
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganizing multivalency in carbohydrate recognition
Christian
Müller
,
Guillaume
Despras
* and
Thisbe K.
Lindhorst
*
Otto Diels Institute of Organic Chemistry, Christiana Albertina University of Kiel, Otto-Hahn-Platz 3/4, D-24118 Kiel, Germany. E-mail: tklind@oc.uni-kiel.de; Fax: +49 431-8807410
First published on 5th May 2016
Abstract
The interactions of cell surface carbohydrates as well as of soluble glycoconjugates with their receptor proteins rule fundamental processes in cell biology. One of the supramolecular principles underlying and regulating carbohydrate recognition is multivalency. Many multivalent glycoconjugates have therefore been synthesized to study multivalency effects operative in glycobiology. This review is focused on smaller multivalent structures such as glycoclusters emphasizing carbohydrate-centered and heteromultivalent glycoconjugates. We are discussing primary, secondary and tertiary structural aspects including approaches to organize multivalency.
 Christian Müller | Christian Müller was born in Bad Oldesloe, Germany, in 1984. He graduated from Christiana Albertina University of Kiel in chemistry. During his diploma thesis he dealt with the synthesis of trivalent cluster glycosides for the investigation of carbohydrate recognition on surfaces. Currently he is finishing his PhD thesis in the research group of Prof. Lindhorst, which is focused on the synthesis of photoswitchable homo- and heteroglycoclusters. |
 Guillaume Despras | Guillaume Despras was born in Ajaccio, France, in 1982. He obtained an engineering degree in chemistry in 2006 at the Ecole Nationale Supérieure des Ingénieurs en Arts Chimiques et Technologiques in Toulouse. He received his PhD degree in organic chemistry at the University Pierre et Marie Curie, Paris, in 2011 under the supervision of Dr Jean-Maurice Mallet. He worked on the synthesis of functional bioactive oligosaccharides, especially chondroitin sulfate analogues. The same year, he joined the group of Prof. Jean-Marie Beau at Université Paris-Sud as a postdoctoral fellow, working on chitooligomers. In 2014 he was a researcher in the Mallet group and in 2015 joined the Lindhorst group at Christiana Albertina University of Kiel with the aim to start an independent carrier. |
 Thisbe K. Lindhorst | Thisbe Lindhorst studied chemistry at the Universities of München and Münster. She obtained her doctorate with Prof. J. Thiem at the University of Hamburg and performed a postdoctoral training with Prof. S. G. Withers at UBC (Vancouver, Canada). Thisbe Lindhorst started her own independent research in the field of the glycosciences at the University of Hamburg and was appointed full professor at Christiana Albertina University of Kiel in 2000. Her research is focused on glycomimetics synthesis, glycoarrays, and the biological role of the glycocalyx. She is the author of the text book “Essentials in Carbohydrate Chemistry and Biochemistry”. In 2016 and 2017 Thisbe Lindhorst serves as elected president of the German Chemical Society (GDCh). Thisbe has two grown-up children. |
1. Introduction
Carbohydrates, beside nucleic acids and proteins, constitute a class of biopolymers that govern the molecular complexity and individual diversity of organisms.1 Conjugated in various forms, such as in glycoproteins and glycolipids for example, they make up a thick layer, called glycocalyx,2 which covers all cell surfaces. A cell's glycocalyx connects the inside of a cell with the outside, thus being involved in numerous biological processes including cell–cell communication.3 However, the investigation of carbohydrate-related cell biology is complicated by the enormous structural complexity of glycocalyx components, which can be hardly overlooked. Moreover, the supramolecular interplay between cell surface components still lies in the dark.A striking characteristic of cell surface glycoconjugates is their multivalent nature. Glycoconjugates typically assemble plenty carbohydrate constituents in multiple copies and various branches. On the cell surface they interact with other glycoconjugates,4 and with various lectins. Lectins are proteins that bind carbohydrates specifically to initiate or mediate certain biological responses.2c,5 The multivalent nature of many carbohydrate–lectin interactions has been recognized back in the 1970's through the seminal work of Y. C. Lee.6 Since then it has become clear that frequently multiple carbohydrate ligands interact with multiple complementary carbohydrate binding sites on lectin receptors, resulting in specific biological effects, which have been intensively explored.7 At the same time it became evident that there is not only one multivalent principle governing carbohydrate recognition in a complex cellular environment, but that many different multivalency effects are operated and cooperating in cell–cell interactions.
In light of this, it is obvious that chemists have come up with a plethora of synthetic multivalent glycoconjugates to facilitate the study of the various multivalency effects related to carbohydrate recognition in biological systems. Almost every possible molecular architecture, which allows multivalent assembly of carbohydrate ligands, has been implemented and evaluated. The multitude of related chemical structures was compiled in several publications and various useful approaches to a systematic classification were chosen in reviewing the field of multivalent glycoconjugates.8–19 Focusing on more recently published work, we add here to the existing overviews a selection of rather small multivalent glycoconjugates, often called glycoclusters or cluster glycosides, respectively. In addition, we propose a categorization of glycoclusters following the terminology of protein structures, in order to organize functional aspects of glycoclusters. Thus, we report on multivalent glycoconjugates with primary, secondary and tertiary structural features.
2. Getting started
The significance of glycoclusters for glycobiological research can be easily appreciated with a brief retrospect to multivalency effects in carbohydrate recognition as introduced by the benchmark work of Y. C. Lee since the 1970's. A series of papers published by Reiko T. Lee and Yuan C. Lee has not only demonstrated how multivalent cluster glycosides can be synthesized, but also how the spatial arrangement and the valency of cluster glycosides can be varied based on simple scaffold molecules.20 Their concept of clustering carbohydrate ligands for lectin binding on molecules such as tris(hydroxymethyl)aminomethane (TRIS) and branched peptides was thereafter followed closely by many other researchers. In Fig. 1 some representative examples of the cluster glycosides, prepared by Lee and Lee, are depicted. As in Fig. 1, in this review arbitrary graphical abbreviations such as spheres will be used to represent carbohydrate residues where appropriate. Equally, graphical elements will be used to symbolize linker moieties or other structural elements of a multivalent glycoconjugate.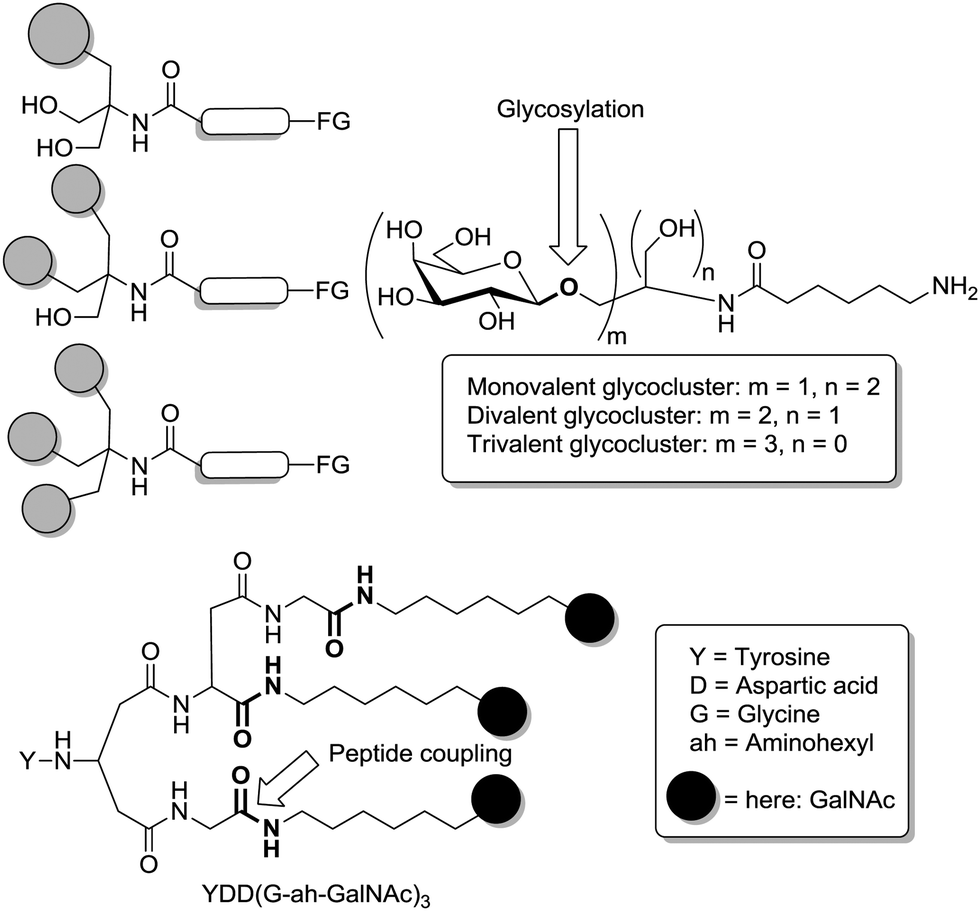 | ||
| Fig. 1 The seminal work by Y. C. Lee and co-workers involved the synthesis of cluster glycosides with different valencies and varied spatial distribution of the sugar epitopes. Cluster glycosides were achieved by glycosylation of TRIS (top) and also by peptide coupling of amino-functionalized glycosides with branched peptide scaffold molecules (bottom). Sugars are depicted as spheres and can represent different carbohydrates such as glucose (Glc), galactose (Gal), L-fucose (Fuc), N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), lactose (Lac), or mannose (Man). FG = functional group. | ||
Notably, also a sensible nomenclature was introduced by Y. C. Lee. The naming of a peptide-based trivalent GalNAc cluster as “YDD(G-ah-GalNAc)3” clearly indicated the comprised building blocks (Fig. 1). In Lee's work, the nature of the sugar ligands was altered and the valency of the glycoclusters was varied from 1 to 4. Glucosides, galactosides, lactosides as well as clusters equipped with GalNAc or GlcNAc residues were prepared along analogous synthetic pathways to investigate interactions with the asialoglycoprotein receptor (ASGPR) on the surface of hepathocytes.20c It was known that mammalian hepatic receptors have a strong affinity toward clustered galactosyl and GalNAc residues as found in desialylated serum glycoproteins with affinities of bi- and triantennary glycans being up to 103- and 106-fold higher, respectively, than of the monovalent glycosides. Then it was shown that this effect, which was termed “cluster effect”, could be reproduced with the synthetic cluster glycosides when the valency and the overall structure of the respective glycoside was appropriate. These findings prompted, however after some delay, a flood of follow-up work, which led to important insights of which some were already suggested by Y. C. Lee earlier on. In his 1995 review he stated farsighted: “… the degree of dependency of affinity increase on valency varie[s] from lectin to lectin. A strong glycoside cluster effect obviously requires two partners: a lectin with clustered sugar binding sites and a multivalent ligand that can present sugars with proper orientation and spacing.”20c Since Y. C. Lee's seminal work, the study of cluster or multivalency effects, respectively, ruling carbohydrate–protein interactions has emerged as a momentous topic in glycoscience. Despite the continuous refining of the related knowledge through the design of potent multivalent glycoconjugates with high avidity and the accurate determination of many lectin structures,5,19 the exact way in which carbohydrate recognition and binding events occur in a biological environment is still uncertain.
In view of Y. C. Lee's work as well as of the research of his successors, the content of this account will be structured into three main sections in order to highlight and discover some of the multifaceted aspects of multivalency in carbohydrate recognition. We will discuss primary, secondary and tertiary aspects of glycocluster structures in analogy to the common terminology used in structural biology of proteins. Naturally, this classification is not meant to strictly trisect the multitude of known glycoclusters. But it suggests a systematic approach to the various structural features of multivalent glycoconjugates, which collectively govern carbohydrate–protein interactions in a supramolecular environment. In reviewing primary structures of glycoclusters, we emphasize the basic structural elements such as the nature of the central core moiety (scaffold), linkers, carbohydrate epitopes, extra functional elements and the connectivity of these building blocks. Within this section we will focus on two special structural aspects of glycoclusters, (i) the use of carbohydrates as core molecules, and (ii) on heteromultivalency as resembled by glycoconjugates in which more than one type of carbohydrate epitope is assembled.
The second main section is focused on aspects of the three-dimensional arrangement of sugar epitopes within a glycocluster as it is governed by the nature of the scaffold moiety and in particular by the nature of the employed linkers. It will be discussed how carbohydrate recognition and the related affinities vary depending on the conformational availability of the sugar epitopes within a glycocluster. The third section, which is dedicated to what we have called tertiary aspects of glycocluster structures, will include supramolecular effects on surfaces, for example, as well as dynamic effects. These can be triggered and regulated through modifications that allow structural responsiveness towards external stimuli such as biochemical, chemical or physical parameters. It will be outlined how responsive glycoclusters can be utilized to study orientational effects in carbohydrate recognition on surfaces.
3. Highlighting primary structural features
In this section, the principal structural architecture of glycoclusters is discussed. In analogy to the primary structure of peptides and proteins, we focus on the connectivity and the nature of the principle components of a glycocluster, the selected scaffold, the employed ligation elements and the nature of the attached carbohydrate ligands. Because much of the related chemistry has been reviewed earlier,8–17 two especially interesting aspects of the primary glycocluster structure will be highlighted, namely carbohydrate scaffolds on the one hand and on the other hand heteroglycoclusters, which assemble different carbohydrate ligands in one molecule.15Glycocluster synthesis has been frequently based on a number of main molecular scaffolds as depicted in Fig. 2, poly(amidoamine) (PAMAM) dendrimers,21 aryl derivatives,13c TRIS (tris(hydroxymethyl)aminomethane), the Newkome-type dendron,22 pentaerythritol, oligonucleotides,12 peptides,11 and carbohydrates or glycosides,10 respectively. Dendrimers like the PAMAM dendrimers have been used to make glycodendrimers. These are multivalent glycoconjugates, which are decorated with carbohydrate ligands in the periphery of the dendrimer. The field of glycodendrimers has been extensively reviewed9 and will not be further discussed here. However, it is worthwhile to mention that a PAMAM core does not offer a broad range of possibilities to introduce heterogeneity in the primary structure of a glycocluster. This can be regarded as a disadvantage of PAMAM-based glycodendrimers. The use of aromatic cores for the construction of multivalent glycoconjugates13c also bears a limitation for the design of glycoclusters as the planarity of an aryl scaffold restricts the spatial variation of attached carbohydrate ligands, which is an important aspect of the secondary structure of glycoclusters.
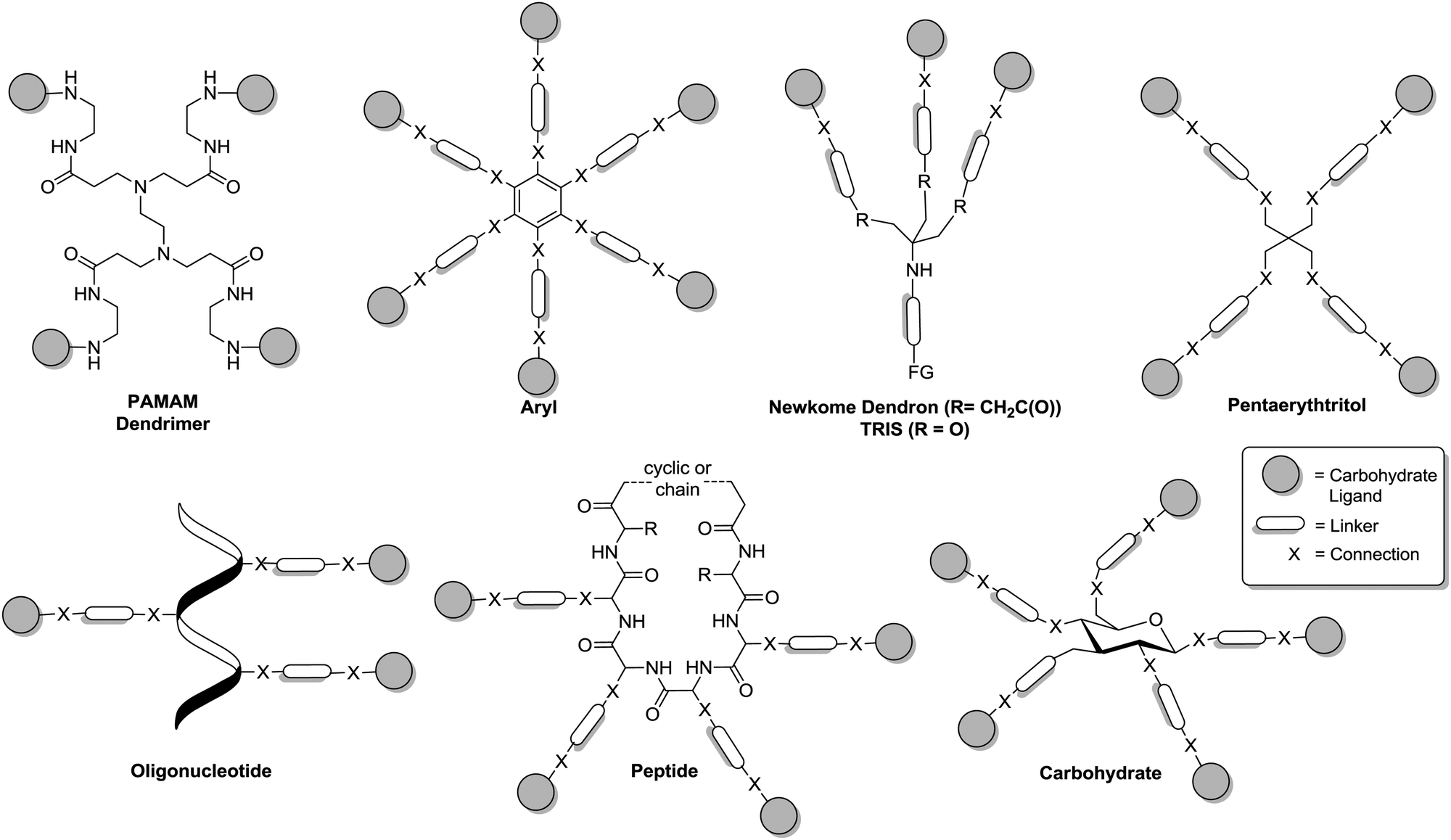 | ||
| Fig. 2 Schematic structures of glycocluster as based on frequently used molecular scaffolds. Carbohydrate ligands are depicted as grey spheres. FG = functional group. | ||
Both, the Newkome-type dendron (di-tert-butyl-4-amino-4-[2-(tert-butoxycarbonyl)ethyl]-heptanedioate), which can be obtained by triple Michael addition of tert-butyl acrylate to nitromethane21 and TRIS, which was used as scaffold for glycocluster synthesis already by Y. C. Lee, offer the intriguing possibility to directly access glycoclusters of an AB3-type. Thus, the orthogonal functional group at the focal point of these two scaffold molecules can be employed to attach a trivalent cluster glycoside to further multivalent molecules or to a surface, respectively. Alternatively, it can be decorated with a carbohydrate ligand of a second type to achieve heteroglycoclusters.
Glycoclusters based on the Newkome dendron or on TRIS, respectively, have been variously functionalized at their focal point (Fig. 3). Thus, several homoglycoclusters could be conjugated with biotin (e.g.1),23 porphyrins (e.g.2),24 rhodamine fluorophores (e.g.3),25 peptides (e.g.4),26 or hydrophobic chains (e.g.5).27 In the latter case, Pucci et al. also performed synthetic sequences suitable for the introduction of two distinct ligands. Alternatively, methallyl dichloride (3-chloro-2-chloromethyl-1-propene) has been employed as core molecule for the synthesis of glycodendrons.28 These carry a double bond at the focal point which can be used for postsynthetic functionalization to make glycolipid mimetics (e.g.6).29
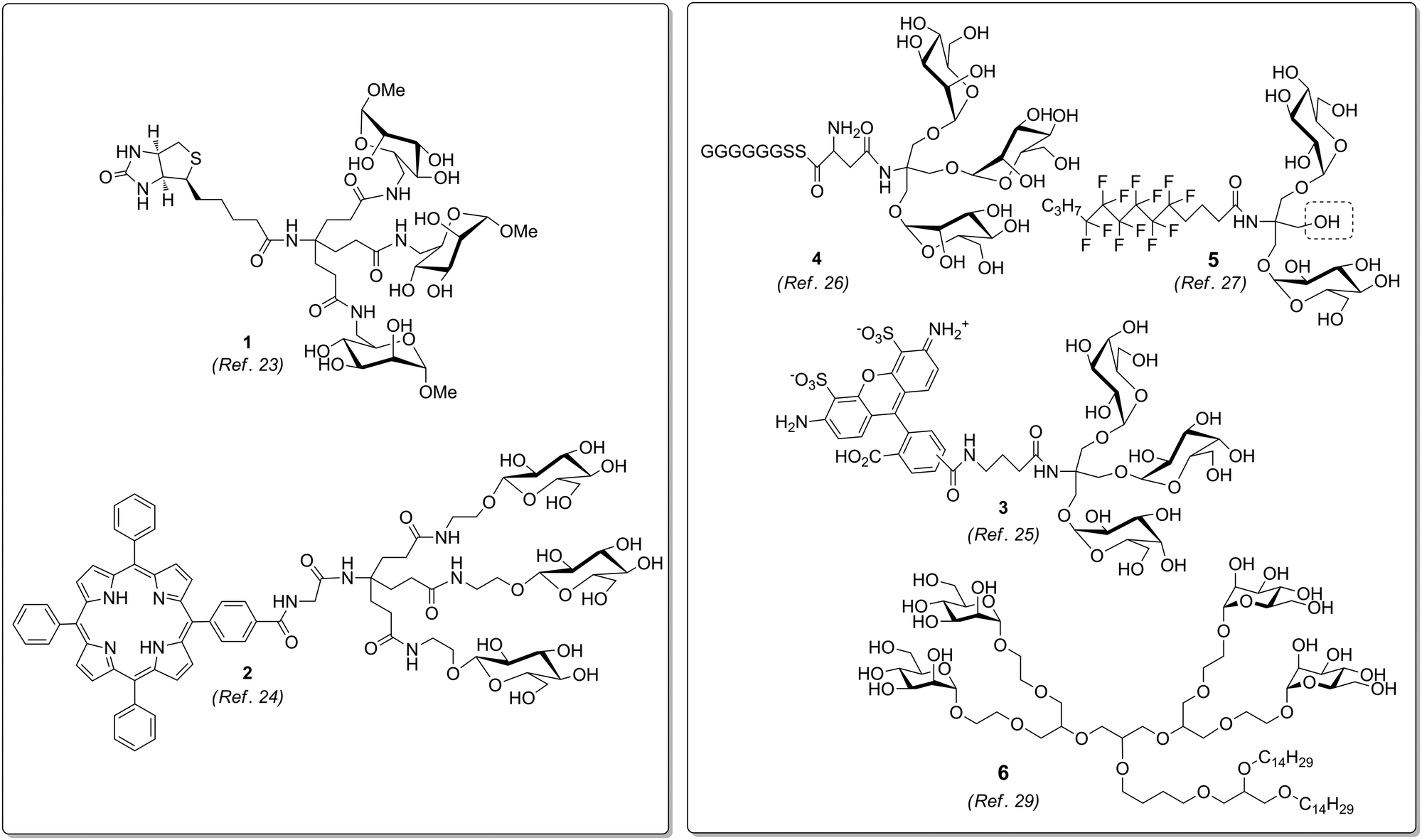 | ||
| Fig. 3 Glycoclusters prepared on the basis of the Newkome dendron (left) or a TRIS scaffold (right), respectively. They can be easily equipped with a functional moiety at the focal point of the molecule. | ||
Pentaerythritol (PE) has frequently been employed for the synthesis of tetravalent cluster glycosides.30 But, PE can also be turned into a scaffold molecule of the AB3-type even though it resembles a highly symmetrical structure with four equivalent hydroxy functional groups. This is due to the fact that further functionalization of a threefold modified PE derivative is especially hard because of increasing steric bulk and because of the neopentyl character of the remaining hydroxy group, which hampers further substitution. Thus, a tri-O-allylated PE derivative (Scheme 1) can be easily achieved to form the starting point for the synthesis of a heterovalent glycocluster as shown by García Fernández and colleagues.31 In this work, acetyl-protected glycosyl thiols of three different sugars (Man, Glc, Lac) were used for conjugation via thiol–ene reaction (Scheme 1). It was possible to install two sugar moieties of the same type in the first ligation step by controlling the amount of used glycosyl thiols (7, 8, or 9) to achieve 10, for example. The third allyl function remained free for further coupling with a different ligand. When a thiolactoside derivative was used, mainly the monovalent product 14 was obtained. The free primary alcohol function of the PE scaffold, which remained free after the attachment of three ligands, was next turned into an isothiocyanato function to afford the heterotrivalent glycodendrons 13 and 17, which can be further conjugated. These heteroglycoclusters were employed in binding studies with the lectins concanavalin A (ConA) and peanut agglutinin (PNA) (cf. Section 5).
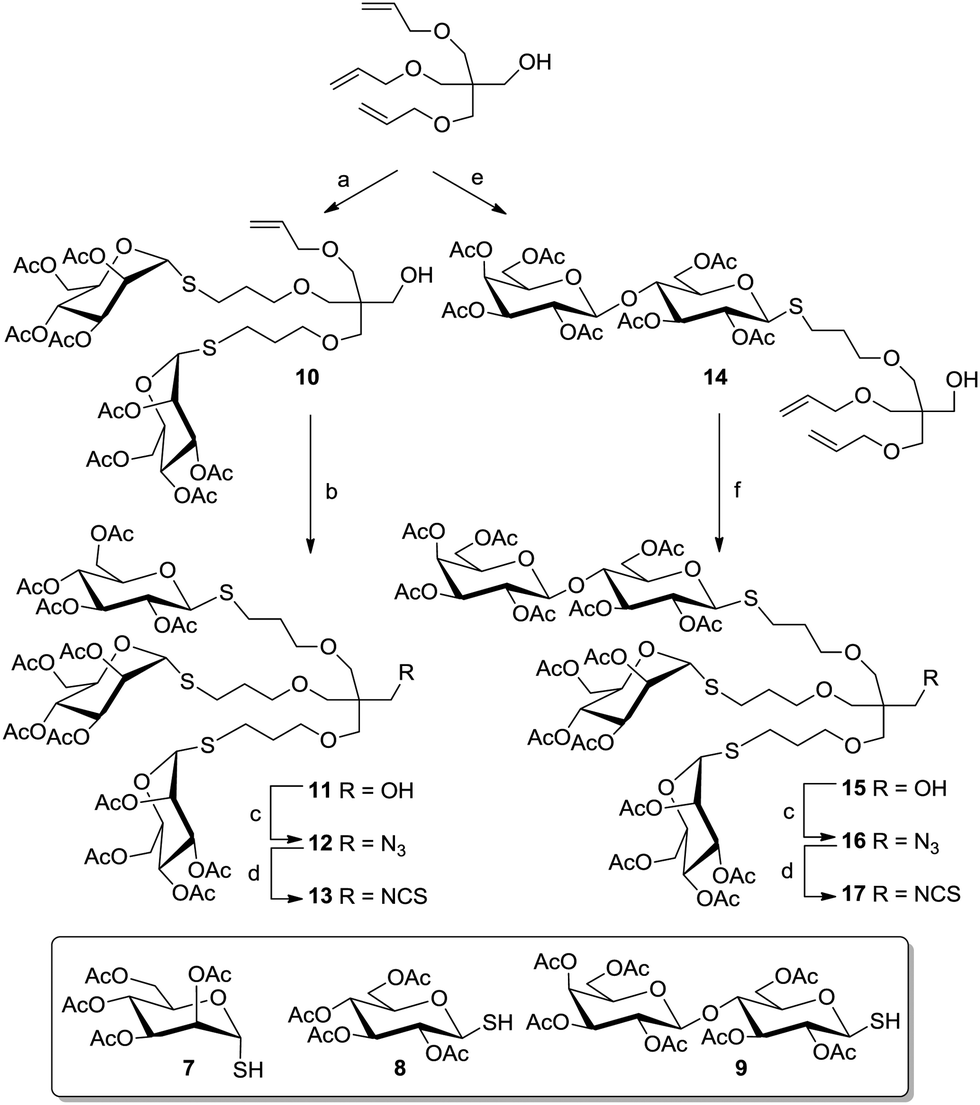 | ||
| Scheme 1 Selective functionalization of tri-O-allylated pentaerythritol. Reagents and conditions: (a) 7 (3 eq.), hν (250 nm), MeOH, rt, 72%; (b) 8 (1.5 eq.), hν (250 nm), MeOH, rt, 68%; (c) (i) Tf2O, DCM/pyridine, −25 °C; (ii) NaN3, DMF, rt, 79% for 12, 71% for 16; (d) PPh3, CS2, dioxane, rt, 80% for 13, 87% for 17; (e) 9 (1.35 eq.), AIBN, dioxane, 75 °C, 71%; (f) 7 (2 eq.), AIBN, dioxane, 75 °C, 35%. | ||
Lönnberg et al. developed a set of approaches involving peptide chemistry on solid support to prepare heteroglyclusters bearing three distinct ligands.32 These strategies were based on the preliminary desymmetrization of pentaerythritol. In a first study, the authors started from the 2,2-bis(bromomethyl)-1,3-propanediol of which one primary alcohol function was selectively protected with a 4-methoxytrityl group (MMT) (Scheme 2). Then, the two bromide functions were substituted to afford the respective diazide, which could be reduced to the monoamino derivative using sodium borohydride and 1,3-propanedithiol. The free amine was Boc-protected, then Staudinger reduction of the remaining azido function and a subsequent carbamoylation step afforded an N-allyloxycarbonyl (Alloc) function. The free primary alcohol function was then converted into an N-phthalimido derivative in a Mitsunobu reaction, the methoxytrityl group was cleaved subsequently and the resulting free hydroxy group oxidized under Jones conditions. Finally, the phthalimide function was replaced by a Fmoc group after a cleavage step in the presence of ethylenediamine. The resulting scaffold molecule 18 was finally attached on a solid support via its carboxylic acid function to afford orthogonally protected 19 (Scheme 2). This allowed sequential deprotection and coupling steps involving pentafluorophenyl-armed glycoamino acids to afford heterotrivalent cluster glycosides such as 20.
