
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
(Hetero)aromatics from dienynes, enediynes and enyne–allenes
Carlotta
Raviola
,
Stefano
Protti
,
Davide
Ravelli
 and
Maurizio
Fagnoni
*
and
Maurizio
Fagnoni
*
PhotoGreen Lab, Department of Chemistry, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: fagnoni@unipv.it; Fax: +39 0382 987323; Tel: +39 0382 987198
First published on 6th June 2016
Abstract
The construction of aromatic rings has become a key objective for organic chemists. While several strategies have been developed for the functionalization of pre-formed aromatic rings, the direct construction of an aromatic core starting from polyunsaturated systems is yet a less explored field. The potential of such reactions in the formation of aromatics increased at a regular pace in the last few years. Nowadays, there are reliable and well-established procedures to prepare polyenic derivatives, such as dienynes, enediynes, enyne–allenes and hetero-analogues. This has stimulated their use in the development of innovative cycloaromatizations. Different examples have recently emerged, suggesting large potential of this strategy in the preparation of (hetero)aromatics. Accordingly, this review highlights the recent advancements in this field and describes the different conditions exploited to trigger the process, including thermal and photochemical activation, as well as the use of transition metal catalysis and the addition of electrophiles/nucleophiles or radical species.
 Left to right: Davide Ravelli, Maurizio Fagnoni, Stefano Protti and Carlotta Raviola | Carlotta Raviola received her PhD from the University of Pavia in 2015 (supervisor: Prof. A. Albini) and spent part of this period at TUM (Germany) in the group of Prof. T. Bach. She is currently a post-doc at the University of Pavia and her research interests focus on the photogeneration of aryl cations. |
Stefano Protti completed his PhD (supervisor: Prof. M. Fagnoni) from the University of Pavia in 2006. He was a post-doctoral fellow at the LASIR laboratory (Lille, France), at the iBitTec-S Laboratory (CEA Saclay). He is currently a researcher at the University of Pavia. His research work is focused on photochemical arylations under metal-free conditions. |
Davide Ravelli obtained his PhD in 2012 from the University of Pavia (Prof. A. Albini as the supervisor). Since November 2015, he is a fixed term researcher at the same University and his main research interests focus on the generation of radical intermediates through photocatalyzed Hydrogen Atom Transfer reactions. |
Maurizio Fagnoni is currently an Associate Professor of the Department of Chemistry at the University of Pavia. His research interests are mainly focused on the photoinduced or photocatalytic generation of reactive intermediates such as (bi)radicals, (phenyl) cations and radical ions and their application in eco-sustainable synthesis. |
1 Introduction
Benzenes, polycyclic aromatic hydrocarbons (PAHs) and heteroaromatics are key classes of compounds with diverse roles in many branches of chemistry, due to their use in the preparation of electroactive organic materials and molecular devices, as well as building blocks in organic synthesis and medicinal chemistry.1–4 Accordingly, the construction and functionalization of aromatic rings have become key objectives for organic chemists. However, whereas a plethora of strategies has been developed for the functionalization of pre-formed rings, the available strategies to build an aromatic core starting from (poly)unsaturated systems is yet a quite unexplored field.5–7 The most used strategies include: (1) the ring-closing metathesis (RCM) of non-conjugated trienes (Scheme 1, path a)8 and enynes;9–12 (2) intra- or intermolecular [3+3] benzannulation reactions;13 and (3) cycloaddition processes involving o-quinodimethanes.14,15 In such cases, a multi-step process is involved and the aromatization occurs after the construction of one or more σ bonds. One-pot strategies for the preparation of benzocondensed derivatives have been likewise reported, such as the photoinduced oxidative cyclization of stilbenes16 introduced by Mallory in 1964 (path b) and the electrophilic cyclization of suitable arene-containing propargyl alcohols and 1-aryl-3-alkyn-2-ones.17–19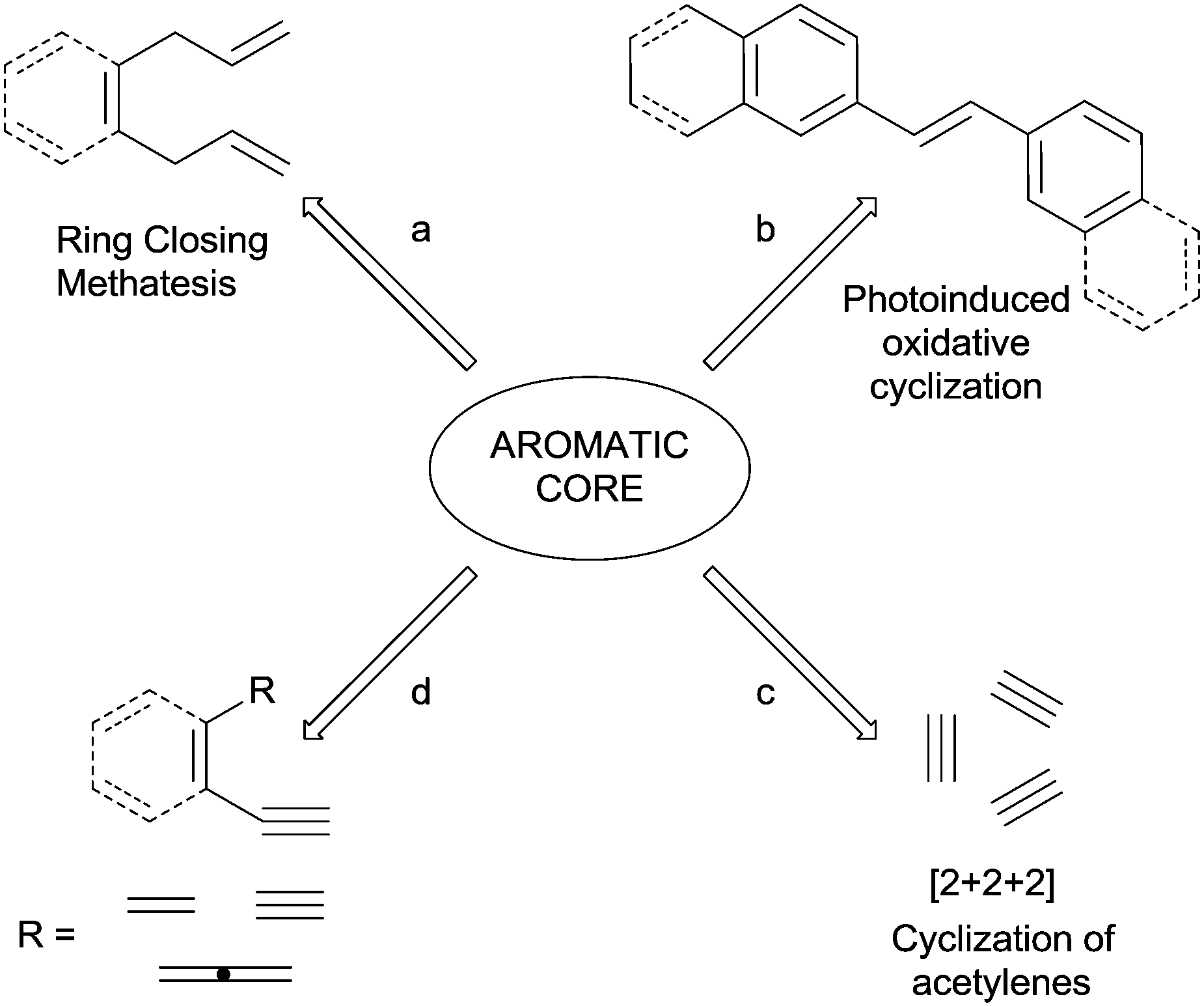 | ||
| Scheme 1 Selected strategies for the construction of aromatic rings. | ||
However, only a few strategies where both the cyclization and the aromatization processes take place in a single step have been reported. This is the case of transition-metal mediated [2+2+2] cycloaddition reactions of triple bonds (Scheme 1, path c).20–25 These are atom-efficient processes and the high tolerance to several functional groups (such as alcohols, amines and unsaturated moieties), as well as the high regio- and chemoselectivities,5 makes them an almost “universal synthetic tool for the synthesis of benzene, pyridine, and other cyclic derivatives”.20
An appealing alternative is the construction of (hetero)aromatic rings from polyenes (dienynes, enediynes, enyne–allenes or their hetero-analogues; path d). The main advantage of this one-pot approach is that several conditions (involving different intermediates, mostly biradicals) can be adopted to trigger the cyclization event, as summarized in Scheme 2.26
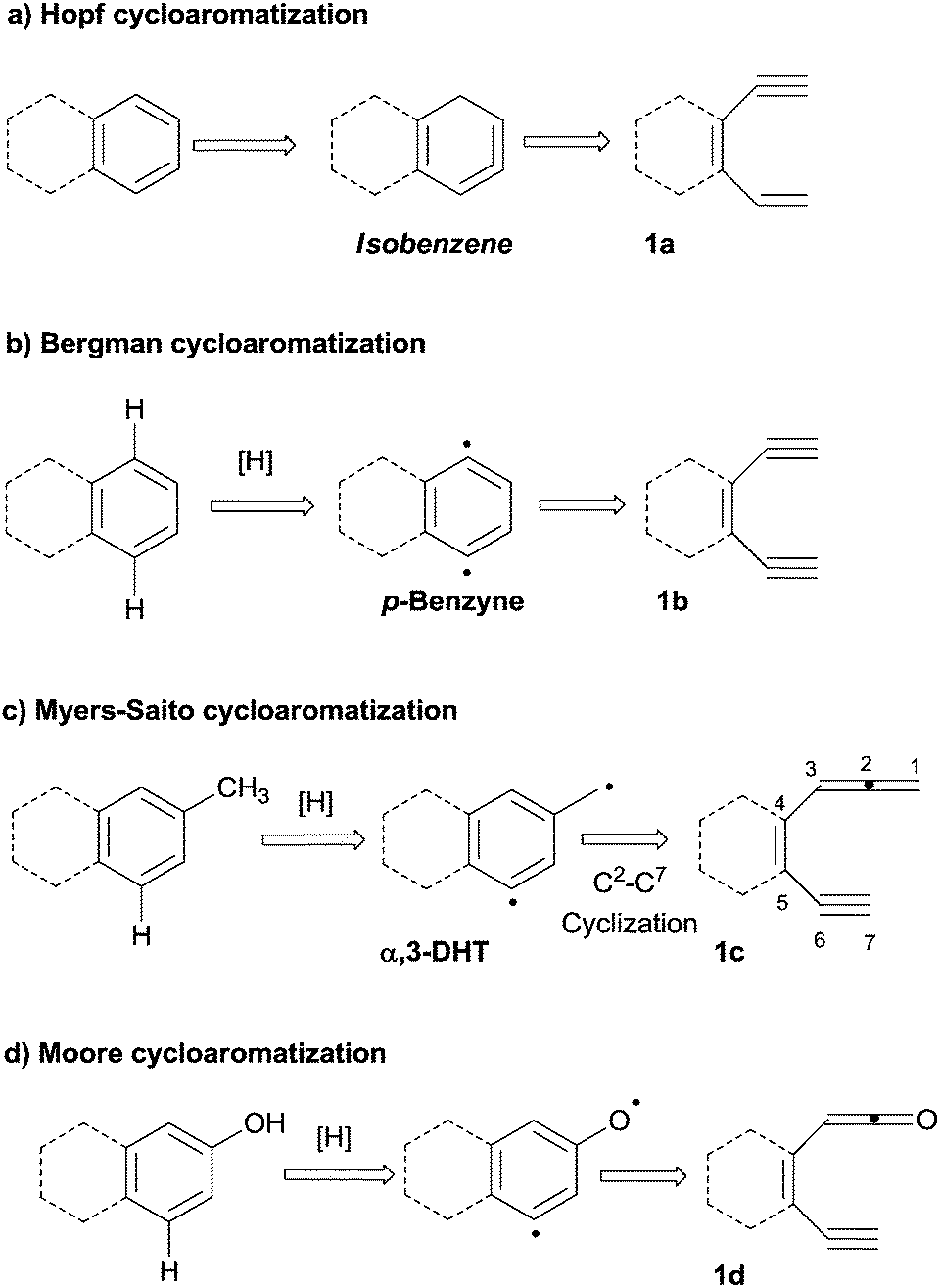 | ||
| Scheme 2 Main approaches for the cycloaromatization of polyenes. | ||
In the so-called Hopf cyclization, the electrocyclization of hexa-1,3-dien-5-ynes (e.g.1a) gives arenes via a hydrogen shift in cyclohexa-1,2,4-trienes (also known as isobenzenes, Scheme 2a).27 However, the first systematic investigation on polyenes as aromatic precursors was authored by Bergman, who described the thermal (200 °C) cyclization of (Z)-3-hexene-1,5-diynes (e.g.1b) to give 1,4-didehydrobenzene biradicals (p-benzynes, Scheme 2b). In the latter case, two radical centers and a new σ bond are formed at the expense of two π-orbitals in a process driven by the aromatic stabilization acquired in the intermediate.28 A related cycloaromatization was later reported by the groups of Myers and Saito, who described the cycloaromatization of enyne–allenes (e.g.1c) via a C2–C7 mode to generate the corresponding α,3-didehydrotoluenes (α,3-DHTs, Scheme 2c).29–31 Heteroatom-substituted enyne–allene analogues undergo similar cyclizations, as in the case of the aza-Myers reaction32 or in the Moore cyclization of enyne–ketenes (1d, Scheme 2d).33 Similar processes take place in enyne–isocyanates,34 enyne–ketenimines35 and enyne–carbodiimides.36 The Schmittel reaction (occurring in enyne–allenes via a C2–C6 mode) gives a non-aromatic biradical,37–40 but several examples describe a subsequent intramolecular reaction involving a phenyl-substituent, likewise leading to benzocondensed aromatics.
20 years ago Wang excellently resumed the synthetic potentialities of cascade radical cyclizations via biradicals (Bergman, Moore and Myers–Saito reactions) for the preparation of complex structures.41 Some synthetic applications have also been reported by Grissom et al. in their review focused on the reactivity of enediynes and analogues, published in the same year.26
While several reviews devoted to alkyne trimerization have been published over the last few decades,20–25 the number of reports focused on the cyclization of conjugated polyenes is still limited. In fact, apart from the synthetic significance of these processes, the biradicals formed in these reactions have been claimed as the key intermediates to explain the chemotherapeutic activity of natural antibiotics containing polyenic moieties (e.g. enediynes).42,43 Accordingly, much attention has been paid to elucidate the cyclization mechanism,44–46 to predict the reactivity of the intermediates,47–54 and to the synthesis of artificial polyene precursors with potential biological activity.42,43 The synthesis of polynuclear aromatics from dienynes has been sparsely reviewed,55,56 but the main focus has been on mechanistic aspects of such cycloaromatizations and on the different approaches to accelerate the reactions.
On the basis of the above, the aim of the present review is to provide an overview of the most recent advances in the construction of aromatic and heteroaromatic rings starting from (in situ generated) conjugated polyenes, such as dienynes, enediynes and enyne–allenes. This review collects representative results (mainly) published since 1996 and has been organized according to the nature and the size of the (hetero)aromatics formed, preceded by a summary of the typical modes used to induce the cycloaromatization event.
2 Mode of activation of cycloaromatization
The great advantage of exploiting the cyclization of polyenes for synthetic purposes is that the cycloaromatization can be induced by heating, light irradiation, transition-metal catalysis or via the addition of electrophiles, nucleophiles or radicals, as summarized below.2.1 Thermal activation
High temperatures (typically above 200 °C) are commonly required for the Hopf cyclization of acyclic hexa-1,3-dien-5-ynes.27 Cycloaromatization of 11- and 10-membered carbocycles bearing a hexa-1,3-dien-5-yne moiety, however, took place at 100 °C and room temperature, respectively.57 By contrast, for larger rings (up to 14-membered rings), the cyclization was inefficient or completely inhibited.57The Bergman cycloaromatization is traditionally performed by heating the polyene moiety in the presence of an excess of either a hydrogen (1,4-cyclohexadiene – CHD – or methanol) or halogen (CCl4) atom donor, depending on the desired product. The reactivity of (Z)-enediynes is influenced by different factors including, among others: (1) the distance between the two terminal acetylenic carbon atoms (generally speaking, the smaller the distance, the faster the cyclization; a value of 3.2–3.3 Å has been suggested as the limit to allow a cyclization at room temperature);58 (2) the difference in strain energy between the enediyne and the transition state (the higher the strain relief in the transition state, the more reactive the enediyne);59 and (3) substituent effects (the presence of substituents on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond heavily affects the cyclization efficiency).60 Accordingly, acyclic enediynes usually undergo cycloaromatization at a high temperature (150–200 °C), whereas milder conditions are required for cyclic analogues.61 The most significant drawback of the thermal cycloaromatization of enediynes (mainly acyclic) and related derivatives is thus the high temperature required that is often incompatible with organic substrates.
C double bond heavily affects the cyclization efficiency).60 Accordingly, acyclic enediynes usually undergo cycloaromatization at a high temperature (150–200 °C), whereas milder conditions are required for cyclic analogues.61 The most significant drawback of the thermal cycloaromatization of enediynes (mainly acyclic) and related derivatives is thus the high temperature required that is often incompatible with organic substrates.
Indeed, a shortening in the distance between the alkyne units in acyclic enediynes bearing chelating moieties (e.g. bisphosphane groups) can be induced by complexation with a metal ion, thus increasing the cycloaromatization rate.62,63
Interestingly, while the cyclization of (Z)-3-hexene-1,5-diyne was found to be endothermic (+13 kcal mol−1),64 the Myers–Saito cycloaromatization of the parent acyclic enyne–allene was found to be exothermic (−15 kcal mol−1),65 thus taking place at ambient or even sub-ambient temperature. In many cases, since enyne–allenes can be troublesome to handle due to their instability, they were generated in situ via the rearrangement of the corresponding enediynes.
The Myers–Saito reaction is generally the most energetically favorable process occurring in enyne–allenes. In substrates where the alkyne terminal hydrogen is replaced by a bulky moiety (such as the tert-butyl or trimethylsilyl groups), however, steric hindrance makes the enyne–allene more stable, thus favoring the Schmittel closure rather than the cycloaromatization.39
As for the Moore reaction, the intrinsic rate of cyclization could not be determined due to the high reactivity of enyne–ketenes, and the substrates are usually generated in situ by heating the corresponding 4-alkynylcyclobutenones (see for instance compound 2 in Scheme 3).66
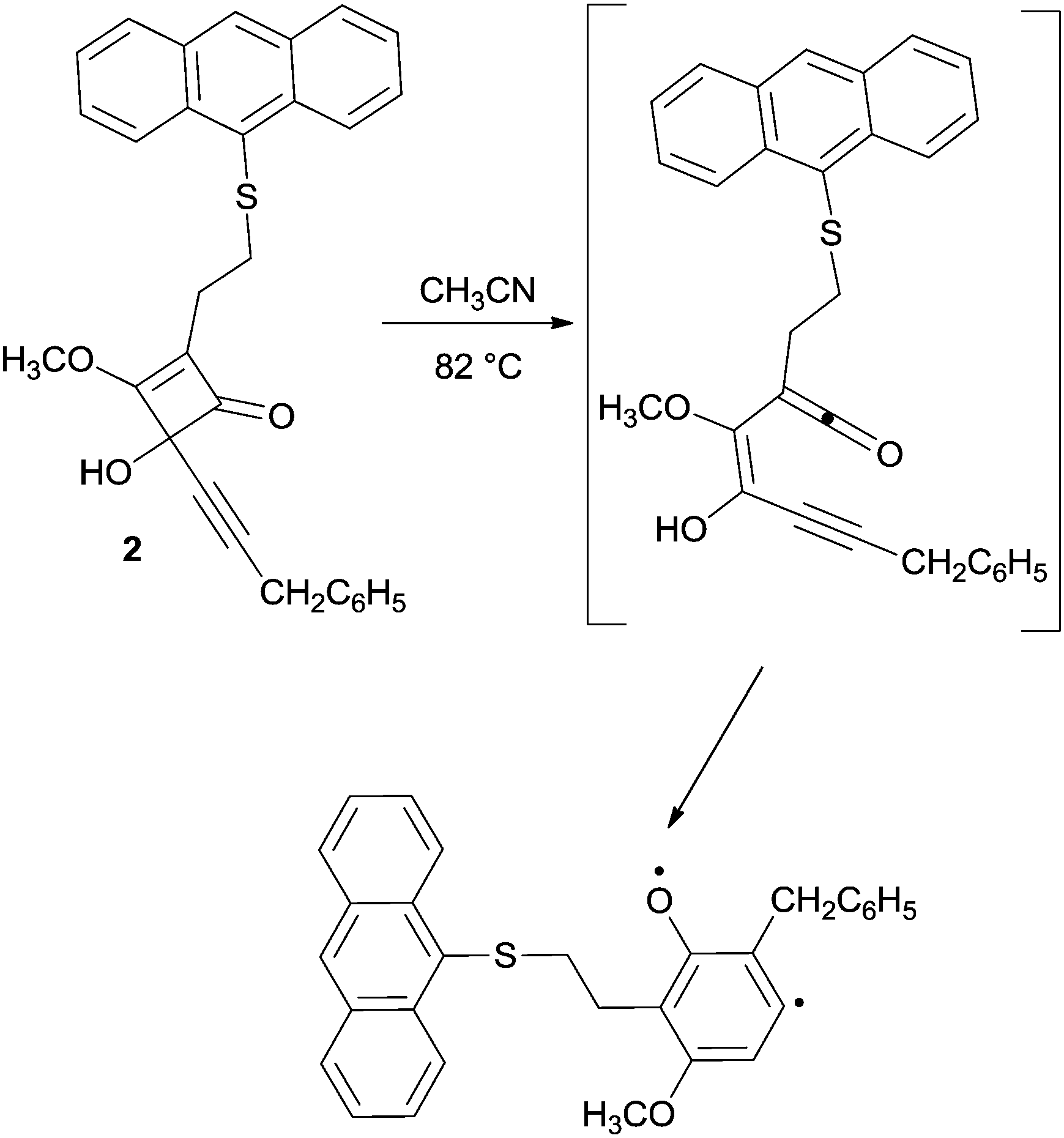 | ||
| Scheme 3 Moore cyclization via a thermally generated enyne–ketene. | ||
2.2 Photochemical activation
The quantitative conversion of cis-1,3-hexadiene-5-yne vapors in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of benzene and fulvene is the first example of photoinduced Hopf cycloaromatization. The process, however, occurred with a low quantum yield (Φ ∼ 0.01).67 By contrast, the photocyclization took place efficiently when dienynes were irradiated in aprotic solvents in the presence of air.56 The substitution pattern of the terminal alkene (e.g. the presence of a methyl group in position 2) was found to play a key role in allowing dienynes 3a,b to exist in the less stable syn conformation, the actual photoreactive conformer precursor of naphthalenes 4a,b (Scheme 4).68
:1 mixture of benzene and fulvene is the first example of photoinduced Hopf cycloaromatization. The process, however, occurred with a low quantum yield (Φ ∼ 0.01).67 By contrast, the photocyclization took place efficiently when dienynes were irradiated in aprotic solvents in the presence of air.56 The substitution pattern of the terminal alkene (e.g. the presence of a methyl group in position 2) was found to play a key role in allowing dienynes 3a,b to exist in the less stable syn conformation, the actual photoreactive conformer precursor of naphthalenes 4a,b (Scheme 4).68
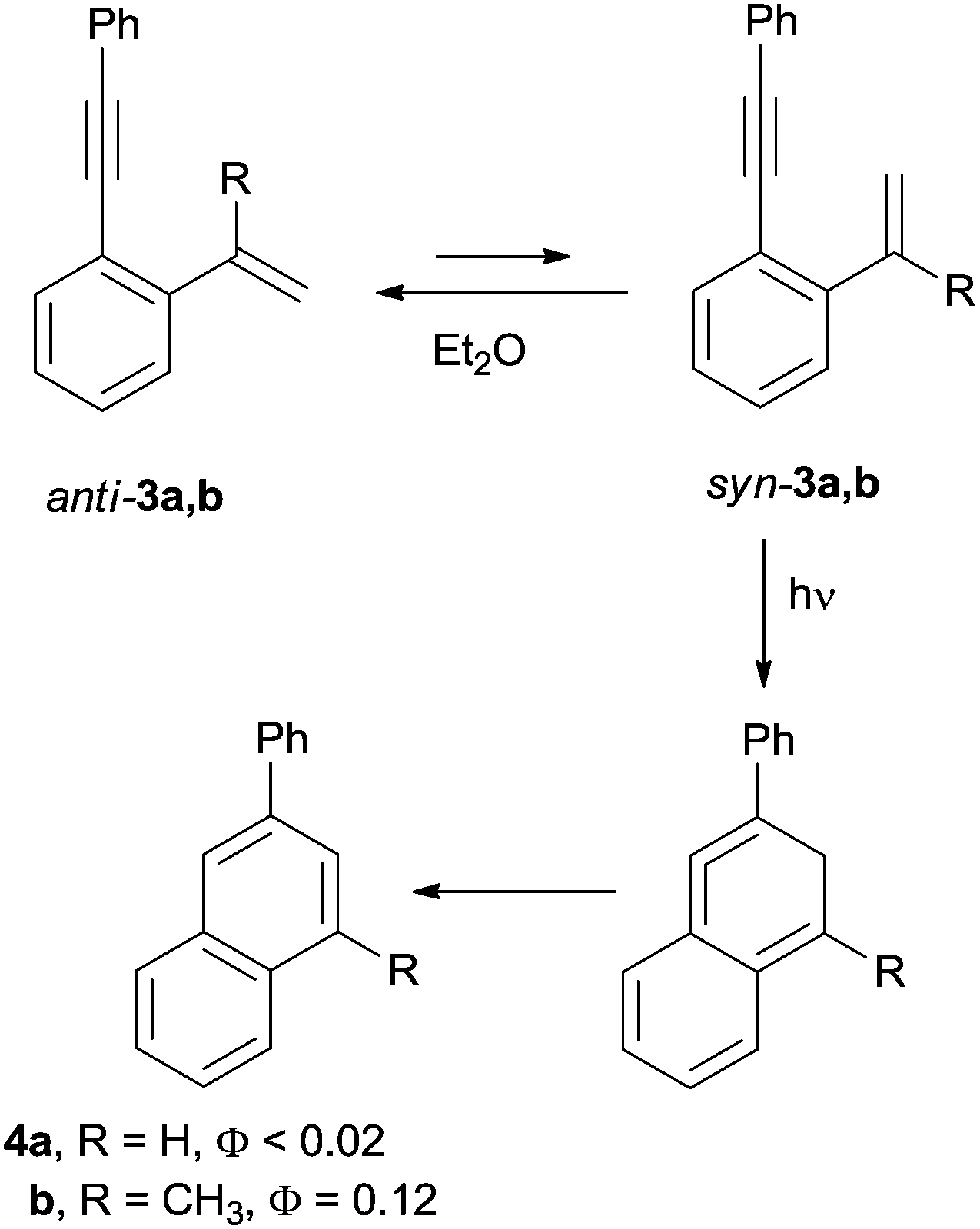 | ||
| Scheme 4 Photoinduced Hopf cycloaromatization. | ||
In a related example, a photo-Hopf cyclization was used for the construction of the naphthalene core upon photocyclization/aromatizing fragmentation of benzannelated dienynes.69
The chance of performing Bergman cyclizations by having recourse to a photochemical approach has been sparsely investigated,70–73 with particular attention to the biological application of photoactivatable enediynes.49,74 The approach was particularly efficient with strained ortho-dialkynylarenes (e.g.5, Scheme 5) that underwent cycloaromatization to 6 in quantitative yield upon 3 h exposure to sunlight.71
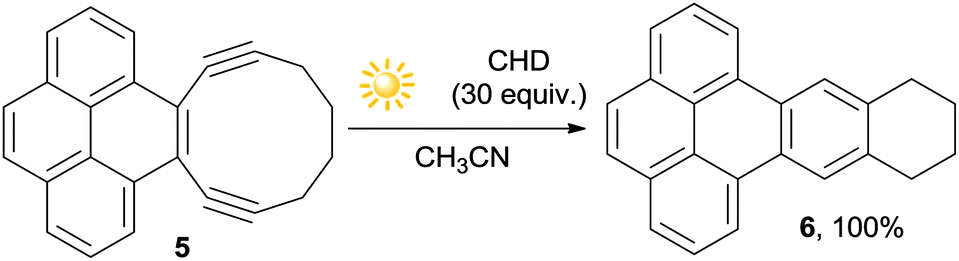 | ||
| Scheme 5 Sunlight-induced Bergman cyclization of strained ortho-dialkynylarenes. | ||
Recently, photochemical variants of the Myers–Saito and the Schmittel cyclizations have also been developed.75,76 As an example, photolysis at 300 nm in toluene of enyne–allene 7 in the presence of CHD (100 equiv.) led to a mixture of 8 (12% yield) and 9 (24% yield) arising from the Schmittel and the Myers–Saito cyclizations, respectively (Scheme 6).75
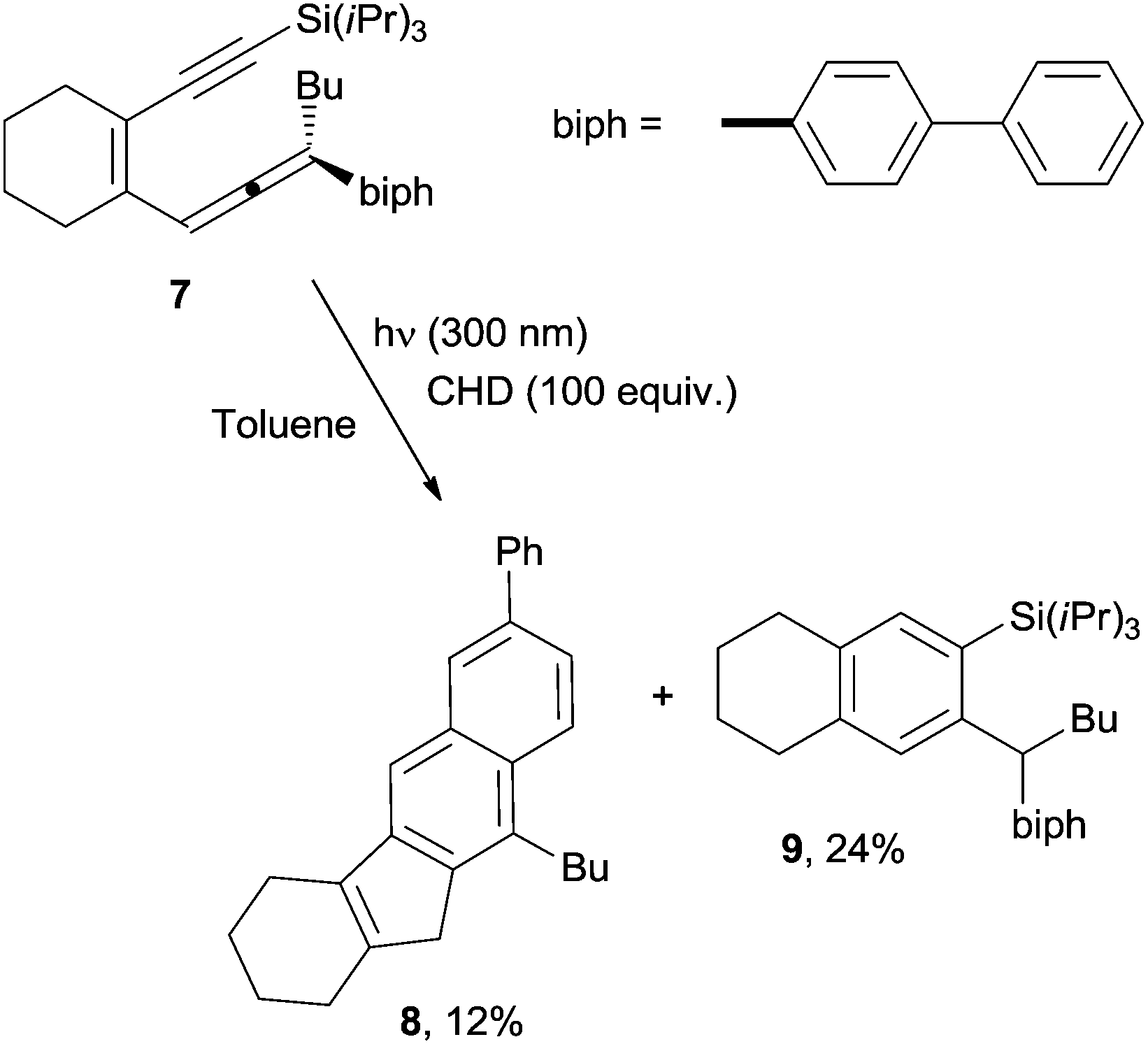 | ||
| Scheme 6 Competitive Schmittel and Myers–Saito cyclizations in the photochemical reaction of enyne–allene 7. | ||
Photoinduced cyclizations, however, are often characterized by a low quantum yield. Thus, in most cases light was instead used to trigger the release of highly reactive enediynes or enyne–allenes from photoactive compounds. As an example, this strategy has been adopted for substrates such as 10, which, upon photoinduced decarbonylation of the cyclopropenone moiety, afforded the corresponding enyne–allene 11 and the α,3-DHT 12 from it (Scheme 7).77
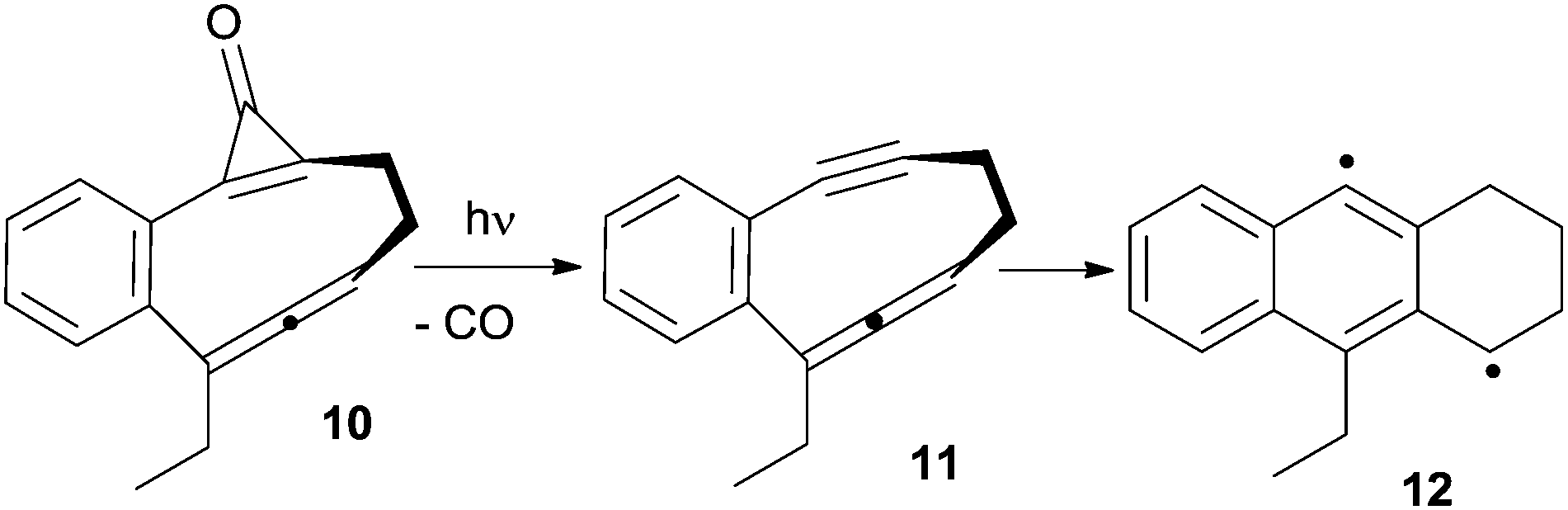 | ||
| Scheme 7 Myers–Saito reaction via photogenerated enyne–allenes. | ||
2.3 Activation by metal catalysis or via metal complexes
The coordinating properties of double and triple bonds of dienynes and enediynes towards metal ions and complexes have been widely exploited to promote cycloaromatization processes under mild conditions.78 As an example, Ruthenium and Rhodium species (e.g. the RhI(Cl)(iPr3P)2 complex) promoted the cyclization of enediynes bearing at least a terminal triple C–C bond (13 in Scheme 8) via vinylidene intermediate 14 that evolved, through a Myers–Saito cyclization, into the corresponding biradical 15.79,80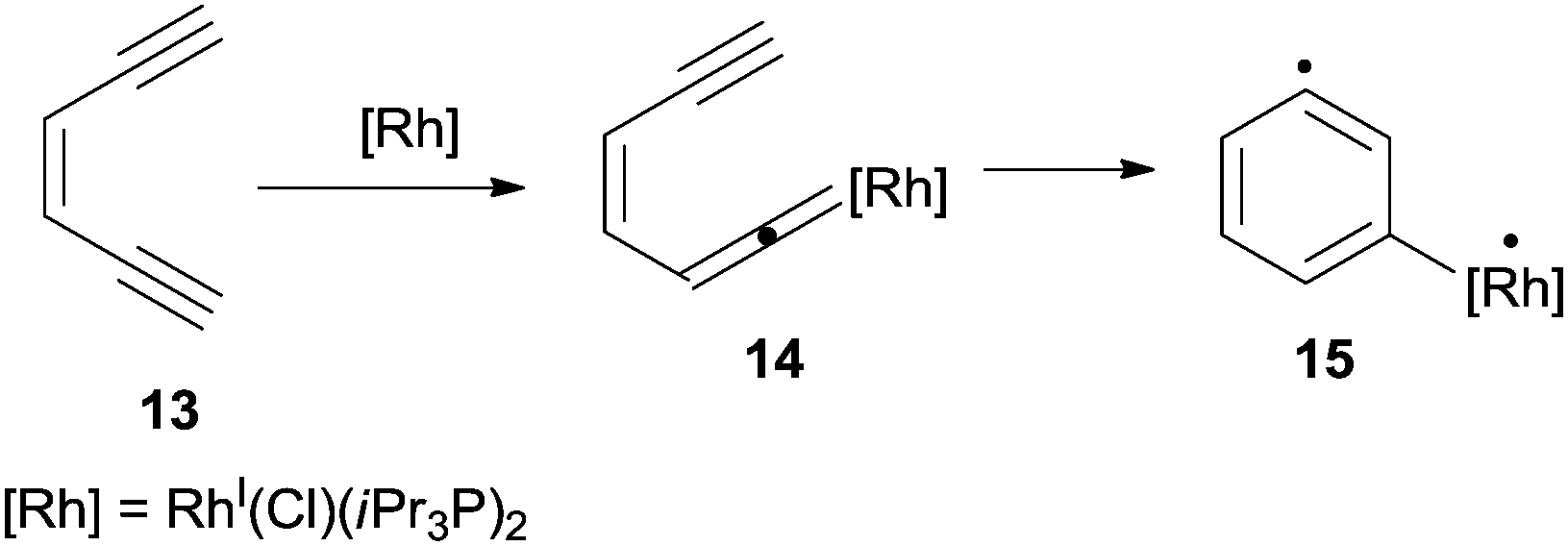 | ||
| Scheme 8 Rhodium-catalyzed cyclization of enediyne 13. | ||
AuI,81 PdII82,83 and FeII84 complexes have been likewise exploited to promote Bergman reactions. As in the case of thermal activation, the effect of metal catalysis can depend as well on the structure of the starting enediyne. Thus, cycloaromatization of strained-ring benzoenediynes was efficiently promoted by RuII(μ5-C5Me5)(CH3CN)3+, whereas the process did not occur in the case of strain-free acyclic derivatives.78
Less efforts have been devoted to the development of metal-catalyzed enyne–allene cyclizations. As an example, the Myers–Saito cycloaromatization took place in the presence of catalytic amounts of AgISbF6 and AuI(Cl)(Ph3P).85 In another instance, the Pauson–Khand cyclocarbonylation and the Schmittel cyclization were induced, respectively, by the presence of a stoichiometric amount of Mo0(CO)3(CH3CN)386 and SmIII2.87
2.4 Activation by electrophilic, nucleophilic or radical species
The π-system of the alkyne in dienynes is an appealing target for metal and non-metal Lewis and Brønsted acids (E+) that act as electrophiles and promote the formation of alkyne complexes, such as 17+ from 16 (Scheme 9). Intramolecular 6-endo-dig attack of the terminal alkene followed by aromatization of the resulting carbocation 18+ affords the desired aromatic ring (19). Protons arising from strong acids, as well as iodine, are commonly used as electrophiles. When metal derivatives (E+ = Pd, Pt, Au, In-based species) are employed, a protodemetallation is commonly involved as the final step, leading to the desired product.56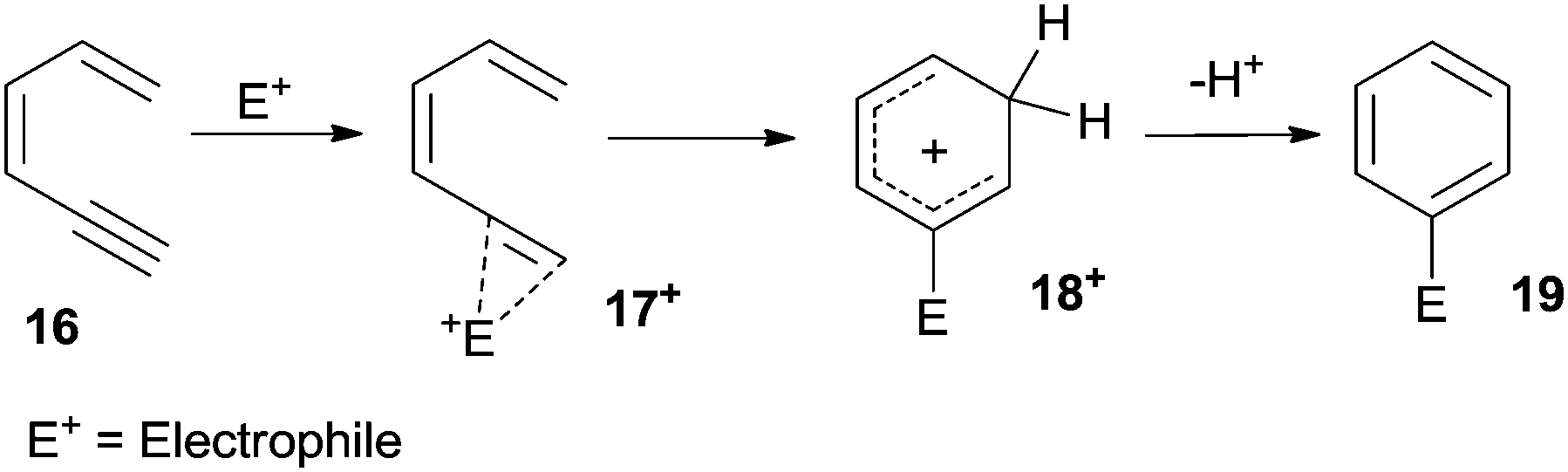 | ||
| Scheme 9 Electrophile-induced cycloaromatization of dienynes. | ||
A peculiar case is the Lewis acid catalyzed [4+2] benzannulation of enynals.88–90 As an example, the electrophilic character of the alkyne moiety in compound 20 (Scheme 10) was enhanced by coordination with AuIIICl3, thus inducing a nucleophilic attack of the carbonyl oxygen to form the highly reactive ate complex 21. This complex underwent a hetero Diels–Alder reaction with a dienophile (either an alkyne88 or a benzyne89,90) and rearrangement of the so-formed adduct 22 led to the desired product 23.
