
Molecular determinants for selective C25-hydroxylation of vitamins D2 and D3 by fungal peroxygenases†
Fátima
Lucas‡
*a,
Esteban D.
Babot‡
b,
Marina
Cañellas‡
ac,
José C.
del Río
b,
Lisbeth
Kalum
d,
René
Ullrich
e,
Martin
Hofrichter
e,
Victor
Guallar
af,
Angel T.
Martínez
g and
Ana
Gutiérrez
*b
aJoint BSC-CRG-IRB Research Program in Computational Biology, Barcelona Supercomputing Center, Jordi Girona 29, E-08034 Barcelona, Spain. E-mail: fatima.lucas@bsc.es
bInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, Reina Mercedes 10, E-41012 Seville, Spain. E-mail: anagu@irnase.csic.es; Fax: +32 954624002; Tel: +32 954624711
cAnaxomics Biotech, Balmes 89, E-08008 Barcelona, Spain
dNovozymes A/S, Krogshoejvej 36, 2880 Bagsvaerd, Denmark
eTU Dresden, Department of Bio- and Environmental Sciences, Markt 23, 02763 Zittau, Germany
fICREA, Passeig Lluís Companys 23, E-08010 Barcelona, Spain
gCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain
First published on 7th August 2015
Abstract
Hydroxylation of vitamin D by Agrocybe aegerita and Coprinopsis cinerea peroxygenases was investigated in a combined experimental and computational study. 25-Monohydroxylated vitamins D3 (cholecalciferol) and D2 (ergocalciferol), compounds of high interest in human health and animal feeding, can be obtained through a reaction with both fungal enzymes. Differences in conversion rates, and especially in site selectivity, were observed. To rationalize the results, diffusion of D2 and D3 on the molecular structure of the two enzymes was performed using the PELE software. In good agreement with experimental conversion yields, simulations indicate more favorable energy profiles for the substrates' entrance in C. cinerea than for A. aegerita enzyme. On the other hand, GC-MS analyses show that while a full regioselective conversion of D2 and D3 into the active C25 form is catalyzed by C. cinerea peroxygenase, A. aegerita yielded a mixture of the hydroxylated D3 products. From the molecular simulations, relative distance distributions between the haem compound I oxygen atom and H24/H25 atoms (hydrogens on C24 and C25, respectively) were plotted. Results show large populations for O–H25 distances below 3 Å for D2 and D3 in C. cinerea in accordance with the high reactivity observed for this enzyme. In A. aegerita, however, cholecalciferol has similar populations (below 3 Å) for O–H25 and O–H24, which can justify the hydroxylation observed in C24. In the case of ergocalciferol, due to the bulky methyl group in position C24, very few structures are found with O–H24 distances below 3 Å and thus, as expected, the reaction was only observed at the C25 position.
Introduction
Selective oxygenations of aliphatic compounds are among the most challenging reactions in organic chemistry for the regio- and/or stereospecific synthesis of pharmaceuticals and fine chemicals. Monooxygenases catalyzing such hydroxylation reactions include cytochrome P450, a family of haem proteins playing a variety of physiological roles but often requiring an auxiliary flavoenzyme (or a flavin-containing module) and a source of reducing power to be activated by O2 – two facts that limit their biotechnological applicability.Recently, a new peroxidase type, which shares the active site architecture and reaction mechanism of cytochrome P450, but has the advantage of being activated directly by H2O2, was isolated from Agrocybe aegerita,45 and later identified in a variety of sequenced basidiomycete genomes including that of Coprinopsis cinerea.13 Due to the above characteristics, these unspecific peroxygenases (EC 1.11.2.1) have huge biotechnological potential as self-sufficient monooxygenases9,19 for hydroxylation of both aromatic2,3,24–27,42–44 and aliphatic compounds.4,15,33
The A. aegerita enzyme has been the most widely investigated basidiomycete peroxygenase, but recent studies have shown that the C. cinerea enzyme has comparative advantages related to its high conversion yield/selectivity for some hydroxylation reactions and its production as a recombinant protein in an industrial expression host (by Novozymes, Bagsvaerd, Denmark).5,6
Hydroxylation of vitamin D for the selective production of its active C25-hydroxylated derivatives is one of the reactions where the C. cinerea peroxygenase can be of biotechnological use (Fig. 1).6 Supplementation with 25-hydroxyvitamin D has a positive effect on different human diseases,11,20,21,28,41 and also raises considerable interest for feeding broiler chickens14,17,23,32 and other farm animals40 to reduce skeleton problems caused by rapid growth and reduced mobility. Therefore, the use of peroxygenase in vitamin D hydroxylation represents an attractive alternative to the chemical synthesis.