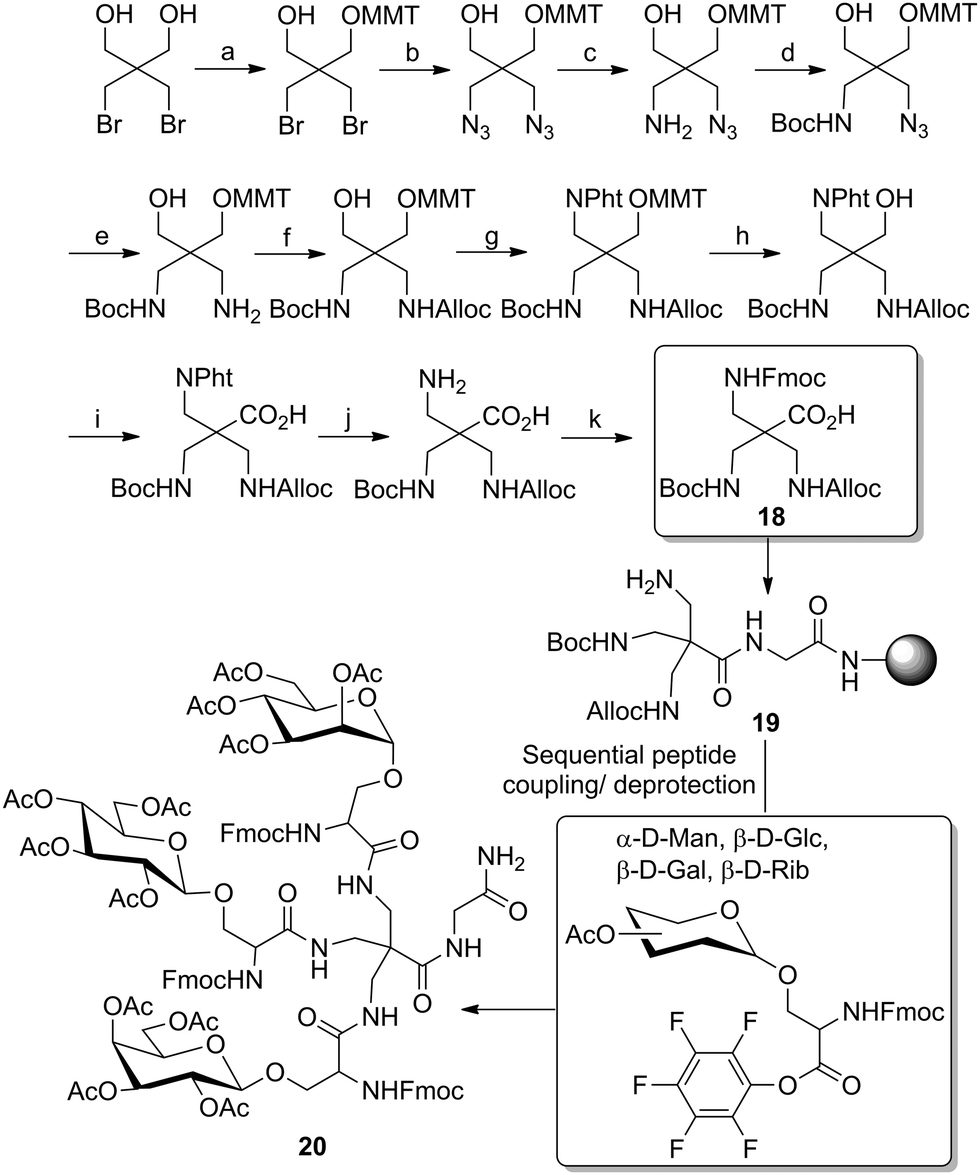 | ||
| Scheme 2 Solid phase-supported synthesis of a trivalent heteroglycocluster based on a pentaerythrityl scaffold. Reagents and conditions: (a) MMT-Cl, pyridine, 94%; (b) NaN3, LiCl, DMF, pyridine, 84%; (c) NaBH4, HS(CH2)3SH, Et3N, iPrOH, 45%; (d) Boc2O, NaOH, MeCN, 99%; (e) PPh3, aq. NH3, 82%; (f) AllocCl, Et3N, dioxane, 78%; (g) phthalimide, PPh3, DEAD, THF, 66%; (h) I2, MeOH, DCM, 72%; (i) CrO3,H2SO4, H2O, acetone, 54%; (j) NH2NH2, allyl alcohol, DMF, dioxane, 71%; (k) FmocCl, K2CO3, H2O, MeCN, 70%. | ||
More recently, the same authors improved this approach to yield biotinylated heteromultivalent glycoclusters in a more straightforward way.32d Here, the branching unit was synthesized starting from 2,2-bis(bromomethyl)-1,3-propanediol, which was first converted into a diamine derivative (Scheme 3). Then, after Boc-protection of both amino groups, the primary alcohol functions were mesylated and nucleophilic displacement then gave the respective diazide. Selective Staudinger reduction led to a monoamine, which was protected as allyl carbamate (Alloc), and then both Boc-protected amino functional groups were set free to result the scaffold molecule 21. This was preliminary anchored on an aldehyde-functionalized solid support via reductive amination to achieve 22 carrying a free a primary amino function. A peptide coupling/deprotection sequence involving protected glycosides, functionalized with a short carboxylic linker, then yielded the desired heteromultivalent clusters such as 23. In order to obtain the biotin conjugate 24, the glycocluster was cleaved from the solid support and condensed with levulinic acid at the liberated secondary amino function. Thus, an aminooxy-linked biotin derivative was finally reacted with the ketone bound to the levulinic tether to furnish the immobilizable conjugate.
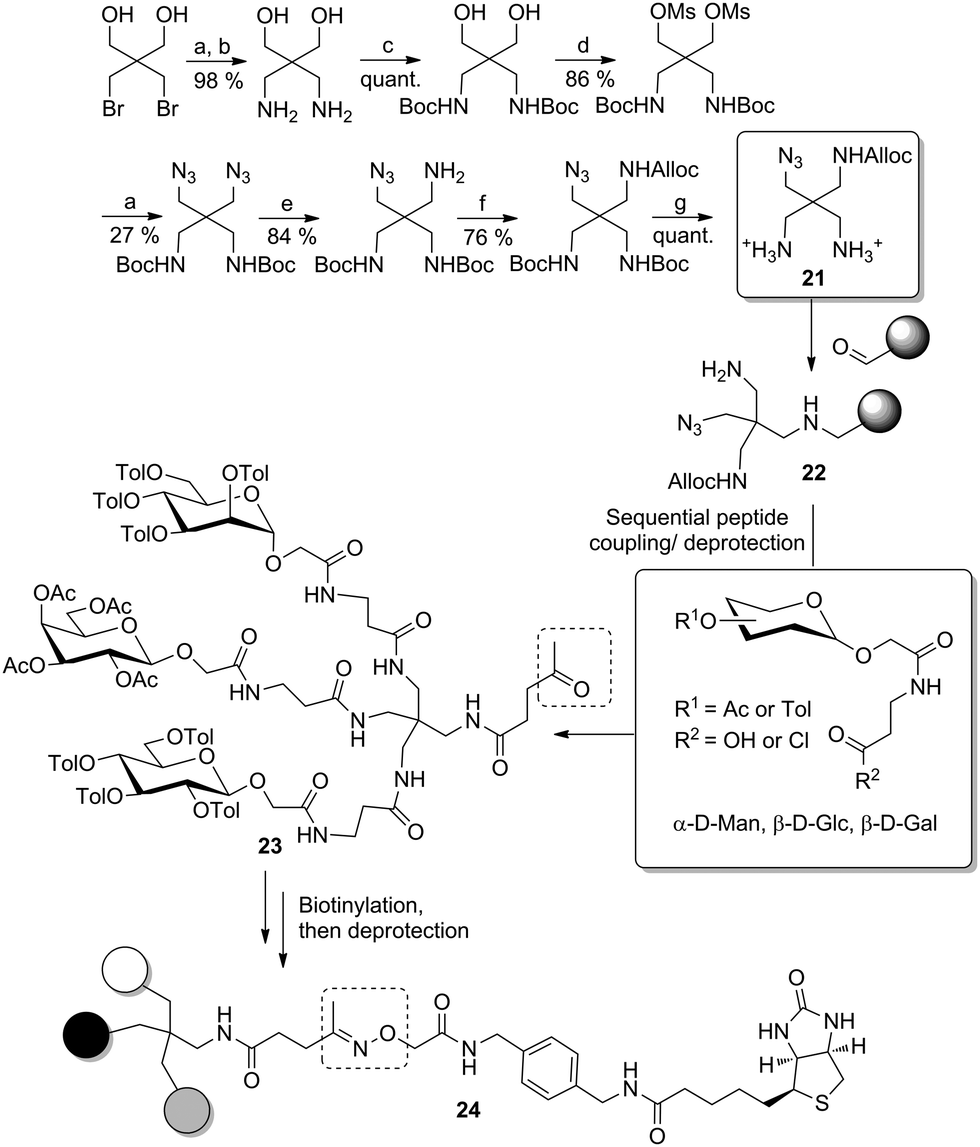 | ||
| Scheme 3 Preparation of a biotinylated trivalent heteroglycocluster. Reagents and conditions: (a) NaN3, DMF; (b) H2, Pd/C, EtOH, over two steps; (c) Boc2O, NaOH, H2O, MeCN, quant.; (d) MsCl, pyridine; (e) (i) PPh3, THF, (ii) NH4OH, THF; (f) AllocCl, Et3N, dioxane; (g) TFA, DCM. | ||
Other attractive molecules for organizing multivalency are cyclic peptides since they can be readily built by automated synthesis and their side chains can be easily orthogonally protected. In a pioneer work, Danishefsky's group prepared an anticancer vaccine candidate exhibiting three different epitopes along a short peptidic chain.33 Wittmann's group extensively used cyclopeptide-based homomultivalent structures to investigate binding of GlcNAc ligands to wheat germ agglutinin (WGA).34 Recently, Renaudet et al. selectively installed up to four orthogonal functions, namely aldehyde, allyl, azide, and chloroacetyl groups at specific positions of a cyclopeptide in order to obtain heterovalent clusters displaying a well-defined distribution of the carbohydrate ligands (Fig. 4).35 By successive application of oxime ligation, thiol–ene reaction, copper-catalyzed azide–alkyne cycloaddition (CuAAC),36 and nucleophilic substitution the heterotetravalent cluster 27 was yielded.35c Notably, one lysine residue of the scaffold peptide could be used for further ligation using thiourea bridging or peptide coupling, to increase valency and/or complexity of the glycoconjugates. Also, using a di-functionalized core ready for oxime chemistry and CuAAC coupling, a library of heteromultivalent glycoclusters with varying proportion of the respective ligands was generated.35b
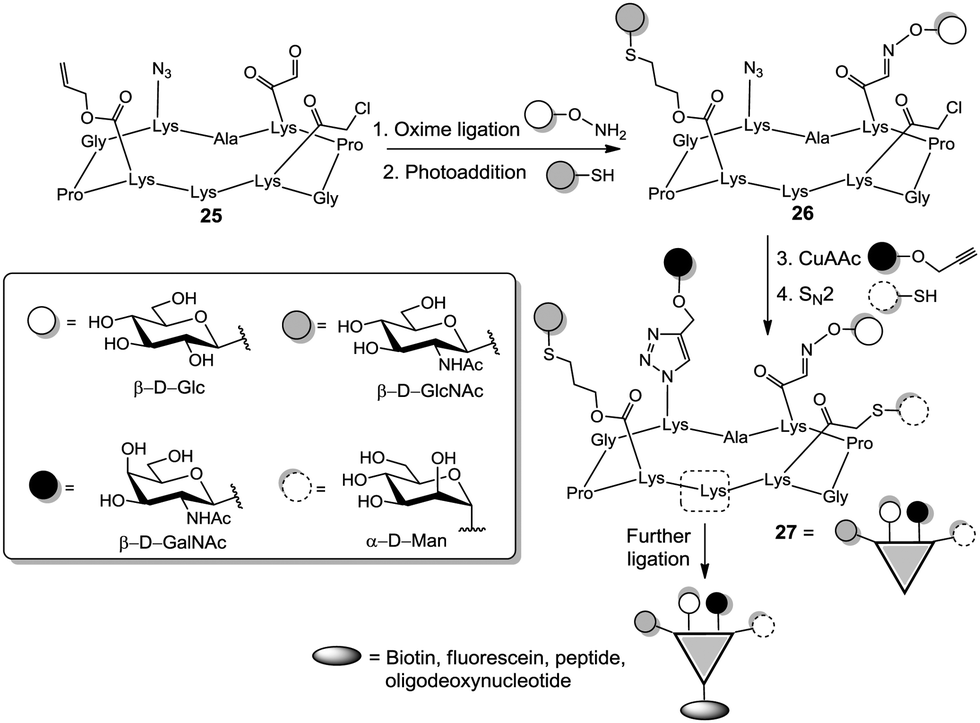 | ||
| Fig. 4 Cyclopeptide-based heteroglycoclusters suitable for orthogonal ligation reactions. | ||
Aiming at the design of new inhibitors for fucosyltransferase, which takes GDP-L-fucose as its natural substrate, van Boom et al. also used a peptide-based approach to provide libraries of glycoclusters equipped with one L-fucose unit, a second sugar moiety different from L-fucose, and thirdly, a guanosine portion (Fig. 5).37 The respective trifunctional clusters were centered on an orthogonally protected lysine derivative and prepared by solid phase synthesis. Structural diversity was introduced by varying the length of peptide linkers, which were installed between the lysine core and the peripheral sugar and nucleoside moieties, respectively.
 | ||
| Fig. 5 Libraries of trivalent heteroglycoclusters centered on a lysine core in order to achieve inhibitors of fucosyltransferase. G = guanosine. | ||
The contribution of DNA chemistry to the design of heteroglycoclusters is noteworthy, since oligonucleotide glycoconjugates bear the potential for immobilization on a surface via hybridization with complementary oligonucleotide strains.12 In a first paper, Dubber and Fréchet exploited automated DNA synthesis to make multivalent glycoconjugates.38 Next, Morvan et al. exploited the phosphoramidite method combined with automated solid phase synthesis to afford short oligophosphates or oligophosphoramidates.39 Phosphoramidite-armed branching units, derived from 1,1,1-tris(hydroxymethyl)ethane, were prepared and functionalized with a propargyl or a bromohexyl group (Scheme 4). The remaining alcohol functional groups served for oligomerization. Thus, the two key building blocks 31 and 32 were alternately attached onto an oligonucleoside moiety to give the scaffold 33, then the first ligand (azido-functionalized) was coupled via CuAAC yielding the conjugate 34. A final coupling step involving the second ligand (alkyne-functionalized) was achieved after displacement of the bromo substituent by azide, hence affording the heteroglycocluster 35.
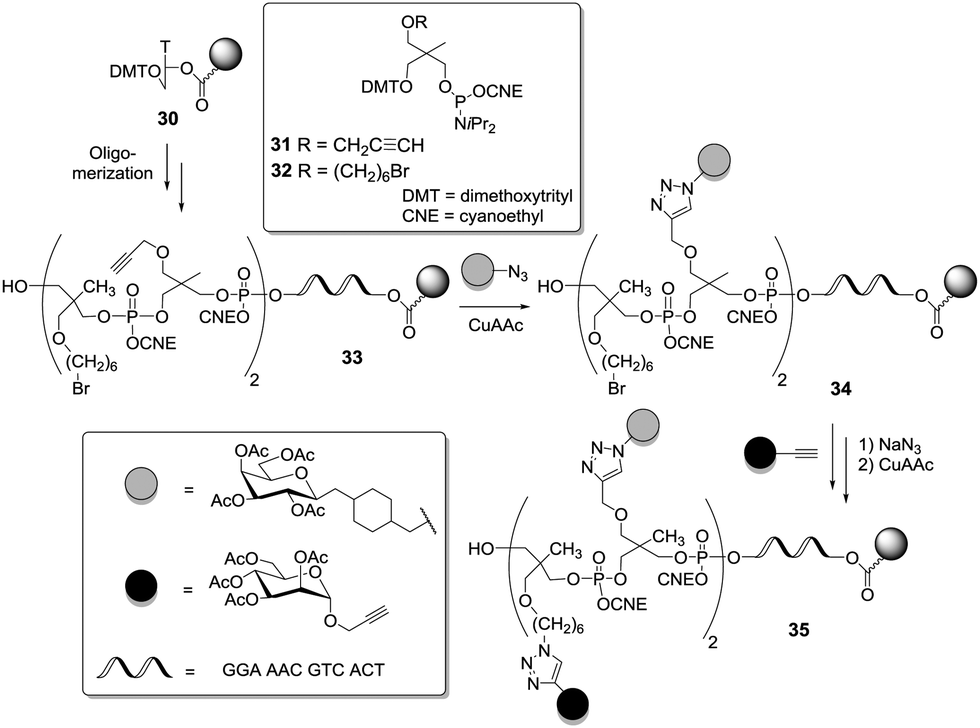 | ||
| Scheme 4 Construction of heterovalent oligonucleotide glycoconjugates by combining phosphoramidite and CuAAC chemistries. | ||
In a similar approach, the two distinct ligands were directly linked to the phosphotriester core 37 (Scheme 5) via sequential or convergent synthesis. CuAAC ligation was employed to obtain 38 and next 39 was yielded after nucleophilic substitution of bromoacetylated glycosides.39d
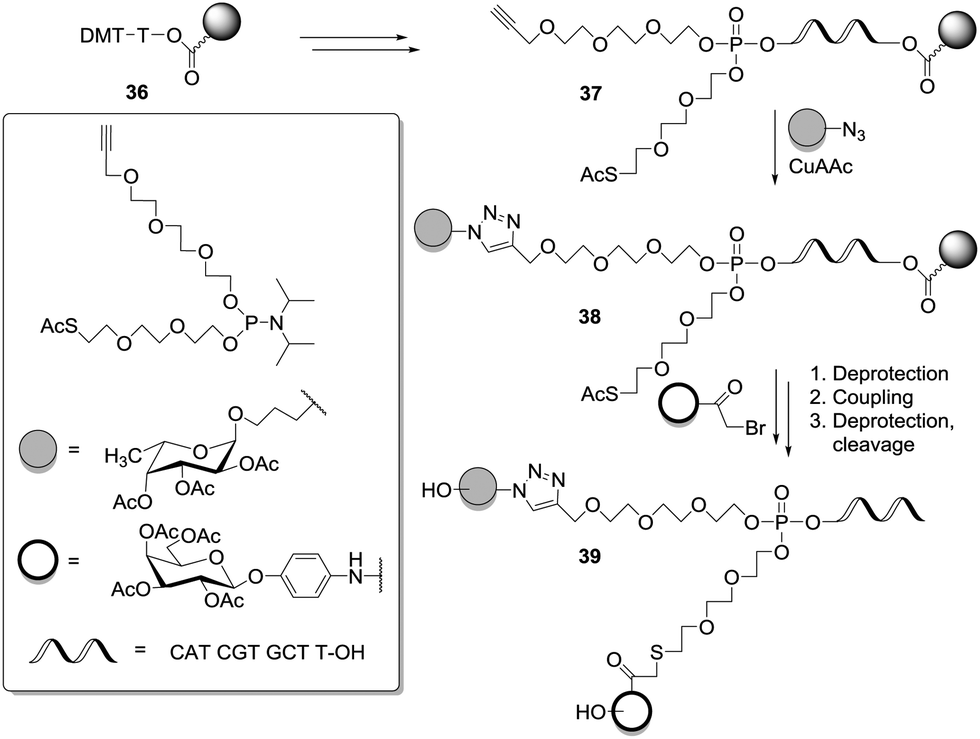 | ||
| Scheme 5 Divalent heteroglycocluster centered on a phosphotriester scaffold. | ||
Lönnberg and co-workers also reported the use of phosphoramidite ligation to provide DNA-functionalized heteroglycoclusters (Scheme 6). In the following synthesis, the O,O-methoxymethylene derivative of diethyl 2,2-bis(hydroxymethyl)malonate 40 was desymmetrized by a selective aminolysis reaction. The resulting hydroxypropyl tether was next protected. A following analogous sequence afforded the orthogonally protected product 42. The key phosphoramidite building block 44 was obtained after cleaving the acetal, selective protection of the primary alcohol and a final reaction with methyl N,N-diisopropylphosphoramidite chloride. After attachment of 44 onto a solid support via a short oligonucleoside, the scaffold 46 was sequentially condensed with three distinct toluyl-protected 6-O-phosphoramidite glycosides (45 as Man, Gal, or Glc) to yield the heterotrivalent cluster 47.
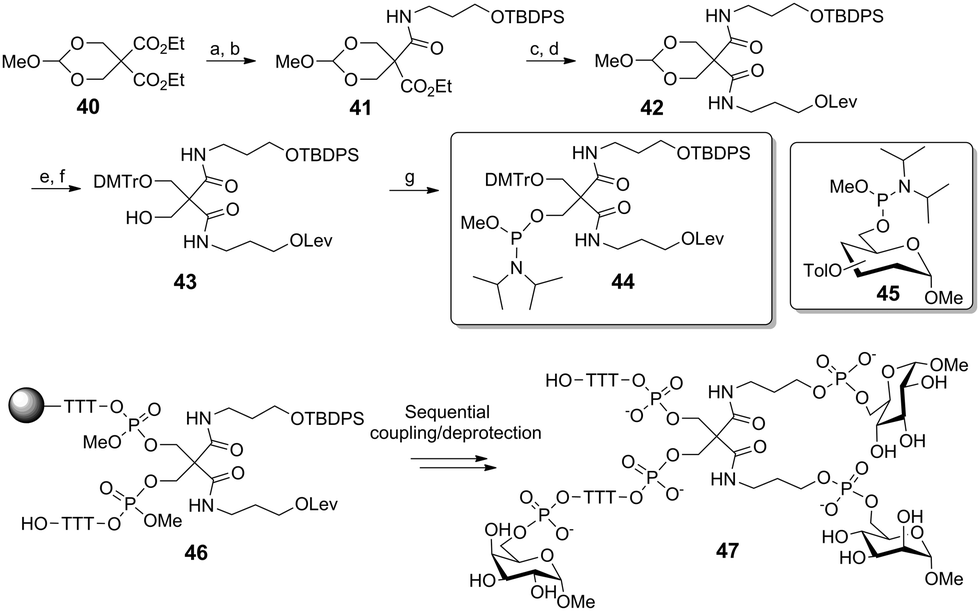 | ||
| Scheme 6 Synthesis of a heterotrivalent glycocluster with phosphate tethers. Reagents and conditions: (a) 3-aminopropanol, THF, 36%; (b) TBDPSCl, imidazole, DMF, 74%; (c) 3-aminopropanol, 79%; (d) levulinic acid, DCC, DMAP, pyridine, dioxane, 80%; (e) 80% AcOH, 91%; (f) DMTrCl, pyridine, 51%; (g) methyl N,N-diisopropylphosphoramidite chloride, Et3N, DCM, 80%. | ||
The so far reviewed interesting approaches to access orthogonally protected scaffold molecules for the synthesis of heteromultivalent glycoclusters suffer from two drawbacks. First, the synthetic pathways starting with TRIS, pentaerythritol or scaffolds of the Newkome-type involve a rather large number of steps. Second, preparation of glycoclusters based on peptide or oligonucleotide solid phase synthesis is difficult to scale up. These obstacles can be circumvented by means of carbohydrate chemistry providing valuable alternatives and options in several regards. Carbohydrates are readily available and cheap materials from natural resources and can be easily functionalized with orthogonal groups thanks to the distinct reactivity of the several positions on the sugar ring (Fig. 6).10c Additionally, the group of carbohydrates represents a large collection of scaffold molecules for variation of the three-dimensional presentation of ligands as they are characterized by defined combinations of stereocenters as related to the molecular diversity of saccharides.
 | ||
| Fig. 6 Special features of carbohydrates. Top box: reactivity pathways of the anomeric position and the primary alcohol. Bottom box: selective protection of a carbohydrate ring, exemplified with D-galactose. | ||
First of all, carbohydrates, in their cyclic forms (pyranose or furanose), possess a hemiacetal function at the C-1 position, the anomeric position. Consequently, this function can be independently activated to give an intermediate oxocarbenium species which can react with various nucleophiles such as alcohols, thiols, amines, or halogen anions (Fig. 6). Moreover, the free anomeric alcohol can be selectively deprotonated to further attack electrophilic molecules. Finally, the anomeric carbon is also prone to selective radical additions to form various products such as C-glycosides, for example.
The second main feature of many common carbohydrates is the presence of a primary hydroxy group at the C-6 position, which can be easily discriminated from the other secondary alcohol functions of the hexopyranose ring. In addition, the reactivity of the secondary hydroxy groups can also be graded as it depends on the respective stereochemistry. For instance, the axial 4-hydroxy group in galactose is less reactive than the OH groups at position 2 and 3 and can thus be kept intact during functionalisation of OH-2 and -3 (Fig. 6). Notably, nature also provides hexosamine sugars which can be advantageously used as scaffolds, thanks to the presence of the secondary amino function at the C-2.40 However, in spite of their great potential as orthogonally functionalized core molecules, hexosamines have not been exploited for fabrication of heteromultivalent glycoclusters.
Pioneer work on carbohydrate scaffolds was achieved by Lindhorst and co-workers.41 In a first report, the authors described the straightforward preparation of a mannosylcluster based on a carbohydrate core derived from per-allylated α-D-glucoside 48 (Scheme 7). This was submitted to a photochemically driven exhaustive addition of cysteamine hydrochloride to the alkene functions. Then, a pentavalent glycocluster was obtained after reacting the amino-functionalized scaffold with acetyl-protected α-mannosyl isothiocyanate 49. Related chemistry could be extended to further carbohydrate scaffolds, such as trehalose, melibiose and raffinose, and the resulting carbohydrate-centered glycoclusters were named as “octopus glycosides” owing to the “multi-arm” nature of the structural architecture.10b,42
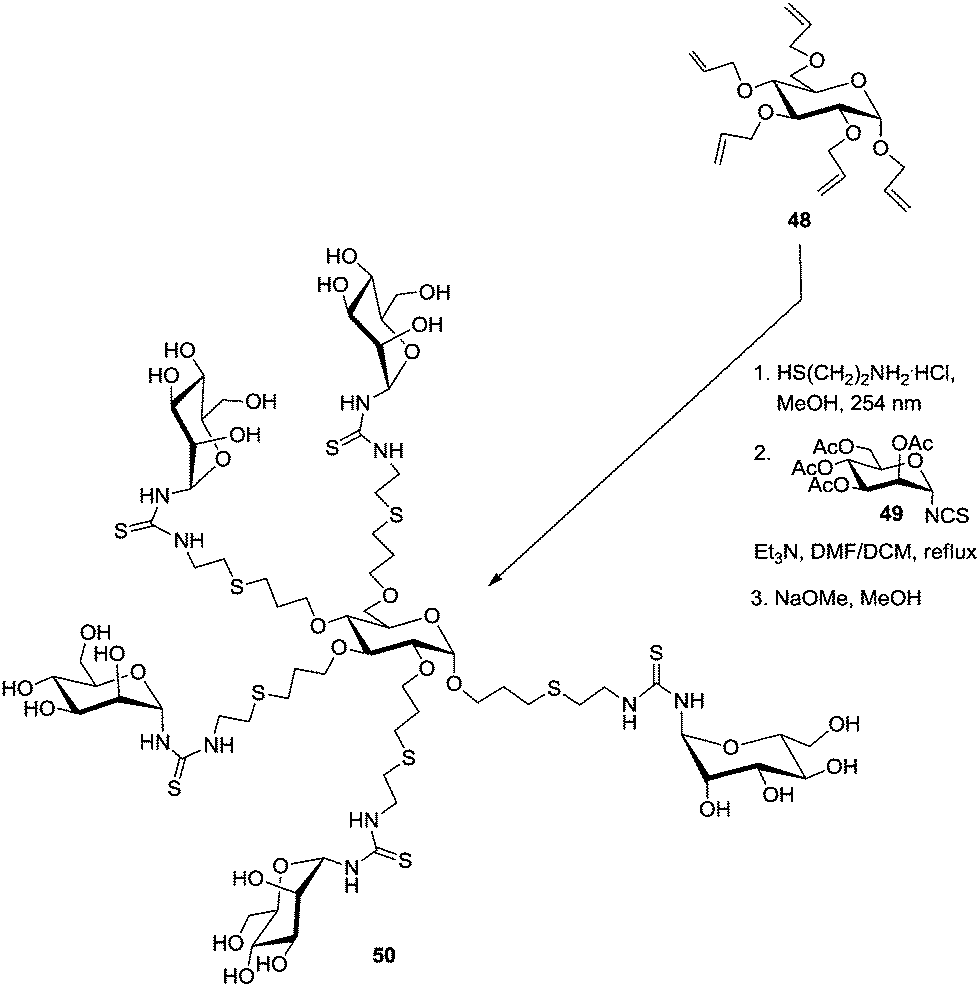 | ||
| Scheme 7 First “octopus glycoside” prepared by the Lindhorst group. | ||
The versatility of the allyl group was further advantageously exploited by the Lindhorst group in ozonolysis, reductive amination or hydroboration reactions to provide scaffolds bearing alcohol, aldehyde or amine linkers of different lengths (Scheme 8).43 Additionally, the allyl moiety could be transformed into an epoxide,44 by oxidation with m-CPBA, and also into carbosilane derivatives by hydrosilylation followed by the double addition of allyl Grignard to the dichlorosilane intermediate (Scheme 8).45 Among the aforementioned approaches, hydroboration is probably the most interesting entry for further functionalization of the scaffold. Indeed, it is an efficient reaction and it could even be successfully applied for the functionalization of per-allylated di- and trisaccharides.42,46 The resulting polyols could be directly glycosylated42,46a or alternatively, converted into polyamines to allow further ligation chemistry (Scheme 8).46b,47
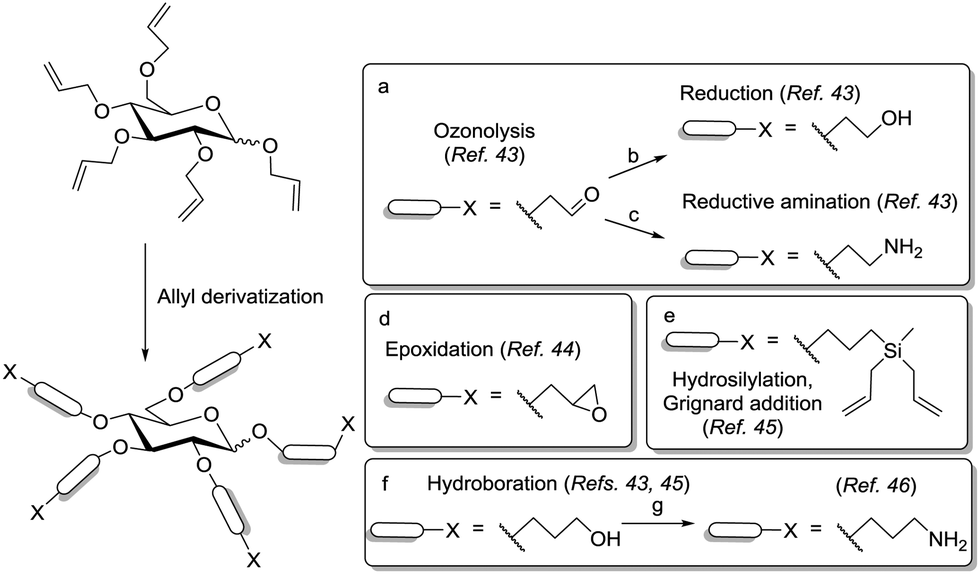 | ||
| Scheme 8 Versatile derivatization of the allyl group. Reagents and conditions: (a) O3, NaHCO3, DCM/MeOH, −78 °C, then PPh3, 60%; (b) O3, NaHCO3, DCM/MeOH, −78 °C, then NaBH4, 85%; (c) (i) Bn2NH, NaHB(OAc)3, AcOH, THF, −10 °C; (ii) Pd/C, NH4HCO2, MeOH, reflux, 66% over two steps; (d) m-CPBA, CHCl3, 60 °C, 70%; (e) (i) HSiMeCl2, H2PtCl6·6 H2O/i-PrOH, THF; (ii) allylMgBr, Et2O, 52% over two steps; (f) 9-BBN, THF, reflux, 97%; (g) Pathway 1: (i) CBr4, PPh3, THF; (ii) KPhthN, DMF; (iii) NH2NH2, THF, 36% overall; Pathway 2: (i) I2, PPh3, DMF, 80 °C, 68%; (ii) NaN3, DMF, 90 °C, 79%; (iii) 1,3-propanedithiol, Et3N, MeOH, 41%, 22% overall. | ||
Other streamlined pathways to install connective linkers at a carbohydrate scaffold were reported by Toth's group,48 by Jensen and coworkers,49 and by the Lindhorst group50 (Scheme 9). In the first case, the Michael addition of the hydroxy groups of galactosyl-β-azide on acrylonitrile yielded the corresponding cyanoethyl derivative 51 which was reduced and protected in situ to give the aminopropylated AB4-type scaffold 52. In a different approach, methyl galactoside was functionalized with aminooxyacetyl (Aoa) groups by carbodiimide-mediated reaction with N,N-di-Boc-protected aminooxyacetic acid to yield 53, followed by deprotection to 54. Very recently it was shown that the mannoside 55 can be favourably derivatized into the AB4-type azide 56 and in the next step into the analogous isothiocyanate 57 (Scheme 9).