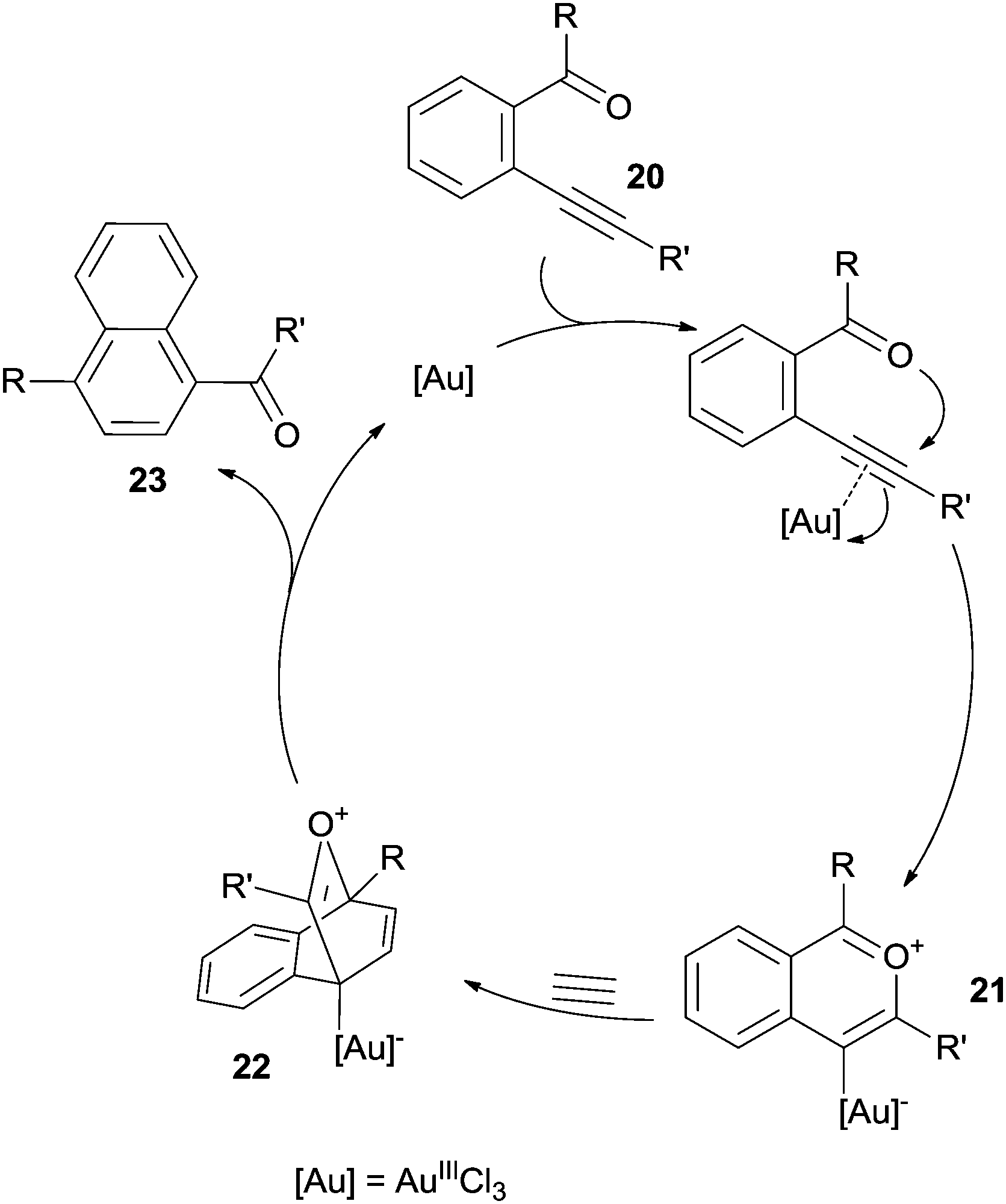 | ||
| Scheme 10 AuIII-catalyzed [4+2] benzannulation of enynals. | ||
Typical electrophiles, such as iodine91 and metal Lewis acids (AuI(Cl)(Ph3P)),92 can also promote the cyclization of enediynes. Electrophilic activation, however, often led to a diverted chemistry with respect to the thermal approach and pentafulvenes could be obtained in place of the expected aromatics.93,94
When sufficiently activated, the terminal alkene in dienynes56 or one of the triple bonds in enediynes95 can likewise act as an electrophile and undergo nucleophilic attack (by a methoxide ion). Indeed, the use of nucleophiles to activate enediynes for the synthesis of heterocycles has been recently reviewed.96
Finally, the generation of a heteroatom-based radical (e.g. Bu3Sn˙) in the presence of dienynes97 and enediynes can be exploited to induce a radical cascade useful for the construction of complex (poly)aromatic structures.98
3 Synthesis of benzenes
Only a few cases in the literature deal with the cyclization of dienynes for the preparation of benzenes.99–103 The ruthenium-catalyzed (by (Cp)RuII(Ph3P)2(Cl), 5 mol%; Cp = C5H5−) coupling of 2-(phenylethynyl)cyclohex-1-enecarbaldehyde with cyclopentanone under basic conditions gave a dienyne that smoothly cyclized to give an indanone derivative.99,100 The cycloaromatization of dienynes was likewise accomplished under gold catalysis (by AuI(Ph3P)NTf2, 5 mol%) and allowed the preparation of tetrahydronaphthalenes. The reaction was initiated by the coordination of the gold complex to the triple bond.101In the field of gold catalysis, the preparation of phenols starting from captodative dienyne carboxylic acids is another interesting case (Scheme 11).102 Thus, a AuI complex coordinated the triple bond of 24a–c leading to 25a–c, which are stabilized by the presence of the alkoxy substituent through resonance structures 26a–c. Thus, cis–trans isomerization followed by intramolecular cyclization promoted by the nucleophilic attack of the COOH group afforded an eight-membered intermediate represented by resonance structures 27a–c and 28a–c, which further evolved into bicyclic structures 29a–c. Aromatization of 29a–c promoted the four-membered ring opening, in turn allowing the formation of the corresponding phenols 30a–c (ca. 70% yield). The reaction was particularly efficient when R in 24 was an aromatic ring.102 When the MeO group in the starting substrate was substituted with an aromatic ring, however, the reaction course completely changed and a 1,6-cyclization/decarboxylation sequence led to biphenyl or m-terphenyl derivatives.102
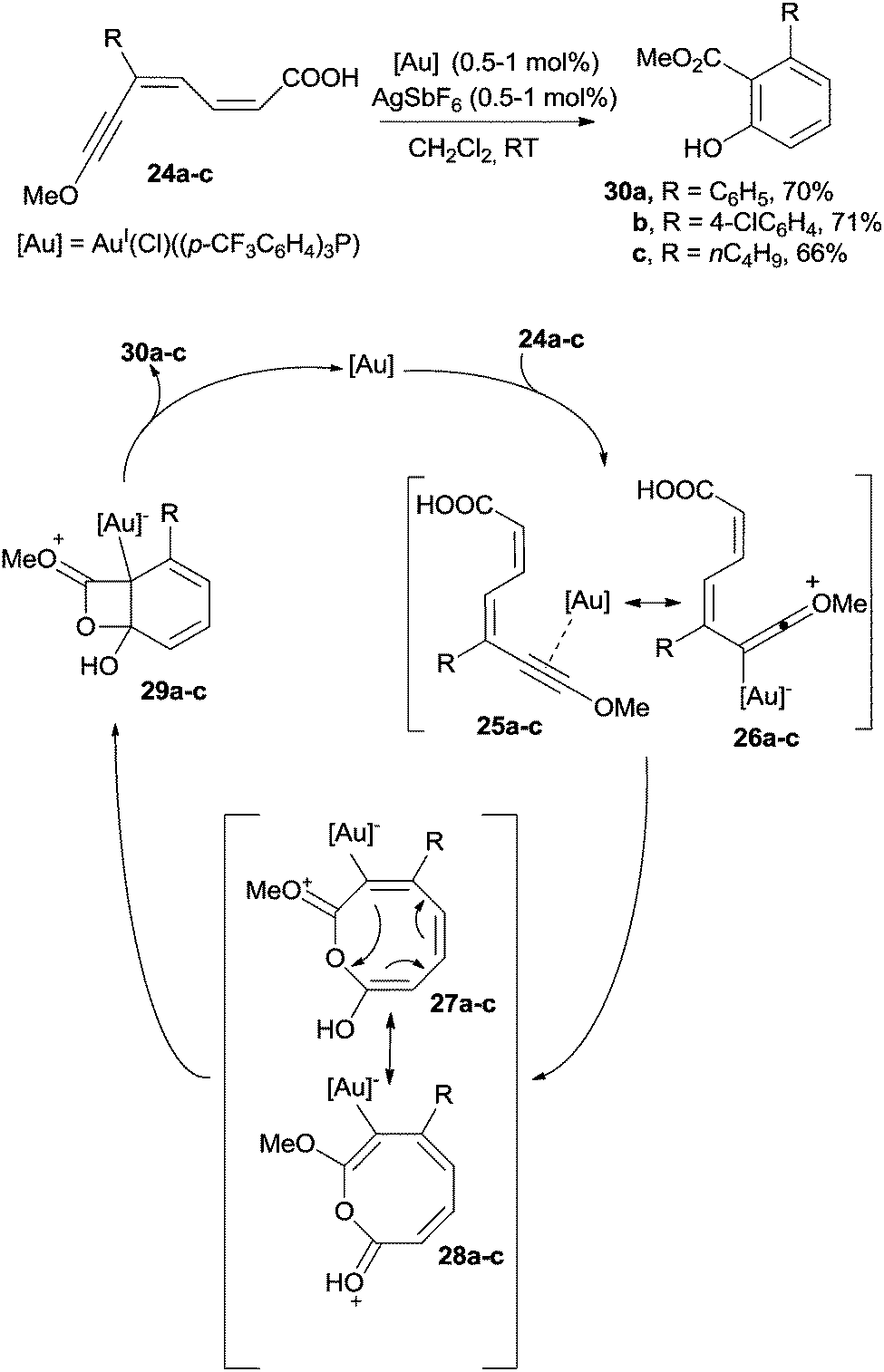 | ||
| Scheme 11 Preparation of phenols by the metal-catalyzed reaction of captodative dienyne carboxylic acids. | ||
Another interesting example is the synthesis of iodoarenes from dienynes through a cycloaromatization step promoted by an electrophile.103 Thus, enyne–dioxinones 31a–d underwent the addition of ICl and were converted into iodotrienyl cations 32+a–d that, upon cyclization to 33+a–d and subsequent aromatization, gave the desired iodobenzenes 34a–d (Scheme 12). The yields of this reaction were satisfactory (50–95% yield) for a wide range of substituted aromatic rings. Derivatives bearing cyclopropyl and thienyl substituents have also been efficiently synthesized.103
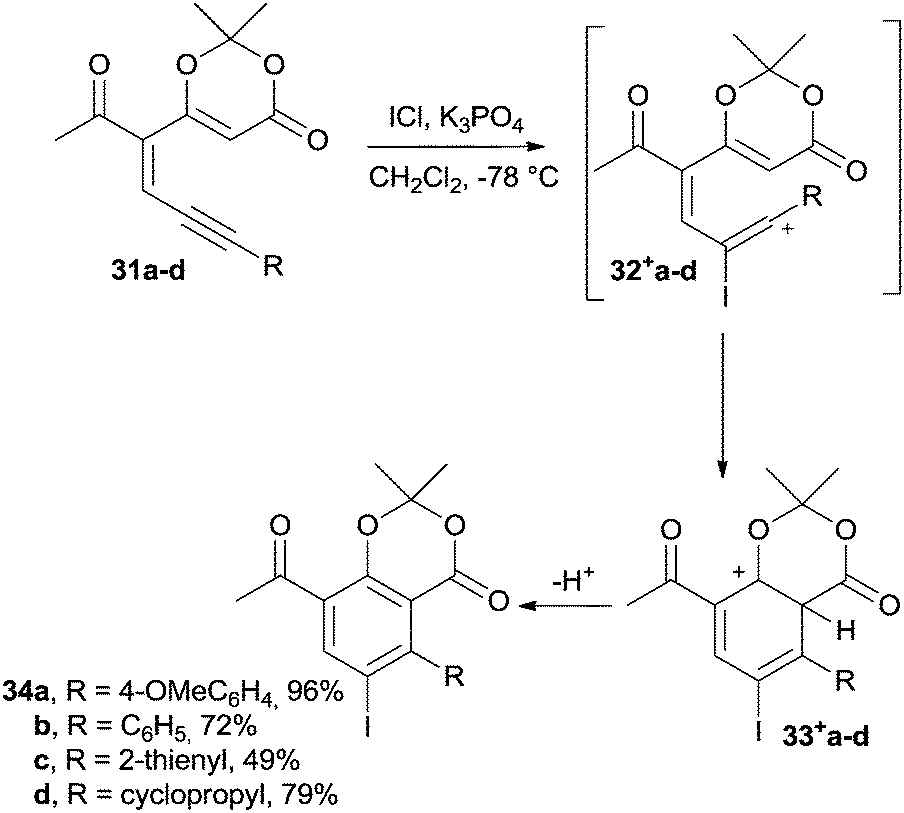 | ||
| Scheme 12 Synthesis of iodoarenes from enyne–dioxinones. | ||
The Bergman cyclization is widely used for the preparation of substituted benzenes. The reaction can be induced either thermally, as reported in the synthesis of cyclophane derivatives,104 or photochemically, where the enediyne was photogenerated in situ and used to prepare tetrahydroanthracene-1,4-diones.105
The metal-catalyzed cycloaromatization of enediynes (via the intermediacy of a metal-alkyne species) has recently found widespread use in the synthesis of arenes. Under refluxing conditions, the ruthenium catalyst (Tp)RuII(Ph3P)(CH3CN)2PF6 (Tp = tris(1-pyrazolyl)borate)80 was able to bind selectively to the more electron-rich triple bond of enediyne 35 leading to ruthenium π-alkyne complex 36+ (Scheme 13). In the presence of a nucleophile (methyl acetoacetate), a ligand exchange with CH3CN took place to give 37a+ and then vinyl ruthenium complex 38a. After 6-endo-dig cyclization to 39a and aromatization, compound 40a was obtained in 60% yield. In the case of pyrrole nucleophiles, a direct attack on 36+ to give 38b and then 40b (78% yield) was invoked, due to the poor ligand properties of pyrrole. The process was compatible with a wide range of nucleophiles (water, alcohols, acetylacetone, dimethyl malonate, anilines, and pyrroles) and a plethora of aromatics, including phenols, alkoxybenzenes and diphenylamines, could be prepared, accordingly.80
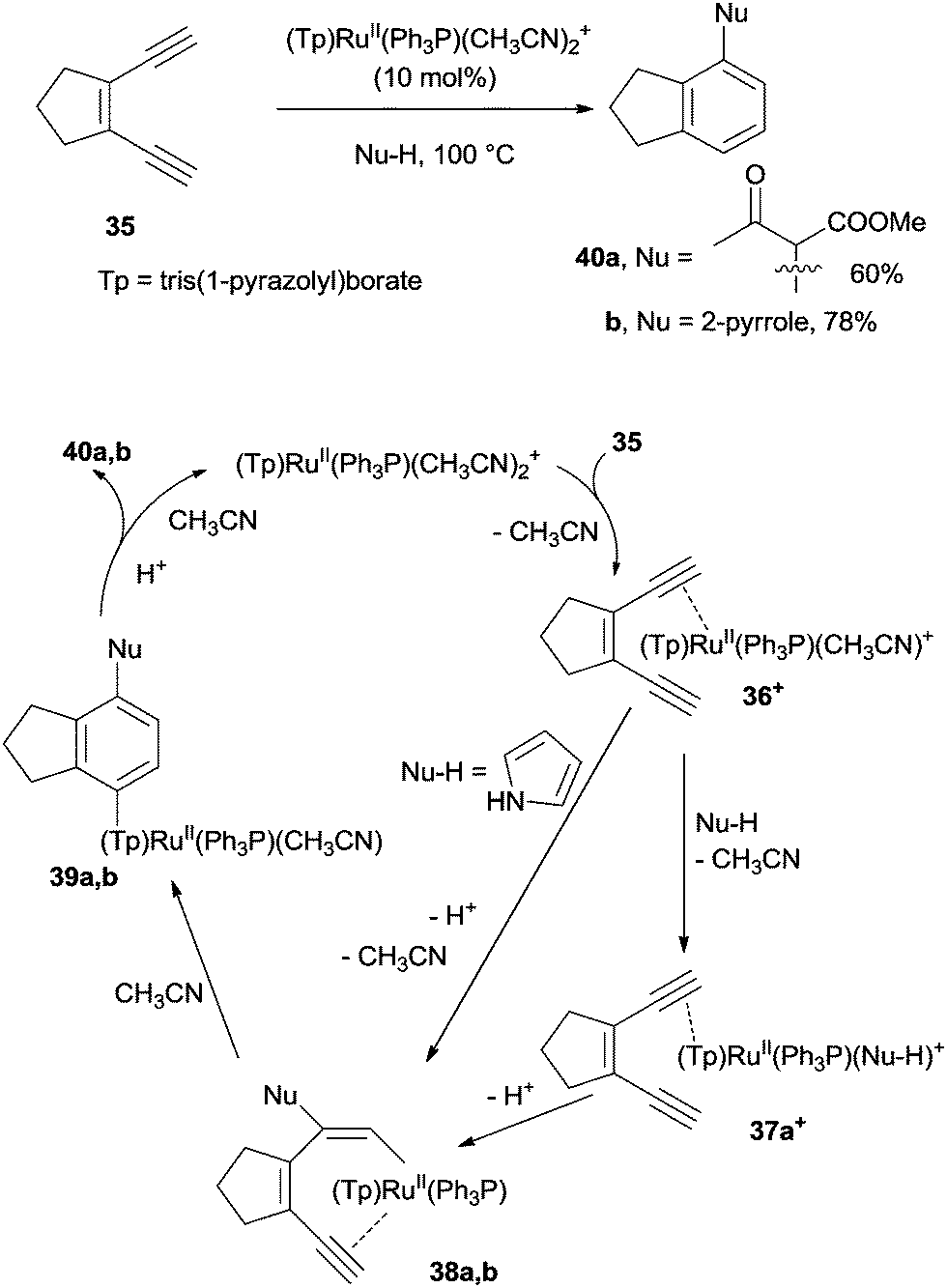 | ||
| Scheme 13 Ruthenium-catalyzed cycloaromatization of enediynes. | ||
In a different application, the Bergman cyclization was catalyzed by PtIICl2. For instance, the benzene core of 46a–c was obtained through the cycloaromatization of enediynes 41a–c under platinum-catalyzed conditions.83 Thus, PtII and 41a–c gave π-alkyne complexes 42a–c which cyclized to biradicals 43a–c (also existing as the carbenoid forms 44a–c). These carbenoid intermediates underwent insertion onto the side alkyl chain to give derivatives 45a–c that, after release of the catalyst, led to the desired benzenes 46a–c in ca. 75% yield (Scheme 14). This approach could be likewise extended to the preparation of polycyclic derivatives.83
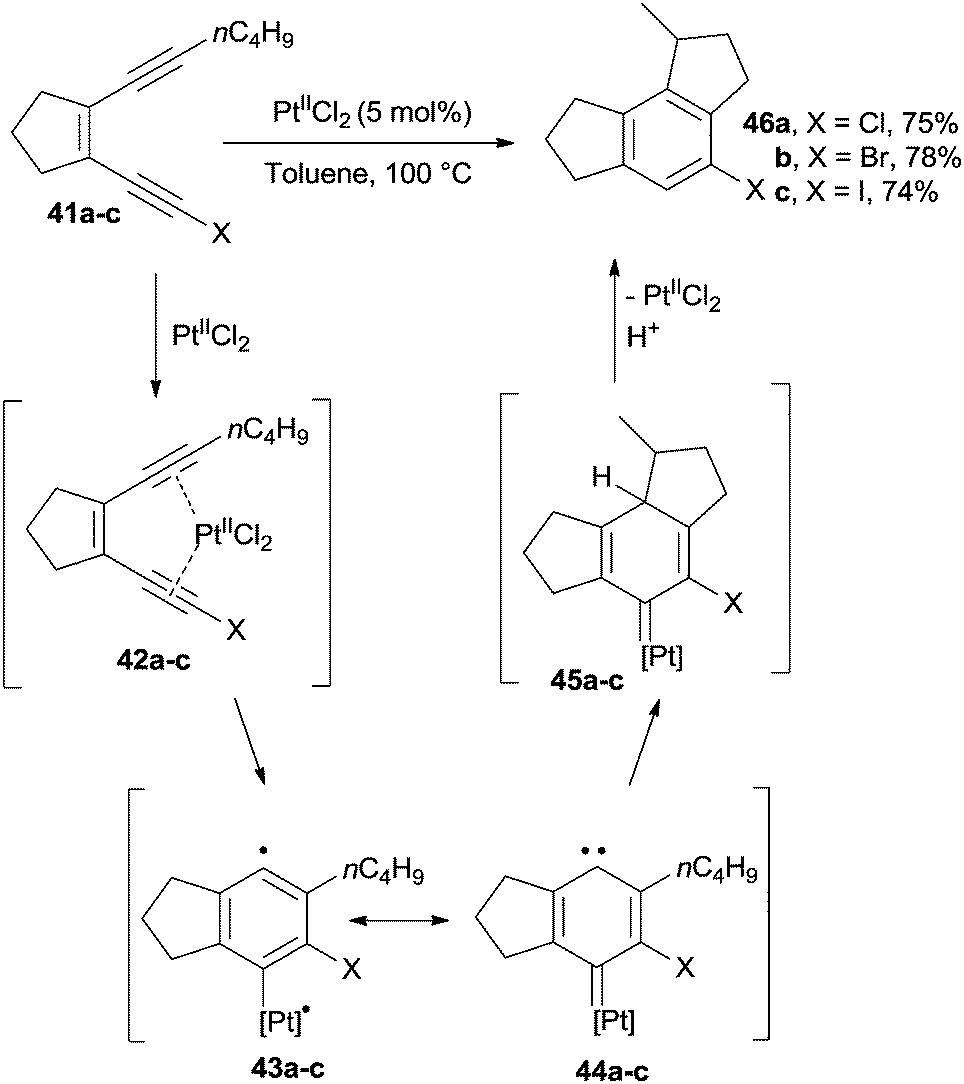 | ||
| Scheme 14 PtIICl2 as a catalyst for the preparation of indanes. | ||
In another instance, a bis(pyridine)enediyne was activated in the presence of MgIICl2.106 In fact, the substituents of the alkyne moieties were able to chelate MgII and this non-covalent interaction induced a change of conformation that favored the aromatization process.106
A smooth synthesis of arenes was achieved via the use of cyclopentadienyl metal complexes, such as (η5-C5Me5)MII(CH3CN)3+ (M = FeII, 47a; RuII, 47b). Indeed, alicyclic (48) and acyclic (49) enediynes cyclized at room temperature in the presence of 47a,b and γ-terpinene 50 as radical trap to give the corresponding complexes 51a,b and 52a,b, respectively.78,84 The so-formed arene ligands were then photochemically released leading to aromatic compounds 53 and 54 in excellent yields (Scheme 15). The well known photochemical interconversion of (E)- and (Z)-enediynes allowed an extension of this reaction also to (E)-enediynes.78,84
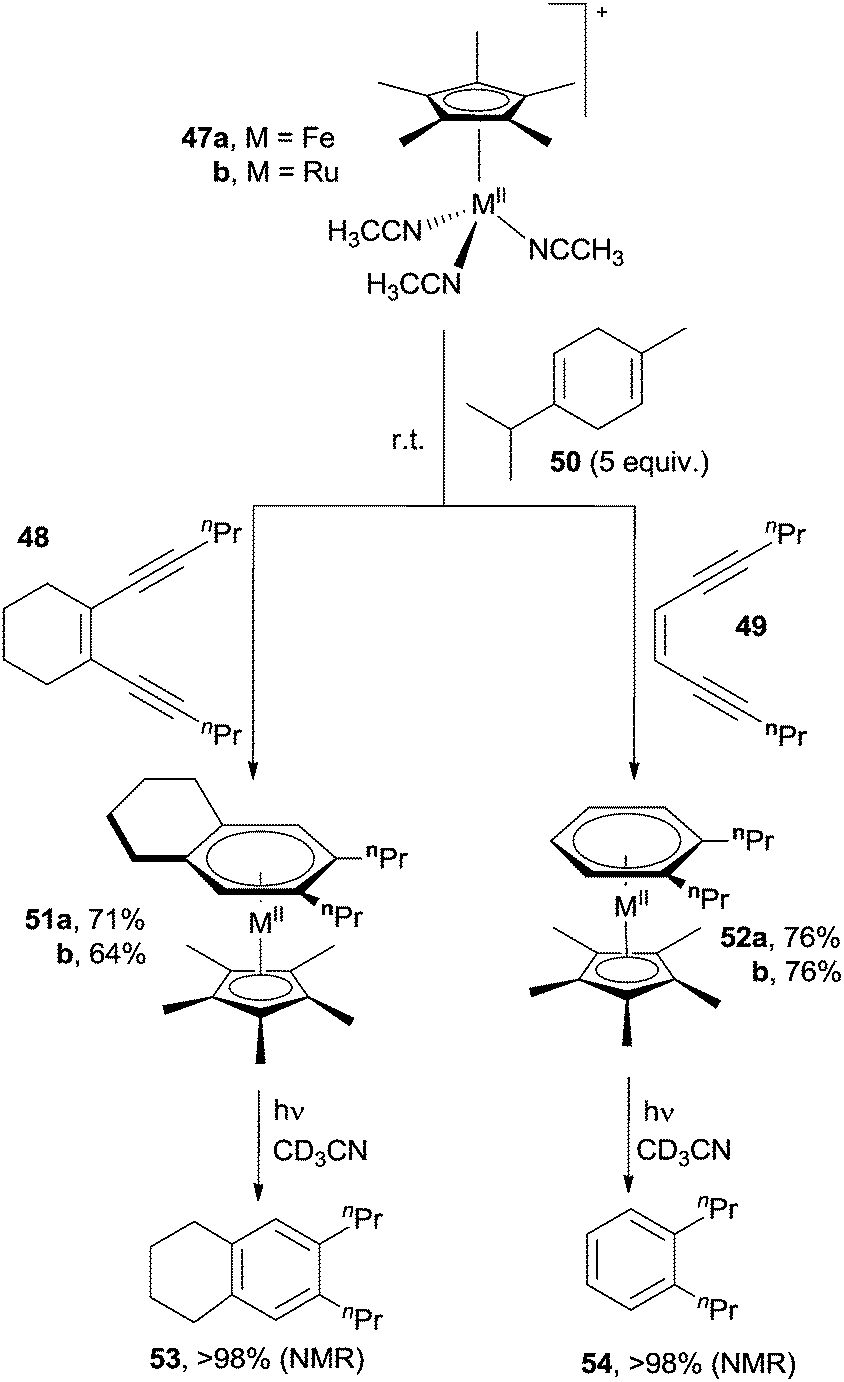 | ||
| Scheme 15 Aromatics from cyclopentadienyl metal complexes. | ||
The Bergman cyclization was also applied to the synthesis of compounds with pharmaceutical interest under metal-free conditions, where the presence of a base (nucleophile) was in most cases required. A typical case is the base-induced ring opening of the lactam ring in N-fused lactendiynes to prepare aromatic β-amino acids.107
A nucleophilic attack onto the polyenic system was also adopted. For instance, phenanthridones or benzo[c]phenanthridones 56a,b were obtained by treating benzonitriles containing an enediyne moiety 55a,b with sodium methoxide (Scheme 16). The reaction was efficient in methanol mixed with 10% of a polar aprotic solvent (e.g. DMSO, HMPA, and THF). The methoxide anion was added to the cyano group triggering the anionic cascade cycloaromatization.108 In contrast, the course of the reaction changed in the presence of tetrabutylammonium iodide since methoxide attacked the C2 of the acetylenic moiety and induced a cyclization to the corresponding biphenyls 57a,b (Scheme 16).
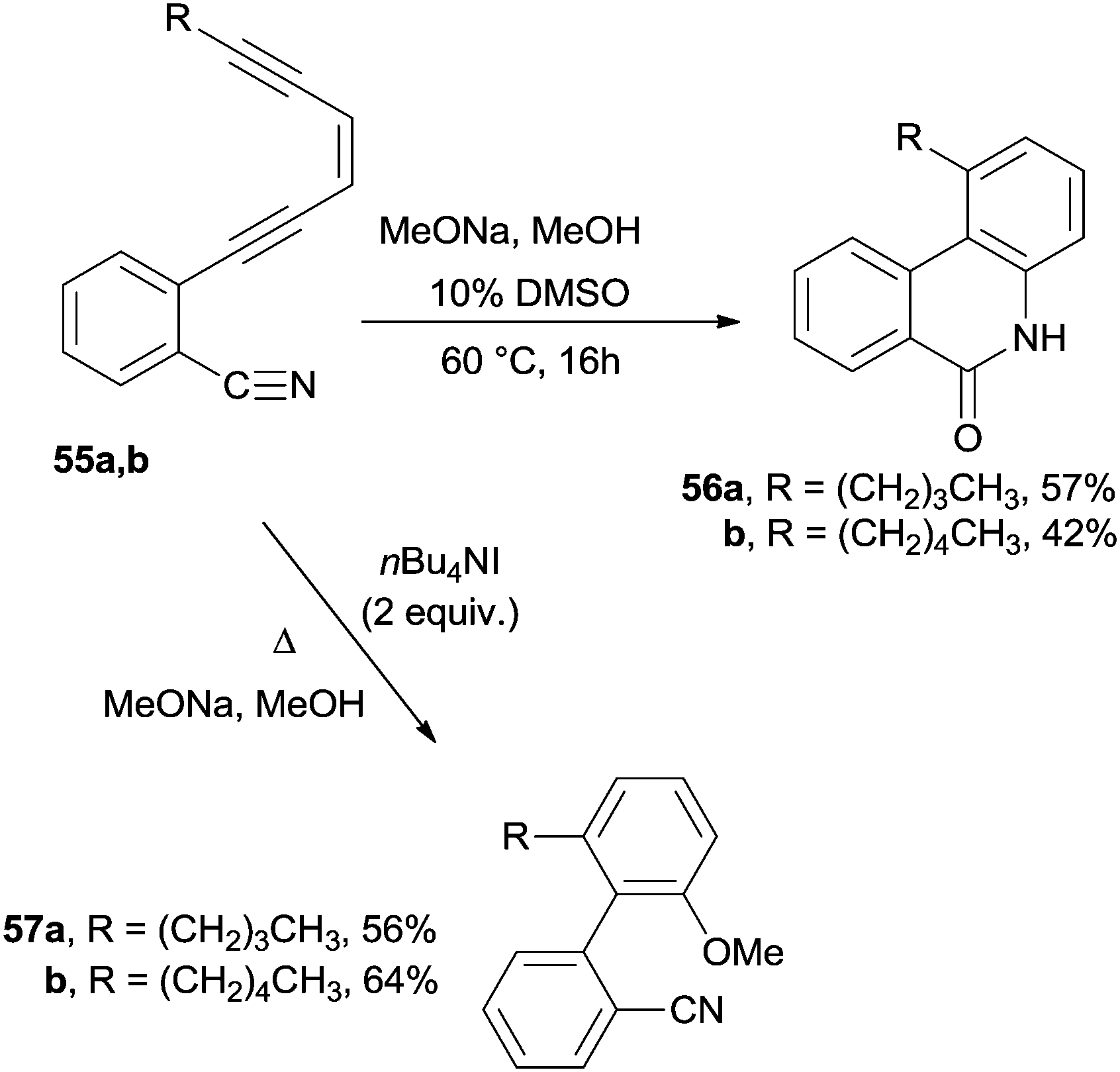 | ||
| Scheme 16 Different pathways in the nucleophile-induced Bergman cyclization. | ||
An alternative route made use of basic conditions (tBuOK) to generate a bicyclic trienediyne system prone to aromatization, giving a key intermediate for the preparation of cyanosporasides.109,110
In some instances, enyne–allenes were the reactive unsaturated system(s). As an example, various enyne–allenes gave access in refluxing benzene to compounds having the tetracyclic steroidal skeleton.111,112
Enediynes could be converted into the corresponding enyne–allenes by acid catalysis. Indeed, when treated with TFA, compound 58 gave enyne–allene acetal 59 that underwent a Myers–Saito cyclization to biradical 60. Different aromatic products 61–63, depending on the reaction conditions (presence of hydrogen atom donors, oxygen, etc.), have been obtained, as illustrated in Scheme 17.113
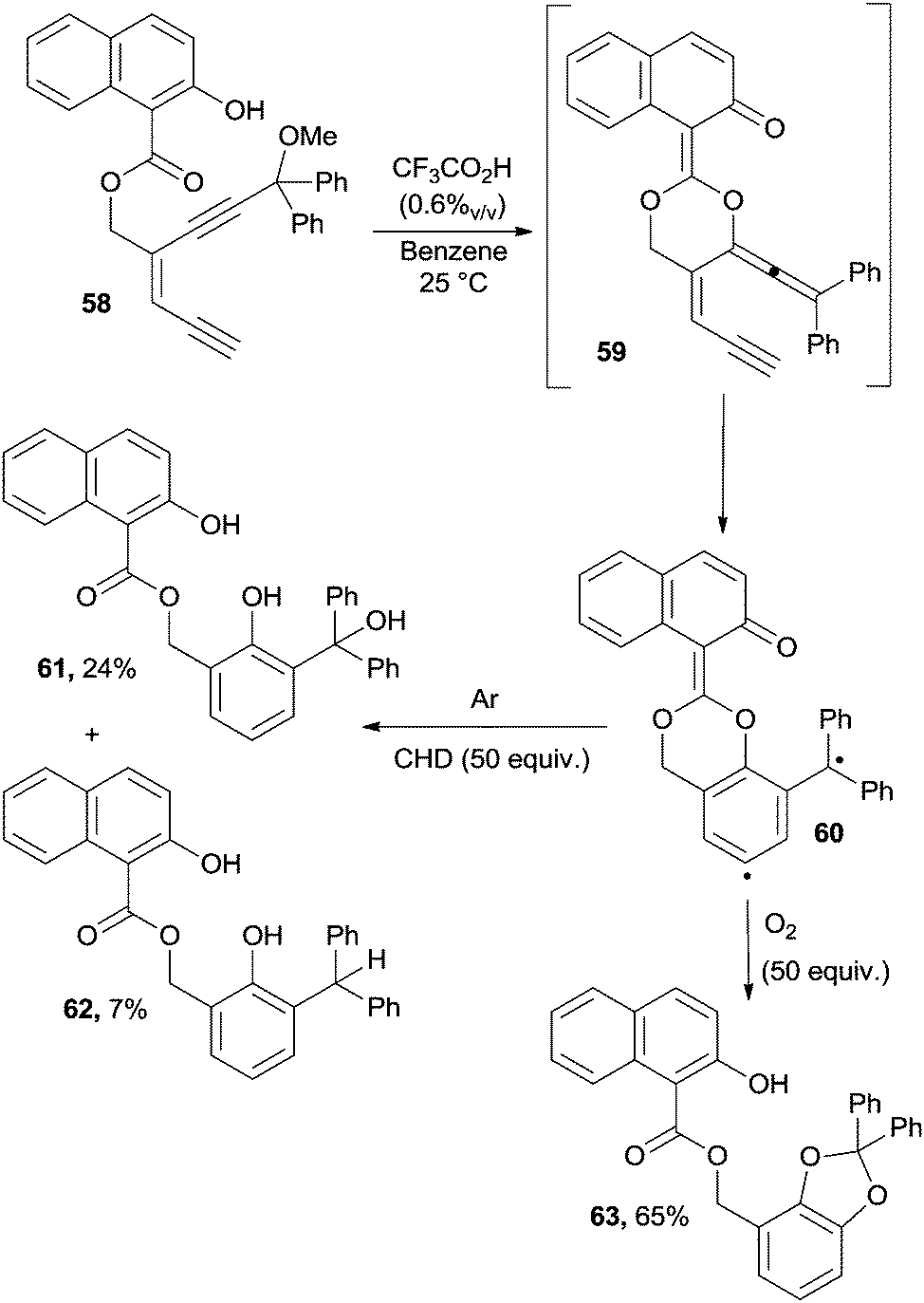 | ||
| Scheme 17 Enedyines as a source of enyne–allenes for Myers–Saito cyclizations. | ||
Enyne–allenes can be considered as good precursors of substituted benzenes, including phenols. It was found that treating acetoxy-substituted enyne–allene 64 with MeLi gave the corresponding oxyanion (upon deprotection of the acetate group). This in turn cyclized (at −10 °C) to the corresponding phenol 65 in 30 minutes (Scheme 18).114 The fate of the reaction, however, depended on the ring size and on the nature of the terminal substituents, the Schmittel cyclization being an important competitive pathway.