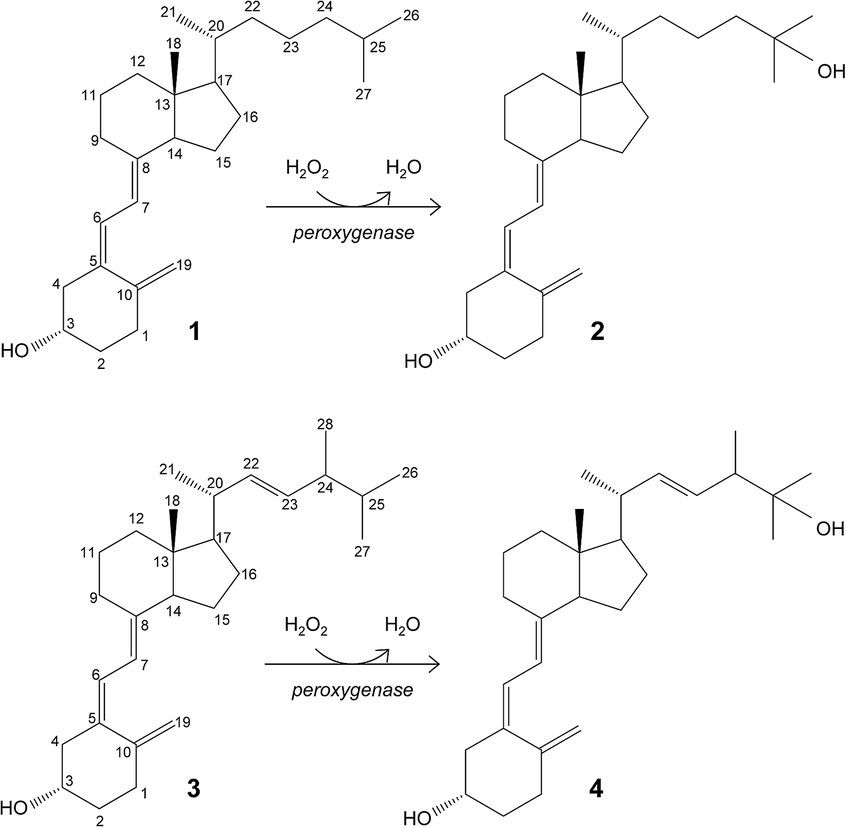 | ||
| Fig. 1 Enzymatic conversion of cholecalciferol (vitamin D3; 1) and ergocalciferol (vitamin D2; 3) into their bioactive 25-hydroxylated derivatives (2 and 4) by basidiomycete peroxygenases. | ||
The goal of the present work is to rationalize the differences observed in cholecalciferol and ergocalciferol's (vitamins D3 and D2, respectively) conversion rates and site selectivity by the A. aegerita and C. cinerea peroxygenases. The work presented here consists of first, the enzymatic conversion of these compounds by the two peroxygenases under the same reaction conditions, and gas chromatography-mass spectrometry (GC-MS) analyses to identify all reaction products. Then, the energy profiles and binding modes of vitamins D2 and D3 were determined by structure-based computational simulations, using the PELE software.10,12 Finally, differences in site selectivity were investigated through the analysis of the most favorable binding orientations in the active site. Results show that molecular simulations can effectively discriminate experimentally observed differences in conversion rates and site selectivity.
Results and discussion
Experimental hydroxylation reactions
The conversion of cholecalciferol and ergocalciferol was experimentally determined for both the A. aegerita and C. cinerea peroxygenases. In the case of cholecalciferol (0.1 mM), GC-MS analyses of the reaction mixture revealed that this compound was completely (100%) converted by the C. cinerea enzyme within 60 min reaction (Fig. 2C) as compared with the control reaction without peroxygenase (Fig. 2A). In the A. aegerita peroxygenase reaction, up to 90% conversion was observed (Fig. 2B). In the case of ergocalciferol (Fig. 2D–F), a conversion of 85% was produced by C. cinerea while in A. aegerita reaction, 81% conversion was observed. When higher substrate concentrations were tested (e.g. 0.5 mM), higher differences in the conversion rates by the two enzymes were observed (40–50% and 20% conversions by the C. cinerea and A. aegerita peroxygenases, respectively).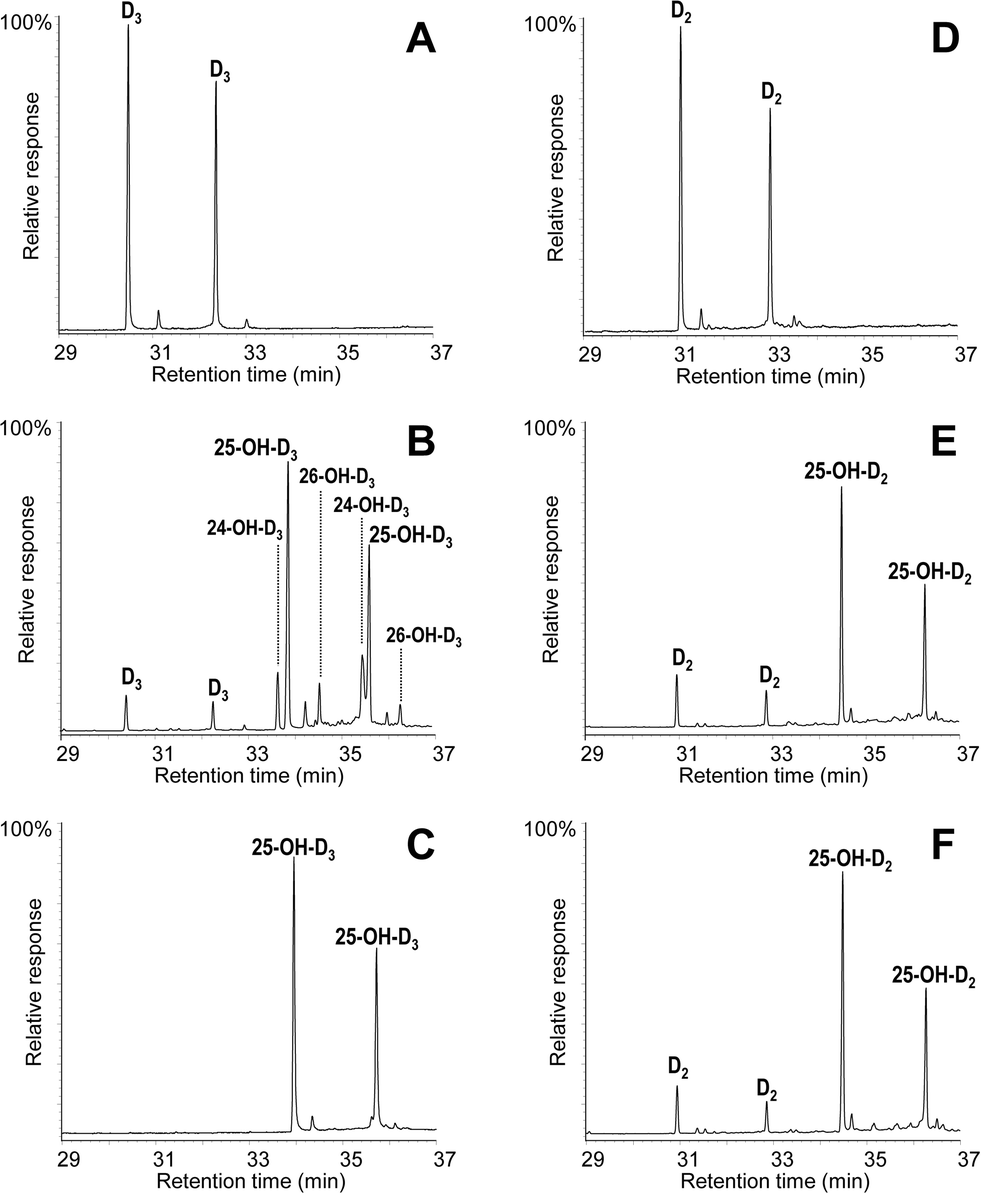 | ||
| Fig. 2 GC-MS analyses of cholecalciferol (left) and ergocalciferol (right) hydroxylation by the A. aegerita (B, E) and C. cinerea peroxygenases (C, F), compared with a control without enzyme showing the substrate peaks (A, D), as TMS derivatives from 60 min reactions. In all cases, the isopyro (left) and pyro (right) isomers from the secosteroid thermal rearrangement are obtained (two small peaks in A and D correspond to minor additional isomers). | ||
Moreover, cholecalciferol and ergocalciferol conversion by the C. cinerea peroxygenase showed a strict site selectivity since it gave exclusively 25-hydroxycalciferol. Likewise, conversion by the A. aegerita enzyme of ergocalciferol yielded exclusively 25-hydroxyergocalciferol but for cholecalciferol, a mixture of products was observed. The products include 25-hydroxycholecalciferol (64% of the initial substrate) together with 24-hydroxycholecalciferol (21%) and 26/27-hydroxycholecalciferol (7%), as confirmed by comparison with true standards showing identical retention times and mass spectra. The double peaks observed in the chromatograms for both the substrate and the product (Fig. 2) correspond