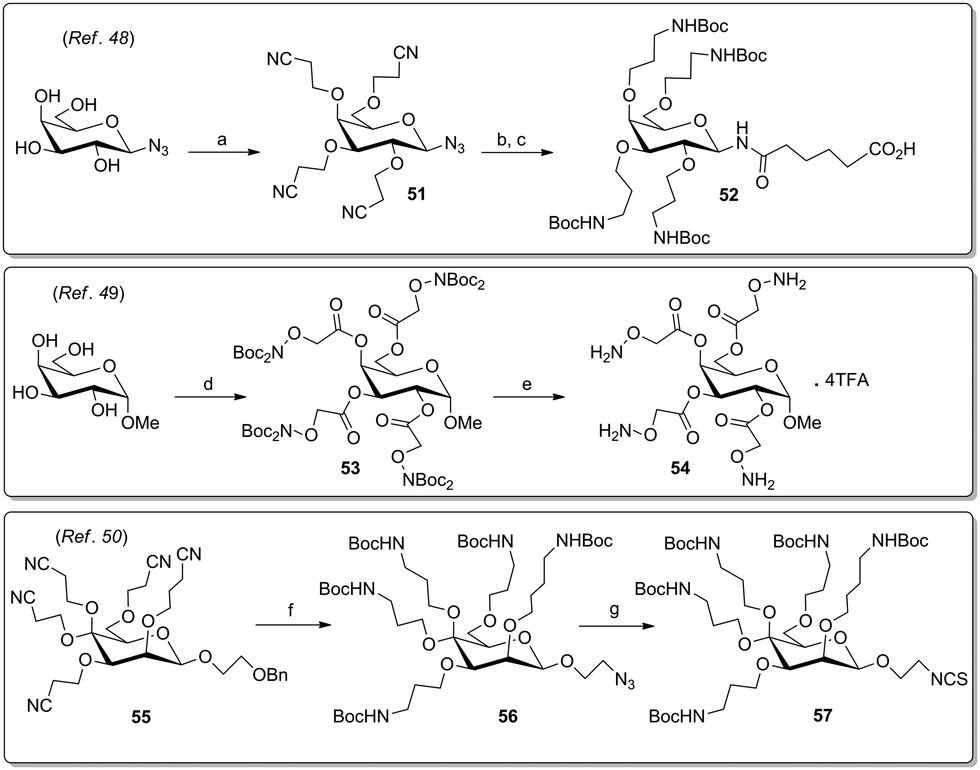 | ||
| Scheme 9 Orthogonal multivalent carbohydrate scaffolds. Reagents and conditions: (a) acrylonitrile, DBU, acetonitrile, rt, 64%; (b) (i) H2, Pd/C, THF, rt; (ii) adipic acid benzyl ester, HBTU, DIPEA, THF, rt, 55% over two steps; (c) (i) NaBH4, CoCl6·6H2O, Boc2O, methanol, 0 °C to rt; (ii) LiOH, methanol/water, rt, 46% over two steps; (d) Boc2-Aoa-OH, DIC, DMAP, pyridine/DCM, 84%; (e) TFA, DCM, quant.; (i) H2, Pd/C, RANEY®-Ni, methanol–NH3, 100 bar, 5 d, 50 °C, (ii) Boc2O, Et3N, methanol, rt, 16 h, (iii) TES, Pd–C, methanol, (iv) PPh3, DIAD, DPPA. THF; (f) CS2, PPh3, CHCl3, rt, 16 h. | ||
The development of the CuAAC36 also opened alternative ways to prepare multivalent structures (Fig. 7). In this context, alkyne-armed scaffolds 58 and 59 were prepared mainly by direct propargylation of glycosides under Williamson's conditions.51 Moreover, Vidal and Nifantiev reported the synthesis of cyclic oligoglucosamine derivatives (60) on which pentynyl tethers were installed via an amide linkage.40b Morvan and co-workers also attached phosphate-linked pentynyl moieties on various glycosides.52 Notably, they used phosphoramidite derivatives bearing one or two pentynyl linkers (61) in order to increase the density of ligands.
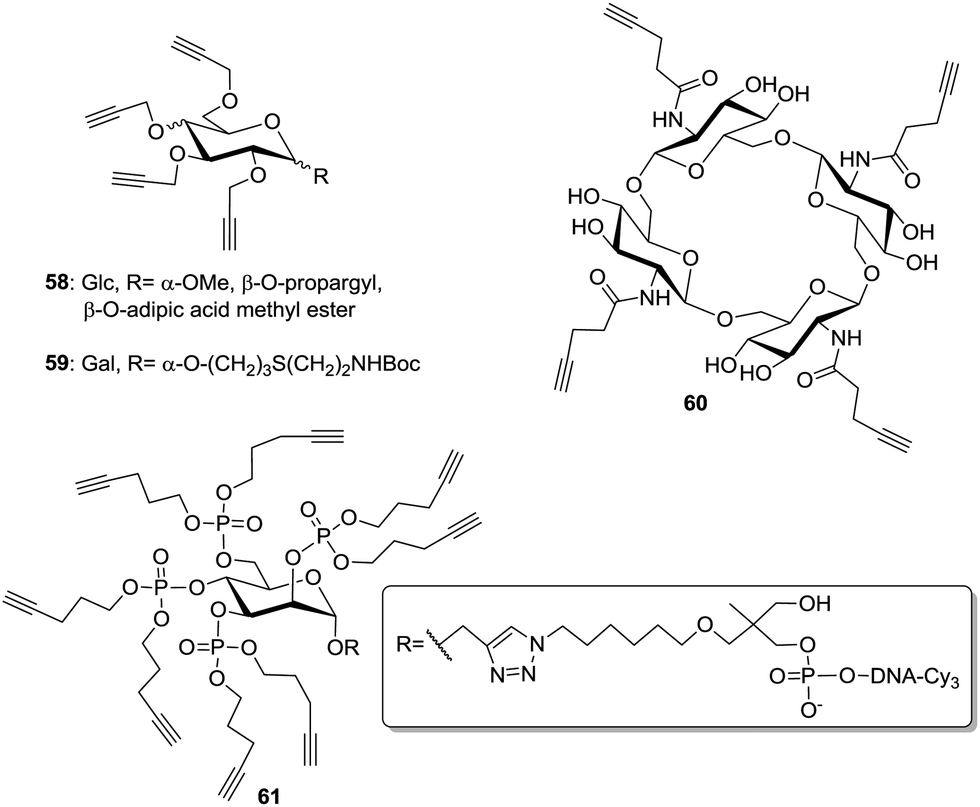 | ||
| Fig. 7 Various alkyne-functionalized multivalent carbohydrate scaffolds for CuAAC. | ||
As previously mentioned, one main advantage of carbohydrates is the facile modification of the anomeric position according to various ways. Thus a high degree of structural complexity is easily gained since such a functionalized scaffold glycoside can be used as a dendron and further conjugated. For instance, Lindhorst et al. prepared a tetravalent mannosyl cluster with a bromohexyl tether at the C-1 (Scheme 10, compound 62).46a
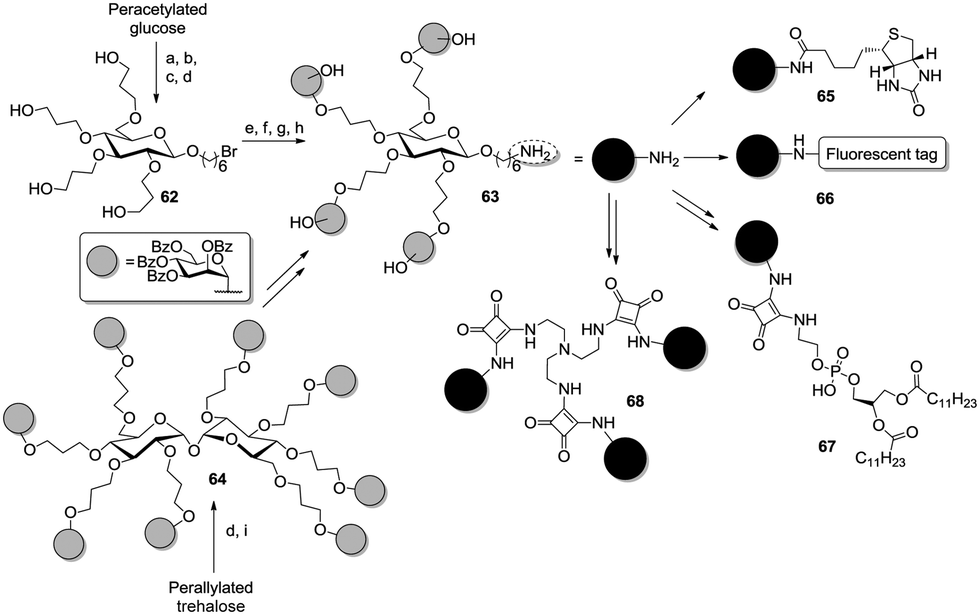 | ||
| Scheme 10 Conjugatable tetravalent homoglycoclusters. Reagents and conditions: (a) BF3·Et2O, 6-bromohexanol, DCM, rt, 36%; (b) NaOMe, MeOH, rt, quant.; (c) allyl bromide, NaOH, tetrabutylammonium bromide, rt, 42%; (d) 9-BBN, THF, rt, 54% for 62, 83% for 64; (e) TMSOTf, DCM, 95%, rt; (g) NaOMe, MeOH/THF, 84%, rt; (h) NaN3, DMF, 97%, 60 °C; (i) H2, Pd/C, 97%, rt; (i) TMSOTf, acetonitrile, 65 °C, then rt, 95%. | ||
Interestingly, the glucoside core was modified in two different ways. On the one hand, the authors applied hydroboration to an allylated 6-bromohexyl glucoside. On the other hand, a per-allylated trehalose derivative was first submitted to the hydroboration/oxidation sequence to obtain the respective octaol and then glycosylated with the mannose ligands (64). In a subsequent step, the interglycosidic linkage was cleaved in presence of 6-bromohexanol to yield the respective glucoside. The bromo substituent was next switched into an amino function, then the cluster 63 was conjugated with biotin (65), fluorophores (66), sepharose or lipidic chains (67).53 Additionally, the same amino-functionalized glycocluster was conjugated to squaric diester to enable a further coupling with tris(2-aminoethyl)amine, hence affording the dodecavalent glycocluster 68.
Roy and co-workers recently reported an efficient strategy based on orthogonal thiol couplings for the preparation of dendritic structures (Scheme 11).54 It involved the derivatization of per-allylated 2-azidoethyl β-D-glucoside (Scheme 11) to afford the N-chloroacetamide 69, which could be selectively linked to a thiol-functionalized core molecule via a nucleophilic substitution. Then, the allyl groups on the sugar ring of 70 could be reacted with thiol-armed ligands in thiol–ene reactions, thus affording galactosyl (71) or polyols (74) dendrimers.
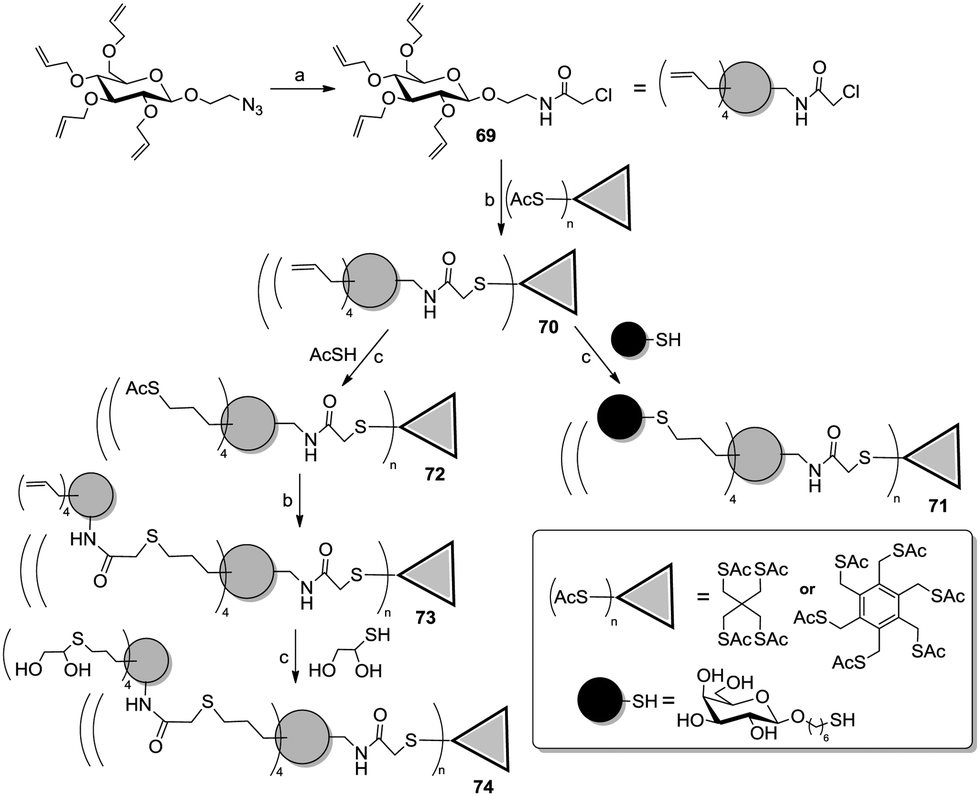 | ||
| Scheme 11 Orthogonal SN2/thiol–ene approach for the preparation of dendrimers involving carbohydrate scaffolds. Reagents and conditions: (a) (i) PPh3, THF/water, rt; (ii) ClCH2COCl, DIPEA, DCM, rt; 64% over two steps; (b) MeONa, MeOH, 69, rt, 73 to 90%; (c) α,α-dimethoxy-α-phenylacetophenone, 365 nm, methanol, rt, 52 to 72%. | ||
The preparation of carbohydrate scaffolds bearing several distinct ligands was also extensively explored by the Lindhorst group using a modular approach (Scheme 12).55 It was based on the selective differentiation of the anomeric position and the primary 6-hydroxy group of several glycoside cores to afford AB2- or ABC-type scaffolds. In a first report, a glucoside was first glycosylated either with bromoethanol (75) for further transformation into an amino-functionalized linker, or with hydroxymethylglutarate (79) to provide two carboxyl-armed tethers. Then the primary alcohol was regioselectively converted into a tosyl group, which was subsequently displaced by azide (76 and 80). This could be reduced into a free amine, then reacted with methyl acrylate to attach propanoate linkers (77). Finally, several divalent clusters were obtained after saponification of the methyl esters and peptide coupling with unprotected aminoethyl mannoside to afford 81 and 82. Following the same strategy, the methyl ester-armed cluster glycoside 84 could be prepared from 77 in order to increase valency.
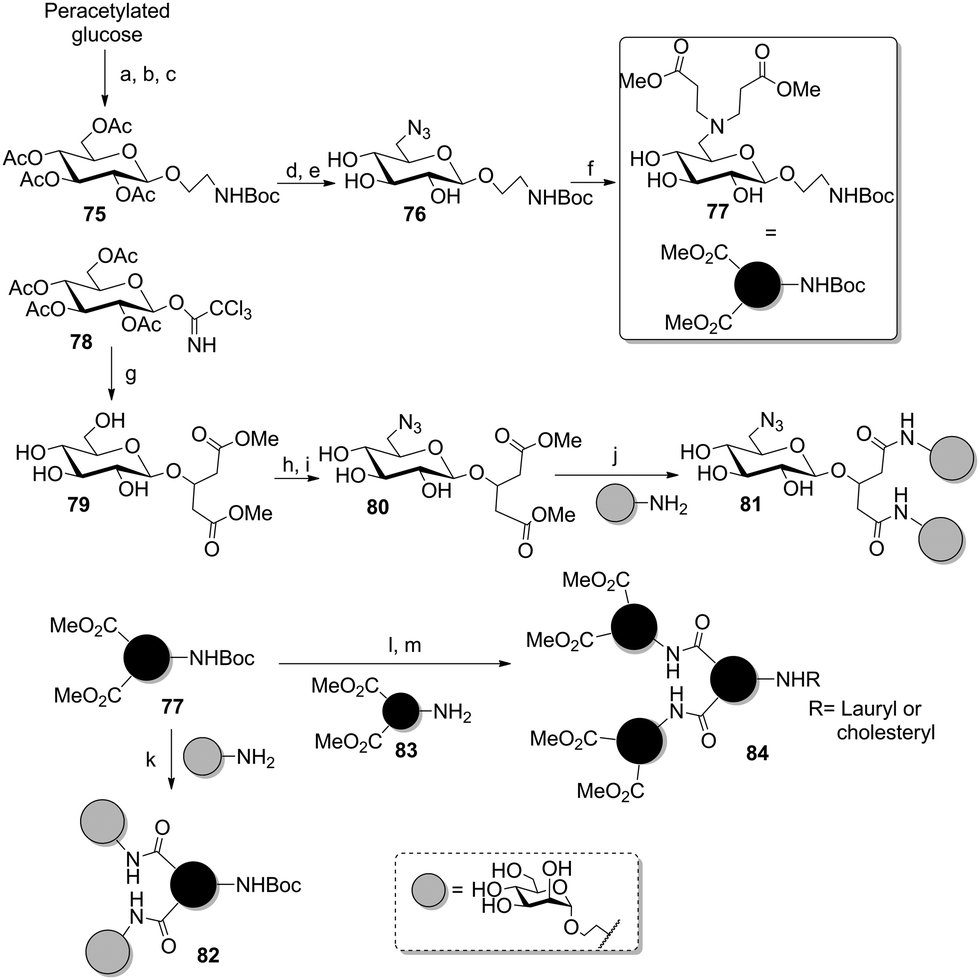 | ||
Scheme 12 AB2-type building blocks and peptide coupling for the synthesis of various glycoclusters. Reagents and conditions: (a) 2-bromoethanol, BF3·Et2O, DCM, 60%; (b) NaN3, n-Bu4NI, DMF, 87%; (c) Pd/C, H2, Boc2O, ethyl acetate, 90%; (d) NaOMe, MeOH; then TsCl, pyridine, 70%; (e) NaN3, n-Bu4NI, DMF, 81%; (f) Pd/C, H2, MeOH; then methyl acrylate, MeOH, 90%; (g) 3-hydroxydimethyl glutarate, TMSOTf, DCM, then NaOMe/MeOH, 82%; (h) TsCl, pyridine, 68%; (i) NaN3, DMF, 65%; (j) LiOH, MeOH/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), then TBTU, DIPEA, 1-HOBt, DMF, 73%; (k) LiOH, MeOH/H2O (2:1); then EEDQ, DMAc, 43%; (l) LiOH, MeOH/H2O (1:1); then HATU, DIPEA, 79%; (m) (i) TFA/SMe2 (2:1), 0 °C, 84%; (ii) lauric or cholic acid, HATU, DIPEA, respectively 58% and 66%. :1), then TBTU, DIPEA, 1-HOBt, DMF, 73%; (k) LiOH, MeOH/H2O (2:1); then EEDQ, DMAc, 43%; (l) LiOH, MeOH/H2O (1:1); then HATU, DIPEA, 79%; (m) (i) TFA/SMe2 (2:1), 0 °C, 84%; (ii) lauric or cholic acid, HATU, DIPEA, respectively 58% and 66%. | ||
Applying a similar approach, useful ABC-type building blocks were synthesized based on the initial selective mono-Michael addition of the free amine, derived from 85, to methyl acrylate to yield 86 (Scheme 13). Then, the aminopropanoate moiety in 86 was first condensed with a p-(α-D-mannosyloxy)methylbenzoic acid (87) under peptide coupling conditions to yield 88, and the carboxylic acid resulting after saponification reaction was reacted with the aminoethyl fucoside 89 to give the divalent glycocluster 90. After unmasking of the amino function at the anomeric spacer, a final coupling step involving a NCS-armed lactoside (91) furnished the final trivalent heteroglycocluster 92.
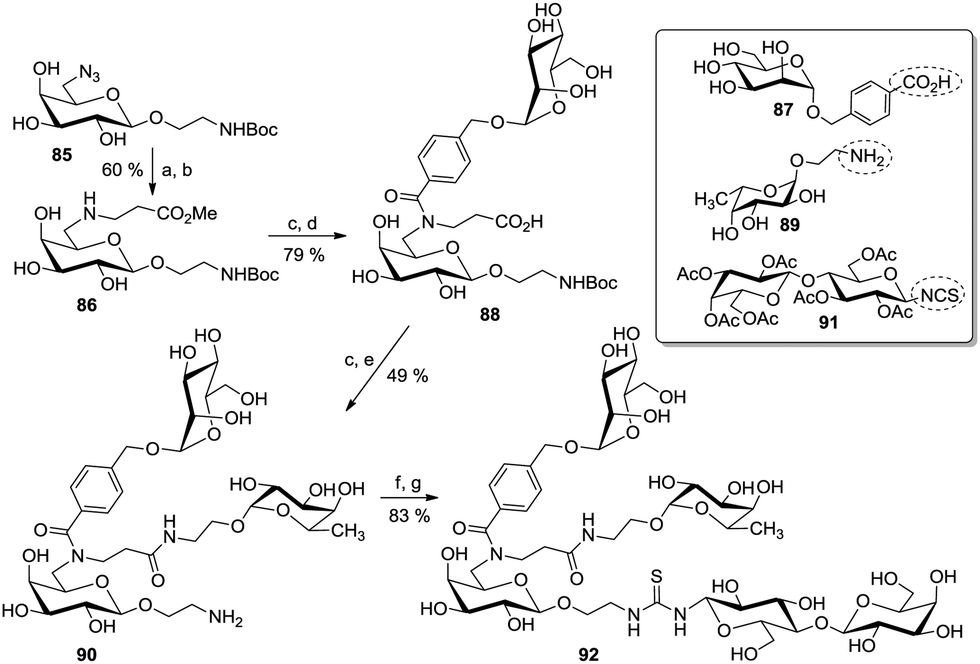 | ||
| Scheme 13 ABC-galactoside core for the preparation of heteroglycoclusters. Reagents and conditions: (a) Pd/C, H2, MeOH, rt; (b) methyl acrylate, MeOH, rt to 40 °C; (c) 87, respectively 89, HATU, DIPEA, DMF, rt to 45 °C; (d) LiOH·H2O, MeOH/H2O (2:1), 0 °C; (e) TFA/SMe2 (2:1), 0 °C; (f) 91, DMF, DIPEA, 50 °C; (g) NH3, MeOH, 0 °C. | ||
An elegant entry to AB3-type scaffolds was based on the azido glucuronic acid derivative 93 which was coupled with a Newkome-type branching unit (94) to yield 95 (Scheme 14).55f Anomeric Staudinger ligation with an Fmoc-protected amino acid to yield 96, followed by peptide coupling with the aminoethyl mannoside 97 allowed the synthesis of the glycocluster 98.
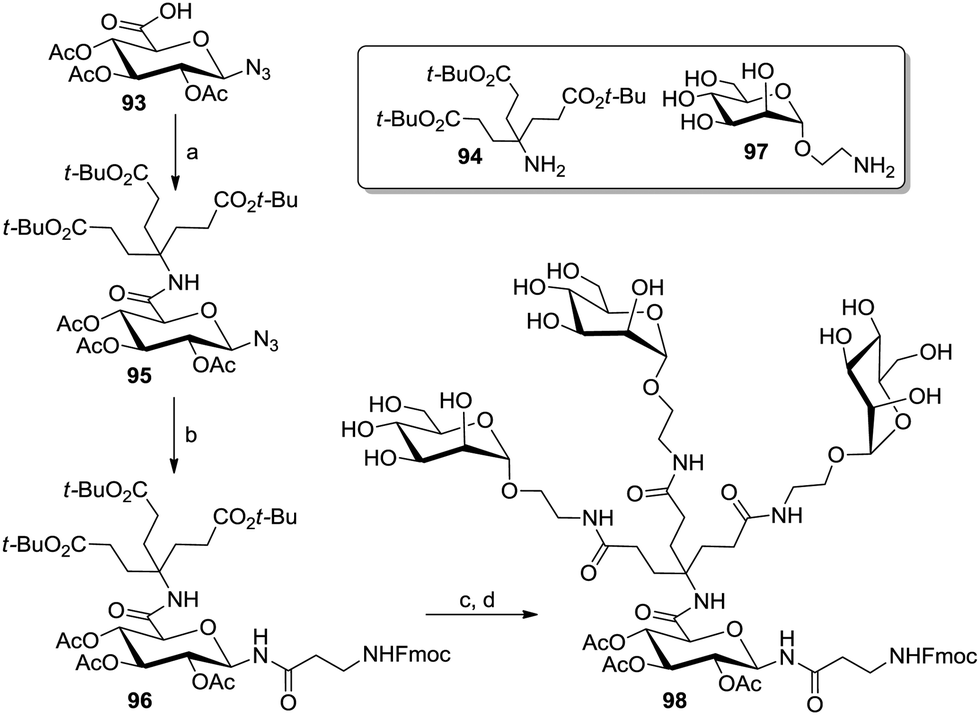 | ||
| Scheme 14 Preparation of an AB3-type glycocluster from glucuronic acid. Reagents and conditions: (a) 94, DIC, HOBt, DCM, 0 °C to rt, 79%; (b) Fmoc-β-Ala-OH, DIC, HOBt, PMe3, THF, 0 °C to rt, 68%; (c) TFA, dichloroethane, rt, 79%; (d) 97, HATU, DIPEA, DMF, rt, 50%. | ||
In the context of fluorescent switches, Xie et al. described the preparation of an AB3-type methyl α-glucoside bearing two different fluorescent groups (Scheme 15).56 This was achieved by protecting the primary 6-hydroxy group with a trityl function and following propargylation of the remaining hydroxy groups to yield 99 (Scheme 15). A first microwave-assisted CuAAC reaction with 100 then targeted positions 2, 3, and 4 to give glucoside 101. Following installation of an azido function at the 6-position (102) and a second CuAAC coupling with the molecular switch 103 yielded the targeted difunctional photoswitch 104.
