
Synthesis of cyclic monothiocarbonates via the coupling reaction of carbonyl sulfide (COS) with epoxides†
M.
Luo
ab,
X.-H.
Zhang
*a and
D. J.
Darensbourg
 *b
*b
aMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xhzhang@zju.edu.cn
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: djdarens@chem.tamu.edu
First published on 17th August 2015
Abstract
Two guanidine bases were used as organocatalysts for the synthesis of cyclic monothiocarbonates via the coupling reaction of carbonyl sulfide (COS) and epoxides. The systems proved to be efficient single-component, metal-free catalysts for the reaction of simple (propylene oxide, 1,3-butene oxide) or activated epoxides (epichlorohydrin, glycidyl phenyl ether) with COS under solvent-free and mild reaction conditions to selectively afford the corresponding cyclic monothiocarbonates. The yield of this reaction is generally high, thereby providing ready means for pure product isolation.
Introduction
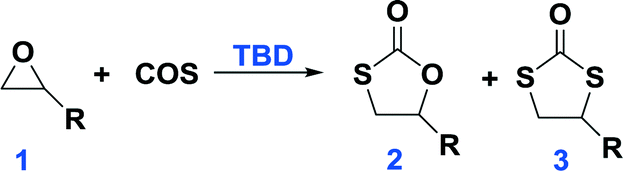
Inasmuch as there are numerous reports of the catalytic preparation of cyclic carbonates from a wide variety of epoxides and CO2, there are no reports of the synthesis of cyclic monothiocarbonates via a similar process utilizing carbonyl sulfide.1 Cyclic monothiocarbonates (MTCs) are important synthetic intermediates in organic2 and polymer sciences.3 A few synthetic methods have been developed for the preparation of MTCs, such as the carbonylation of β-hydroxy thiol with phosgene, the reaction of oxiranes with sulfur and carbon monoxide in the presence of sodium hydride, the base-catalyzed cyclization of the imidazolide derivative prepared by the treatment of epoxy alcohol with thiocarbonyl diimidazole, the oxidation of 2-alkoxy-1,3-oxathiolane, and the acid-assisted cyclization of 2-hydroxyethyl thiocarbonate.2–6 All these methods have their disadvantages: (i) the use of poisonous phosgene, carbon monoxide and odorous thiol (ii) multiple component reaction system, making the purification of the product difficult, and (iii) harsh reaction condition. In addition, the decarboxylation of MTCs occasionally occurs under these reaction conditions, and thiirane can be formed as a byproduct.6In this article, we report on the development of an efficient and easy-handling method for the synthesis of MTC by the coupling reaction of epoxides with carbonyl sulfide (COS) catalyzed by a single compound and metal-free catalyst, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), under solvent-free and mild reaction conditions. The selectivity of this reaction is very high, no byproduct is produced, and following the full conversion of the starting epoxide, minor purification is needed, the only impurity being a trace amount of catalyst.