to the isopyro (19β, 9β) and pyro (19α, 9α) isomers formed by a thermal rearrangement involving ring-B closure that vitamin D and its hydroxylated derivatives undergo due to the temperature at which GC-MS EI(+) is carried out. Indeed, the presence of the two isomers during GC separation is a useful indication that a secosteroid of the vitamin D type was injected into the system.31 The position of the hydroxyl group at the target C25 position was established by the MS of the TMS derivative. The spectrum shows a prominent ion from the C24–C25 bond cleavage, with characteristic fragment at m/z 131 and molecular ion at m/z 544. Additionally, characteristic fragments at m/z 529 ([M-15]+), m/z 454 ([M-90]+), m/z 439 ([M-90-15]+), m/z 349 ([M-90-90-15]+) and m/z 413 were also present. Therefore, both the chromatographic profiles and the mass spectra correspond to the isomerized secosteroids.
Peroxygenase structure
A superimposition of the general structure and the haem pocket residues in A. aegerita and C. cinerea peroxygenases is shown in Fig. 3. The main differences are the two longer loops (Fig. 3A, arrows) and the substitution of Phe69 by Met69 (Fig. 3B, red label) in the C. cinerea enzyme. All other haem pocket residues, including the proximal cysteine acting as the fifth ligand of the haem iron (Cys36) and the distal glutamic acid and arginine involved in haem activation by H2O2 (Glu169 and Arg189),34 are conserved in the two enzymes. Moreover, differences in the channel providing access to the haem cofactor and the neighbor residues at the channel entrance are shown in Fig. 4.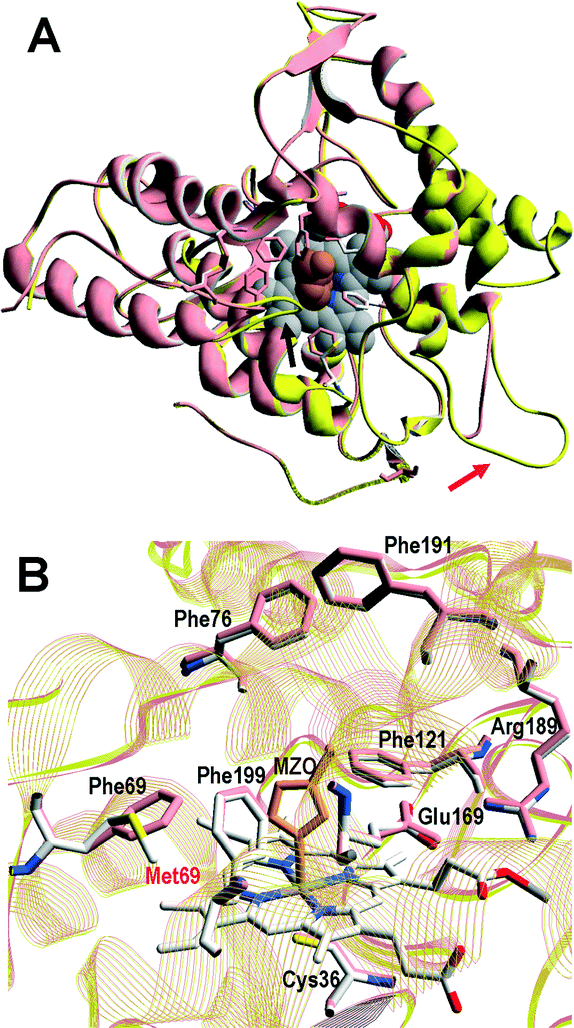 | ||
| Fig. 3 Comparison of peroxygenase molecular structures. A) General superimposition with the A. aegerita and C. cinerea proteins as pink and yellow ribbons, respectively. The haem group (CPK-colored spheres) and several haem pocket residues (pink- and CPK-colored sticks, respectively) are shown, and two larger loops in the second enzyme are indicated by arrows. B) Haem pocket residues in the A. aegerita (pink sticks) and C. cinerea (CPK-colored sticks) peroxygenases (red label indicates non-conserved methionine in position 69 of the latter enzyme). A hypothetical product molecule identified as 4-hydroxymethylimidazole is shown in A (brown-colored vdW spheres) and B (MZ0, brown-colored sticks).34 From A. aegerita PDB 2YOR, and C. cinerea homology model (provided in the ESI†). | ||
 | ||
| Fig. 4 Solvent access surfaces showing differences in the size of the haem (CPK-colored sticks) access channel in the peroxygenases of A. aegerita (A) and C. cinerea (B). Several neighbor residues are shown as CPK-colored vdW spheres, including Gly323 and Ser324 contributing to occlude the haem channel in the C. cinerea enzyme. | ||
Computational modeling
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O· porphyrin cation radical complex). The substrate was placed close to the entrance of the haem-access channel of the proteins prepared at the optimal pH for peroxygenase activity (pH 7). This initial location was identified using SiteMap,39 and from there, the ligand was spawned inside the protein by PELE,10 following the distance between the reactive O atom in the haem compound I and the cholecalciferol/ergocalciferol's C25 atom (Fig. 1).