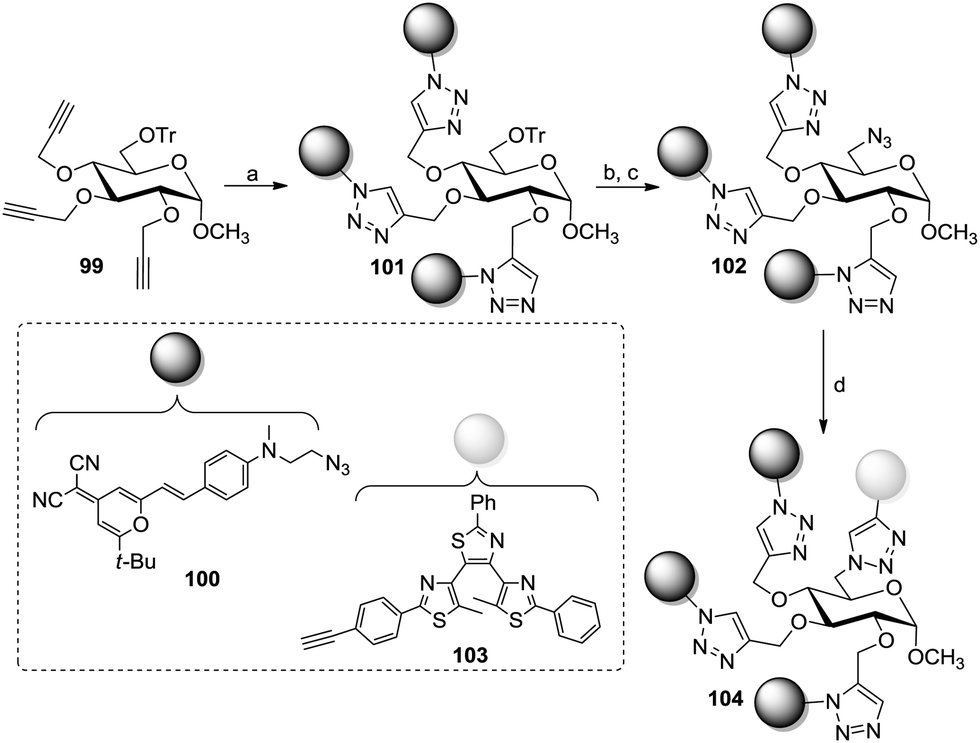 | ||
| Scheme 15 Fluorescent photoswitch on an AB3-type glucoside core. Reagents and conditions: (a) 100, CuSO4, sodium ascorbate, DMF, MW 70 °C, 5 min, then 130 °C, 30–45 min, 65%; (b) AcCl, MeOH/DCM, 74%; (c) (i) MsCl, Et3N, DCM, 0 °C to rt; (ii) NaN3, DMF, MW 110 °C, 15 min, 75% over two steps; (d) 103, CuSO4, sodium ascorbate, DMF, 130 °C, 30–45 min, 66%. | ||
The relative stereochemistry of two vicinal hydroxy groups of a glycoside can be utilized for discrimination of these two positions after formation of a cyclic acetal such as benzylidene or isopropylidene in the adjacent ring positions. This was applied by Santoyo-Gonzales and co-workers to glucoside, galactoside, and trehaloside cores (Scheme 16).51b After acetalization, propargyl groups were installed at the remaining free alcohol functions, then the acetal protecting group was cleaved and chloro-substituted diethylene glycol spacers were installed to afford A2B2-type scaffold glycosides such as 106 and 108. Then, a preliminary click reaction with an acetylated azidoethyl manoside (109) gave clusters of type 110 and after nucleophilic substitution of the chloro substituents with azide, a second CuAAC coupling involving an alkyne glycoside (111) was carried out. An ultimate deprotection step under Zemplén conditions gave the final heteroglycoclusters of the type 112.
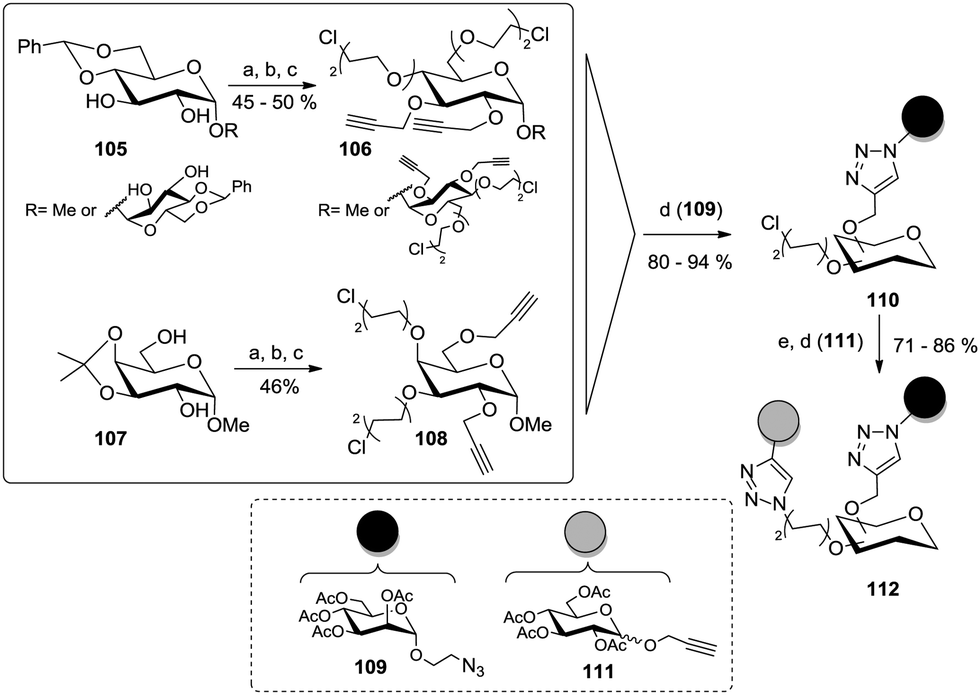 | ||
| Scheme 16 The use of acetal protecting groups for the synthesis of A2B2-type carbohydrate scaffolds. Reagents and conditions: (a) propargyl bromide, NaH, DMF, rt; (b) AcOH/H2O (7:3), 50 °C; (c) n-Bu4NHCO3, NaOH, bis(2-chloroethyl) ether, H2O, rt; (d) (EtO)3P·CuI, toluene or toluene/THF (1:1), reflux; (e) NaN3, n-Bu4NI, DMF, 75–80 °C. | ||
The Lindhorst group prepared mannoside scaffolds by differentiating positions 3 and 6 with allyl groups in a bis(tributyltin)oxide-mediated57 one-pot fashion (114, Scheme 17).58 After protecting the free hydroxy groups as benzoic esters, the resulting product was further converted into the branching glycosyl donor 115 with a view to build up dendritic structures such as 119.
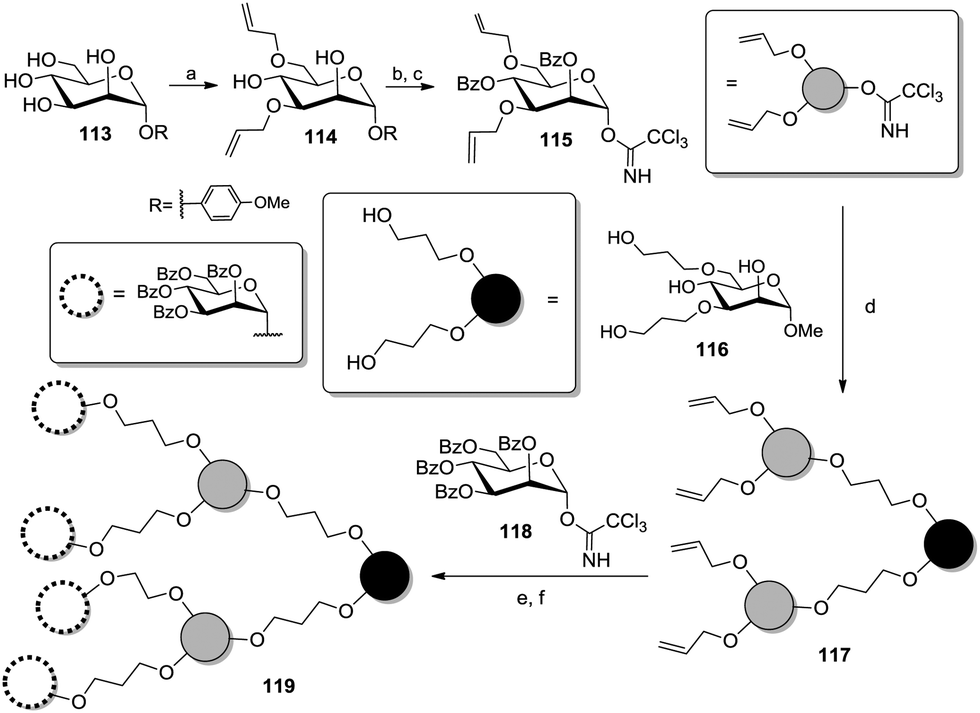 | ||
| Scheme 17 Regioselective di-allylation of a mannoside scaffold for the construction of a dendrimer. Reagents and conditions: (a) (i) bis(tributyltin)oxide, toluene, reflux; (ii) AllBr, TBABr, 80 °C, 51%; (b) BzCl, pyridine, 0 °C to rt, quant.; (c) (i) CAN, MeCN/H2O (4:1), 0 °C to rt; (ii) trichloroacetonitrile, DBU, DCM, 0 °C to rt, 53% over two steps; (d) TMSOTf, DCM, rt, 79%; (e) (i) 9-BBN, THF, reflux; (ii) 3M NaOAc (aq.), H2O2 (30%), 0 °C to rt, 40%; (f) TMSOTf, DCM, rt, 54%. | ||
Compain et al. designed an interesting approach based on the simultaneous protection of both the anomeric and the primary alcohol function via the formation of the 1,6-anhydro glucoside 120 (Scheme 18).59 After functionalizing the remaining hydroxy groups with trimethylsilyl-protected propargyl groups, the anhydro ring was stereoselectively opened in presence of trimethylsilyl azide and a catalytic amount of trimethylsilyl trifluoromethanesulfonate. The resulting α-configured glycosyl azide 121 was next conjugated via CuAAC with the benzylated glucoside ligand 122, bearing a propargyl group at position 6. The TMS-propargyl groups of the conjugate 123 were then unmasked applying fluoride anions and a second CuAAC coupling step with the protected α-glycosyl azide 124 afforded the heterodivalent glycocluster 125.
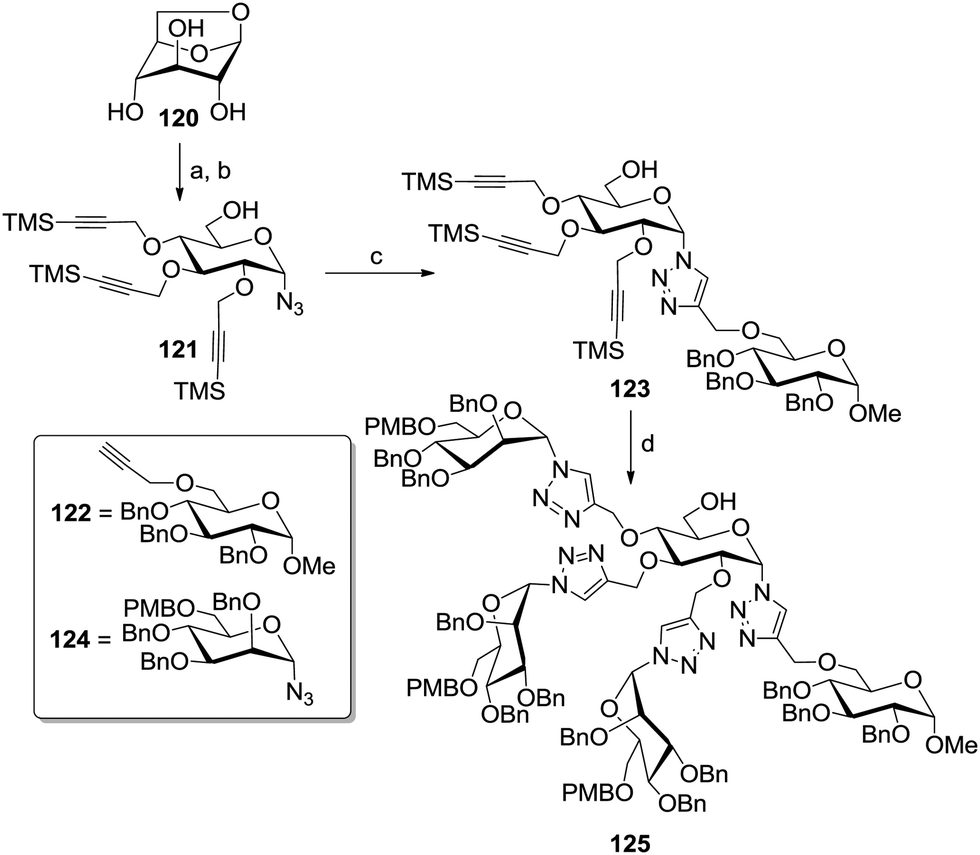 | ||
| Scheme 18 Differentiation of positions 1 and 6 via a 1,6-anhydro motif. Reagents and conditions: (a) propargyl bromide, NaH, DMF, rt, quant.; (b) TMSN3, TMSOTf, acetonitrile, rt, 82%; (c) 122, CuSO4·5H2O, sodium ascorbate, Na2CO3, THF/H2O (1:1), rt, 69%; (d) 124, TBAF, CuSO4·5H2O, sodium ascorbate, Na2CO3, DMF/H2O (4:1), MW 100 °C, 1 h, 63%. | ||
In this section various strategies were displayed which enable the synthesis of relatively complex heteromultivalent glycocluster architectures. However, biological testing results were not reviewed so far. In the following section, the biological properties of cluster glycosides will be reported and the influence of the spatial orientation of carbohydrate ligands as well as of both the length and the flexibility of the linker moieties will be discussed.
4. Highlighting secondary structural features
This section is dedicated to highlighting the importance of the three-dimensional presentation of sugar epitopes in multivalent glycoconjugates, the relevance of distances between carbohydrate ligands and their conformational availability. All of these aspects influence the biological properties of the prepared multivalent glycoconjugates. They are greatly related to the nature of the linkers, which conjugate the sugar ligands and also to the choice of the multivalent core molecule (scaffold) used for glycocluster synthesis. Naturally, the design of a specific spatial arrangement is demanding and requires information about the structure of the lectin which is targeted. When the structure of the lectin is known, computer-assisted matching of the architecture of a multivalent glycoconjugate can be used in order to complex several carbohydrate binding sites of a multivalent lectin.5f,60Famous target lectins with medical relevance are the bacterial toxins of the AB5-type, a research field that has been recently reviewed by Turnbull and colleagues.8f Kitov and Bundle et al. had the idea to employ a carbohydrate core, as introduced by the Lindhorst group,41,43,45 to match the homopentameric lectin B5.61 Each B-subunit contains three binding sites for the globotrioside of the glycolipid GB3 (Galα1,4Galβ1,4Glcβ-Cer), however of unequal affinity for the carbohydrate ligand. Based on this, Kitov and Bundle reasoned that if a divalent globotrioside dendron is attached in five copies onto a glucose scaffold, a perfect match with the B5 subunit of the E. coli Shiga-like toxin could be achieved. The employed spacers were designed based on the crystal structure of the B5 subunit. The resulting decavalent glycoconjugate 126 (Fig. 8), which was called “Starfish” according to its shape, indeed resulted in an almost one million-fold enhancement of binding as compared to the monovalent ligand. However, this success was not the result of the lectin-ligand match, the authors were aiming for. Instead, a different complex was formed as revealed by X-ray crystallography. A hamburger-shaped 2:1 toxin–Starfish complex was received in which the two globotriose units per glycodendron occupy carbohydrate binding sites not of the same but different lectin units.61
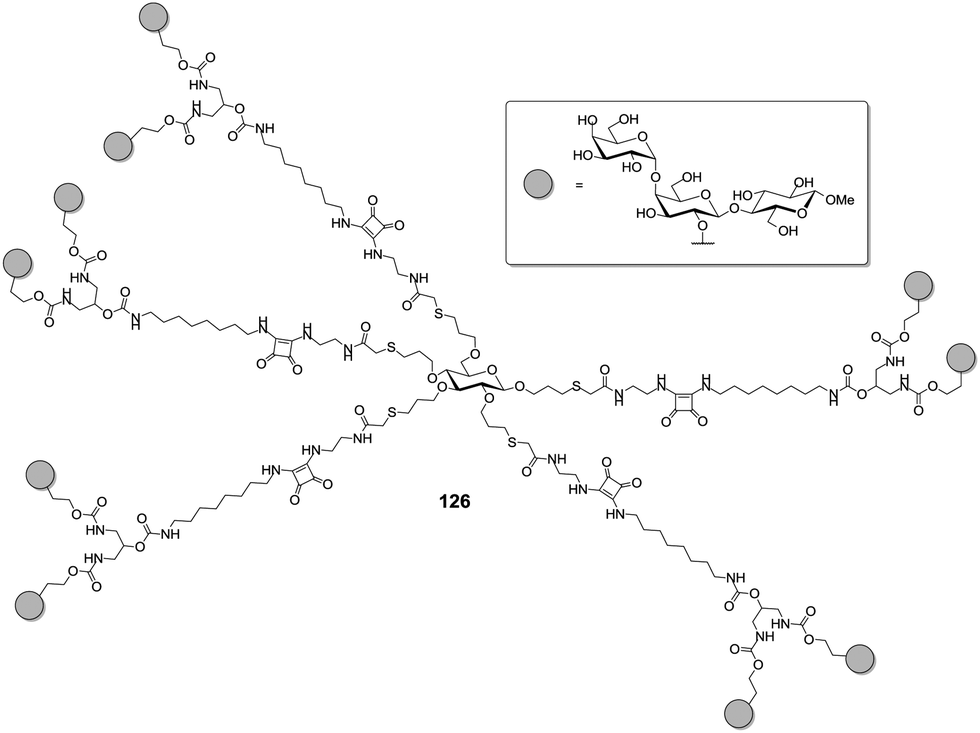 | ||
| Fig. 8 Structure of the “Starfish” ligand for the B5 subunit of Shiga-like toxin. Ten globotrioside ligands were assembled on a glycoside scaffold. | ||
This result shows that even when a lectin's structure is known, the design of a matching multivalent carbohydrate ligand does not always lead to the expected results. This multifaceted problem of ligand design has been discussed in detail in comprehensive publications by Wittmann and Pieters.19b,c In these contributions it is made clear, that the attempt to target multivalency by using large flexible linkers does not necessarily arrive at the desired chelate effects. Chelating binding, that is simultaneous binding of multiple sugar ligands to multiple binding sites, is desired as it leads to high avidities. Considering the length of the linkers used in the Starfish molecule 126, it is not that surprising the original idea of the chosen design did not exactly come true.
Alternatively, more rigid linkers can be employed successfully as shown for LecA (also called PA-IL) which is a soluble adhesin and important virulence factor of Pseudomonas aeruginosa. LecA is a tetramer and specific for galactosides. The closest proximity between two carbohydrate binding sites is 26 Å and these two were thus targeted with divalent ligands. To achieve chelating binding, Pieters and co-workers aimed at the design of rigid glycoclusters employing glucose-triazole repeating units to be able to adjust the distance between the two attached galactose ligands according to a molecular ruler (Fig. 9).62 The glucose moieties within the molecular ruler portion served to enhance its water solubility. ELISA testing with LecA showed that even subtle variations of the linker had an important influence on the affinity. Whereas in the divalent ligand 127 the binding potency was decreased in comparison to the monovalent reference compound, the slightly modified divalent ligands 128 and 129 were able to enhance the binding potency 545-fold (272-fold per sugar unit) and a 7555-fold (3780-fold per sugar unit), respectively. Both ligands, 128 and 129, are suited to complex LecA in a chelating binding mode. Ligand 127 on the other hand is too short to bridge two carbohydrate binding sites of the lectin.
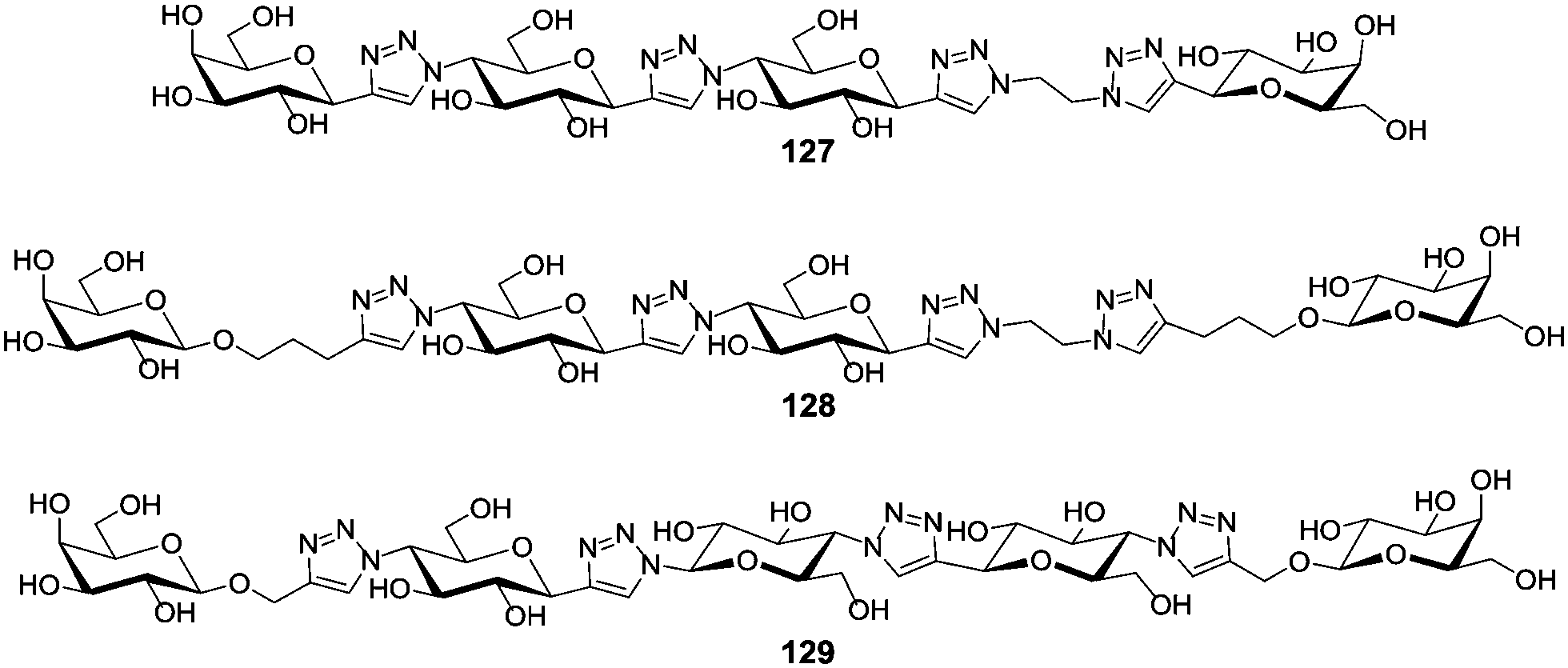 | ||
| Fig. 9 Molecular rulers built from glucose and triazole units were used to adjust the distance between two galactose ligands to achieve chelating binding in LecA. | ||
Another work on bacterial lectins of Pseudomonas aeruginosa and Burkholderia ambifaria bacterial lectins by Ligeour et al. showed the importance of topology for glycocluster binding.63 The P. aeruginosa lectins LecA (PAIL) and LecB (PAIIL) are both homotetrameric lectins with one binding site per monomer. LecA is galactose-specific, LecB is L-fucose-specific. To gain more insight into the effect of topology on the affinity of glycoclusters beside valency, two very different molecular scaffolds were used for clustering, namely an acyclic azido-mannitol and furanosidic nucleoside derivatives (Fig. 10). Both could be assembled into multimeric phosphodiester-linked clusters via their azido functional group, whereas the hydroxy groups, which are spatially distributed in different ways, were used to attach the sugar ligands through various linkers. This way, a large collection of differently shaped oligonucleotide-type glycoclusters were obtained and tested with all three lectins. The obtained data demonstrated that the topology and the nature of the ligands were the predominant factors for high avidity.
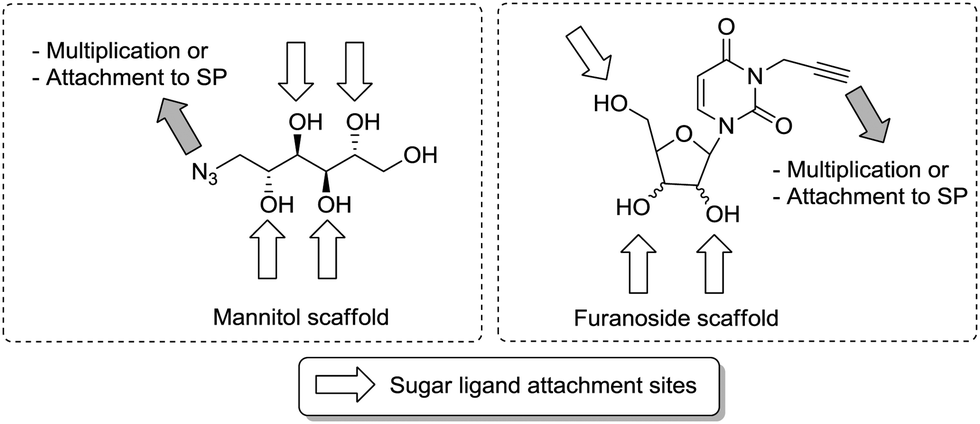 | ||
| Fig. 10 Glycoclusters with different topologies were obtained using a mannitol scaffold (left) or various nucleoside scaffolds (right) resulting in very different specific avidities in lectin binding assays. SP: solid phase. | ||
Another toxin of the AB5-type is cholera toxin, causing cholera.64 The B5 subunit of cholera toxin constitutes a nontoxic pentameric receptor for the GM1 glycolipid which is found on the cell surface of the host cells. Turnbull and colleagues recently reported on a modified protein scaffold which exactly matched the cholera toxin B5 subunit.65 Conjugation with five GM1 oligosaccharide units led to a pentavalent neoglycoprotein reaching an inhibitory potency in the picomolar range. On the other hand, Pieters et al. have previously synthesized a tetravalent inhibitor of the cholera toxin B5 with subnanomolar potency using a classical glycocluster approach.66 More recently, they synthesized a pentavalent version of this inhibitor and compared it with the tetravalent analogue.67 Surprisingly, the pentavalent geometry did not yield major benefits, rather it performed a little worse. This result shows that ligand receptor systems may adopt more than one matching geometry, further complicating ligand design.
The influence of linker length and scaffolding on inhibition of type 1 fimbriae-mediated adhesion of E. coli bacteria, which is α-D-mannopyranoside-specific, has been tested with different cluster mannosides, which were based on TRIS or the Newkome dendron (cf. Section 3), such as 130,26131,68 and 13268 (Fig. 11). Whereas the anomerically linked cluster mannosides 130 and 131 showed favourable inhibitory potencies, the situation in case of the cluster mannoside 132, which is linked via the 6-position, is different. First, a high inhibitory potency was reported,68 later it became clear, that this was a false positive result, which could not be repeated. Even nonavalent clusters, which were made based on 132 did not show any inhibitor effect.69 Owing to the crystal structure of FimH70 it becomes clear that clustering via the 6-position of a mannoside cannot lead to effective ligands. Thus, with FimH the situation is different as for the Starfish inhibitor of the Shiga-like toxin, where the globotriosyl carbohydrate ligands could be scaffolded via the 2′-position of the middle galactosyl residue (Fig. 8).
 | ||
| Fig. 11 Anomerically linked trivalent cluster mannosides (130, 131) are effective inhibitors of type 1 fimbriae-mediated bacterial adhesion, whereas when the mannoside moieties are linked to the scaffold via the 6-position (132), they lose their inhibitory potential towards FimH. | ||
The cluster mannoside 131, which was based on the Newkome-type scaffold, was used to explore the influence of linker properties on the affinity and inhibitory potential of the respective glycoclusters. The choice of linkers can affect the conformational preorganization of bound carbohydrate ligands as well as their conformational flexibility.71 Thus, mannosides 133–135 equipped with amino-functionalized aglycone portions of different length (Scheme 19) were ligated with the Newkome scaffold via an EEDQ (2-ethoxy-N-ethoxycarbonyl-1,2-dihydroquinoline)-mediated peptide coupling reaction to yield the cluster glycosides 136–138. When tested as inhibitors of type 1 fimbriae-mediated adhesion of E. coli bacteria by ELISA (enzyme-linked immunosorbent assay), the inhibitory potency of 131 was found to be 250 times higher than that of the reference inhibitor methyl α-D-mannoside; 136 was 18-fold better, 137 19-fold, and 138 32-fold more potent than MeMan. Thus, the cluster mannoside 131 prepared first (Fig. 11), showed the best inhibitory potency and among the amide-linked clusters (136–137), the most flexible one, 138, was the leading one.