Results and discussion
Initially, we examined the reaction of the simple aliphatic terminal epoxide, propylene oxide (PO, 1a in Table 1), with carbonyl sulfide (COS) in the presence of TBD at 60 °C. After 12 hours, 26% of the PO was consumed on the basis of the 1H NMR spectrum of the crude product (Fig. S1†). The cyclic monothiocarbonate, 5-methyl-1,3-oxathiolan-2-one (2a in Table 1), was produced, which was confirmed by NMR, IR, and Gas Chromatography Mass Spectrometry (GC-MS) (Fig. S1–4†). The crude product was removed from the reactor and no further purification was undertaken. The yield of product was calculated after the evaporation of the unreacted PO. In order to accelerate the reaction, it was carried out at higher temperatures (entries 2–5, Table 1). Interestingly, higher reaction temperature initiated oxygen/sulfur scrambling in the reaction process. An additional cyclic dithiocarbonate, 4-methyl-1,3-dithiolan- 2-one (3a in Table 1), was produced. 3a was similarly determined by NMR, IR and GC-MS (Fig. S5–8†). In our previous report on the copolymerization of COS with PO, 2a and 3a have been observed as reaction byproducts at elevated reaction temperatures.7Fig. 1 depicts the 1H NMR spectra of the coupling reaction products obtained at various temperatures. These spectra clearly illustrate the relationship between reaction temperature and cyclic product distribution, with an increase in the percentage of 3a with increasing temperature. Hence, it is possible by selecting temperature and/or catalyst to selectively synthesize either 2a or 3a. That is, using TBD as catalyst at 60 °C only 2a is produced, whereas, at 120 °C predominantly 3a (>94%) is afforded. Alternatively, employing MTBD as catalyst, the process is highly selective for producing 2a at all temperatures investigated.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Catalyst | T (°C) | t (h) | Conv.b (%) | Yieldc (%) | 2a (%) | 3a (%) | 4a (%) |
