction product, high DME selectivities of up to 68% together with a low content in methanol (<1%) and hydrocarbons (<1%) were reached for catalyst CZA-5 under test conditions. The product distribution agreed well with the syngas to DME conversion described by reaction eqn (4) for CO-rich synthesis gas (66% DME, 33% CO2). Similar results were also reported for physical mixtures of the commercial methanol and dehydration catalysts.4a Recently, a DME selectivity of 92% was reported for a STD catalyst, where 5 nm Cu/ZnO-based nanoparticles were sputtered on a HZSM-5 dehydration catalyst (T = 250 °C, P = 50 bar, H2:CO ratio 2:1).29 In this case, the authors suggested that the CO2 conversion to methanol exceeded the CO2 production from the water gas shift reaction, with an active Cu/Zn nanoalloy layer promoting CO2 conversion to some extent. In the case of catalyst CZA-1, some methanol (6%) remained in the reaction product. A partial covering of active sites on the γ-Al2O3 dehydration catalyst after nanoparticle immobilization (CuZn-NP-1) together with a low content in γ-Al2O3 may have limited methanol dehydration to DME here, which will be the subject of future work with respect to a more balanced spatial distribution and interplay of the two active components in the hybrid catalyst. For catalyst CZA-2, the same nanoparticles were not directly supported on γ-Al2O3 but individually treated and physically mixed with the dehydration catalyst. Hence, the blocking of active sites on the dehydration catalyst should be well reduced, which accounts for an increased DME selectivity of 64% together with a lower methanol content (approx. 1%). The overall CO conversion and reaction rate on the Cu nanoparticles were 11% and 6.30 × 10−3 mmolCO s−1 gCu−1, respectively, most probably a consequence of the larger size of the Cu nanocrystallites (30 nm). Furthermore, the CO conversion is not only affected by the methanol active component (i.e., the Cu nanoparticles), but also by the dehydration catalyst.4a
Table 4 Catalytic performance of bifunctional catalysts (reaction conditions: T = 250 °C, P = 51 bar, TOS = 2.6 h, H2:CO ratio = 1:1)
| Catalyst |
XCO (%) |
Reaction rate (mmolCO s−1 gCu−1) |
S
DME (%) |
S
MeOH (%) |
S
CO2 (%) |
S
CH (%) |
|
CO conversion below detection limit.
|
| CZA-1 |
24.4 |
6.43 × 10−3 |
58.7 |
6.2 |
31.8 |
3.4 |
| CZA-2 |
11.1 |
6.30 × 10−3 |
64.3 |
1.2 |
30.7 |
3.9 |
| CA-3 |
—a |
| CZA-4 |
7.1 |
1.56 × 10−3 |
67.0 |
1.1 |
29.9 |
2.0 |
| CZA-5 |
19.9 |
6.71 × 10−3 |
68.3 |
0.9 |
30.2 |
0.7 |
Fig. 7 displays exemplary the CO conversion and the product distribution with time on stream for the hybrid catalyst CZA-1. The CO conversion slightly decreased from 24% to 21% during the first 2.6 h on stream, a general trend which was observed for all of the catalysts. When the catalysts were isolated after the reduction and the initial screening of catalytic performance, the XRD measurements revealed an increase in the size of the Cu particles (as compared to the supported CuO particles) and a sintering behaviour, which was dependent on the composition of the bifunctional catalyst (Table 3). A long-term study (115 h TOS) was performed for CZA-5, which revealed a progressive and significant catalyst deactivation, as indicated by a decrease in CO conversion to 40% of the initial CO conversion, whereas DME selectivity remained constant and high. After 115 h TOS, the size of the Cu crystallites was 15 nm (as determined by XRD analysis), indicating no further particle growth. A decrease in catalyst activity, which could reach up to 60%, has been previously reported by others. Coke deposition on the active sites of the metallic and acidic functions of the catalyst has been suggested as one of the reasons for the deactivation of bifunctional STD catalysts.8a,30 The coke presumably originated from degradation of reaction intermediates (i.e., methoxy species), which subsequently transformed into higher molecular weight structures. It was assumed to initially block the metallic sites. Coke deposition on the acidic functions was reported to be noticeable for longer time on stream. Details concerning the mechanism of deactivation of the nanoparticle-derived catalysts will need to be considered as a subject of future studies. Although the nanoparticle-derived catalysts did not reach the same reaction rates and long-term stability, which were previously reported for some admixed systems of γ-Al2O3 and methanol catalyst (e.g., flame-made methanol catalyst),4c the initial catalytic tests revealed a high DME selectivity. Hence, those nanoparticle-derived systems provide model kits, which will enable the future rational and individual tuning of the catalytic components in bifunctional STD catalysts.
 |
| | Fig. 7 CO conversion and product selectivity of the bifunctional catalyst CZA-1 with time on stream. | |
Conclusions
Well-defined Cu/Zn-based nanoparticles, 6 nm in size, were initially obtained by reacting Cu(acac)2 with Zn(C2H5)2. Additionally, 11 nm Cu nanoparticles were synthesized by thermal decomposition of Cu(acac)2 in OA and used to prepare a monometallic reference catalyst. Further addition of Zn(C2H5)2 and its subsequent hydrolysis yielded Cu/Zn-based nanoparticles with different Cu/Zn ratios. The colloidal nanoparticles were used to prepare a series of bifunctional STD catalysts, where the nanoparticles were either directly supported on the dehydration catalyst (γ-Al2O3) or integrated into the STD catalyst by physical mixing. By using this approach, active catalysts for the STD reaction with CO conversions of up to 24% and selectivities of up to 68% were obtained. Several material parameters were shown to affect the sintering behavior and the catalytic characteristics of the final STD catalyst. Consequently, well-defined nanoparticle building units will open an encouraging avenue for the preparation of bifunctional STD catalysts, enabling future systematic studies of synthesis–structure–function relationships and a systematic balancing of the two catalytic entities in the final bifunctional STD catalyst.
Acknowledgements
The authors would like to kindly thank the following colleagues, Sarah Essig and Hermann Köhler for technical assistance, Dr. Henning Lichtenberg and Dr. Stefan Mangold for support during XAS measurements, Thilo Henrich for discussions, Doreen Neumann-Walter and Dr. Thomas Otto for support with BET and TPR analysis. Acknowledgement is further given to the ANKA synchrotron radiation source (KIT, Karlsruhe) for providing beamtime at the XAS beamline. M. G. acknowledges financial support of the Helmholtz Research School Energy-Related Catalysis.
Notes and references
-
N. Dahmen, E. Dinjus and E. Henrich, in Renewable Energy, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 83–87 Search PubMed.
- J. G. van Bennekom, R. H. Venderbosch, J. G. M. Winkelman, E. Wilbers, D. Assink, K. P. J. Lemmens and H. J. Heeres, Chem. Eng. Sci., 2013, 87, 204–208 CrossRef CAS.
- J. Sun, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2014, 4, 3346–3356 CrossRef CAS.
-
(a) M. Stiefel, R. Ahmad, U. Arnold and M. Döring, Fuel Process. Technol., 2011, 92, 1466–1474 CrossRef CAS;
(b) N.-K. Park and T. Lee, Korean J. Chem. Eng., 2011, 28, 2076–2080 CrossRef CAS;
(c) R. Ahmad, M. Hellinger, M. Buchholz, H. Sezen, L. Gharnati, C. Wöll, J. Sauer, M. Döring, J.-D. Grunwaldt and U. Arnold, Catal. Commun., 2014, 43, 52–56 CrossRef CAS.
-
(a) J. W. Bae, S.-H. Kang, Y.-J. Lee and K.-W. Jun, Appl. Catal., B, 2009, 90, 426–435 CrossRef CAS;
(b) Q. Ge, Y. Huang, F. Qiu and S. Li, Appl. Catal., A, 1998, 167, 23–30 CrossRef CAS;
(c) Y. J. Lee, M. H. Jung, J.-B. Lee, K.-E. Jeong, H.-S. Roh, Y.-W. Suh and J. W. Bae, Catal. Today, 2014, 228, 175–182 CrossRef CAS;
(d) R. Ahmad, D. Schrempp, S. Behrens, J. Sauer, M. Döring and U. Arnold, Fuel Process. Technol., 2014, 121, 38–46 CrossRef CAS.
- H. Jiang, H. Bongard, W. Schmidt and F. Schüth, Microporous Mesoporous Mater., 2012, 164, 3–8 CrossRef CAS.
-
(a) S.-H. Kang, J. Bae, H.-S. Kim, G. Dhar and K.-W. Jun, Energy Fuels, 2010, 24, 804–810 CrossRef CAS;
(b) J.-H. Fei, M.-X. Yang, Z.-Y. Hou and X.-M. Zheng, Energy Fuels, 2004, 18, 1584–1587 CrossRef CAS.
-
(a) J. Abu-Dahrieh, D. Rooney, A. Goguet and Y. Saih, Chem. Eng. J., 2012, 203, 201–211 CrossRef CAS;
(b) M. Xu, J. H. Lunsford, D. W. Goodman and A. Bhattacharyya, Appl. Catal., A, 1997, 149, 289–301 CrossRef CAS.
-
(a) G. Yang, N. Tsubaki, J. Shamoto, Y. Yoneyama and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 8129–8136 CrossRef CAS PubMed;
(b) G. Yang, D. Wang, Y. Yoneyama, Y. Tan and N. Tsubaki, Chem. Commun., 2012, 48, 1263–1265 RSC;
(c) R. Phienluphon, K. Pinkaew, G. Yang, J. Li, Q. Wei, Y. Yoneyama, T. Vitidsant and N. Tsubaki, Chem. Eng. J., 2015, 270, 605–611 CrossRef CAS.
- K. Sun, W. Lu, F. Qiu, S. Liu and X. Xu, Appl. Catal., A, 2003, 252, 243–249 CrossRef CAS.
-
(a) A. García-Trenco, A. Vidal-Moya and A. Martínez, Catal. Today, 2012, 179, 43–51 CrossRef;
(b) A. García-Trenco and A. Martinez, Appl. Catal., A, 2015, 493, 40–49 CrossRef.
-
(a) C. Kiener, M. Kurtz, H. Wilmer, C. Hoffmann, H.-W. Schmidt, J.-D. Grunwaldt, M. Muhler and F. Schüth, J. Catal., 2003, 216, 110–119 CrossRef CAS;
(b) B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem. – Eur. J., 2003, 9, 2039–2052 CrossRef CAS PubMed.
- C.-J. Jia and F. Schüth, Phys. Chem. Chem. Phys., 2011, 13, 2457–2487 RSC.
-
(a) M. Buck and R. Schaak, Angew. Chem., Int. Ed., 2013, 52, 2–27 CrossRef PubMed;
(b) H. Bönnemann and R. Richards, Eur. J. Inorg. Chem., 2001, 2455–2480 CrossRef;
(c) J. Park, J. Joo, S. Kwon, Y. Jang and T. Hyeon, Angew. Chem., 2007, 119, 4714–4745 CrossRef.
-
(a) M. A. Sliem, S. Turner, D. Heeskens, S. B. Kalidindi, G. V. Tendeloo, M. Muhler and R. A. Fischer, Phys. Chem. Chem. Phys., 2012, 14, 8170–8178 RSC;
(b) M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS PubMed;
(c) S. Schimpf, A. Rittermeier, X. Zhang, Z.-A. Li, M. Spasova, M. W. E. van den Berg, M. Farle, Y. Wang, R. A. Fischer and M. Muhler, ChemCatChem, 2010, 2, 214–222 CrossRef CAS;
(d) A. Rittermeier, S. Miao, M. K. Schröter, X. Zhang, M. W. E. van den Berg, S. Kundu, Y. Wang, S. Schimpf, E. Löffler, R. A. Fischer and M. Muhler, Phys. Chem. Chem. Phys., 2009, 11, 8358–8366 RSC;
(e) S. Vukojevic, O. Trapp, J.-D. Grunwaldt, C. Kiener and F. Schüth, Angew. Chem., Int. Ed., 2005, 44, 7978–7981 CrossRef CAS PubMed;
(f)
S. Wang, Copper Colloid-Based Catalysts for Methanol Synthesis, PhD Thesis, Ruhr-Universität Bochum, Fakultät für Chemie und Biochemie, 2012 Search PubMed;
(g) K. Schütte, H. Meyer, C. Gemel, J. Barthel, R. Fischer and C. Janiak, Nanoscale, 2014, 6, 3116–3126 RSC.
- S. U. Son, I. K. Park, J. Park and T. Hyeon, Chem. Commun., 2004, 778–779 RSC.
- J.-D. Grunwaldt, S. Hannemann, J. Göttlicher, S. Mangold, M. Denecke and A. Baiker, Phys. Scr., 2005, T115, 769–772 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
- W. M. Alley, I. K. Hamdemir, K. A. Johnson and R. G. Finke, J. Mol. Catal. A: Chem., 2010, 315, 1–27 CrossRef CAS.
-
(a) J. Hambrock, M. K. Schröter, A. Birkner, C. Wöll and R. Fischer, Chem Mater., 2003, 15, 4217–4222 CrossRef CAS;
(b) T. Bollermann, C. Gemel and R. A. Fischer, Coord. Chem. Rev., 2012, 256, 537–555 CrossRef CAS.
- K. Fischer, K. Jonas, P. Misbach, R. Stabba and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 943–953 CrossRef.
-
(a) M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef CAS;
(b) T. Fujitani and J. Nakamura, Catal. Lett., 1998, 56, 119–124 CrossRef CAS;
(c) J.-D. Grunwaldt, A. Molenbroek, N.-Y. Topsøe, H. Topsøe and B. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- B. Lindström, L. J. Pettersson and P. Govind Menon, Appl. Catal., A, 2002, 234, 111–125 CrossRef.
-
(a) S. J. Gentry and P. J. Walsh, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 1515–1523 RSC;
(b) E. Matulewicz, M. de Keijser, J. Mol and F. Kapteijn, Thermochim. Acta, 1984, 72, 111–116 CrossRef CAS;
(c) B. Lindström, L. Pettersson and P. Menon, Appl. Catal., A, 2002, 234, 111–125 CrossRef.
- S. Zander, E. Kunkes, M. Schuster, J. Schumann, G. Weinberg, D. Teschner, N. Jacobsen, R. Schlögl and M. Behrens, Angew. Chem., Int. Ed., 2013, 52, 6536–6540 CrossRef CAS PubMed.
- D. Sung, Y. Kim, E. Park and J. Yie, Res. Chem. Intermed., 2010, 36, 653–660 CrossRef CAS.
-
(a) M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed;
(b) P. Hansen, J. Wagner, S. Helveg, J. Rostrup-Nielsen, B. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed;
(c) F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- J. Sun, G. Yang, Q. Ma, I. Ooki, A. Taguchi, T. Abe, Q. Xie, Y. Yoneyama and N. Tsubaki, J. Mater. Chem. A, 2014, 2, 8637–8643 CAS.
-
(a) A. Aguayo, J. Eren, I. Sierra, M. N. Olazar and J. Bilbao, Catal. Today, 2005, 106, 265–270 CrossRef CAS;
(b) J. Eren, I. Sierra, M. Olazar, A. Gayubo and A. Aguayo, Ind. Eng. Chem. Res., 2008, 47, 2238–2247 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01043h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.