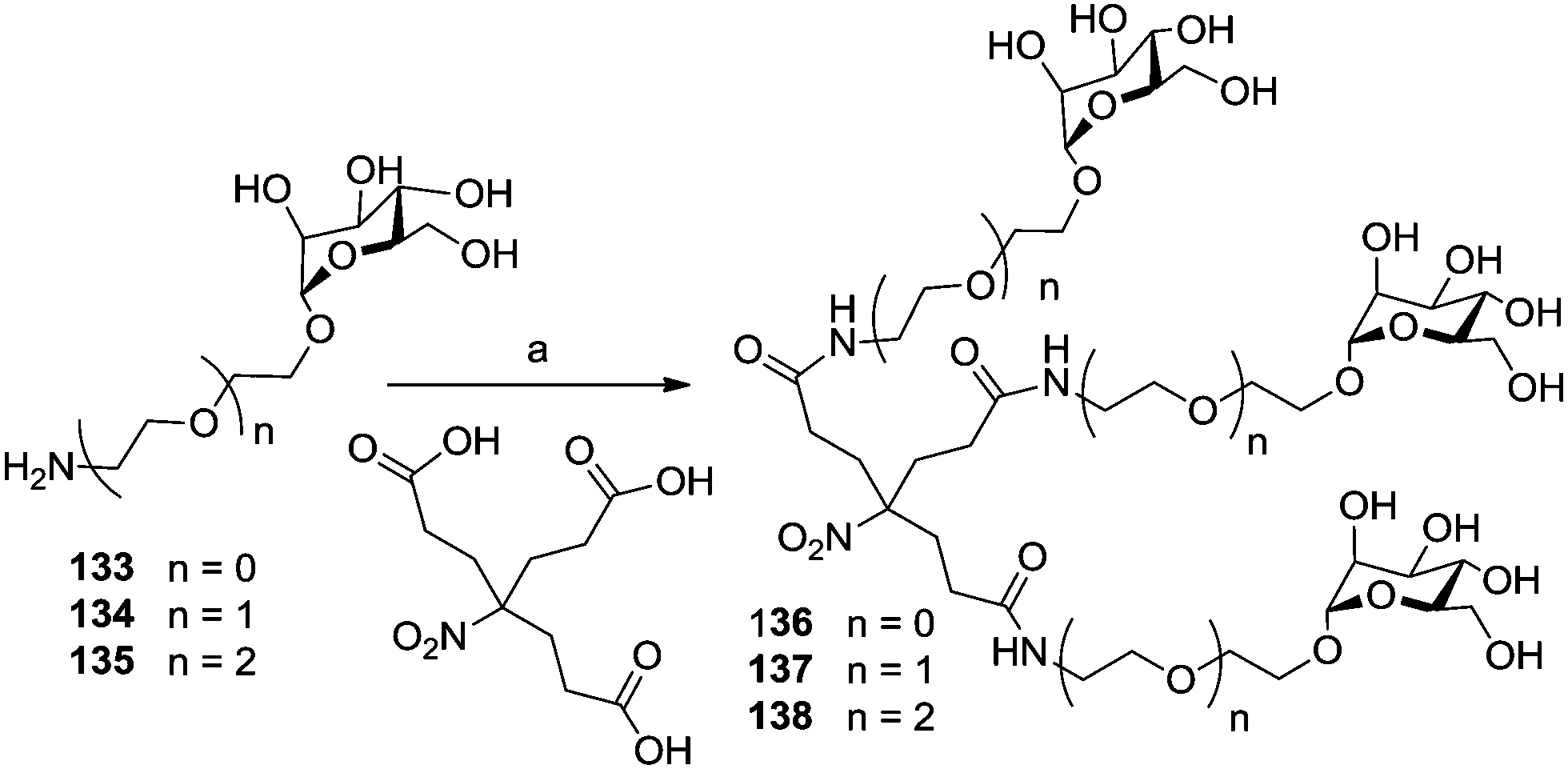 | ||
| Scheme 19 Reagents and conditions: (a) 3.3 eq. amine (133, 134, or 135), EEDQ, DMA, 60 °C, 4 d, 74% for 136, 69% for 137, 59% for 138. | ||
As often, when the structure of cluster glycosides is varied arbitrarily, these testing results are difficult to interpret. The bacterial adhesin FimH is a monovalent lectin and in spite of this, cluster effects have been measured with many multivalent mannosyl conjugates.19a Apparently, the nature of the linkers can play a critical role in FimH binding, but this is not always the case. It has been discussed on the basis of detailed thermodynamic and kinetic studies of carbohydrate–protein interactions that multivalency effects could arise for a “bind and jump” mechanism72 in analogy to binding of protein ligands to DNA, in which bind and slide or bind and hop mechanisms have been discussed.
An addition, it has been shown that intramolecular interactions of linkers or of aglycon portions, respectively, can restrict the conformational availability of the attached carbohydrate ligands and thus compromise lectin binding. For a series of cluster mannosides and mannose-terminated glycodendrimers this has been recently substantiated by molecular dynamics simulations showing the effect of intramolecular ππ stacking.73
The influence of linker hydrophilicity could be elegantly addressed by oxidation of thioether-linked carbohydrate dendrimers, which were prepared in analogy to the dendritic growth concept discussed earlier (cf.Scheme 17). Carbohydrate-scaffolded cluster mannoside 139 could be treated with magnesium monoperoxyphthalate (MMPP) to oxidize the sulfides to corresponding sulfones and result in glycocluster 140 (Scheme 20).58 For both clusters the inhibitory potencies were tested by ELISA and the IC50 values determined. Each of them provides seven mannosyl moieties. They just differ in their linkers, sulfides versus sulfones, and this had a remarkable effect on their inhibitory power. While 139 showed an IC50 value of 0.055 mmol and a relative inhibition potency (RIP) of 110 based on methyl α-D-mannopyranoside, for compound 140 an increased IC50 value of 0.23 mmol and a corresponding RIP of 25 was observed. Thus, after oxidation of the linkers, the inhibitory power of the respective glycocluster is diminished by approx. fourfold.
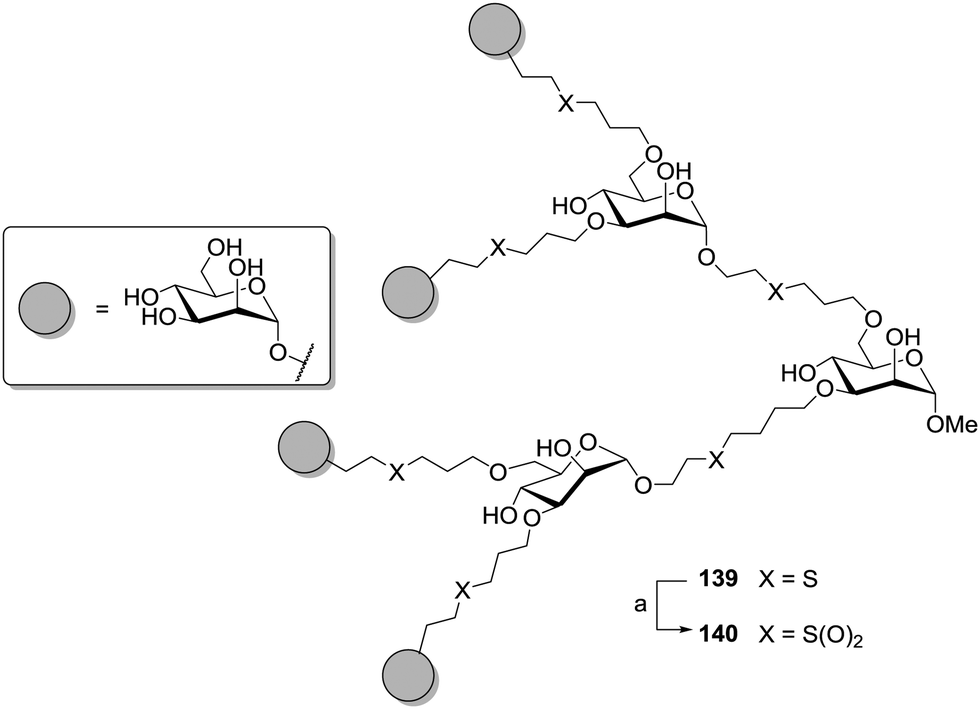 | ||
| Scheme 20 Disulfide-linked cluster mannosides could be oxidized to the respective sulfones to vary the hydrophilicity of the linker moieties. (a) MMPP, H2O/MeOH, overnight, rt, 67%. | ||
The influence of ligand spacing and the rigidity of the scaffold was evaluated by Gabius and coworkers using a series of more or less flexible glycoclusters (Fig. 12).74 Divalent cluster mannosides were prepared based on glycocyclophane and terephthalamide and tested towards their inhibitory potency in a medically relevant system. Two different lines of human cancer cells were evaluated to show that macrocyclic derivatives as well as terephthalamide derivatives served as effective ligand-presenting scaffold molecules. The intra-mannosyl distance and the respective cluster type showed to be influential for lectin binding. The decrease of inherent flexibility had a favourable effect, in particular on cells with high ligand density.
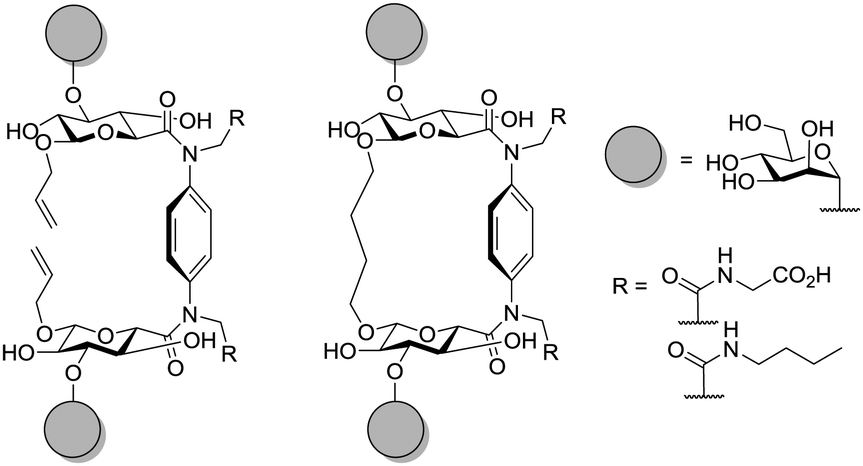 | ||
| Fig. 12 Schematic depiction of cyclophane scaffolds with differing rigidity. Divalent cluster mannosides with more (left) and less flexibility (right) are shown. | ||
The importance of the spatial arrangement of sugar epitopes for lectin binding is also evident from research reported by Moni et al.,75 where linear, antenna- and calixarene-based trivalent glycoclusters, all ligated to galactose residues, were compared (Fig. 13). Biological testing was performed with lectins from P. aeruginosa and Ricinus communis revealing that the spatial arrangement of sugars can be more important than sugar valency of a glycocluster. The chosen molecular architecture was also suited for ligation with oligonucleotides and subsequent hybridization on DNA chips.
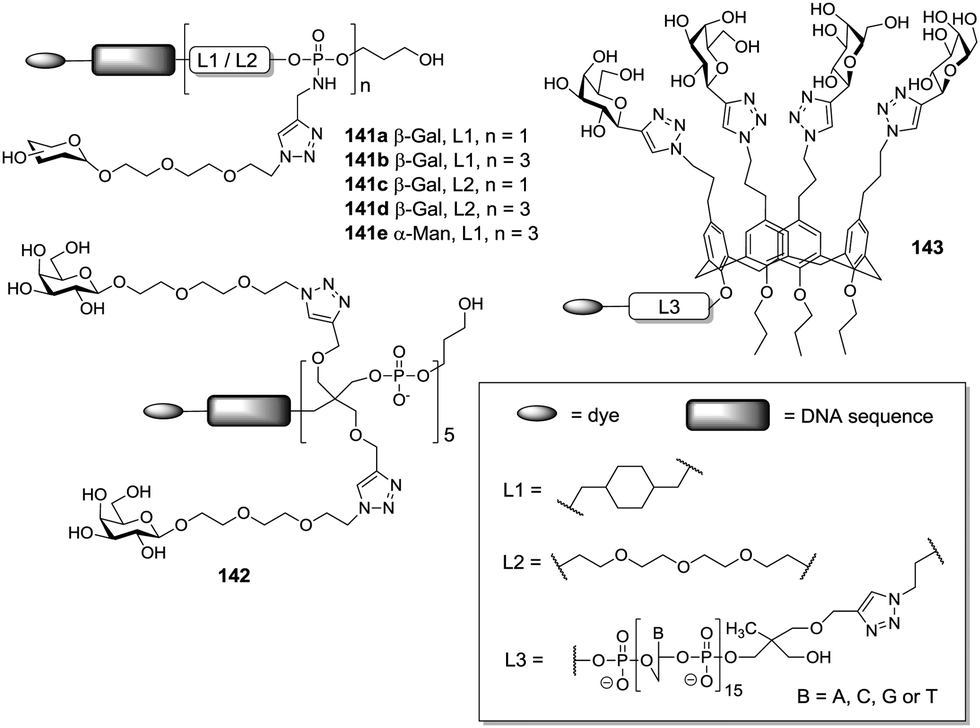 | ||
| Fig. 13 Different scaffolding of lectin ligands (according to 141, 142, or 143) revealed the importance of spatial arrangement for lectin binding. | ||
Immobilization through DNA hybridization was also used by Gerland et al. to test the influence of different three-dimensional architectures on lectin binding.52b Again the L-fucose-specific lectin LecB from P. aeruginosa was used for testing and thus fucosylated glycoclusters of different shape were synthesized (Fig. 14). Linear, antenna-like and crown-like shaped glycoclusters were immobilized through DNA hybridization to gain carbohydrate microarrays. For the tests a library of 16 glycomimetics were synthesized where the number of ligands, the three-dimensional arrangement, the stereochemistry of the carbohydrate core and the linker lengths were varied. This work revealed that the binding affinity was highest for the antenna-like cluster with four fucosyl residues attached and for the mannose-centered glycocluster having four fucose moieties attached through the longest of all tested linkers. Again it was shown that spatial distribution is crucial for lectin binding. However, in this case, linkers were too short to achieve chelating binding.
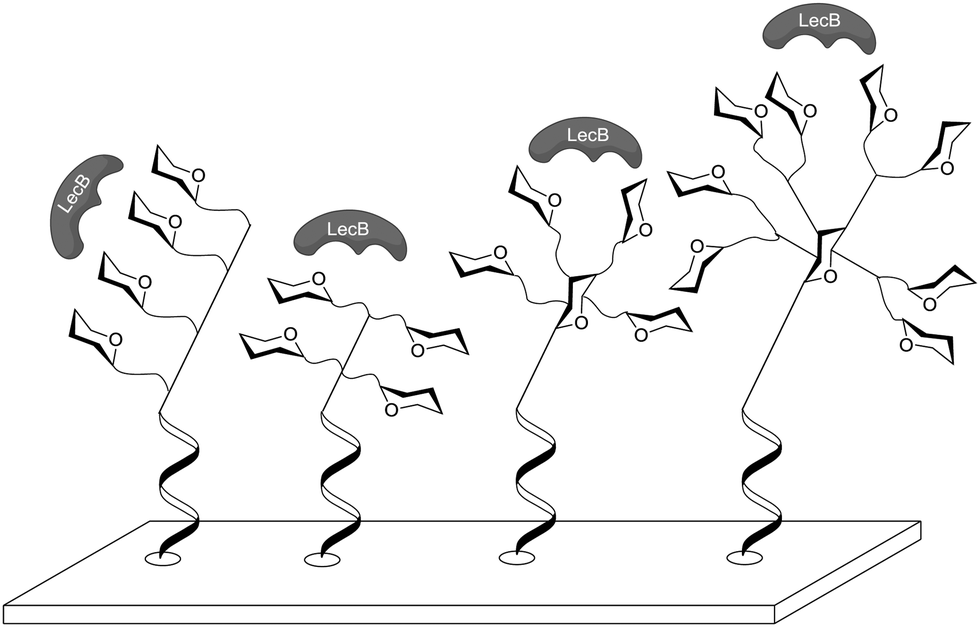 | ||
| Fig. 14 Fucosylated linear, antenna-like and crown-like glycoclusters as ligands of the LecB. | ||
Grünstein et al. in 2011 presented the synthesis of multivalent bacteria sensors, based on a fluorescent core template.76 This template is derived from [Ru(bipy)2]Cl2 with two, four and six adamantyl residues attached. The adamantyl moieties self-assemble with heptamannosylated β-cyclodextrin scaffolds via host–guest interactions (Fig. 15). The authors investigated the influence of mannose density on binding of ConA by surface plasmon resonance (SPR) spectroscopy. High ConA concentrations were found to stabilize binding with dimeric and tetrameric supramolecular complexes. The hexameric complex did not bind ConA at all. It was assumed that the pattern of presented mannosyl units in this case is not suited to reach the binding sites of the surface-immobilized lectin in a favourable manner. These results show again, how important spatial orientation of sugar ligands is for lectin binding and that it can overcompensate the effects of multivalency.
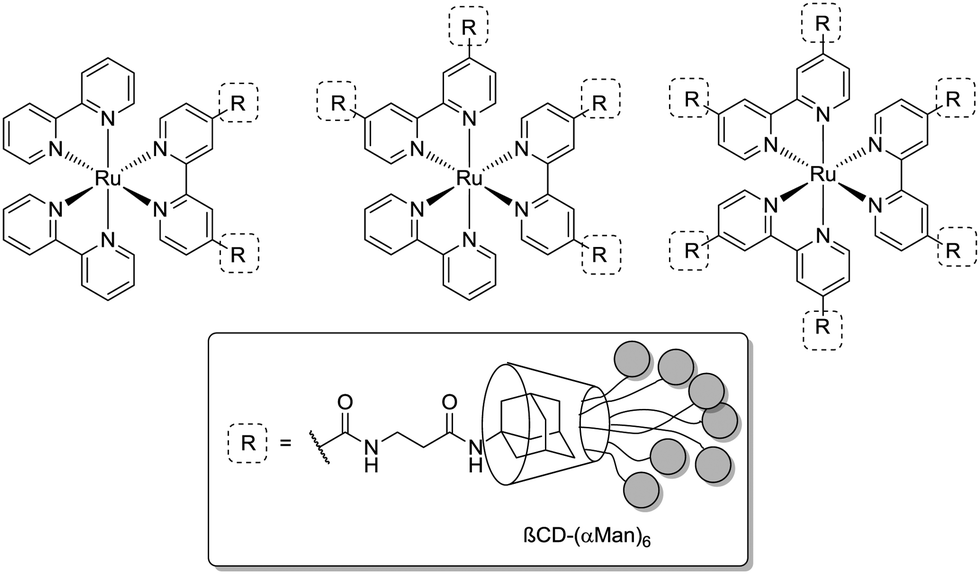 | ||
| Fig. 15 Fluorescent mannosylated dimeric, tetrameric and hexameric supramolecular complexes, prepared for binding assays with ConA and for sensing E. coli. | ||
In another setup, the hexameric mannosylated complex (Fig. 15) was incubated with the E. coli strain ORN178, which expresses the bacterial wild type lectin FimH. Binding was investigated by confocal laser microscopy which was facilitated by the observable red fluorescence of the [Ru(bipy)2]Cl2 core and the bacteria labeled in blue through staining with DAPI dye. A star-shaped distinctive surrounding of the Ru(II)-containing complexes by bacterial clusters was obtained, hence suggesting that the FimH-mediated binding of bacteria is defined and restricted by the spatial arrangement of mannoside ligands in the multivalent complex.
Thus, for the orientation of carbohydrate ligands, the nature of the scaffold on which a multivalent glycoconjugate is based, can be critical. Surprisingly, in a pentavalent glucoside-based cluster mannoside even the anomeric configuration of the sugar scaffold led to affinity differences for ConA. This was shown by Köhn et al. with glycoclusters 144 and 145 (Fig. 16).47 When tested in an ELLA with ConA the α-anomer showed a 6.4-fold increased affinity to ConA as compared to methyl α-D-mannopyranoside, and for the β-anomer-based glycocluster a 5.5-fold enhancement was measured. This small difference was highly reproducible in a series of experiments in which a series of structurally varied glycoclusters was compared. On the other hand, cluster mannoside 146 and 147, which are also based on a carbohydrate core, showed basically the same potency when tested as inhibitors of type 1 fimbriae-mediated bacterial adhesion, in spite of the fact, that the linker length was altered significantly.42
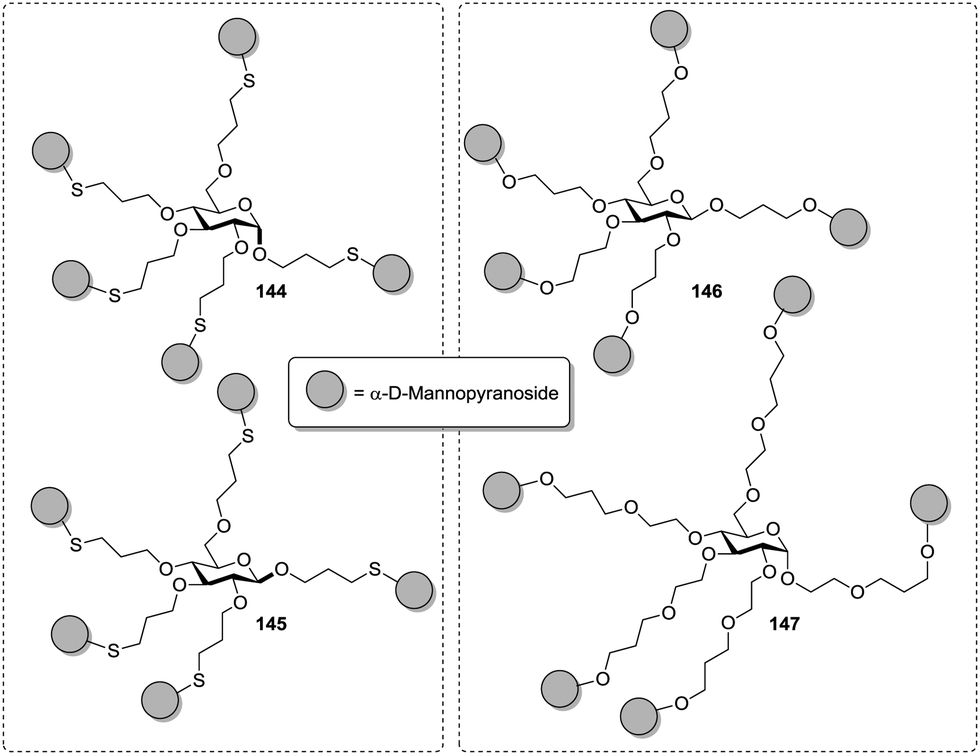 | ||
| Fig. 16 Carbohydrate-centered glycoclusters differing in the anomeric position (left) and in spacer length (right). | ||
It will be interesting to see if carbohydrate scaffolds will get further explored in the future to evaluate their potential to arrange carbohydrate epitopes in space. An obviously advantageous option for spatial arrangement of carbohydrate ligands is the use of DNA constructs, a field which has very recently been reviewed (cf. Section 3).77 Also peptides have been greatly explored as scaffolds for a defined presentation of multiple carbohydrate ligands. The field has been pioneered by the Wittmann group.34a Libraries of conformationally restricted glycoclusters were obtained by a split and mix synthesis of cyclic peptides to which identical copies of carbohydrate ligands were attached (Fig. 17). The authors stated that this synthetic approach is suited to present any carbohydrate ligand in varying valency and distance.
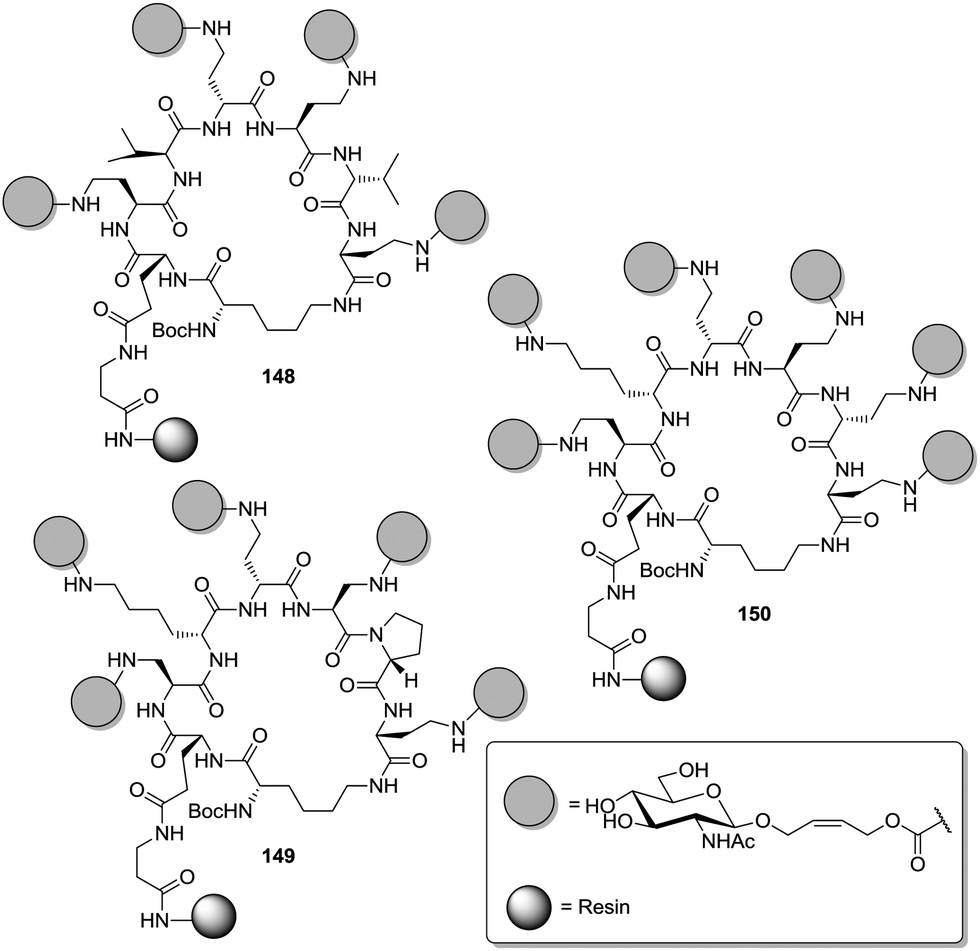 | ||
| Fig. 17 Glycoclusters by combinatorial chemistry of cyclic peptide scaffolds on solid phase; representative examples with different sugar valencies are depicted. | ||
A library consisting of almost 19500 glycoclusters was screened with WGA (wheat germ agglutinin) in an enzyme-linked lectin binding assay (ELLA).34b WGA is a dimeric plant lectin specific for GlcNAc and presenting a total of eight carbohydrate binding sites. This screening revealed that the spatial presentation of GlcNAc residues on the peptide scaffold was critical for WGA binding. For the GlcNAc clusters 148, 149, and 150 a 200- to 600-fold enhancement of the binding potency as compared to GlcNAc was detected.
Very recently, the Wittmann group has refined the work on ligand design for the lectin WGA.78 Di- and trivalent N,N′-diacetylchitobiose derivatives were synthesized according to a one-pot procedure for Cu(II)-catalyzed diazo transfer and Cu(I)-catalyzed azide–alkyne cycloaddition of the chitobiose derivative 151 (Fig. 18). The obtained glycoclusters were tested as inhibitors of WGA binding to GlcNAc-coated microtiter plates. IC50 values were in the low micromolar to high nanomolar range, depending on the linker between the two disaccharide units. Molecular modeling studies, in which the chitobiose moieties were fitted into the WGA carbohydrate binding sites as known from X-ray studies, correlate the measured binding enhancement of specific multivalent ligands with their ability to bind to the protein in a chelating mode. The best WGA ligand was the trivalent cluster 157 (cf.Fig. 18) with an IC50 value of 220 nM.
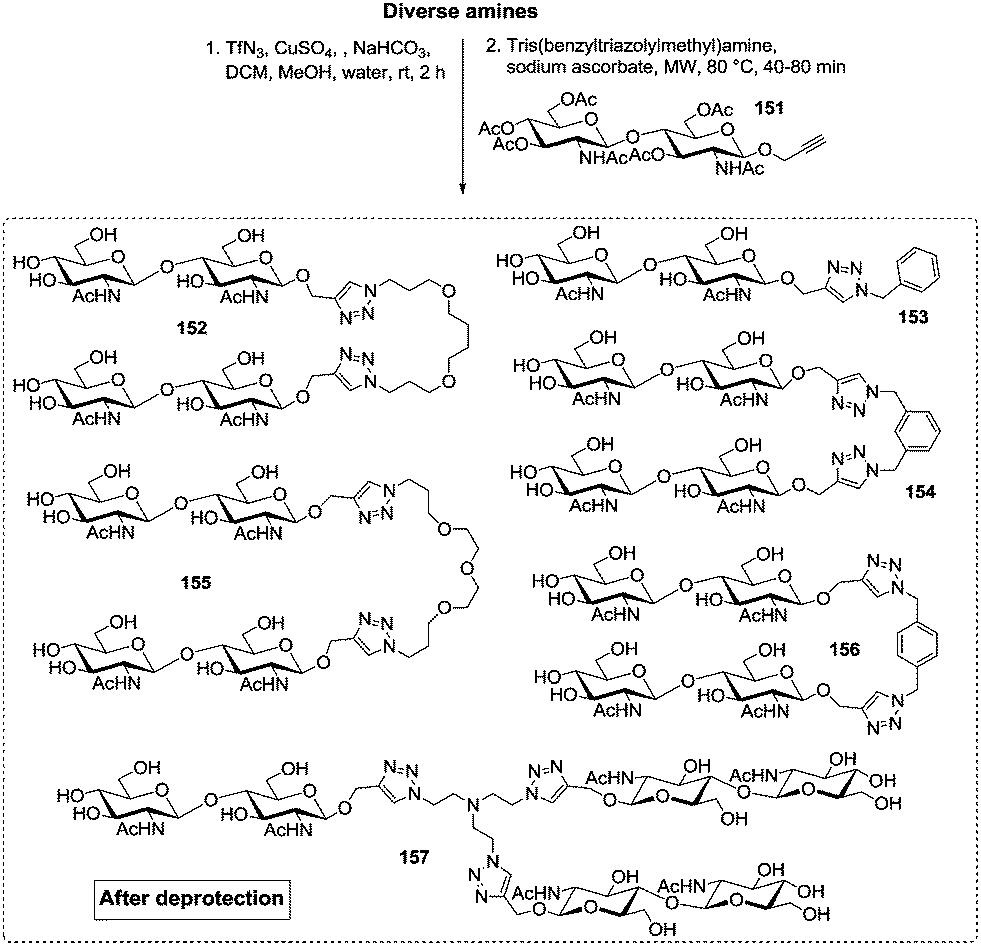 | ||
| Fig. 18 Ligands for the lectin WGA; the trivalent glycocluster 157 was most efficient. | ||
An obvious idea is to use linear peptides to assemble carbohydrate ligands in a defined way. A related work was performed by Schierholt et al. who synthesized di- and trivalent glycopeptides equipped with α-D-mannosyl residues.79 Six mannose-containing glycopeptides were made in which the number of presented carbohydrate ligands was varied, the nature of the aglycone and the spatial arrangement of ligated sugar moieties as well (Fig. 19). Variations of the peptide backbone were obtained via solid phase peptide synthesis (SPPS) followed by HATU-mediated peptide coupling of appropriately functionalized mannosides. Adhesion-inhibition assays were performed with mannose-specific E. coli. The results of this testing clearly showed that variation of the three-dimensional orientation of sugar ligands matters for the inhibitory potency in addition to valency and nature of the aglycone moiety.