O· porphyrin cation radical complex). The substrate was placed close to the entrance of the haem-access channel of the proteins prepared at the optimal pH for peroxygenase activity (pH 7). This initial location was identified using SiteMap,39 and from there, the ligand was spawned inside the protein by PELE,10 following the distance between the reactive O atom in the haem compound I and the cholecalciferol/ergocalciferol's C25 atom (Fig. 1).
PELE simulations were done in two stages: first, the substrate is perturbed to reduce the C25–O distance and, when this distance is below 5 Å, the substrate is free to explore the active site cavity (at the haem distal side) with a 15 Å restrain. In the first step, the ligand is perturbed with a combination of large and small translations and rotations, ranging from 0.5 to 1.5 Å for translations, and 0.05 to 0.25 radians for rotations. However, during the second stage, the translation range is reduced (0.75–0.25 Å) to perform a finer active site exploration. The plots shown in Fig. 5 correspond to three 48 h simulations, each with 80 processors, and show the substrate-C25 to haem-O distance vs. the interaction energy between the protein and the substrate at each of the different poses explored.
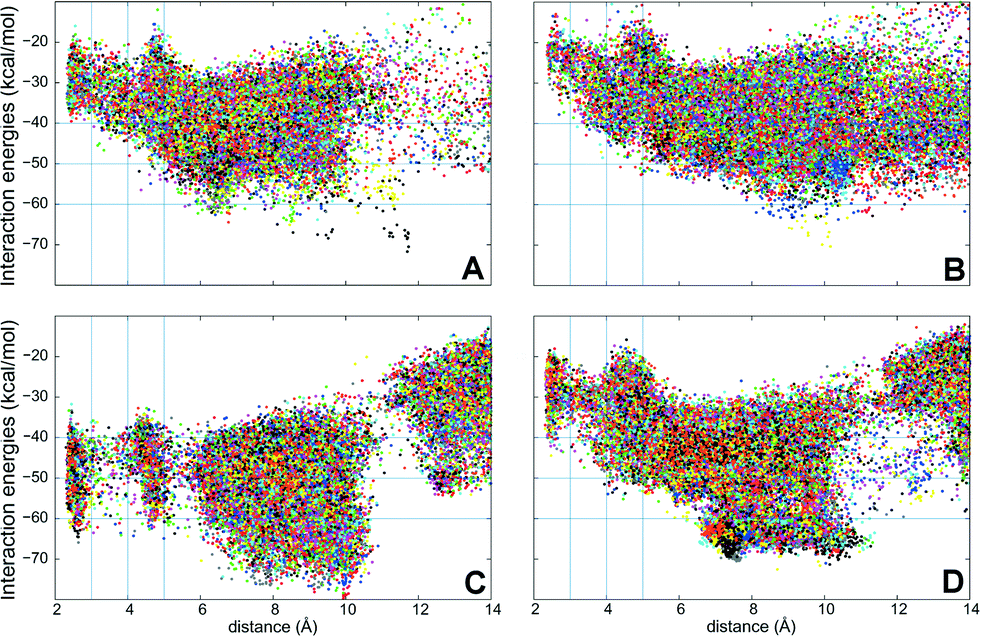 | ||
| Fig. 5 Interaction energies vs. ligand distances from PELE simulations for cholecalciferol (left) and ergocalciferol (right) entrance by the C25 end in the peroxygenases from A. aegerita (A, B) and C. cinerea (C, D). The distances shown (Å) are between the reactive O atom in the haem compound I and the calciferol C25 (for substrate numbering see Fig. 1). | ||
Simulations show that for the A. aegerita peroxygenase, the entrance of the substrates from the surface of the protein is quite favorable but then, the access to the activated haem is obtained against an uphill potential (Fig. 5A and B). For the C. cinerea enzyme, the entrance is less open than in the A. aegerita peroxygenase and, for this reason, we observed a constrained access to the protein (at around 12 Å). However, the overall energy profile is more favorable, in particular for cholecalciferol (Fig. 5C). Once inside the protein, the ligand must surpass smaller barriers to reach the haem. From Fig. 5, it is clearly seen that C. cinerea peroxygenase has the most favorable energy profiles and well-defined minima in the active site (with C25–O distance around 3 Å). Fig. 5A and C show that the binding of cholecalciferol at the haem site is more favorable in the C. cinerea peroxygenase (−65 kcal mol−1) than in the A. aegerita enzyme (−40 kcal mol−1). This difference comes from the fact that the first protein has a tighter binding pocket (see below). In the case of ergocalciferol in Fig. 5B and D, the binding is also more favorable in the C. cinerea (−42 kcal mol−1) than in the A. aegerita enzyme (−30 kcal mol−1). These differences in the energy profiles, which indicate a more favorable protein–ligand interaction in C. cinerea, can explain the higher conversion rates observed for the two compounds in this peroxygenase.