| a The reactions were performed in neat PO (0.5 ml, 2.64 mmol; 1.5 MPa COS was added; catalyst/epoxides = 1/1000 in molar ratio) in a 10 ml autoclave. b Determined by 1H NMR spectrum of the crude product. c The crude product was removed from autoclave to a clean vial, and the isolated yield was calculated after the evaporating of the PO. The yield is for the total cyclic products. d The molar ratio of 2a and 3a was determined by 1H NMR spectrum of the crude product. e In entries 8 and 9, besides 2a and 3a, another cyclic product 4a: 4-methyl-1,3-oxathiolan-2-one was produced. | ||||||||
| 1 | TBD | 60 | 12 | 26 | 25 | 100 | 0 | 0 |
| 2 | TBD | 70 | 24 | >99 | 96 | 75 | 25 | 0 |
| 3 | TBD | 80 | 24 | >99 | 96 | 63 | 37 | 0 |
| 4 | TBD | 100 | 12 | >99 | 98 | 34 | 66 | 0 |
| 5 | TBD | 120 | 12 | >99 | 98 | 6 | 94 | 0 |
| 6 | MTBD | 70 | 72 | 43 | 40 | 100 | 0 | 0 |
| 7 | MTBD | 80 | 48 | 45 | 41 | 100 | 0 | 0 |
| 8e | MTBD | 100 | 24 | 41 | 38 | 95 | 0 | 5 |
| 9e | MTBD | 120 | 48 | >99 | 95 | 93 | 0 | 7 |
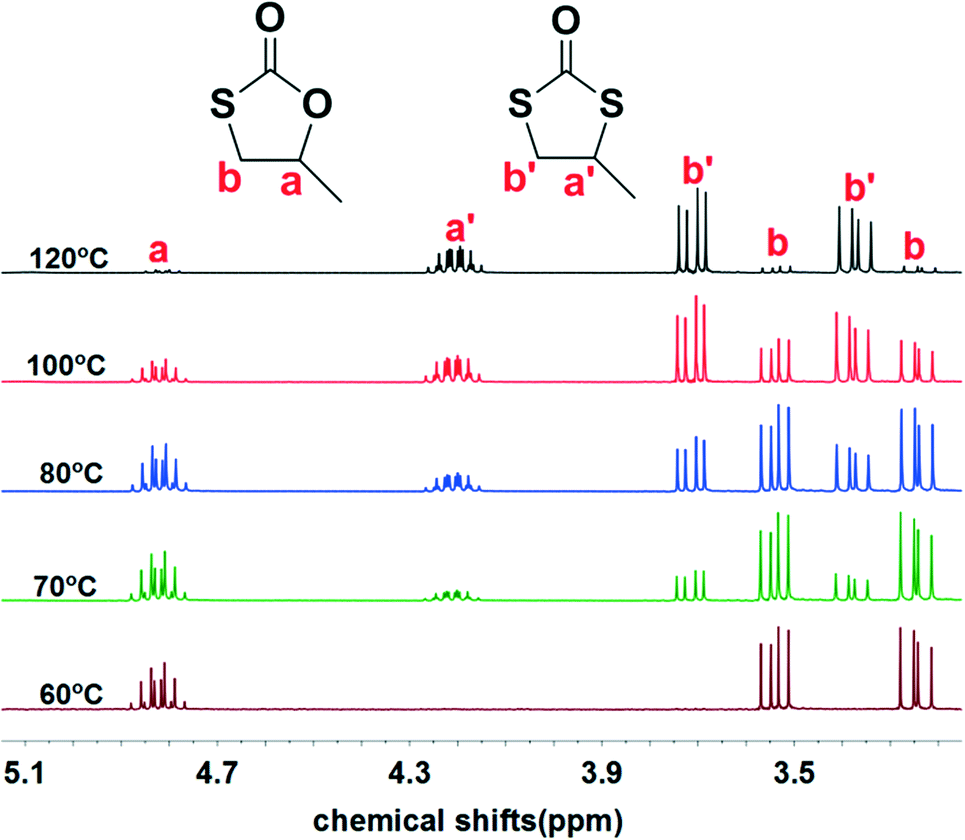 | ||
| Fig. 1 1H NMR spectra of the products from PO/COS coupling at different temperatures. 1H NMR signals for methyl hydrogens are further upfield and not shown here, see Fig. S28–30.† | ||
The plausible reaction pathways to the formation of 2a and 3a are shown in Schemes 1 and 2, respectively. As shown in Scheme 1, the coordination of TBD with COS forms a transiently stable six-membered ring with the oxygen atom of COS interacting with the N–H proton of TBD via a hydrogen bond. A similar pathway has been presented by Cantat and coworkers for the organocatalyzed reductive functionalization of CO2 in the presence of TBD.8 A crystal structure of the CO2 adduct of TBD has been reported, where a zwitterionic structure is seen with N⋯O distance in the N–H⋯O unit of 2.535(2) Å.9 This is followed by sulfur attack of the PO on the less hindered carbon leading to an intermediate where an oxygen anion is formed. 