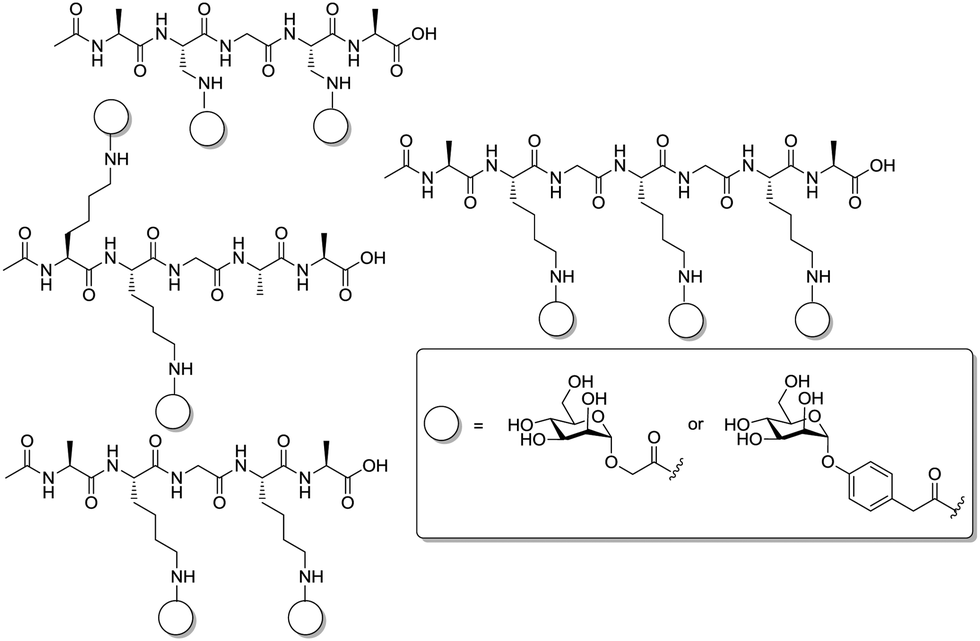 | ||
| Fig. 19 Multivalent glycopeptides with variations of valency, linker length inter-ligand distance and aglycon moiety. | ||
In a more recent approach introduced by the Hartmann group,80 not only the distance between several carbohydrate ligands was chosen and varied. In addition, carbohydrate ligands were installed at defined positions of a multivalent peptide backbone, that is tailor-made by SPPS. In this work, a protocol was employed allowing for the successive installation of different carbohydrate ligands in certain distances to obtain defined heteromultivalent glycooligomers. Three kinds of building blocks were employed, first an alkyne-functionalized building block at solid phase (Scheme 21), second, an azido-functionalized carbohydrate building blocks for CuAAC, and thirdly a building block (EDS) that serves as a spacer to adjust the distance between carbohydrate ligands.81
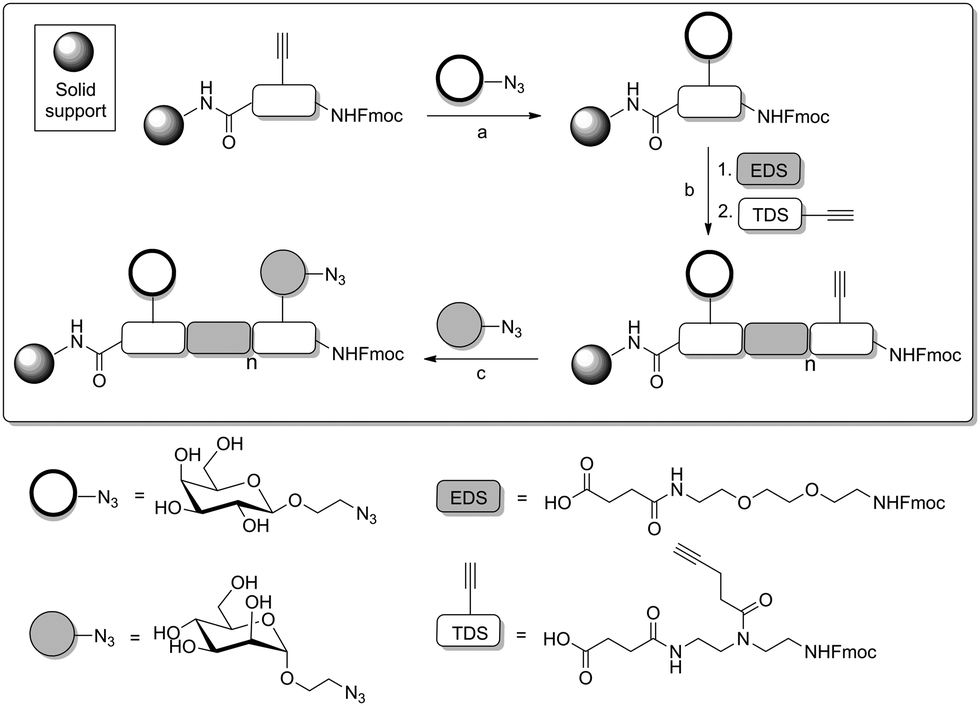 | ||
| Scheme 21 Sugar assembly on tailor-made peptides. (a) On-resin CuAAC reaction with the first alkyne group; (b) continued solid phase synthesis; (c) second on-resin CuAAC o couple a second sugar moiety. EDS = 2,2′-(ethylenedioxy)bis(ethylamine); TDS = 1-(fluorenyl)-3,11-dioxo-7-(pent-4-ynoyl)-2-oxa-4,7,10-triazatetradecan-14-oic acid. | ||
Using this methodology, easily monodisperse sequences of glycooligomers can be obtained with control over all structural parameters (number, spacing, position and type of sugar ligands). When homomultivalent as well as heteromultivalent glycopeptides were tested as ligands of ConA, the results confirmed that the composition of a glycopeptide is critical for its binding affinity. It was shown that a trivalent glycopeptide, carrying three Man residues at positions 1, 3 and 5 (Man-1,3,5) showed less affinity for ConA values than the analogous ManGalMan-1,3,5 oligomer. This is an important example of a heteromultivalency effect. Interestingly, saturation transfer difference nuclear magnetic resonance (STD-NMR) and dual-focus fluorescence correlation spectroscopy (2fFCS) measurements suggested a change in binding mechanism for trivalent glycooligomers presenting mannose or combinations of mannose and galactose residues.
For the investigation of the relationship between structural characteristics of homo- as well as heteroglycoclusters and anti-adhesion activity Li et al. synthesized a series of homomultivalent and heteromultivalent cluster glycosides which were tested in a static cell-based adhesion assay.82 It has previously been shown that multivalent lactosides can serve as valuable inhibitors for the adhesion of leukocytes to endothelial cells.83 For targeting the responsible integrin CD11b on the surface of leukocytes, Li and co-workers compared divalent, tetravalent, hexavalent, and octavalent glycoclusters comprised of different combinations of glucose, galactose, mannose, cellobiose and lactose residues. The results showed that neither high valencies nor the nature of the sugar ligand were determining factors for anti-adhesion activity. Homo- and heteroglycoclusters provided similar biological results. However, the linker moiety by which divalent building blocks were combined to higher valent glycoclusters had a particular influence on affinity. Here, a flexible L-glutamic acid linker was more beneficial compared to rigid aromatic linkers.
A remarkable effect on affinity in lectin binding could also be observed by employment of supramolecular systems for assembly of sugar ligands, as shown by Vincent and co-workers.84 They synthesized [2]rotaxane systems consisting of a pillar[5]arene scaffold having fucosyl or galatosyl residues attached, and a sugar-bearing axle (Scheme 22). The resulting heteroglycoclusters were tested against the two bacterial lectins LecA and LecB from Pseudomonas aeruginosa. In this study, the [2]rotaxane system offers the possibility to control spatial organization of sugars. Ten copies of one type of sugar ligand were attached to the macrocyclic core and a different sugar type was used as stopper ligand at the axle and provided a binding site for a second lectin. This architecture resulted in significantly increased binding affinities. The axle was obtained by CuAAC ligation of alkyne-functionalized sugar derivatives with an alkyl azide. Inclusion of the axle component into pillar[5]arene systems was carried out in chloroform at low temperature using high concentrations of the starting material.
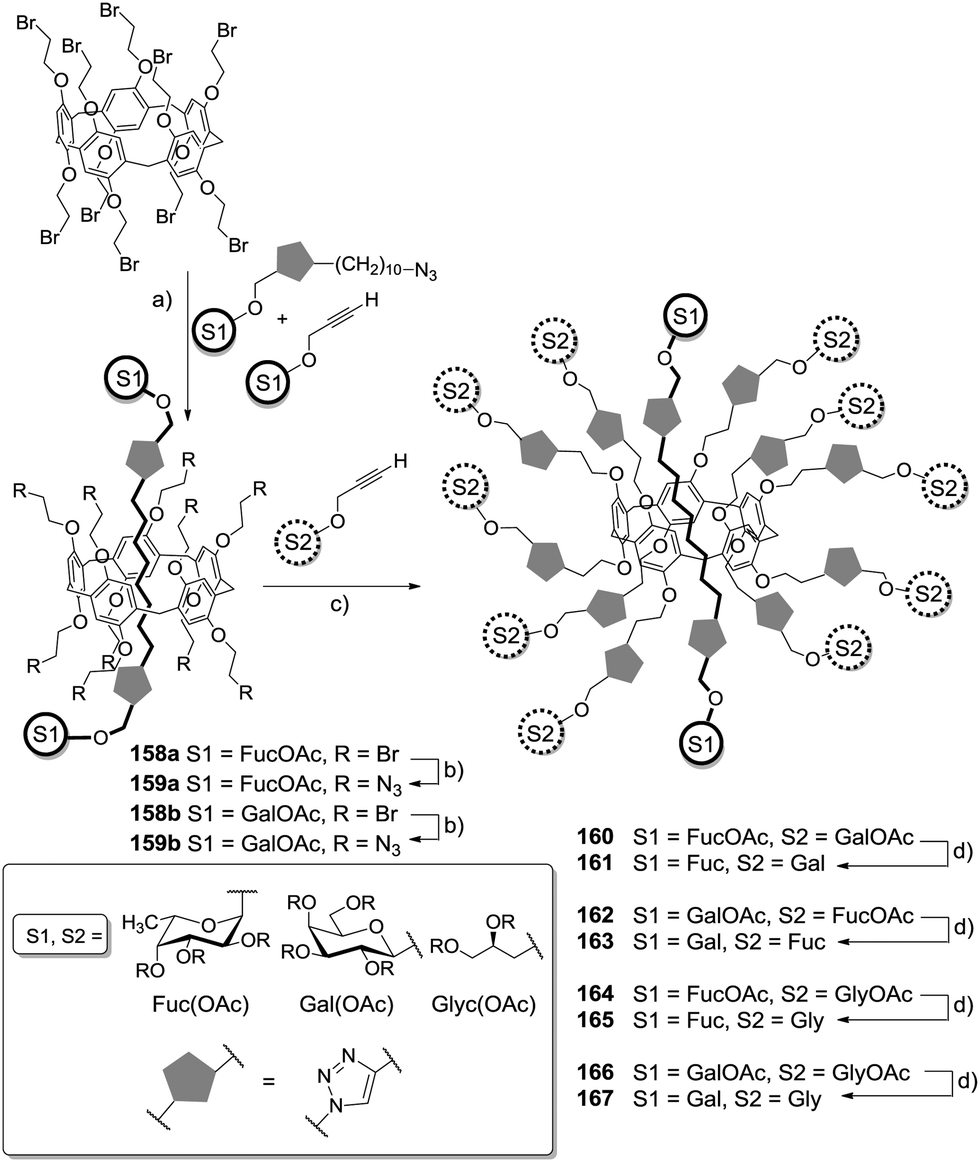 | ||
| Scheme 22 Pillar[5]arene-containing [2]rotaxane-based heteroglycoclusters. (a) CuBr·SMe2, CHCl3, −20 °C to rt (158a: 32%, 158b: 26%); (b) NaN3, DMF, rt, (159a: 85%, 159b: 84%); (c) CuSO4·5 H2O, sodium ascorbate, CH2Cl2/H2O, rt (160: 85%, 162: 82%, 164: 65%, 166: 79%); (d) MeOH, MeONa, rt (161: 86%, 163: 91%, 165: 89%, 167: 88%). | ||
1H NMR spectroscopic analysis after several days did not show any evolution of signals. Thus, the association between macrocycle and axle compound was proven to be stable. For the synthesis of heteroglycoclusters first the inclusion of a sugar-functionalized axle moiety was carried out, then the bromo-functionalized-macrocycle was converted into an azide-armed pillar[5]arene derivative. After deacetylation, the resulting unprotected rotaxanes could be obtained in high yields. This molecular architecture resulted in unprecedented affinities to both lectins.
The examples discussed in this section demonstrate how quickly this research field is accumulating data about new structure–activity relationships in multivalency studies, revealing secondary structural effects. It appears that researchers have indeed entered a next level of understanding how the different threedimensional parameters of a multivalent architecture play together to govern avidity. In the next section we are discussing further considerations of multivalency which go beyond the primary and secondary structural aspects discussed so far.
5. Highlighting tertiary structural features
The degree of complexity rises significantly when glycoclusters are employed in a supramolecular context such as on a surface. Glycoclusters of an AB3-type can be directly attached onto surfaces via the functional group “A” at the focal point of the molecule to obtain glycoarrays.6d,18 This focal point functionality also enables further conjugation with larger entities such as proteins, polymers, or cyclodextrins for example. Then again, the resulting multivalent glycosystem is characterized by primary and secondary structural properties of the comprised glycocluster components, as discussed in Sections 3 and 4. Additionally, a third feature level can be assigned to such complex glycocluster assemblies, which we call “tertiary”. Accordingly, the tertiary structure of multivalent glycomimetics indicates properties and characteristics of the molecular system which lie beyond the connectivity and three-dimensional quality of a glycocluster. Moreover, we suggest to consider also those structural aspect as “tertiary”, which allow to control ligand orientation and ligand density, thus facilitating the control of multivalency.In spite of the fact that so much research has been dedicated to the study of multivalency effects in carbohydrate recognition, a conclusive understanding of the underlying principles has still not been reached. This is mainly due to the structural complexity of the glycosylated cell which is involved in the majority of carbohydrate-related recognition events. The glycocalyx in fact has a significant dimension with a width of ∼100 nm,2 it is highly microheterogenic and apparently unordered. It remains a mystery to scientists how specific biological events are orchestrated in this seemingly chaotic background of glycoproteins, proteoglycans, glycolipids and glycosylphosphatidylinositol (GPI) anchors. Nevertheless, much thought and effort have been spent on how carbohydrate binding is governed on the cell surface. It seems to be obvious that glycocalyx characteristics are modified by glycosylation as well as deglycosylation, both catalyzed by specific enzymes, glycosyltransferases and glycosidases, respectively. Also, it has been postulated that the cell coat could undergo some local conformational reorientation to achieve structural matching in carbohydrate recognition.2c,4a,20c,85 Dynamic supramolecular reorientation could also affect the local carbohydrate density of a given glycocalyx area. Likewise its heterogeneity could be modified by supramolecular switching or segregation effects, thus altering lectin binding (Fig. 20).
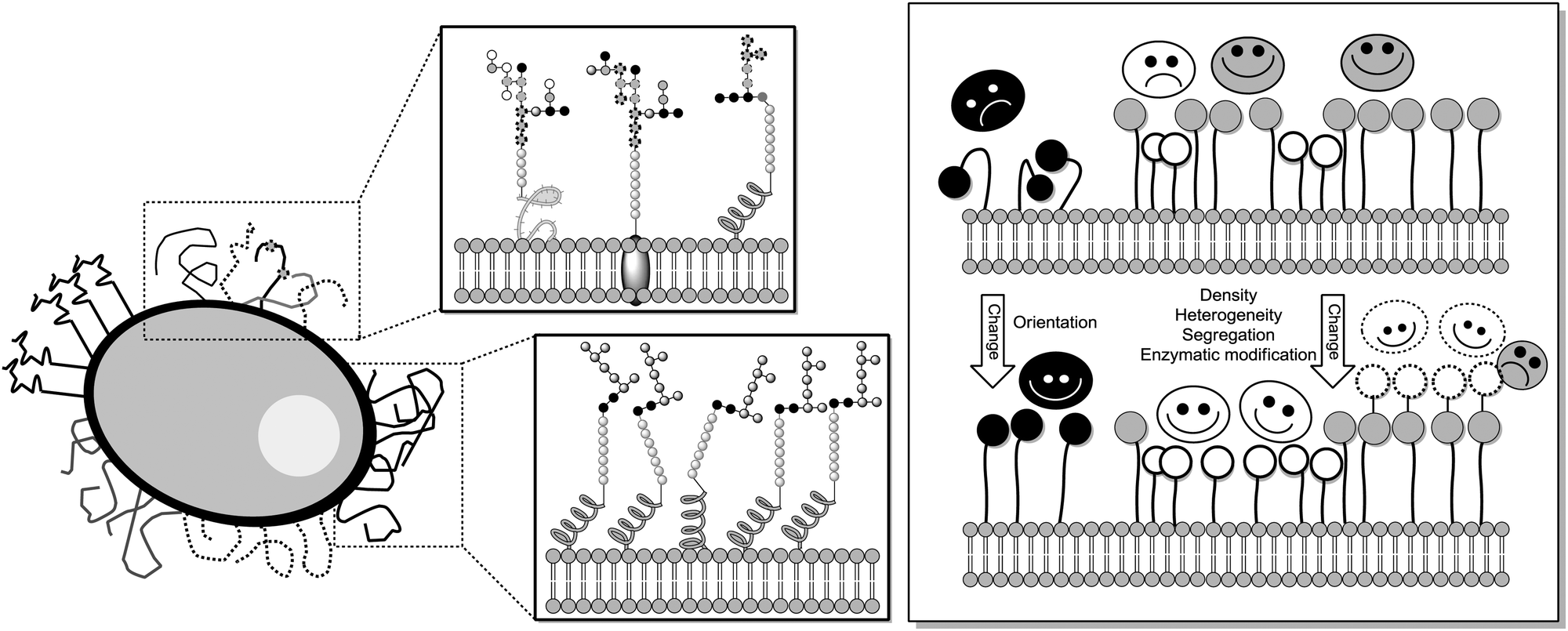 | ||
| Fig. 20 Schematic representation of the glycocalyx highlighting its inherent structural complexity as well the potential for microenvironment formation by supramolecular “switching” or segregation effects. Right: Dynamic structural changes of the glycocalyx layer toward switching protein binding. Spheres represent sugar ligands and respective smileys their specific protein receptors (lectins). | ||
Thus, structural variations within the glycocalyx might even conduct and finetune protein interactions with the cell surface. In pioneer work, Kahne et al. used surface plasmon resonance (SPR) to compare the binding of Bauhinia purpurea (BP) lectin to two different disaccharide ligands18a clustered as self-assembled monolayers (SAMs).86 Two slightly different disaccharide ligands were studied and their density varied in SAMs as detailed in Fig. 21. It was found that the lectin interacted with one disaccharide at low densities whereas binding to the other disaccharide ligand was preferred at high densities. Thus it was possible to “switch” lectin binding between two related saccharide ligands through adjustment of the sugar density of the surface. To explain the observed “on–off” switching of the binding specificity of the BP lectin, the authors hypothesized secondary interactions between the immobilized ligands and also between the lectins interacting with the SAM. It was concluded that the modification of the structure and density of the glycocalyx would also allow to switch protein binding from one “on” state to another “on” state.
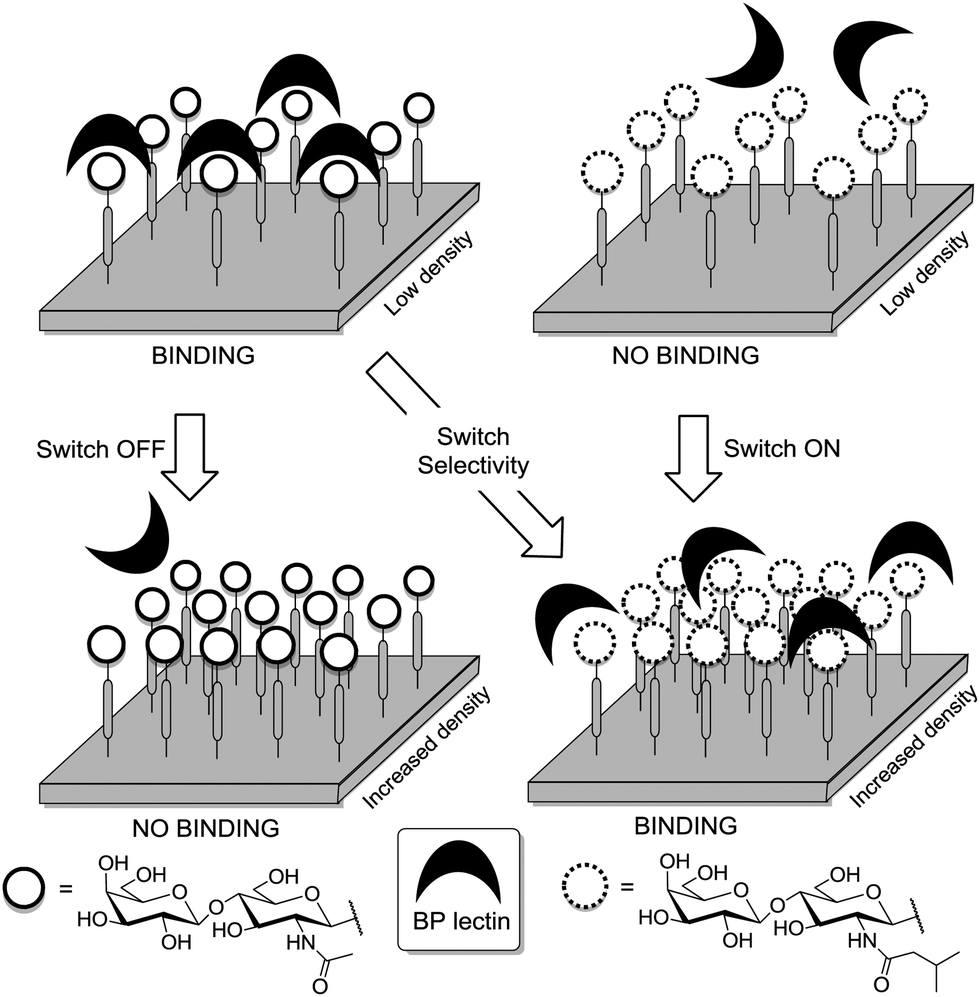 | ||
| Fig. 21 Lectin binding can occur with two distinct ligands, the binding switches according to the surface density of the anchored epitopes. | ||
In continuation of Kahne's work, García Fernández and colleagues added an important contribution in which they addressed how modification of the sugar composition at the cell surface might modulate carbohydrate–protein interactions.31 They designed a synthetic strategy (cf. Section 3) allowing the preparation of both homo- and heteromultivalent branching units which can be further attached to a β-cyclodextrin scaffold. Hence, the authors prepared several glycoclusters bearing up to 21 sugar ligands with different sugar densities. In heteroglycoclusters Man/Glc and Man/Lac combinations were realized (Fig. 22). The resulting heteromultivalent glycoconjugates were evaluated by enzyme-linked lectin assaying (ELLA), isothermal titration microcalorimetry (ITC), SPR and turbidity assays towards their interaction with concanavalin A (ConA) and peanut agglutinin (PNA). Whereas ConA has a specificity for mannose and glucose, PNA is lactose-specific. In accordance with the studies reported in the literature, highest binding affinities were observed with those homoglycoclusters that carried the respective carbohydrate ligands in high density. But unexpectedly highly dense heteroglycoclusters exhibited a stronger affinity than the respective homoclusters with the same content of the specific sugar epitope. This phenomenon was not observed with low-density heteroclusters, as further reported by Santoyo-Gonzales and co-workers.51c Moreover, the turbidity assays showed a similar behaviour of the highly-dense heteroclusters, as it was observed with the specific homoglycoclusters.
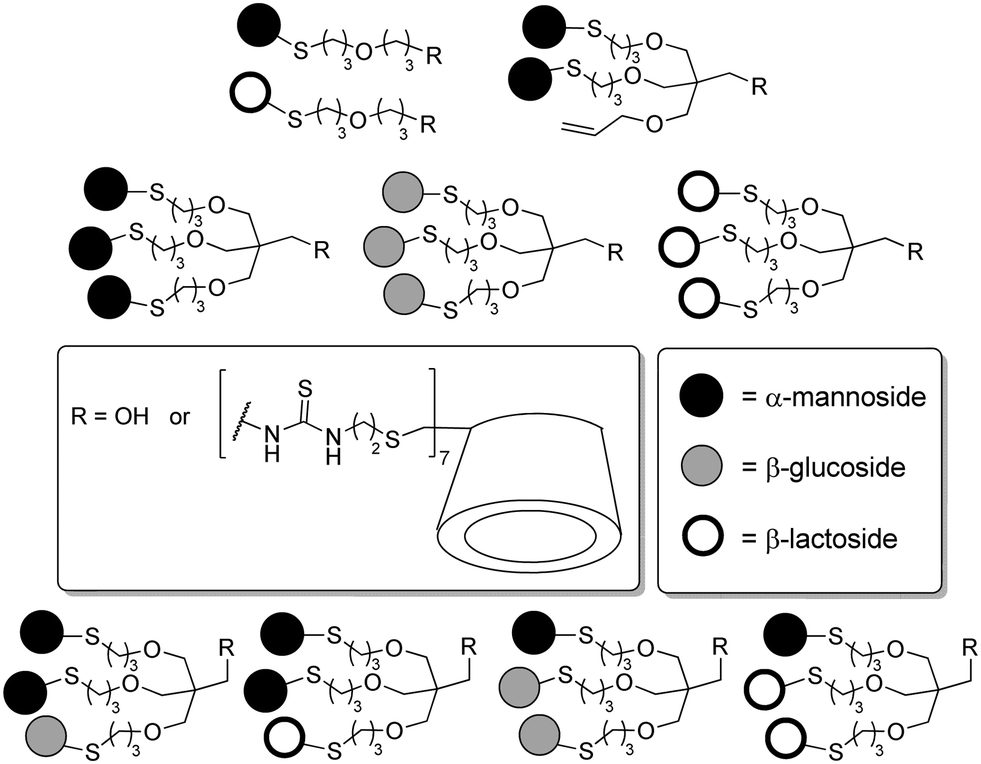 | ||
| Fig. 22 Glycoclusters with varied density and heterogeneity as prepared by Gómez-García and co-workers. Low-density clusters (1 to 3 epitopes) correspond to the functionalized branching unit bearing a primary alcohol at the focal point; high-density clusters (7 to 21 epitopes) were obtained by scaffolding the branching unit on a β-cyclodextrin. | ||
At first, the authors assumed that this “heterocluster effect” could be explained in terms of secondary interactions by the non-specific carbohydrate with subsites of the lectin. Finally, according to the ITC results, the authors suggested a possible thermodynamic origin of this phenomenon. Actually, the calculated free enthalpies of binding were nearly identical when ConA was tested with the heteroclusters or the respective 21-Man cluster.31a,b But strikingly, the measured enthalpies of binding and the resulting entropic terms were significantly different. Whilst the enthalpy related to the 21-Man cluster was the highest, the entropic penalty upon the interaction with the lectin was also maximal among the tested glycoclusters. The opposite trend was observed with the heterovalent glycoassemblies. Thus, it has been deduced that the binding of a specific ligand to its receptor could be entropically favoured in a heterogeneous environment, following a bind and slide model.72 Indeed, the entropy–enthalpy compensation, in the presence of the secondary ligand, would promote the sliding of the lectin along the glycosylated area, hence resulting in improved binding.