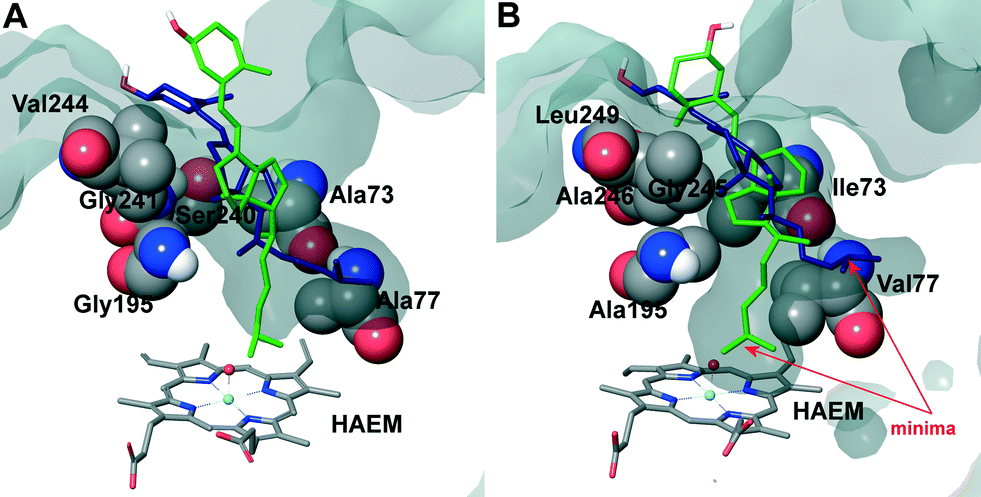 | ||
| Fig. 6 Superposition of cholecalciferol main active site positions (as liquorice) obtained in PELE simulations on the A. aegerita (A) and C. cinerea (B) peroxygenases. Haem cofactor also in liquorice and selected residues as vdW spheres. The protein's active site access is shown as a surface. | ||
The two orientations in Fig. 6B correspond to the two minima observed in cholecalciferol's diffusion in the C. cinerea peroxygenase (Fig. 5C). One of the minima (in green) is found at the binding distance to the haem O, and a second (in blue), about 5 Å away from the haem oxygen (Fig. 5C). The ligand's position observed in the C. cinerea peroxygenase is also found in the A. aegerita enzyme (Fig. 6A), although they do not correspond to the energy minima (Fig. 5A). Likewise, ergocalciferol can adopt two main positions in the binding pocket, although the minima (Fig. 5D) are less marked than that found for cholecalciferol (Fig. 5C).
When cholecalciferol is in an optimal reacting position, it is held in place by C. cinerea/A. aegerita peroxygenase Val77/Ala77, Phe121/Phe121, Thr192/Thre192, Ala195/Gly195, Phe199/Phe199 and Glu196/Glu196 (Fig. S1†). It is worthy to note that Phe69 also contributes to substrate positioning in A. aegerita peroxygenase, but the homologous Met69 of the C. cinerea enzyme (Fig. 3B) is placed away from cholecalciferol and does not appear to have any effect on its position in the active site. Ergocalciferol, however, with an extra methyl group in position C24 and with a C22–C23 double bond (Fig. 1), is positioned in a slightly different manner in the active site, and residues in position 69 (phenylalanine and methionine) now interacts with the ligand (red arrows in Fig. S2†). This extra constraint could be responsible for the lower reactivity observed for D2 in both peroxygenases. The influence of the side-chain structure (alkyl substituent and unsaturation presence) on the activity of the A. aegerita and C. cinerea peroxygenases was evidenced in the reaction of these enzymes with different sterols (Table S1†).
Inspection of the haem entrance when both ligands are in the active site reveals a better wrapping of the protein around the ligands in C. cinerea peroxygenase, compared with the A. aegerita enzyme. This is mainly due to the larger loop where Gly323 is located, which is smaller in the A. aegerita peroxygenase (Fig. 3A, black arrow). These are better illustrated in the haem-access channels of both peroxygenases in Fig. 4, where the position of the above Gly323 is shown. The narrower access to the haem in C. cinerea is also the result of several hydrophobic amino acid substitutions. Replacements to larger side chains in C. cinerea are dominant with: Ala73/Ile73, Ala77/Val77, Gly195/Ala195, Gly241/Ala246 and Val244/Leu249; and only one to a smaller amino acid, Ser240/Gly245. Due to the larger entrance cavity of the A. aegerita peroxygenase, both substrates remained solvent exposed at the C2 end. In contrast, more favorable interactions are established at the final substrate position inside the tighter channel of the C. cinerea peroxygenase, which presents extra interactions on the protein surface. The combination of the surface interactions, along with a tighter haem cavity, result in the improved interaction energies seen for D2 and D3 in this protein.
O–H36 was computed for the different reactive positions in the cholecalciferol and ergocalciferol side-chains (Fig. S3†).
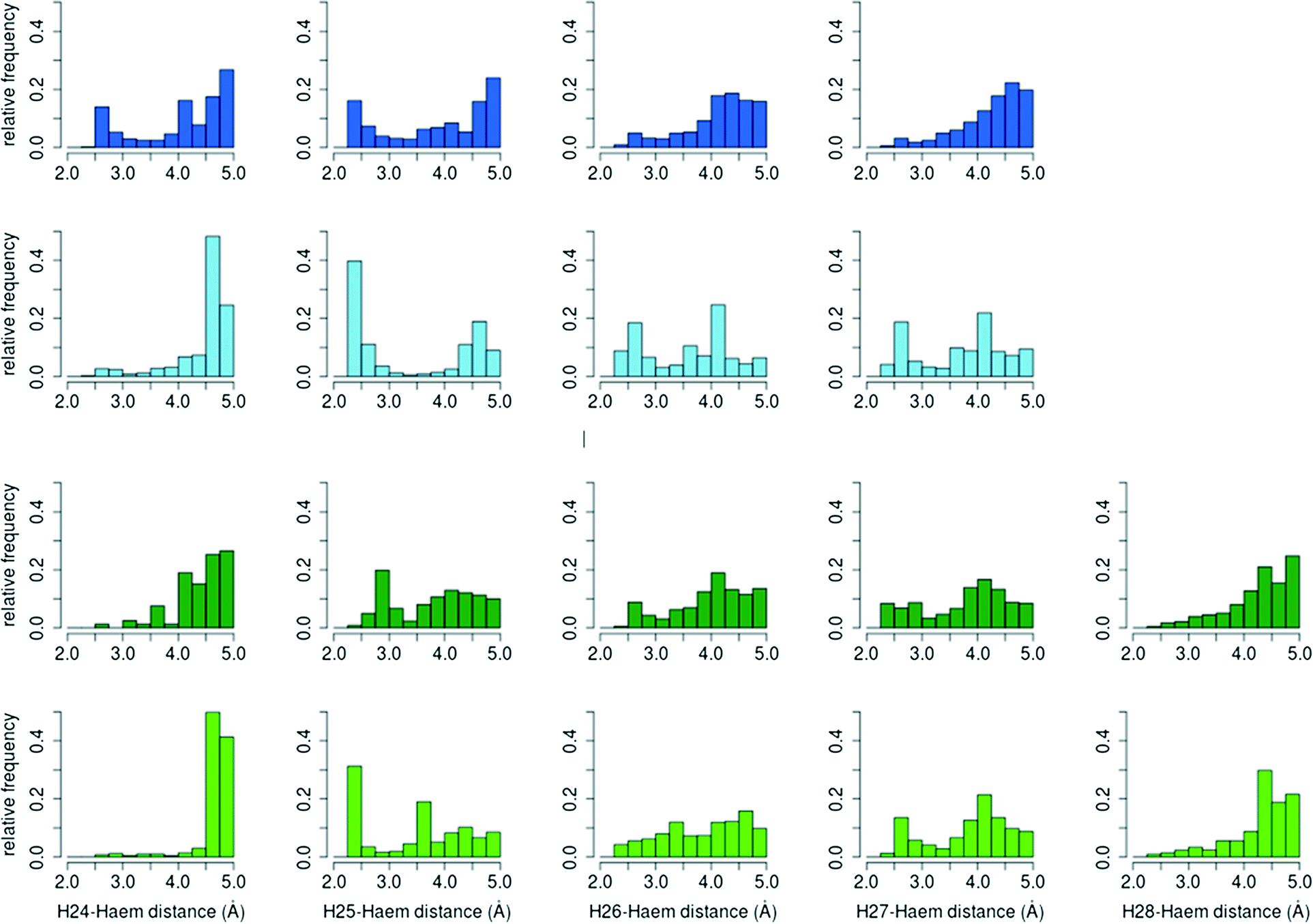 | ||
| Fig. 7 Relative distance distributions of cholecalciferol (blue) and ergocalciferol (green) reactive hydrogen atoms to haem compound I oxygen atom. Histograms for A. aegerita are in dark colours, and in light colours for C. cinerea. | ||
The percentage of O–H25 distances below 3 Å in C. cinerea is 54.5% for D3 and 36.2% for D2, whereas in A. aegerita it is 27.4% and 25.7%, respectively. Moreover, in the D3 simulations in A. aegerita, the fraction of structures with O–H24 below 3 Å is 19.3%, which is quite high when compared to the other cases. In fact, the fraction of O–H25 distances is only 1.4 times superior to O–H24 while for the other systems, it ranges between 10 to 20 times. This higher fraction of reactive O–H24 distances can explain the observed formation of C24 hydroxylated products in cholecalciferol. To sum up, and in agreement with experimental results, from the relative frequencies in C. cinerea peroxygenase, and also for ergocalciferol in A. aegerita, we expect a completely regioselective reaction in C25. In contrast, for cholecalciferol in A. aegerita enzyme, it seems that hydroxyl addition is possible not only in C25 position, but also in other carbons (C24 and C26/C27).
Conclusions
Atomic level simulations have been used here to rationalize the differences observed for ergocalciferol and cholecalciferol's conversion rates and site selectivity in two peroxygenases. The overall improved energy profiles in C. cinerea, the presence of favorable minima, and a high fraction of favorable O–H25 distances agrees well with experimentally higher conversion rates.The main structural differences between the A. aegerita and C. cinerea peroxygenases that modify the access of cholecalciferol to the activated haem are the changes Ala73/Ile73, Ala77/Val77, Gly195/Ala195, Gly241/Ala246 and Val244/Leu249, all to larger amino acids, with only Ser240/Gly245 to a smaller one. These larger hydrophobic side chains augment the interaction of D2 and D3 substrates with C. cinerea. The better conversion rates observed for C. cinerea peroxygenase do not originate in the active site itself (where Phe69 is replaced by Met69), but in the ligand access to the active site instead, especially in the entrance to the protein. In particular, the larger loop hosting Gly323 creates a barrier that reduces the size of the entrance channel in the C. cinerea peroxygenase. This, along with a tighter cavity, is reflected in the more favorable interaction energies.
Although the improved reactivity of C. cinerea does not appear to be affected by the larger Phe69 side chain, it does hinder the entrance of ergocalciferol relative to cholecalciferol. The presence of an extra methyl group that interacts directly with position 69 in both proteins lowers the reactivity of this compound. Finally, the computed relative frequency of the distances between the activated haem oxygen and the hydrogen atoms in C24 and C25 show that, despite the more favourable hydroxylation on tertiary carbons, the reaction at cholecalciferol C24 occurs when the ratio of favourable O–H25/O–H24 distances decreases.
Materials and methods
Enzymes and chemicals
Peroxygenase (isoform II) was isolated from A. aegerita DSM 22459 grown in soybean medium using a combination of SP-Sepharose chromatography and Mono-P chromatofocusing.45A. aegerita DSM 22459 is deposited at the Deutsche Stammsammlung für Mikroorganismen und Zellkulturen Braunschweig (Germany). C. cinerea peroxygenase was provided by Novozymes A/S (Bagsvaerd, Denmark). The enzyme corresponds to the protein model 7249 from the sequenced C. cinerea genome available at the JGI (http://genome.jgi.doe.gov/Copci1), which was expressed in Aspergillus oryzae and purified using a combination of S-Sepharose and SP-Sepharose ion-exchange chromatography (patent WO/2008/119780). One peroxygenase unit is defined as the amount of enzyme oxidizing 1 μmol of veratryl alcohol to veratraldehyde (ε310 9300 M−1 cm−1) in 1 min at 24 °C, pH 7, after addition of 0.5 mM H2O2 in the C. cinerea peroxygenase reactions and 2.5 mM H2O2 in those with the A. aegerita peroxygenase.Vitamin D3, also known as calciol or cholecalciferol ((5Z,7E)-(3S)-9,10-seco-5,7,10(19)-cholestatrien-3-ol; Fig. 1, structure 1), was tested as substrate of the A. aegerita and C. cinerea peroxygenases. 25-Hydroxyvitamin D3, also known as calcidiol or 25-hydroxycholecalciferol ((5Z,7E)-(3S)-9,10-seco-5,7,10(19)-cholestatriene-3,25-diol; Fig. 1 structure 2), was used as standard for gas chromatography-mass spectrometry (GC-MS) analyses. Vitamin D2, also known as ercalciol or ergocalciferol ((5Z,7E,22E)-(3S)-9,10-seco-5,7,10(19),22-ergostatetraen-3-ol; Fig. 1, structure 3), was also tested as substrate of the two peroxygenases. 25-Hydroxyvitamin D2, also known as ercalcidiol or 25-hydroxyergocalciferol ((5Z,7E,22E)-(3S)-9,10-seco-5,7,10(19),22-ergostatetraene-3,25-diol; Fig. 1, structure 4), was also used as standard for GC-MS analyses. All compounds were from Sigma-Aldrich.
Enzymatic reactions
Reactions of cholecalciferol and ergocalciferol (0.1 and 0.5 mM) with the A. aegerita and C. cinerea peroxygenases (1 U) were performed in 5 mL of 50 mM sodium phosphate, pH 7, at 40 °C for 60 min, in the presence of 0.5 mM H2O2. The substrates were previously dissolved in acetone, and added to the buffer (the acetone concentration in the reaction was 40%). In control experiments, the substrates were treated under the same conditions (including H2O2) but without enzyme. After the enzymatic reactions, the products were recovered by liquid–liquid extraction with methyl tert-butyl ether, dried under N2, and redissolved in chloroform for GC-MS analyses. Bis(trimethylsilyl)trifluoroacetamide (Supelco) in the presence of pyridine was used to prepare trimethylsilyl (TMS) derivatives. An internal standard was added after the enzymatic reactions to determine product yields.GC-MS analyses
GC-MS analyses were performed with a Shimadzu QP2010 Ultra equipment, using a fused-silica DB-5HT capillary column (30 m × 0.25 mm internal diameter, 0.1 μm film thickness) from J&W Scientific.16 The oven was heated from 120 °C (1 min) to 300 °C (15 min) at 5 °C min−1. The injection was performed at 300 °C, the transfer line was kept at 300 °C, and helium was used as carrier gas. Compounds were identified by mass fragmentography and by comparing their mass spectra with standards, and quantitation was obtained from total-ion peak area using molar response factors obtained from cholecalciferol, ergocalciferol, 25-hydroxycholecalciferol and 25-hydroxyergocalciferol standards. The two latter compounds were also used as external standards for calculation of product yields.PELE and other computational analyses
The starting structures for PELE simulations were the A. aegerita peroxygenase crystal (2YOR)34 and a homology model for the C. cinerea peroxygenase structure obtained using 2YOR as the template.8 As the optimum pH for peroxygenase activity is 7, the structures were prepared accordingly using Schrodinger's Protein Preparation Wizard35 and H++ web server.1 Histidines were δ-protonated, with the exception of His82 (ε-protonated) and His118 and His251 (double protonated). All acidic residues were deprotonated, except for Asp85 which was kept in its protonated state. The ergocalciferol and cholecalciferol molecules were optimized with Jaguar37 at the DFT/M06 level with the 6-31G** basis and a PBF implicit solvent in order to obtain their electrostatic potential atomic charges. Finally, the haem site was modeled as thiolate-ligated compound I after being fully optimized in the protein environment with quantum mechanics/molecular mechanics (QM/MM) using QSite.38 The electronic calculations show three unpaired electrons: two located on the oxoiron group and a third on the heme and less than 1% spin contamination.Once the initial protein structure was prepared and ligands optimized, these were placed manually in identical positions at the entrance of the haem-access channel and PELE simulations were performed.10 PELE is a Monte Carlo-based algorithm that produces new configurations through a sequential ligand and protein perturbation, side chain prediction and minimization steps, freely available at https://pele.bsc.es. New configurations are then filtered with a Metropolis acceptance test, where the energy is described with an all-atom OPLS force field22 and a surface generalized Born solvent.7 In this way, it is possible to locate and characterize local and global minima structures for the most favorable protein–ligand interactions. PELE has been successfully used in a number of ligand migration studies with both small and large substrates.18,29,30
Distance and angle distributions were computed after screening all structures from PELE simulations by using the interaction energy and distance to the compound I oxygen atom. The structures which showed interaction energies below −20 kcal mol−1 for ergocalciferol and −30 kcal mol−1 for cholecalciferol were selected. Reactive hydrogen atoms were considered to be below 5 Å from the compound I oxygen atom for the distance analysis and 3Å for the angle study (angles are much more sensitive and thus only near attack conformations were selected). All the selected structures were then used to compute the relative frequencies of the ligand's hydrogen atoms at reactive distances to the haem oxygen and FeO–H and O–H–C angles.
Acknowledgements
This work was supported by the INDOX (KBBE-2013-7-613549) and PELE (ERC-2009-Adg 25027) EU projects, and by the BIO2011-26694 and CTQ2013-48287 projects of the Spanish Ministry of Economy and Competitiveness.Notes and references
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- E. Aranda, M. Kinne, M. Kluge, R. Ullrich and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 82, 1057–1066 CrossRef CAS PubMed.
- E. Aranda, R. Ullrich and M. Hofrichter, Biodegradation, 2010, 21, 267–281 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, M. Cañellas, F. Sancho, F. Lucas, V. Guallar, L. Kalum, H. Lund, G. Gröbe, K. Scheibner, R. Ullrich, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Appl. Environ. Microbiol., 2015, 81, 4130–4142 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, Biotechnol. Bioeng., 2013, 110, 2332 CrossRef PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, ChemCatChem, 2015, 7, 283–290 CrossRef CAS.
- D. Bashford and D. A. Case, Annu. Rev. Phys. Chem., 2000, 51, 129–152 CrossRef CAS PubMed.
- L. Bordoli, F. Kiefer, K. Arnold, P. Benkert, J. Battey and T. Schwede, Nat. Protoc., 2009, 4, 1–13 CrossRef CAS PubMed.
- S. Bormann, A. G. Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 CAS.
- K. W. Borrelli, A. Vitalis, R. Alcantara and V. Guallar, J. Chem. Theory Comput., 2005, 1, 1304–1311 CrossRef CAS.
- N. R. Buck, W. Claerhout, B. H. Leuenberger, E. Stoecklin, K. Urban and S. Wolfram, US Pat., US20130210782 A1, 2013 Search PubMed.
- B. P. Cossins, A. Hosseini and V. Guallar, J. Chem. Theory Comput., 2012, 8, 959–965 CrossRef CAS PubMed.
- D. Floudas, M. Binder, R. Riley, K. Barry, R. A. Blanchette, B. Henrissat, A. T. Martínez, R. Otillar, J. W. Spatafora, J. S. Yadav, A. Aerts, I. Benoit, A. Boyd, A. Carlson, A. Copeland, P. M. Coutinho, R. P. de Vries, P. Ferreira, K. Findley, B. Foster, J. Gaskell, D. Glotzer, P. Górecki, J. Heitman, C. Hesse, C. Hori, K. Igarashi, J. A. Jurgens, N. Kallen, P. Kersten, A. Kohler, U. Kües, T. K. A. Kumar, A. Kuo, K. LaButti, L. F. Larrondo, E. Lindquist, A. Ling, V. Lombard, S. Lucas, T. Lundell, R. Martin, D. J. McLaughlin, I. Morgenstern, E. Morin, C. Murat, M. Nolan, R. A. Ohm, A. Patyshakuliyeva, A. Rokas, F. J. Ruiz-Dueñas, G. Sabat, A. Salamov, M. Samejima, J. Schmutz, J. C. Slot, F. St.John, J. Stenlid, H. Sun, S. Sun, K. Syed, A. Tsang, A. Wiebenga, D. Young, A. Pisabarro, D. C. Eastwood, F. Martin, D. Cullen, I. V. Grigoriev and D. S. Hibbett, Science, 2012, 336, 1715–1719 CrossRef CAS PubMed.
- C. A. Fritts and P. W. Waldroup, J. Appl. Poult. Res., 2003, 12, 45–52 CrossRef CAS.
- A. Gutiérrez, E. D. Babot, R. Ullrich, M. Hofrichter, A. T. Martínez and J. C. del Río, Arch. Biochem. Biophys., 2011, 514, 33–43 CrossRef PubMed.
- A. Gutiérrez, J. C. del Río, F. J. González-Vila and F. Martín, J. Chromatogr., 1998, 823, 449–455 CrossRef.
- J. M. Hernández, US Pat., US20130137662 A1, 2013 Search PubMed.
- A. Hernández-Ortega, F. Lucas, P. Ferreira, M. Medina, V. Guallar and A. T. Martínez, J. Biol. Chem., 2011, 286, 41105–41114 CrossRef PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- G. Jean, J. C. Terrat, T. Vanel, J. M. Hurot, C. Lorriaux, B. Mayor and C. Chazot, Nephrol., Dial., Transplant., 2008, 23, 3670–3676 CrossRef CAS PubMed.
- G. Jones, Annu. Rev. Nutr., 2013, 33, 23–44 CrossRef CAS PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- S. Käppeli, E. Frohlich, S. G. Gebhardt-Henrich, A. Pfulg, H. Schäublin, R. Zweifel, H. Wiedmer and H. H. Stoffel, Arch. Gefluegelkd., 2011, 75, 179–184 Search PubMed.
- A. Karich, M. Kluge, R. Ullrich and M. Hofrichter, AMB Express, 2013, 3, 5 CrossRef PubMed.
- M. Kinne, C. Zeisig, R. Ullrich, G. Kayser, K. E. Hammel and M. Hofrichter, Biochem. Biophys. Res. Commun., 2010, 397, 18–21 CrossRef CAS PubMed.
- M. Kluge, R. Ullrich, C. Dolge, K. Scheibner and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 81, 1071–1076 CrossRef CAS PubMed.
- M. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Green Chem., 2012, 14, 440–446 RSC.
- G. A. Leichtmann, J. M. Bengoa, M. J. G. Bolt and M. D. Sitrin, Am. J. Clin. Nutr., 1991, 54, 548–552 CAS.
- D. Linde, R. Pogni, M. Cañellas, F. Lucas, V. Guallar, M. C. Baratto, A. Sinicropi, V. Sáez-Jiménez, C. Coscolín, A. Romero, F. J. Medrano, F. J. Ruiz-Dueñas and A. T. Martínez, Biochem. J., 2015, 466, 253–262 CrossRef PubMed.
- M. F. Lucas and V. Guallar, Biophys. J., 2012, 102, 887–896 CrossRef CAS PubMed.
- H. L. J. Makin and D. B. Gower, Steroid analysis, Springer, NY, 2010 Search PubMed.
- M. Michalczuk, D. Pietrzak, J. Niemiec and J. Mroczk, Pol. J. Food Nutr. Sci., 2010, 60, 121–126 CAS.
- S. Peter, M. Kinne, X. Wang, R. Ulrich, G. Kayser, J. T. Groves and M. Hofrichter, FEBS J., 2011, 278, 3667–3675 CrossRef CAS PubMed.
- K. Piontek, E. Strittmatter, R. Ullrich, G. Grobe, M. J. Pecyna, M. Kluge, K. Scheibner, M. Hofrichter and D. A. Plattner, J. Biol. Chem., 2013, 288, 34767–34776 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- J. C. Schöneboom, S. Cohen, H. Lin, S. Shaik and W. Thiel, J. Am. Chem. Soc., 2004, 126, 4017–4034 CrossRef PubMed.
- Schrödinger. Jaguar 7.8, LCC, New York, 2011 Search PubMed.
- Schrödinger. QSite 5.7, LCC, New York, 2011 Search PubMed.
- Schrödinger. SiteMap 2.5, LCC, New York, 2011 Search PubMed.
- C. Simoes-Nunes and G. M. Weber, EP Pat., EP1516540 B1, 2004 Search PubMed.
- M. D. Sitrin and J. M. Bengoa, Am. J. Clin. Nutr., 1987, 46, 1011–1015 CAS.
- R. Ullrich, C. Dolge, M. Kluge and M. Hofrichter, FEBS Lett., 2008, 582, 4100–4106 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular model for C. cinerea peroxygenase obtained using A. aegerita's crystal structure as the template. See DOI: 10.1039/c5cy00427f |
| ‡ These three authors equally contributed to this work |
| This journal is © The Royal Society of Chemistry 2016 |