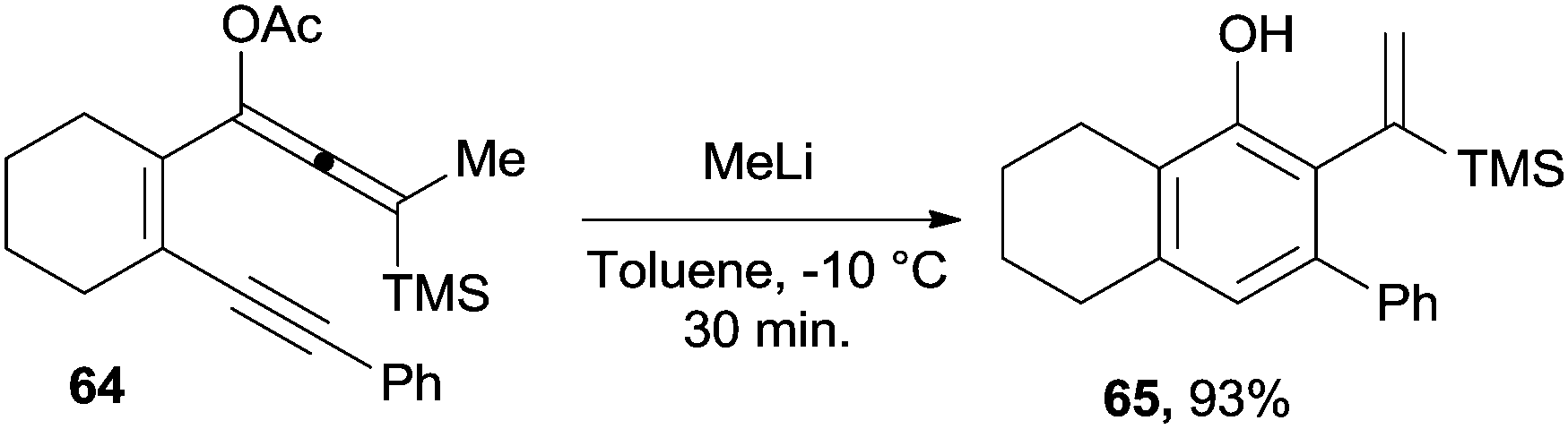 | ||
| Scheme 18 Methyl lithium-induced cycloaromatization of enyne–allenes. | ||
The intervention of a metal-catalyst can induce the formation of a vinylidene complex. For example, a modest variety of substituted benzenes were prepared by heating enediynes 66a,b at 50 °C in the presence of a catalytic amount of RhI(Cl)(iPr3P)2.79 The thus formed vinylidene-rhodium complexes 67a,b underwent a Myers–Saito cyclization to the corresponding benzenoid radicals 68a,b. A 1,5-hydrogen shift gave biradicals 69a,b and intermediates 70a,b (path a) after radical coupling. Compounds 72a,b were then formed in good yields (50–70%) by a syn elimination of the β-hydride followed by reductive elimination (Scheme 19). An alternative mechanism was also postulated, where intermediates 71a,b were formed by a second 1,5-hydrogen shift in 69a,b (from the alkyl chain to the rhodium-centered radical, path b).
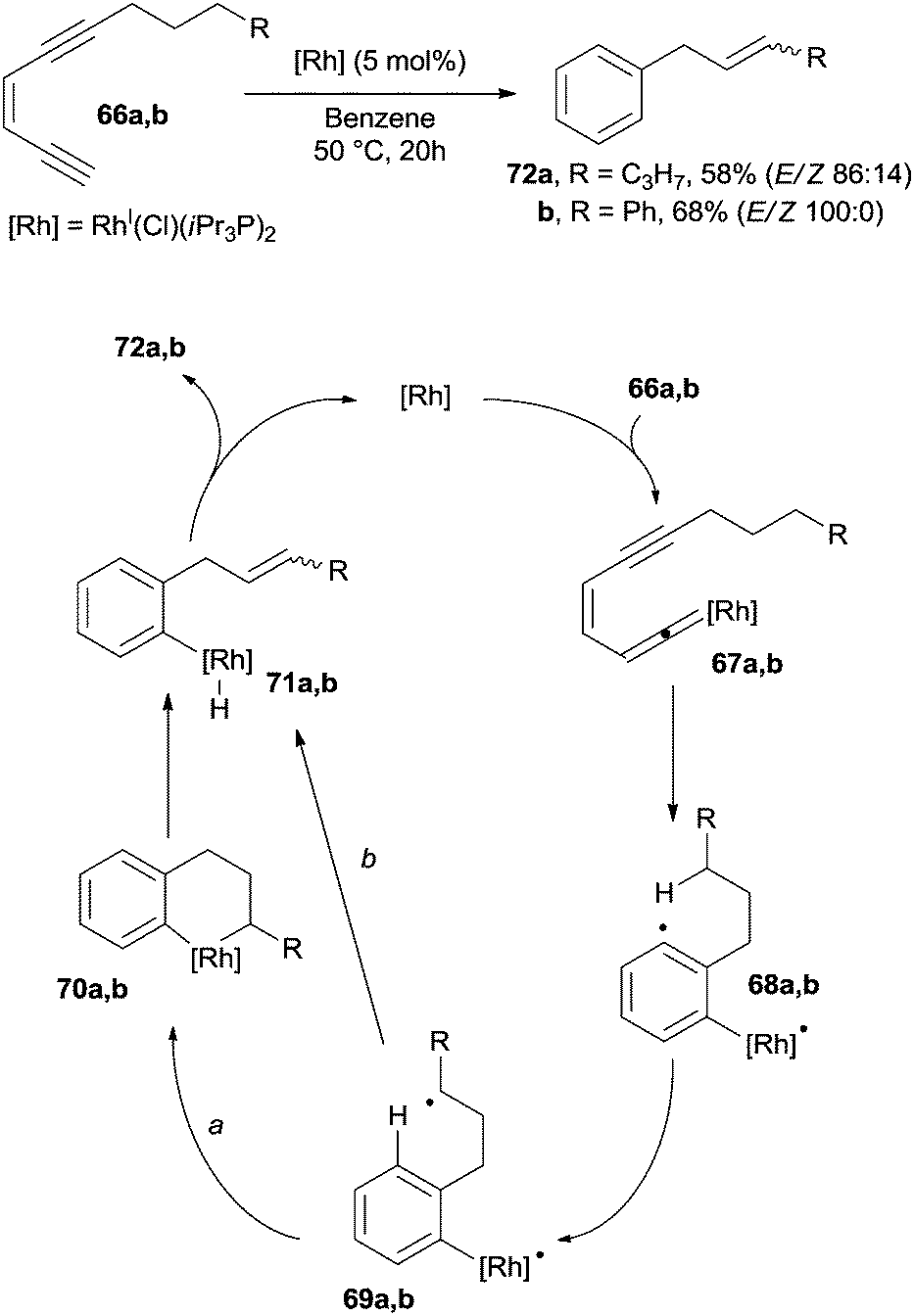 | ||
| Scheme 19 Rhodium-catalyzed synthesis of allyl aromatics. | ||
Another interesting example deals with the use of enyne–ketenes. The reaction of 73 with the chromium-carbene complex 74 led to enyne–ketene 75 that underwent a Moore cyclization to heterobiradical 76, and a complex mixture containing 77 and 78 was then obtained. Treatment of the final mixture with a strong acid (TsOH), allowed the formation of benzofurans 79 and 80 (Scheme 20).115 Moore cyclization of enyne–ketene intermediates (in turn prepared via the thermally induced ring opening of the corresponding 4-alkynylcyclobutenones) was also exploited as a key step in the synthesis of naturally occurring alkaloids pyrrolophenanthridine and assoanine.116
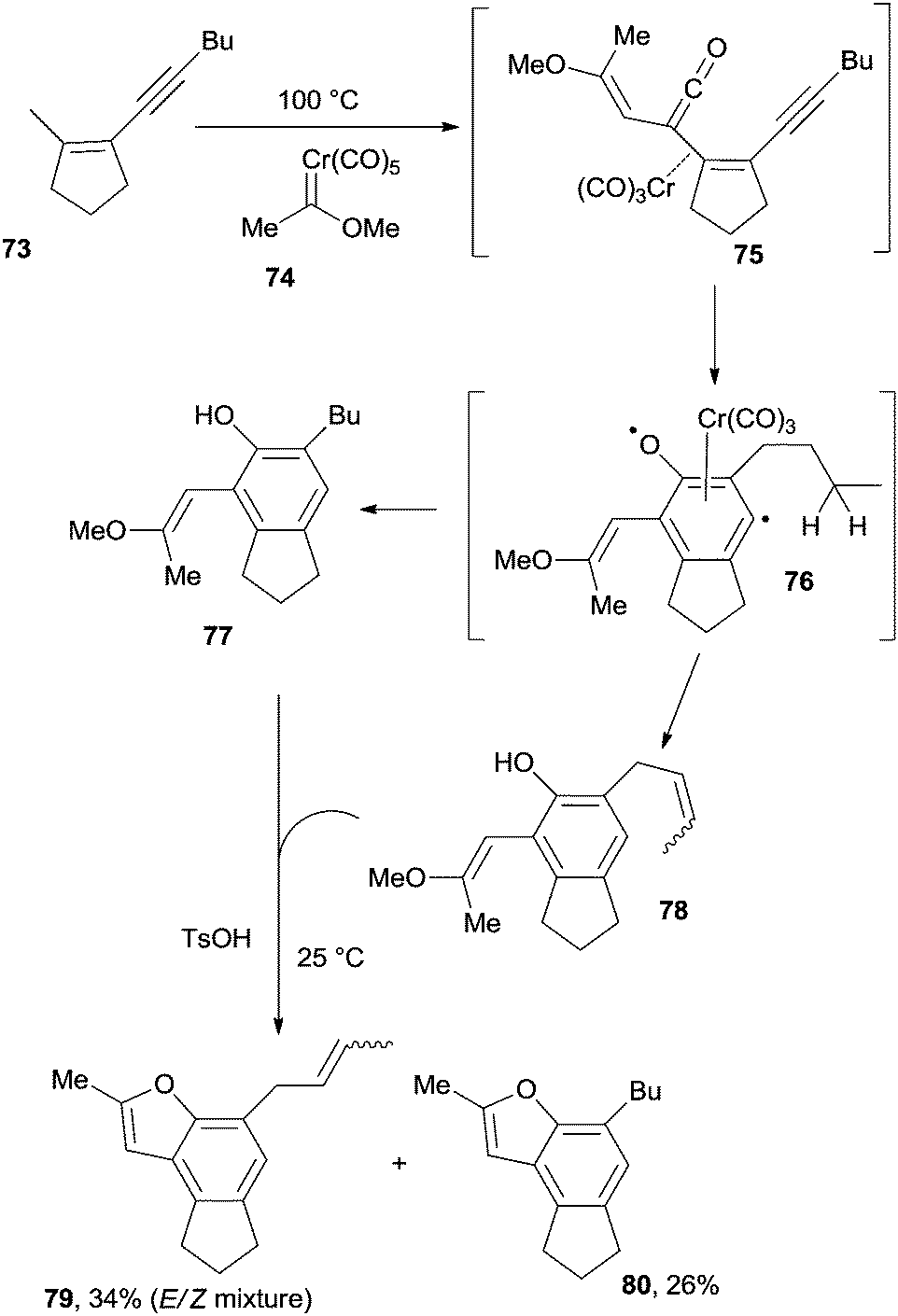 | ||
| Scheme 20 Synthesis of aromatics via enyne–ketenes. | ||
4 Synthesis of naphthalenes
Benzofused dienynes and enediynes are widely used to prepare naphthalene derivatives. The substrates can cycloaromatize directly or, in the case of enediynes, undergo rearrangement to the corresponding (and more reactive) enyne–allenes.Despite the fact that fluorinated naphthalenes can be obtained via direct fluorination of the aromatic ring, the reaction often lacks selectivity and fluoronaphthalene isomers are difficult to separate. Accordingly, these derivatives were obtained from the base-catalyzed (DBU was used) cyclization of aromatic fluorodienynes in refluxing N-methyl-2-pyrrolidone (NMP).117
The electrocyclization of aromatic dienynes has also been achieved by having recourse to the (Tp)RuII(Ph3P)(CH3CN)2(PF6) complex, in turn causing the formation of a metal–vinylidene intermediate.118,119 Analogous results were obtained when using the W0(CO)5(THF) complex as the catalyst.119 Cyclization of silyl enol ethers 81a–c was found to take place efficiently in the presence of Rh, Pd and Pt-based catalysts. The desired naphthalenes 82a–c were obtained in good yields, independent of the aromatic substituent (Scheme 21a).120 Substituted naphthalenes 84a,b were also obtained via a tin-mediated radical cyclization/fragmentation reaction occurring on dienynes 83a,b (Scheme 21b).121,122
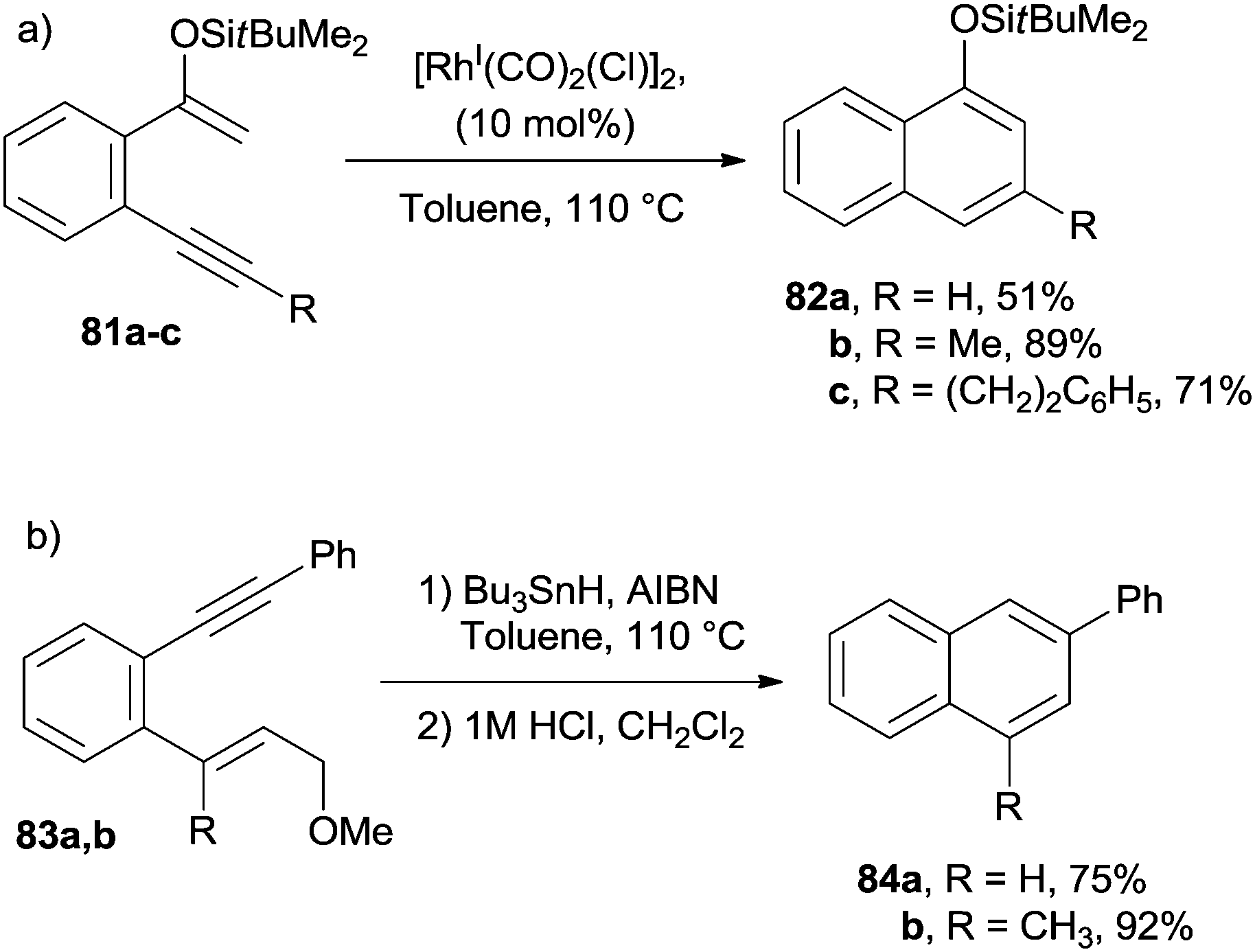 | ||
| Scheme 21 Strategies for the synthesis of the naphthalene core from dienynes. | ||
Benzofused enynals and enynones are valuable substrates in benzannulation processes under Lewis acid catalysis. Thus, naphthyl ketone 87 was prepared by the reaction of ortho-alkynyl benzaldehyde 85 with phenyl acetylene 86 in the presence of a catalytic amount of AuIIICl3 (Scheme 22a).123 The same process could be likewise carried out by replacing AuIIICl3 with CuII(OTf)2 in the presence of an equivalent of a strong acid (e.g. CF2HCO2H).88 More recently, [4+2] benzannulations between enynals and alkynes were carried out under heterogeneous catalysis (by using nanoporous gold materials), despite the fact that a high temperature (150 °C) was required.124
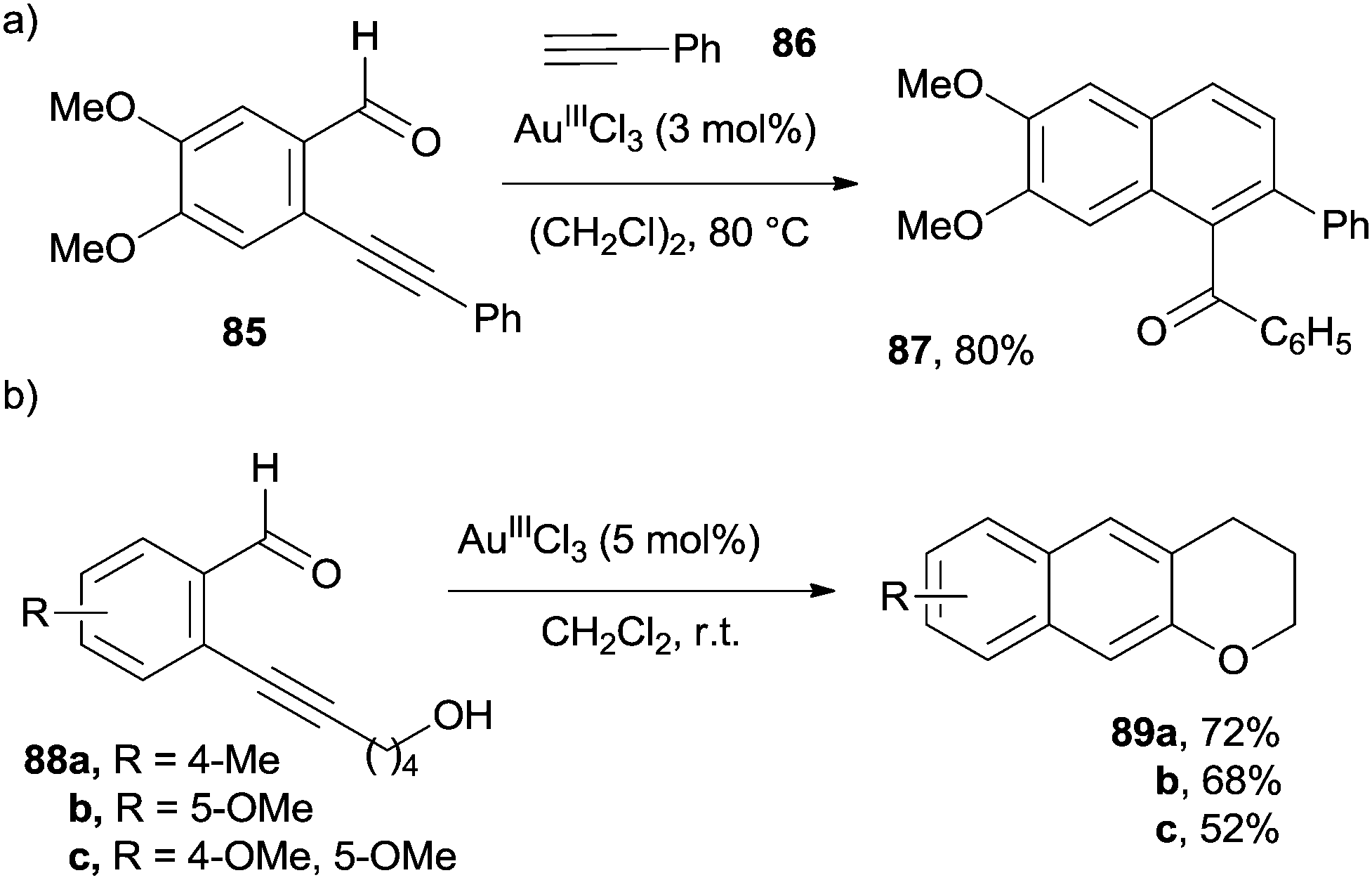 | ||
| Scheme 22 Gold-catalyzed preparation of naphthalenes from ortho-alkynyl benzaldehydes. | ||
Indeed, an intramolecular variant of the process opened the way to substituted 2,3-dihydrophenanthrenones.125 In a similar manner, 1-acyl-4-alkoxy and 1-acyl-4-alkylsulfanyl naphthalenes were prepared by treating ortho-ethynyl benzoates and benzothioates, respectively, with vinyl ethers in the presence of a catalytic amount of PtIICl2.126 Substituted benzochromanes 89a–c have been synthesized in high yield via the AuIIICl3-catalyzed rearrangement of the corresponding 2-(ynol)aryl aldehydes 88a–c (Scheme 22b).127
Polyfluorinated naphthalenes, such as 91a,b, were synthesized in high yields via the Bergman cyclization of benzocondensed enediyne precursors 90a,b (in turn obtained via an efficient Sonogashira cross-coupling) in the presence of a large excess of CHD as a hydrogen atom donor (Scheme 23).128 Both 1- and 2-nitronaphthalene were obtained in 70% yield from the corresponding benzannelated enediynes.129
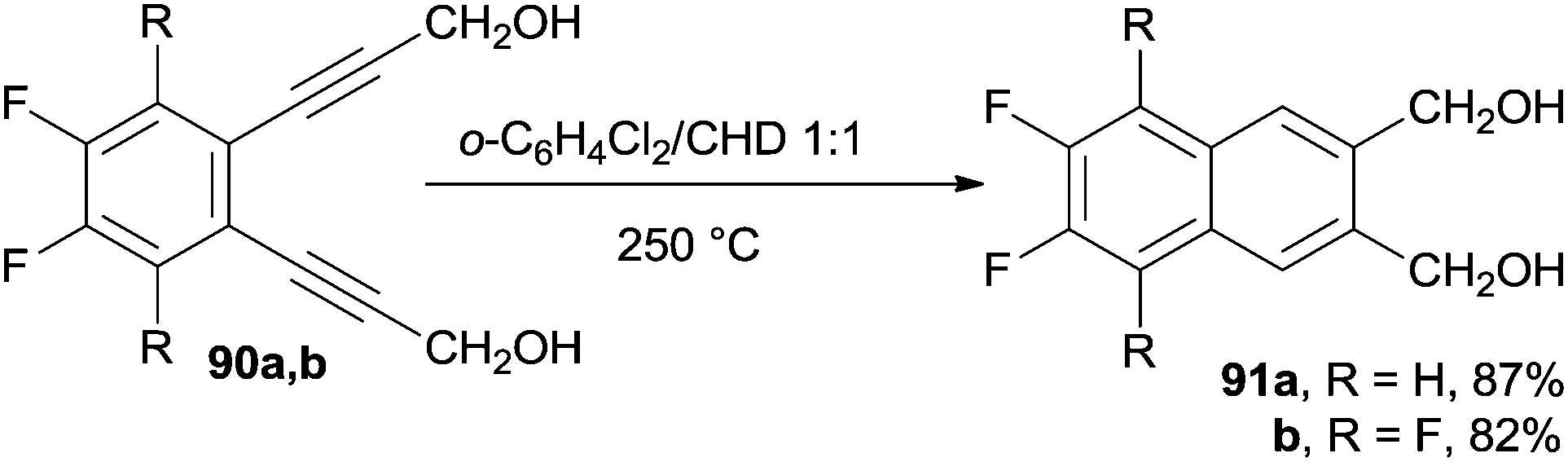 | ||
| Scheme 23 Polyfluorinated naphthalenes 91a,bvia thermal cyclization. | ||
2-(Phenylethynyl)naphthalene and 2-naphthyl butyl sulfide were thermally obtained in discrete yield from the corresponding enetriyne130 and butyl ortho-diethynylbenzene sulfide, respectively.131
Recently, the thermal cyclization (at 250 °C) of porphyrin–enediyne hybrids to give the corresponding naphthyl-porphyrins in modest yields has been described.132 Notably, traces of the cyclized product were also observed at a lower temperature (140 °C) under concomitant visible light irradiation (419 nm) of the starting solution.132
Enediyne-aminoacid conjugates are important substrates for biological applications. As an example, photoactivated lysine–enediyne conjugates caused double-stranded DNA damage and the amino acid residue played a key role in directing the selectivity of DNA cleavage.133 Some of these derivatives, however, also have interesting synthetic applications. The Bergman cycloaromatization taking place in 11-membered cyclic enediyne-containing aminoacid moieties (such as 92, prepared from homopropargylglycine) afforded the corresponding naphthalenes (93), though a high temperature (100–150 °C) and long reaction times (up to 9 days) were required (Scheme 24).134 Microwave irradiation was otherwise exploited to promote the cyclization of this class of substrates.135
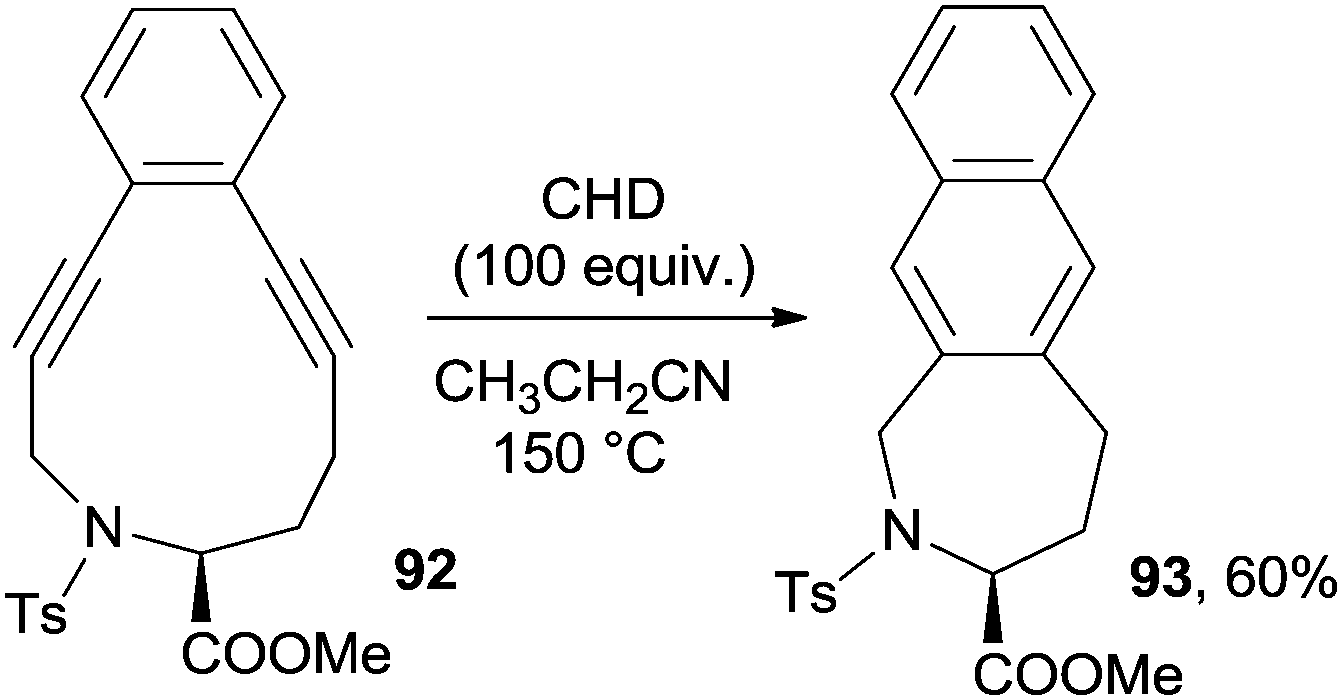 | ||
| Scheme 24 Bergman cycloaromatization of 11-membered cyclic aminoacid-derived enediyne 92. | ||
On the other hand, when using acyclic aminoacid-derived enediynes 94a,b, the formation of the naphthalene core was followed by an intramolecular SN2 substitution to give the corresponding 2,3-dihydrobenzo[f]isoindoles 95a,b, although in low yields (Scheme 25).136
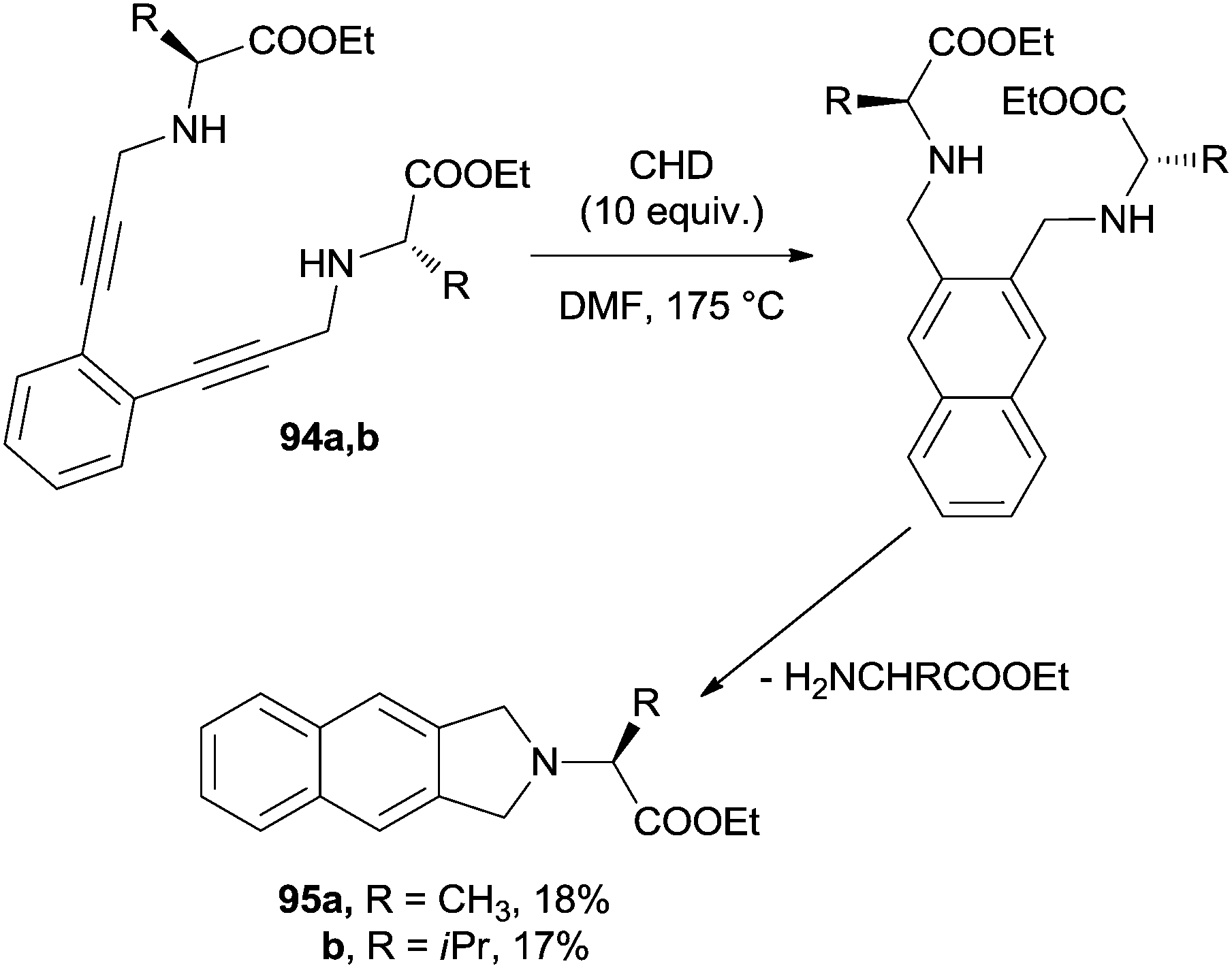 | ||
| Scheme 25 Synthesis of 2,3-dihydrobenzo[f]isoindoles 95a,b. | ||
Biradicals arising from cycloaromatization reactions have been exploited as key intermediates in cascade radical cyclizations. Tandem enediyne radical cyclizations were developed in the early 90s and applied, when selective, to the synthesis of different benzocondensed derivatives.137 As an example, the thermal activation of enediyne 96 in the presence of CHD as a hydrogen atom donor resulted in the quantitative formation of 97, obtained as a mixture of two diastereoisomers (Scheme 26).137,138
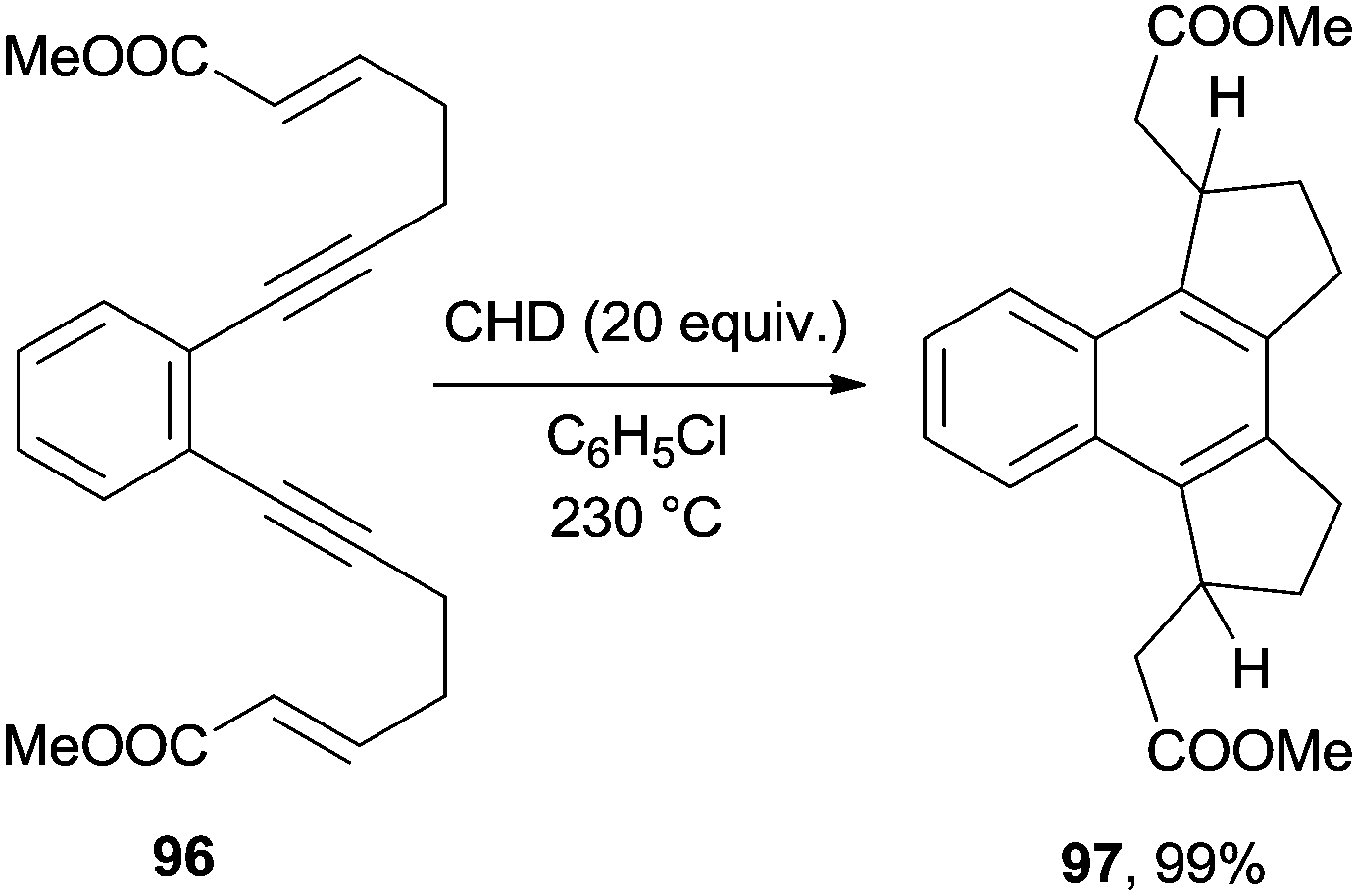 | ||
| Scheme 26 Cascade radical cyclizations triggered by a Bergman cycloaromatization. | ||
As hinted above, the synthetic significance of cycloaromatization reactions applied to acyclic enediynes is often limited by the high temperatures (typically >180 °C) required for the reaction and by the use of high amounts of (expensive) hydrogen atom donors (CHD). In some instances, however, photochemical activation can be a convenient alternative. An example deals with a photoinduced ring contraction, in turn triggering a thermal Bergman cyclization. Thus, irradiation (350 nm) in isopropanol of 11-membered enediyne 98, incorporating a 2-diazo-1,3-diketone moiety, resulted in a Wolff rearrangement via the corresponding ketenes 99a,b to produce strained, highly reactive, 10-membered enediynes 100a,b, which afforded a mixture of dihydroanthracene derivatives 101a (54%) and 101b (31%), along with small amounts of diketone 102 (<10%; Scheme 27).139,140 Analogously, the photochemical activation of a 9-membered ring enediyne precursor, with one of the triple bonds masked as cyclopropenone, resulted in the CO loss and in the final formation of the corresponding 1,4-didehydronaphthalene biradical, which yielded, after hydrogen atom abstraction from the reaction medium (2-propanol), the corresponding benzo[f]indanol as the only product (cycloaromatization yield not reported).141
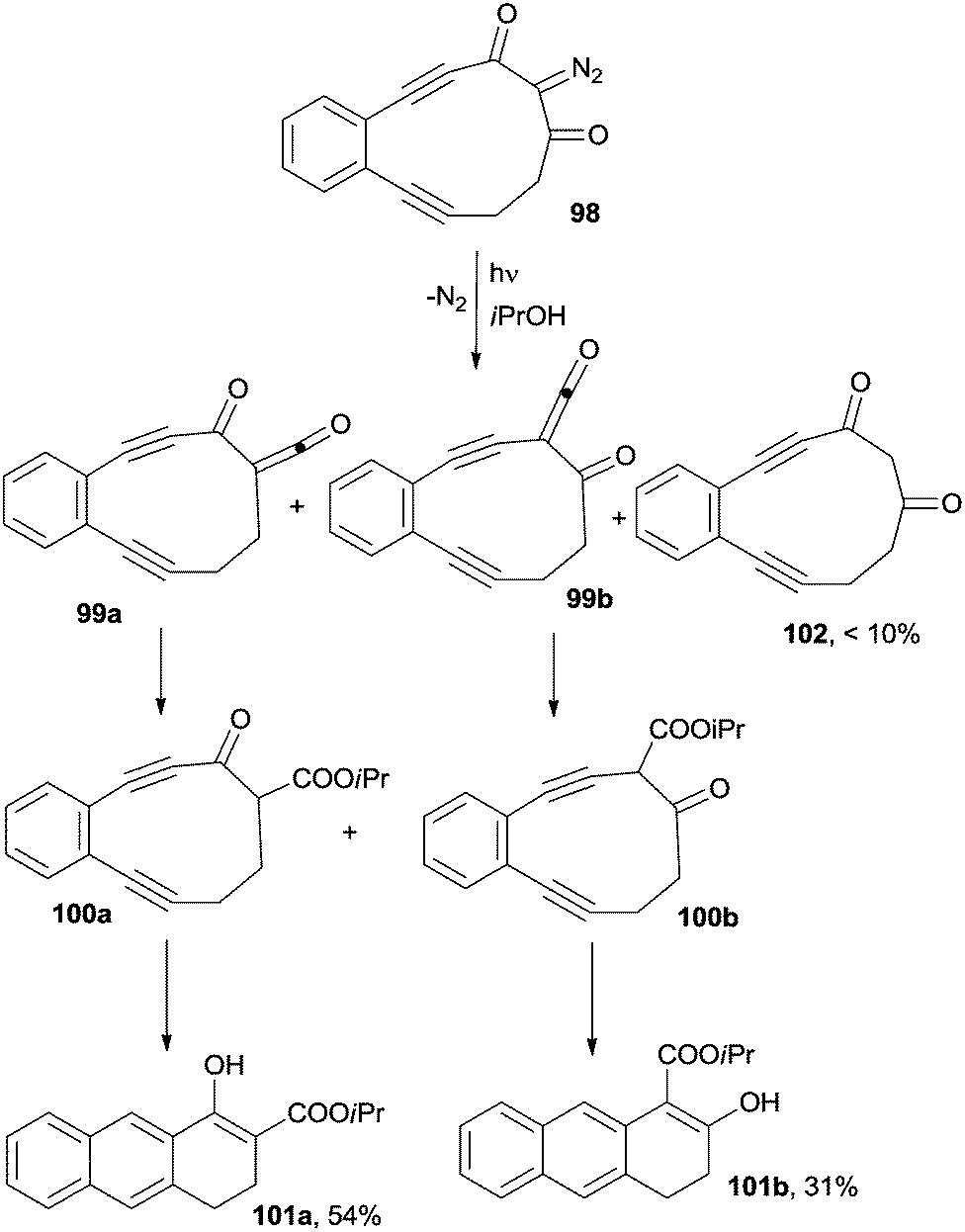 | ||
| Scheme 27 Synthesis of naphthalenes via a photoinduced Wolff rearrangement. | ||
The intervention of an organometallic or metallic species often allows for milder conditions to be adopted to promote the desired process. For example, gold complexes have found large application in this field. Coordination of arene-1,2-diyne 103 by AuI complex 104 (used in catalytic amount) resulted in the formation of intermediate 105.142–145 When benzene was used as the solvent, trapping of 105 by the medium143 to afford the corresponding β-phenyl naphthalene 106 took place as the preferred path (Scheme 28, path a; the α-isomer was also formed in 2% yield).146 Cyclization of substituted 1,2-bis-(iodoethynyl)-benzenes in the presence of IPrAuINTf2 (104, 5 mol%) in benzene afforded the desired 1-phenyl-2,4-diiodonaphthalene.147 The scope of this reaction has been widely explored and the gold-catalyzed synthesis of cyclobutenes 107a–c from 103 in the presence of several alkenes (that acted both as reactants and solvents, Scheme 28, path b) has been reported.148
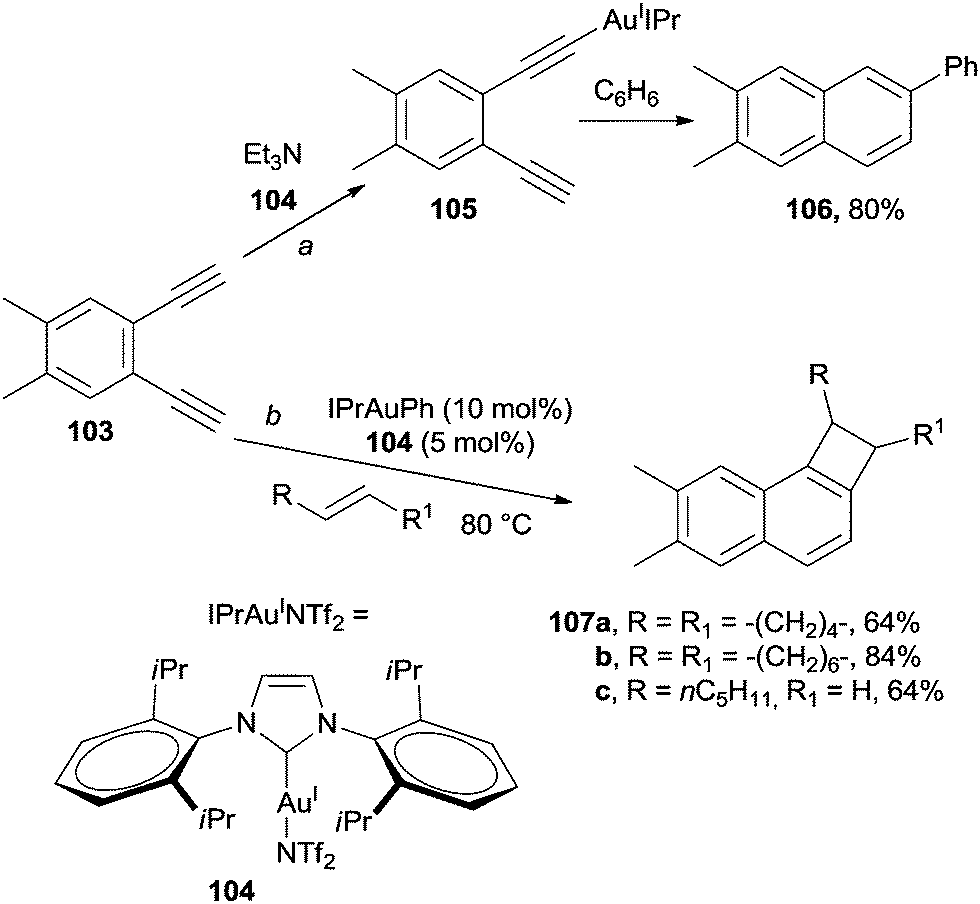 | ||
| Scheme 28 Gold-catalyzed synthesis of substituted naphthalenes. | ||
AuI-catalyzed cycloaromatization of benzofused enediyne 108 in the presence of various neutral N- and O-based nucleophiles (e.g. N-methyl aniline) afforded a wide range of naphthalene cores (e.g.109) functionalized at the 1-position (Scheme 29a).149 Interestingly, in the presence of a RuII-complex, a different substitution pattern of the final naphthalene ring (see products 110a–c in Scheme 29b) was observed.80
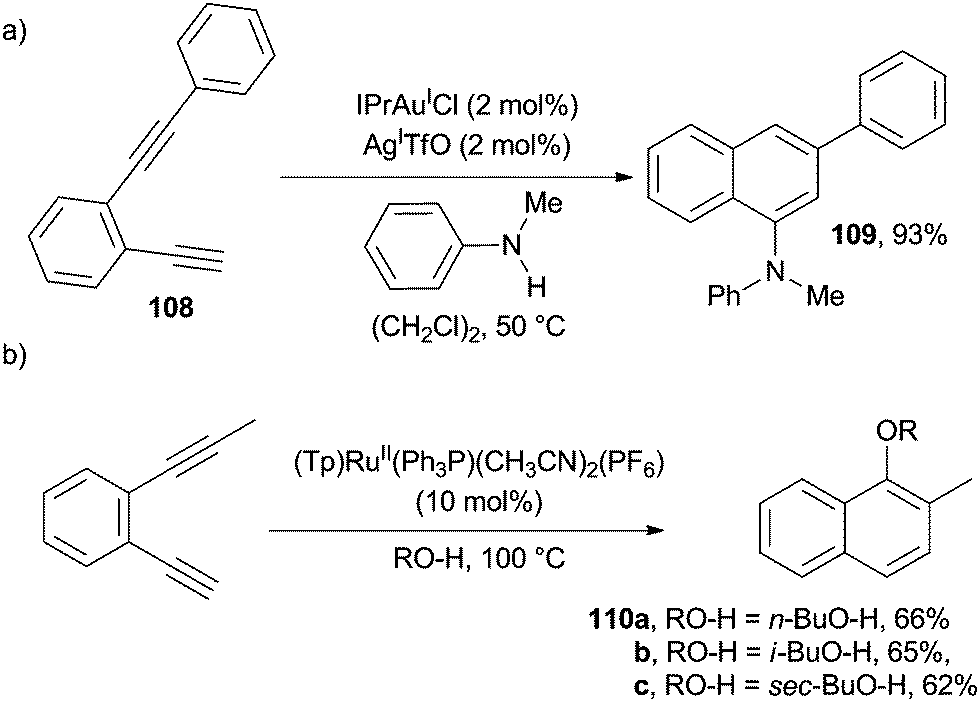 | ||
| Scheme 29 Synthesis of 1-substituted naphthalenes. | ||
Desilylation/cycloaromatization of arenediynes 111a–c to give 112a–c took place in good yields at 40 °C in the presence of stoichiometric amounts of Te0 and ultrasound irradiation. Notably, the reaction was repeated for substrate 111a on a 5 g scale, with a similar yield (68%), and on differently substituted substrates. Comparable results were consistently obtained when the mixture was irradiated with visible light (Scheme 30).150
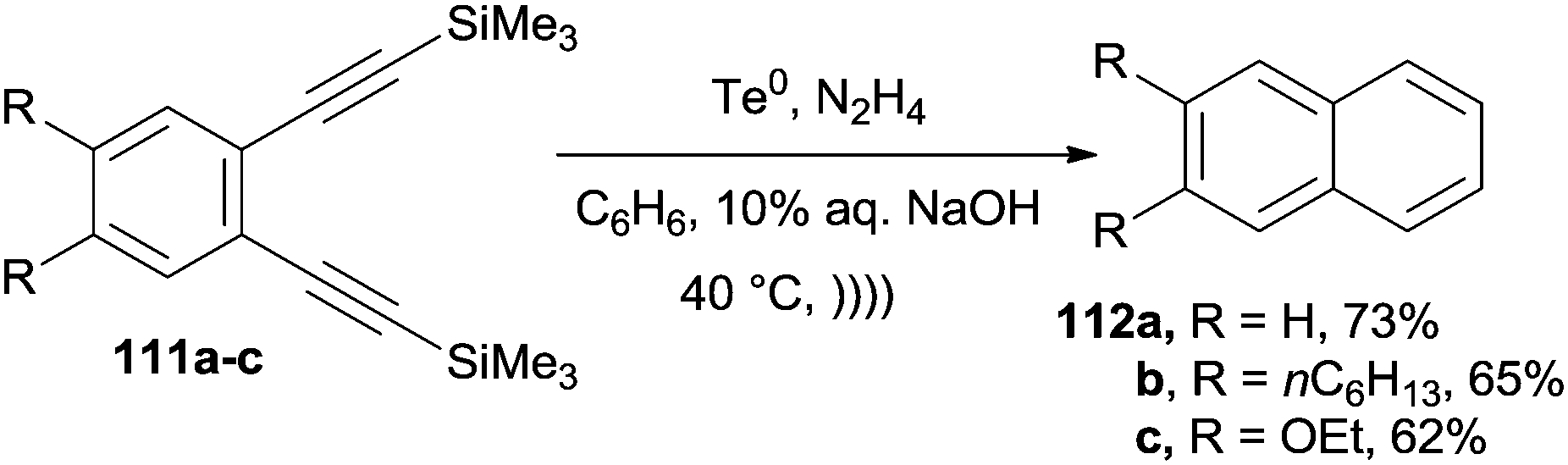 | ||
| Scheme 30 Te-catalyzed cyclization of silylated arenediynes. | ||
The highly coordinating PtIICl2 species is an efficient catalyst for promoting the cyclization of enediyne moieties having a tethered alkyl chain (e.g.113a–c). Under these conditions, the cycloaromatization process was followed by a C–H bond insertion to afford the corresponding naphthalenes 114a–c (Scheme 31).83
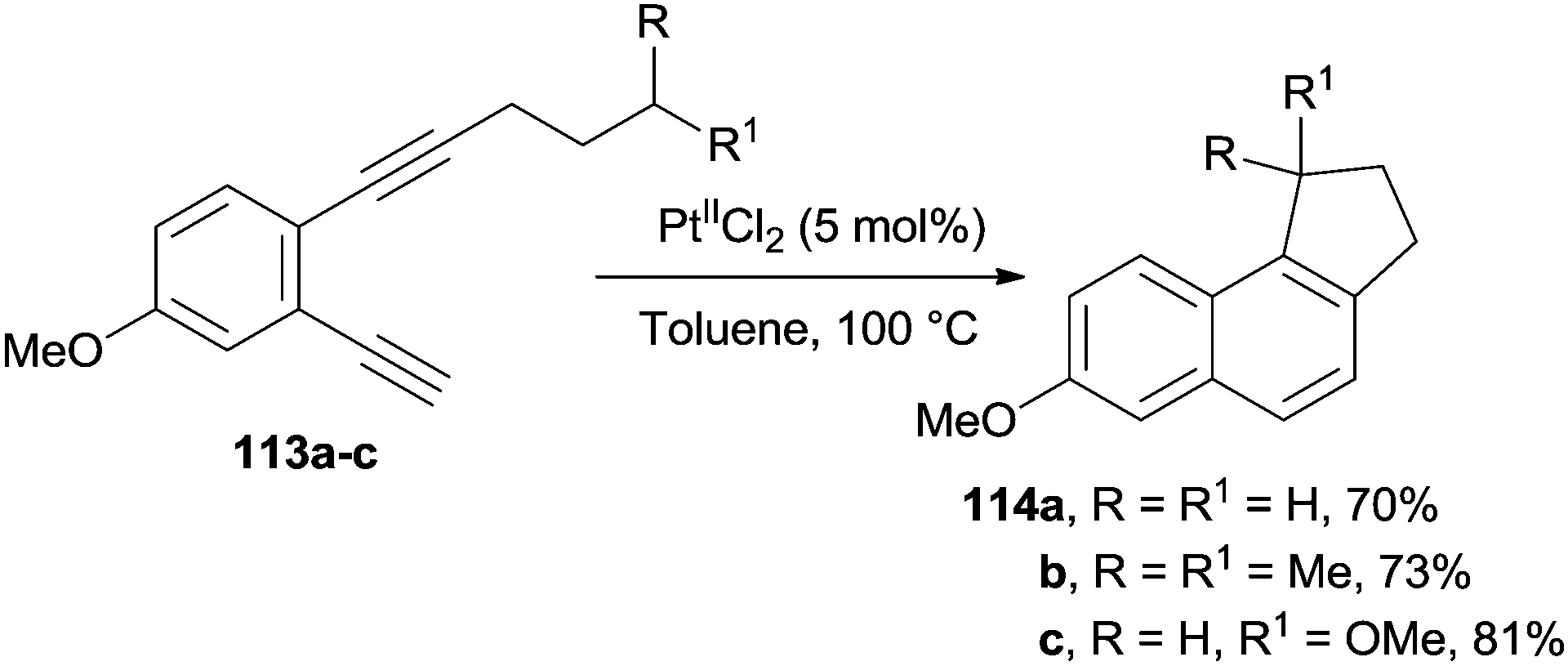 | ||
| Scheme 31 Pt-catalyzed synthesis of methoxynaphthalenes. | ||
Similarly, the PtIICl2-catalyzed cyclization cascade of enediyne–enone systems has been recently described.151 By following this approach, tetracyclic chromenones were obtained in excellent yields through two consecutive, highly regioselective, 6-endo-dig cyclizations, where two new C–C bonds and two new rings were formed. The proposed mechanism involved the initial coordination of the catalyst by one of the triple bonds of 115a–c to afford complexes 116a–c. Then, 6-endo-dig cyclization of the enone moiety occurred followed by deprotonation leading to intermediates 117−a–c. Protodemetallation of 117−a–c to 118a–c and ensuing coordination of the remaining triple bond by the catalyst afforded 119a–c. A second 6-endo-dig cyclization of the electron rich C5 pyran carbon onto the activated alkyne gave intermediates 120−a–c, which further underwent protodemetallation to give the final products 121a–c (Scheme 32).151 The same catalyst has been employed in the synthesis of benzo- and naphthopyranones from the corresponding enediynes.152 Treatment of 1,2-bis(ethynyl)arenes with hydrohalic acids in the presence of 5–10 mol% of PtIICl2 led to the formation of the corresponding 1-halonaphthalenes.153 As for metal-free activation procedures, 1,2-bis(alkynyl)arenes reacted with highly electrophilic boranes, yielding boryl-functionalized naphthalenes.154 Benzofused dihydroisoquinolones were obtained via TsOH-promoted cascade cyclization of diyne-tethered ynamides in the presence of AuI(CH3CN)[(2-biphenyl)(tBu)2P]SbF6 as the catalyst.155 The nucleophilic cycloaromatization of 1-aryl-3-hexen-1,5-diynes promoted by the methoxide anion was proposed as an efficient route to phenanthridinone and benzo[c]phenanthridinone derivatives.108
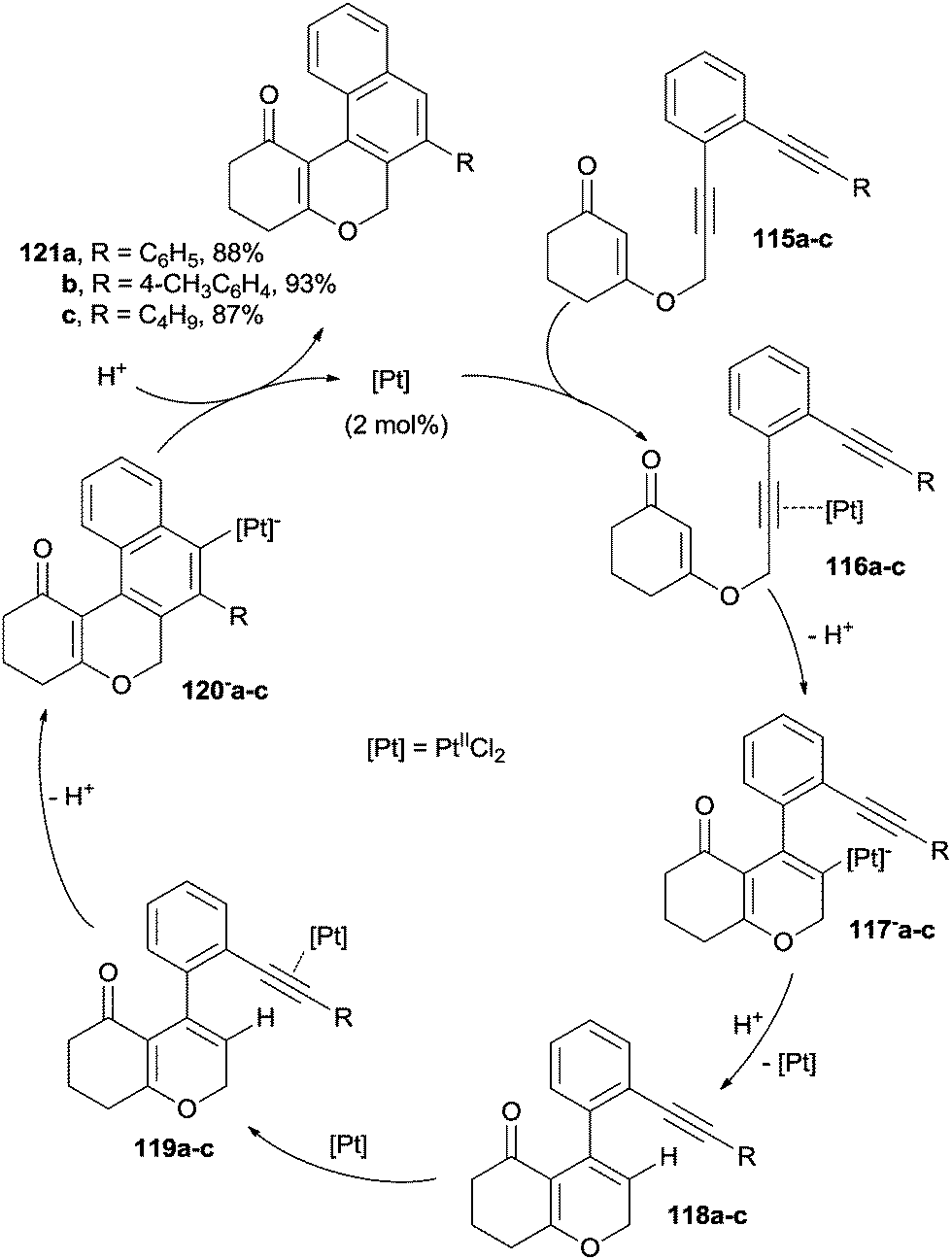 | ||
| Scheme 32 Pt-catalyzed synthesis of tetracyclic chromenones. | ||
Naphthocyclobutenes 125a–c were obtained by the treatment of the corresponding benzene-bridged bis(propargyl alcohols) 122a–c with Ph2PCl, in the presence of a base, via formation of 1,2-bis(phosphinyl)allenes 123a–c, which were in turn converted to the corresponding ortho-quinodimethane intermediates 124a–c (Scheme 33).156
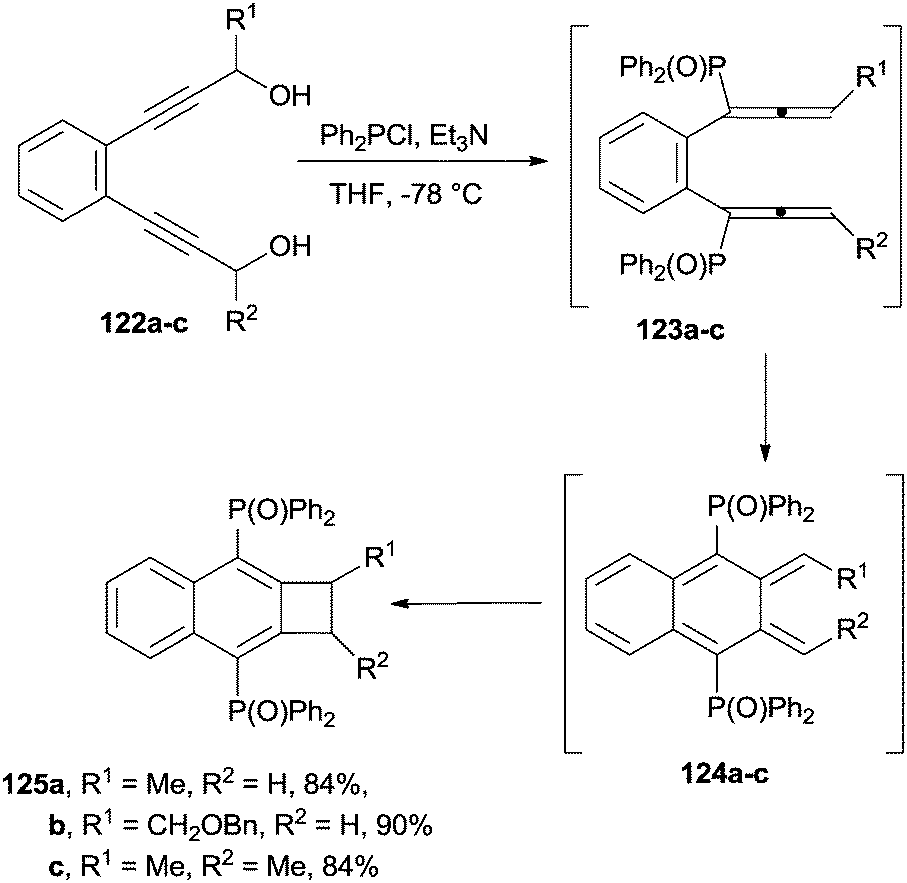 | ||
| Scheme 33 1,2-Bis(phosphinyl)allenes as intermediates in the synthesis of naphthalenes. | ||
The cascade rearrangement of glycine derivatives containing an enediyne moiety took place via a base-catalyzed 1,3-proton shift, followed by the Myers–Saito cyclization of the obtained enyne–allene. 1,5-Hydrogen atom transfer and intramolecular coupling of the resulting biradical led to the formation of tri- and tetracyclic heterocycles with a quaternary stereogenic center with a very high level of stereoselectivity. Scheme 34 shows the case of 126 that was converted in 5 h at 65 °C (99% conversion according to 1H NMR) to 127 as a single stereoisomer (82% yield).157
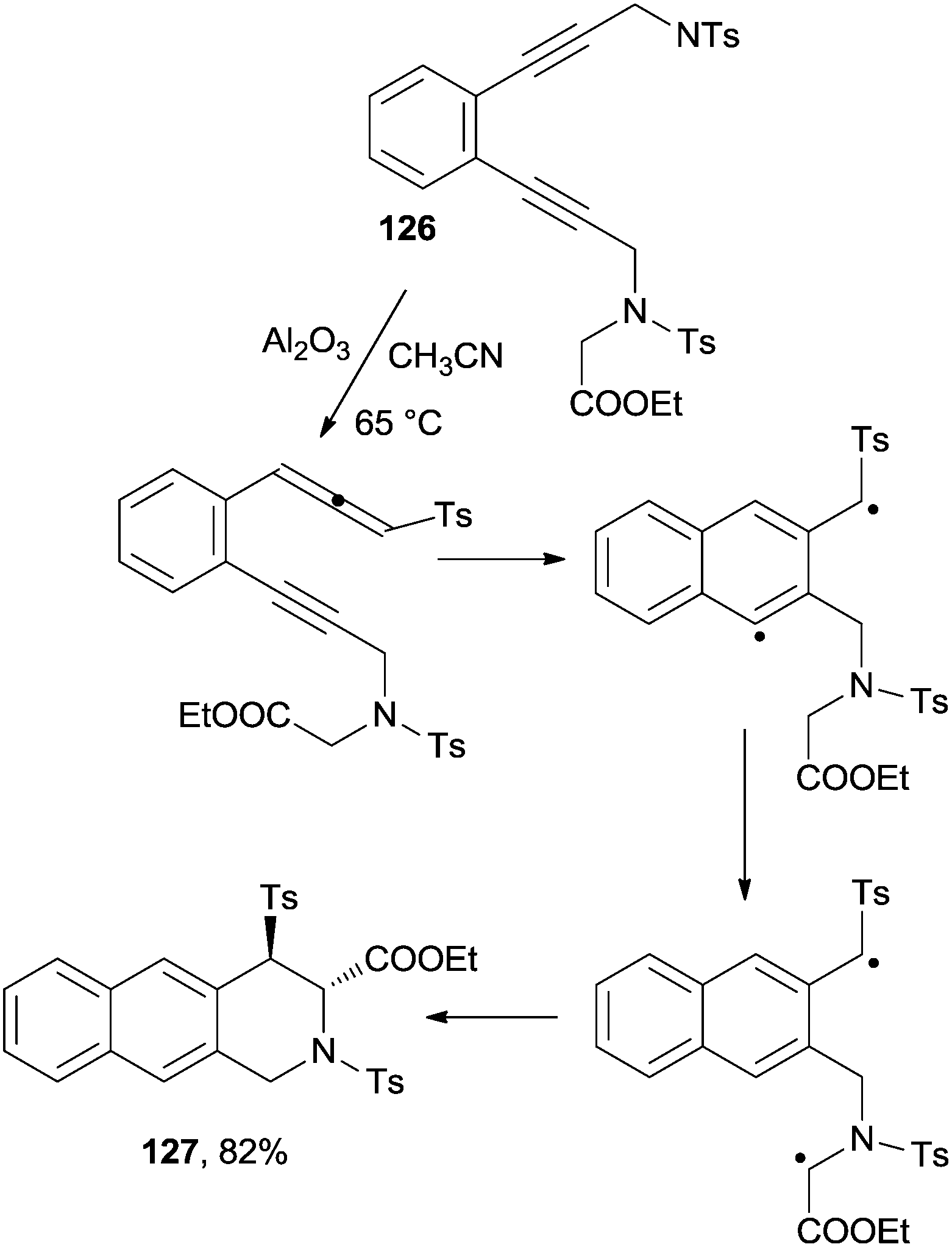 | ||
| Scheme 34 Formation of tricyclic heterocycle 127. | ||
Recently, the chance to apply a memory of chirality (MOC) approach to the Myers–Saito cycloaromatization has been debated. Six- and seven-membered ring α-aminoesters were obtained in a stereoselective fashion through a tandem Crabbé homologation-radical rearrangement of terminal enediyne precursors.158,159 Thus, upon reflux in dioxane in the presence of a secondary amine (Cy2NH), CuII and paraformaldehyde, enediyne 128 was converted into the corresponding enyne–allene 129 that underwent a spontaneous Myers–Saito cycloaromatization to α,3-didehydrotoluene 130. The latter evolved through a 1,5-hydrogen atom shift from the captodative position, leading to the formation of biradical 131 in a stereoselective fashion. Recombination of the two radical sites afforded cyclized product 132 (Scheme 35a).158,159
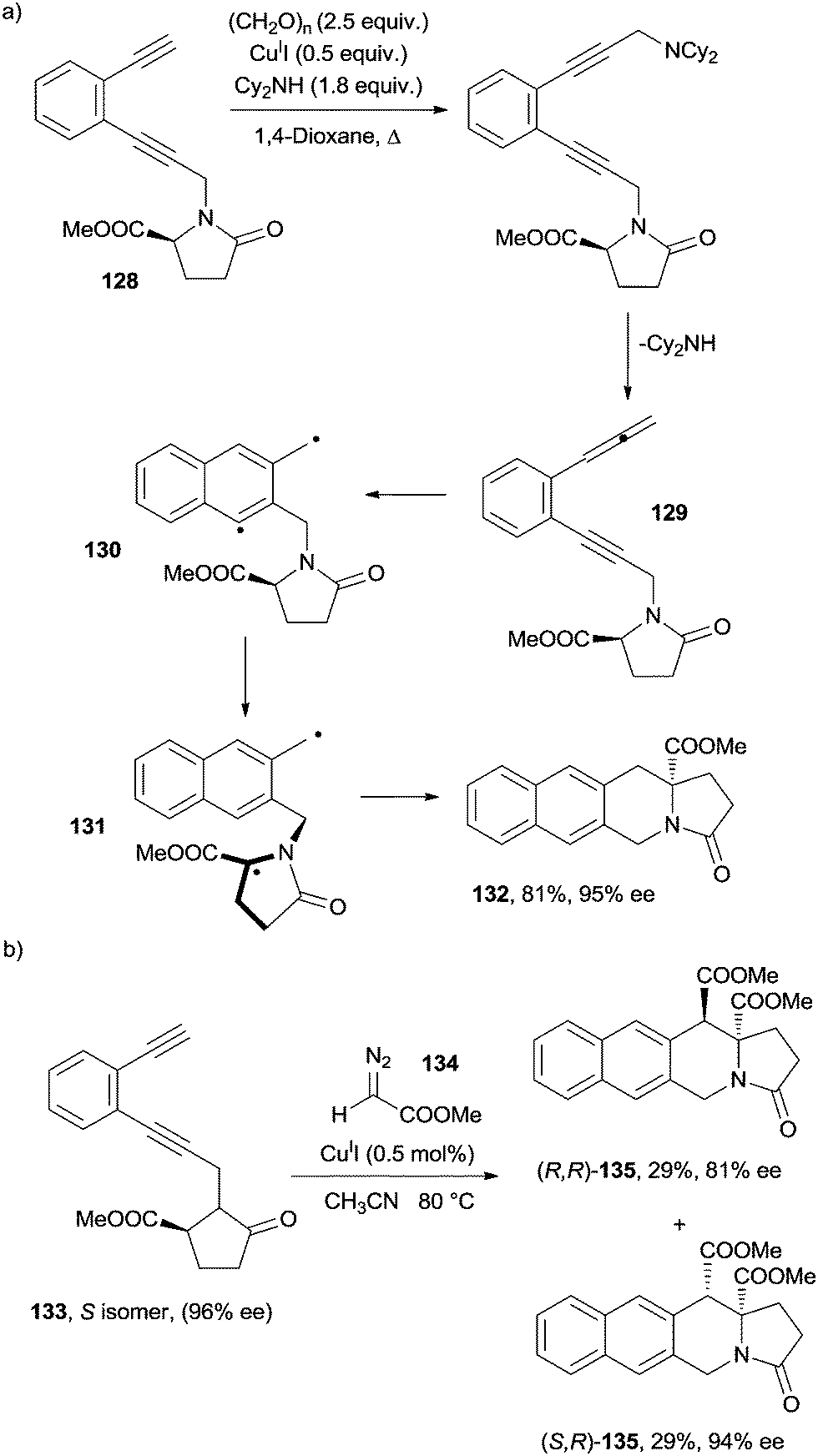 | ||
| Scheme 35 Enantioselective cycloaromatizations exploiting the memory of chirality approach. | ||
Two contiguous tetrasubstituted stereocenters were formed in compounds 135via the CuI-catalyzed coupling of enediyne 133 with diazo ester 134 (Scheme 35b).160 Recently, a mesoporous silica grafted with a tertiary amine was used as a basic nanocatalyst in the synthesis of 127 and 135 analogues from the corresponding enediynes.161
Cycloadditions ([2+2], [3+2] and [4+2]) taking place on alkynylchromium Fischer carbene complexes can trigger cascade reactions, in turn leading to the formation of functionalized polyaromatics. As an example, treating 136 with either 2,3-dihydrofuran (Scheme 36, path a) or nitrone 137 (path b) afforded adducts 138 and 139 that were in turn converted in discrete yields to the silylketene-containing naphthalene 140 and naphthoisooxazole 141, respectively (Scheme 36).162
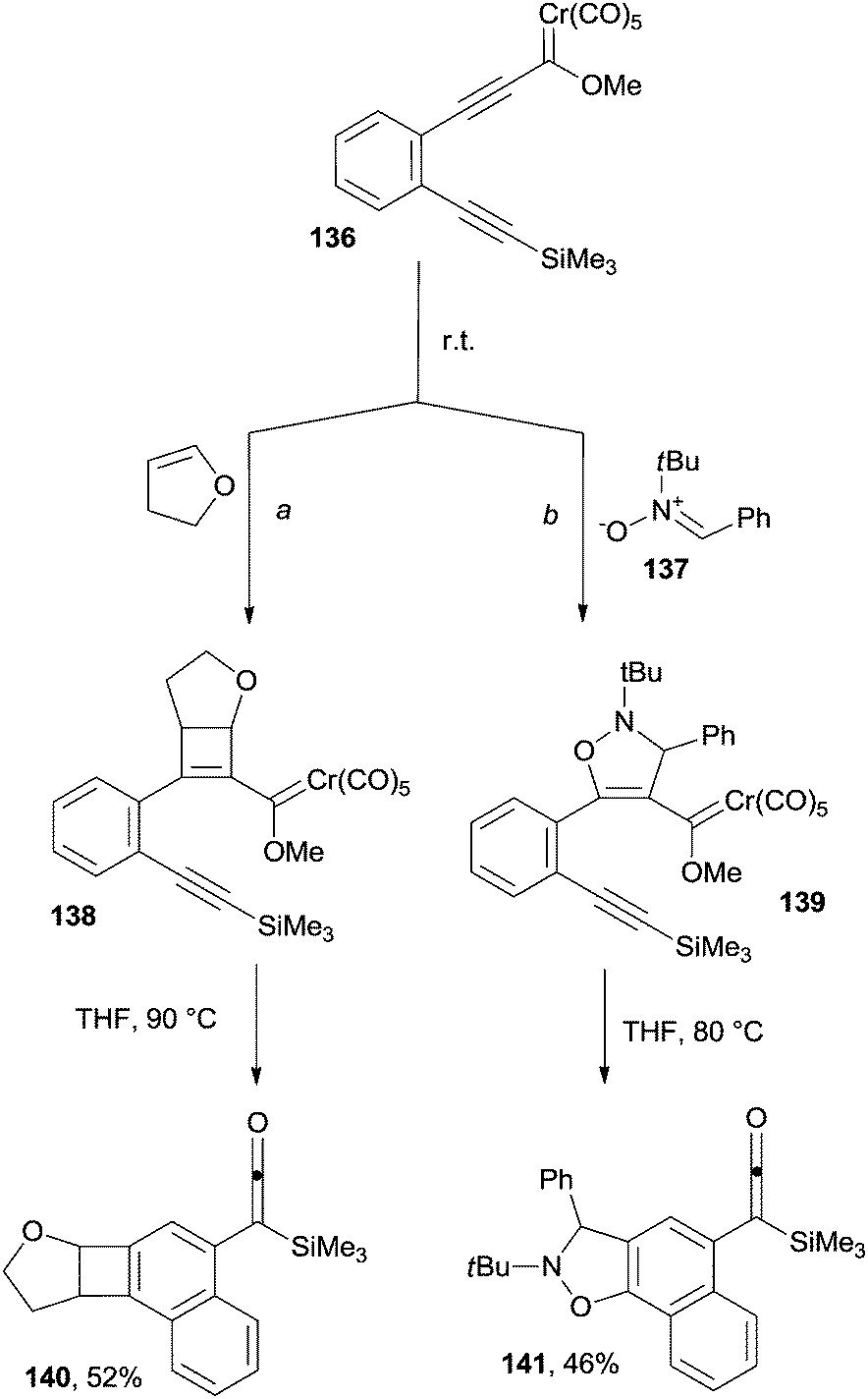 | ||
| Scheme 36 Fischer carbene complexes as reactants for the synthesis of substituted naphthalenes. | ||
With respect to the Bergman cycloaromatization, the adoption of transition-metal catalyzed methods in the related Myers–Saito cyclization has received considerably less attention. The synthesis of aromatic ketones 144a–c through the in situ generation of enyne–allene precursors is a representative example (Scheme 37).85 Enyne–allenes were obtained via an AgI-catalyzed sigmatropic rearrangement of an alkyne group. Thus, coordination of the AgI species to the triple bond of propargyl esters 142a–c resulted in the formation of enyne–allenes 143a–c through a [3,3]-sigmatropic rearrangement (Scheme 37, cycle A). The involvement of 143a–c has been confirmed by isolation and characterization of these intermediates. Ensuing coordination of the residual alkyne moiety by the AgI species promoted a 6-endo-dig addition of the allenyl acetate to afford, after protodemetallation, 144a–c in good yields (cycle B).
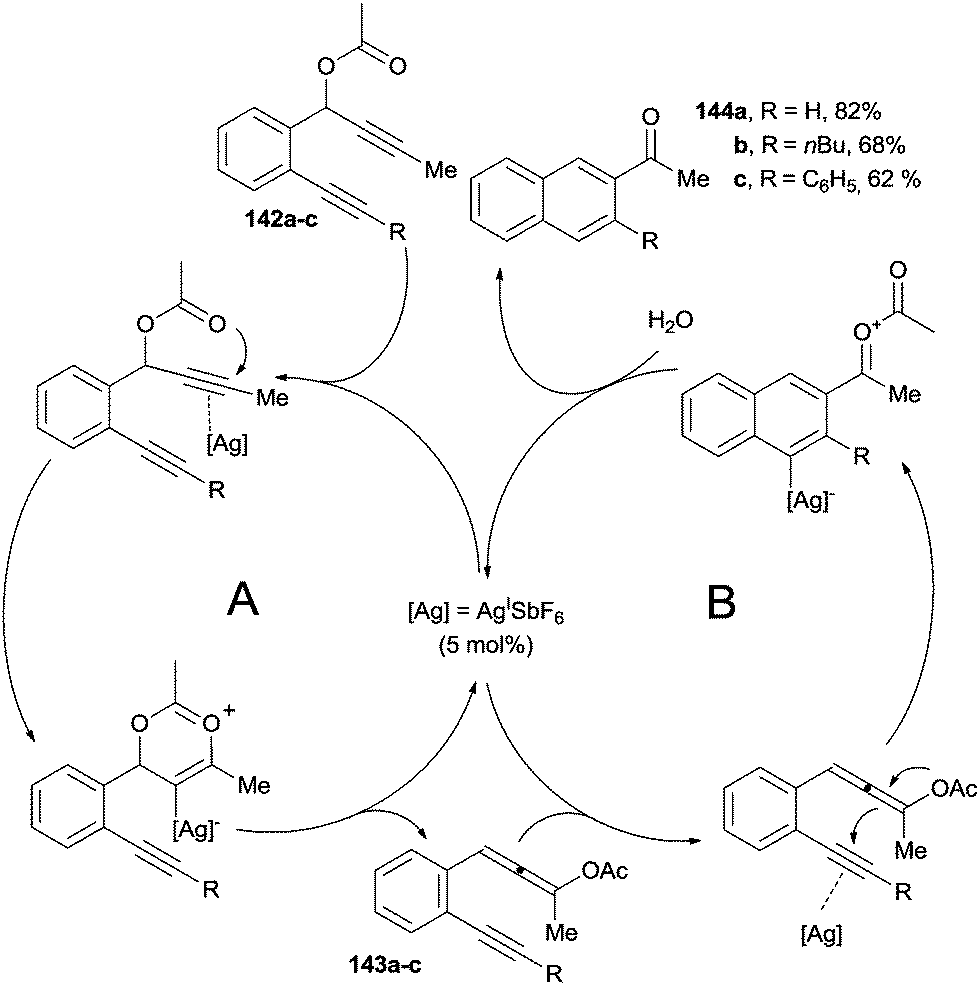 | ||
| Scheme 37 Acyl naphthalenes from the silver-catalyzed reaction of enyne–allenes. | ||
The base-promoted [2,3]-sigmatropic shift taking place on the enediyne moiety in derivatives 145a–c led to the formation of enyne–allenes 146a–c and substituted benzo[e]indanes 147a–c from them via α,3-didehydrotoluene biradicals (Scheme 38).138
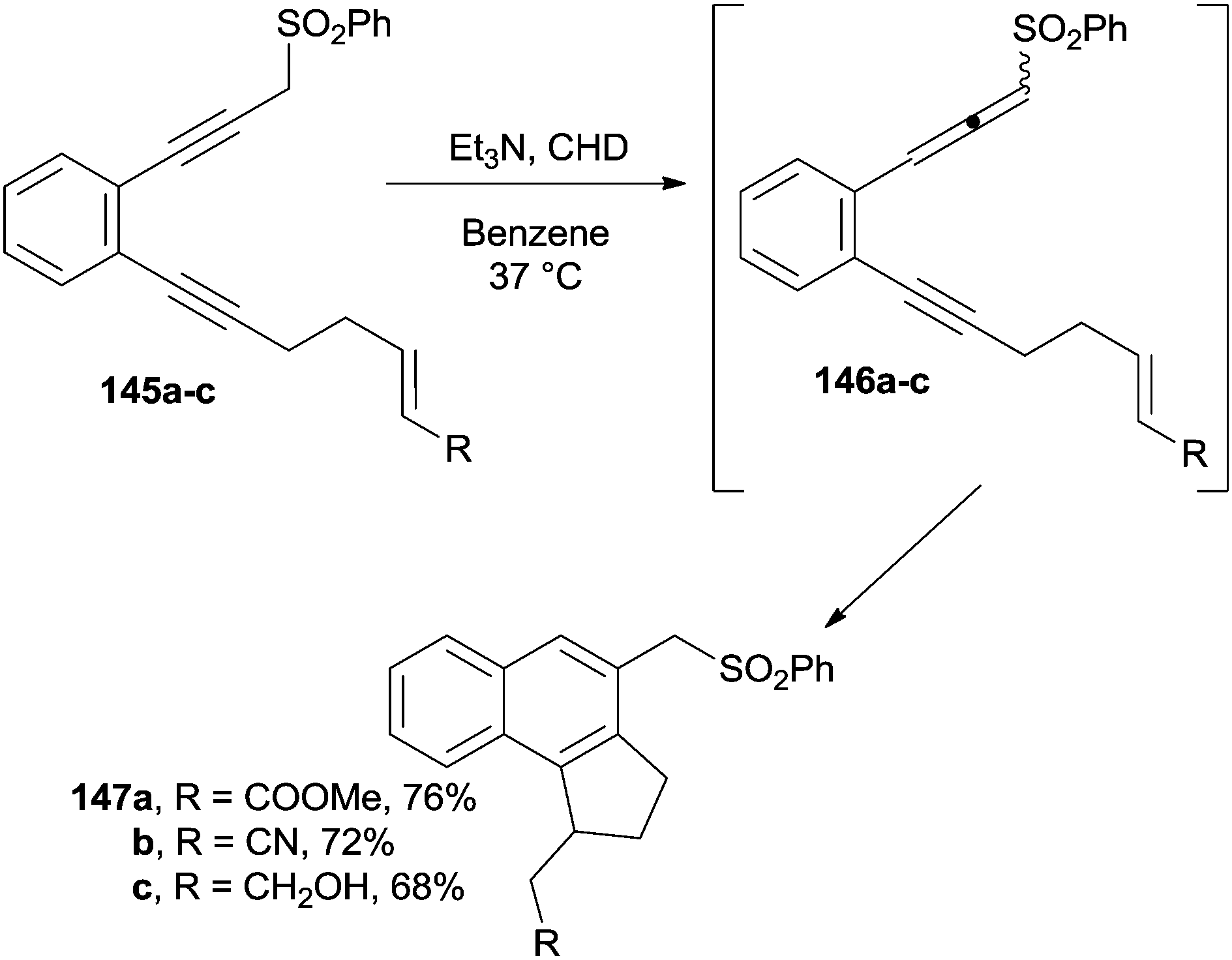 | ||
| Scheme 38 Smooth preparation of benzo[e]indanes. | ||
The Schmittel cyclization, a competing pathway for Myers–Saito cycloaromatization, has been exploited as well in the synthesis of polycondensed aromatic rings. Thus, naphthyl (see compound 152 in Scheme 39) and binaphthyl derivatives were obtained by treatment of the corresponding enediynes (e.g.148) with potassium tert-butoxide in refluxing toluene.163 A 1,3-prototropic rearrangement was responsible for the in situ formation of enyne–allene 149 which in turn underwent a Schmittel cyclization to generate biradical 150. An intramolecular radical–radical coupling finally afforded the corresponding benzo[b]fluorene derivative 152 upon hydrogen shift occurring in 151. A small amount of compound 153 was likewise found as a by-product (Scheme 39). This approach has been also exploited in the two-step synthesis of bowl-shaped polycyclic aromatic hydrocarbons (see Section 5).164
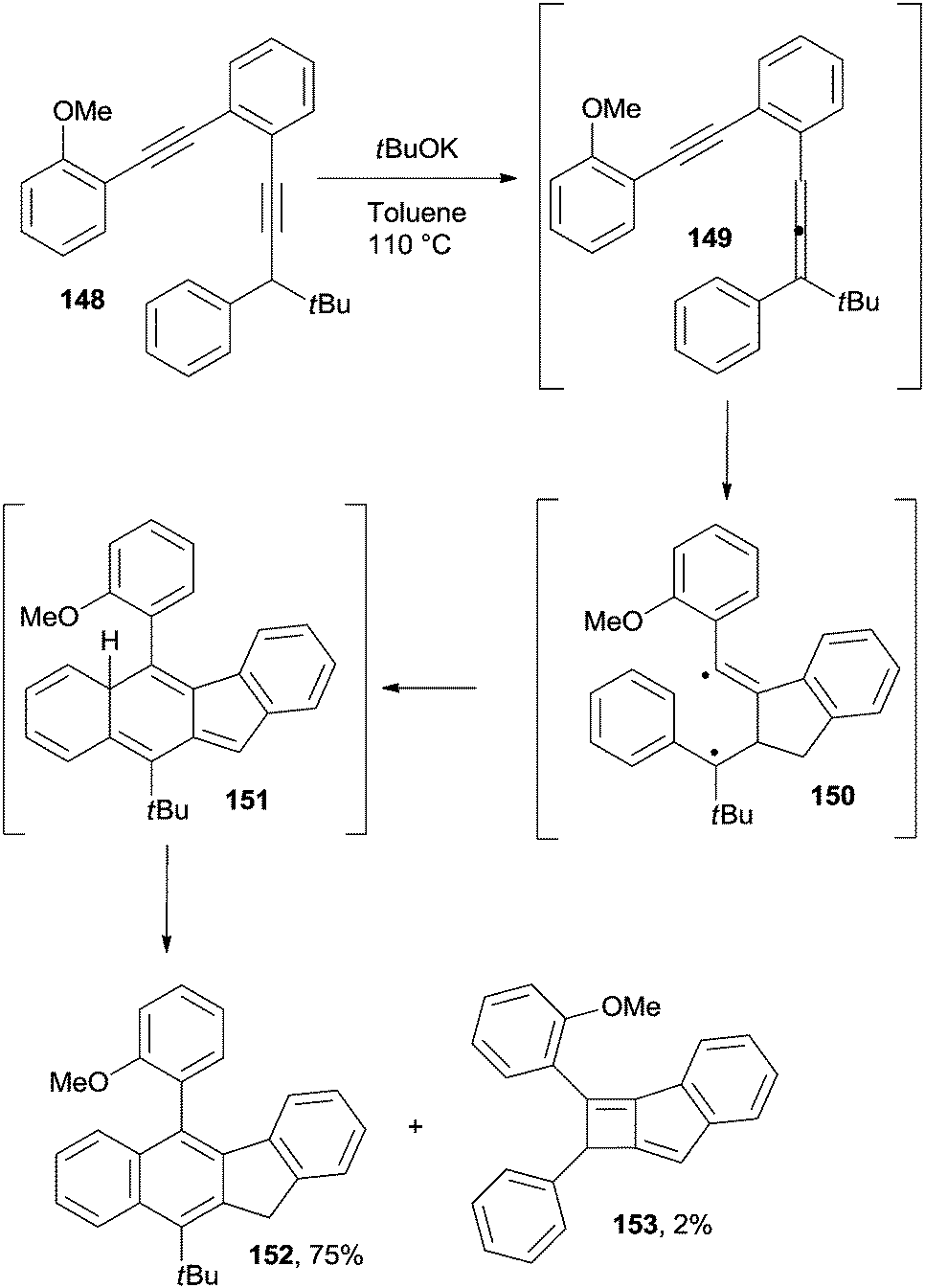 | ||
| Scheme 39 Base-induced reactions on enediynes to give benzo[b]fluorene derivatives. | ||
The triazole–AuI (TA–AuI) catalyzed propargyl vinyl ether rearrangement taking place at room temperature in compounds 154a–c (Scheme 40) triggered a Schmittel cyclization and the subsequent formation of the substituted naphthalene core in 155a–c. Notably, the mild conditions employed avoided the need for sterically hindered groups (such as a tBu) on the allene moiety to maintain a satisfactory selectivity.165
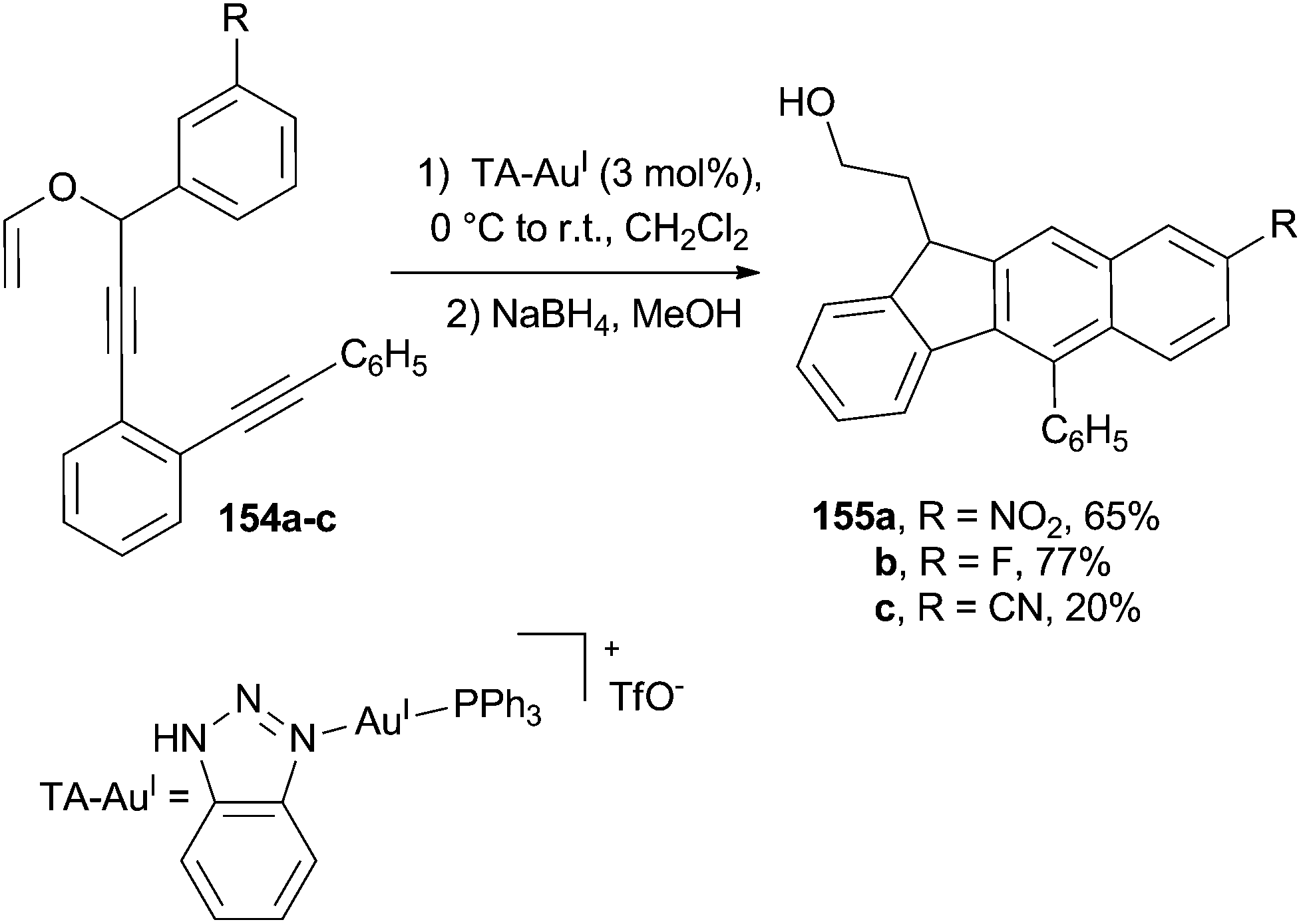 | ||
| Scheme 40 Schmittel cyclization as the key step for the formation of a substituted naphthalene core. | ||
1,2-Ethynylbenzenes bearing one unsubstituted and one triarylmethyl-substituted alkyne (156a–c) were efficiently converted into substituted benzofluorenones 158a–c through a one-pot reaction involving a gold-catalyzed cyclization to indenones 157a–c followed by a photochemical cyclization/oxidation sequence (Scheme 41).166
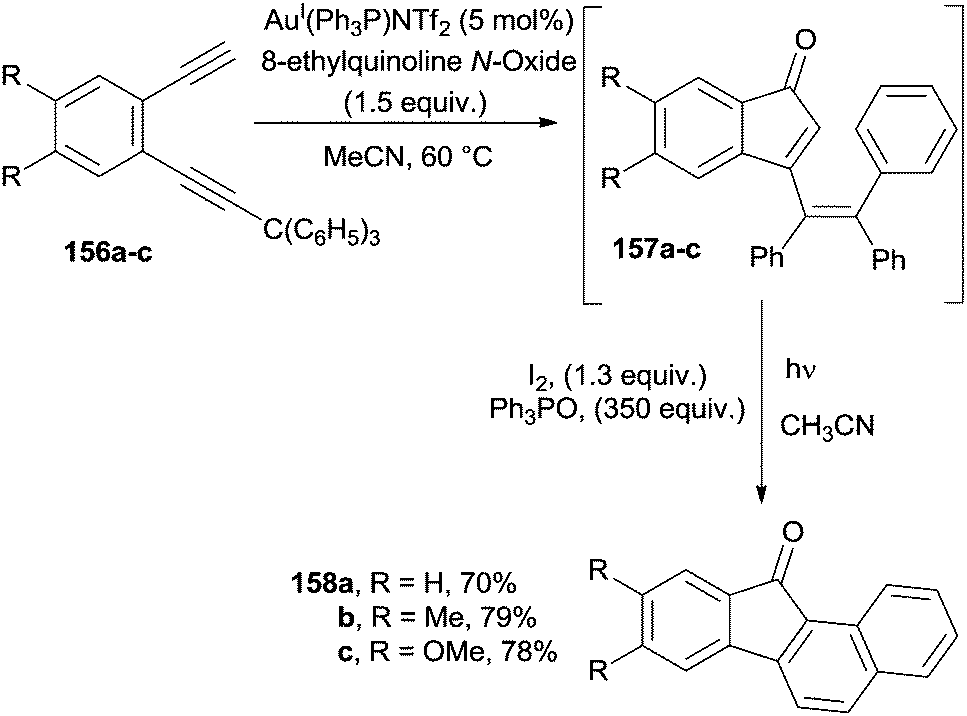 | ||
| Scheme 41 Benzofluorenones by a two step process from 1,2-ethynylbenzenes. | ||
5 Synthesis of benzocondensed derivatives
Highly extended aromatic cores have recently gained increasing attention, particularly due to their application in the chemistry of materials. This section deals with the synthesis of aromatic nuclei larger than naphthalene (see Section 4) and the selected examples have been classified according to the size of the final aromatic nucleus.Anthracene and its derivatives are linear polycyclic aromatic compounds showing high potential for use in materials science, particularly due to their photophysical properties (e.g. fluorescence probing, photochromic systems, and electroluminescence).167 Indeed, the substitution pattern plays a fundamental role in determining the actual properties.
The preparation of anthracene derivatives bearing a ketone group at the 9-position (160a,b) has been carried out under AuICl-catalyzed conditions, starting from o-alkynyl(oxo)benzenes 159a,b in the presence of benzenediazonium 2-carboxylate. The process occurred under mild conditions and good yields of the desired compounds were obtained (Scheme 42; for the mechanism, see Scheme 10).89
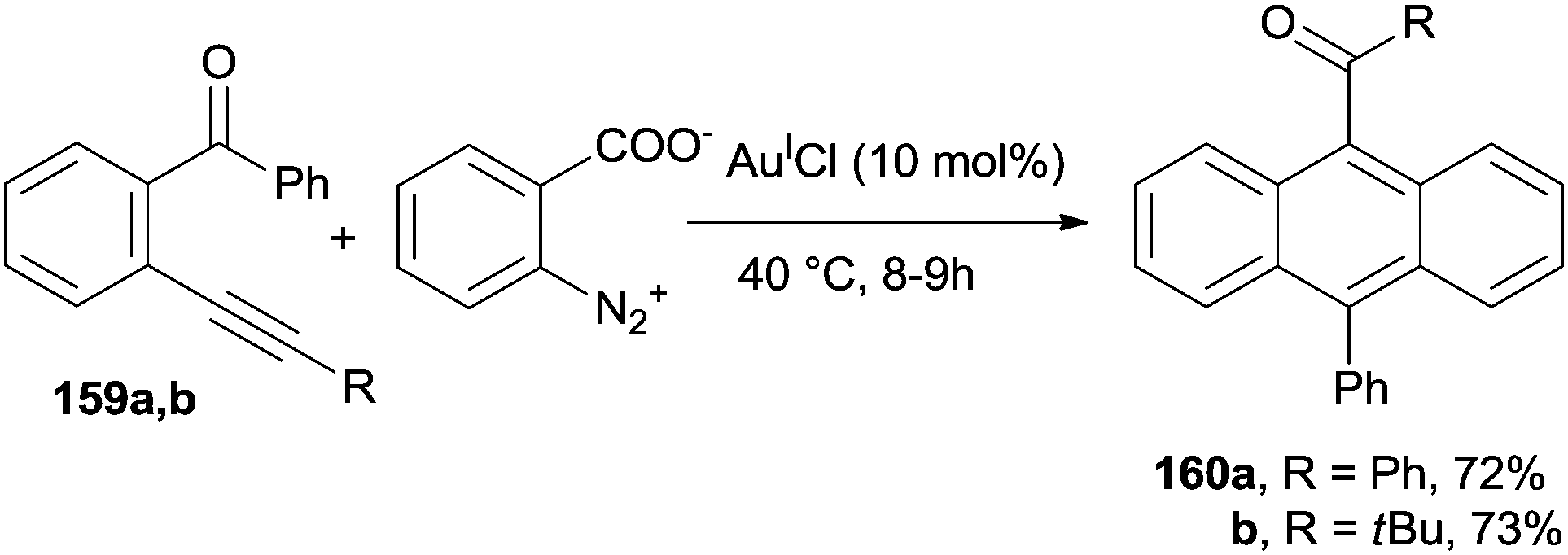 | ||
| Scheme 42 AuI-catalyzed conversion of o-alkynyl(oxo)benzenes into anthracene derivatives. | ||
In a different example, anthracenes bearing substituents on the outer rings (a non-trivial synthetic target) have been prepared by a cycloaromatization process involving 2,3-bis(bromoethynyl)naphthalene 162 (Scheme 43). This substrate has been in turn easily prepared via the desilylative halogenation of the corresponding trimethylsilyl derivative 161. Heating a solution of the so prepared enediyne in a 10:1v/v benzene/CHD mixture at 180 °C in a steel bomb for 2 h led to 2,3-dibromoanthracene 163 in a good yield.168 Further alkynylation of the resulting haloaromatic compound to afford the corresponding 2,3-diethynyl anthracene 164 generated a new enediyne system, homologated by one aromatic ring. This iterative methodology was also applied to the synthesis of unsubstituted naphthacenes.168
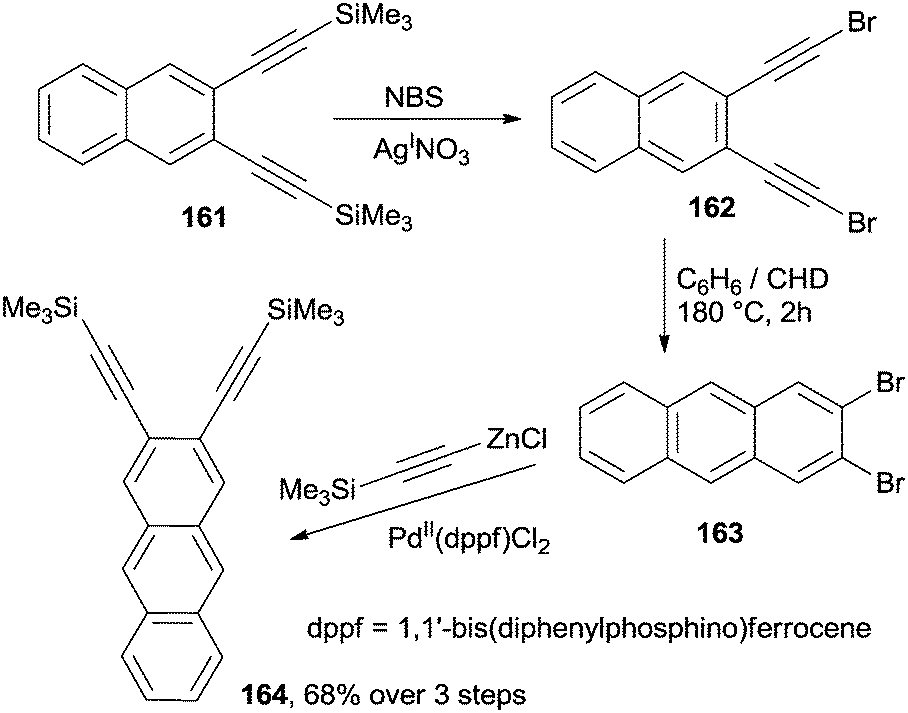 | ||
| Scheme 43 Multistep synthesis of 2,3-disubstituted anthracenes via Bergman cyclization. | ||
Similar conditions were applied to the synthesis of 2,3,6,7-tetrabromoanthracene from the corresponding 1,2,4,5-tetrakis(bromoethynyl)benzene.169
Apart from acenes, polyenes have been likewise used for the synthesis of angularly fused derivatives. Phenanthrenes have been prepared adopting a photochemical approach starting from the so-called Y-enynes, viz. cross-conjugated enediynes, such as 165 (Scheme 44). Thus, irradiation of 165 at 350 nm promoted the photocyclization to 167 through the allene intermediate 166. Interestingly, the mechanism was demonstrated to depend on the solvent polarity, with a 1,5-H shift or protonation at the central allene carbon occurring in non-polar (benzene and cyclohexane) and polar (methanol) solvents, respectively.170,171
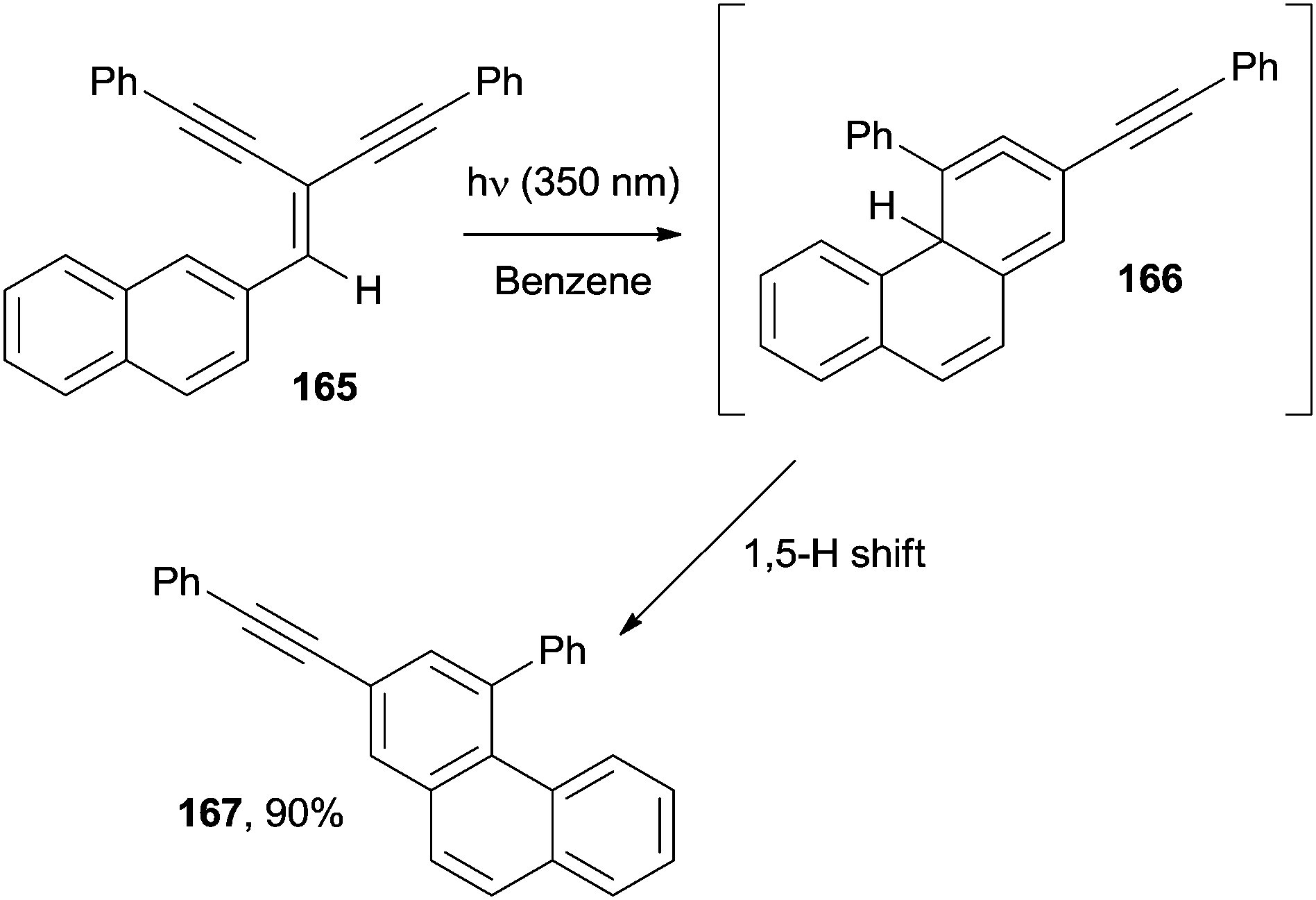 | ||
| Scheme 44 Photoinduced preparation of substituted phenanthrenes. | ||
In a different approach, phenanthrenes have been synthesized via a C2–C6 cyclization occurring in enyne–allenes. In particular, this strategy gave access to 4,5-diarylphenanthrenes (174 in Scheme 45) having a helical twist. The reaction involved treatment of the precursor (168) with tBuOK in refluxing toluene for up to 10 h, depending on the substitution pattern. The transformation proceeded through benzannulated enyne–allene 169, in turn formed via a prototropic isomerization. A Schmittel cyclization ensued, giving non-aromatic biradical 170 and then intermediate 171 upon reaction with the central benzene ring. The same sequence was repeated through intermediates 172 and 173, finally leading to product 174 upon tautomerization (Scheme 45).172 The structure of 174 was demonstrated via X-ray analysis to bear the two phenyl substituents markedly bent away from each other, with the central aromatic system severely distorted.172 Interestingly, highly congested diindenophenanthrenes containing four phenyl groups could be likewise synthesized by having recourse to this approach.173 Similar strategies have been adopted to synthesize diindeno-fused 4H-cyclopenta[def]phenanthren-4-ones174 and a set of angularly fused polycyclic aromatic hydrocarbons containing the phenanthrene core.175–177
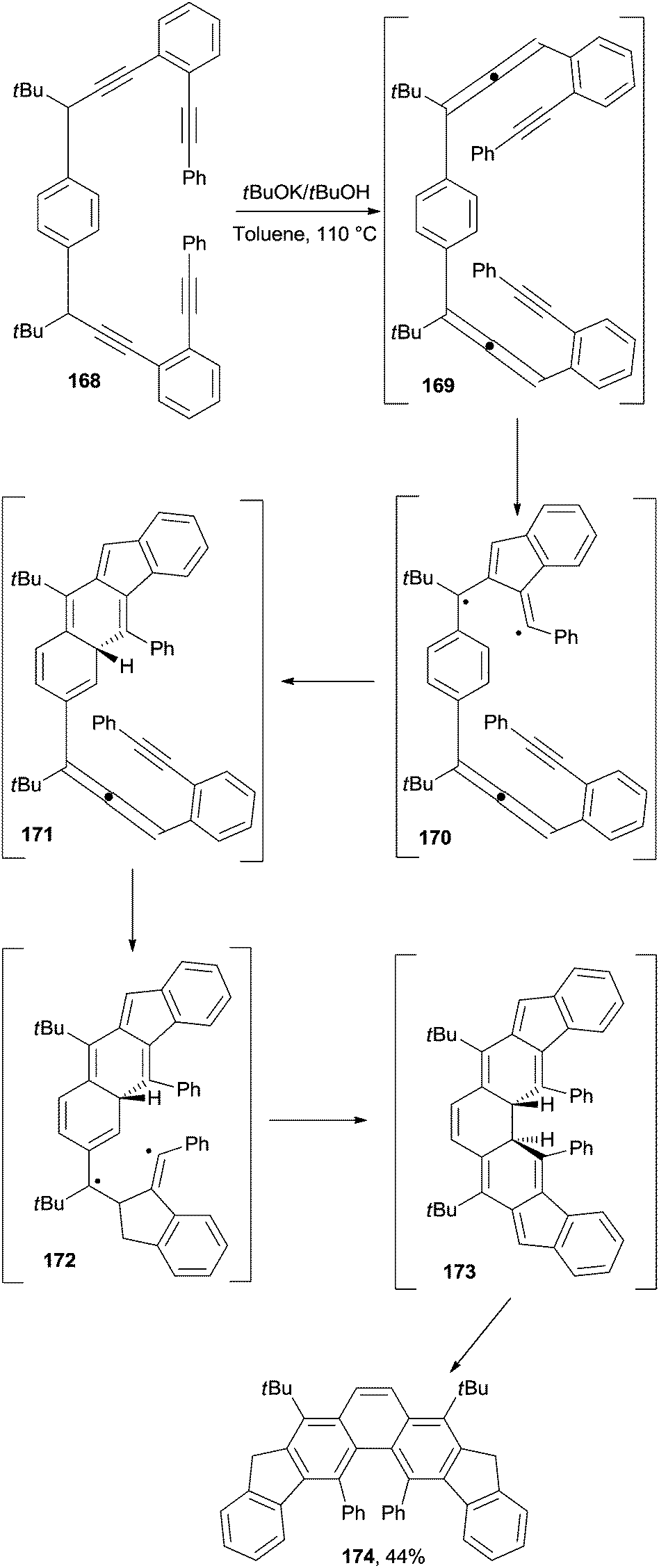 | ||
| Scheme 45 Pathway for the base-induced synthesis of diindenophenanthrenes. | ||
Phenanthrenes can be likewise synthesized from biphenyl aryl acetylenes (175a–c) under radical conditions, in the presence of a tin hydride derivative (Bu3Sn-H) and an initiator (azobisisobutyrronitrile, AIBN). Thus, 175a–c were treated in refluxing toluene to give the corresponding stannylated phenanthrenes 176a–c (Scheme 46). Interestingly, the Sn-substituent could be removed via acid hydrolysis to give 177a–c in good yields.178
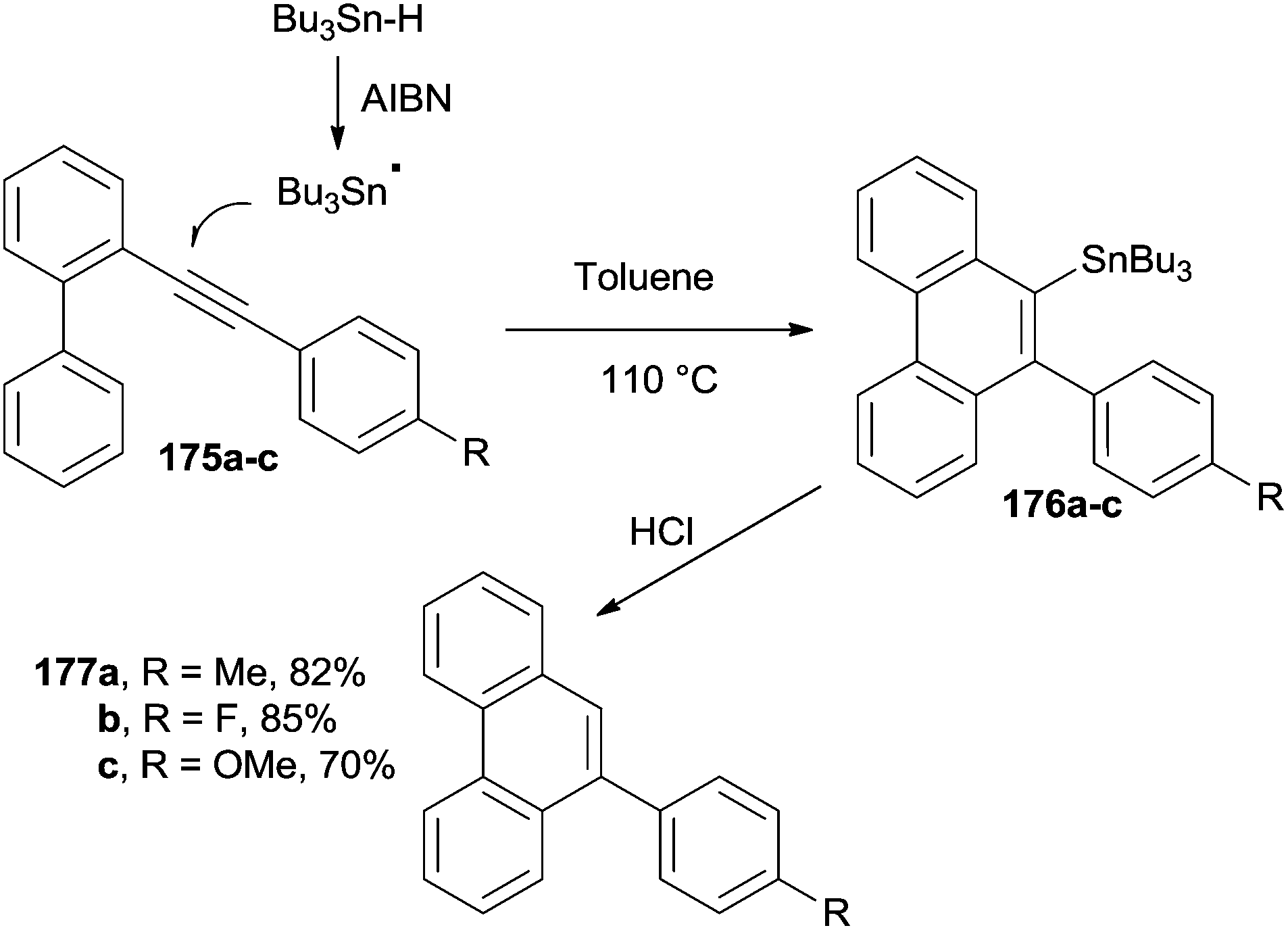 | ||
| Scheme 46 Radical initiated cycloaromatization. | ||
Turning to aromatic hydrocarbons bearing 4 nuclei, a large number of isomeric structures are possible. Tetraphene derivatives are important due to the presence of this structural motif in landomycins, a class of antibiotics with potent antitumor and antibacterial activity. During a synthetic study intended to the synthesis of the structural motif contained in landomycinone 178 (Scheme 47), tetraphene 180 was synthesized in 25% yield starting from enediyne 179 through a Masamune–Bergman cyclization followed by the undesired elimination of the OH group (Scheme 47).179
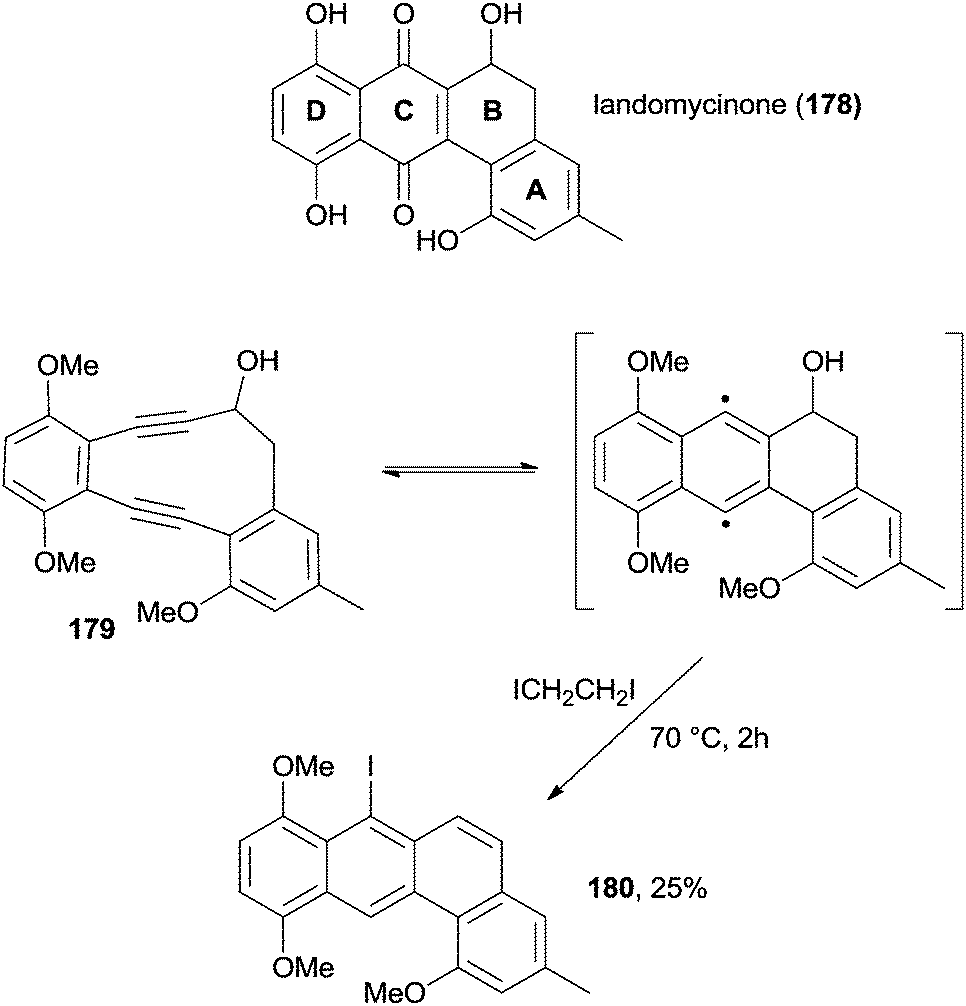 | ||
| Scheme 47 Synthesis of the tetraphene core of landomycins. | ||
[4]Helicenes differ for the arrangement of the fused aromatic rings and can exist in two enantiomeric forms, due to steric hindrance that forces the molecule to adopt a helical-like structure.
Acyclic diaryl ketones can be exploited for the synthesis of [4]helicenes through a four-step reaction sequence, involving a Corey–Fuchs olefination, a Sonogashira coupling, a desilylation step and a RuII-catalyzed annulation process. Thus, benzophenone 181 was treated with CBr4 and Ph3P to yield dibromide 182 that was in turn converted to enyne 183 in the presence of PdII(Ph3P)2Cl2. After the removal of the two Me3Si-groups promoted by the fluoride ion, 184 was treated with a catalytic amount (ca. 30 mol%) of RuII(Ph3P)(η6-p-cymene)Cl2 to afford [4]helicene 185 in a good yield (60% over the last two steps; Scheme 48). The mechanism proposed for the 184 to 185 conversion was postulated to involve the initial formation of a vinylidene–Ru intermediate (see Scheme 8).180 Interestingly, (Tp)RuII(Ph3P)(CH3CN)2PF6 was likewise found to be very active in a plethora of catalytic benzannulation reactions involving the same strategy. Indeed, different polycyclic aromatic hydrocarbons, such as phenanthrene and various substituted coronene derivatives, could be synthesized.181
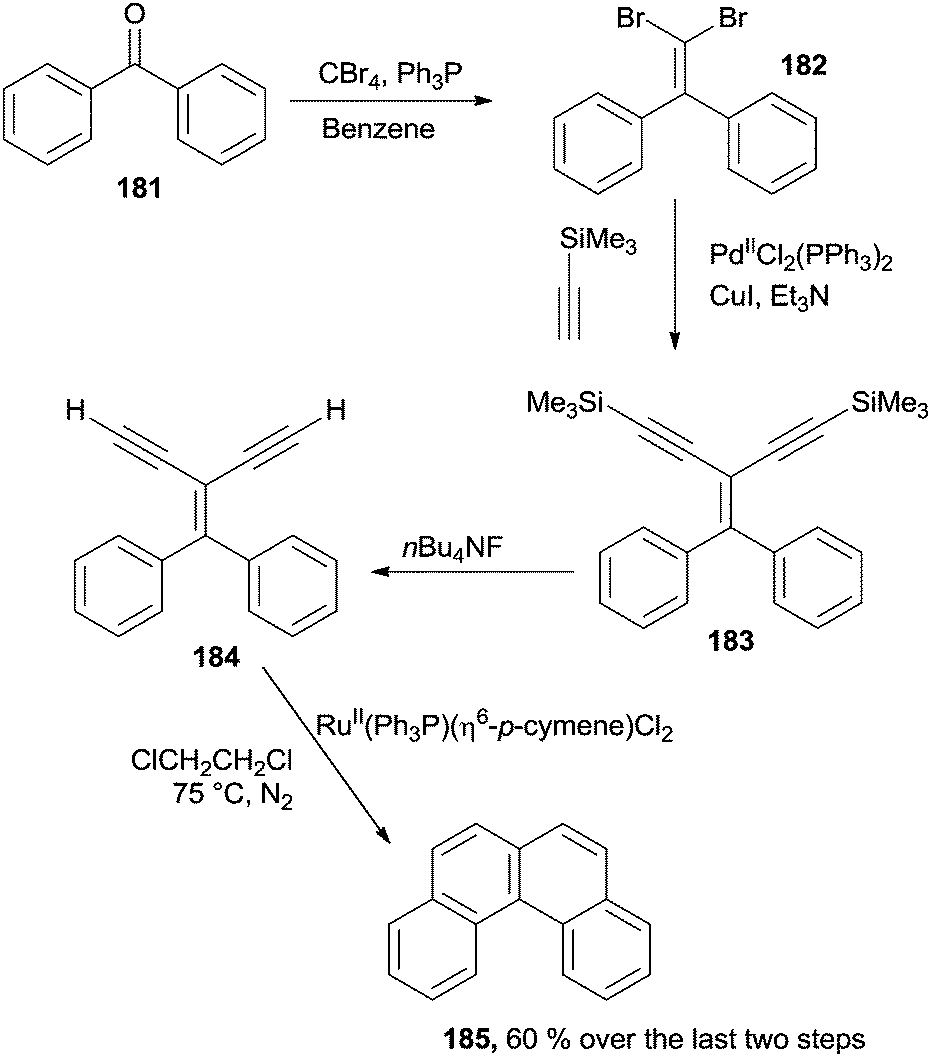 | ||
| Scheme 48 RuII-catalyzed synthesis of [4]helicenes. | ||
A general method for the synthesis of [4]helicenes is based on a Bergman cyclization applied to alkenyl enediynes (Scheme 49). Thus, compounds 186a–d were heated at 90 °C in DMSO for 10–38 h to afford the corresponding (substituted) [4]helicenes in good yields. The proposed mechanism, also supported by deuterium labeling experiments by using d6-DMSO as the solvent, claimed the involvement of three different biradical species. Biradicals 187a–d arose from the initial Bergman cyclization and then underwent an intramolecular addition onto the lateral aromatic ring to give 188a–d and then 189a–d upon intramolecular hydrogen atom abstraction. Finally, radical quenching by the solvent ensued to give 190a–d. Aromatization via elimination of a hydrogen molecule led to [4]helicenes 191a–d (Scheme 49).182
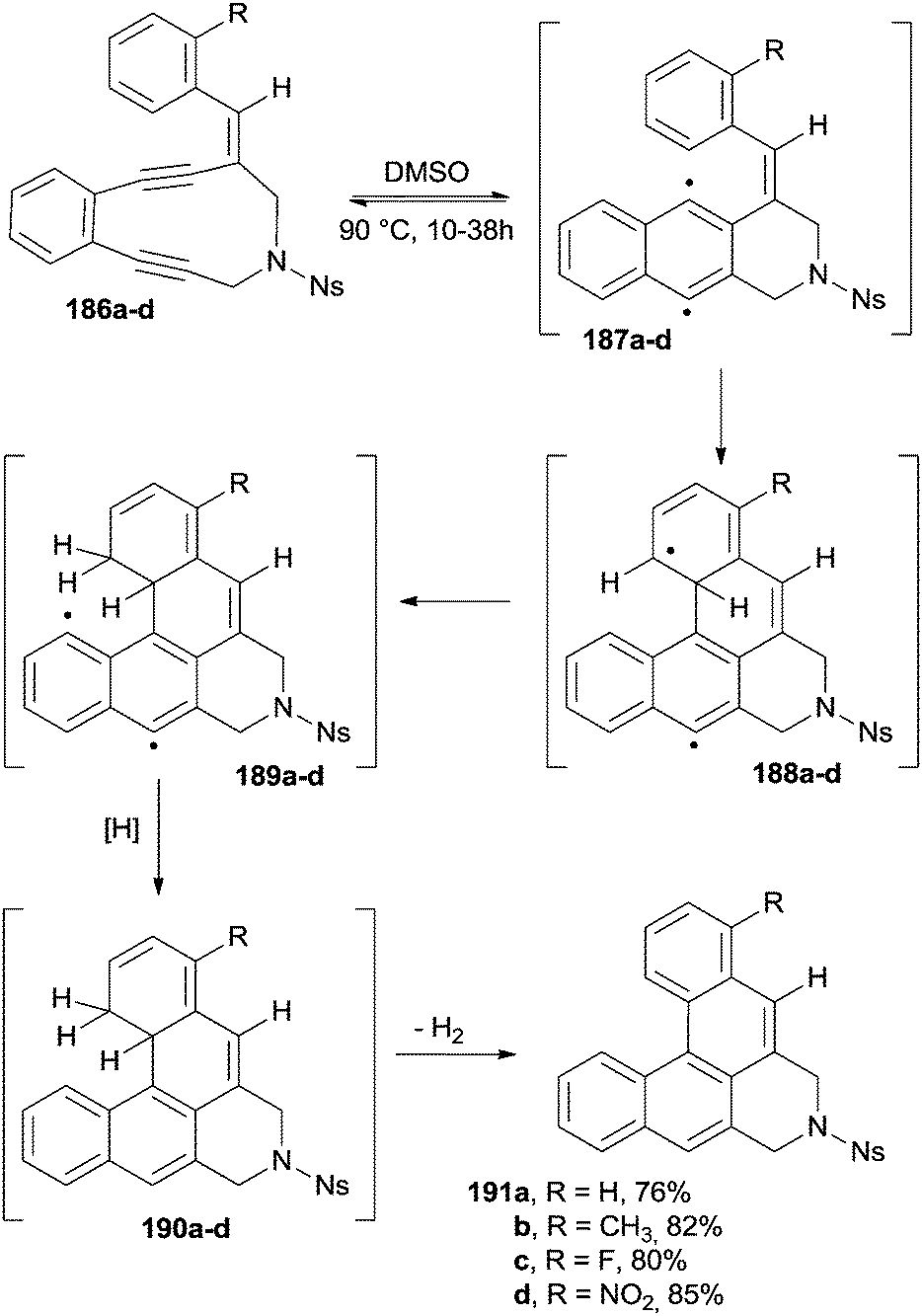 | ||
| Scheme 49 Radical cascade in the synthesis of [4]helicenes 191a–d (Ns = 4-NO2C6H4). | ||
Interestingly, chiral amino acids could be introduced into the skeleton of the starting material, imparting some diastereoselectivity to the process (the best results were obtained with L-leucine appended enediynes).183 The same synthetic strategy could be extended also to the synthesis of [5]helicenes and was based on the use of naphthyl-substituted alkenyl enediynes.183
The same base-promoted approach reported above for the synthesis of phenanthrenes (Scheme 45) has been successfully extended to [4]helicenes (192, Scheme 50a)175 and [5]helicenes (193, Scheme 50b).175
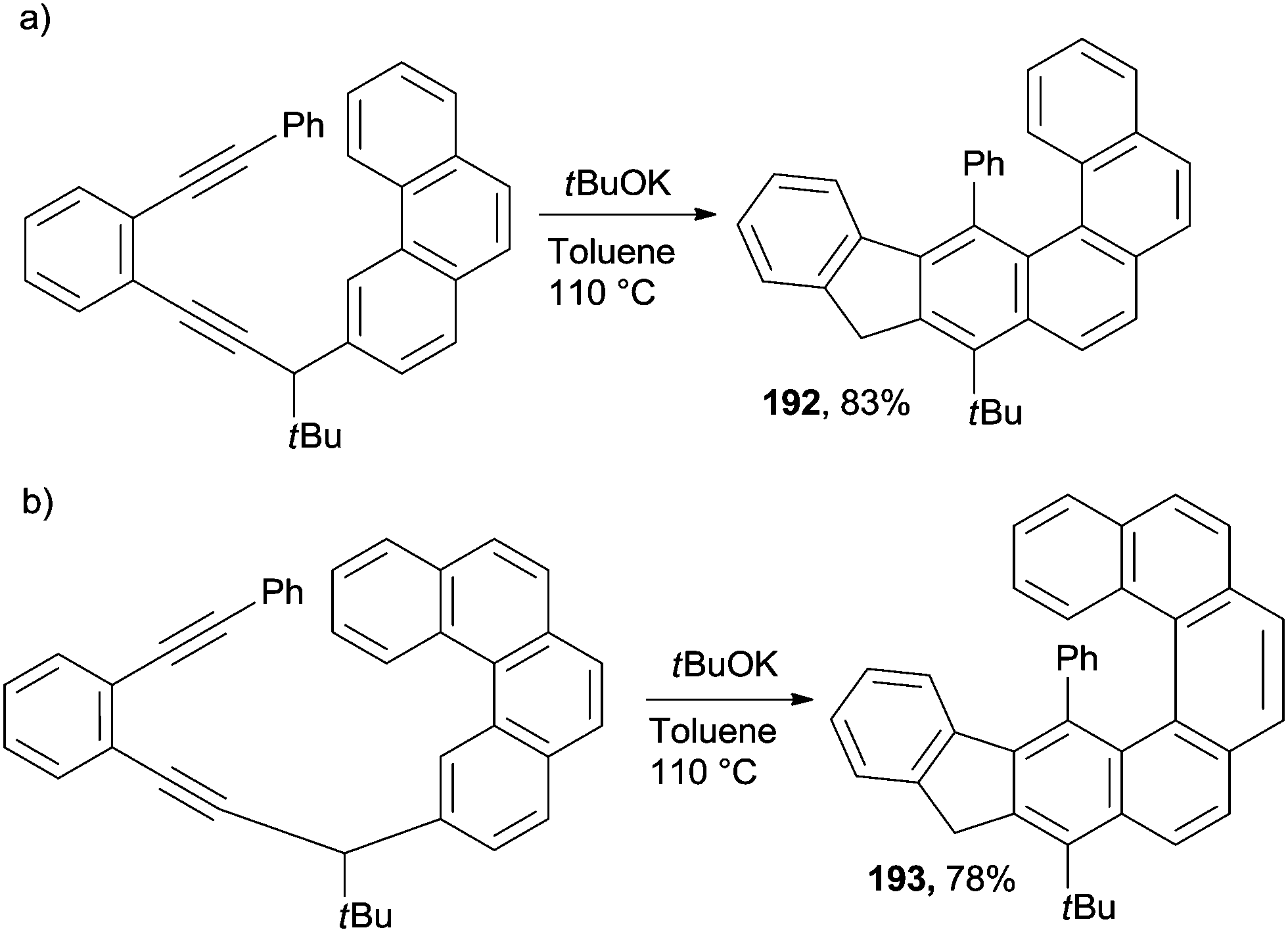 | ||
| Scheme 50 Enediynes as precursors for the preparation of [4]helicenes and [5]helicenes. | ||
Recently, a general procedure for the synthesis of polyaromatic systems based on the use of oligoalkynes was proposed, making use of a highly selective radical cascade.98,184 In one instance, the process occurred with the aid of a traceless directing group, lost at the end of the reaction. As an example, stannylated chrysene 198 was synthesized from tris-alkyne 194 using a radical chain process initiated by Bu3Sn-H/AIBN in toluene at 110 °C through intermediates 195–197 (Scheme 51). The resulting Sn-containing product 198 could then be functionalized by treatment with a variety of electrophiles. In the simplest case, the Bu3Sn group was eliminated via protodestannylation of compound 198 to give 199.184 [5]Helicenes could be obtained as well, when starting from the corresponding tetra-alkynes.184
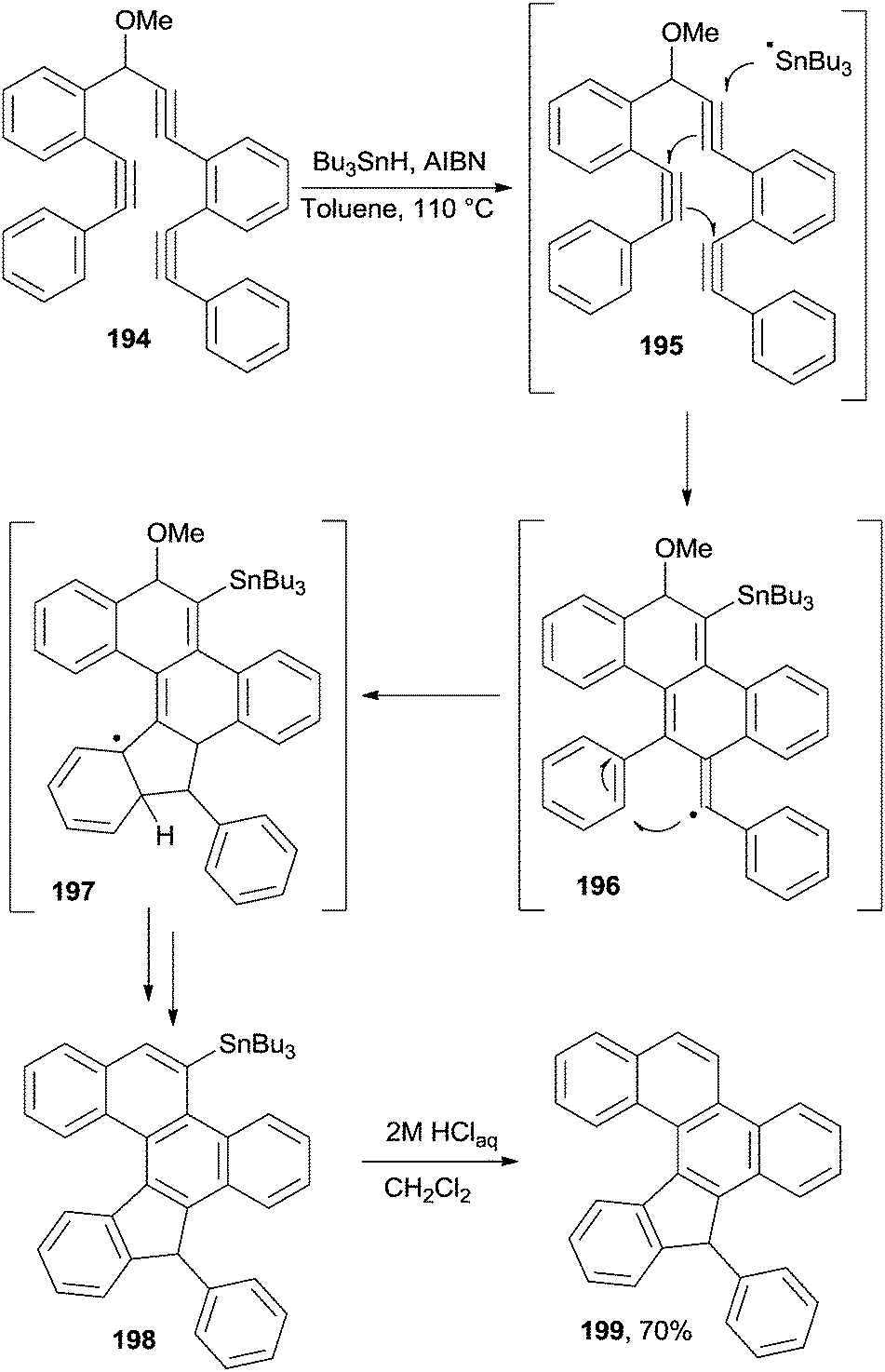 | ||
| Scheme 51 Radical cascade in the synthesis of stannylated chrysene 198. | ||
The cycloaromatization of polyenynes was widely applied for the synthesis of polynaphthalene networks.185–188 Perylene 201 was formed in 66% yield from a solution of the corresponding tetraethynylbiphenyl 200 in a 10:1V/V benzene/CHD mixture heated to 180 °C in a sealed steel bomb. Binaphthyl 202 was likewise obtained as a minor product in 10% yield (Scheme 52).189 Interestingly, when the same process was carried out on bulky derivatives (R1 = tBu), the cycloaromatization took place exclusively on the unsubstituted enediyne core, and naphthalene 203 was obtained in 20% yield along with a large amount of by-products (Scheme 52).189
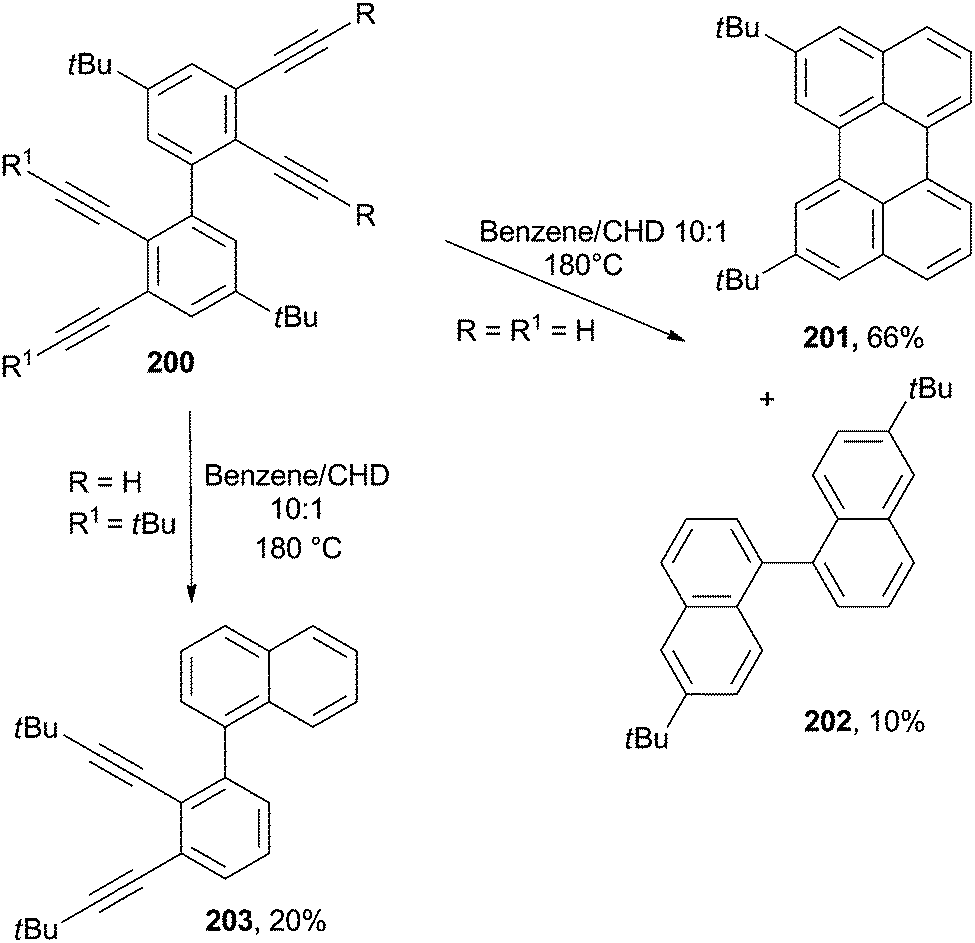 | ||
| Scheme 52 Different pathways in the cycloaromatization of tetraethynylbiphenyls. | ||
6 Synthesis of heteroaromatics
Heteroaromatic derivatives can be obtained by a cyclization step starting from azadienynes, via a (photochemical) Bergman reaction involving enediynes or starting from ortho-alkynylaryl isocyanides and eneallene-isonitriles. The ring formed, however, may either contain a heteroatom or be fused with a pre-existing heteroaromatic system.A nice example is the synthesis of naphtho[1,2-b]benzofurans 206a–c by AuI-assisted 6-endo-dig cyclization of benzofurans 204a–c carried out at 150 °C in o-xylene (Scheme 53). The cyclization was initiated by the activation of the alkyne moiety by the AuI species that allowed the addition of the triple bond onto the benzofuran (acting as the nucleophile). Compounds 206a–c were then formed in a very good yield (mostly >90%) by proto-deauration of the cyclized intermediates 205−a–c, also allowing the regeneration of the AuI species.92 Milder conditions were later applied (60 °C) to the synthesis of substituted carbazoles starting from (Z)-2-(enynyl)indoles by using AgISbF6 (5 mol%) and AuI(Ph3P)Cl (5 mol%) as the catalytic system.190
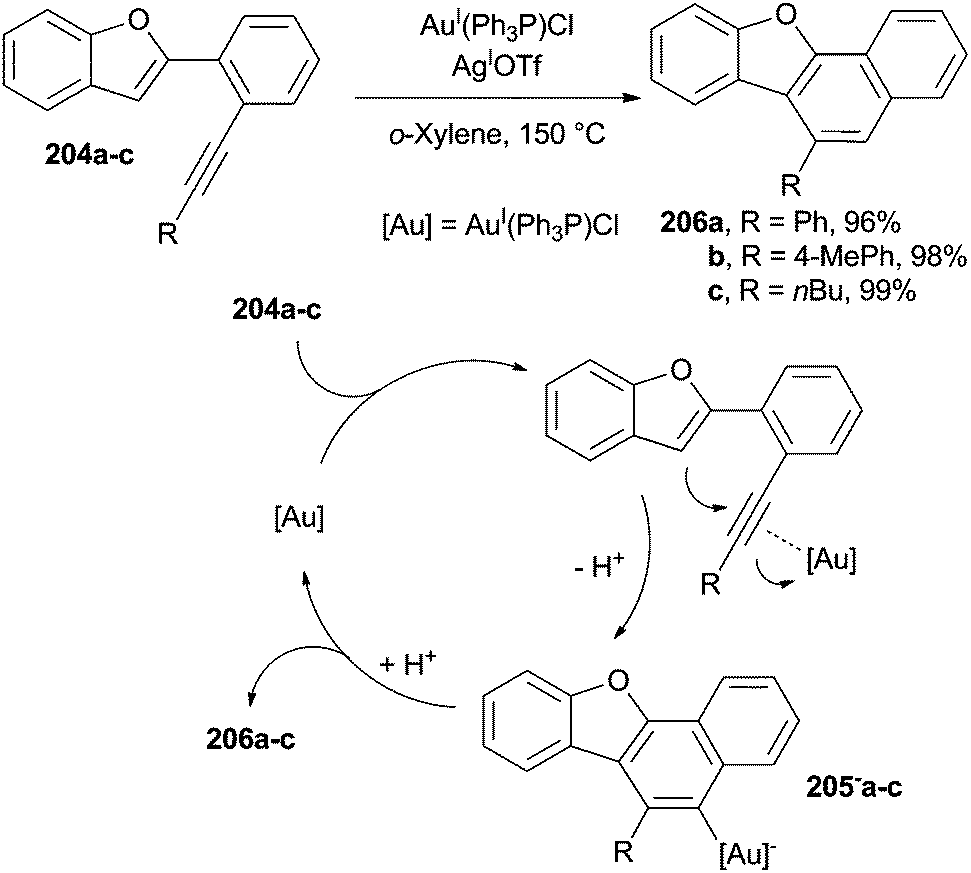 | ||
| Scheme 53 Gold-catalyzed preparation of naphtho[1,2-b]benzofurans. | ||
Dienylalkynes cyclized even by using RuII(η6-p-cymene)(Ph3P)Cl2 (ca. 5 mol%) as the catalyst in the presence of NH4PF6via vinylidene intermediates. As an example, 2-ethynyl-1-(2-furyl)cyclohexene was easily converted into 6,7,8,9-tetrahydronaphtho[1,2-b]furan in 89% yield in refluxing dichloromethane under stirring for 10 h.191
PdII catalysts are able to promote the flexible synthesis of furo[2,3-b]pyridines 209a–c starting from substituted enediyne-imides 207a–c (Scheme 54). The reaction was promoted by the coordination of the alkyne moieties in 207a–c by the PdII catalyst, in turn inducing a trans-oxypalladation to give imidates 208a–c. A cycloisomerization ensued to afford substituted furopyridines 209a–c in a very good yield upon tosyl group loss.192
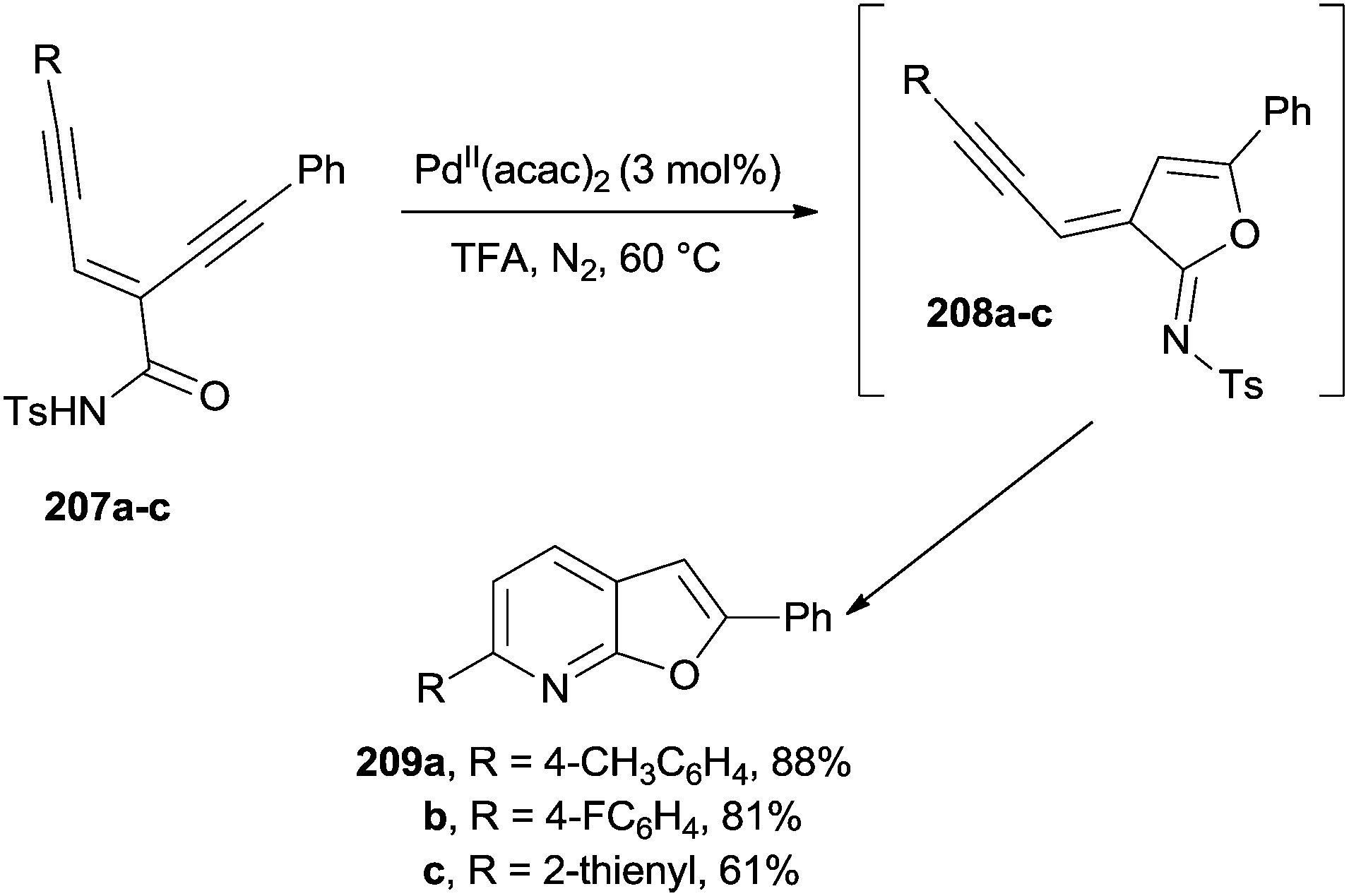 | ||
| Scheme 54 PdII-catalyzed conversion of enediyne-imides into furo[2,3-b]pyridines. | ||
Peri-alkynyl-9,10-anthraquinones 210a–c (Scheme 55) were efficiently employed as building blocks for the synthesis of aporphinoid alkaloid analogues 7H-dibenzo[de,h]quinolin-7-ones 211a–c, upon heating in molten urea.193
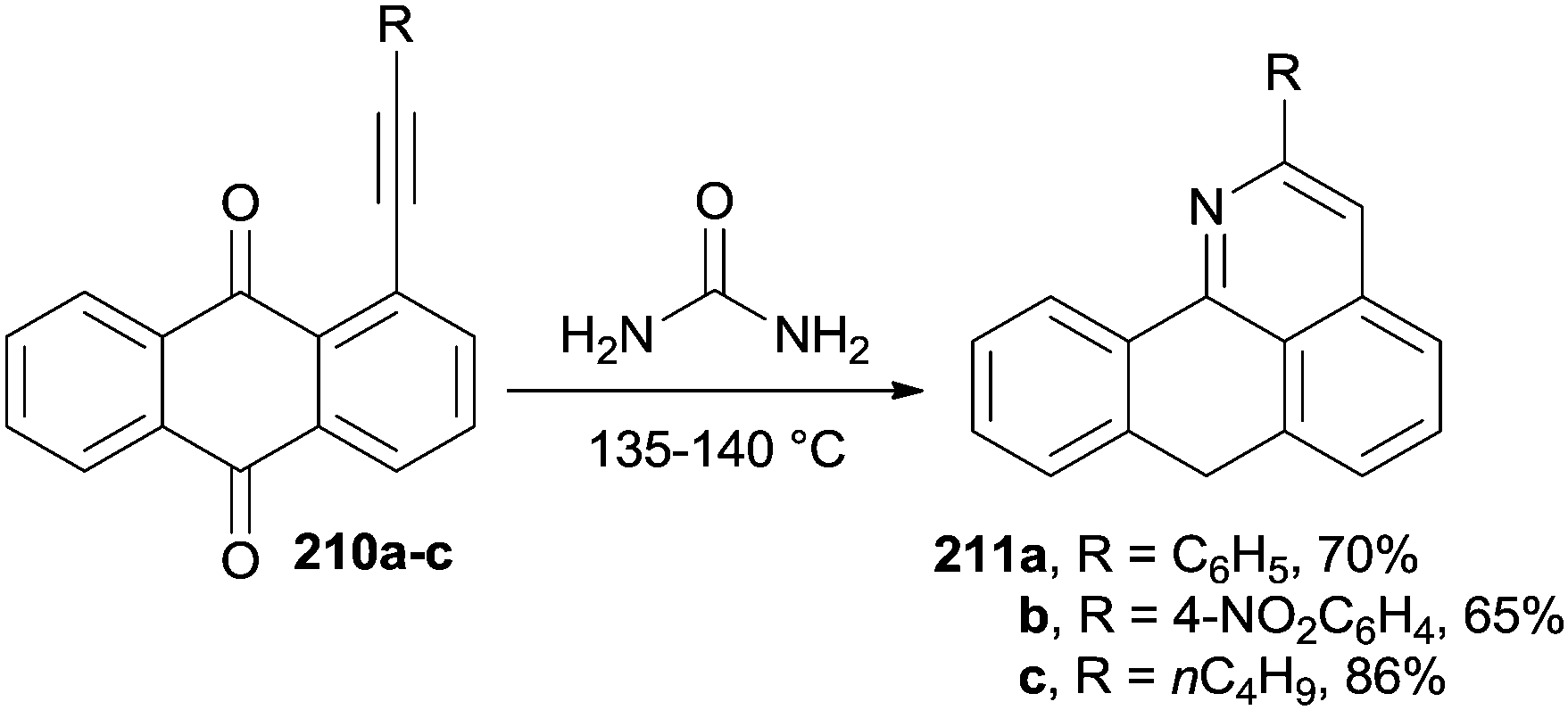 | ||
| Scheme 55 Synthesis of 7H-dibenzo[de,h]quinolin-7-ones via cycloaromatization. | ||
Interestingly, the intermolecular version of the reaction involving the reductive dimerization of acetylenic anthraquinones in the presence of guanidine gave access both to 2H-dibenzo[de,h]isoquinoline-3,7-diones (main product) along with tetrabenzo[a,de,j,mn]tetracene-4,13-diones.194
The construction of the pyridine ring was likewise accomplished via the RuII-catalyzed protodesilylation and cycloisomerization of C6-trimethylsilyl 3-azadienynes 212a–c to give the corresponding aza-heterocycles 213a–c (Scheme 56). The catalytic system made use of the RuII(Cp)(Ph3P)2Cl complex, along with the 2-dicyclohexyl-phosphino-2′,6′-dimethoxy-1,1′-biphenyl (SPhos) ligand in the presence of NH4PF6 as an additive.195 Substituted phenylquinolines 213a–c were then isolated in very good yields (Scheme 56).
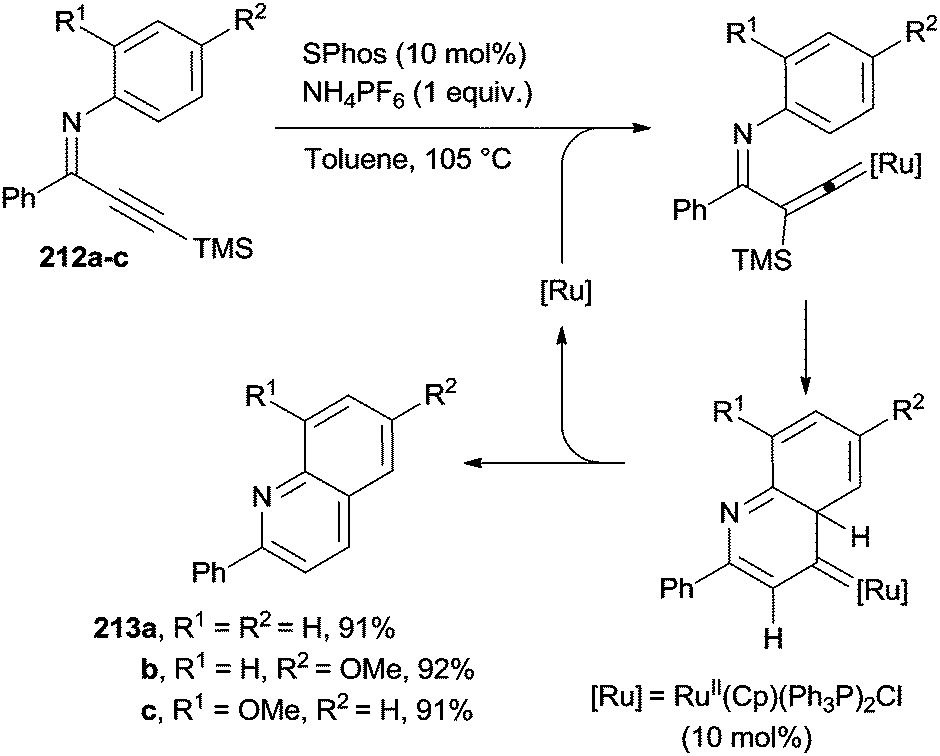 | ||
| Scheme 56 Smooth entry to phenylquinolines via cycloaromatization of azadienynes. | ||
The best approach for the synthesis of heterocycles is, however, by having recourse to a Bergman cyclization starting from cyclic enediynes. Scheme 57 illustrates a representative example where quinazolines 215a,b were prepared either under thermal or photochemical conditions from 10-membered pyrimidine enediynes 214a,b.196 Interestingly, alcohol 214a cyclized in iPrOH both by heating under reflux or under irradiation at 40 °C (overall yield >80%). The presence of a ketone moiety in compound 214b, however, inhibited the formation of quinazoline 215b upon irradiation (Scheme 57). This was probably due to the involvement of a triplet state.†
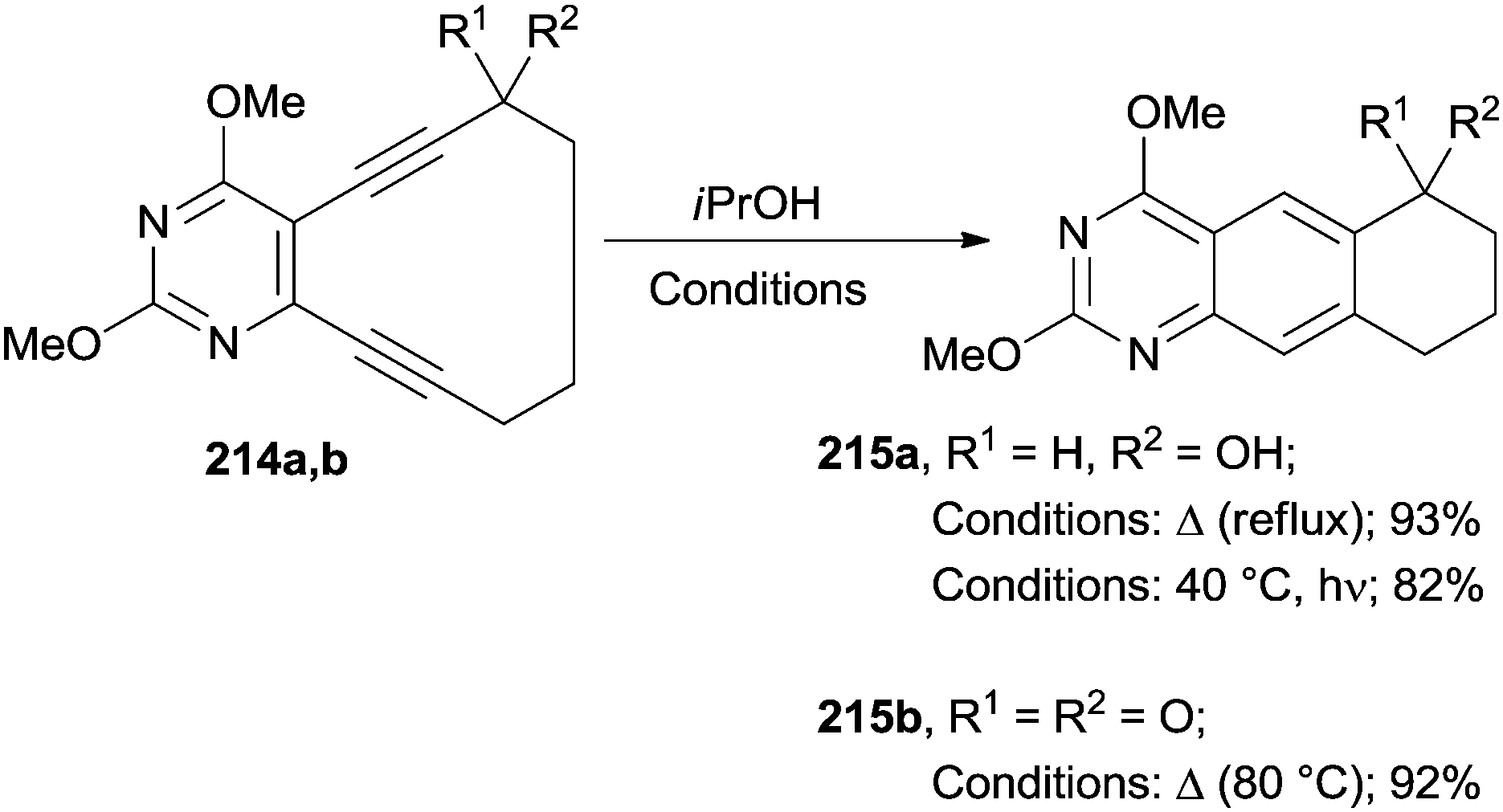 | ||
| Scheme 57 Thermal and photoinduced Bergman cyclizations. | ||
Analogously, irradiation of 11-membered 4,5-bis-(alkyn-1-yl)imidazoles gave the corresponding cycloaromatized products. The overall yields were satisfactory only when the reaction was applied to conformationally rigid derivatives and strongly depended on the solvent used.197 A smooth procedure for the synthesis of 10-membered cinnoline-fused cyclic enediynes was recently reported.198 These compounds were prone to cyclization under mild conditions (75 °C, 24 h) in iPrOH to afford tetrahydrodibenzo[c,g]cinnolines via a Bergman cyclization. In particular, cinnolino[5,4-c]cyclodeca-4-ene-2,6-diyn-1-ol was shown to be four times more reactive than the benzo-fused analogue.198
Acyclic enediynes require harsh conditions to cyclize. A typical case is that of imidazole-fused (Z)-3-ene-1,5-diynes which required 145 °C to be converted into the corresponding benzimidazoles.199 The cyclization rate, however, changed according to the substitution pattern of the nitrogen atom in the imidazole ring. Thus, N-aryl substitution was able to enhance the rate by up to seven-times with respect to the corresponding N-alkyl derivatives, although the reasons were not fully proven.199
A smooth way to induce the cycloaromatization in acyclic enediynes is by adding an electrophile (e.g. iodine), as summarized in Scheme 58. The process took place at room temperature within 2 h. Thus, treatment of N,N-dimethyl 2-[2-(2-ethynylphenyl)ethynyl]anilines 216a–c with a slight excess of I2 caused an initial cyclization to form iodinated indoles 217a–c. Further addition of iodine followed by iodide attack onto cations 218+a–c formed the desired benzo[a]carbazoles 219a–c in high yield, provided no bulky substitutents on the terminal alkyne (e.g. a tBu group) or electron-withdrawing groups on the aniline ring were present.91 A related process was used for the preparation of benzo[b]naphtha[2,1-d]thiophenes (X = SMe in compounds 216a–c) under uncatalyzed200 or catalyzed conditions (AuI(Ph3P)Cl in combination with AgISbF6 as the catalyst).201 Iodine, as well as N-iodosuccinimide, was used here as the electrophile.200–202 Iodine-free carbazole derivatives were similarly prepared from substituted ethynylanilines (X = NH2 in 216a–c), again making use of Au/Ag catalysts in a process initiated by an intramolecular 5-endo-dig hydroamination cascade.202
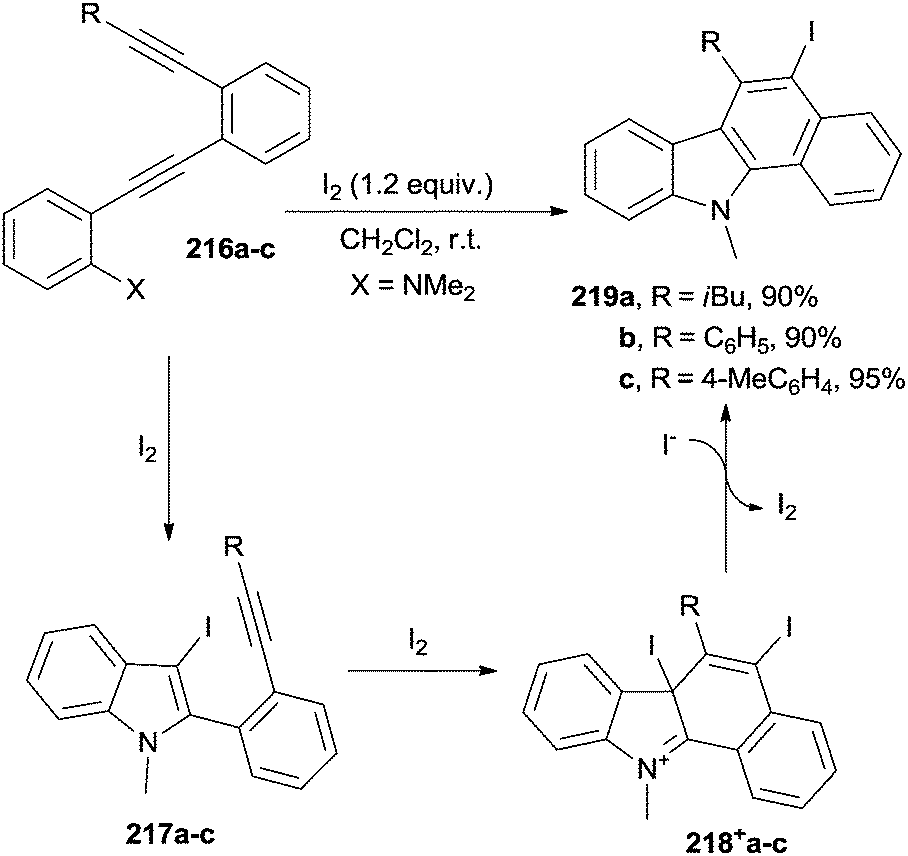 | ||
| Scheme 58 Preparation of benzo[a]carbazoles. | ||
Carbazoles featuring a silylketene moiety have been obtained from the corresponding indole-based Fischer carbene complexes by following the same approach described in Scheme 36.162
The preparation of chlorinated analogues of 219a–c has been accomplished by the treatment of compounds 216a–c with PdIICl2 (10 mol%) in refluxing THF in the presence of an excess (2 equiv.) of CuIICl2.82 In alternative, a stronger electrophile, such as (C6F5)3B, was used to initiate the cyclization of N-heterocyclic 1,2-bis-(trimethylsilylethynyl)arenes via 1,1-carboboration. The method is versatile since highly substituted quinolines, benzothiophenes and carbazoles were accessed.203
A particular case is the cyclization of compound 220 that, under basic conditions, cyclized to a halogen-free indole fused with a cyclopenta[b]pyridine unit (221, Scheme 59). It is noteworthy that the latter compound exhibited a significant antifungal activity against T. mentagrophytes and T. rubrum.202 Bergman cyclization was likewise promoted under basic conditions by the treatment of a methanolic solution of various enediynylphenyl tert-butyldimethylsilyl ethers with sodium methoxide under reflux for 16 h to give a series of 5-substituted dibenzofurans.204
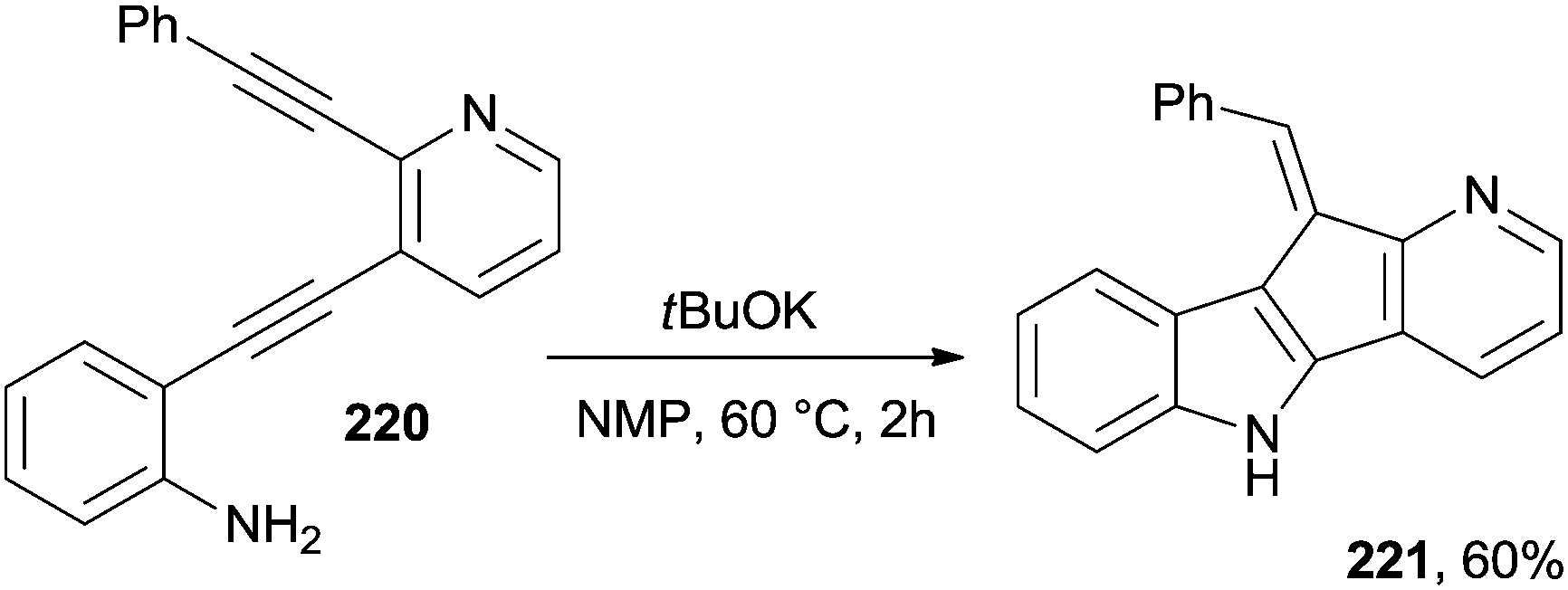 | ||
| Scheme 59 Base-induced preparation of heteroaromatics. | ||
Addition of nucleophiles (e.g. the azide anion) onto enediynes is another viable approach for the construction of heterocyclic systems, as in the tandem cyclization of (Z)-1-aryl-3-hexen-1,5-diynes 222a–c (Scheme 60). The heterocyclization was initiated by the formation of triazole anions 223−a–c followed by the attack of the charged nitrogen atom onto the neighboring triple bond to generate anions 224−a–c and [1,2,3]triazolo[1,5-a]pyridines 225a–c from them.205 The approach has been extended to the synthesis of [1,2,3]triazolo[1′,5′;1,2]pyrido[3,4-b]pyrazines.206
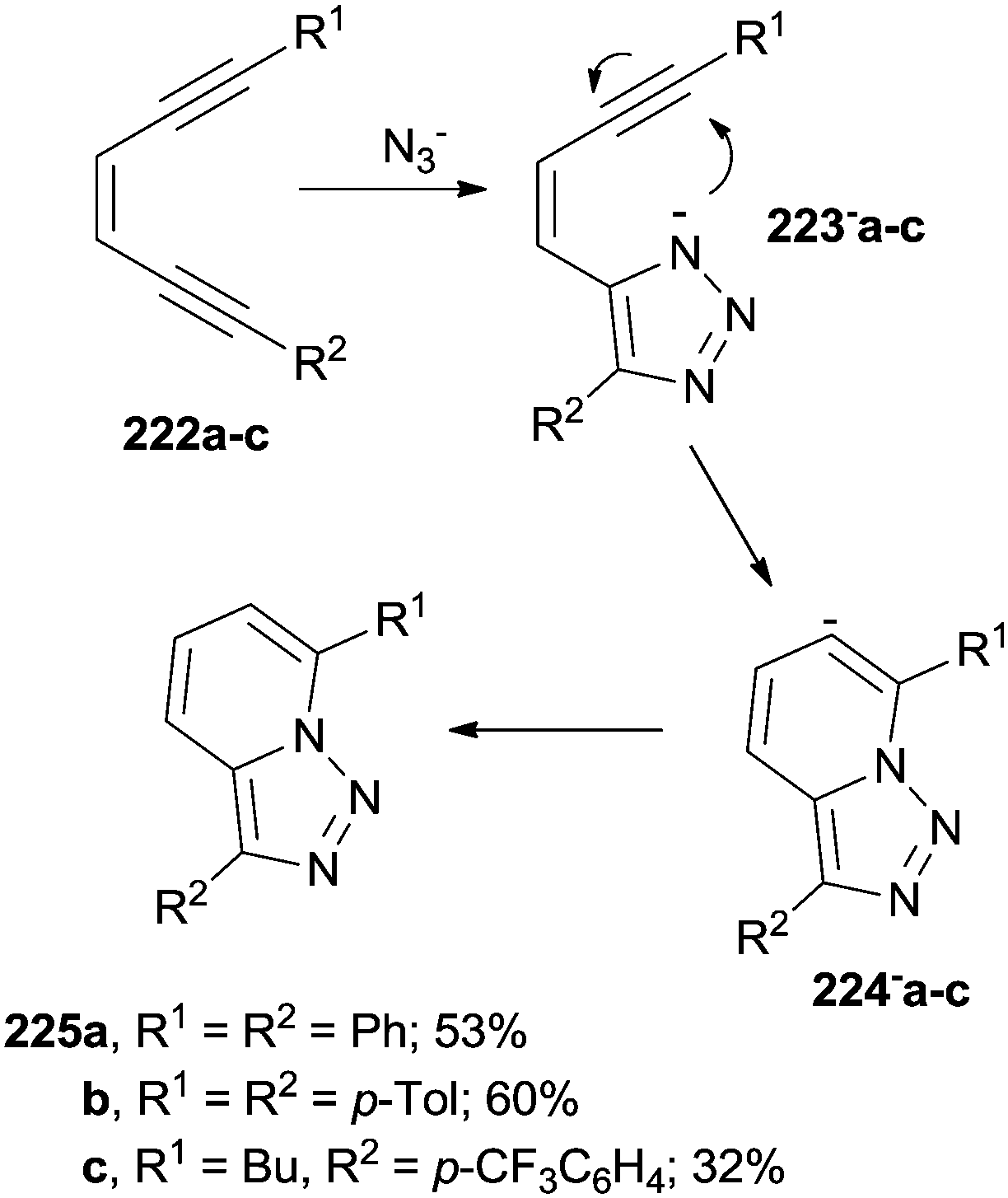 | ||
| Scheme 60 Azide addition onto enediynes to give [1,2,3]triazolo[1,5-a]pyridines. | ||
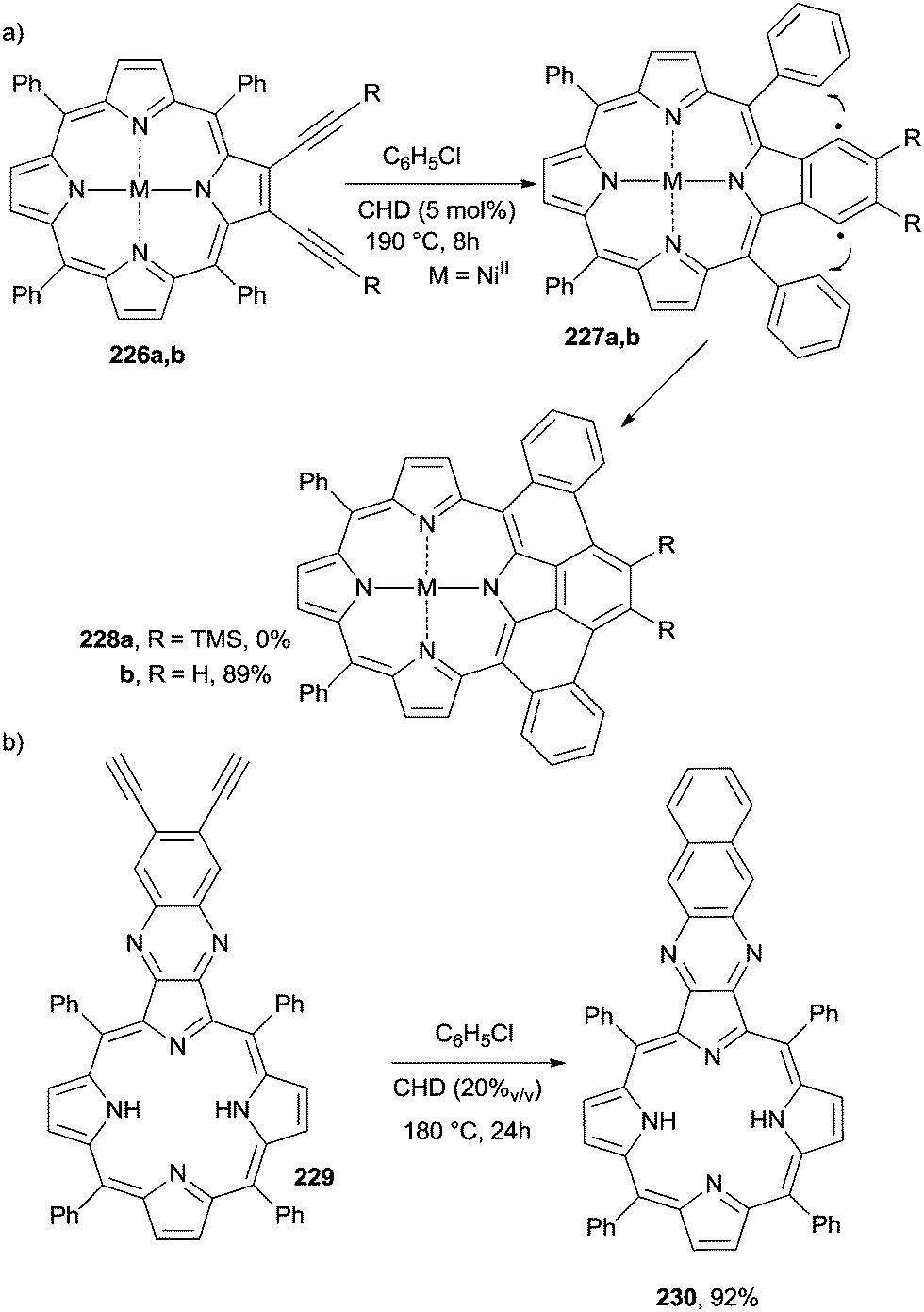
The wide application of porphyrin derivatives led to the development of procedures intended to extend the π-system in order to tune the electronic properties of the resulting derivatives. A first report dealt with the thermal annulation of NiII 2,3-dialkynyl-5,10,15,20-tetraphenylporphyrins 226a,b to form picenoporphyrins 228a,b (Scheme 61a).207 The conditions were quite prohibitive, however, since the Bergman cyclization was carried out in a sealed Schlenk tube in chlorobenzene at 190 °C in the presence of CHD as a hydrogen atom donor. The reaction was very sensitive to the presence of substituents on the triple bonds and was particularly efficient only when using unsubstituted derivatives.207 Arenoporphyrin 1,4-biradicals 227a,b were postulated as the key intermediates (Scheme 61a). In the case of 226b, the addition of 2 equiv. of DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone), which plays the role of a hydrogen atom acceptor, markedly accelerated the process and allowed for milder reaction conditions (25 °C), despite the fact that a lower yield (30–40%) of picenoporphyrin 228b was obtained.208 Bergman cyclization took place even when starting from ZnII tetraphenylporphyrin 226b (M = ZnII), again in the presence of CHD under thermal (120 °C, 70%) or photochemical (λ ≥ 395 nm) conditions. In the latter case, however, only partial conversion of the starting enediyne resulted.209 The presence of CHD was again required in the conversion of metal-free tetraphenyl diethynylquinoxalino[2,3-b]-porphyrin 229 into tetraphenylbenzoquinoxalino[2,3-b]porphyrin 230 (92%) under heating (Scheme 61b).210
 | ||
| Scheme 61 Synthesis of arenoporphyrins. | ||
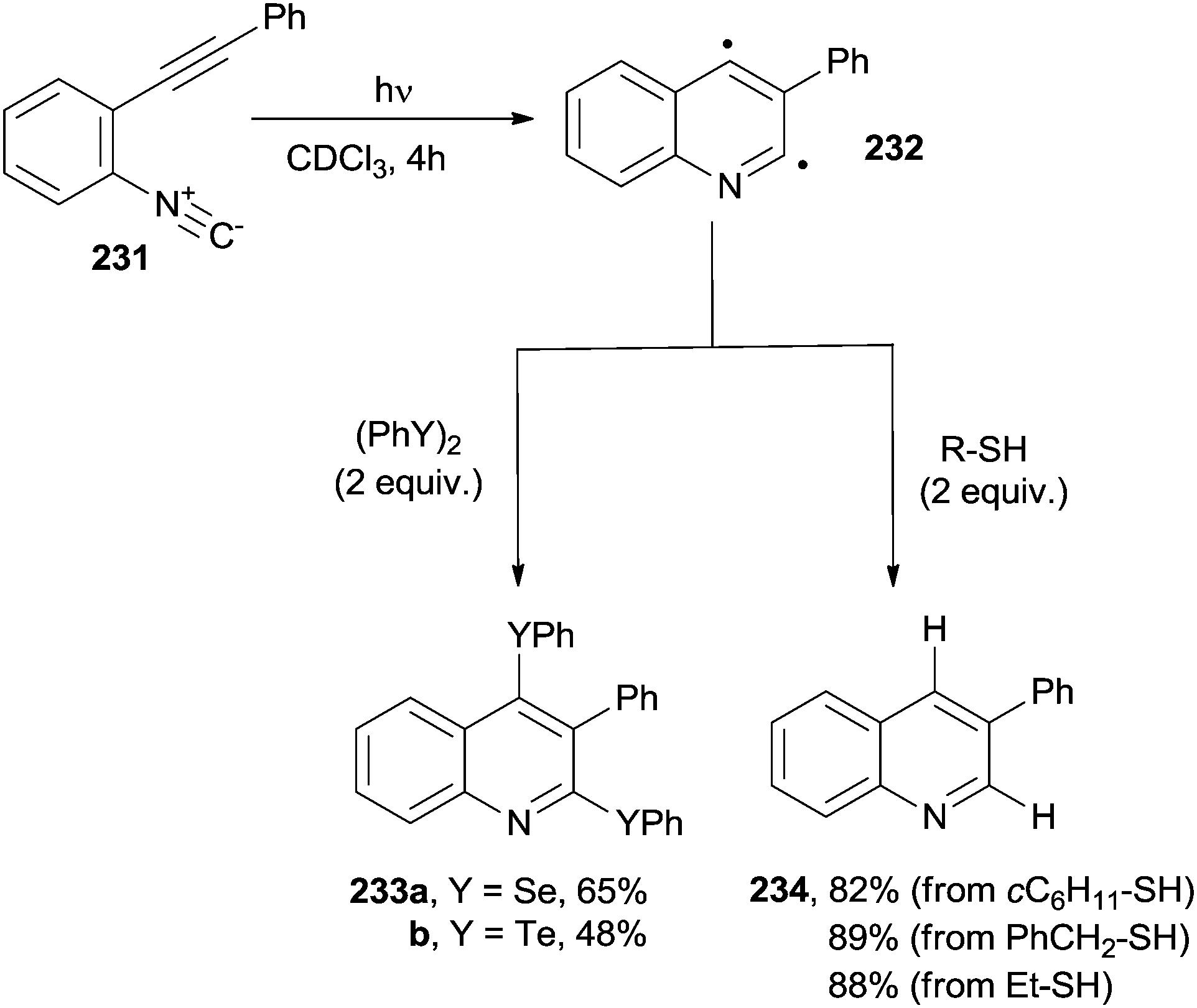
Unsaturated aromatic isonitriles have been sparsely used for the synthesis of nitrogen-containing heterocycles. A typical case is the photocyclization of ortho-alkynylaryl isocyanides (231, Scheme 62). Thus, the photochemical (λ ≥ 300 nm) addition of 231 onto organic dichalcogenides (diselenides or ditellurides) induced the formation of 2,4-bischalcogenated quinolines 233a,bvia the intermediacy of biradical 232. The reaction did not take place, however, when using diphenyl disulfide as dichalcogenide.211 As an alternative, 3-phenylquinoline 234 was smoothly prepared when the reaction was carried out in the presence of a hydrogen atom transfer reagent, such as a thiol (Scheme 62).211 Substituted quinolines also arose from a radical cascade reaction involving o-alkenyl arylisocyanides and boronic acids in the presence of 3 equiv. of MnIII(acac)3.212
 | ||
| Scheme 62 Photocyclization of ortho-alkynylaryl isocyanides. | ||
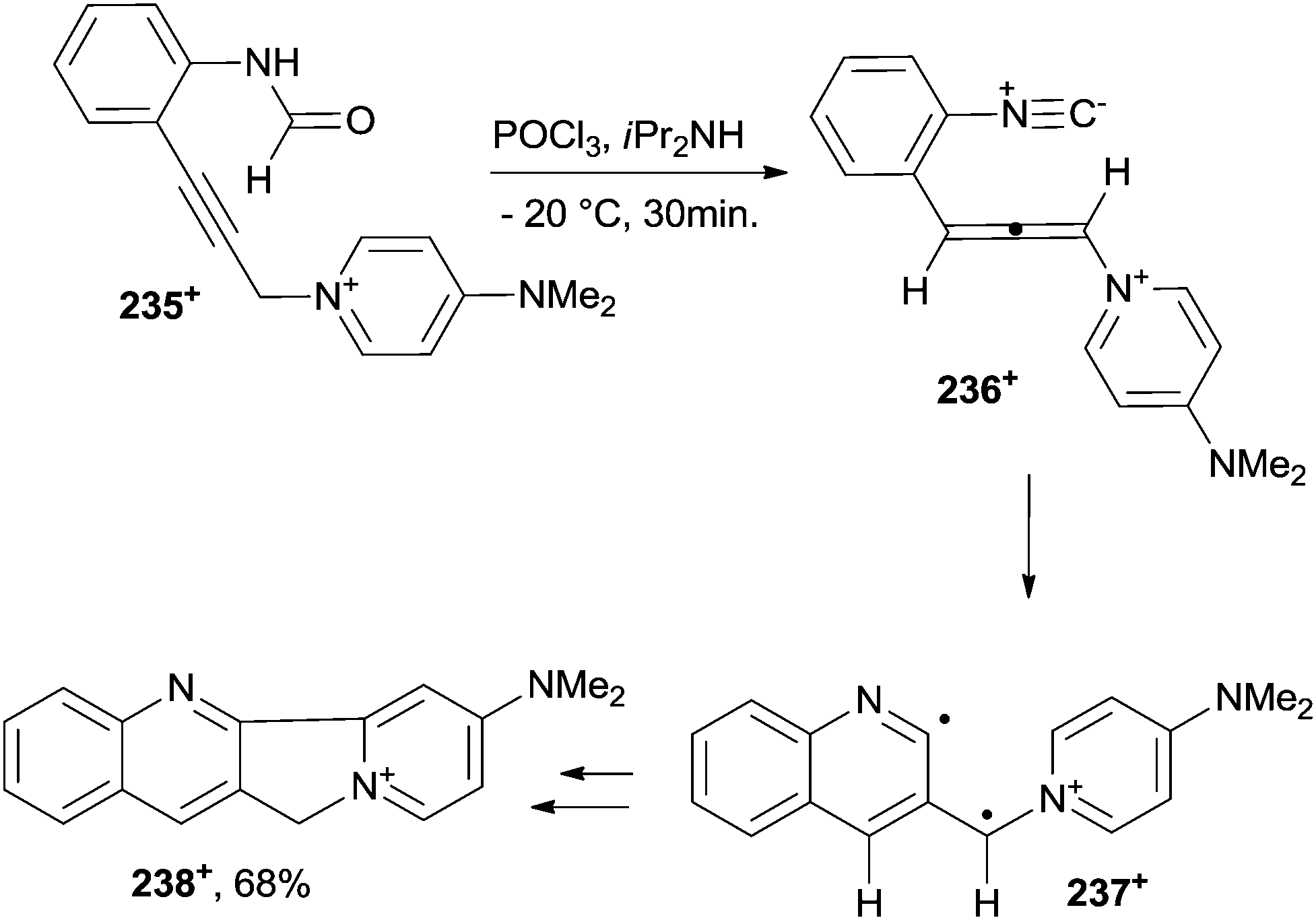
A nitrogen-containing six-membered ring was likewise obtained by the cycloaromatization of eneallene-isonitrile 236+, in turn easily obtained in situ from pyridinium 235+, as the mesylate salt. In such a way, a quinoline biradical 237+ was generated that, upon intramolecular cyclization (via a formal [4+1] cycloaddition), afforded the end 11H-indolizino[1,2-b] quinolin-10-ium 238+ in a good yield (Scheme 63).213 A different path was observed instead when substituting a MeO group for the NMe2 group in 235+, where a 2-substituted indole was formed in place of the quinoline. This behaviour was attributed to the lower donation capability of the alkoxy group in intermediate 236+. At any rate, in favourable cases this approach allowed for the construction of the heteroaromatic ring core of the antitumor agent camptothecin (as in the case of 238+). Substituted 6H-indolo[2,3-b]quinolines and tetrahydropyrimido[4,5-b]quinolines were obtained from the corresponding enyne–carbodiimides via thermal C2–C6 Schmittel214 and C2–C7 Myers–Saito215 reactions, respectively.
 | ||
| Scheme 63 Easy route to 11H-indolizino[1,2-b]quinolin-10-ium. | ||
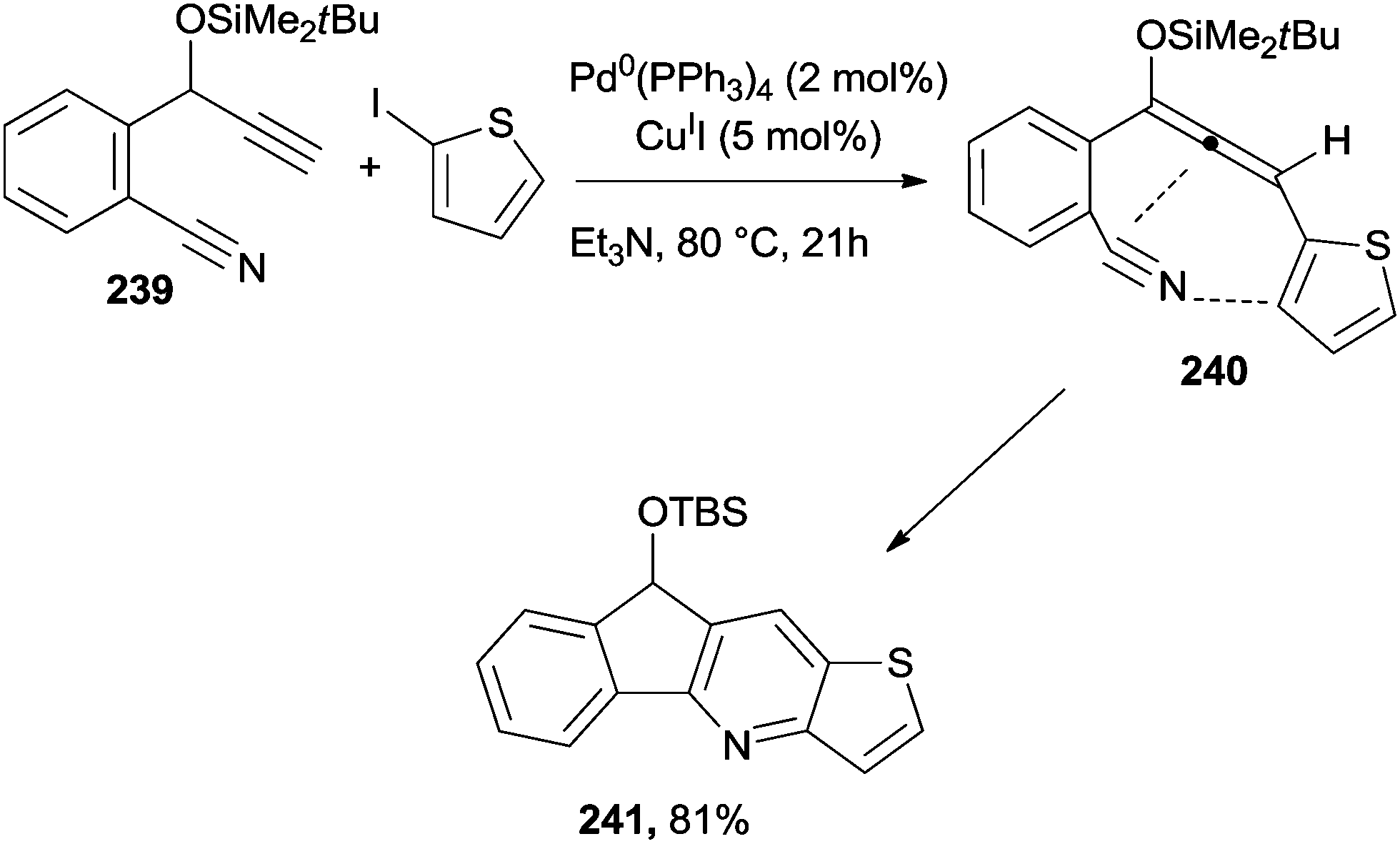
Pyridine-fused compounds (e.g.241) were easily prepared by making use of a cyano-Schmittel cyclization (Scheme 64). Thus, a Sonogashira coupling between benzonitrile 239 and 2-iodothiophene, followed by a base-induced propargyl–allenyl isomerization, afforded cyano-allene 240 that easily cyclized to 241.216
 | ||
| Scheme 64 Cyano-Schmittel cyclization for the synthesis of 241. | ||
7 Conclusions
The purpose of this Review was to demonstrate that the cyclization of dienynes, enediynes and enyne–allenes is a versatile method for the preparation of (hetero)aromatics. The main advantage of this approach, in comparison with other established benzannulation reactions, is that the cyclization event can be induced in different ways. The thermal cyclization of dienynes and enediynes (resulting in Hopf and Bergman cycloaromatizations, respectively) takes place under harsh conditions (T mainly >150 °C), unless the unsaturated system has the right geometry (often encountered in cyclic polyenic derivatives). In contrast, enyne–allenes are very reactive and difficult to handle and they easily undergo the cycloaromatization process (Myers–Saito reaction). For this reason such polyenes are conveniently generated in situ from enediynes via rearrangement under basic conditions.The photochemical approach allows the use of mild conditions for the activation of the polyenic substrate, but it has been applied so far only in rare instances to promote an efficient cycloaromatization. Addition of electrophiles or radicals onto the polyenic system is a valuable alternative since the incorporation of the promoting agent into the structure of the end product(s) is an added bonus of the reaction.97,184 Metal-catalysis, however, is at present a widely applied strategy of activation and it is the main reason for the recent growth of these cycloaromatizations in modern organic synthesis. However, the strategies summarized in this review often lack generality, since different substrates usually require different modes of activation to achieve the desired target. Nonetheless, the use of dienynes, enediynes, enyne–allenes and hetero-analogues may open the way for innovative synthetic strategies for the preparation of (hetero)aromatic building blocks and polycyclic aromatic hydrocarbons (PAHs) useful in medicinal chemistry and in the preparation of electroactive organic materials and molecular devices.
Acknowledgements
This work was supported by the Fondazione Cariplo (Grant 2011-1839). We acknowledge Prof. A. Albini (University of Pavia) for fruitful discussions.Notes and references
- Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed.
- Palladium Reagents and Catalysts, ed. J. Tsuji, John Wiley & Sons, Chichester, UK, 2004 Search PubMed.
- Transitions Metal Reagents and Catalysts, ed. J. Tsuji, John Wiley & Sons, Chichester, UK, 2000, ch. 3, pp. 27–108 Search PubMed.
- M. Beller, A. Zapf and W. Maegerlein, Chem. Eng. Technol., 2001, 24, 575–582 CrossRef CAS.
- S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS PubMed.
- I. Ojima, M. Tzamarioudaki, Z. Li and R. J. Donovan, Chem. Rev., 1996, 96, 635–662 CrossRef CAS PubMed.
- C. B. de Koning, A. L. Rousseau and W. A. L. van Otterlo, Tetrahedron, 2003, 59, 7–36 CrossRef CAS.
- T. J. Donohoe, A. J. Orr and M. Bingham, Angew. Chem., Int. Ed., 2006, 45, 2664–2670 CrossRef CAS PubMed.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS.
- S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317–1382 CrossRef CAS PubMed.
- W. A. L. Van Otterlo and C. B. De Koning, Chem. Rev., 2009, 109, 3743–3782 CrossRef CAS PubMed.
- H. Villar, M. Frings and C. Bolm, Chem. Soc. Rev., 2007, 36, 55–66 RSC.
- A. R. Katritzky, J. Li and L. Xie, Tetrahedron, 1999, 55, 8263–8293 CrossRef CAS.
- Y. Ito, M. Nakatsuka and T. Saegusa, J. Am. Chem. Soc., 1982, 104, 7609–7622 CrossRef CAS.
- S. Kuroda, M. Oda, S. Zuo, K. Kanayama, S. I. M. Shah, S. Furuta, R. Miyatake and M. Kyogoku, Tetrahedron Lett., 2001, 42, 6345–6348 CrossRef CAS.
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef PubMed.
- G. W. Kabalka, Y. Ju and Z. Wu, J. Org. Chem., 2003, 68, 7915–7917 CrossRef CAS PubMed.
- R. Balamurugan and V. Gudla, Org. Lett., 2009, 11, 3116–3119 CrossRef CAS PubMed.
- Q. Huang and R. C. Larock, Org. Lett., 2002, 4, 2505–2508 CrossRef CAS PubMed.
- P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS.
- S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741–4767 CrossRef CAS.
- J. A. Varela and C. Sa, Chem. Rev., 2003, 103, 3787–3801 CrossRef CAS PubMed.
- T. Shibata and K. Tsuchikama, Org. Biomol. Chem., 2008, 6, 1317–1323 CAS.
- D. Peña, D. Pérez and E. Guitàn, Chem. Rec., 2007, 7, 326–333 CrossRef PubMed.
- K. P. C. Vollhardt, Acc. Chem. Res., 1977, 10, 1–8 CrossRef CAS.
- J. W. Grissom, G. U. Gunawardena, D. Klingberg and D. Huang, Tetrahedron, 1996, 52, 6453–6518 CrossRef CAS.
- H. Hopf and H. Musso, Angew. Chem., Int. Ed. Engl., 1969, 8, 680–685 CrossRef CAS.
- R. G. Bergman, Acc. Chem. Res., 1973, 6, 25–31 CrossRef CAS.
- A. G. Myers, P. S. Dragovich and E. Y. Kuo, J. Am. Chem. Soc., 1992, 114, 9369–9386 CrossRef CAS.
- A. G. Myers, N. S. Finney and E. Y. Kuo, Tetrahedron Lett., 1989, 30, 5747–5750 CrossRef CAS.
- R. Nagata, H. Yamanaka, E. Okazaki and I. Saito, Tetrahedron Lett., 1989, 30, 4995–4998 CrossRef CAS.
- L. Feng, D. Kumar, D. M. Birney and S. M. Kerwin, Org. Lett., 2004, 6, 2059–2062 CrossRef CAS PubMed.
- H. W. Moore and B. R. Yerxa, Chemtracts, 1992, 273–313 Search PubMed.
- H. Li, H. Yang, J. L. Petersen and K. K. Wang, J. Org. Chem., 2004, 69, 4500–4508 CrossRef CAS PubMed.
- C. Spöler and B. Engels, Chem. – Eur. J., 2003, 9, 4670–4677 CrossRef PubMed.
- H. Li, J. L. Petersen and K. K. Wang, J. Org. Chem., 2003, 68, 5512–5518 CrossRef CAS PubMed.
- M. Schmittel, M. Strittmatter and S. Kiau, Tetrahedron Lett., 1995, 36, 4975–4978 CrossRef CAS.
- M. Schmittel, M. Strittmatter, K. Vollmann and S. Kiau, Tetrahedron Lett., 1996, 37, 999–1002 CrossRef CAS.
- M. Schmittel, S. Kiau, T. Siebert and M. Strittmatter, Tetrahedron Lett., 1996, 37, 7691–7694 CrossRef CAS.
- B. Engels, C. Lennartz, M. Hanrath, M. Schmittel and M. Strittmatter, Angew. Chem., Int. Ed., 1998, 37, 1960–1963 CrossRef CAS.
- K. K. Wang, Chem. Rev., 1996, 96, 207–222 CrossRef CAS PubMed.
- K. C. Nicolau and A. L. Smith, Acc. Chem. Res., 1992, 25, 497–503 CrossRef.
- Polyenes: Synthesis, Properties and Applications, ed. G. Guanti, L. Banfi, A. Basso and R. Riva, Taylor & Francis Group, Boca Raton, FL, 2006, ch. 19, pp. 454–492 Search PubMed.
- P. W. Peterson, R. K. Mohamed and I. V. Alabugin, Eur. J. Org. Chem., 2013, 2505–2527 CrossRef CAS.
- P. R. Schreiner, A. Navarro-Vazquez and M. Prall, Acc. Chem. Res., 2005, 38, 29–37 CrossRef CAS PubMed.
- E. Kraka and D. Cremer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 285–324 CrossRef CAS.
- H. Hopf, H. Berger, G. Zimmennann, U. Niichter, P. G. Jonesa and I. Dix, Angew. Chem., Int. Ed. Engl., 1997, 36, 1187–1190 CrossRef CAS.
- K. Gilmore and I. V. Alabugin, Chem. Rev., 2011, 111, 6513–6556 CrossRef CAS PubMed.
- M. Kar and A. Basak, Chem. Rev., 2007, 107, 2861–2890 CrossRef CAS PubMed.
- F. S. Amegayibor, J. J. Nash, A. S. Lee, J. Thoen, C. J. Petzold and H. I. Kentta, J. Am. Chem. Soc., 2002, 124, 12066–12067 CrossRef CAS PubMed.
- M. E. Cremeens, T. S. Hughes and B. K. Carpenter, J. Am. Chem. Soc., 2005, 127, 6652–6661 CrossRef CAS PubMed.
- M. E. Cremeens and B. K. Carpenter, Org. Lett., 2004, 6, 2349–2352 CrossRef CAS PubMed.
- C. Raviola, D. Ravelli, S. Protti and M. Fagnoni, J. Am. Chem. Soc., 2014, 136, 13874–13881 CrossRef CAS PubMed.
- S. Protti, D. Ravelli, B. Mannucci, A. Albini and M. Fagnoni, Angew. Chem., Int. Ed., 2012, 51, 8577–8580 CrossRef CAS PubMed.
- G. Zimmermann, Eur. J. Org. Chem., 2001, 457–471 CrossRef CAS.
- D. M. Hitt and J. M. O'Connor, Chem. Rev., 2011, 111, 7904–7922 CrossRef CAS PubMed.
- M. Prall, A. Krüger, P. R. Schreiner and H. Hopf, Chem. – Eur. J., 2001, 7, 4386–4394 CrossRef CAS.
- K. C. Nicolaou, A. L. Smith and E. W. Yue, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5881–5888 CrossRef CAS.
- P. Magnus, P. Carter, J. Elliott, R. Lewis, J. Harling, T. Pitterna, W. E. Bauta and S. Fortt, J. Am. Chem. Soc., 1992, 114, 2544–2559 CrossRef CAS.
- T. P. Lockhart, P. B. Comita and R. G. Bergman, J. Am. Chem. Soc., 1981, 103, 4082–4090 CrossRef CAS.
- K. C. Nicolaou, G. Zuccarello, Y. Ogawa, E. J. Schweiger and T. Kumazawa, J. Am. Chem. Soc., 1988, 110, 4866–4868 CrossRef CAS.
- B. P. Warner, S. P. Millar, R. D. Broene and S. L. Buchwald, Science, 1995, 269, 814–816 CrossRef CAS PubMed.
- A. Basak, S. Mandal and S. Sekhar Bag, Chem. Rev., 2003, 103, 4077–4094 CrossRef CAS PubMed.
- P. G. Wenthold and R. R. Squires, J. Am. Chem. Soc., 1994, 116, 6401–6412 CrossRef CAS.
- P. G. Wenthold, S. G. Wierschke, J. J. Nash and R. R. Squires, J. Am. Chem. Soc., 1993, 115, 12611–12612 CrossRef CAS.
- R. W. Sullivan, V. M. Coghlan, S. A. Munk, M. W. Reed and H. W. Moore, J. Org. Chem., 1994, 59, 2276–2278 CrossRef CAS.
- L. Kaplan, S. P. Walch and K. E. Wilzbach, J. Am. Chem. Soc., 1968, 90, 5644–5646 CrossRef.
- M. C. Sajimon and F. D. Lewis, Photochem. Photobiol. Sci., 2005, 4, 629–636 CAS.
- R. K. Mohamed, S. Mondal, K. Jorner, T. Faria Delgado, V. V. Lobodin, H. Ottosson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 15441–15450 CrossRef CAS PubMed.
- N. J. Turro, A. Evenzahav and K. C. Nicolaou, Tetrahedron Lett., 1994, 35, 8089–8092 CrossRef CAS.
- R. L. Funk, E. R. R. Young, R. M. Williams, M. F. Flanagan and T. L. Cecil, J. Am. Chem. Soc., 1996, 118, 3291–3292 CrossRef CAS.
- R. K. Mohamed, K. Kaya and I. V. Alabugin, Photochemical Bergman Cyclization and Related Reactions, in Arene Chemistry, ed. J. Mortier, Wiley CH, 2016, pp. 869–887 Search PubMed.
- I. V. Alabugin and S. V. Kovalenko, J. Am. Chem. Soc., 2002, 124, 9052–9053 CrossRef CAS PubMed.
- A. Evenzahav and N. J. Turro, J. Am. Chem. Soc., 1998, 120, 1835–1841 CrossRef CAS.
- M. Schmittel, A. A. Mahajan and G. Bucher, J. Am. Chem. Soc., 2005, 127, 5324–5325 CrossRef CAS PubMed.
- G. Bucher, A. A. Mahajan and M. Schmittel, J. Org. Chem., 2009, 74, 5850–5860 CrossRef CAS PubMed.
- A. V. Kuzmin and V. V. Popik, Chem. Commun., 2009, 5707–5709 RSC.
- J. M. O'Connor, S. J. Friese and M. Tichenor, J. Am. Chem. Soc., 2002, 124, 3506–3507 CrossRef.
- K. Ohe, M. Kojima, K. Yonehara and S. Uemura, Angew. Chem., Int. Ed. Engl., 1996, 35, 1823–1825 CrossRef CAS.
- A. Odedra, C.-J. Wu, T. B. Pratap, C.-W. Huang, Y.-F. Ran and R.-S. Liu, J. Am. Chem. Soc., 2005, 127, 3406–3412 CrossRef CAS PubMed.
- Y. Wang, A. Yepremyan, S. Ghorai, R. Todd, D. H. Aue and L. Zhang, Angew. Chem., Int. Ed., 2013, 125, 7949–7953 CrossRef.
- C.-C. Chen, L.-Y. Chin, S.-C. Yang and M.-J. Wu, Org. Lett., 2010, 12, 5652–5655 CrossRef CAS PubMed.
- B. P. Taduri, Y.-F. Ran, C.-W. Huang and R.-S. Liu, Org. Lett., 2006, 8, 883–886 CrossRef CAS PubMed.
- J. M. O'Connor, S. J. Friese and B. L. Rodgers, J. Am. Chem. Soc., 2005, 127, 16342–16343 CrossRef PubMed.
- J. Zhao, C. O. Hughes and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 7436–7437 CrossRef CAS PubMed.
- S. Datta and R.-S. Liu, Tetrahedron Lett., 2005, 46, 7985–7988 CrossRef CAS.
- M. Schmittel and M. Strittmatter, Tetrahedron, 1998, 54, 13751–13760 CrossRef CAS.
- N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921–10925 CrossRef CAS PubMed.
- N. Asao and K. Sato, Org. Lett., 2006, 8, 5361–5363 CrossRef CAS PubMed.
- K. Sato, Menggenbateer, T. Kubota and N. Asao, Tetrahedron, 2008, 64, 787–796 CrossRef CAS.
- C.-C. Chen, S.-C. Yang and M.-J. Wu, J. Org. Chem., 2011, 76, 10269–10274 CrossRef CAS PubMed.
- P. M. Byers, J. I. Rashid, R. K. Mohamed and I. V. Alabugin, Org. Lett., 2012, 14, 6032–6035 CrossRef CAS PubMed.
- P. R. Schreiner, M. Prall and V. Lutz, Angew. Chem., Int. Ed., 2003, 42, 5757–5760 CrossRef CAS PubMed.
- A. V. Gulevskaya and R. Y. Lazarevich, Chem. Heterocycl. Compd., 2013, 49, 117–139 CrossRef.
- M.-J. Wu, C.-F. Lin and S.-H. Chen, Org. Lett., 1999, 1, 767–768 CrossRef CAS PubMed.
- A. V. Gulevskaya and A. S. Tyaglivy, Chem. Heterocycl. Compd., 2012, 48, 82–94 CrossRef CAS.
- R. K. Mohamed, S. Mondal, B. Gold, C. J. Evoniuk, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 6335–6349 CrossRef CAS PubMed.
- P. M. Byers and I. V. Alabugin, J. Am. Chem. Soc., 2012, 134, 9609–9614 CrossRef CAS PubMed.
- C.-H. Yang and R.-S. Liu, Tetrahedron Lett., 2007, 48, 5887–5889 CrossRef CAS.
- J.-J. Lian, C.-C. Lin, H. K. Chang, P. C. Chen and R. S. Liu, J. Am. Chem. Soc., 2006, 128, 9661–9667 CrossRef CAS PubMed.
- P. Garcia-Garcia, A. Martinez, A. M. Sanjuan, M.-A. Fernandez-Rodriguez and R. Sanz, Org. Lett., 2001, 13, 4970–4973 CrossRef PubMed.
- P. Garcia-Garcia, M.-A. Fernandez-Rodriguez and E. Aguilar, Angew. Chem., Int. Ed., 2009, 48, 5534–5537 CrossRef CAS PubMed.
- P. A. Brookes and A. G. M. Barrett, J. Org. Chem., 2014, 79, 8706–8714 CrossRef CAS PubMed.
- I. Dix, L. Bondarenko, P. J. Jones, L. Ernst, K. Ibrom, J. Grunenberg, R. Boese and H. Hopf, Chem. – Eur. J., 2014, 20, 16360–16376 CrossRef CAS PubMed.
- A. Poloukhtine and V. V. Popik, J. Org. Chem., 2006, 71, 7417–7421 CrossRef CAS PubMed.
- D. S. Rawat and J. M. Zaleski, J. Am. Chem. Soc., 2001, 123, 9675–9676 CrossRef CAS PubMed.
- L. Banfi and G. Guanti, Eur. J. Org. Chem., 1998, 1543–1548 CrossRef CAS.
- M.-J. Wu, C.-F. Lin and W.-D. Lu, J. Org. Chem., 2002, 67, 5907–5912 CrossRef CAS PubMed.
- K. Yamada, M. J. Lear, T. Yamaguchi, S. Yamaschita, I. D. Gridnev, Y. Hayaschi and M. Hirama, Angew. Chem., Int. Ed., 2014, 53, 13902–13906 CrossRef CAS PubMed.
- C. L. Perrin, B. L. Rodgers and J. M. O'Connor, J. Am. Chem. Soc., 2007, 129, 4795–4799 CrossRef CAS PubMed.
- K. K. Wang, Z. Wang, A. Tarli and P. Gannett, J. Am. Chem. Soc., 1996, 118, 10783–10791 CrossRef CAS.
- J. W. Andemichael, J. Huang and K. J. Wang, J. Org. Chem., 1993, 58, 1651–1652 CrossRef.
- I. Suzuki, Y. Naoe, M. Bando, H. Nemoto and M. Shibuya, Tetrahedron Lett., 1998, 39, 2361–2364 CrossRef CAS.
- M. K. Waddell, T. Bekele and M. A. Lipton, J. Org. Chem., 2006, 71, 8372–8377 CrossRef CAS PubMed.
- J. W. Herndon and H. Wang, J. Org. Chem., 1998, 63, 4562–4563 CrossRef CAS.
- Y. Xiong and H. W. Moore, J. Org. Chem., 1996, 61, 9168–9177 CrossRef CAS.
- Y. Wang, J. Xu and D. J. Burton, J. Org. Chem., 2006, 71, 7780–7784 CrossRef CAS PubMed.
- H.-C. Shen, S. Pal, J.-J. Lian and R.-S. Liu, J. Am. Chem. Soc., 2003, 125, 15762–15763 CrossRef CAS PubMed.
- K. Maeyama and N. Iwasawa, J. Org. Chem., 1999, 64, 1344–1346 CrossRef CAS.
- J. W. Dankwardt, Tetrahedron Lett., 2001, 42, 5809–5812 CrossRef CAS.
- S. Mondal, B. Gold, R. K. Mohamed, H. Phan and I. V. Alabugin, J. Org. Chem., 2014, 79, 7491–7501 CrossRef CAS PubMed.
- S. Mondal, B. Gold, R. K. Mohamed and I. V. Alabugin, Chem. – Eur. J., 2014, 20, 8664–8669 CrossRef CAS PubMed.
- N. Asao, K. Takahashi, S. Lee, T. Kasahara and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 12650–12651 CrossRef CAS PubMed.
- N. Asao, Menggenbateer, Y. Seya, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Synlett, 2012, 66–69 CrossRef CAS.
- N. Asao, K. Sato, Menggenbateer and Y. Yamamoto, J. Org. Chem., 2005, 70, 3682–3685 CrossRef CAS PubMed.
- H. Kusama, H. Funami, J. Takaya and N. Iwasawa, Org. Lett., 2004, 6, 605–608 CrossRef CAS PubMed.
- L.-P. Liu and G. B. Hammond, Org. Lett., 2010, 12, 4640–4643 CrossRef CAS PubMed.
- C. M. Kane, T. B. Meyers, X. Yu, M. Gerken and M. Etzkorn, Eur. J. Org. Chem., 2011, 2969–2980 CrossRef CAS.
- T. A. Zeidan, S. V. Kovalenko, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2006, 71, 962–975 CrossRef CAS PubMed.
- J. D. Spence, M. L. Lackie, N. A. Clayton, S. A. Toscano, M. A. Farmer, E. Popova and M. M. Olmstead, Tetrahedron Lett., 2014, 55, 1569–1572 CrossRef CAS.
- K. D. Lewis, M. P. Rowe and A. J. Matzger, Tetrahedron, 2004, 60, 7191–7196 CrossRef CAS.
- J. D. Spence, A. E. Hargrove, H. L. Crampton and D. W. Thomas, Tetrahedron Lett., 2007, 48, 725–728 CrossRef CAS.
- W.-Y. Yang, B. Breiner, S. V. Kovalenko, C. Ben, M. Singh, S. N. LeGrand, Q. X. A. Sang, G. F. Strouse, J. A. Copland and I. V. Alabugin, J. Am. Chem. Soc., 2009, 131, 11458–11470 CrossRef CAS PubMed.
- J. Kaiser and B. C. J. van Esseveldt, Org. Biomol. Chem., 2009, 7, 695–705 CAS.
- Y. Du, C. J. Creighton, Z. Yan, D. A. Gauthier, J. P. Dahl, B. Zhao, S. M. Belkowski and A. B. Reitz, Bioorg. Med. Chem., 2005, 13, 5936–5948 CrossRef CAS PubMed.
- M. Gredičak, I. Matanović, B. Zimmermann and I. Jerić, J. Org. Chem., 2010, 75, 6219–6228 CrossRef PubMed.
- J. W. Grissom, T. L. Calkins and M. Egan, J. Am. Chem. Soc., 1993, 115, 11744–11752 CrossRef CAS.
- J. W. Grissom, D. Klingberg, D. Huang and B. J. Slattery, J. Org. Chem., 1997, 62, 603–626 CrossRef CAS PubMed.
- G. V. Karpov and V. V. Popik, J. Am. Chem. Soc., 2007, 129, 3792–3793 CrossRef CAS PubMed.
- N. G. Zhegalovaa and V. V. Popik, J. Phys. Org. Chem., 2011, 24, 969–975 CrossRef.
- D. R. Pandithavidana, A. Poloukhtine and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 351–356 CrossRef CAS PubMed.
- L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed.
- A. S. K. Hashmi, I. Braun, P. Nçsel, J. Schidlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nçsel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS.
- A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. Højer Vilhelmsen, M. Rudolph and F. Rominger, Chem. – Eur. J., 2013, 19, 1058–1065 CrossRef CAS PubMed.
- A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644–661 CrossRef CAS.
- P. Nösel, V. Müller, S. Mader, S. Moghimi, M. Rudolph, I. Braun, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2015, 357, 500–506 CrossRef.
- A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 10633–10637 CrossRef CAS PubMed.
- S. Naoe, Y. Suzuki, K. Hirano, Y. Inaba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2012, 77, 4907–4916 CrossRef CAS PubMed.
- C. A. Landis, M. M. Payne, D. L. Eaton and J. E. Anthony, J. Am. Chem. Soc., 2004, 126, 1338–1339 CrossRef CAS PubMed.
- M. Sivaraman and P. T. Perumal, Org. Biomol. Chem., 2014, 12, 1318–1327 CAS.
- W.-R. Chang, Y.-H. Lo, C.-Y. Lee and M.-J. Wu, Adv. Synth. Catal., 2008, 350, 1248–1252 CrossRef CAS.
- C.-Y. Lo, M. P. Kumar, H.-K. Chang, S.-F. Lush and R.-S. Liu, J. Org. Chem., 2005, 70, 10482–10487 CrossRef CAS PubMed.
- R. Liedtke, M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Org. Lett., 2012, 14, 1448–1451 CrossRef CAS PubMed.
- S. Nayak, N. Ghosh, B. Prabagar and A. K. Sahoo, Org. Lett., 2015, 17, 5662–5665 CrossRef CAS PubMed.
- S. Kitagaki, Y. Okumura and C. Mukai, Tetrahedron Lett., 2006, 47, 1849–1852 CrossRef CAS.
- M. Nechab, D. Campolo, J. Maury, P. Perfetti, N. Vanthuyne, D. Siri and M. P. Bertrand, J. Am. Chem. Soc., 2010, 132, 14742–14744 CrossRef CAS PubMed.
- S. Mondal, M. Nechab, N. Vanthuyne and M. P. Bertrand, Chem. Commun., 2012, 48, 2549–2551 RSC.
- A. Gaudel-Siri, D. Campolo, S. Mondal, M. Necha, D. Siri and M. P. Bertrand, J. Org. Chem., 2014, 79, 9086–9093 CrossRef CAS PubMed.
- S. Mondal, M. Nech, D. Campolo, N. Vanthuyne and M. P. Bertrand, Adv. Synth. Catal., 2012, 354, 1987–2000 CrossRef CAS.
- M. Nechab, E. Besson, D. Campolo, P. Perfetti, N. Vanthuyne, E. Bloch, R. Denoyel and M. P. Bertrand, Chem. Commun., 2011, 47, 5286–5288 RSC.
- J. Barluenga, M. Fañanàs-Mastral, F. Andina, F. Aznar and C. Valdés, Organometallics, 2008, 27, 3593–3600 CrossRef CAS.
- Y.-H. Wang, J. F. Bailey, J. L. Petersen and K. K. Wang, Beilstein J. Org. Chem., 2011, 7, 496–502 CrossRef CAS PubMed.
- D. Kim, J. L. Petersen and K. K. Wang, Org. Lett., 2006, 8, 2313–2316 CrossRef CAS PubMed.
- Q. Wang, S. Aparaj, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Lett., 2012, 14, 1334–1337 CrossRef CAS PubMed.
- P. Nösel, S. Moghimi, C. Hendrich, M. Haupt, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 3755–3760 CrossRef.
- H.-D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- D. M. Bowles and J. E. Anthony, Org. Lett., 2000, 2, 85–87 CrossRef CAS PubMed.
- C. Schäfer, F. Herrmann and J. Mattay, Beilstein J. Org. Chem., 2008, 4 DOI:10.3762/bjoc.4.41.
- B. R. Kaafarani and D. C. Neckers, Tetrahedron Lett., 2001, 42, 4099–4102 CrossRef CAS.
- B. R. Kaafarani, B. Wex, J. A. Krause Bauer and D. C. Neckers, Tetrahedron Lett., 2002, 43, 8227–8230 CrossRef CAS.
- H. Li, J. L. Petersen and K. K. Wang, J. Org. Chem., 2001, 66, 7804–7810 CrossRef CAS PubMed.
- W. Dai, J. L. Petersen and K. K. Wang, Org. Lett., 2004, 6, 4355–4357 CrossRef CAS PubMed.
- X. Han, Y. Zhang and K. K. Wang, J. Org. Chem., 2005, 70, 2406–2408 CrossRef CAS PubMed.
- Y. Zhang, J. L. Petersen and K. K. Wang, Tetrahedron, 2008, 64, 1285–1293 CrossRef CAS.
- H. Yang, J. L. Petersen and K. K. Wang, Tetrahedron, 2006, 62, 8133–8141 CrossRef CAS.
- Y. Yang, W. Dai, Y. Zhang, J. L. Petersen and K. K. Wang, Tetrahedron, 2006, 62, 4364–4371 CrossRef CAS.
- K. Pati, C. Michas, D. Allenger, I. Piskun, P. S. Coutros, G. dos Passos Gomes and I. V. Alabugin, J. Org. Chem., 2015, 80, 11706–11717 CrossRef CAS PubMed.
- S. Yamaguchi, H. Tanaka, R. Yamada, S. Kawauchi and T. Takahashi, RSC Adv., 2014, 4, 32241–32248 RSC.
- P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2004, 126, 3108–3112 CrossRef CAS PubMed.
- H.-C. Shen, J.-M. Tang, H.-K. Chang, C.-W. Yang and R.-S. Liu, J. Org. Chem., 2005, 70, 10113–10116 CrossRef CAS PubMed.
- S. Roy, A. Anoop, K. Biradha and A. Basak, Angew. Chem., Int. Ed., 2011, 50, 8316–8319 CrossRef CAS PubMed.
- S. Roy and A. Basak, Tetrahedron, 2013, 69, 2184–2192 CrossRef CAS.
- K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165–1180 CrossRef CAS PubMed.
- D. W. Smith Jr, D. A. Babb, R. V. Snelgrove, P. H. Townsend III and S. J. Martin, J. Am. Chem. Soc., 1998, 120, 9078–9079 CrossRef.
- J. A. John and J. M. Tour, Tetrahedron, 1997, 53, 15515–15534 CrossRef CAS.
- C. Miao, J. Zhi, S. Sun, X. Yang and A. Hu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2187–2193 CrossRef CAS.
- J. P. Johnson, D. A. Bringley, E. E. Wilson, K. D. Lewis, L. W. Beck and A. J. Matzge, J. Am. Chem. Soc., 2003, 125, 14708–14709 CrossRef CAS PubMed.
- S.-Y. Chow, G. J. Palmer, D. M. Bowles and J. E. Anthony, Org. Lett., 2000, 2, 961–963 CrossRef CAS PubMed.
- C. Praveen and P. T. Perumal, Synlett, 2011, 521–524 CAS.
- C. A. Merlic and M. E. Pauly, J. Am. Chem. Soc., 1996, 118, 11319–11320 CrossRef CAS.
- Z. Li, F. Ling, D. Cheng and C. Ma, Org. Lett., 2014, 16, 1822–1825 CrossRef CAS PubMed.
- D. S. Baranov, S. F. Vasilevsky, B. Gold and I. V. Alabugin, RSC Adv., 2011, 1, 1745–1750 RSC.
- S. F. Vasilevsky, D. S. Baranov, V. I. Mamatyuk, D. S. Fadeev, Y. V. Gatilov, A. A. Stepanov, N. V. Vasilieva and I. V. Alabugin, J. Org. Chem., 2015, 80, 1618–1631 CrossRef CAS PubMed.
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592–4593 CrossRef CAS PubMed.
- N. Choy, B. Blanco, J. Wen, A. Krishan and K. C. Russell, Org. Lett., 2000, 2, 3761–3764 CrossRef CAS PubMed.
- Z. Zhao, J. G. Peacock, D. A. Gubler and M. A. Peterson, Tetrahedron Lett., 2005, 46, 1373–1375 CrossRef CAS.
- O. V. Vinogradova, I. A. Balova and V. V. Popik, J. Org. Chem., 2011, 76, 6937–6941 CrossRef CAS PubMed.
- Z. Zhao, Y. Peng, N. K. Dalley, J. F. Cannon and M. A. Peterson, Tetrahedron Lett., 2004, 45, 3621–3624 CrossRef CAS.
- G. Ferrara, T. Jin, M. Akhtaruzzaman, A. Islam, L. Han, H. Jiang and Y. Yamamoto, Tetrahedron Lett., 2012, 53, 1946–1950 CrossRef CAS.
- C.-C. Chen, C.-M. Chen and M.-J. Wu, J. Org. Chem., 2014, 79, 4704–4711 CrossRef CAS PubMed.
- K. Hirano, Y. Inaba, N. Takahashi, M. Shimano, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 1212–1227 CrossRef CAS PubMed.
- R. Liedtke, F. Tenberge, C. G. Daniliuc, G. Kehr and G. Erker, J. Org. Chem., 2015, 80, 2240–2248 CrossRef CAS PubMed.
- M.-J. Wu, C.-Y. Lee and C.-F. Lin, Angew. Chem., Int. Ed., 2002, 41, 4077–4079 CrossRef CAS.
- A. V. Gulevskaya, A. S. Tyaglivy, A. F. Pozharskii, J. I. Nelina-Nemtseva and D. V. Steglenko, Org. Lett., 2014, 16, 1582–1585 CrossRef CAS PubMed.
- A. V. Gulevskaya, S. Van Dang, A. S. Tyaglivy, A. F. Pozharskii, O. N. Kazheva, A. N. Chekhlov and O. A. Dyachenko, Tetrahedron, 2010, 66, 146–151 CrossRef CAS.
- H. Aihara, L. Jaquinod, D. J. Nurco and K. M. Smith, Angew. Chem., Int. Ed., 2001, 40, 3439–3441 CrossRef CAS.
- M. Nath, J. C. Huffman and J. M. Zaleski, Chem. Commun., 2003, 858–859 RSC.
- M. Nath, M. Pink and J. M. Zaleski, J. Am. Chem. Soc., 2005, 127, 478–479 CrossRef CAS PubMed.
- J. D. Spence, A. C. Rios, M. A. Frost, C. M. McCutcheon, C. D. Cox, S. Chavez, R. Fernandez and B. F. Gherman, J. Org. Chem., 2012, 77, 10329–10339 CrossRef CAS PubMed.
- T. Mitamura, K. Iwata, A. Nomoto and A. Ogawa, Org. Biomol. Chem., 2011, 9, 3768–3775 CAS.
- C. J. Evoniuk, M. Ly and I. V. Alabugin, Chem. Commun., 2015, 51, 12831–12834 RSC.
- X. Lu, J. L. Petersen and K. K. Wang, Org. Lett., 2003, 5, 3277–3280 CrossRef CAS PubMed.
- C. Shi, Q. Zhang and K. K. Wang, J. Org. Chem., 1999, 64, 925–932 CrossRef CAS PubMed.
- A. Rana, M. E. Cinar, D. Samanta and M. Schmittel, Beilstein J. Org. Chem., 2016, 12, 43–49 CrossRef CAS PubMed.
- X. You, X. Xie, H. Chen, Y. Li and Y. Liu, Chem. – Eur. J., 2015, 21, 18699–18705 CrossRef CAS PubMed.
Footnote |
| † A referee pointed out that a low energy triplet state could be involved thus interfering with the usual triplet Bergman cyclization (per se not an unfavorable process), see I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2003, 125, 4495–4509. |
| This journal is © The Royal Society of Chemistry 2016 |