Ravoo and co-workers proposed a supramolecular approach to assess the influence of ligand packing on carbohydrate–lectin binding.87 They designed a tool based on amphiphilic β-cyclodextrins that can self-assemble to form bilayers and cyclodextrin vesicles (CDV). These can host adamantane–glycoside conjugates. This way it was possible to fabricate glycocalyx models in which the softness and fluidity of the cell membrane were reproduced with the possibility to systematically vary its glycocoating (Fig. 23). In fact, homo- and heteromultivalent glycoassemblies were prepared using maltose and/or lactose ligands and the resulting sugar-coated CDVs tested towards their interaction with ConA or PNA. Optical density measurements, dynamic light scattering and ITC measurements were performed. Selective agglutination of ConA was observed in the presence of a homomaltose CDV while the respective homolactose CDV exhibited a specific agglutination with PNA lectin. Interestingly, the experiments carried out with the heteromultivalent CDVs revealed that lectin aggregation is ligand density-dependent and its rate scales linearly with both the lectin and the covering ligand concentrations. More precisely, high densities were required to induce a significant aggregation, since the critical surface coating is about 50% in the case of the maltose–ConA interaction while this value is about 75% for the lactose–PNA interaction. Based on these data and on the known distance separating the effective binding sites of ConA (3.6 to 4.3 nm), the authors suggested that the corresponding distance would be smaller for PNA. Hence, their CDV model could be used as a means to estimate such unknown distances between carbohydrate binding sites as the spatial distribution of carbohydrate constituents within a CVD's sugar can easily be controlled and measured.
 | ||
| Fig. 23 Amphiphilic β-cyclodextrins self-assemble to form vesicles (CDVs) then adamantane–glycoside conjugates could anchor into the external layer via inclusion complex. The PNA lectin was specifically recognized to form aggregates with the CDVs in an epitope density-dependent manner. | ||
In a recent report, Lindhorst et al. studied the influence of glycoarray density towards type 1-fimbriated E. coli adhesion.88 Mono-, di- and trivalent cluster mannosides were prepared then first tested as inhibitors of bacterial adhesion in solution according to a standard adhesion–inhibition assay. As expected, the observed inhibition increased while the cluster valency increased. They further used the conjugatable clusters to fabricate arrays displaying different mannose concentration, in order to measure bacterial adhesion. In that case, preferential binding was observed with arrays bearing the monovalent mannoside moiety at the highest concentration while the polyvalent clusters exhibited a better adhesion on diluted surfaces (Fig. 24). Hence, it appeared that at high densities a cluster effect would be hampered by the steric bulk whereas such effect would rather occur at low densities, when the glycoarray provides high local concentration of the sugar ligand.
 | ||
| Fig. 24 Investigation of the influence of density on glycoarrays towards bacterial adhesion. The density was modulated by the use of varied valency epitopes. Monovalent arrays exhibited an increased adhesion while the epitope concentration increased; the opposite trend was observed when polyvalent clusters were used for glycoarray fabrication. | ||
As glycolipids can be clustered into the cell bilayer in association with cholesterol and glycoproteins, it has been postulated that the so-called “lipid rafts” could play a major role in carbohydrate–protein recognition. In order to investigate this, Webb and Flitsch designed relevant model systems based on the anchoring of pyrene-perfluoroalkyl glycosides into artificial membrane bilayers prepared from dimyristoyl phosphatidylcholine (DMPC).89 In a first report,89a they assessed the ability of mannosylated lipids to bind to ConA when dispersed over the surface or clustered via the formation of lipid rafts (Fig. 25). In the case of the dispersed glycolipids, the interaction with the lectin was weaker than in solution at low membrane loading (1% mol glycolipid/mol DMPC), while increasing the loading increased the affinity. Nonetheless, clustering the mannolipids into artificial lipid rafts at a loading of 1% mol/mol did not improve binding compared to the dispersed glycolipids at the same loading. It was assumed that the formation of lipid rafts could disturb the membrane arrangement, thus leading to a reduced availability of the mannoside ligands. In a further study,89b the same authors applied an analogous approach to study the enzymatic galactosylation of N-acetylglucosaminolipids (Fig. 25). Here, a significant positive effect was observed with clustered glycolipids (1% mol/mol). Enzymatic galactose transfer was 9-fold faster showing a lower Michaelis constant than in the dispersed condition. Again, the authors assumed possible secondary interactions, statistical rebinding or boundary effects to explain this cluster effect. Notably, unlike observed by García Fernández and colleagues, the construction of mixed microdomains containing both the specific substrate and the non-substrate galactolipid led to a slower enzymatic reaction.
 | ||
| Fig. 25 Clustering of carbohydrate epitopes within artificial phospholipid bilayers by formation of lipid rafts (top box). The galactosylation of N-acetylglucosamine residues by bovine β-(1,4)-galactosyltransferase is favoured in the clustered microdomains (bottom box). | ||
In consideration of the research discussed in the last paragraph, it appears to be crucial to design multivalent systems which not only provide improved binding affinities but moreover, -perhaps more importantly-, lead to reliable data which support our understanding of glycocalyx function. Following this direction, a new trend has emerged over the last decade, the conception of responsive architectures able to undergo dynamic modifications. This is related to the glycosylated cell surface, which has to be recognized as a dynamic supramolecular assembly.
Regarding the design of dynamic glycoassemblies, one main bias is to build structures able to modify the orientation or conformation of the active ligand triggered by an external stimulus (physical, chemical, biological). In that sense, light-responsive materials constitute an attractive means to achieve such a goal, as first proven by Willner et al. by the input of photoisomerizable moieties on ConA.90 Especially, the well known azobenzene derivatives appeared to be molecules of choice to probe dynamics of carbohydrate recognition, as they are easy to synthesize, responsive to light of different wavelength (from UV to red light), and are relatively small (the distance between the 4- and 4′-carbon atoms is 9 Å for the E-configured isomer and 5 Å for the Z-isomer).91 In the early 2000 years, Jayaraman et al. introduced the first photoswitchable cluster glycosides.92 Several azobenzene glycoside were prepared with various valencies and sugar moieties (Fig. 26). After preliminar photoisomerization studies, showing a higher stability of the Z-forms in water, the clusters were evaluated towards lectin binding by ITC using ConA or PNA. As expected, multivalent glycoclusters interacted more strongly with the lectins than the monovalent ligands. In addition, the authors reported a higher binding affinity for the Z-configured isomers in comparison to the analogous E-isomers. Despite the fact, that this effect observed for the Z-configured glycoclusters could not be well explained at the time, this fundamental study has opened the way to a new generation of photoresponsive glycoconjugates.
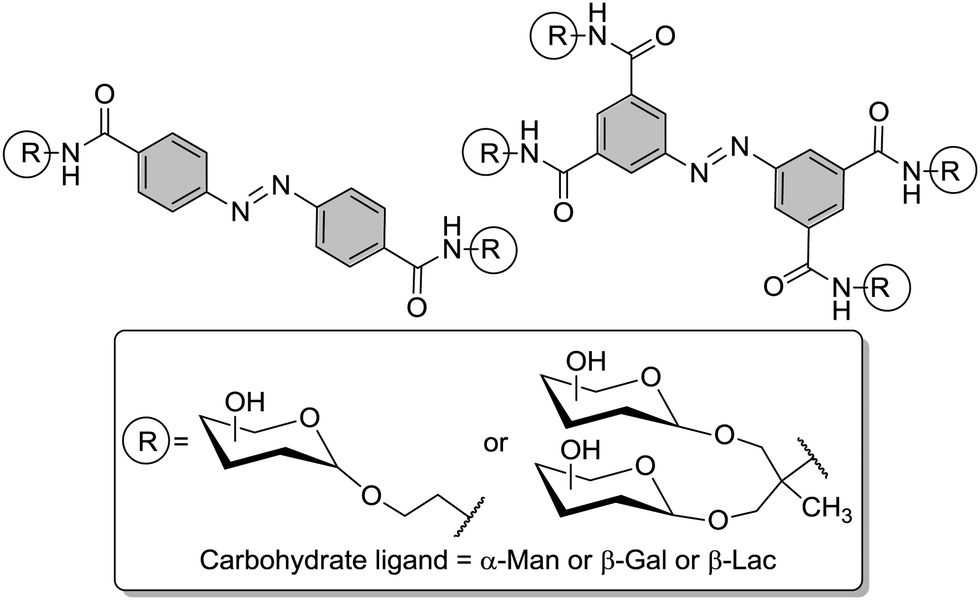 | ||
| Fig. 26 Photoswitchable azobenzene glycoconjugates were first prepared by Jayaraman and co-workers to test carbohydrate–lectin interactions. | ||
Having well understood the importance of Jayaraman's seminal work, Lindhorst and colleagues have commenced a project to develop new photoswitchable glycocalyx mimetics.93 This work falls within the study of bacterial adhesion mediated by fimbrial lectins, especially FimH which specifically recognizes α-D-mannopyranosides. First of all, various azobenzene glycoconjugates were synthesized, including symmetrical, unsymmetrical, and mono- to trivalent cluster mannosides (Fig. 27).93a,c
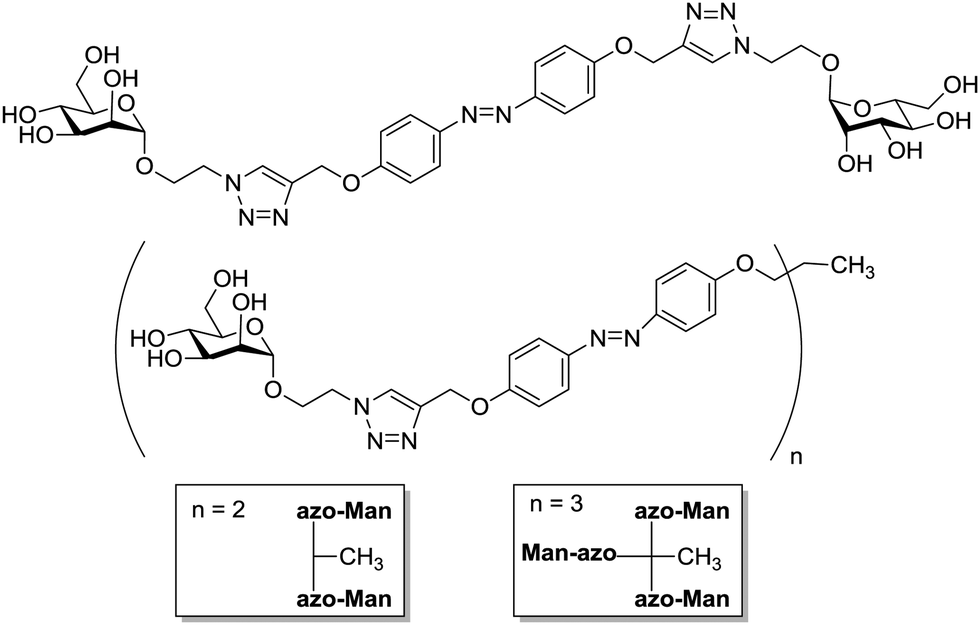 | ||
| Fig. 27 Multivalent azobenzene mannosides introduced as “sweet switches” by the Lindhorst group. | ||
Photochromic properties of the resulting structures were investigated both by UV/Vis and 1H NMR spectroscopic studies to establish the ratio of E- and Z-isomers in the photostationary state (PSS). For instance, E/Z ratios of 99:1 in the ground state (GS) versus >2:98 in the PSS were found, with the Z-form exhibiting half-life for thermal back-isomerization of up to 94 h. In addition, the isomerization was reversible without damaging the structural integrity of the conjugates. The ability to bind to type 1-fimbriated E. coli bacteria in solution was further assessed with monovalently conjugated azobenzene-bearing dimannosides ligands.93b Standard inhibition assays showed similar binding affinities for the two isomers, with a 2-fold increasing efficiency compared to the methyl α-D-mannoside reference. This result was supported by computer-assisted docking studies.
Finally, as an ultimate goal, linker-tethered monovalent azobenzene–mannose conjugates were immobilized on SAMs to investigate how their isomerization could influence FimH-mediated bacterial adhesion. After probing the effectiveness and reversibility of the isomerization on the gold surface by infrared absorption spectroscopy (IRRAS),93d,e the glyco-SAMs were incubated with GFP-expressing type 1-fimriated E. coli bacteria after irradiation of the SAM with UV (365 nm, to effect E → Z isomerization) or visible light (450 nm, for Z → E), respectively (Fig. 28).93e A dramatic decrease of bacterial adhesion (∼80%) was observed in the Z-form of the azobenzeneglyco-SAM, in comparison to the E-form. Full adhesion could be recovered after back-isomerization (Z → E) of the glycoarray over several cycles of light-induced isomerization. In sharp contrast with the result aforementioned, obtained with both the soluble E- or Z-isomers, the position-dependent bacterial adhesion is an evidence that the orientation of the ligand on a surface is a critical parameter for a successful binding event.
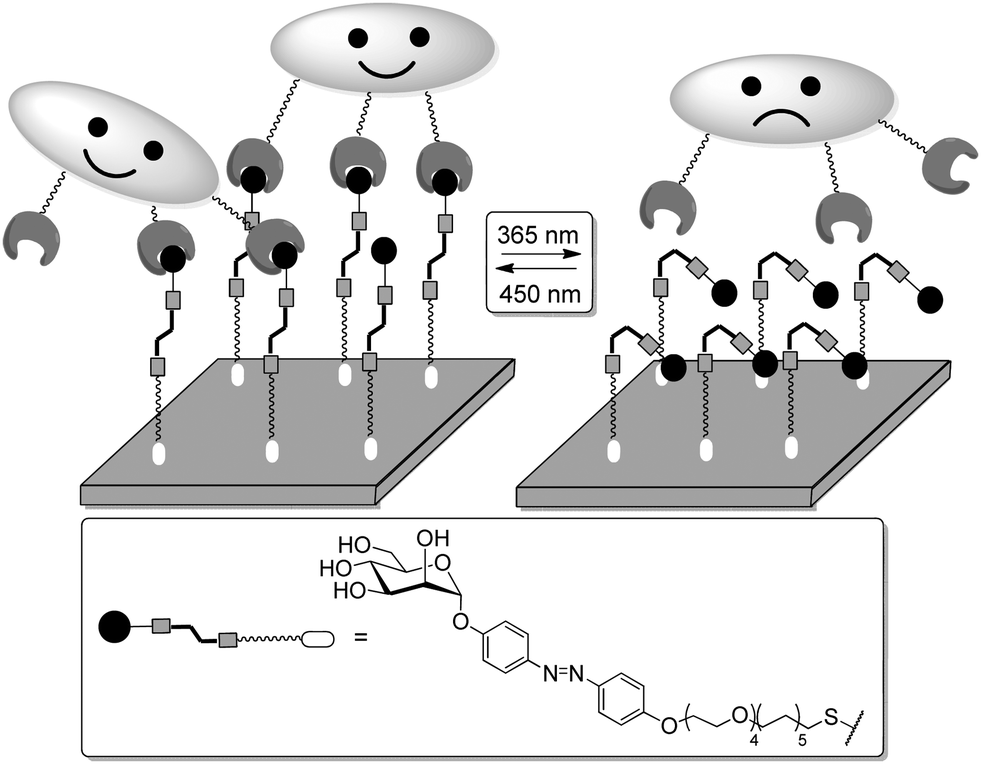 | ||
| Fig. 28 Probing the importance of epitope orientation on lectin binding through the use of photoswitchable glyco-SAMs. Adhesion of type 1-fimbriated E. coli bacteria can be reversibly switched between two states (high and low adhesion) by re-orientation of an azobenzene-conjugated carbohydrate ligand through photochemical E/Z isomerization of the azobenzene hinge. | ||
Importantly, this approach has been transposed from artificial SAMs to human cells surfaces (using human microvascular endothelial cells).94 Hence, metabolic oligosaccharide engineering (MOE) was employed to functionalize the cell surface with azide-bearing sugars. It was furthermore possible to attach an alkyne-armed mannose–azobenzene conjugate via CuAAC coupling. In accordance with the previous study, E → Z photoswitching showed a significantly decreased adhesion in flow-based experiments (about 50%).
Related to this work on orientational control of cell adhesion, is a report of the Ravoo group on a smart light-driven system, which is capable to catch and release lectins in a reversible fashion.95 Taking advantage of the cyclodextrin vesicles (CDVs) described by the same group (vide supra), here the authors replaced the adamantane moiety of the sugar conjugates (maltose or lactose) by an azobenzene unit which served as a photosensitive switch (Fig. 29). It is well established that E-configured azobenzene strongly complexes with the α- or β-CD cavities whereas the corresponding Z-form is unable to fit into the hydrophobic pocket of cyclodextrins, due to its bent structure and higher polarity. CDVs were first associated with E-azobenzene glycoconjugates and then incubated with ConA or PNA lectins in aqueous solution at physiological pH. The resulting aggregate solutions were then irradiated with UV light (350 nm) to effect E → Z isomerization of the azobenzene anchors, thus leading to the dissociation of the ternary CDV–glycoconjugate–lectin complex. It is noteworthy that the ternary complex could be recovered after irradiation with visible light (455 nm) and this reversibility of the complexation/decomplexation process was in full function over four cycles. Finally, the authors claimed that such reversible and biocompatible assembly could be used as a versatile system for targeted protein delivery.
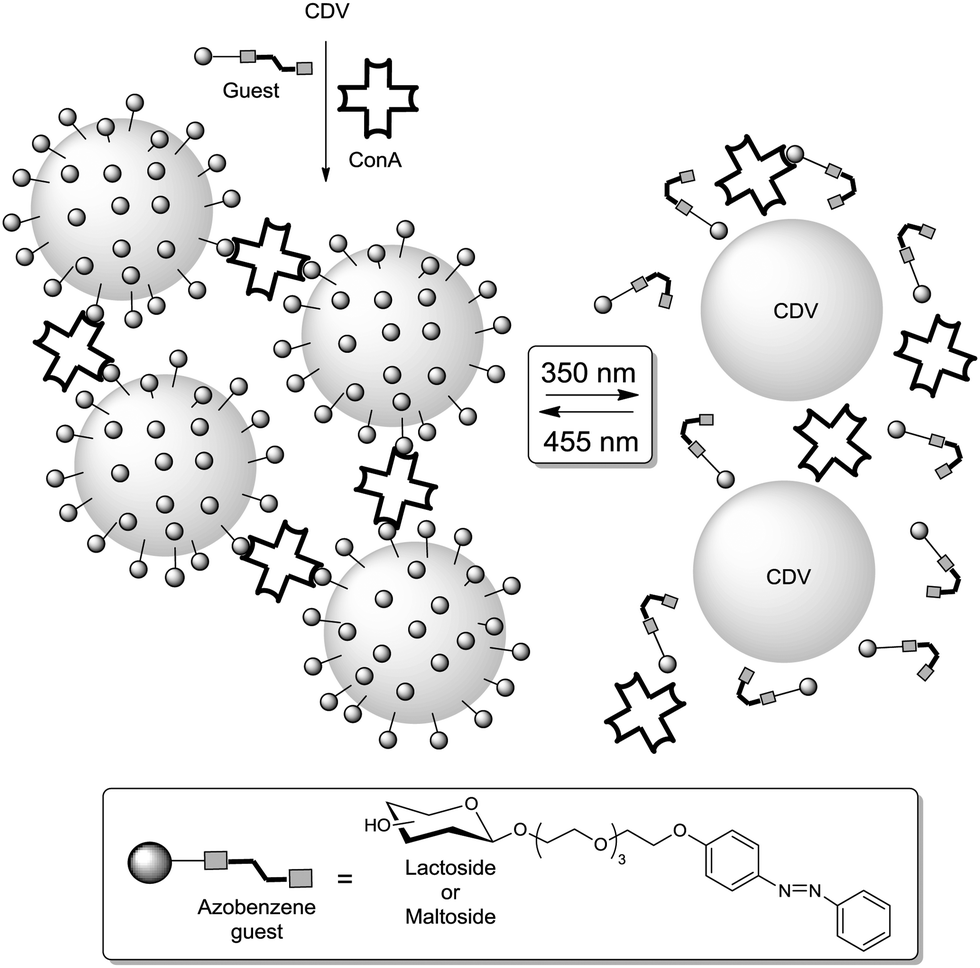 | ||
| Fig. 29 Lectin catch by CDV–glycoazobenzene supramolecular complex and light-driven release by switching of the azobenzene guest into its Z-form (irradiation at 350 nm). The processes is reversible by irradiating the system with visible light (455 nm). | ||
Before the recent photoisomerizable glyco-SAMs reported by Lindhorst and colleagues, a supramolecular switch was jointly designed by Djedaïni-Pilard and García Fernández in order to mimic the allosteric modification of a glycoprotein.96 Here, the authors conjugated mono- or divalently branched trimannosides (ConA-specific) to β-CD via a linker equipped with a tyrosine moiety. This is able to form a host–guest complex with the CD cavity (Fig. 30). This last key feature enabled the possibility to put the ligand moiety into a position close to the CD scaffold, hence hampering the recognition by the lectin (“off” state). NMR studies confirmed the inclusion of the tyrosine residue then the conjugate was evaluated toward ConA binding by ELLA, in comparison with the analogous conjugate devoid of the tyrosine part. According to the authors' hypothesis, the tyrosine-armed conjugate was not recognized by the lectin (IC50 up to 1 mM) unlike the other one, exhibiting an IC50 of 21 μM. The reversibility of the system was further assessed, firstly by getting the ligand available for recognition (“on” state), then by going back to the “off” state. To achieve this goal, the self-inclusive tyrosine was removed from the CD cavity by the addition of an excess of adamantane-1-carboxylate (AD), leading to the “on” state with a measured IC50 of 22 μM. The “off” state could be recovered by the addition of an AD-scavenger, showing the efficiency of such a system to simulate a ligand reorientation in view of a recognition event.
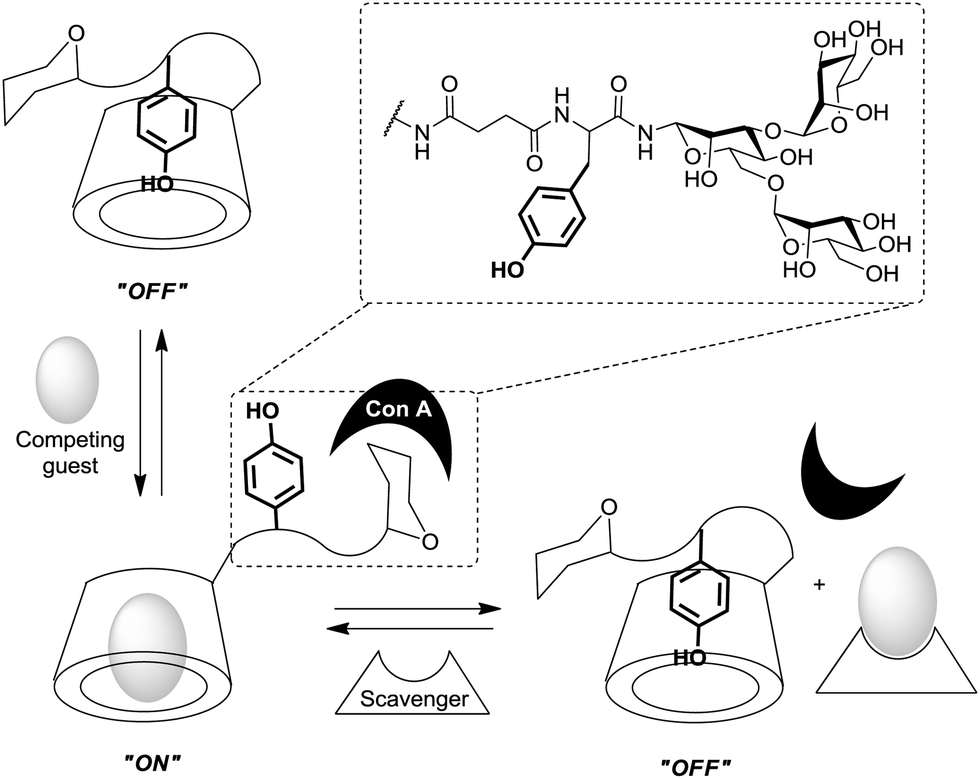 | ||
| Fig. 30 Changing of an epitope orientation via a supramolecular switch. The tyrosine part of the linker spontaneously self-includes into the CD cavity to put the system into the “OFF” state. The system can be turned “ON” by addition of a competing guest; the “OFF” state can be recovered by addition of a scavenger chelating the competing guest. | ||
Recently, an additional parameter for the organization of carbohydrate ligands has been introduced by the Lindhorst group.97 The chosen approach is based on the restriction of the conformational flexibility of carbohydrate ligands via functional linkers. Thus mannoside ligands were attached onto thymine rings, which are known to undergo a [2+2] photocycloaddition upon irradiation. A first study involving amphiphilic glycothymidine derivatives was carried out in order to determine the potential of such a system for relevant biological studies. Amphiphilic glycothymidine conjugates (Fig. 31) were incorporated into sodium dodecyl (SDS) micelles, as a mimic of a glycosylated cell surface. Then these micelles were irradiated with light of 295 nm. The efficiency of the dimerization was proven by 1H NMR and NMR diffusion experiments, thus validating the proof of concept.
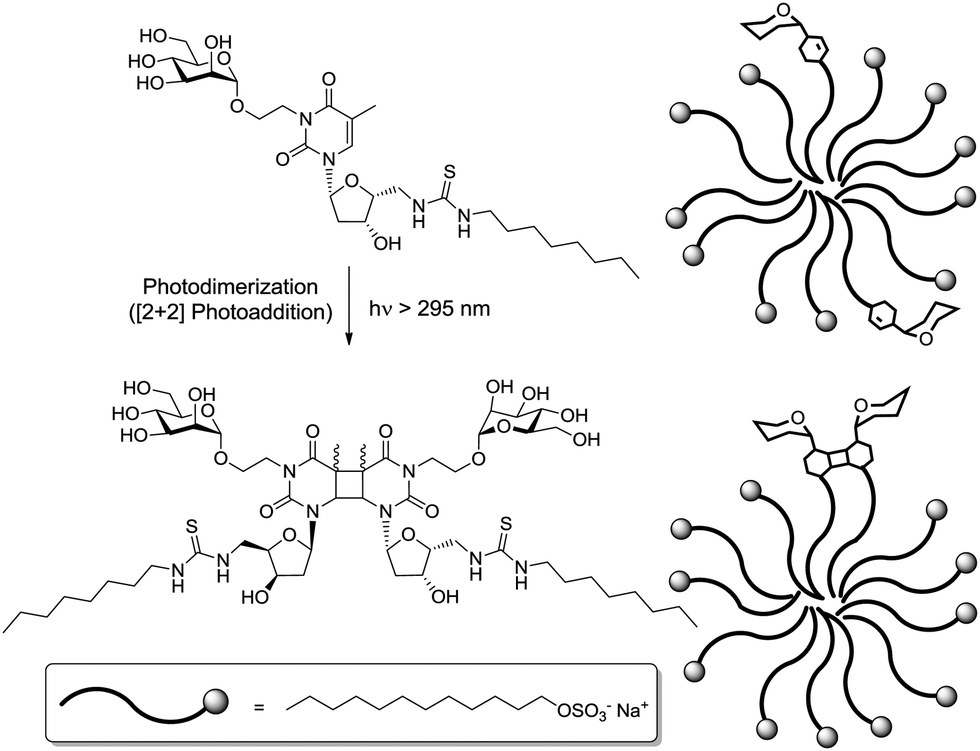 | ||
| Fig. 31 Photodimerizable amphiphilic glycothymidine to lock the conformational arrangement of sugar epitopes. The mannothymidines were incorporated into SDS micelle to assess whether the cycloaddition could occur into a supramolecular system. | ||
In a very recent paper, the same authors described the synthesis of divalent glycoclusters where the lectin-relevant α-mannoside ligand were anchored on a mannoside scaffold via a thymine-based linker.98 In such a scaffolded system, the photoaddition should only occur in an intramolecular pathway, which was confirmed by NMR after irradiation by UV light (hν > 290 nm) in a water/acetone mixture. Furthermore, the light-responsive cluster has been armed at the anomeric position with a conjugatable linker, making it amenable to surface immobilization (Fig. 32).
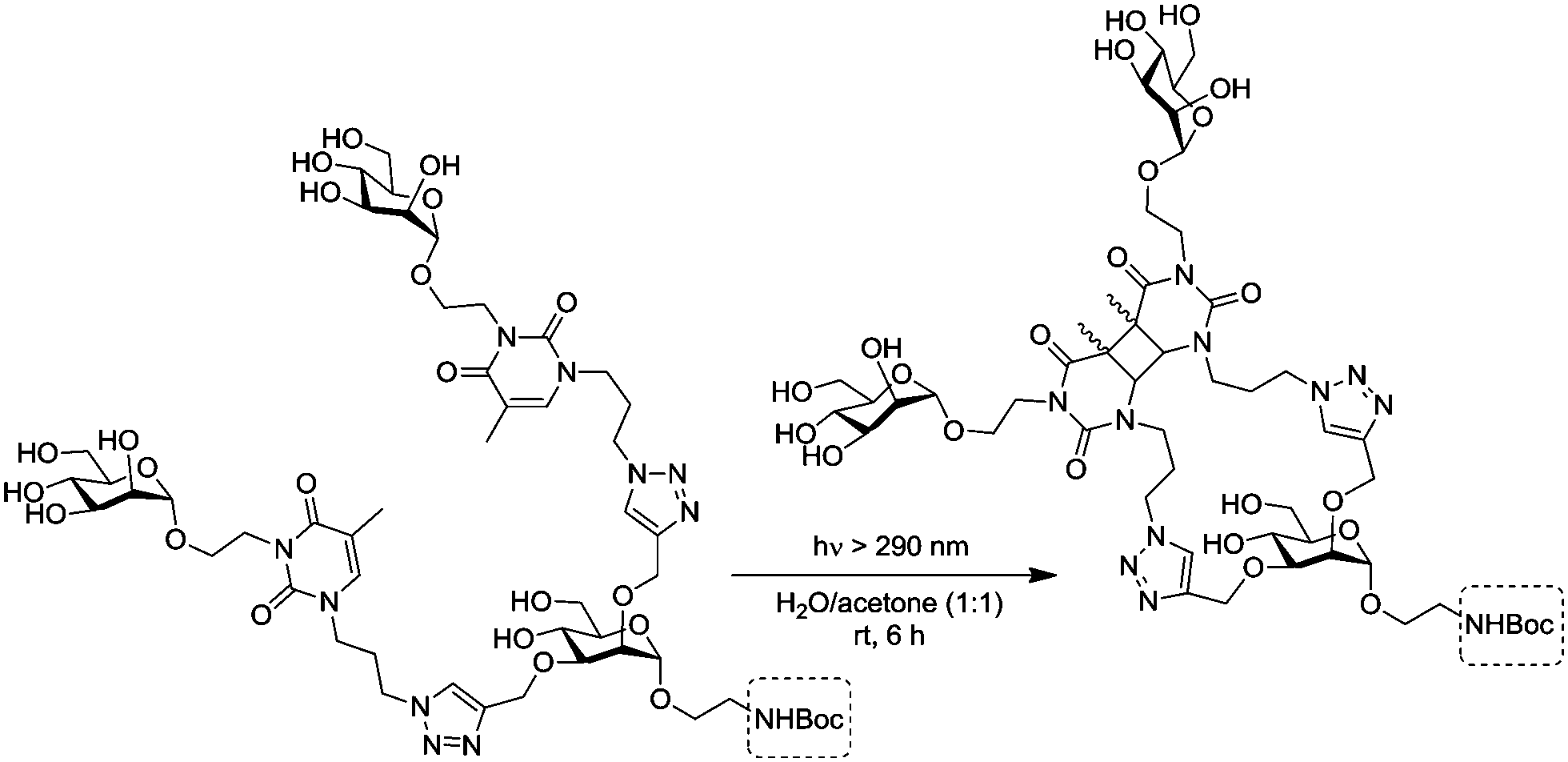 | ||
| Fig. 32 Conformational locking of mannosyl ligands attached on a mannoside cluster via thymine photodimerization. The cluster is equipped with a masked amino tether for further immobilization. | ||
6. Conclusions
This account falls into the collection of reviews on multivalent glycoconjugates which have been published earlier. We have focussed our report on rather small glycoclusters and at the same time made an attempt to point out the more recent work in the field. We are aware that we have also omitted a huge amount of science which would be worthwhile to tell. Rather then to be comprehensive we have highlighted three different aspects of glycocluster research, which we have called primary, secondary and tertiary, in analogy to the terminology used for protein structures. As a conclusive theory of multivalency effects in carbohydrate recognition has not been reached as yet, this is our suggestion for a systematic as well as inspiring approach to understanding of the various parameters which govern multivalent interactions between sugars and their receptors. In particular, we hope that the last section on “tertiary” considerations of multivalency has provided a new perspective about the way multivalency can be organized. The herein reviewed studies have supplied some key concepts to define how multivalent systems can be manipulated and controlled in a supramolecular context. Obviously, much remains to be done in order to unravel the “glycocalyx enigma”. Reasonably, the next step of this quest would be to refine and, maybe, to favourably combine the smart multivalency tools which have been introduced by glycoscientists. It will be important to further explore density, heterogeneity and orientational effects in carbohydrate recognition and identify further parameters providing specificity of biological effects as a consequence of carbohydrate–protein interactions.Acknowledgements
We acknowledge support by Christiana Albertina University and by the DFG, in particular in the frame of the collaborative network SFB 677.Notes and references
- A. Varki and J. B. Lowe, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, NY, 2009, 2nd edn, ch. 6, pp. 75–88 Search PubMed.