2a is generated by the backbiting of the oxygen anion on the carbonyl carbon. For the dithiocarbonate 3a, an oxygen/sulfur scrambling may occur during the reaction as shown in Scheme 2. At high reaction temperatures, the oxygen anion can attack another COS with the formation of a sulfur anion. Subsequently, the sulfur anion attacks the methine carbon of PO to form a new sulfur anion with release of CO2. 3a is generated by the backbiting of the sulfur anion on the carbonyl carbon. In order to verify the credibility of this pathway, a reaction of CO2 and PO with TBD was carried out at 120 °C. After 24 hours only trace amount of cyclic carbonate was observed, with the conversion of PO being less than 1%. Thus, CO2 does not effectively react with PO under the condition of TBD as in the presence of COS. This explains why no cyclic carbonate was observed in entries 2–5.
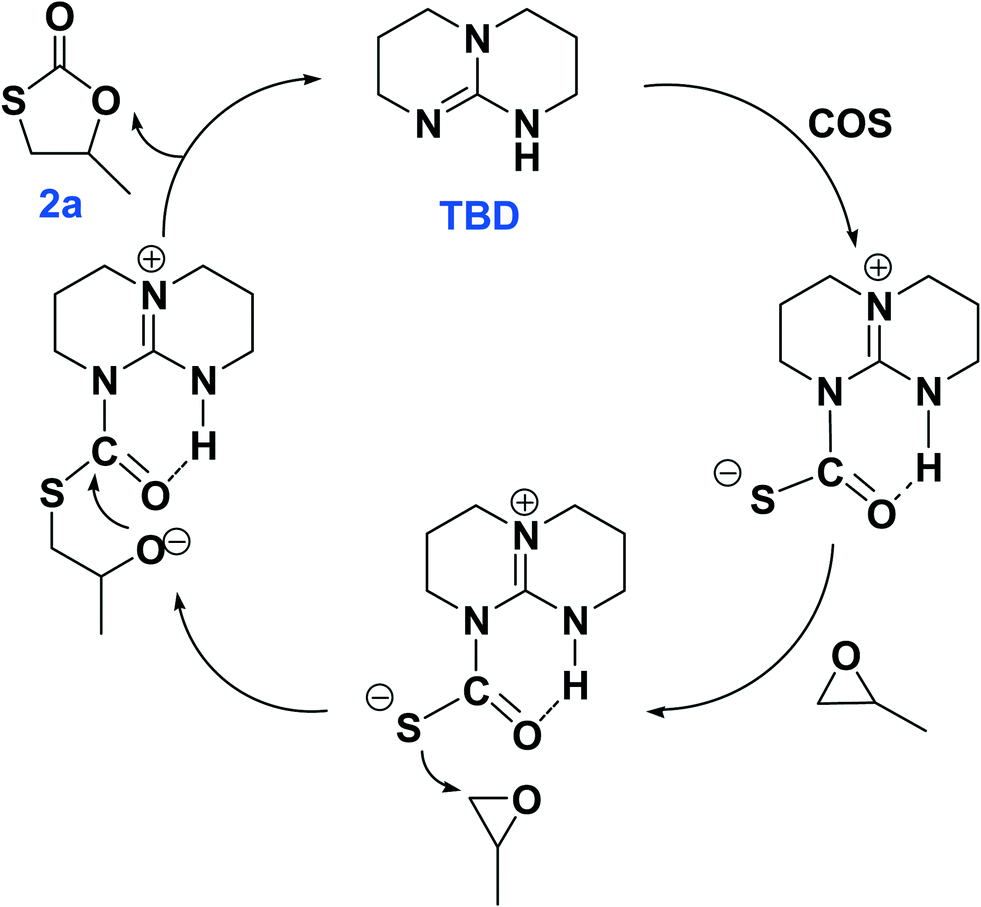 | ||
| Scheme 1 The plausible reaction pathway to the generation of 2a catalyzed by TBD. | ||
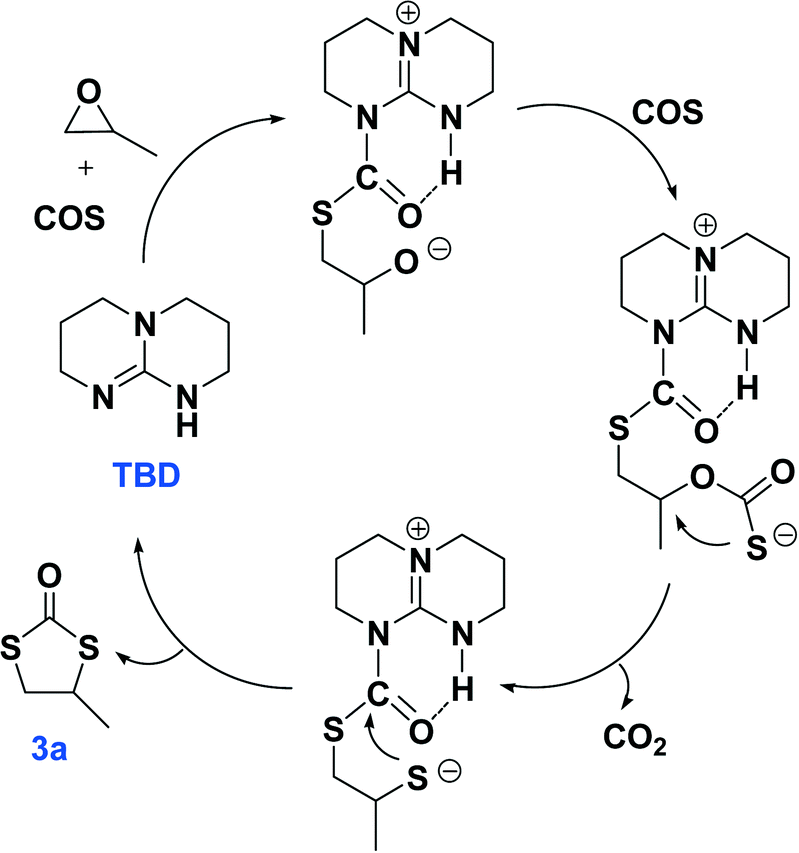 | ||
| Scheme 2 The plausible reaction pathway to the generation of 3a catalyzed by TBD. | ||
The two pathways shown in Schemes 1 and 2 rely on the N–H group of TBD to stabilize the orientation of COS and support subsequent reaction processes.10 Hence, if indeed this is a necessary requirement for oxygen/sulfur exchange, this scrambling process should be eliminated upon removing the N–H function from the organocatalyst. In order to support this hypothesis, a derivative of TBD, 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD), was employed as catalyst for the coupling reaction. The results of these reactions are provided in Table 1 (entries 6–9).
As is evident in Table 1, MTBD is less active as a catalyst relative to TBD, however it is more selective. For example, at the lower temperatures of 70 °C and 80 °C the only cycle product observed was 2a based on 1H NMR spectroscopy. However, at the higher temperatures of 100 °C and 120 °C, although no 3a product was found which is indicative of no oxygen/sulfur scrambling; trace quantities of the other isomer of 2a, 4-methyl-1,3-oxathiolan-2-one (4a), were produced. 4a was identified by 1H NMR and GC-MS analysis (Fig. S11–13†). Although, 2a and 4a have the same mass, they afford different mass fragments in the GC-MS. Only 5 and 7% of 4a were produced at 100 °C and 120 °C (entries 8 and 9), respectively. 4a results from a difference in the site of the PO ring-opening step, where in this instance cleavage occurs at the sterically hindered methine carbon center. Scheme 3 represents proposed mechanistic pathways for the COS/PO coupling reaction as catalyzed by MTBD. Presumably, reaction pathway B has a higher activation barrier.
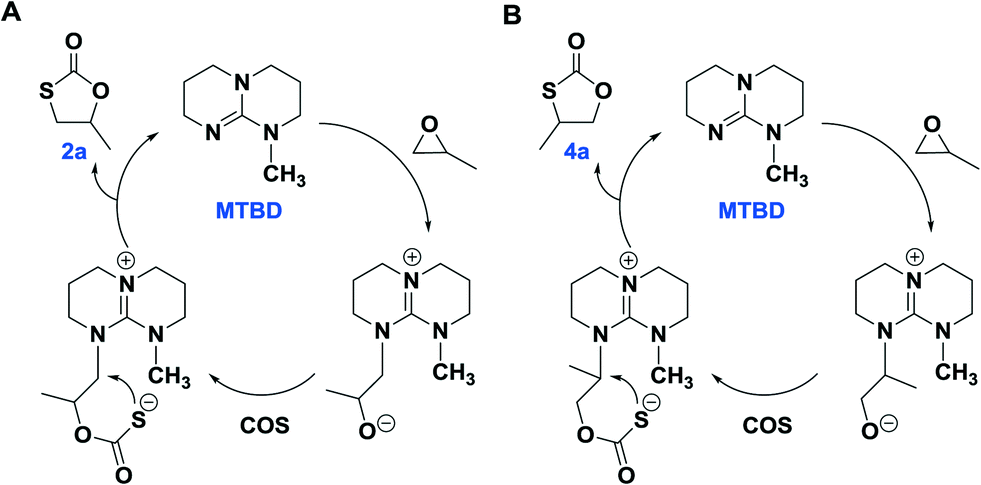 | ||
| Scheme 3 The plausible reaction pathway for the generation of 2a (pathway A) and 4a (pathway B) catalyzed by MTBD. | ||
In order to examine the effect of substituents on the epoxide substrate when coupling with carbonyl sulfide on the cyclic thiocarbonate product(s), we carried out studies with a variety of epoxides, and the results are summarized in Table 2. The aliphatic 1,2-butene oxide (1b, entries 1 and 2 in Table 2) behaves similarly to propylene oxide, providing cyclic monothiocarbonate (2b) with 100% selectively at 60 °C, and the product was well-defined (Fig. S14–17†). Upon raising the temperature to 70 °C, about 6% of cyclic dithiocarbonate (3b) was produced as a result of oxygen/sulfur exchange. However, for the halogen-substituted epoxide, epichlorohydrin (1c, entries 3–5 in Table 2), the selectivity for the cyclic monothiocarbonate (2c) was not influenced by reaction temperatures. Over the temperature range of 70 to 90 °C the selectivity remained 100%, and the product was well-defined (Fig. S18–21†). Notably, this coupling reaction occurred efficiently with phenyl glycidyl ether (1d, entry 6 in Table 2), where the coupling of phenyl glycidyl ether with COS was completed in 12 hours at 60 °C providing selectively 100% cyclic monothiocarbonate (2d), and the product was well-defined (Fig. S22–25†). For the vinyl-substituted epoxide, 1,3-butadiene oxide (1e, entries 7, 8 in Table 2), no reaction occurred at 60 °C, but at a temperature of 70 °C, 47% of the epoxide converted to cyclic monothiocarbonate (2e) in 12 hours (Fig. S26†). However, because of the reactivity of the vinyl group on the cyclic product, the thiocarbonate resulted in formation of a cross-linked compound which was insoluble in most organic solvents such as chloroform, tetrahydrofuran, and dimethyl sulphoxide. The phenyl-substituted styrene oxide (1f, entries 9, 10 in Table 2) underwent oxygen/sulfur scrambling at 70 °C, and both monothiocarbonate (2f) and dithiocarbonate (3f) were observed in the crude product. In addition, the reaction showed no activity at the lower temperature (60 °C). Therefore, it was impossible to achieve a good selectivity for the product by adjusting temperature in the coupling of styrene oxide with COS. Furthermore, utilizing MTBD as catalyst, there was no reaction between styrene oxide and COS. In addition, the disubstituted epoxide, cyclohexene oxide, did not undergo reaction with COS in the presence of TBD at 80–110 °C. This is consistent with our earlier report, where the cyclohexene oxide/COS reaction catalyzed by a zinc–cobalt double metal cyanide complex provided exclusively copolymer at 110 °C.11
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Epoxide 1: R | T (°C) | t (h) | Conv.b (%) | Yieldc (%) | Productd (%) |
| a The reactions were performed in neat epoxide (0.5 ml at R.T.; 1.5 MPa COS was added; TBD was used as catalyst, catalyst/epoxides = 1/1000 in molar ratio) in a 10 ml autoclave. b Determined by using 1H NMR spectroscopy. c The crude product was removed from autoclave to a clean vial, and the isolated yield was calculated after the evaporating of the unreacted epoxide. d The molar ratio of cyclic products were determined by 1H NMR spectrum of the crude product. | ||||||
| 1 | 1b: C2H5 | 60 | 24 | 33 | 30 | 2b: 100% |
| 2 | 1b: C2H5 | 70 | 12 | 63 | 59 | 2b: 94% 3b: 6% |
| 3 | 1c: CH2Cl | 70 | 12 | 27 | 25 | 2c: 100% |
| 4 | 1c: CH2Cl | 80 | 12 | 77 | 75 | 2c: 100% |
| 5 | 1c: CH2Cl | 90 | 12 | 78 | 76 | 2c: 100% |
| 6 | 1d: CH2OPh | 60 | 12 | 99 | 98 | 2d: 100% |
| 7 | 1e: CHCH2 | 70 | 12 | — | — | — |
| 8 | 1e: CHCH2 | 80 | 12 | 47 | 40 | Cross-linked product |
| 9 | 1f: Ph | 60 | 24 | — | — | — |
| 10 | 1f: Ph | 70 | 12 | 69 | 60 | 2f: 43% 3f: 57% |
Conclusions
In summary, we have reported a new method for synthesizing cyclic monothiocarbonate via the coupling reactions of carbonyl sulfide with epoxides catalyzed by two guanidine bases TBD and MTBD at mild reaction conditions. These organocatalysts have proven to be efficient single-compound and metal-free catalyst for this reaction. The yield of cyclic thiocarbonate is generally high, thereby, no purification is needed upon full conversion of the epoxides. For aliphatic epoxides, cyclic monothiocarbonate or dithiocarbonate can be selectively synthesized by adjusting the reaction temperature. However, for epichlorohydrin and phenyl glycidyl ether, the corresponding cyclic monothiocarbonates are the only products.Experimental section
Materials and methods
Unless otherwise specified, all syntheses and manipulations were carried out on a double-manifold Schlenk vacuum line under an atmosphere of argon or in an argon-filled glovebox. Following purification, materials were stored in an argon-filled glovebox prior to use. Epoxides (propylene oxide, 1,2-butene oxide, butadiene monoxide, epichlorohydrin, styrene oxide, phenyl glycidyl ether) were purchased from Acros or TCI and distilled over CaH2 before used. Carbonyl sulfide (99.0%, 0.756 lbs) was purchased from Specialty Gases of America Inc., and used directly. 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) were purchased from Aldrich and dried at 80 °C in vacuum and then transferred into the glovebox. MTBD was dissolved in dry dichloromethane as a solution with a concentration of 10 μl ml−1. Dichloromethane was purified by an MBraun Manual Solvent Purification System packed with Alcoa F200 activated alumina desiccant. 1H and 13C NMR spectra were recorded on Mercury 300 MHz and Inova 300 MHz spectrometers. The peak frequencies were referenced versus the internal standard (TMS) shift at 0 ppm for 1H NMR and against the solvent chloroform-d at 77.0 ppm for 13C NMR.Typical procedure for coupling reaction of carbonyl sulfide and epoxides
The coupling reactions of carbonyl sulfide (COS) and epoxides were performed in a 10 mL autoclave equipped with a magnetic stirrer and a pressure gauge. In a typical experiment, the autoclave was firstly dried in an oven for 24 hours and transferred into the glovebox. 0.5 ml PO and 1 mg TBD were added into the autoclave successively. The autoclave was pressurized to 1.5 MPa pressure with COS and then placed in an oil bath of 60 °C, and the reaction mixture was stirred for 6 hours. After reaction, the reactor was cooled in an ice bath to room temperature and COS was slowly released. An aliquot was taken from the crude product for the determination of the conversion rate of epoxide and the percentage of different cyclic products by 1H NMR spectra. Subsequently, the crude product was removed and placed into a clean vial, and upon evaporation of the unreacted epoxide, the product was weighed in order to calculate the yield. The obtained pure product was analyzed by 1H NMR, 13C NMR, IR and GC-MS.Characterization of the products
Acknowledgements
We gratefully acknowledge the financial support of the National Science Foundation (CHE-1057743) and the Robert A. Welch Foundation (A-0923). The China Scholarship Council and National Science Foundation of the People's Republic of China (no. 21274123 and 21474083) are also acknowledged for their support.Notes and references
- (a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; (b) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed; (c) R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef CAS PubMed; (d) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC; (e) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (f) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- W. R. Roush and D. Gustin, Tetrahedron Lett., 1994, 35, 4931–4934 CrossRef.
- P. C. Wang, Heterocycles, 1986, 24, 329–369 CrossRef CAS.
- (a) Y. Nishiyama, C. Katahira and N. Sonoda, Tetrahedron, 2006, 62, 5803–5807 CrossRef CAS; (b) Y. Taguchi, K. Yanagiya, I. Shibuya and Y. Suhara, Bull. Chem. Soc. Jpn., 1988, 61, 921–926 CrossRef CAS; (c) R. Feinauer, M. Jacobi and K. Hamann, Chem. Ber., 1965, 98, 1782–1788 CrossRef CAS.
- (a) D. D. Reynolds, M. K. Massad, D. L. Fields and D. L. Johnson, J. Org. Chem., 1961, 26, 5122–5124 CrossRef CAS; (b) D. D. Reynolds, J. Am. Chem. Soc., 1957, 79, 4951–4952 CrossRef CAS.
- S. Hata, H. Goto, S. s and A. Oku, J. Appl. Polym. Sci., 2003, 90, 2959–2968 CrossRef CAS.
- M. Luo, X. H. Zhang, B. Y. Du, Q. Wang and Z. Q. Fan, Macromolecules, 2013, 46, 5899–5904 CrossRef CAS.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef PubMed.
- C. Villiers, J.-P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–34680 CrossRef CAS PubMed.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- M. Luo, X. H. Zhang, B. Y. Du, Q. Wang and Z. Q. Fan, Polymer, 2014, 55, 3688–3695 CrossRef CAS.
- S. Sakai, Y. Fujimura and Y. Ishii, J. Org. Chem., 1970, 35, 2344–2347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental spectroscopic data. See DOI: 10.1039/c5cy00977d |
| This journal is © The Royal Society of Chemistry 2016 |