- (a) H. S. Benett, J. Histochem. Cytochem., 1963, 11, 14 CrossRef; (b) S. Reitsma, D. W. Slaaf, H. Vink, M. A. M. J. van Zandvoort and M. G. A. oude Egbrink, Pflugers Arch., 2007, 454, 345 CrossRef CAS PubMed; (c) H.-J. Gabius, S. André, J. Jiménez-Barbero, A. Romero and D. Solis, Trends Biochem. Sci., 2011, 36, 298 CrossRef CAS PubMed.
- (a) B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 2014, 6th edn, ch. 15, pp. 813–888 Search PubMed; (b) M. E. Taylor and K. Drickamer, Curr. Opin. Cell Biol., 2007, 19, 572 CrossRef CAS PubMed; (c) A. A. Khalili and M. R. Ahmad, Int. J. Mol. Sci., 2015, 16, 18149 CrossRef CAS PubMed.
- (a) S. Hakomori, Arch. Biochem. Biophys., 2004, 426, 173 CrossRef CAS PubMed; (b) A. G. Todeschini and S. Hakomori, Biochim. Biophys. Acta, 2008, 1780, 421 CrossRef PubMed.
- (a) J. M. Rini, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 551 CrossRef CAS PubMed; (b) S. Elgavish and B. Shaanan, Trends Biochem. Sci., 1997, 22, 462 CrossRef CAS PubMed; (c) H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637 CrossRef CAS PubMed; (d) N. Sharon and H. Lis, Glycobiology, 2004, 14, 53R CrossRef CAS PubMed; (e) J. Arnaud, A. Audfray and A. Imberty, Chem. Soc. Rev., 2013, 42, 4798 RSC; (f) S. André, H. Kaltner, J. C. Manning, P. V. Murphy and H.-J. Gabius, Molecules, 2015, 20, 1788 CrossRef PubMed.
- (a) S. Chipowsky, Y. C. Lee and S. Roseman, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2309 CrossRef CAS PubMed; (b) M. J. Krantz, N. A. Holtzman, C. P. Stowell and Y. C. Lee, Biochemistry, 1976, 15, 3963 CrossRef CAS PubMed; (c) P. H. Weigel, E. Schmell, Y. C. Lee and S. Roseman, J. Biol. Chem., 1978, 253, 330 CAS; (d) Y. C. Lee, Carbohydr. Res., 1978, 67, 509 CrossRef CAS.
- (a) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef; (b) C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357 CrossRef CAS PubMed; (c) J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555 CrossRef CAS PubMed; (d) L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348 CrossRef CAS PubMed; (e) C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472 CrossRef CAS PubMed.
- (a) R. Roy, Top. Curr. Chem., 1997, 187, 241 CrossRef CAS; (b) T. K. Lindhorst, Top. Curr. Chem., 2002, 218, 201 CrossRef CAS; (c) N. Jayaraman, Chem. Soc. Rev., 2009, 38, 3463 RSC; (d) Y. M. Chabre and R. Roy, Adv. Carbohydr. Chem. Biochem., 2010, 63, 165 CrossRef CAS PubMed; (e) T. K. Lindhorst, in Proceedings of the 2nd International Beilstein Symposium on Glyco-Bioinformatics 2011, ed. M. G. Hicks and C. Kettner, Logos-Verlag, Berlin, 2012, pp. 147–164 Search PubMed; (f) T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613 RSC; (g) A. Bernardi, et al. , Chem. Soc. Rev., 2013, 42, 4709 RSC; (h) S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525 CrossRef CAS PubMed.
- (a) N. Röckendorf and T. K. Lindhorst, Top. Curr. Chem., 2001, 217, 201 CrossRef; (b) W. B. Turnbull and J. Fraser Stoddart, Rev. Mol. Biotechnol., 2002, 90, 231 CrossRef CAS; (c) K. Hatano, K. Matsuoka and D. Terunuma, Chem. Soc. Rev., 2013, 42, 4574 RSC; (d) M. Gingras, Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4823 RSC; (e) D. Appelhans, B. Klajnert-Maculewicz, A. Janaszewska, J. Lazniewska and B. Voit, Chem. Soc. Rev., 2015, 44, 3968 RSC.
- (a) L. L'Haridon and J.-M. Mallet, in Carbohydrate Chemistry, ed. A. P. Rauter, T. K. Lindhorst and Y. Queneau, The Royal Society of Chemistry, Cambridge, vol. 40, 2014, pp. 257–267 Search PubMed; (b) T. K. Lindhorst and M. Dubber, Carbohydr. Res., 2015, 403, 90 CrossRef CAS PubMed; (c) R. Roy and T. C. Shiao, Chem. Soc. Rev., 2015, 44, 3924 RSC.
- (a) M. C. Galan, P. Dumy and O. Renaudet, Chem. Soc. Rev., 2013, 42, 4599 RSC; (b) J.-L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814 RSC; (c) G. C. Daskhan, N. Berthet, B. Thomas, M. Fiore and O. Renaudet, Carbohydr. Res., 2015, 405, 13 CrossRef CAS PubMed.
- N. Spinelli, E. Defrancq and F. Morvan, Chem. Soc. Rev., 2013, 42, 4557 RSC.
- (a) A. Dondoni and A. Marra, Chem. Rev., 2010, 110, 4949 CrossRef CAS PubMed; (b) F. Sansone and A. Casnati, Chem. Soc. Rev., 2013, 42, 4623 RSC; (c) Y. M. Chabre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657 RSC.
- (a) N. Jayaraman, K. Maiti and K. Naresh, Chem. Soc. Rev., 2013, 42, 4640 RSC; (b) A. Martinez, C. Ortiz Mellet and J. M. García Fernández, Chem. Soc. Rev., 2013, 42, 4746 RSC; (c) M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693 CrossRef CAS PubMed.
- J. L. Jiménez Blanco, C. Ortiz Mellet and J. M. García Fernández, Chem. Soc. Rev., 2013, 42, 4518 RSC.
- P. Bojarová, R. R. Rosencrantz, L. Elling and V. Křen, Chem. Soc. Rev., 2013, 42, 4774 RSC.
- M. Marradi, F. Chiodo, I. García and S. Penadés, Chem. Soc. Rev., 2013, 42, 4728 RSC.
- (a) R. Liang, L. Yan, J. Loebach, M. Ge, Y. Uozumi, K. Sekanina, N. Horan, J. Gildersleeve, C. Thompson, A. Smith, K. Biswas, W. C. Still and D. Kahne, Science, 1996, 274, 1520 CrossRef CAS PubMed; (b) T. Feizi and W. Chai, Nat. Rev. Mol. Cell Biol., 2004, 5, 582 CrossRef CAS PubMed; (c) N. Laurent, J. Volgmeir and S. Flitsch, Chem. Commun., 2008, 4400 RSC; (d) T. Horlacher and P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 1414 RSC.
- (a) A. Imberty, Y. M. Chabre and R. Roy, Chem. – Eur. J., 2008, 14, 7490 CrossRef CAS PubMed; (b) V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492 RSC; (c) V. Wittmann, Curr. Opin. Chem. Biol., 2013, 17, 982 CrossRef CAS PubMed.
- (a) R. T. Lee and Y. C. Lee, Glycoconjugate J., 1987, 4, 317 CrossRef CAS; (b) J. J. Hangeland, J. T. Levis, Y. C. Lee and P. O. Ts'o, Bioconjugate Chem., 1995, 6, 695 CrossRef CAS; (c) Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321 CrossRef CAS; (d) M. S. Quesenberry, R. T. Lee and Y. C. Lee, Biochemistry, 1997, 36, 2724 CrossRef CAS PubMed; (e) R. T. Lee and Y. C. Lee, Bioconjugate Chem., 1997, 8, 762 CrossRef CAS PubMed.
- (a) B. P. Devlin and D. A. Tirell, Macromolecules, 1986, 19, 2466 CrossRef; (b) A. D. Meltzer, D. A. Tirell, A. A. Jones, P. T. Inglefield, D. M Hedstrand and D. A. Tomalia, Macromolecules, 1992, 25, 4541 CrossRef CAS.
- G. R. Newkome, R. K. Behera, C. N. Moorefield and G. R. Baker, J. Org. Chem., 1991, 56, 7162 CrossRef CAS.
- T. K. Lindhorst, S. Kötter, U. Krallmann-Wenzel and S. Ehlers, J. Chem. Soc., Perkin Trans. 1, 2001, 823 RSC.
- S. Ballut, D. Naud-Martin, B. Loock and P. Maillard, J. Org. Chem., 2011, 76, 2010 CrossRef CAS PubMed.
- O. Khorev, D. Stokmaier, O. Schwardt, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2008, 16, 5216 CrossRef CAS PubMed.
- H. A. Shaikh, F. D. Sönnichsen and T. K. Lindhorst, Carbohydr. Res., 2008, 343, 1665 CrossRef CAS PubMed.
- (a) M. Wathier, A. Polidori, K. Ruiz, A.-S. Fabiano and B. Pucci, New J. Chem., 2001, 25, 1588 RSC; (b) M. Abla, G. Durand and B. Pucci, J. Org. Chem., 2011, 76, 2084 CrossRef CAS PubMed.
- M. M. K. Boysen, K. Elsner, O. Sperling and T. K. Lindhorst, Eur. J. Org. Chem., 2003, 4376 CrossRef CAS.
- T. K. Lindhorst and K. Elsner, Beilstein J. Org. Chem., 2014, 10, 1482 CrossRef PubMed.
- (a) P. Langer, S. J. Ince and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1998, 3913 RSC; (b) T. K. Lindhorst, M. Dubber, U. Krallmann-Wenzel and S. Ehlers, Eur. J. Org. Chem., 2000, 2027 CrossRef CAS.
- (a) M. Gómez-García, J. M. Benito, D. Rodríguez-Lucena, J.-X. Yu, K. Chmurski, C. Ortiz Mellet, R. Gutiérrez Gallego, A. Maestre, J. Defaye and J. M. García Fernández, J. Am. Chem. Soc., 2005, 127, 7970 CrossRef PubMed; (b) M. Gómez-García, J. M. Benito, R. Gutiérrez-Gallego, A. Maestre, C. Ortiz Mellet, J. M. García Fernández and J. L. Jimenez-Blanco, Org. Biomol. Chem., 2010, 8, 1849 RSC; (c) M. Gómez-García, J. M. Benito, A. P. Butera, C. Ortiz Mellet, J. M. García Fernández and J. L. Jimenez-Blanco, J. Org. Chem., 2012, 77, 1273 CrossRef PubMed.
- (a) J. Katajisto, T. Karskela, P. Heinonen and H. Lönnberg, J. Org. Chem., 2002, 67, 7995 CrossRef CAS PubMed; (b) P. Virta, M. Leppänen and H. Lönnberg, J. Org. Chem., 2004, 69, 2008 CrossRef CAS PubMed; (c) J. Katajisto, P. Heinonen and H. Lönnberg, J. Org. Chem., 2004, 69, 7609 CrossRef CAS PubMed; (d) M. Karskela, M. von Usedom, P. Virta and H. Lönnberg, Eur. J. Org. Chem., 2012, 6594 CAS.
- (a) J. R. Allen, C. R. Harris and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 1890 CrossRef CAS PubMed; (b) G. Ragupathi, D. M. Coltart, L. J. Williams, F. Koide, E. Kagan, J. Allen, C. Harris, P. W. Glunz, P. O. Livingston and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13699 CrossRef CAS PubMed; (c) S. J. Keding and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11937 CrossRef CAS PubMed.
- (a) V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2000, 39, 4348 CrossRef CAS; (b) V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 900 CrossRef CAS PubMed; (c) D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 555 CrossRef PubMed.
- (a) O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243 CrossRef CAS PubMed; (b) B. Thomas, M. Fiore, I. Bossu, P. Dumy and O. Renaudet, Beilstein J. Org. Chem., 2012, 8, 421 CrossRef CAS PubMed; (c) B. Thomas, M. Fiore, G. C. Daskhan, N. Spinelli and O. Renaudet, Chem. Commun., 2015, 51, 5436 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tørnoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 74, 3057 CrossRef.
- D. V. Filippov, H. van den Helst, C. M. Tromp, G. A. van der Marel, C. A. A. van Boeckel, H. S. Overkleeft and J. H. van Boom, Synlett, 2004, 773 CAS.
- M. Dubber and J. M. J. Fréchet, Bioconjugate Chem., 2003, 14, 239 CrossRef CAS PubMed.
- (a) G. Pourceau, A. Meyer, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 1218 CrossRef CAS PubMed; (b) A. Meyer, G. Pourceau, J.-J. Vasseur and F. Morvan, J. Org. Chem., 2010, 75, 6689 CrossRef CAS PubMed; (c) Y. Chevolot, J. Zhang, A. Meyer, A. Goudot, S. Rouanet, S. Vidal, G. Pourceau, J.-P. Cloarec, J.-P. Praly, E. Souteyrand, J.-J. Vasseur and F. Morvan, Chem. Commun., 2011, 47, 8826 RSC; (d) A. Meyer, M. Noël, J.-J. Vasseur and F. Morvan, Eur. J. Org. Chem., 2015, 2921 CrossRef CAS.
- (a) D. J. Lee, S.-H. Yang, G. M. Williams and M. A. Brimble, J. Org. Chem., 2012, 77, 7564 CrossRef CAS PubMed; (b) M. L. Gening, D. V. Titov, S. Cecioni, A. Audfray, A. G. Gerbst, Y. E. Tsvetkov, V. B. Krylov, A. Imberty, N. E. Nifantiev and S. Vidal, Chem. – Eur. J., 2013, 19, 9272 CrossRef CAS PubMed.
- C. Kieburg, M. Dubber and T. K. Lindhorst, Synlett, 1997, 1447 CAS.
- M. Dubber, O. Sperling and T. K. Lindhorst, Org. Biomol. Chem., 2006, 4, 3901 CAS.
- M. Dubber and T. K. Lindhorst, Carbohydr. Res., 1998, 310, 35 CrossRef CAS.
- D. Solomon, P. I. Kitov, E. Paszkiewicz, G. A. Grant, J. M. Sadowska and D. R. Bundle, Org. Lett., 2005, 7, 4369 CrossRef CAS PubMed.
- M. M. K. Boysen and T. K. Lindhorst, Org. Lett., 1999, 1, 1925 CrossRef CAS.
- (a) M. Dubber and T. K. Lindhorst, J. Org. Chem., 2000, 65, 5275 CrossRef CAS PubMed; (b) M. Dubber and T. K. Lindhorst, Org. Lett., 2001, 3, 4019 CrossRef CAS PubMed.
- M. Köhn, J. M. Benito, C. Ortiz Mellet, T. K. Lindhorst and J. M. García Fernández, ChemBioChem, 2004, 5, 771 CrossRef PubMed.
- R. P. McGeary, I. Jablonkai and I. Toth, Tetrahedron, 2001, 57, 8733 CrossRef CAS.
- J. Brask and K. J. Jensen, J. Pept. Sci., 2000, 6, 290 CrossRef CAS PubMed.
- T.-E. Gloe, A. Müller, A. Ciuk, T. M. Wrodnigg and T. K. Lindhorst, Carbohydr. Res., 2016, 425, 1–9 CrossRef CAS PubMed.
- (a) M. Ortega-Muñoz, J. Morales-Sanfrutos, F. Perez-Balderas, F. Hernandez-Mateo, M. D. Giron-Gonzales, N. Sevillano-Tripero, R. Salto-Gonzales and F. Santoyo-Gonzales, Org. Biomol. Chem., 2007, 5, 2291 RSC; (b) F. Perez-Balderas, J. Morales-Sanfrutos, F. Hernandez-Mateo, J. Isac-García and F. Santoyo-Gonzales, Eur. J. Org. Chem., 2009, 2441 CrossRef CAS; (c) M. Ortega-Muñoz, F. Perez-Balderas, J. Morales-Sanfrutos, F. Hernandez-Mateo, J. Isac-García and F. Santoyo-Gonzales, Eur. J. Org. Chem., 2009, 2454 CrossRef; (d) A. A. Kulkarni, A. A. Weiss and S. S. Iyer, Anal. Chem., 2010, 82, 7430 CrossRef CAS PubMed; (e) V. Fagan, I. Toth and P. Simerska, Beilstein J. Org. Chem., 2014, 10, 1741 CrossRef PubMed.
- (a) B. Gerland, A. Goudot, G. Pourceau, A. Meyer, S. Vidal, E. Souteyrand, J.-J. Vasseur, Y. Chevolot and F. Morvan, J. Org. Chem., 2012, 77, 7620 CrossRef CAS PubMed; (b) B. Gerland, A. Goudot, G. Pourceau, A. Meyer, V. Dugas, S. Cecioni, S. Vidal, E. Souteyrand, J.-J. Vasseur, Y. Chevolot and F. Morvan, Bioconjugate Chem., 2012, 23, 1534 CrossRef CAS PubMed.
- O. Sperling, M. Dubber and T. K. Lindhorst, Carbohydr. Res., 2007, 342, 696 CrossRef CAS PubMed.
- N. Kottary, Y. M. Chabre, T. C. Shiao, R. Rej and R. Roy, Chem. Commun., 2014, 50, 1983 RSC.
- (a) C. Kieburg, K. Sadalapure and T. K. Lindhorst, Eur. J. Org. Chem., 2000, 2035 CrossRef CAS; (b) K. Sadalapure and T. K. Lindhorst, Angew. Chem., Int. Ed., 2000, 39, 2010 CrossRef CAS; (c) A. Patel and T. K. Lindhorst, J. Org. Chem., 2001, 66, 2674 CrossRef CAS PubMed; (d) A. Patel and T. K. Lindhorst, Eur. J. Org. Chem., 2002, 79 CrossRef CAS; (e) M. Dubber, A. Patel, K. Sadalapure, I. Aumüller and T. K. Lindhorst, Eur. J. Org. Chem., 2006, 5357 CrossRef CAS; (f) N. Röckendorf and T. K. Lindhorst, J. Org. Chem., 2004, 69, 4441 CrossRef PubMed.
- S. Maisonneuve, R. Métivier, P. Yu, K. Nakatani and J. Xie, Beilstein J. Org. Chem., 2014, 10, 1471 CrossRef PubMed.
- (a) T. Ogawa and M. Matsui, Carbohydr. Res., 1977, 56, C1 CrossRef CAS; (b) T. Ogawa and M. Matsui, Carbohydr. Res., 1978, 62, C1 CrossRef CAS; (c) S. Hanessian, Preparative Carbohydrate Chemistry, Marcel Dekker, New York, 1997, pp. 73–74 Search PubMed.
- C. D. Heidecke and T. K. Lindhorst, Chem. – Eur. J., 2007, 13, 9056 CrossRef CAS PubMed.
- M. L. Lepage, A. Bodlenner and P. Compain, Eur. J. Org. Chem., 2013, 1963 CrossRef CAS.
- (a) A. M. Wu, T. Singh, J.-H. Liu, M. Krzeminski, R. Russwurm, H.-C. Siebert, A. M. J. J. Bonvin, S. André and H.-J. Gabius, Glycobiology, 2007, 17, 165 CrossRef CAS PubMed; (b) P. Liu, L. Chen, J. K. C. Toh, Y. L. Ang, J.-E. Jee, J. Lim, S. S. Lee and S.-G. Lee, Chem. Sci., 2015, 6, 450 RSC.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669 CrossRef CAS PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008 RSC.
- C. Ligeour, L. Dupin, A. Angeli, G. Vergoten, S. Vidal, A. Meyer, E. Souteyrand, J.-J. Vasseur, Y. Chevolot and F. Morvan, Org. Biomol. Chem., 2015, 13, 11244 CAS.
- (a) E. A. Merritt and W. G. Hol, Curr. Opin. Struct. Biol., 1995, 5, 165 CrossRef CAS PubMed; (b) D. Vanden Broeck, C. Horvath and M. J. S. DeWolf, Int. J. Biochem. Cell Biol., 2007, 39, 1771 CrossRef CAS PubMed.
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323 CrossRef CAS PubMed.
- A. V. Pukin, H. M. Branderhorst, C. W. Sisu, C. A. G. M. Weijers, M. Gilbert, R. M. J. Liskamp, G. M. Visser, H. Zuilhof and R. J. Pieters, ChemBioChem, 2007, 8, 1500 CrossRef CAS PubMed.
- A. V. Pukin, H. C. Q. van Ufford, T. R. Branson, D. M. E. Thies-Weesie, W. B. Turnbull, G. M. Visser and R. J. Pieters, ChemistryOpen, 2015, 4, 471 CrossRef PubMed.
- S. Kötter, U. Krallmann-Wenzel, S. Ehlers and T. K. Lindhorst, J. Chem. Soc., Perkin Trans. 1, 1998, 2193 RSC.
- A. Patel and T. K. Lindhorst, Carbohydr. Res., 2006, 341, 1657 CrossRef CAS PubMed.
- S. Vanwetswinkel, A. N. Volkov, Y. G. J. Sterckx, A. Garcia-Pino, L. Buts, W. F. Vranken, J. Bouckaert, R. Roy, L. Wyns and A. J. van Nuland, J. Med. Chem., 2014, 57, 1416 CrossRef CAS PubMed.
- T. K. Lindhorst, S. Kötter, U. Krallmann-Wenzel and S. Ehlers, J. Chem. Soc., Perkin Trans. 1, 2001, 823 RSC.
- (a) T. K. Dam and C. F. Brewer, Biochemistry, 2008, 47, 8470 CrossRef CAS PubMed; (b) T. K. Dam, T. A. Gerken and C. F. Brewer, Biochemistry, 2009, 48, 3822 CrossRef CAS PubMed; (c) B. Turnbull, Nat. Chem., 2011, 3, 267 CrossRef PubMed.
- C.-W. von der Lieth, M. Frank and T. K. Lindhorst, Rev. Mol. Biotechnol., 2002, 90, 311 CrossRef CAS PubMed.
- S. Andre, T. Velasco-Torrijos, R. Leyden, S. Gouin, M. Tosin, P. V. Murphy and H.-J. Gabius, Org. Biomol. Chem., 2009, 7, 4715 CAS.
- L. Moni, G. Pourceau, J. Zhang, A. Meyer, S. Vidal, E. Souteyrand, A. Dondoni, F. Morvan, Y. Chevolot, J.-J. Vasseur and A. Marra, ChemBioChem, 2009, 10, 1369 CrossRef CAS PubMed.
- D. Grünstein, M. Maglinao, R. Kikkeri, M. Collot, K. Barylyuk, B. Lepenies, F. Kamena, R. Zenobi and P. H. Seeberger, J. Am. Chem. Soc., 2011, 133, 13957 CrossRef PubMed.
- A. Novoa and N. Winssinger, Beilstein J. Org. Chem., 2015, 11, 707 CrossRef CAS PubMed.
- H. S. G. Beckmann, H. M. Möller and V. Wittmann, Beilstein J. Org. Chem., 2012, 8, 819 CrossRef CAS PubMed.
- A. Schierholt, M. Hartmann and T. K. Lindhorst, Carbohydr. Res., 2011, 346, 1519 CrossRef CAS PubMed.
- (a) D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845 CrossRef CAS PubMed; (b) F. Wojcik, A. G. O'Brien, S. Götze, P. H. Seeberger and L. Hartmann, Chem. – Eur. J., 2013, 19, 3090 CrossRef CAS PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008 CrossRef CAS PubMed.
- Q. Li, T.-T. Yan, S. Niu, Y.-T. Zhao, X.-B. Meng, Z.-H. Zhao and Z.-J. Li, Carbohydr. Res., 2013, 379, 78 CrossRef CAS PubMed.
- (a) Q. Li, M. S. Cai and Z. J. Li, Chem. J. Chin. Univ., 2000, 21, 70 CAS; (b) X. B. Meng, L. D. Yang, H. Li, Q. Li, T. M. Cheng, M. S. Cai and Z. J. Li, Carbohydr. Res., 2002, 337, 977 CrossRef CAS PubMed; (c) W. Zhang, Q. Li, X. B. Meng, Z. H. Zhao and Z. J. Li, J. Chin. Pharm. Sci., 2007, 16, 128 CAS.
- S. P. Vincent, K. Buffet, I. Nierengarten, A. Imberty and J.-F. Nierengarten, Chem. – Eur. J., 2016, 22, 88 CrossRef CAS PubMed.
- N. Strömberg, P.-G. Nyholm, I. Pascher and S. Normark, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9340 CrossRef.
- N. Horan, L. Yan, H. Isobe, G. M. Whitesides and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11782 CrossRef CAS.
- (a) J. Voskuhl, M. C. A. Stuart and B. J. Ravoo, Chem. – Eur. J., 2010, 16, 2790 CrossRef CAS PubMed; (b) R. V. Vico, J. Voskuhl and B. J. Ravoo, Langmuir, 2011, 27, 1391 CrossRef CAS PubMed.
- J. W. Wehner, M. Hartmann and T. K. Lindhorst, Carbohydr. Res., 2013, 371, 22 CrossRef CAS PubMed.
- (a) G. T. Noble, S. L. Flitsch, K. P. Liem and S. J. Webb, Org. Biomol. Chem., 2009, 7, 5245 RSC; (b) G. T. Noble, F. L. Craven, J. Volgmeir, R. Sardzik, S. L. Flitsch and S. J. Webb, J. Am. Chem. Soc., 2012, 134, 13010 CrossRef CAS PubMed.
- I. Willner and S. Rubin, J. Am. Chem. Soc., 1992, 114, 3150 CrossRef CAS.
- F. Hamon, F. Djedaini-Pilard, F. Barbot and C. Len, Tetrahedron, 2009, 65, 10105 CrossRef CAS.
- (a) O. Srinivas, N. Mitra, A. Suriola and N. Jayaraman, J. Am. Chem. Soc., 2002, 124, 2124 CrossRef CAS PubMed; (b) O. Srinivas, N. Mitra, A. Suriola and N. Jayaraman, Glycobiology, 2005, 15, 861 CrossRef CAS PubMed.
- (a) V. Chandrasekaran and T. K. Lindhorst, Chem. Commun., 2012, 48, 7519 RSC; (b) V. Chandrasekaran, K. Kolbe, F. Beiroth and T. K. Lindhorst, Beilstein J. Org. Chem., 2013, 9, 223 CrossRef CAS PubMed; (c) V. Chandrasekaran, E. Johannes, H. Kobarg, F. D. Sönnichsen and T. K. Lindhorst, ChemistryOpen, 2014, 3, 99 CrossRef CAS PubMed; (d) V. Chandrasekaran, H. Jacob, F. Petersen, K. Kathirvel, F. Tuczek and T. K. Lindhorst, Chem. – Eur. J., 2014, 20, 8744 CrossRef CAS PubMed; (e) T. Weber, V. Chandrasekaran, I. Stamer, M. B. Thygesen, A. Terfort and T. K. Lindhorst, Angew. Chem., Int. Ed., 2014, 53, 14583 CrossRef CAS PubMed.
- L. Möckl, A. Müller, C. Bräuchle and T. K. Lindhorst, Chem. Commun., 2016, 52, 1254 RSC.
- A. Samanta, M. C. A. Stuart and B. J. Ravoo, J. Am. Chem. Soc., 2012, 134, 19909 CrossRef CAS PubMed.
- N. Smiljanic, V. Moreau, D. Yockot, J. M. Benito, J. M. García Fernández and F. Djedaïni-Pilard, Angew. Chem., Int. Ed., 2006, 45, 5465 CrossRef CAS PubMed.
- K. Schwekendiek, H. Kobarg, L. Daumlechner, F. D. Sönnichsen and T. K. Lindhorst, Chem. Commun., 2011, 47, 9399 RSC.
- A. K. Ciuk and T. K. Lindhorst, Beilstein J. Org. Chem., 2015, 11, 668 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |