
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical and semisynthetic approaches to study and target deubiquitinases
Pushparathinam
Gopinath
,
Shimrit
Ohayon
,
Mickal
Nawatha
and
Ashraf
Brik
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology Haifa, 3200008, Israel. E-mail: abrik@technion.ac.il
First published on 6th April 2016
Abstract
Ubiquitination is a key posttranslational modification, which affects numerous biological processes and is reversed by a class of enzymes known as deubiquitinases (DUBs). This family of enzymes cleaves mono-ubiquitin or poly-ubiquitin chains from a target protein through different mechanisms and mode of interactions with their substrates. Studying the role of DUBs in health and diseases has been a major goal for many laboratories both in academia and in industry. However, the field has been challenged by the difficulties in obtaining native substrates and novel reagents using traditional enzymatic and molecular biology approaches. Recent advancements in the synthesis and semisynthesis of proteins made it possible to prepare several unique ubiquitin conjugates to study various aspects of DUBs such as their specificities and structures. Moreover, these approaches enable the preparation of novel activity based probes and assays to monitor DUB activities in vitro and in cellular contexts. Efforts made to bring new chemical entities for the selective inhibition of DUBs based on these tools are also highlighted with selected examples.
 Pushparathinam Gopinath | P. Gopinath received his BSc Chemistry from Sacred Heart College and MSc Chemistry from Bharathiar University, India. He completed his PhD with Prof. Muraleedharan in 2013 from Indian Institute of Technology (IIT-Madras) and moved to Prof. Herbert Waldmann group (with Dr Kamal Kumar) at Max-Planck Institute of Molecular Physiology, Germany, as a postdoctoral fellow. Since 2014, he has been working as a postdoctoral fellow in Prof. Brik Lab, in a project related to developing novel inhibitors for deubiquitinases at Schulich Faculty of Chemistry, Technion – Israel Institute of Technology. |
 Shimrit Ohayon | Shimrit Ohayon received her BSc in 2010 and MSc in 2012 in Chemistry from the Ben Gurion University of the Negev. She is currently a PhD student in Prof. Brik laboratory at the Schulich Faculty of Chemistry in the Technion – Israel Institute of Technology, working on developing chemical approaches for studying and targeting deubiquitinases. |
 Mickal Nawatha | Mickal Nawatha received her BSc in Medical Laboratory Sciences, a second BSc in Biology in 2013 and MSc in 2015 in Biology from the Technion – Israel Institute of Technology. She is currently a PhD student in Prof. Brik laboratory at the Schulich Faculty of Chemistry in the Technion, working on finding inhibitors against deubiquitinases. |
 Ashraf Brik | Ashraf Brik is a Professor of Chemistry at the Schulich Faculty of Chemistry in the Technion. Brik received his BSc in Chemistry from the Ben-Gurion University of the Negev and his MSc degree in Chemistry in 1998 from the Technion. He obtained his PhD in 2001 from the Faculty of Chemistry in the Technion. From 2002 to 2006, Brik was a Research Associate in the Scripps Research Institute. In 2007, Brik joined the Department of Chemistry in the Ben-Gurion University of the Negev as an Assistant Professor and moved to Technion in 2015. Brik is the recipient of the Bessel Award of the Humboldt Foundation for 2015, the 11th Hirata Award, the 2013 Teva Award for Excellence in memory of Eli Hurvitz, the 2013 Tetrahedron Young Investigator Award in Bioorganic and Medicinal Chemistry and the 2011 Israel Chemical Society prize for Outstanding Young Chemist. |
1. Introduction
The function and cellular localization of most proteins are regulated by numerous types of posttranslational modifications (PTMs), which are carried out, after the translation step, by various enzymes (e.g. kinases and methyltransferases).1 Ubiquitination is an example of one of the highly used PTMs in eukaryotes and involves the attachment of a ubiquitin (Ub) monomer or a poly-Ub chain to a target protein. Three enzymes, namely E1, E2 and E3, efficiently catalyze this process in an ATP dependent manner.2,3 Ubiquitination has been mainly studied in protein degradation,2,4–6 however several recent studies have also documented the involvement of ubiquitination in various non-proteolytic signals such as in chromatin remodeling and DNA repair.7–10 Remarkably, Ub chains are assembled through the attachment of any of the seven Lys residues in Ub via an isopeptide bond with the consecutive Ub, hence forming various types of homogeneous, heterogeneous or mixed chains depending on the Lys residue(s), which is involved in isopeptide bond formation.11,12 In addition, linear chains where the N-terminal amine of Ub is linked to the C-terminal Gly of the subsequent Ub via an amide bond are also known. Another layer of complexity that expands the Ub landscape is different possibilities of anchoring sites on the target protein, which leads to the formation of various types of Ub conjugates,11,12e.g. mono-ubiquitination, multiple mono-ubiquitination and poly-ubiquitination. The formation of various types of Ub chains with different lengths and connectivities underscores the diverse regulation of many biological processes in health and disease.Like many PTMs, ubiquitination is a reversible process and tightly regulated by a family of enzymes called deubiquitinases (DUBs).13–15 DUBs play multiple roles in Ub signaling and are involved, among the many functions, in maintaining a pool of stable free Ubs in cells (Fig. 1).15 Once the target protein undergoes ubiquitination, it could then take various possible pathways by the action of DUBs; namely (i) editing, where ubiquitination and deubiqutination work in concert to alter the Ub signal and change the fate of the substrate, for example, from a degradative to a non-degradative process or vice versa;16 (ii) rescuing the protein from the specific fate (e.g. degradation) by cleaving Ub or Ub chains;17 (iii) preventing poly-Ub chains from degradation along with the substrate during proteasomal degradation;17 (iv) processing pro-Ub – as a mono-Ub or linear chain – to active Ub;17 and (v) recycling trapped Ub by undesired reactions with cellular nucleophiles such as glutathione into the cellular Ub pool.18
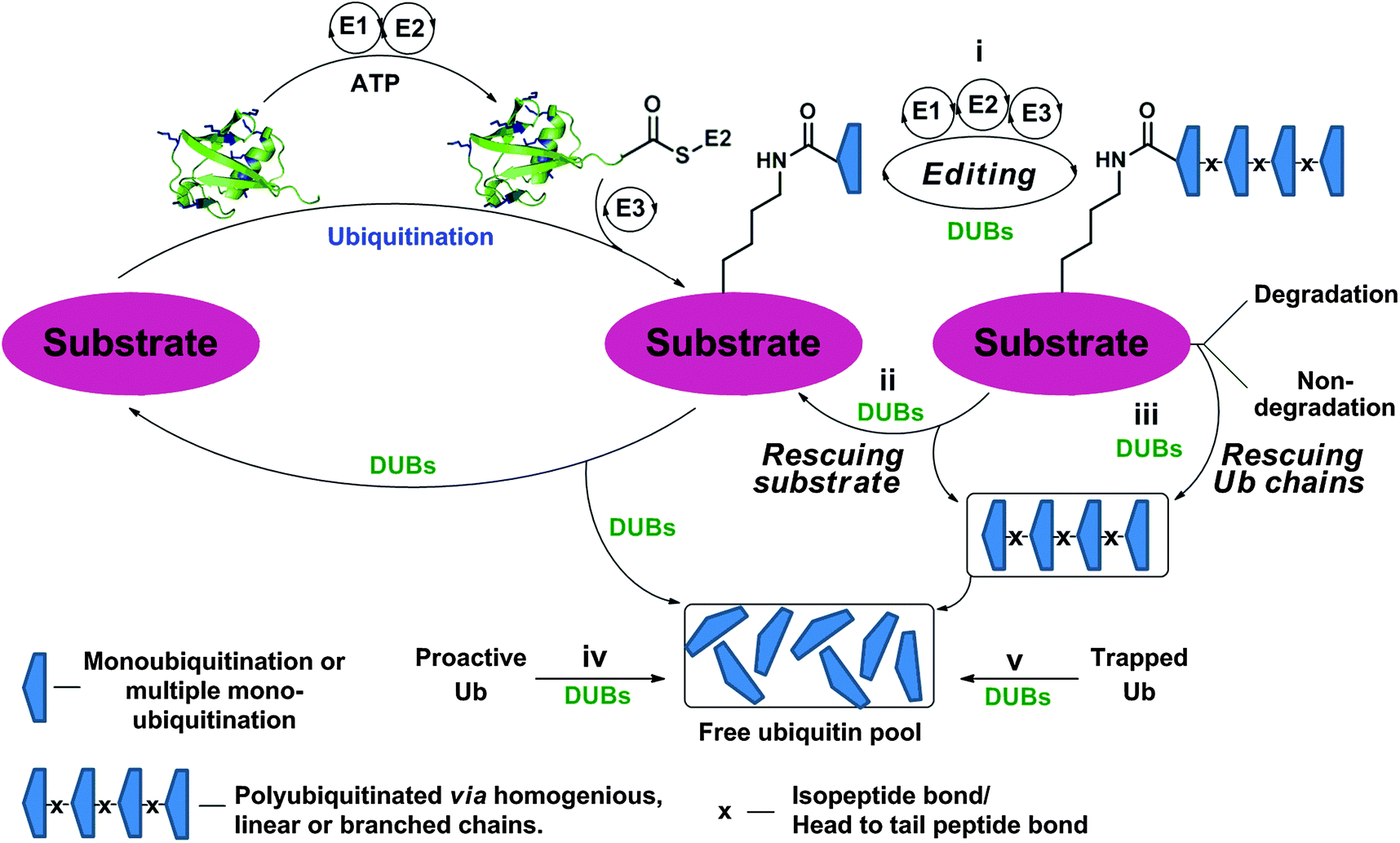 | ||
| Fig. 1 Ubiquitination of the substrate via E1, E2 and E3 in an ATP dependent manner and the different roles of DUBs by acting on Ub chains and ubiquitinated proteins at different levels. | ||
1.1 Classification of DUBs
Approximately 100 putative DUBs are known and broadly classified into five different subclasses, namely Ub C-terminal hydrolase (UCH), Ub specific protease (USP), ovarian tumor protease (OTU), Josephin/Machado–Joseph disease protease (MJD) and JAB1/MPN/MOV34 metalloenzyme (JAMM).13–15 This classification is based on sequence homology, the mechanism of action and the structure of the catalytic domain.19 The majority of DUBs fall under the USP family, which contains ∼70 members with variable sizes (300–800 residues). The UCH family is composed of four DUBs known as UCH-L1, UCH-L3, UCH-L5/UCH37 and BAP1. Similarly, the MJD family also has four members known as Ataxin3, Ataxin3L, JOSD1 and JOSD2, each containing ∼180 residues. There are 15 members in the OTU family, where DUBs from this family contain ∼180 residues. The JAMM metalloprotease family is composed of eight DUBs known as POH1, AMSH, AMSH-LP, BRCC36, CSN5, MYSM1, PRPF8 and MPND.DUBs operate in two different mechanisms, in which four out of the five families are papain like Cys-proteases, while the fifth belongs to zinc metalloproteases. In Cys-proteases, the catalytic triad consists of two (Cys and His) or three amino acids (Asn/Asp), which are involved in the mechanism of hydrolysis.15,20 Here, the catalytic process is initiated by proton abstraction from the catalytic Cys by the His side-chain, thereby facilitating the nucleophilic attack of the Cys on the carbonyl carbon, resulting in a thioester intermediate (Fig. 2a). This step is followed by the formation of an oxyanion hole bearing negative charge on the carbonyl oxygen, which is often stabilized by hydrogen bond donating residues (Asn/Asp). Finally, the thioester is hydrolyzed by a water molecule to complete the catalytic cycle. Structural insights into the mechanism of the hydrolysis of Lys63-linked di-Ub by JAMM were recently elucidated (Fig. 2b).21 Here, in the initial step of hydrolysis, the catalytic zinc is coordinated by two His residues: Asp and a water molecule. Abstraction of the proton from the zinc-bound water facilitates its nucleophilic attack to form an oxyanion hole, which is stabilized by hydrogen bonding with the nearby Ser residue. Finally, this intermediate collapses leading to the hydrolysis of the isopeptide bond.
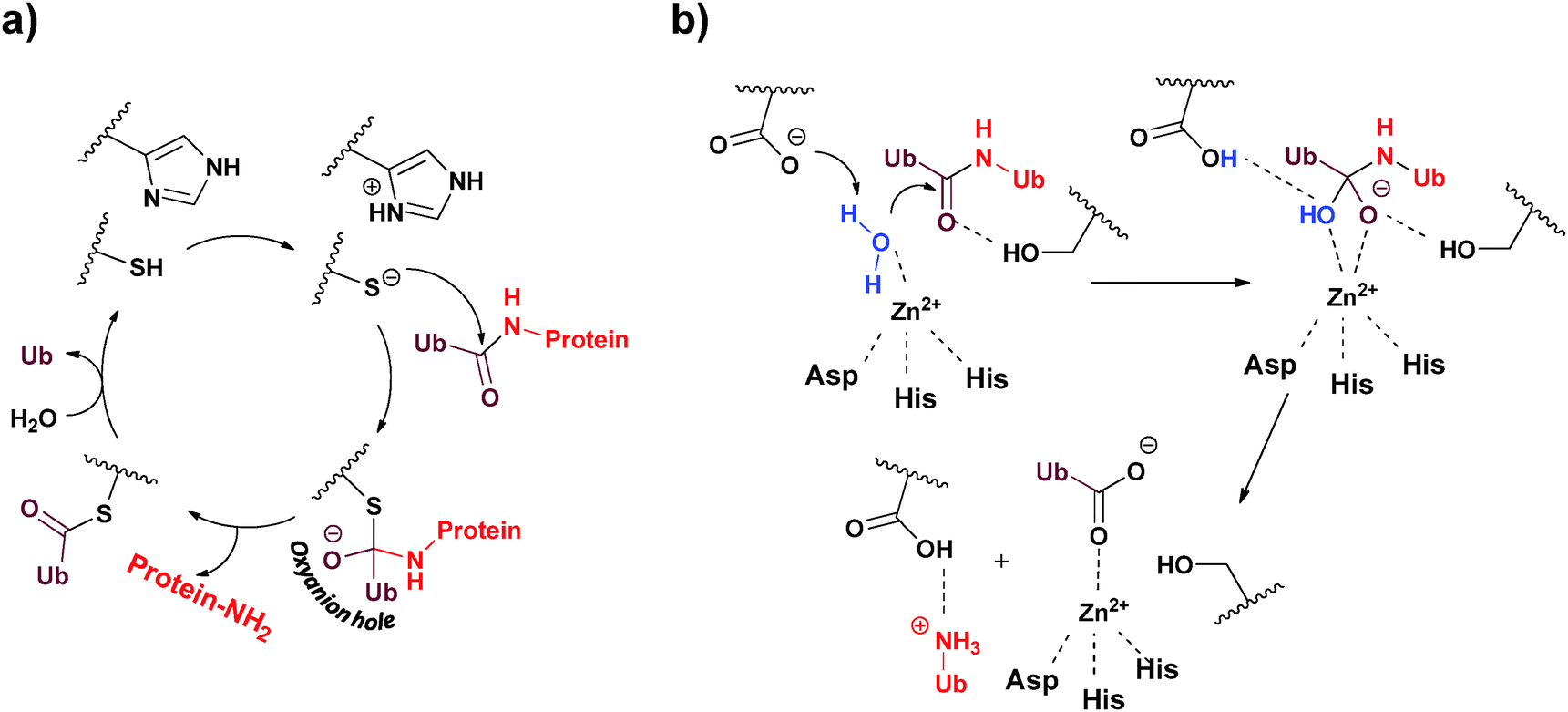 | ||
| Fig. 2 The two different mechanisms employed by DUBs to hydrolyze their substrates. (a) Cys-protease based mechanism and (b) metalloprotease based mechanism. | ||
1.2 Structural aspects of DUBs
DUBs are unique in the sense that they bind to large surfaces via their Ub binding domains (UBDs).22–24 In addition, major alterations in their conformations occur during the binding step with their substrates.17 DUBs from the UCH family cleave mainly small adducts from the C-terminus of Ub.18,25 One of the most studied enzymes in this class is UCH-L3 whose structural features are presented in Fig. 3a. The active site of UCH-L3 is covered by a loop, which dictates the size of the substrate to be processed.26 Upon binding to the Ub substrate, major conformational changes occur in the loop that covers the active site, thus ensuring substrate selectivity (Fig. 3a).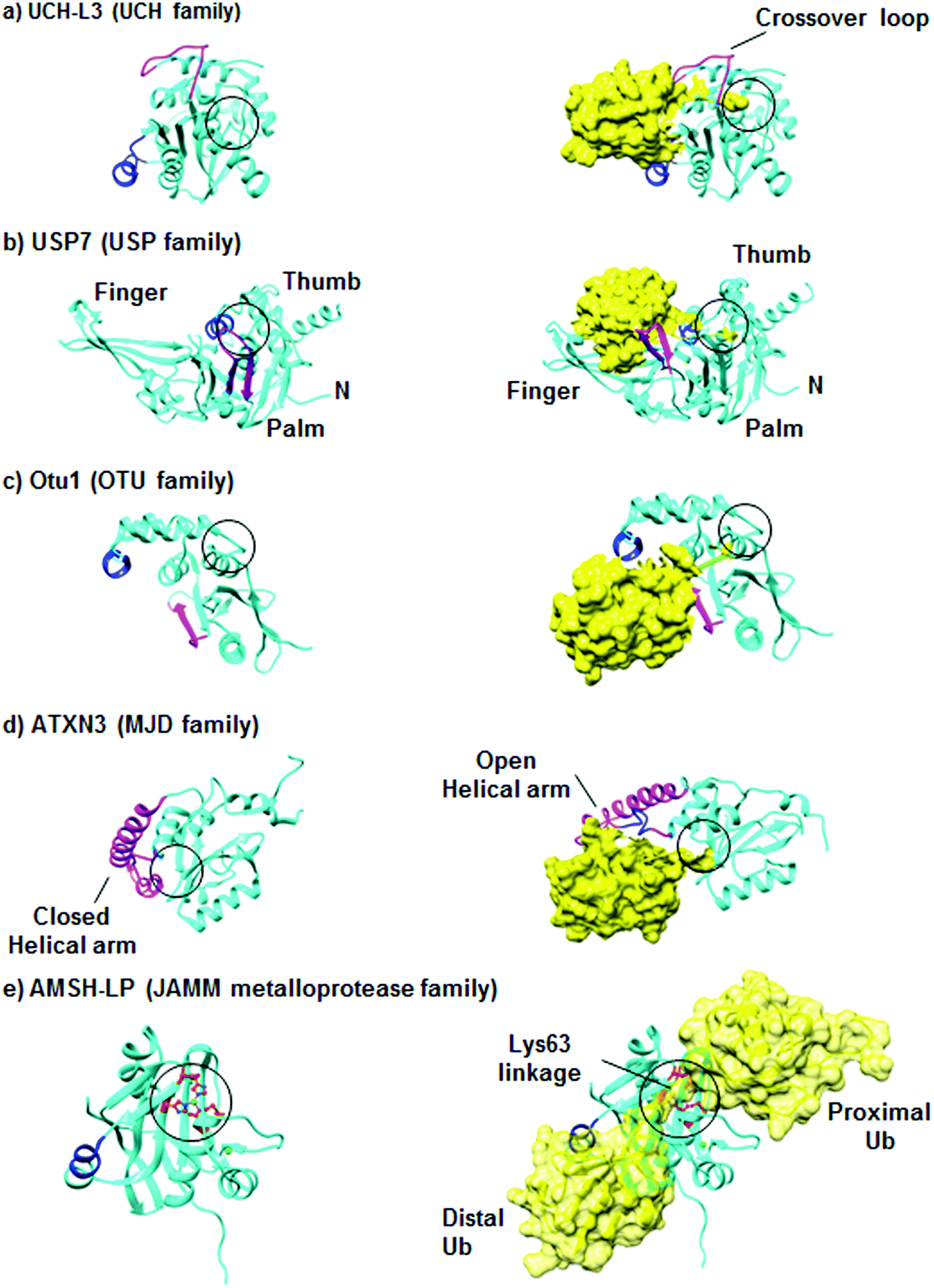 | ||
| Fig. 3 Representative structures of DUBs in their inactive and active conformations. DUBs are shown in a cartoon representation and colored in cyan, where Ub is shown in yellow. The active sites are indicated in black circles and Ile44-interacting motifs are shown in blue. The structural elements, which become ordered upon Ub binding, are shown in pink. (a) The structures of inactive (PDB: 1UCH)31 and active (PDB: 1XD3)25 forms of UCH-L3. (b) The structures of inactive (PDB: 1NB8) and active (PDB: 1NBF) forms of USP7.28 (c) The structures of Otu1 in the absence and presence of Ub (PDB: 3BY4).29 (d) The solution structures of ATXN3 in the absence (PDB: 2AGA)32 and in the presence of Ub (PDB: 2JRI).33 (e) The structures of the catalytic domain of AMSH-LP in the absence of Ub (PDB: 2ZNR) and in a complex with Lys63-linked di-Ub (PDB: 2ZNV).21 Figures were generated using Chimera. | ||
The overall structure of USP is divided into three domains, namely the palm, thumb and finger. The structure of USP7 with and without Ub is presented in Fig. 3b. The active site lies between the palm and thumb domains, while the finger domain is responsible to hold the distal Ub. Some USP members are found in nonproductive conformation in the free form.14 However, upon Ub binding they undergo major conformational changes, thus shifting from the inactive to the active conformation. Such conformational behavior could also lead to changes in the pKa of the catalytic Cys as was reported for USP7.27,28
The OTU core domain consists of five β-sheets surrounded by two helical domains, in which their sizes differ among the family members.17 These DUBs contain the conserved Cys, His, and Asp residues, which define the putative catalytic triad of Cys-proteases. The crystal structure of the yeast Otu1 catalytic domain reveals two globular parts, which come together to form the Ub binding site (Fig. 3c).29 Like the USPs, OTUs also undergo conformational changes from the inactive to active conformation upon Ub binding. On the other hand, this family has a high degree of Ub chain linkage specificity compared to the USP family.30
Among the four members of the MJD family, Ataxin3 (ATXN3) and ATXN3L, each contains two tandem Ub interacting motifs (UIMs) in their C-terminal region, in addition to a third UIM in the ATXN3.34 ATXN3 is the best studied member in this family, while the other members remain poorly characterized. The structure of ATXN3 features an extended helical arm that is proposed to regulate the access of the substrate to the active site (Fig. 3d).33 Furthermore, structural studies of ATXN3 bound to Ub revealed that it can bind two Ub molecules simultaneously.33
The fifth family consisting of the JAMM proteases represents the metalloproteases, which contain two zinc ions with a highly conserved His-X-His-X7-Ser-X2-Asp motif that binds Zn2+, where X is non-conserved amino acid.35 The zinc ion activates water molecules to attack the isopeptide bond between the Ub and its substrate. The crystal structure of AMSH-LP bound to Lys63-linked di-Ub represents the first example of such a structure for DUBs in general and reveals the molecular basis of the specificity of this DUB towards this chain (Fig. 3e).21
1.3 Substrate recognition and specificity
The structures of DUBs include several UBDs, such as UIM and UBA, which regulate the activity and specificity towards different substrates.22–24 The interactions between Ub and DUBs are mediated by the hydrophobic patch in Ub (Leu8, Ile44 and Val70) and the surrounding residues (Fig. 4a). Another important factor that plays a key role in the specificities of DUBs is the conformation of the Ub chains, which are known to adopt different conformations varying from an open to a closed conformation (Fig. 4a).36 For example, a Lys48-linked di-Ub chain adopts a closed conformation, where the hydrophobic patches of the Ub monomers interact with each other to form a compact structure.37,38 On the other hand, the Lys63-linked di-Ub chain adopts mainly an open conformation, where the Ubs in the chain do not interact with each other (Fig. 4a).39 These differential linkages can be recognized by different UBDs, which dictate the specificity of DUBs. For example, UBA is specific for the Lys48 linkage while UIM is specific for the Lys63 linkage (Fig. 4b).40–42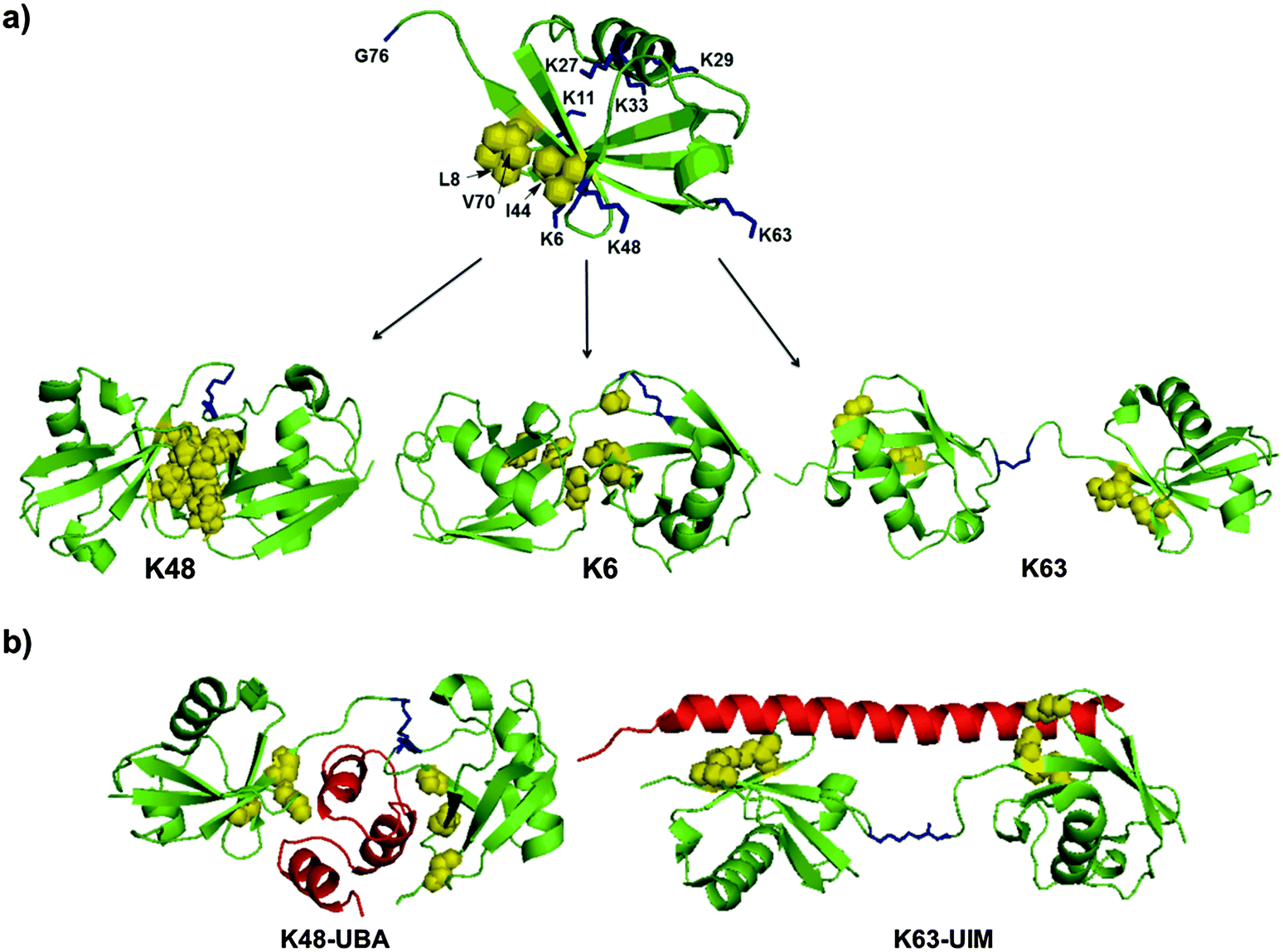 | ||
| Fig. 4 The structural elements in Ub and Ub chains that dictate DUB specificities. (a) The structures of Ub (PDB: 1UBQ)43 and various di-Ub chains having different conformations (PDBs: 2PEA, 2XK5, 2JF5).44–46 (b) Examples for specific interactions of the Ub chain with UBD (in red) (PDBs: 1ZO6, 3A1Q).42,47 Lys residues are highlighted in blue and the hydrophobic patch is highlighted in yellow. | ||
There are several examples of DUBs where UBDs determine the specificity toward different substrates. For example, through NMR studies, the ankyrin repeat domain – an extended site of an OTU domain in TRABID enzymes – was found to serve as a Ub interacting site (AnkUBD), where the hydrophobic surface of ankyrin interacts with Ile44 from the hydrophobic patch of Ub.48 In the absence of AnkUBD, TRABID exhibits efficient activities toward Lys29-, Lys33-, Lys48- and Lys63-linked di-Ub chains in addition to a weak activity towards Lys6- and Lys11-linked di-Ub chains. The addition of the AnkUBD domain leads to specific hydrolysis of Lys29- and Lys33-linked di-Ub chains, which highlight the importance of UBD for the observed specificity.
1.4 The roles of DUBs in cellular pathways
The significant roles of DUBs in various cellular processes are now well documented and new functions continue to emerge from several academic and industrial research groups. DUBs are involved in degradative and non-degradative signaling such as protein trafficking, gene transcription, DNA repair and replication.49 The involvement of DUBs in protein degradation is attributed to the presence of three different DUBs in the proteasome machinery: USP14, UCH-L5/UCH37 and Rpn11.50–52 Thus, targeting any of the three DUBs could have a direct influence on the fate of proteins. The role of DUBs in non-degradative signaling is illustrated with a representative example as follows. USP1 is important in the DNA damage response (DDR) pathway, where it acts on two DNA damage related proteins, namely proliferating cell nuclear antigen (PCNA) and fanconi anemia group D2 protein (FANCD2).53 The knockdown of USP1 in cells leads to the accumulation of mono-ubiquitinated FANCD2, further resulting in DNA damage repair.54The two DUBs, namely CYLD (a member of USP family) and A20 (metalloproteases), are known to be involved in regulating the NF-κB signaling.55,56 Mutations in CYLD lead to the dysregulated action of NF-κB, whereas A20 is needed for the termination of the NF-κB activation signal. Considering the role of NF-κB in innate and adaptive immune response, it is not surprising that CYLD and A20 are involved in its regulation.49
It is now clear that the aberration in DUB functions leads to various diseases. For example, different studies have shown that DUBs, especially from the USP family, are regulating various important oncogenic proteins such as the tumor suppressor p53 and its ligases MDM2 and MDMX.57 For example, USP7 deubiquitinates p53 as well as its ligases (MDM2 and MDMX) and serves as a regulator for p53 levels directly by acting on p53 itself or indirectly on MDM2/MDMX.58 On the other hand, USP2 regulates the p53 pathway by acting only on MDM2.59 This is supported by studies where the knockdown of USP2 in prostate cancer cell lines leads to the accumulation of the p53 protein.59 Together, these studies support that the inhibition of these DUBs can be a novel approach to target cancer.
The involvement of DUBs in the survival of viruses also makes them possible targets in viral diseases.60 For example, Epstein Barr nuclear antigen 1 (EBNAI), the regulator of both transcription and replication of the Epstein-Barr virus (EBV), interacts with USP7 in a similar way USP7 does with p53, thus preventing the deubiquitination of p53.61 As a result, the degradation of p53 is facilitated thereby preventing the apoptosis of virus-infected cells.
The role of DUBs in neurodegenerative diseases is also well documented. For example, UCH-L1 is highly abundant in brain and found to be connected to several neurodegenerative diseases (e.g. Alzheimer’s and Parkinson's diseases).62 It has been shown that UCH-L1 exhibits the Ub ligase activity and promotes the aggregation of α-synuclein – a protein that is associated with several neurodegenerative diseases.63 Moreover, like α-synuclein, UCH-L1 was also found to be localized in Lewy bodies, the hallmark for Parkinson’s disease.64
The association of a large number of DUBs in various cellular processes undoubtedly illustrates the high importance of DUBs in human health. Hence, DUBs are emerging as attractive targets, as can also be learned in dedicated reviews related to cancer,65–68 neurodegenerative69,70 and antiviral therapeutic areas.60
1.5 Challenges in studying and targeting DUBs
Despite various impressive developments in the ubiquitination and deubiquitination fields, a comprehensive understanding of these complex processes is still lacking. Moreover, the characterization of the cellular substrates for DUBs is still in its infancy, which creates more challenges when attempting to target a particular DUB. Even when knowledge exists on a specific substrate, in many cases we lack full understanding of the detailed interactions with its DUB. It is also mandatory to have knowledge on DUB levels in normal and abnormal cells in order to understand more about their role in disease states. Additionally, DUBs are regulated by various PTMs, which further increases the complexity in studying and targeting them.71,72 For example, the activity of the NF-κB associated DUB, CYLD, is regulated by both phosphorylation73 and SUMOylation.74 Finally, developing new drugs against DUBs in the presence of a plethora of various other proteases, which are largely investigated in various diseases, requires understanding the molecular basis of DUB-inhibitor interactions.One of the main challenges to address several of these questions has been the lack of homogeneous Ub based conjugates in high purity and large quantities. These could serve as novel reagents for different studies such as determining the specificities of DUBs and to monitor their activities as well as to determine their structures with cellular substrates. This review highlights the recent developments in the chemical and semisynthesis of well-designed Ub based reagents to assist in studying DUBs. Emphasis will be given on the synthesis of novel reagents and how they are aiding detailed analyses of the effect of the linkage types and lengths of the Ub chains on the specificity and recognition of DUBs. Moreover, we cover the development of activity-based probes useful in various studies such as discovering new DUBs and examining the cellular effect of a specific inhibitor on a particular DUB. We also cover the development of assays useful in finding novel inhibitors for this group of enzymes. A brief survey of the current inhibitors for DUBs that are associated with different diseases is also included.
2. Synthesis and semisynthesis of Ub conjugates
2.1 Challenges and motivation
Probing the mode of action, recognition and inhibition of DUBs is an important step for better understanding their roles in biological processes and ultimately in disease states. The preparation of Ub conjugates in order to study the different aspects of DUBs is thus required. Such conjugates can be prepared either enzymatically or by using chemical and semisynthetic approaches. Enzymatic synthesis of Ub chains and ubiquitinated proteins requires the judicious choice of E2/E3 enzyme pairs, which are not known or available for many of these targets. Even if a particular enzymatic pair is known and available, it is quite difficult in vitro to control the length of the growing Ub chain and the site of ubiquitination in the protein substrate. This often leads to a mixture of Ub conjugates, which makes it very difficult to understand various aspects of the Ub signal at the molecular level. One approach to overcome this, when using enzymes, is by mutating native Lys residues in Ub to Arg, which could affect the properties of the native protein. Hence, the accessibility of Ub conjugates (e.g. poly-Ub, Ub-based probes) in high purity, homogeneity and workable quantities became the limiting step in various studies.In order to overcome these limitations, organic chemists and chemical biologists have developed a variety of tools to enable the preparation of several types of Ub conjugates. These approaches not only enable the access to native structures but also for novel analogues labelled with fluorophores and unique functionalities. Hence, Ub conjugates with high complexity such as activity-based probes, and free or anchored poly-Ub chains to protein substrates could also be prepared. These developments are being used to understand the function, specificity, structure and inhibition of DUBs.
2.2 Ub analogues
Access to Ub and its conjugates was achieved mainly through three approaches, namely transpeptidation, intein and total chemical synthesis. Transpeptidation was for a while the method of choice to prepare Ub analogues via selective cleavage of Ub at Arg74 and the incorporation of C-terminally modified Gly-ester for subsequent manipulation to generate a C-terminally modified Ub (Fig. 5a).75 In the intein based approach, a Ub mutant (Gly76Cys) is fused to intein, which is immobilized on chitin beads. N–S acyl transfer of Ub mutant occurs upon activation (Fig. 5b).76 This intermediate can then be trapped with various nucleophiles such as 2-mercaptoethane sulfonate (MES) to generate Ub-MES, which can serve as a reactive precursor for the synthesis of Ub conjugates. The third approach relies on the total chemical protein synthesis employing solid phase peptide synthesis (SPPS) and ligation approaches (Fig. 5c). SPPS is mainly effective for the preparation of relatively short peptides (30–40 residues).77 In order to overcome the size limitation in polypeptide synthesis, chemoselective ligation approaches were developed.78,79 Native chemical ligation (NCL) is considered the method of choice when attempting the ligation of unprotected peptides.80 In NCL, a chemoselective reaction occurs between the C-terminal thioester of one peptide and the N-terminal Cys of the second peptide to form the native amide bond. The need for a Cys residue at the N-terminal peptide has limited the scope of NCL for the synthesis of proteins that lacks Cys residues or has non-strategically placed Cys, which is known to be one of the rare amino acids in nature. In order to expand the scope of NCL for the synthesis of proteins, various thiol-modified amino acids were developed to enable ligation, which upon desulfurization results in the unmodified ligation junction.81,82 With more than 11 amino acids modified at β, γ or δ positions with a thiol handle, it has been possible to assemble the protein of choice with less synthetic barriers.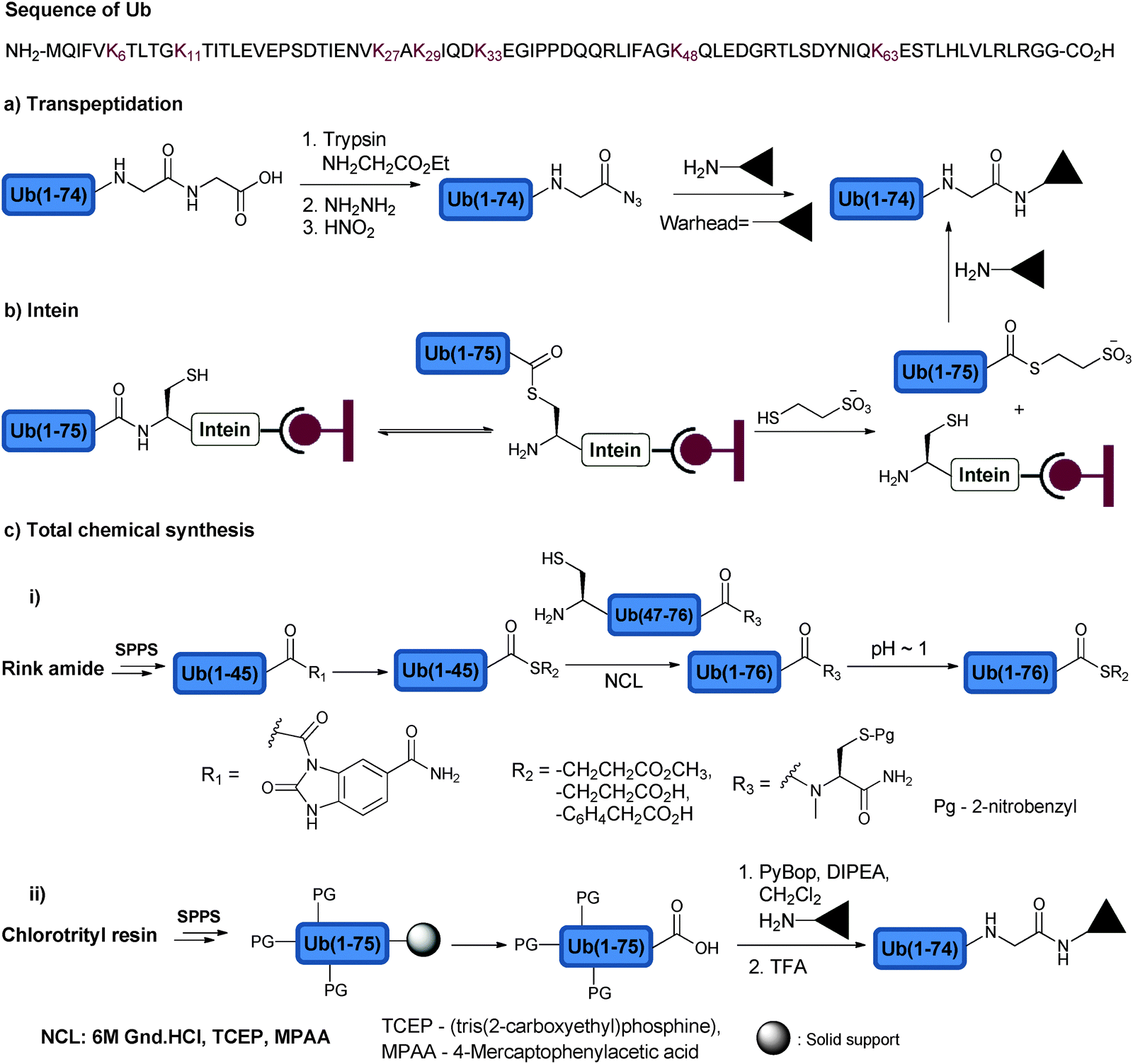 | ||
| Fig. 5 Approaches to synthesize the C-terminal modified Ub using (a) transpeptidation, (b) intein and (c) total chemical synthesis approaches. The Ub sequence is shown at the top of the figure highlighting the Lys residues, which undergo ubiquitination for the chain assembly. | ||
Another important development in NCL based methods is related to the installation of a C-terminal thioester functional group by using Fmoc-SPPS, which is limited due to the basic conditions employed during the Fmoc deprotection steps. Various approaches to prepare this synthetic intermediate via Fmoc-SPPS were reported; some of these are routinely being used in protein synthesis.83 One such approach is based on the use of a 3-Fmoc-4-diaminobenzoic acid (Fmoc-DBZ) linker, which can be coupled to the resin and elongated with the target peptide.84 Once the synthesis is completed, the DBZ can be converted to an excellent leaving group, N-acyl-benzimidazolinone (NBZ), which by thiolysis can be converted to the thioester functionality.
These advancements made it possible to synthesize Ub and its analogues either via a fragment approach or full synthesis using Fmoc-SPPS. Ramage and coworkers reported the first total synthesis of Ub using Fmoc-SPPS.85,86 The one pot NCL approach developed by Kent and coworkers enabled the synthesis of Ub from three fragments.87 Despite the advancements in the total synthesis of Ub, access to C-terminal modifications demanded to device new strategies to obtain the required Ub conjugates in milligram quantities for various biochemical studies. For example, the synthesis of Ub thioesters was achieved through two peptide fragments and by employing N-methylcysteine as a latent thioester group.88 The required fragments, Ub(1–45)-thioester and Cys-Ub(47–76)-N-methylcysteine, were synthesized using Fmoc-SPPS. While the Ala46Cys mutation in the N-terminal fragment enabled NCL with Ub(1–45)-thioester to give Ub(1–76)-N-methylcysteine, the C-terminal N-methylcysteine enabled thioester formation by employing mercaptopropionic acid (MPA) under acidic conditions. Improved synthesis of the full length Ub was also made using the Fmoc-SPPS method on Wang, chlorotrityl or Rink amide resins.89,90 In order to make C-terminal analogues, for example, Ub was synthesized on the acid labile trityl resin. Taking advantage of the mild deprotection conditions (HFIP/DCM), the peptide was cleaved from the resin while keeping the protecting groups intact.89 This made it possible to couple the free C-terminus acid with the desired nucleophiles, which was followed by global deprotection using trifluoroacetic acid (TFA). Using the above-described approaches, novel Ub analogues, highly relevant to DUB studies, were prepared and will be discussed in the following sections.
2.3 Activity based probes
In order to study DUBs in natural systems, one has to develop various tools to study their function. In this context, activity based probes (ABPs) have been developed to study DUBs in cellular contexts among several other studies.91,92 ABPs mimic the natural substrates of DUBs, however bearing a reactive moiety nearby the scissile bond, which can react with the catalytic Cys of the targeted DUB and form a covalent adduct. The key advantage of activity-based protein profiling over traditional methods, besides avoiding the isolation of the target protein, is the reliance on studying the target enzyme based on its catalytic activity and not on its expression level.92In the design of an ABP three main parts must be considered: (1) the targeting element, (2) the warhead and (3) the recognition tag (Fig. 6a).93 In order to achieve the specificity of DUBs in a pool of proteins, Ub has been used as the targeting element since it is a major part of the natural substrate. In addition, the warhead, which is generally a reactive functional group, has to be appropriately positioned in order to react with a nucleophilic thiol. The recognition tag is used to visualize the trapped DUBs.
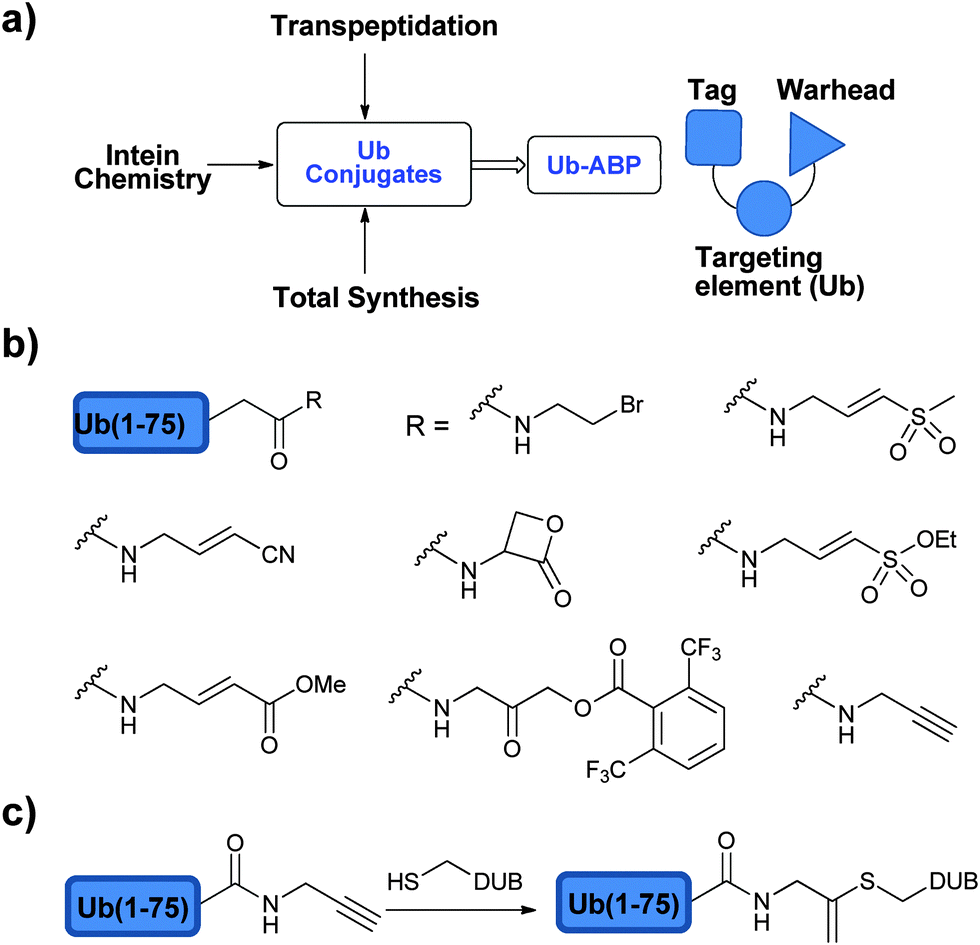 | ||
| Fig. 6 ABPs to study DUBs: (a) approaches to prepare ABPs and their components. (b) Selected warheads used in the design of ABPs. (c) Ub-Prg probes. | ||
While probing the mechanism of UCH, it was found that Ub-aldehyde (Ubal) forms an irreversible complex with the protein and inhibits its action.94 The 1,2 addition of the thiol group to aldehyde resulted in a stable protein complex, which prompted others to utilize Ubal as an ABP.95 During previous studies to understand the editing role of isopeptidase, 125I-labelled Ub-nitrile was first used as an ABP.96 This successful inhibition with the nitrile-based probe, via the 1,2 addition of catalytic Cys on the nitrile group, triggered the search for even more efficient ABPs.
Borodovsky et al. explored the synthesis and utility of Ub vinyl sulfone (UbVS) as an ABP to label DUBs to shed light on the association of USP14 with the 26S proteasome.50 The required C-terminal modified Ub was made using the transpeptidation approach. Trypsin digestion of Ub followed by purification and treatment with hydrazine gave Ub(1–75)-NHNH2, which was further oxidized using nitrous acid leading to Ub(1–75)-N3. Treatment with Gly-vinyl sulfone led to the formation of the desired probe. The ABP was enriched with 125I (radioiodinated with Na125I) for the direct visualization of the trapped DUBs in the crude cell extract. The probe is mechanism based leading to an irreversible mode of labelling of DUBs by the reaction of catalytic Cys with the vinyl sulfone. This leads to the formation of a stable thioether linkage, which is compatible under routine SDS-PAGE conditions, in contrast to the product obtained with the Ubal. Using this probe, 6 out of 17 putative DUBs from a yeast cell extract were labelled. On the other hand, the labelling pattern in the mammalian cell extract reflected a larger number of USPs and UCHs than yeast. As expected, the labelling of DUBs exhibited different patterns depending on the used tissue source, where the expression and the activation levels of DUBs can be different. In this study, it was also found that the labelling of USP14 is inversely proportional to the proteasome activity, which provided further evidence of the connection between USP14 and the proteasome.
In a later study, the same group used the hemagglutinin (HA)-tag to enable visualization.76 The shift from audioradiography to the HA-tag not only made the visualization of the labelled DUBs more convenient but also was used to perform immunoprecipitation to identify the labelled DUBs. In order to improve the labelling efficiency of the probes, warhead groups with varying electrophilicity were introduced and the labelling propensity of DUBs was studied.76 Hence, the required HA-UbMES was made using the more efficient intein approach and several other probes with different C-terminal warheads were synthesized, such as bromoethyl (HA-UbBr2), vinyl methyl sulfone (HA-UbVS), vinyl methyl ester (HA-UbVME) and vinyl cyanide (HA-UbVCN) (Fig. 6b). The labelling efficiency of each probe was tested against cell extracts from EL4 mouse thymoma cell lines. HA-UbVME labelled USP11, 12, 13, 25, 28 and CYLD1, in addition to those that were labelled by HA-UbVS, making it the most efficient probe. The application of this ABP was further demonstrated by the identification of M48(USP) DUB, which does not have sequence similarity with other known DUBs.97
In order to decrease the complexity of the ABP probes, peptides with varying lengths derived from the C-terminal of Ub were tested.98 Though 12-mer peptide (Z-STLHLVLRLRGG-VS) was found to be reactive with UCH-L3, the full Ub structure was needed for efficient and selective recognition by DUBs.
Increasing the electrophilicity of the warhead could lead to the labelling of more DUBs in the cell extract. Thus, second generation ABPs were designed and prepared using the intein approach, which included Ub-vinylethoxysulfone (Ub-OEtVS), Ub-β-lactone (Ub-Lac) and Ub-2,6-trifluoromethylbenzyloxymethylketone (Ub-TF3BOK) (Fig. 6b).99 A comparative assessment of these probes was made with HA-UbVME, which showed a broader labelling pattern from the first generation ABP. These results indicated that HA-UbVME is still the most efficient probe for DUB profiling. The increased electrophilicity was not reflected in higher reactivity, which might be due to rapid hydrolysis of the more reactive probes during the labelling reaction.99 It is noteworthy to mention that the selectivity of first generation Ub-ABPs towards DUBs over other Ub interacting enzymes like E1 and E2 was observed due to the low affinity of these enzymes for the free Ub.76 However, increasing the electrophilicity of the second generation Ub-ABPs enabled the detection of several Ub interacting enzymes, including HECT domain containing proteins.99
The importance of ABPs has not been limited only to identifying new DUBs but also for the comparison of DUB activation and expression levels in normal and disease states.100 Using HA-UbVME and HA-UbBr2 ABPs, the activity based profiling of DUBs in normal, virus-infected and malignant human cells was studied. Upregulation of several USPs (USP7, 9, 13, 15 and 22) was found in virus infected and mitogen activated cells, which makes them potential drug targets for the development of novel therapeutics.
During the synthesis of propargylated-Ub (Ub-Prg), as a precursor for click chemistry, Ovaa and coworkers surprisingly found that UCH-L3 was inhibited stoichiometrically by treatment with Ub-Prg (Fig. 6c).101 Through X-ray crystallography the group found that the irreversible inhibition is due to the nucleophilic addition of the DUB active site Cys to the β-carbon of the terminal alkyne resulting in the vinyl thioether. The scope of this probe was further extended to all the four families of Cys DUBs. When compared with the labelling efficiency with Ub-VME in cell lysates, Ub-Prg labelled more DUBs under identical conditions. In particular, this probe was able to label A20, which was not reactive with other Ub-based ABP. The Mootz group simultaneously found that when they append the propargylic group instead of C-terminal Gly of SUMO or Ub, a similar observation was made as to the reactivity of the alkyne towards the catalytic Cys residue of SUMO protease, Senp1.102 These results also highlight that terminal alkynes may not be as innocent as they have been often thought when click chemistry is applied in a cellular environment.103
The reactive Ub-thioester prepared via the intein approach is limited in terms of the type of tags attached with the probe. To circumvent this, total chemical synthesis of various ABPs with various modifications at the N- and C-terminal of Ub was demonstrated by Jong et al.104 The incorporation of various tags (e.g. HA and Biotin) and the flexibility to introduce various linkers in the desired position and the conjugation of various electrophilic warheads were also demonstrated.
These ABPs based on mono-Ub were further extended to di-Ub based probes and will be discussed in Section 2.7.
2.4 Strategies to construct the isopeptide bond in Ub conjugates
In order to probe the specificity, mechanism and recognition of DUB native substrates, one has to construct the Ub chains and ubiquitinated peptides/proteins with the native isopeptide bond within the chain and between the substrate and the proximal Ub. The strategies to make this isopeptide bond through semisynthetic and synthetic approaches are referred as non-enzymatic approaches. There are different ways available to make the isopeptide linkage. The first approach that was developed was based on the photolabile auxiliary mediated isopeptide bond formation (Fig. 7a).105 A proof of concept was demonstrated with the 11-mer peptide from the C-terminal of H2B histone. The Lys was acetylated using bromoacetic acid under DIC conditions, which can be displaced by the free amine of the photolabile auxiliary under basic conditions (Fig. 7a). NCL with Ub(1–75)-MES resulted in the desired native isopeptide bond formation and upon exposure to photolytic conditions (UV, 325 nm) it led to the removal of the auxiliary to give the Ub conjugate with the native isopeptide bond. One drawback of this method is the slow rate of the ligation reaction, which required several days to give the desired product in a moderate yield. Recent addition to this auxiliary based approach was reported by Weller et al. where they utilized 2-(aminooxy)ethanethiol as an auxiliary, which can be removed under denaturating conditions in the presence of Zn.106 This approach facilitates NCL to occur within 12 hours and was further expanded to SUMOylation. One interesting aspect of this approach is the possibility to create stable Ub conjugates against the action of DUBs by leaving the auxiliary attached (see further discussion on the stable Ub conjugates in Section 2.6).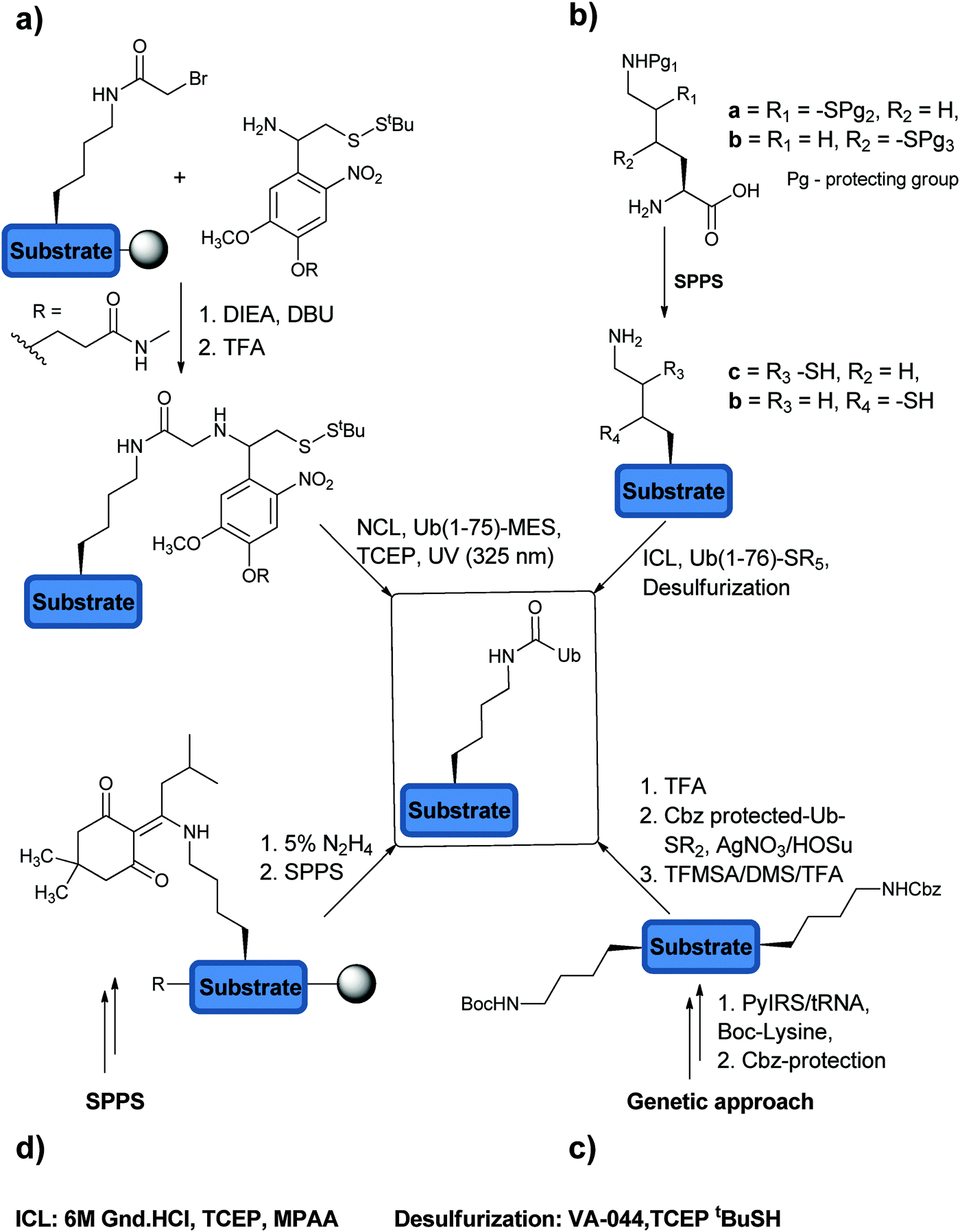 | ||
| Fig. 7 The different methods that are being used to construct the native isopeptide bond. (a) Auxiliary mediated isopeptide formation. (b) Thiol modified Lys. (c) Genetic approach. (d) Expeditious synthesis method. | ||
The second method relies on the usage of thiol-modified Lys (i.e. γ-mercaptolysine and δ-mercaptolysine)89,107–109 to facilitate efficient isopeptide chemical ligation (ICL). Here, orthogonally protected γ/δ-mercaptolysine is incorporated into the desired position during SPPS, which upon side chain deprotection provides the site for ICL (Fig. 7b). Subsequent isopeptide bond formation and desulfurization afford the Ub conjugate in the native form. These synthetic tools have enabled access to highly complex ubiquitinated targets, useful for studying DUBs and the ubiquitin signal in general.
The third method of generating an isopeptide linkage is based on genetically encoded orthogonal protection and activation ligation (GOPAL) (Fig. 7c).45 The site-specific Lys residue required for the isopeptide bond formation is incorporated as tert-butyloxycarbonyl-Lys (Boc-Lys) and the rest of the Lys residues are protected orthogonally with benzyloxycarbamate (Cbz). Removal of the Boc protecting group enables isopeptide bond formation with a Cbz-protected Ub-thioester using AgNO3/HOSu. Global Cbz deprotection results in Ub conjugates in the native form.
The fourth strategy relies on the use of orthogonally protected Fmoc-Lys-(ivDde)-OH (ivDde: 1-[4,4-dimethyl-2,6-dioxo-cyclohexylidene]-3-methylbutyl) (Fig. 7d).110 This method not only enables rapid synthesis of Ub conjugates with a native isopeptide bond but also bypasses the usage of precious building blocks e.g. δ-mercaptolysine. The Fmoc-Lys-(ivDde)-OH is positioned in the specific location during SPPS. The protecting group can be then removed by applying 5% hydrazine in DMF solution on the solid support, which gives rise to the free amine on the side chain of the Lys. This can be then elongated using standard SPPS with the C-terminal fragment of Ub, Ub(47–76), while installing the isopeptide bond on the solid support. Ligation with the complementary fragment Ub(1–45)-thioester affords the desired ubiquitinated peptides.
2.5 Synthesis of ubiquitinated peptides
Studying the efficiency of cleaving a native isopeptide bond in semisynthetic ubiquitinated peptides by DUBs was first demonstrated with UCH-L3, which is known to cleave small adducts from the C-terminal of Ub.18,25 In an earlier study reported by Mishagi et al. Ub conjugated to a 13-mer peptide from the surrounding of Lys48 was prepared.25 The Ub part, which contains an HA tag, was expressed while the peptidic part with a biotin tag was made using SPPS. The formation of the isopeptide bond was carried out using the E1 and E2-25K enzymes. The activity of UCH-L3 was addressed by western blot analysis, which showed efficient hydrolysis of the isopeptide bond. Nevertheless, this method of preparing ubiquitinated peptides with a native isoform is limited due to the low yield of the E1–E2 reaction and the lack of generality of this approach.The above-described synthetic approaches to prepare Ub conjugates with a native isopeptide bond afforded precious and diverse substrates to study DUBs. Our lab, for example, used the expeditious synthesis method to prepare different ubiquitinated conjugates bearing peptides of different lengths to study the preference of UCH-L3 to the peptide size.110 The hydrolysis of the ubiquitinated peptides was monitored by LC-MS in which UCH-L3 showed clear preference for the shorter peptides (up to 20 amino acids).
To explore the activities of DUBs with ubiquitinated peptides, Sixma and coworkers reported the synthesis of 14-mer peptides labelled with tetramethylrhodamine (TAMRA) and linked to Ub employing δ-mercaptolysine.27 These ubiquitinated peptides comprised the close residues for each one of the seven Lys residues in Ub, as an approach to mimic the di-Ub chains. These Ub conjugates were tested for their activities and specificities toward various DUBs and their variants. The study revealed that the ubiquitinated peptides do not exhibit similar behaviors to di-Ub chains highlighting the importance of the entire composition of the substrate when testing the activities and specificities of DUBs.
2.6 Synthesis of di-Ub chains
Identifying the cellular targets of DUBs and gaining more insights into their mode of recognition of Ub chains will assist in further determining the cellular roles of DUBs and for future developments of novel modulators. A major progress in the field has been achieved by expanding the chemical toolbox for the synthesis of conjugates beyond mono Ub. These tools were further exploited in the synthesis of native di-Ub chains and their analogues (Fig. 8).45,89,108,111–115 The access for such constructs made it possible for better understanding the DUB preference to different types of Ub chains, besides the well-studied Lys48- and Lys63-linked Ub chains and to shed light on structural elements that can influence DUB activities.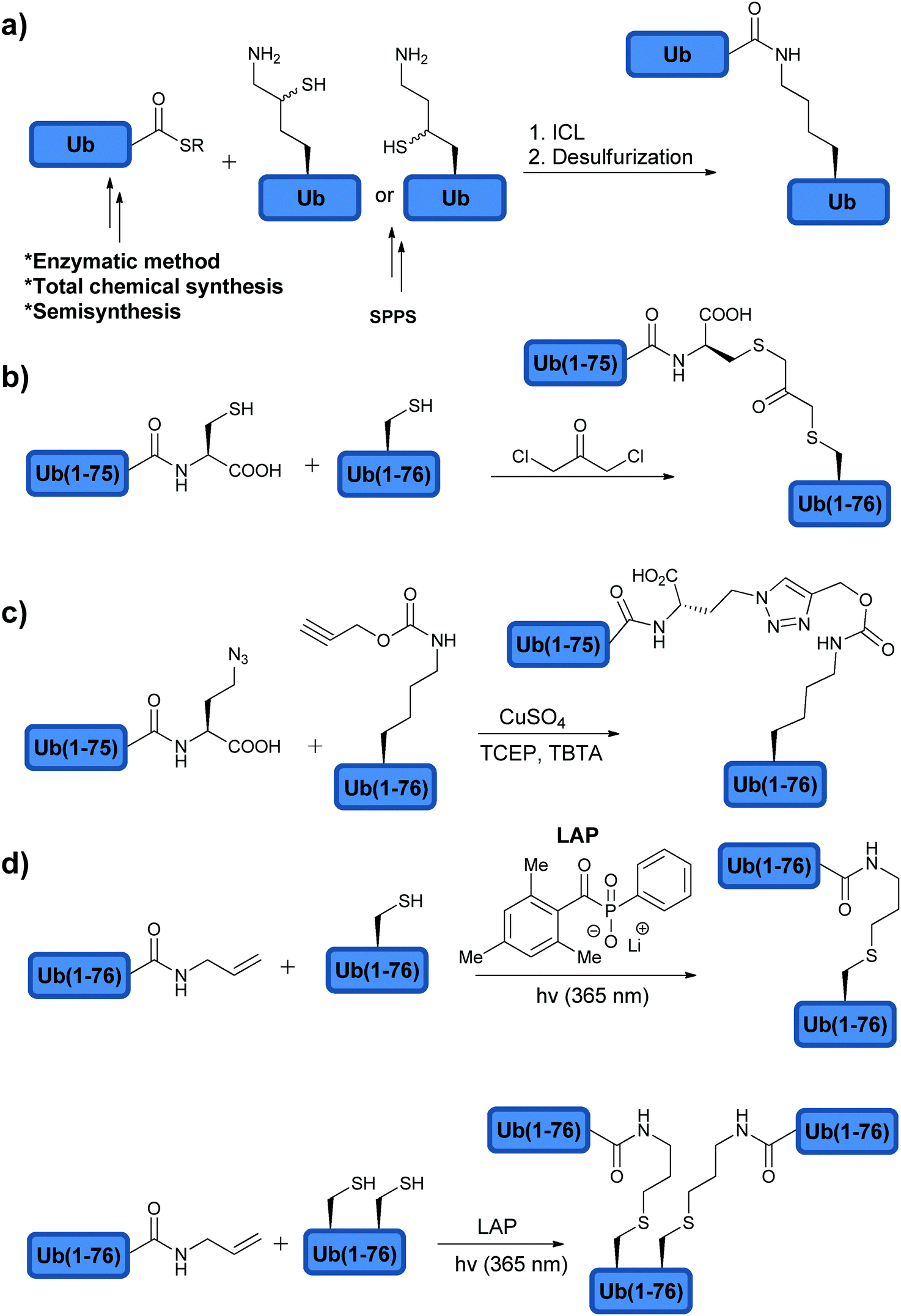 | ||
| Fig. 8 Synthesis of (a) native di-Ub using modified amino acids and (b–d) non-native di-Ub chains. | ||
The synthesis of di-Ub chains has been achieved using different methods, which were developed for constructing the isopeptide bond (Section 2.4). For example, chemical synthesis of Ub having δ-mercaptolysine at the desired position enabled the synthesis of all Ub chains linked through Lys residues by ligating Ub bearing δ-mercaptolysine at a selected position e.g. Lys48 and Ub-thioester.89,112 The Ub-thioester could be obtained chemically, enzymatically or semisynthetically (Fig. 8a).
Chin and coworkers used the GOPAL method in order to synthesize the non-canonical di-Ub chains, linked through Lys6 or Lys29.45 The accessibility for these atypical chains enabled structure determination of the Lys6-linked di-Ub, which exhibited a compact and different structure from the known di-Ub structures. In addition, the authors compared the activity of Lys6- and Lys29-linked di-Ub chains and the well-characterized Lys48- or Lys63-linked di-Ub chains against a panel of eleven DUBs. From this study, TRABID was found to be the selective DUB for the Lys29-linked di-Ub chain.
Sixma and coworkers used synthetic native di-Ub chains to study DUB specificities. Specifically, the authors studied the linkage preference of 12 DUBs from the USP family27 and found that all these DUBs have almost no preference toward any specific linkage type, but rather some changes in the efficiency of the hydrolysis were observed. For example, USP7 exhibited the modest activity with Lys27- or Lys29-linked di-Ub chains compared to other linkage types and no activity with the linear chain. On the other hand, USP16 was found to cleave efficiently all Lys-linked di-Ub chains.27 Although most of the USPs do not exhibit linkage preference, it is known that CYLD prefers the Lys63 linkage116 while USP14 prefers the Lys48 linkage.117
It has been recently shown that Ub undergoes phosphorylation at Ser65 by PINK1 (PTEN induced putative kinase 1), which activates the PARKIN, E3 ligase118–120 and is linked to the clearness of damaged mitochondria (i.e. mitophagy). Activated PARKIN was found to promote the synthesis by increasing the content of Lys6-, Lys11-, Lys48- and 63-linked Ub chains.121 Phospho-Ub was found to be recognized by the E1 and can be activated in a similar manner to Ub to form phosphorylated Ub chains by selected E2 and E3. Phosphorylated Ub chains are made either by the activity of PARKIN that uses phosphorylated Ub or by the activity of PINK1 on Ub chains.121 This has raised the question how phosphorylation on Ub chains affects DUB activities.122–124 In order to shed light on the relation between Ub phosphorylation and the processing of Ub chains by different DUBs, Komander and coworkers prepared Lys63-linked poly-Ub chains, which were enzymatically phosphorylated at Ser65.122 Phosphorylation of the poly-Ub chain was found to inhibit most DUBs that were studied. For example, the activities of USP2, 8, 15 and 30 with the poly-phosphoUb were significantly decreased compared to the unmodified poly-Ub. Similar behavior was found for Ataxin3, a member of the Josephin family. In contrast, USP21 and vOTU hydrolyzed similarly both unmodified and phosphorylated poly-Ub chains.
Using total chemical synthesis, our group prepared in addition to the unmodified Lys63-linked di-Ub chain, three forms of phosphorylated Lys63-linked di-Ub. These included a phosphorylated di-Ub chain at both Ub moieties, the distal or the proximal Ub at Ser65 (Fig. 9).123 The synthesis of the modified di-Ub chain was carried out using SPPS, NCL and ICL (Fig. 9). Five building blocks were synthesized in which the combination of these fragments via NCL afforded four different Ub analogues. Subsequent ligation of the Ub analogues gave the unmodified and the phosphorylated di-Ub chains. Different forms of the phosphorylated di-Ub chains were checked with various DUBs and compared to the unmodified one. Interestingly, the hydrolysis of these chains was found to be dependent on the site of phosphorylation and the tested DUBs. For example, while USP2 and AMSH were both inactive towards the doubly phosphorylated di-Ub, with the singly modified one, AMSH exhibited great sensitivity to phosphorylation at the proximal site and was insensitive to phosphorylation at the distal site. On the other hand, USP2 was sensitive to phosphorylation at the distal site, but not to the proximal Ub. Although no evidence exists yet to support differential phosphorylation of Ub chains in cells, these synthetic analogues can be used as reagents to probe the recognition of DUBs to different Ubs within the chain.
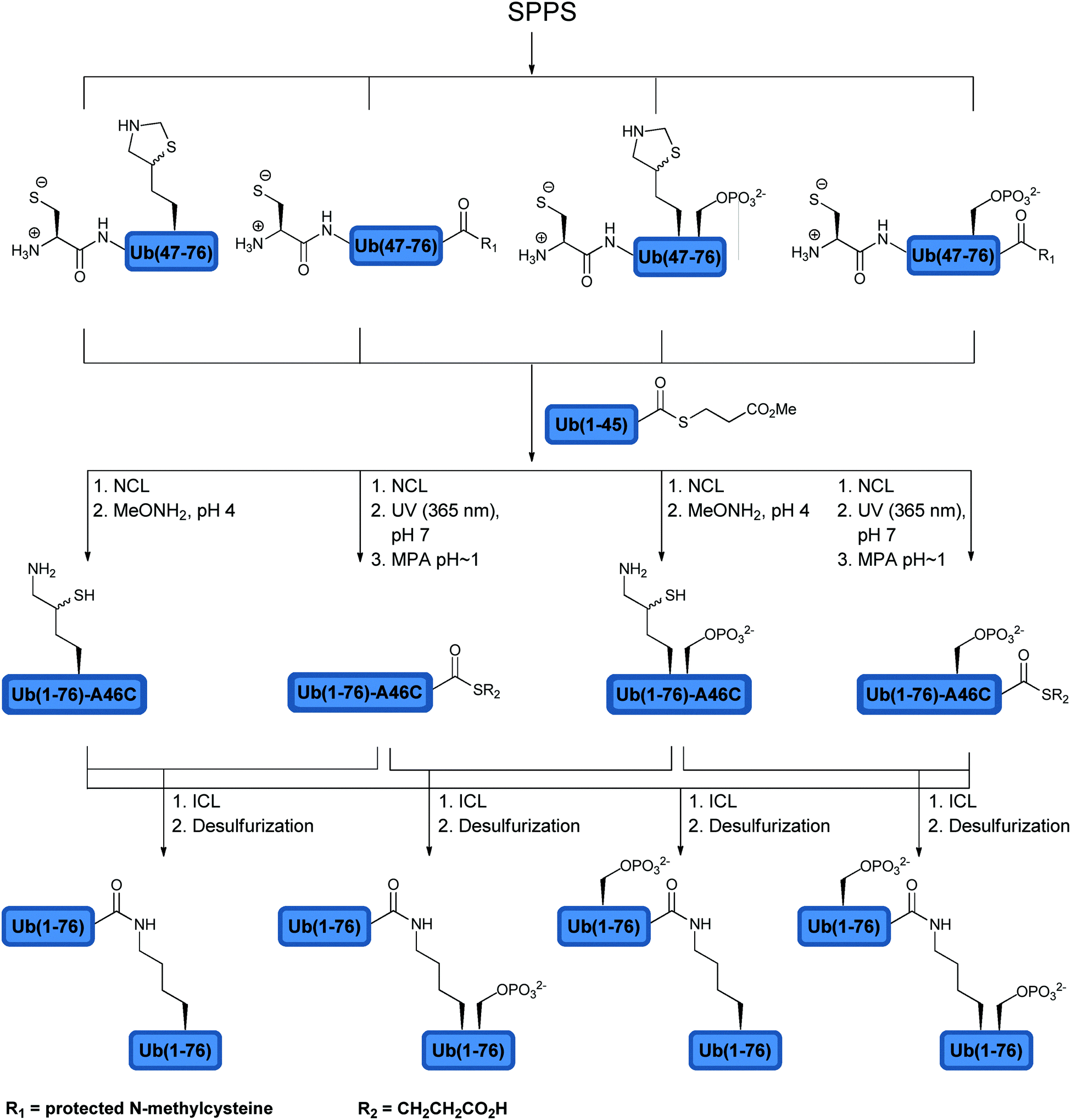 | ||
| Fig. 9 Synthesis of native and phosphorylated di-Ubs to examine their behaviors with various DUBs. | ||
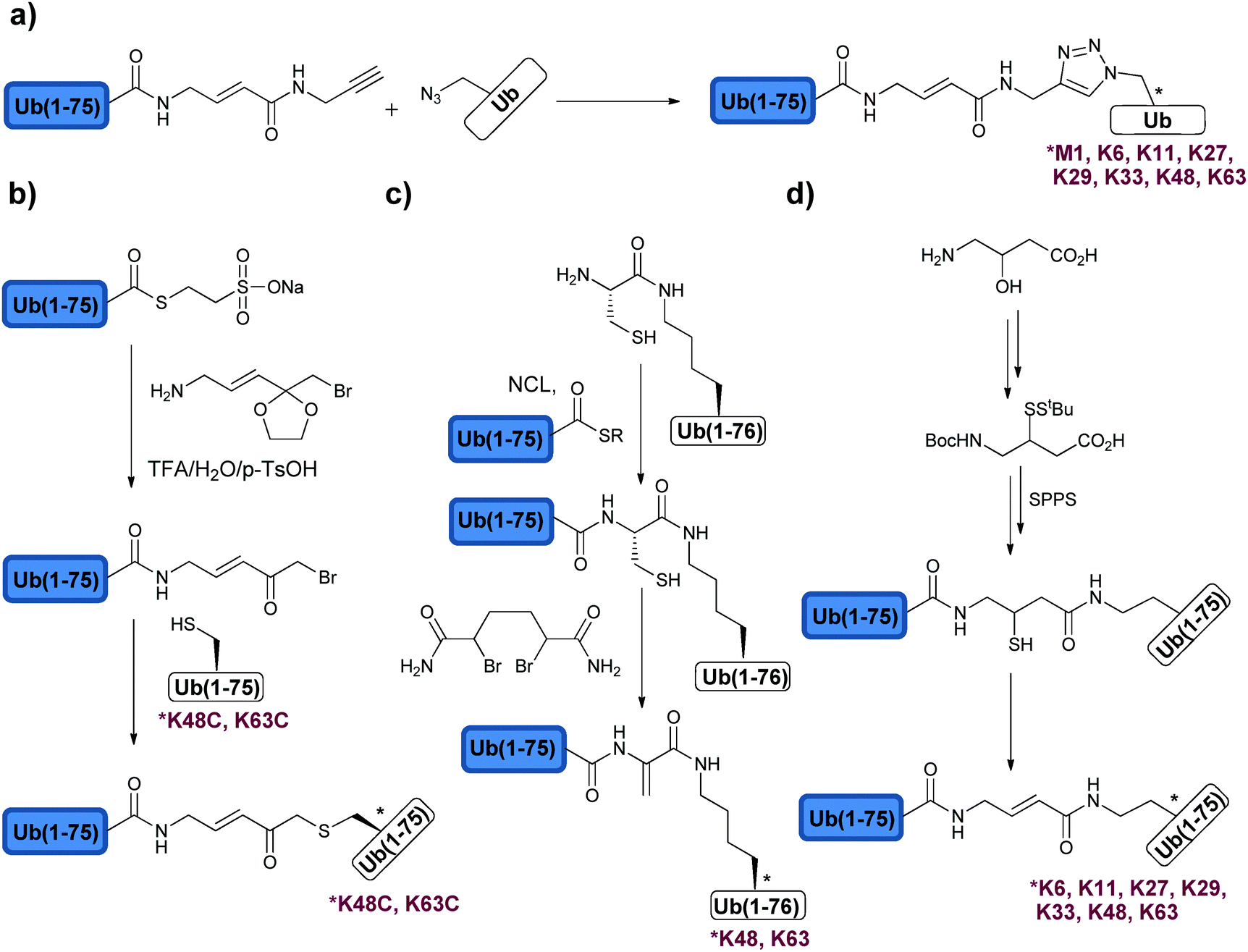
Di-Ub chains, where the isopeptide bond was replaced with the non-native bond, were also prepared and used to interrogate DUBs (Fig. 8). Here the motivation is to simplify the synthesis of these targets and to generate stable analogues that might be useful for various studies, such as structural analyses, binding studies and inhibition of DUBs. The Wilkinson group was the first to synthesize non-hydrolysable di-Ub chains based on the Lys11, Lys29, Lys48, and Lys63 linkages.114 In this work, the Lys side chain at a selected position in the proximal Ub and Gly76 at the distal Ub were replaced by Cys. A reaction with the bi-functional reagent, dichloroacetone (DCA), was carried out to form the non-hydrolysable di-Ub chain (Fig. 8b). These chains were found to inhibit various DUBs differently. For example, Lys29-linked di-Ub inhibited selectively IsoT, whereas the Lys11-, Lys29- or Lys48-linked di-Ub chains inhibited UCH-L3. This method of constructing non-hydrolysable Ub chains was extended to the synthesis of a Lys29-linked tetra-Ub chain that was immobilized to sepharose resin and served to reveal new protein interactors such as the yeast DUB Ubp14 (IsoT in human).125
The interactions of a Ub chain with a particular UBD could lead to significant alterations in the chain conformation, suggesting differential dynamic behavior of Ub chains in the presence of various Ub-binding proteins.126 To shed light on this aspect, Komander and coworkers prepared enzymatically Lys48-, Lys63- and Met1-linked di-Ub chains, which were labelled with fluorescence resonance energy transfer (FRET) dyes at the C- and N-terminus of the proximal and distal Ub, respectively.127 The group used single-molecule FRET to show different states of conformations of the labelled chains in the absence or presence of different Ub binding proteins such as DUBs. For example, the Lys63-linked di-Ub chain, which is known to adopt an open conformation, exhibited an additional population of a semi-compact conformation as also has been shown by the Wolberger group.128 However, upon addition of inactive AMSH-LP, which is known to recognize specifically this chain, this semi-compact conformation shifted to the open state.127 On the other hand, in the case of Lys48-linked di-Ub, which is known to adopt a compact conformation, upon addition of USP21 the conformation of the chain changed from the compact to the semi-compact conformation. Together, these results support a major role of the dynamic changes in these chains on the activity and specificity of DUBs.
Synthesis of di-Ub chains, which undergo cleavage by DUBs, yet bearing minimal modifications in the isopeptide bond linkage was also reported. Recently, Strieter and coworkers demonstrated the use of thiol–ene chemistry for the synthesis of all Lys-linked di-Ub chains.111 To achieve this, the desired site of ubiquitination was replaced with Cys in Ub and allylamine functionality was introduced at the C-terminus of the other Ub using the UCH enzyme. To promote the thiol–ene reaction, the group used lithium acyl phosphinate (LAP) as a radical initiator, which led to the formation of di-Ub bearing a longer isopeptide bond due to the additional sulfur atom (Fig. 8d). Tinkering with the isopeptide bond could affect the dynamics of the chains and its subsequent recognition by DUBs and other Ub binding proteins.129,130 In order to examine the influence of the new isopeptide mimic generated by thiol–ene chemistry, the Strieter group studied the structure and function of the non-native linkage in comparison to the native one. Specifically the group used small angle X-ray scattering (SAXS) and steady state kinetic analyses and showed that both native and non-native di-Ub chains share similar structures and comparable hydrolysis by DUBs.131 In the latter study, the authors tested the modified chains with the known linkage specific DUBs, e.g. A20 (Lys48 specific) and AMSH (Lys63 specific), and found similar activities with these chains when compared to the native ones.
Thiol–ene chemistry was also used to prepare branched Ub chains, a unique form of ubiquitination, which involves different Lys residues in the same Ub molecule (Fig. 8d).111,132 This class of ubiquitination is not well characterized and the knowledge about the parameters that are important for its processing by DUBs (i.e. structure and connectivity) is still in its infancy. The processing of the Lys-Cys(6, 48), Lys-Cys(11, 48) and Lys-Cys(48, 63) branched Ub chains by IsoT, A20 and AMSH was tested. Surprisingly, both Lys-Cys(11, 48) and Lys-Cys(48, 63) were cleaved by A20, whereas no cleavage was observed with the Lys-Cys(6, 48) chain.111 This suggests a possible new layer of selectivity by DUBs and further supports the presence of other unique regulatory mechanisms of the Ub signal in general. Future studies using similar synthetic tools could assist in unraveling these mechanisms and their importance in health and diseases.
Fushman and coworkers reported the preparation of branched and mixed chains using the improved GOPAL approach.133 The extent of the hydrolysis of Lys(11, 33) and Lys(11, 63) branched chains was compared with the mixed chain Lys(11, 33) using Ubp6 (USP14 in human). These chains exhibited similar processing when compared with the di-Ub analogues bearing the same linkages indicating no new mode of recognition of these branched/mixed chains by DUBs. For example, Ubp6 showed efficient hydrolysis of Lys11-linked di-Ub compared to the Lys33-linked di-Ub chain. In the case of Lys(11, 33) branched and mixed chains, the hydrolysis appeared to be efficient as indicated by accumulating di-Ub but to a lesser extent the monomeric Ub. These results suggest that Ubp6 processes the branched chain at Lys11 and much less at Lys33.
Taken together, these studies with the unique forms of Ub chains show the high complexity of the Ub signal and the processing of Ub chains in general and the branched/mixed chains in particular. A better understanding of the processing of these unique forms of chains and their roles might be crucial for future design of inhibitors that target DUBs.
2.7 Synthesis of di-Ub ABPs
The development of various mono-Ub based ABPs has been useful in studying and unravelling new DUBs as described before in Section 2.3. However, these probes are based on mono-Ub and not Ub chains with different linkages, which are essential in the recognition and specificity of DUBs. The structures and the conformations of the Ub chains further dictate the specificity, as for example in the case of the Lys48 and Lys63 linkages, which adopts mainly closed and open conformation, respectively. The mode of cleavage i.e. exo vs. endo cannot also be studied using monoUb based ABPs. In order to address these key issues various di-Ub based ABPs were developed.The first di-Ub ABPs were reported by Kessler and coworkers (Fig. 10a).134 In this approach, the authors modified the proximal Ub with azidohomoalanine, in any of the Lys positions or the N-terminus, while the distal Ub was modified with the alkyne functionality. Using Cu(I)-mediated click chemistry, the 8 di-Ub ABPs were prepared and compared with HA-UbVME and HA-UbPrg in the labelling of DUBs in the cell extract. With the monomeric Ub ABP, a broad labelling spectrum with less selectivity across DUBs was observed. In the case of di-Ub ABPs, the labelling was more specific. It was also found that many DUBs exhibited preference to non-canonical linkages over the canonical one. For example, Ataxin3 was selectively labelled with the Lys29-linked di-Ub probe. One has to note that these types of ABPs have major perturbations in the isopeptide bond, which could affect the labelling patterns and as a result this may lead to incomplete knowledge about the studied systems. This study, nevertheless, promoted the developments of di-Ub based ABPs with minimal perturbations in the isopeptide bond and its vicinity.
 | ||
| Fig. 10 Synthesis of different di-Ub based ABPs using (a) click chemistry, (b) ketal protected amine linker, (c) dehydroalanine chemistry and (d) thiol elimination. | ||
Zhuang and coworkers reported the synthesis of two di-Ub ABPs, where Lys48Cys or Lys63Cys mutations were introduced in the proximal Ub.135 The distal Ub with a reactive handle containing acetyl bromide was made by reacting Ub-MES with a ketal protected amine linker (Fig. 10b). Deprotection of the ketal followed by the displacement of the bromine by Cys48 or Cys63 from the proximal Ub afforded the required di-Ub probes. The reactivity and the selectivity of these probes with USP2, 7, 8, 21, UCH-L1 and OTUB1 were studied and compared with HA-UbVME. The USPs showed similar activities with both di-Ub probes, where UCH-L1 showed a very weak activity and OTUB1 was selectively labelled by the Lys48-based di-Ub probe, but did not react with the Lys63-based di-Ub probe.
Our group reported dehydroalanine (DHA) based di-Ub probes employing total chemical synthesis and taking advantage of a late stage conversion of Cys to DHA using 2,5 dibromohexanediamide (Fig. 10c).136 To achieve this, orthogonally protected Lys with Dde was incorporated at the position 48 or 63 in the proximal Ub. Deprotection of the Dde protecting group followed by incorporation of Cys enabled NCL with the Ub(1–75) thioester. Subsequently, this Cys was converted to DHA to afford the required di-Ub probes. Six DUBs, specifically USP2, IsoT, OTUB1, OTUB2, OTULIN and CYLD, were screened using both di-Ub probes, which reacted with the specific probe according to their known selectivity.
Similar to the DHA approach, the Ovaa group prepared all seven di-Ub probes (Fig. 10d).137 In order to maintain the isopeptide linkage length, mutation of Lys to diaminobutyric acid (Dab) was employed with the side chain protected with the allyloxycarbonyl group (Alloc). After the assembly of proximal Ub on resin, the Alloc group was deprotected and the thiol-incorporated amino acid was coupled to provide the thiol handle and enable NCL with the Ub-thioester (Fig. 10d). The resulting thiol after ligation was eliminated using 2,5-dibromohexanediamide to generate the Michael acceptor along the peptide backbone. Using Lys11- and Lys48-linked di-Ub ABPs, the labelling of USP8 and Cezanne was demonstrated. It was found that both probes label USP8, whereas the selective labelling of Cezanne was observed using the Lys11- over Lys48-linked di-Ub ABPs.
Having the di-Ub ABPs described above, the field is now equipped with novel tools that will lead to new applications and discoveries related to DUBs that might be difficult, or impossible, to achieve with the mono-Ub based ABPs. Notably, using similar approaches one could also generate similar probes based on Ub or Ub chains linked to protein substrates, which is the expected future step in this field.
2.8 The effect of the Ub chain length on DUBs
Different studies, which highlight the importance of the Ub chain length on DUB substrate preference mostly as a recognition element for degradation via the proteasome, have been reported. Morgan and coworkers studied the influence of the chain lengths of Lys48-linked Ub chains on the activity of various DUBs.138 Ubp15 (USP7 in human) was found to cleave efficiently mono- and di-Ub chains but was sluggish towards the longer Ub chains. In order to test if the structure of the longer chain is responsible for the observed slower rate of hydrolysis, the authors mutated Ile44 in the hydrophobic patch of Ub to Ala, which resulted in the disruption of Lys48 poly-Ub chain conformation and subsequently enhanced the hydrolysis of the chain. These results indicate that the conformation adopted by the specific chain, which is influenced by the chain lengths, is a key factor in dictating the hydrolysis efficiency of DUBs and the subsequent biological outcome.Recently, Bavikar et al. reported the synthesis of a tripeptide linked to mono-, di, tri- and tetra-Ub chains, linked through Lys48 or Lys63.90 The authors used these constructs to study systematically UCH-L3 and IsoT preference toward a defined length of the chain. UCH-L3 is known to cleave small adducts from the C-terminal of Ub, yet the effect of the chain length on its activity was not studied before. Interestingly, UCH-L3 was found to cleave the tripeptide in all cases with a slight preference to the shorter chains and for the Lys63- over Lys48-linked Ub chains.
Taken together, these studies showed that the combined effects of the chain length and conformation could influence the hydrolysis by DUBs.
2.9 Synthesis of ubiquitinated proteins
With several advancements in the chemical and semisynthetic approaches to access Ub conjugates, it is now possible to prepare ubiquitinated proteins having Ub chains with defined linkages and lengths in high homogeneity and workable quantities.130,139–148 Such conjugates have enabled various important studies including, for example, proteasomal degradation and how DUBs perform their function at the molecular level.Very recently, the Brik and Wolberger group used synthetic ubiquitinated H2B to study the structure of the DUB module (Ubp8/Sgf11/Sus1/Sgf73) of Spt-Ada-Gcn acetyltransferase (SAGA) bound to the monoubiquitinated nucleosome core particle (NCP-Ub) (Fig. 11a).148 This study provided, at the atomic level, a first glance on the selective removal of Ub from H2B-Lys120 by a SAGA DUB module. To prepare the ubiquitinated histone, H2B-Lys120Cys and Ub-Gly76Cys were expressed and crosslinked using DCA to afford the H2B-Ub having the non-hydrolysable DCA linkage (Fig. 11b). The NCP containing two H2B-Lys120Ub and the SAGA-DUB module was crystallized and solved at 3.9 Å resolution (Fig. 11a). Interestingly, this structure revealed that Ub makes no contacts with the nucleosome by pointing outward in order to be more accessible for the DUB module. Moreover, the conserved acidic patch on H2A/H2B (H2A-Glu61, Glu65 and H2B-Glu107) and the C-terminal helix of H2B in the nucleosome makes contacts with the USP domain of Ubp8 and Sgf11 zinc finger (Arg78, 84 and 91). However, due to the resolution limits the interactions of the Arg side chains of the Sgf11 zinc finger with H2A/H2B-Ub have to be modeled to shed light on the importance of these residues on binding. In addition, in vitro Ala mutations of the Arg cluster in Sgf11 revealed that three out of the six Arg residues (Arg78, 84 and 91) are important for the cleavage of H2B-Ub in the NCP. Moreover, mutations of the acidic path residues of H2A/H2B showed that H2A-Glu65 is the most important residue for the DUB activity. In contrast, mutations in Ubp8 including Tyr233Phe, Arg374Glu or Arg374Ala have minimal effects on the DUB activity, suggesting an upper hand of Sgf11 in recognizing specifically the substrate.
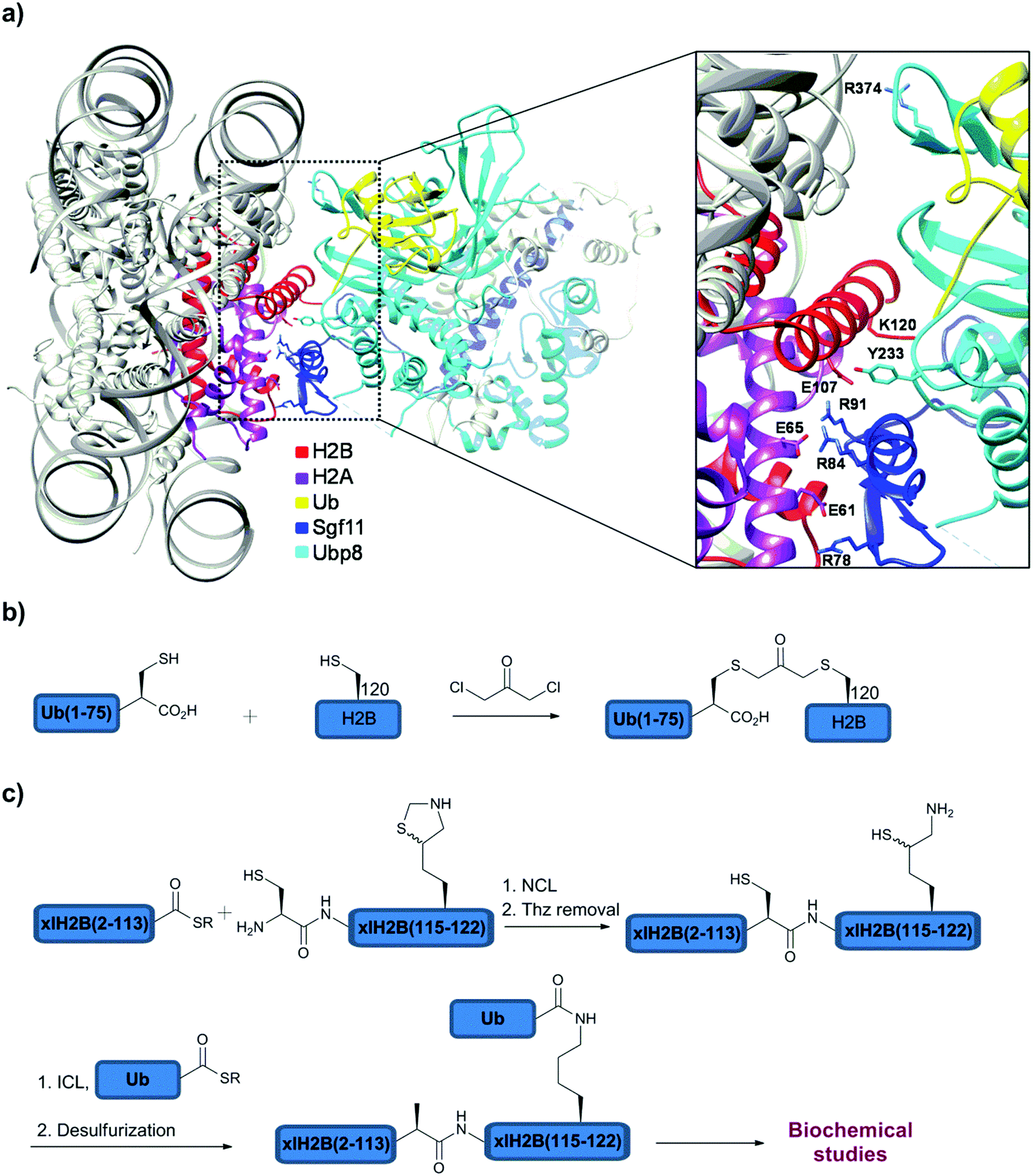 | ||
| Fig. 11 (a) Crystal structure of the SAGA DUB module with NCP containing H2B-Lys120Ub. Synthesis of ubiquitinated H2B with (b) non-native and (c) native linkages. | ||
Notably, these biochemical studies were performed using H2B-Lys120Ub with the native isopeptide linkage that was prepared in large quantities using semisynthesis (Fig. 11c). To prepare this native analogue of ubiquitinated H2B, the expressed H2B(2–113)-thioester was ligated with the complementary part, H2B(114–122), bearing N-terminal Cys and orthogonally protected δ-mercaptolysine at position 120. Unmasking Thz, followed by ICL and desulfurization gave the desired H2B-Lys120Ub with the native isopeptide bond (Fig. 11c). Using this native substrate, it was also found that the chaperon that mediates H2A/H2B dimer eviction and nucleosome assembly, FACT (Facilitates Chromatin Transcription), does not have an effect on the deubiquitination of H2A/H2B-Ub, whether the ubiquitinated heterodimer is part of the nucleosome or not. Taken together, the study showed that deubiquitination of H2B-Ub by the SAGA DUB module could occur at various stages of nucleosome assembly and disassembly.
Until recently it was speculated that in order for the substrate to be degraded by the proteasome, a protein must be ubiquitinated with longer chains having at least four Ubs.149 However, it has been shown that mono-ubiquitination may also target the protein for degradation.139 In this regard, Ciechanover and coworkers studied the hypothesis that mono-Ub can target proteins for proteasomal degradation and tested the relation between the size of the protein that is degraded and the length of the Ub chain.139 Synthesizing ubiquitinated peptides having different lengths, derived from H2B histone, supported that a minimum of 20 residues is required for targeting mono-ubiquitinated peptides by the proteasome. Polypeptides with more than 150 residues were found to be stable for degradation in the case of mono-ubiquitination and required a poly-Ub chain in order to be degraded by the proteasome, both in vitro and in cellular contexts. Deletion of Rpn10, a proteasome associated DUB in yeast, which is known to be involved in the recognition of Ub by the proteasome, was found to be critical for the degradation of mono-ubiquitinated peptides up to 150 residues, but did not influence the stability of larger polypeptides. This gives further support that Ub recognition by the proteasome machinery is the main factor for degradation. Several examples were given, including stable mono-ubiquitinated GFP, small-medium naturally occurring proteins, such as cyclin-dependent kinases regulatory subunit 2 and α-synuclein. The latter was prepared using semisynthesis employing δ-mercaptolysine mediated ICL between α-synuclein and the Ub-thioester.142In vitro results clearly showed the efficient degradation of mono-ubiquitinated α-synuclein, supporting the hypothesis on the proteasome ability to degrade mono-ubiquitinated proteins with up to 150 residues.139
In order to test the effect of different lengths of Ub chains on the proteasomal signaling, our group described the semisynthesis of di- and tetra-ubiquitinated α-synuclein,140 where the ubiquitination takes place in site specific manner using δ-mercaptolysine at the desired position (Lys12) in α-synuclein. Assaying of the degradation of mono-, di- and tetra-ubiquitinated α-synuclein in a crude cell extract, which contains the entire pool of cellular DUBs, exhibited different behavior of the proteasome toward the different lengths of Ub chains. Whereas in the case of mono-ubiquitination the Ub moiety was cleaved by DUBs before degradation, the di- and tetra-ubiquitinated α-synuclein were resistant to the pool of DUBs and underwent proteasomal degradation. This could be as a result of the compact structure of the poly-Ub chains and their resistance to cleavage by DUBs, as previously suggested by Morgan and coworkers,138 hence making the protein with the longer chains more prone to degradation by the proteasome.
3. DUB assays
Having seen the importance of DUBs in cellular contexts and their roles in various signaling processes and diseases, it is now evident that DUBs could be promising drug targets. In order to study the kinetics of DUBs and identify inhibitors for some members of these enzymes one has to develop robust assays, which can be prepared in large quantities as well as enable straightforward high throughput screening (HTS). Toward this goal, novel approaches have been developed and selected examples are documented in this section.The early approaches of monitoring DUB activities started with the Ub-amino-4-methylcoumarin (Ub-AMC) assay, where the C-terminus of Ub is attached with the AMC fluorophore using the transpeptidation approach (Fig. 12a).150 Upon incubation with the specific DUB the fluorophore is cleaved, which enables subsequent readout of the emission of AMC at 460 nm (λex = 380 nm). The field has then geared up and the high utility of this substrate led to its commercialization for various uses, among them is the HTS of libraries of small molecules. One of the main drawbacks in using the Ub-AMC assay is the excitation wavelength (380 nm), which makes it less suitable for compounds that absorb or emit at this range. Moreover the fluorophore is attached at the C-terminal of Ub through an aromatic amide whereas most of DUBs cleave an isopeptide linkage.
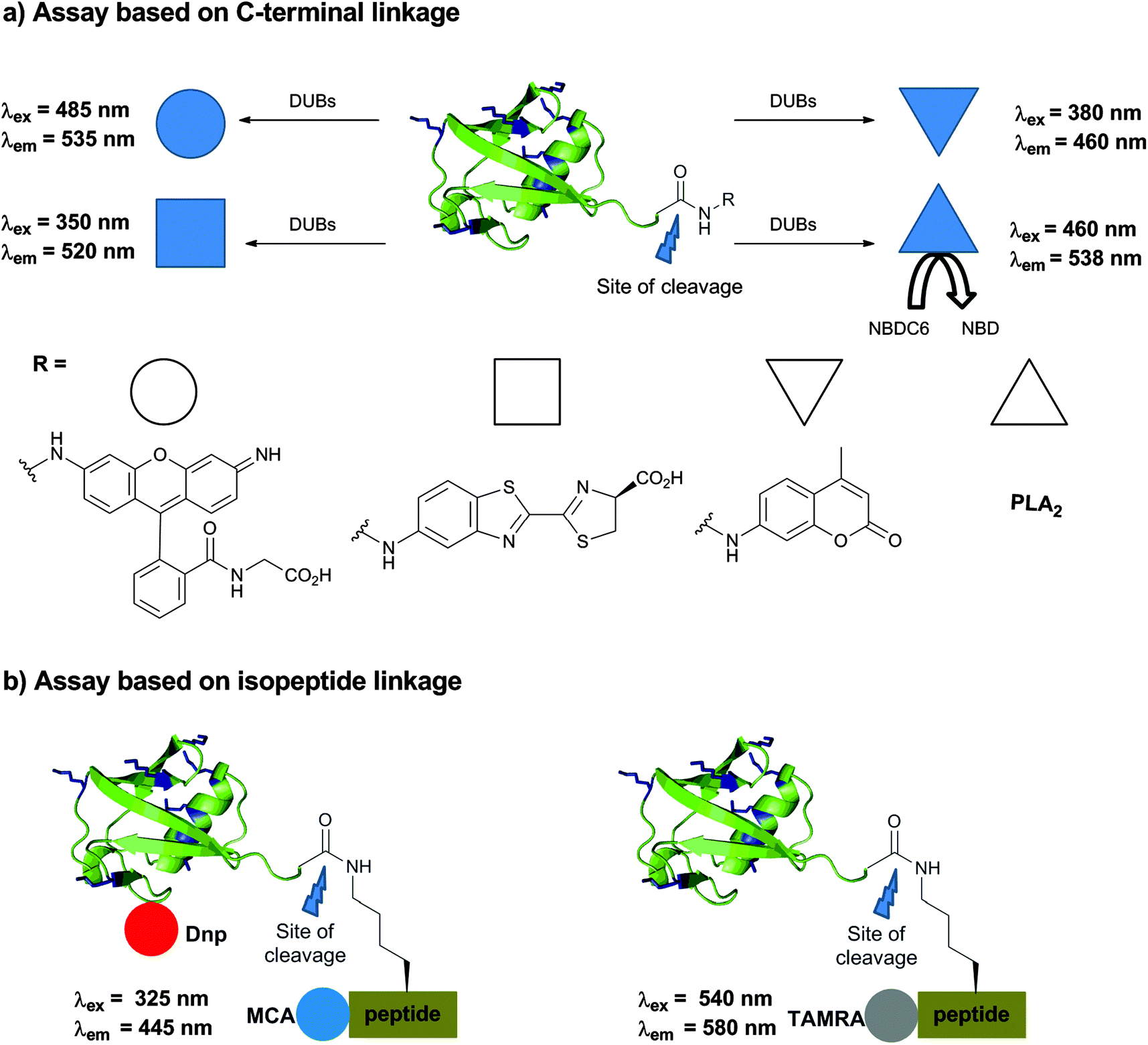 | ||
| Fig. 12 Assays for following DUB activities. (a) Based on a modified Ub at the C-terminus. (b) Based on Ub conjugates having the isopeptide bond. | ||
In order to address these concerns, a rhodamine-based fluorescent polarization technique was employed by linking TAMRA to the Lys α-amine group or the N-terminus of a peptide, linked to Ub.151 Moving from AMC to TAMRA allows shifting of the excitation wavelength from 380 nm to 540 nm (Fig. 12b). To prepare this substrate, a peptide derived from the surrounding residues of Lys48 in Ub was synthesized via SPPS, followed by coupling of the TAMRA fluorophore. Using E1–E2, the TAMRA conjugated peptide or Lys(TAMRA) was ligated to Ub having the Lys48Arg mutation to afford the desired substrate. In order to increase the molecular weight of the substrate and enable a large shift in fluorescence polarization between the cleaved and non-cleaved Ub, sizable tags like NusA proteins were attached to the N-terminus of Ub. Recently, Ovaa and coworkers devised a new synthesis by using δ-mercaptolysine mediated ubiquitination of labelled peptides with the Ub-thioester to overcome the tedious enzymatic approach to prepare this substrate.152 Despite the fact that Lys(TAMRA)-Ub was used to perform HTS, its moderate dynamic range of fluorescence polarization further demanded improvement in developing assays with higher efficacy.
An assay, which utilizes the yellow fluorescent protein and terbium FRET pair, was also reported.153 This approach requires a tedious enzymatic process thus limiting the utility of this method for broad applications. A reporter-based approach was developed for the preparation of new substrates, where the C-terminus of Ub was fused with phospholipase A2 (PLA2) (Fig. 12a).154 In this conjugate, the activity of PLA2 is blocked since it requires the free amine. Short incubation with the DUB releases the active form of PLA2, which then cleaves its substrate NBDC6-HPC (1-hexadecanoyl-sn-glycero-3-phosphocholine) to NBD, leading to the release of a fluorescence signal. The same group utilized this assay for HTS against USP7,155 however the method could not distinguish often whether the compounds inhibited USP7 or the PLA2 itself, which limits its broader applicability.156
Ulrich and coworkers came up with a rhodamine based fluorescence assay, which was demonstrated for USP2 and UCH-L3 (Fig. 12a).157 The required probe was prepared using the intein approach, where a reaction of Ub-MES with bis-glycyl-rhodamine110 gave the desired substrate. Upon DUB cleavage, it leads to the release of a rhodamine-Gly unit that emits fluorescence following irradiation at 485 nm. Since the resulting fluorescence is quite intense, the sensitivity to measure the turnover of a few hundred femtomoles is possible. This further served as a substrate for HTS to identify inhibitors against USP1.158,159
Aminoluciferin (AML) based luminescence substrates were developed by Promega and subsequently improved by the Strickler group to obtain Ub-aminoluciferin (Ub-AML) (Fig. 12a).160 The high sensitivity and clean background makes this assay quite suitable for HTS, though the external addition of luciferase is needed. Aminoluciferin derivatives of SUMO2 and NEDD8 were also demonstrated as sensitive substrates for deSUMOylases and deNEDDylases, respectively; however the system is not yet widely explored.
Various substrates that were used to monitor DUB activities thus far rely on the modification of the C-terminus of Ub (i.e. Ub-AMC, Ub-AML, etc.).150,153,154,157,160 As mentioned before, ubiquitinated peptides labelled with TAMRA having the isopeptide bond in these substrates were explored and prepared semisynthetically.151,152 Our group reported new substrates containing the isopeptide bond in very good yields using our expeditious synthesis approach described in Section 2.4 (Fig. 12b).161 The design of the substrate included the labelling of the ubiquitinated peptide with the quenching pair 7-methoxycoumarin (MCA) as a donor and 2,4-dinitrophenyl (Dnp) as an acceptor. Here, the donor molecule was anchored to the N-terminus of a 6-mer peptide and the acceptor molecule to Ub. For the attachment site on Ub, Asp52 was found to be appropriate since it is in a good distance from the MCA and also in a solvent exposed area. Excitation at 325 nm and the detection of the fluorescence emission at 445 nm allowed the monitoring of the activities of several DUBs.
Despite that the synthetic substrate for the assay can be prepared in tens of milligram quantities, it is still very important to have the most efficient substrate. Further optimization of the substrate was made through Ala screening of the 6-mer peptide, which enabled us to obtain better substrates for USP2 and USP7 with two-fold increase in the binding affinities.162 Despite modest improvement, this allows us to use this synthetic substrate more efficiently in the screening of large libraries of small molecules. Efforts made to optimize this substrate by tinkering with the isopeptide bond and modifying the amide unit with the N-hydroxy group did not lead to improvements in hydrolysis by various DUBs.163
It is noteworthy to mention that the assays developed above can be extrapolated straightforwardly for other Ubl proteins like SUMO and NEDD8, which makes these chemical tools broadly applicable.
4. DUB inhibitors
The emerging knowledge on the role of DUBs in various diseases like cancer, neurodegenerative and infectious diseases has made them promising and attractive drug targets. The various assays, which were developed to monitor DUBs, were also utilized in order to search for potent and selective inhibitors against disease associated DUBs. Various small molecules and Ub based inhibitors are briefly discussed in this section with selected examples. The reader is also encouraged to refer to other excellent reviews dedicated solely to this aspect.164–168UCH-L1
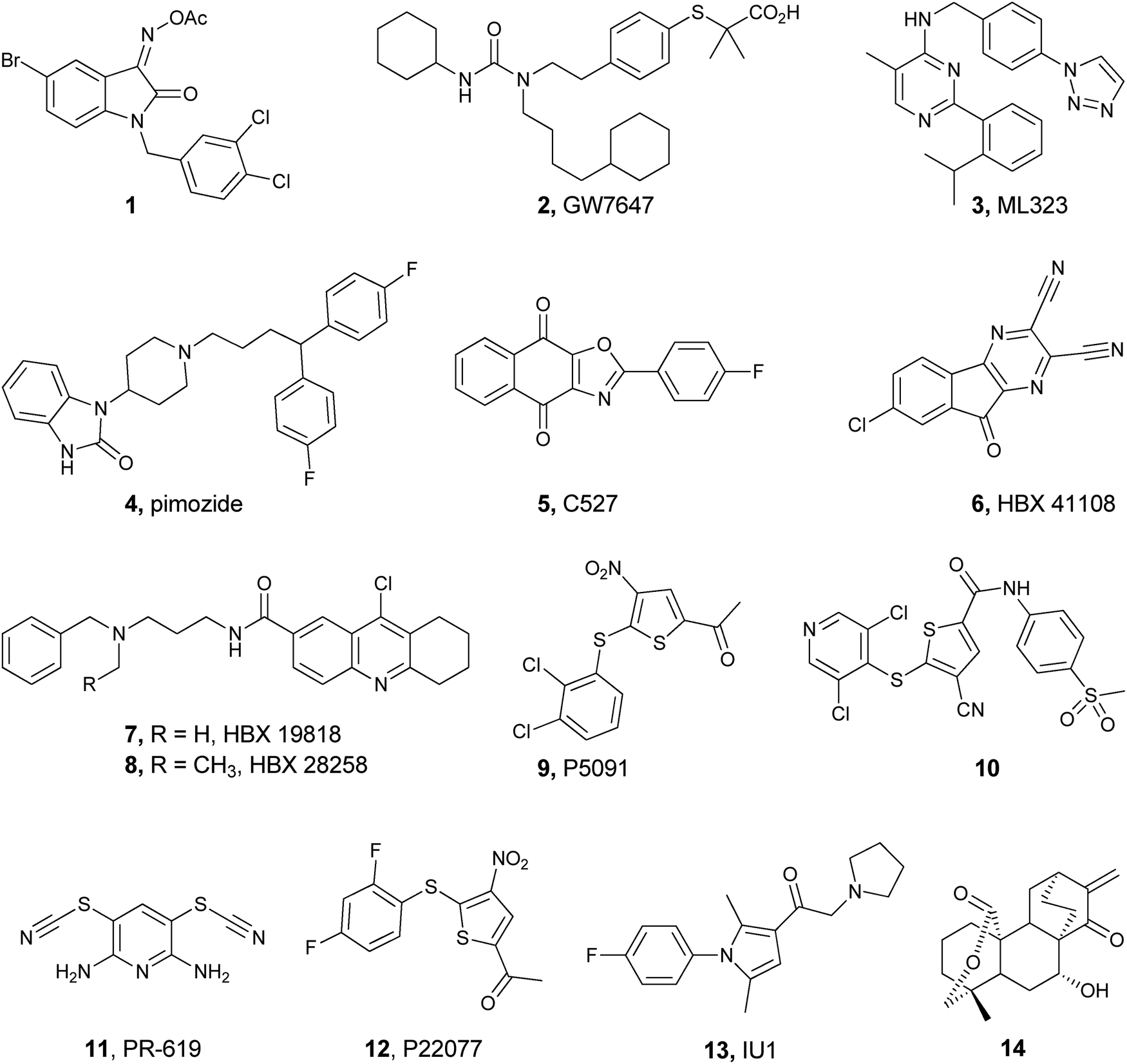
The involvement of UCH-L1 in various neurodegenerative diseases like Parkinson’s makes it a potential drug target. The first successful inhibitor against UCH-L1 was found using a chemical genetics approach by screening 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 drug-like compounds employing the Ub-AMC assay.169 Detailed structure activity relationship (SAR) study of the hits led to the identification of the isatin O-acyl oxime derivative, 1, as the most potent and selective inhibitor for UCH-L1 (Fig. 13). Through various control experiments it was also shown that 1 is a competitive and reversible inhibitor. In particular, it was shown that 1 is a selective inhibitor to UCH-L1 over UCH-L3, despite the fact that these DUBs share 52% sequence homology.
000 drug-like compounds employing the Ub-AMC assay.169 Detailed structure activity relationship (SAR) study of the hits led to the identification of the isatin O-acyl oxime derivative, 1, as the most potent and selective inhibitor for UCH-L1 (Fig. 13). Through various control experiments it was also shown that 1 is a competitive and reversible inhibitor. In particular, it was shown that 1 is a selective inhibitor to UCH-L1 over UCH-L3, despite the fact that these DUBs share 52% sequence homology.
 | ||
| Fig. 13 Selected inhibitors against different DUBs. | ||
USP1
USP1 exerts its biological function as a USP1/UAF1 protein complex. As mentioned earlier, USP1 regulates DDR and hence becomes an attractive target for cancer therapy and prompted the search for potent and selective inhibitors against it.53 Using the Ub-Rho110 assay, 9625 compounds were screened.158 Among the 42 hits, five compounds were chosen based on the additional inhibitory activity of these compounds in the hydrolysis of the Lys63-linked di-Ub chain. In this study, compounds 2 and 4 (Fig. 13) were identified as inhibitors with IC50 values of 2 and 5 μM, respectively, and exhibited good selectivity for USP1/UAF1 compared to USP2, 5, 7, 8, 46/UAF1, UCH-L1 and UCH-L3 (Fig. 13).It was also found that 2 and 4 are reversible and noncompetitive inhibitors. Cellular studies with these inhibitors revealed increased levels of USP1 substrates including mono-ubiquitinated PCNA and FANCD2. The same group further developed compound 3 (Fig. 13) as a highly potent and selective inhibitor for USP1/UAF1. This compound exhibited IC50 values of 76 nM, 174 nM and 820 nM when using Ub-Rho110, Lys63-linked di-Ub and Ub-PCNA assays, respectively.159 In addition, it was found to be a reversible and allosteric inhibitor without disrupting the USP1/UAF1 complex and inhibits deubiquitination of PCNA and FANCD2 in cells. When tested against a panel of 18 DUBs, SENP1, NEDP1 (deNEDDylase), 70 unrelated proteases and 451 kinases, 3 exhibited very weak activity in all these systems. Due to the rapid metabolism of this compound (T1/2 of 15 min) by oxidative removal of the benzyl group and the hydroxylation of the isopropyl group, it requires further development for potential uses as a drug candidate.170
The inhibitor of the DNA binding 1 protein (1D1) is needed for the proliferation of many cancer cells.171 In the presence of USP1, 1D1 is rescued from proteasomal degradation. Hence, inhibition of USP1 could lead to 1D1 degradation, which in turn can kill cancer cells. In this regard, Mistry et al. found the paraquinone based tricyclic compound 5 as an inhibitor of USP1 with IC50 = 0.88 μM (Fig. 13), through HTS of about 150000 compounds.172 Later it was found that the defluoro analogue of 5 is a more potent analogue (IC50 = 0.544 μM), which promotes the degradation of ID1 and is cytotoxic in leukemic cells.
USP7 and USP47
The interactions of USP7 with several viral proteins and its deubiquitination of various substrates like MDM2 and p53 have made USP7 as a promising target for several diseases.58 USP47, a closely related DUB to USP7, is also an emerging target in cancer due to its role in decreasing the DNA polymerase β.156,173 Among the 65092 compounds, which were screened using the Ub-AMC assay and conventional medicinal chemistry optimization, cyano-indenopyrazine, 6, was found to be a potent, uncompetitive and reversible inhibitor with IC50 = 424 nM against USP7 (Fig. 13).174 The same group reported other structurally different USP7 inhibitors (7 and 8, Fig. 13), which were found to achieve their inhibition through a covalent mechanism, yet with a low activity.175
Another study using the HTS Ub-PLA2 reporter assay revealed the trisubstituted thiophene based inhibitor 9 for USP7 with a moderate potency (EC50 = 4.2 μM),155,176 but with very good selectivity for USP7 within the selected DUBs and Cys proteases (Fig. 13). Later studies revealed that it also inhibits USP47 with equal potency.156 The target for the inhibitor was further confirmed in cells by conducting cell labelling studies with Ub-VME and also through USP7 knockout animal model experiments. Detailed in vitro and in vivo studies with compound 9, P5091, suggested that USP7 based inhibitors could offer a potential platform to treat multiple myeloma.155 Further medicinal chemistry optimization of 9 led to the identification of compound 10 (Fig. 13), which exhibited increased potency for USP7 (0.42 μM) and USP47 (1.0 μM).156
The use of ABP to monitor the activity and selectivity of discovered inhibitors175 in cellular contexts has also been shown by the Kessler group.176,177 In this work, the authors developed a competitive assay where cells were incubated with inhibitors 11 or 12 (Fig. 13), followed by labelling the cell extract with HA-UbBr2 or HA-UbVME. Through this study the group demonstrated that compound 11 is a broad selective DUB inhibitor whereas compound 12 is selective for USP7 and USP47 in the cell extract. In addition to the activity and selectivity information one could obtain when using ABP, it is also possible to learn whether the inhibitors are cell permeable and also screen fluorescent inhibitors, which are difficult to study using fluorescent-based assays.
USP14
As mentioned earlier, three DUBs, namely Rpn11, UCH37 and Ubp6 (USP14 in human), are present in the proteasome regulatory part and are associated with its function. It is believed that Rpn11 cleaves the Ub chains from the substrate in the proximal end, whereas UCH37 and Ubp6 are involved in chain trimming by removing mono-Ubs and di/tri-Ubs from the distal end, respectively.6 It has been proposed that Rpn11 exhibits its action only when the substrate is ‘committed’ for degradation whereas the action of the other DUBs can antagonize proteasomal degradation.6 Hence, inhibition of USP14 could lead to efficient substrate degradation. In this context, Lee et al. screened 63052 compounds using the Ub-AMC assay and found the highly substituted pyrrole 13 (Fig. 13) as the potent inhibitor against USP14 (IC50 = ∼5 μM) with high selectivity among the 8 DUBs screened.178 Interestingly, compound 13 is active when USP14 is in the active state i.e. part of the proteasome, whereas it did not show any activity in the absence of the proteasome. When examining the effect of this inhibitor on Tau and TAR DNA binding proteins (TDP-43) (involved in neurodegenerative disease), it was found that in the absence of 13 this leads to the accumulation of these substrates, whereas in the presence of 13 these proteins were degraded by proteasomes.
USP30
Upon ubiquitination of the damaged mitochondria by PARKIN (PARK2) and PINK1, a protein kinase leads to mitophagy, where USP30 is localized and antagonizes this process.179 Hence, it has been found that the overexpression of USP30 leads to the accumulation of damaged mitochondria, which is associated with Parkinson’s disease, making USP30 a potential therapeutic target. Yue and coworkers reported the natural product 15-oxospiramilactone 14 as an inhibitor for USP30, which was selected from a 300 compound library (Fig. 13).180 Upon treatment with 2 μM of 14, 80% of mitochondrial elongation was observed in the mitofusion-1 knockout cells supporting the role of USP30 in this process.Inhibition of DUBs by reactive oxygen species
Recently several studies showed that DUBs are susceptible to oxidation when exposed to exogenous hydrogen peroxide.181–183 It is well known that Cys residues in cellular proteins can be modified by reactive oxygen species (ROS) and undergo oxidation to different levels, such as sulfenic acid (–SOH), sulfinic acid (–SO2H) or sulfonic acid (–SO3H) (Fig. 14a).184 Huang and coworkers demonstrated the oxidation of the active site Cys of several DUBs in cells for the first time.181 Testing USP1 with exogenous hydrogen peroxide led to the accumulation of mono-ubiquitinated PCNA, which plays a critical role in overcoming DNA damage. It has been shown that a cyclic 1,3-dione based probe can detect sulfenic acid in proteins in the cellular environment (Fig. 14b).185,186 Utilizing this probe, the authors found that the catalytic Cys in USP1 was oxidized to sulfenic acid in the cell extract. Furthermore, active site mutation in USP1 did not show any reaction with this probe, and with UbVME ABP, further supporting the site of oxidation as the catalytic Cys. This type of inhibition was further tested and found to be reversible, where the addition of reducing reagents such as dithiothreitol (DTT) regains the activity of the oxidized DUB.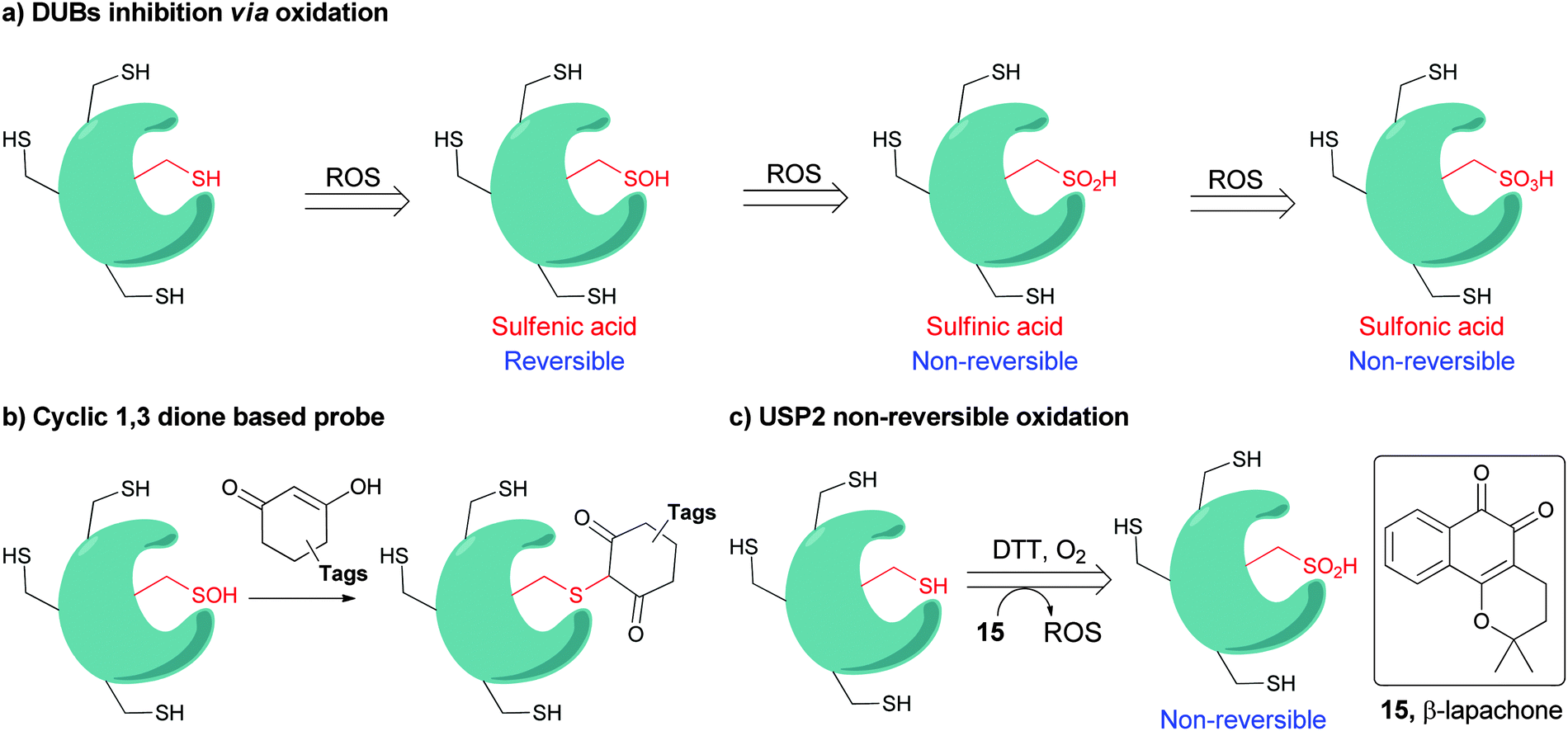 | ||
| Fig. 14 Inhibition of DUBs via oxidation. (a) Different level of Cys residue oxidation. (b) Cyclic 1,2 dione based probes. (c) Inhibition of USP2 by β-lapachone. | ||
In another study by Komander and coworkers it was reported that the active site Cys of A20 from the OTU family undergoes reversible oxidation in vitro and in the cellular environment.101 Here, the reversibility of the oxidation was also demonstrated by incubating A20 with elevated concentrations of DTT, which reduces the sulfenic acid to the active thiol. Interestingly, incubating A20 with a high concentration of hydrogen peroxide led to irreversible oxidation states i.e. sulfinic or sulfonic acid. From the crystallographic studies of A20 in different oxidized forms, the group found that even the unstable sulfenic acid form was stabilized by hydrogen bonds around the catalytic core, which demanded a large excess of hydrogen peroxide to undergo further oxidation.
In a parallel study by Lee et al. it was further demonstrated that the oxidation of Cys to sulfenic acid in DUBs is reversible when treated with DTT.183 In addition, the formation of a reversible cyclic sulphenyl-amide intermediate in USP19CD, formed by the attack of the nearby amide nitrogen on the sulfenic acid, was also observed in mass spectrometry. Based on the previous studies and computational modeling they proposed that Ser510 in USP19CD is the residue that is involved in such an intermediate. Ser510Gly mutation prevented the formation of such an intermediate, which leads to irreversible oxidation of USP19CD upon hydrogen peroxide treatment. Importantly, the susceptibility of DUBs to undergo oxidation was found to be associated with its activity. Some DUBs undergo transition from non-productive to productive conformation upon substrate binding.17 This leads to lowering of the pKa of the active site Cys, hence making it more prone to oxidation. For example, USP7, which is known to adopt a disordered active site in the absence of Ub, did not exhibit sensitivity to oxidation in this conformation. Nevertheless increasing the pH of the reaction led to increased sensitivity of the active Cys to hydrogen peroxide. Similar results were also obtained for UCH-L1.
Using our in-house developed assay161 our group identified β-lapachone, 15 (Fig. 14c), an orthoquinone based natural product, as a potent inhibitor for USP2, which is associated with aggressive prostate cancer.187 Interestingly, β-lapachone is known to generate ROS and has been in clinical trials against several kinds of cancers, however the proposed mode of action has not been associated with the inhibition of any DUB.188 Further studies on the mechanism of action of β-lapachone, including LC-MS analysis, ABP assay, reducing agent treatment and enzyme mutation, revealed that the mode of inhibition is via irreversible oxidation of the USP2 active site Cys to the sulfinic acid form (Fig. 14c).187 β-lapachone was also found to inhibit USP1 and UCH-L3, but to a lesser extent USP7 and UCH-L1, due to their unproductive catalytic state.
The above-mentioned studies depict that DUBs could be modulated and regulated by oxidative stress. The proof of concept that ROS generating compounds can be employed to inhibit DUBs was also exemplified. These new insights could be useful for future development of novel inhibitors against DUBs.
Protein based inhibitors
The development of small molecule based inhibitors against a desired DUB could lead to cross reactivity of closely related DUBs and other enzymes. In order to achieve more selective inhibition, engineering of Ub based inhibitors aided by computational approaches and phage display technologies has also been demonstrated.Using computational macromolecular modeling (Rosetta Design), Corn and coworkers chose seven positions around the β1–β2 loop in Ub (Thr7, Leu8, Ile13, Glu34, Ile36, Leu69 and Leu71) to create Ub variants with high conformational variability (Fig. 15).189 Sixty-nine unique sequences were made using the phage display technology, among those 62 clones were disulfide based and 7 are non-disulfide based Ub variants. U7Ub7 (disulfide) and U7Ub25 (non-disulfide) were obtained as the first hits with binding affinities to catalytically inactive USP7 (USP7-CD*) of 225 nM and 141 nM, respectively. Further affinity maturation studies using U7Ub25 revealed U7Ub25.2540 as the tightest binder of USP7-CD* with a KD of 56 nM (Fig. 15). Within the panel of the screened DUBs both U7Ub7 and U7Ub25.2540 exhibited activity against USP5 and USP7. U7Ub25 inhibited the full length USP7 and USP5 with IC50 values of 250 nM and 373 nM, respectively, whereas U7Ub25.2540 inhibited USP7 and USP5 with 160 nM and 251 nM, respectively. Similar results were also reflected in the cell based studies. Interestingly, truncation of the first two Gly residues in U7Ub25 (U7Ub25ΔGG) completely abolished the activity against USP5, whereas the activity for USP7 was retained. A similar trend was obtained with U7Ub25.2540ΔGG in the cell-based study.
 | ||
| Fig. 15 Inhibitors based on Ub variants generated using phage display. | ||
Sidhu and coworkers also generated a library of Ub mutants using phage display library and found highly potent and selective inhibitors against several DUBs.190 The Ub:USP21 crystal structure revealed that ∼75% of binding interactions falls in the non-conserved region of the USP family. Based on this observation and for the generation of a library containing Ub variants, ∼30 residues were chosen, which make contacts between Ub and USP21 (Fig. 15). The Ub variants obtained exhibited very strong binding affinities with the cognate USP. For example, Ubv.8.2, Ubv.21.4 and Ubv.2.3 showed IC50 values of 4.8, 2.4 and 25 nM for USP8, 7 and 2a, respectively, whereas these Ub variants did not show any activity when screened with other DUBs. In addition to this, they found several Ub variants for the selective binding to specific DUBs. This study was combined with structural analyses of the new Ub variants and the specific inhibited DUB, which explained the acquired specificity at the molecular level. From structural analyses one could see, for example, that the Ub mutants appear to make key contacts with the non-conserved region in the USP family, hence accounting for the observed specificities. We envisaged that such highly specific Ub variants could be utilized in the design of activity based probes, assays, and ubiquitinated substrates for various studies.
5. Summary and outlook
Motivated by the emerging important roles of DUBs in health and disease, various synthetic tools have been developed to support the research community with unique substrates that are difficult to obtain otherwise and assist in unraveling the mysteries of DUBs. The possibility of different anchoring sites in Ub, which results in Ub chains having different lengths and various conformations, not only increases the complexity of ubiquitin signaling but also lays a complex platform for the selective recognition and processing of Ub conjugates by DUBs. The continuous efforts made to understand the significance of DUBs in various cellular functions have validated that the aberration in DUB functions leads to various diseases such as cancer.The substantial growth in the field of protein synthesis and semisynthesis,78,191–193 which allow site selective protein manipulations, made it now possible to prepare a variety of reagents of interest for studying DUBs. In this regard, the synthesis of homogenous free Ub chains as well as proteins that are site specifically modified with Ub or a specific Ub chain with a defined length and linkage has enabled performing several biochemical studies for better understanding the specificity and structure of important DUBs.
Recent discoveries that Ub is also modified by PTMs such as acetylation on Lys6 and Lys48 as well as phosphorylation at multiple sites, such as Ser57 and Ser65, have raised several questions and brought more mysteries to the ubiquitin signal in general and deubiquitination in particular.124 Many of the enzymes that carry out and remove these PTMs are still unknown rendering the enzymatic preparation of related analogues based on Ub difficult. Thus chemical and semisynthesis approaches can be instrumental to prepare these analogues and assist in unraveling unknown aspects of these marks.
Several Ub-based ABP and di-Ub ABP reagents have been developed and are being utilized in identifying new DUBs and unraveling their structures and functions. Moreover, these ABPs can be useful to study the selectivity profile of DUBs in cellular contexts. In addition, the ability to prepare Ub conjugates labelled with a fluorophore at desired positions made it possible to screen a large number of libraries of small molecules and bring novel inhibitors for DUBs with therapeutic potentials. This area is relatively still underdeveloped as evident by the discovery of only a few scaffolds as potential leads and the lack of inhibitors in advanced clinical trials. One of the challenges is to achieve the desired selectivity in a pool of cysteine proteases. Though the road towards the development of drug candidates is far, there are ample opportunities to develop more novel inhibitors against a selected DUB through more rational medicinal chemistry design coupled with the development of unique reagents to study the activities of DUBs in vivo. In addition, the learned lessons from the general field of protein–protein interactions in developing inhibitors that target large surfaces could be also exercised in the design of novel inhibitors against DUBs.194,195
In addition to ubiquitination and deubiquitination, protein modification by Ub like modifiers such as SUMO and NEDD8 also takes place simultaneously.196 In addition to this, mixed forms of Ub and Ubl modifiers have recently also been reported and their biological significance in the signaling process is still relatively unexplored.197 We believe that the tools thus far developed can be further extended to these systems and assist in studying also the reverse process of these modifications similar to DUBs. In all these studies organic chemistry will continue to be an essential part in offering new reagents ranging from small molecules to large proteins.
Acknowledgements
A. Brik is a Neubauer Professor and a Taub Fellow-Supported by the Taub Foundations. We would like to thank the Ministry of Science, Technology and Space (MOST) for funding (2021900).References
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Jr., Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503–533 CrossRef CAS PubMed.
- M. H. Glickman and A. Ciechanover, Physiol. Rev., 2002, 82, 373–428 CrossRef CAS PubMed.
- A. M. Weissman, N. Shabek and A. Ciechanover, Nat. Rev. Mol. Cell Biol., 2011, 12, 605–620 CrossRef CAS PubMed.
- D. Finley, Annu. Rev. Biochem., 2009, 78, 477–513 CrossRef CAS PubMed.
- C. M. Pickart and D. Fushman, Curr. Opin. Chem. Biol., 2004, 8, 610–616 CrossRef CAS PubMed.
- L. Sun and Z. J. Chen, Curr. Opin. Cell Biol., 2004, 16, 119–126 CrossRef CAS PubMed.
- K. Haglund and I. Dikic, EMBO J., 2005, 24, 3353–3359 CrossRef CAS PubMed.
- Y. Kulathu and D. Komander, Nat. Rev. Mol. Cell Biol., 2012, 13, 508–523 CrossRef CAS PubMed.
- L. Spasser and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 6840–6862 CrossRef CAS PubMed.
- Y. Kravtsova-Ivantsiv, T. Sommer and A. Ciechanover, Angew. Chem., Int. Ed., 2013, 52, 192–198 CrossRef CAS PubMed.
- K. R. Love, A. Catic, C. Schlieker and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 697–705 CrossRef CAS PubMed.
- F. E. Reyes-Turcu and K. D. Wilkinson, Chem. Rev., 2009, 109, 1495–1508 CrossRef CAS PubMed.
- D. Komander, M. J. Clague and S. Urbe, Nat. Rev. Mol. Cell Biol., 2009, 10, 550–563 CrossRef CAS PubMed.
- I. E. Wertz, K. M. O'Rourke, H. Zhou, M. Eby, L. Aravind, S. Seshagiri, P. Wu, C. Wiesmann, R. Baker, D. L. Boone, A. Ma, E. V. Koonin and V. M. Dixit, Nature, 2004, 430, 694–699 CrossRef CAS PubMed.
- F. E. Reyes-Turcu, K. H. Ventii and K. D. Wilkinson, Annu. Rev. Biochem., 2009, 78, 363–397 CrossRef CAS PubMed.
- C. M. Pickart and I. A. Rose, J. Biol. Chem., 1985, 260, 7903–7910 CAS.
- Z. M. Eletr and K. D. Wilkinson, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 114–128 CrossRef CAS PubMed.
- W. Zhang, T. Sulea, L. Tao, Q. Cui, E. O. Purisima, R. Vongsamphanh, P. Lachance, V. Lytvyn, H. Qi, Y. Li and R. Menard, Biochemistry, 2011, 50, 4775–4785 CrossRef CAS PubMed.
- Y. Sato, A. Yoshikawa, A. Yamagata, H. Mimura, M. Yamashita, K. Ookata, O. Nureki, K. Iwai, M. Komada and S. Fukai, Nature, 2008, 455, 358–362 CrossRef CAS PubMed.
- L. Hicke, H. L. Schubert and C. P. Hill, Nat. Rev. Mol. Cell Biol., 2005, 6, 610–621 CrossRef CAS PubMed.
- K. Husnjak and I. Dikic, Annu. Rev. Biochem., 2012, 81, 291–322 CrossRef CAS PubMed.
- J. H. Hurley, S. Lee and G. Prag, Biochem. J., 2006, 399, 361–372 CrossRef CAS PubMed.
- S. Misaghi, P. J. Galardy, W. J. N. Meester, H. Ovaa, H. L. Ploegh and R. Gaudet, J. Biol. Chem., 2005, 280, 1512–1520 CrossRef CAS PubMed.
- M. W. Popp, K. Artavanis-Tsakonas and H. L. Ploegh, J. Biol. Chem., 2009, 284, 3593–3602 CrossRef CAS PubMed.
- A. C. Faesen, M. P. A. Luna-Vargas, P. P. Geurink, M. Clerici, R. Merkx, W. J. van Dijk, D. S. Hameed, F. El Oualid, H. Ovaa and T. K. Sixma, Chem. Biol., 2011, 18, 1550–1561 CrossRef CAS PubMed.
- M. Hu, P. Li, M. Li, W. Li, T. Yao, J.-W. Wu, W. Gu, R. E. Cohen and Y. Shi, Cell, 2002, 111, 1041–1054 CrossRef CAS PubMed.
- T. E. Messick, N. S. Russell, A. J. Iwata, K. L. Sarachan, R. Shiekhattar, J. R. Shanks, F. E. Reyes-Turcu, K. D. Wilkinson and R. Marmorstein, J. Biol. Chem., 2008, 283, 11038–11049 CrossRef CAS PubMed.
- T. E. T. Mevissen, M. K. Hospenthal, P. P. Geurink, P. R. Elliott, M. Akutsu, N. Arnaudo, R. Ekkebus, Y. Kulathu, T. Wauer, F. El Oualid, S. M. V. Freund, H. Ovaa and D. Komander, Cell, 2013, 154, 169–184 CrossRef CAS PubMed.
- S. C. Johnston, C. N. Larsen, W. J. Cook, K. D. Wilkinson and C. P. Hill, EMBO J., 1997, 16, 3787–3796 CrossRef CAS PubMed.
- Y. Mao, F. Senic-Matuglia, P. P. Di Fiore, S. Polo, M. E. Hodsdon and P. De Camilli, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12700–12705 CrossRef CAS PubMed.
- G. Nicastro, L. Masino, V. Esposito, R. P. Menon, A. De Simone, F. Fraternali and A. Pastore, Biopolymers, 2009, 91, 1203–1214 CrossRef CAS PubMed.
- G. Nicastro, R. P. Menon, L. Masino, P. P. Knowles, N. Q. McDonald and A. Pastore, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10493–10498 CrossRef CAS PubMed.
- V. Maytal-Kivity, N. Reis, K. Hofmann and H. Glickman Michael, BMC Biochem., 2002, 3, 28 CrossRef PubMed.
- D. Komander, Biochem. Soc. Trans., 2009, 37, 937–953 CrossRef CAS PubMed.
- R. Varadan, O. Walker, C. Pickart and D. Fushman, J. Mol. Biol., 2002, 324, 637–647 CrossRef CAS PubMed.
- Y. Ryabov and D. Fushman, Proteins: Struct., Funct., Bioinf., 2006, 63, 787–796 CrossRef CAS PubMed.
- R. Varadan, M. Assfalg, A. Haririnia, S. Raasi, C. Pickart and D. Fushman, J. Biol. Chem., 2004, 279, 7055–7063 CrossRef CAS PubMed.
- S. Raasi and C. M. Pickart, J. Biol. Chem., 2003, 278, 8951–8959 CrossRef CAS PubMed.
- S. Raasi, R. Varadan, D. Fushman and C. M. Pickart, Nat. Struct. Mol. Biol., 2005, 12, 708–714 CAS.
- Y. Sato, A. Yoshikawa, H. Mimura, M. Yamashita, A. Yamagata and S. Fukai, EMBO J., 2009, 28, 2461–2468 CrossRef CAS PubMed.
- S. Vijay-Kumar, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1987, 194, 531–544 CrossRef CAS PubMed.
- Y. Ryabov and D. Fushman, J. Am. Chem. Soc., 2007, 129, 7894–7902 CrossRef CAS PubMed.
- S. Virdee, Y. Ye, D. P. Nguyen, D. Komander and J. W. Chin, Nat. Chem. Biol., 2010, 6, 750–757 CrossRef CAS PubMed.
- D. Komander, F. Reyes-Turcu, J. D. F. Licchesi, P. Odenwaelder, K. D. Wilkinson and D. Barford, EMBO Rep., 2009, 10, 466–473 CrossRef CAS PubMed.
- R. Varadan, M. Assfalg, S. Raasi, C. Pickart and D. Fushman, Mol. Cell, 2005, 18, 687–698 CrossRef CAS PubMed.
- J. D. F. Licchesi, J. Mieszczanek, T. E. T. Mevissen, T. J. Rutherford, M. Akutsu, S. Virdee, F. El Oualid, J. W. Chin, H. Ovaa, M. Bienz and D. Komander, Nat. Struct. Mol. Biol., 2012, 19, 62–71 CAS.
- S.-C. Sun, Nat. Rev. Immunol., 2008, 8, 501–511 CrossRef CAS PubMed.
- A. Borodovsky, B. M. Kessler, R. Casagrande, H. S. Overkleeft, K. D. Wilkinson and H. L. Ploegh, EMBO J., 2001, 20, 5187–5196 CrossRef CAS PubMed.
- R. Verma, L. Aravind, R. Oania, W. H. McDonald, J. R. Yates, III, E. V. Koonin and R. J. Deshaies, Science, 2002, 298, 611–615 CrossRef CAS PubMed.
- T. Yao, L. Song, W. Xu, G. N. DeMartino, L. Florens, S. K. Swanson, M. P. Washburn, R. C. Conaway, J. W. Conaway and R. E. Cohen, Nat. Cell Biol., 2006, 8, 994–1002 CrossRef CAS PubMed.
- I. Garcia-Santisteban, G. J. Peters, E. Giovannetti and J. A. Rodriguez, Mol. Cancer, 2013, 12, 91 CrossRef CAS PubMed.
- S. M. B. Nijman, T. T. Huang, A. M. G. Dirac, T. R. Brummelkamp, R. M. Kerkhoven, A. D. D'Andrea and R. Bernards, Mol. Cell, 2005, 17, 331–339 CrossRef CAS PubMed.
- A. Kovalenko, C. Chable-Bessia, G. Cantarella, A. Israel, D. Wallach and G. Courtois, Nature, 2003, 424, 801–805 CrossRef CAS PubMed.
- E. G. Lee, D. L. Boone, S. Chai, S. L. Libby, M. Chien, J. P. Lodolce and A. Ma, Science, 2000, 289, 2350–2354 CrossRef CAS PubMed.
- T. DeVine and M.-S. Dai, Curr. Pharm. Des., 2013, 19, 3248–3262 CrossRef CAS PubMed.
- M. Li, D. Chen, A. Shiloh, J. Luo, A. Y. Nikolaev, J. Qin and W. Gu, Nature, 2002, 416, 648–652 CrossRef CAS PubMed.
- L. F. Stevenson, A. Sparks, N. Allende-Vega, D. P. Xirodimas, D. P. Lane and M. K. Saville, EMBO J., 2007, 26, 976–986 CrossRef CAS PubMed.
- H. A. Lindner, Virology, 2007, 362, 245–256 CrossRef CAS PubMed.
- V. Saridakis, Y. Sheng, F. Sarkari, M. N. Holowaty, K. Shire, T. Nguyen, R. G. Zhang, J. Liao, W. Lee, A. M. Edwards, C. H. Arrowsmith and L. Frappier, Mol. Cell, 2005, 18, 25–36 CrossRef CAS PubMed.
- D. M. Maraganote, T. G. Lesnick, A. Elbaz, M.-C. Charrier-Harlin, T. Gasser, R. Krueger, N. Hattori, G. D. Mellick, A. Quattrone, J.-i. Satoh, T. Toda, J. Wang, J. P. A. Ioannidis, M. de Andrade and W. A. Rocca, Ann. Neurol., 2004, 55, 512–521 CrossRef CAS PubMed.
- Y. Liu, L. Fallon, H. A. Lashuel, Z. Liu and P. T. Lansbury, Jr., Cell, 2002, 111, 209–218 CrossRef CAS PubMed.
- J. Choi, A. I. Levey, S. T. Weintraub, H. D. Rees, M. Gearing, L.-S. Chin and L. Li, J. Biol. Chem., 2004, 279, 13256–13264 CrossRef CAS PubMed.
- P. D'Arcy, X. Wang and S. Linder, Pharmacol. Ther., 2015, 147, 32–54 CrossRef PubMed.
- B. Nicholson, J. G. Marblestone, T. R. Butt and M. R. Mattern, Future Oncol., 2007, 3, 191–199 CrossRef CAS PubMed.
- S. Hussain, Y. Zhang and P. J. Galardy, Cell Cycle, 2009, 8, 1688–1697 CrossRef CAS PubMed.
- R. Pfoh, I. K. Lacdao and V. Sarldakis, Endocr.-Relat. Cancer, 2015, 22, T35–T54 CrossRef CAS PubMed.
- S. V. Todi and H. L. Paulson, Trends Neurosci., 2011, 34, 370–382 CrossRef CAS PubMed.
- G. Ristic, W.-L. Tsou and S. V. Todi, Front. Mol. Neurosci., 2014, 7, 1–15 Search PubMed.
- B. M. Kessler and M. J. Edelmann, Cell Biochem. Biophys., 2011, 60, 21–38 CrossRef CAS PubMed.
- D. D. Sahtoe and T. K. Sixma, Trends Biochem. Sci., 2015, 40, 456–467 CrossRef CAS PubMed.
- W. Reiley, M. Zhang, X. Wu, E. Granger and S.-C. Sun, Mol. Cell. Biol., 2005, 25, 3886–3895 CrossRef CAS PubMed.
- T. Kobayashi, K. C. Masoumi and R. Massoumi, Oncogene, 2015, 34, 2251–2260 CrossRef CAS PubMed.
- K. D. Wilkinson, M. J. Cox, A. N. Mayer and T. Frey, Biochemistry, 1986, 25, 6644–6649 CrossRef CAS PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- M. Schnoelzer and S. B. H. Kent, Science, 1992, 256, 221–225 CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- P. E. Dawson, Isr. J. Chem., 2011, 51, 862–867 CrossRef CAS.
- L. R. Malins and R. J. Payne, Curr. Opin. Chem. Biol., 2014, 22, 70–78 CrossRef CAS PubMed.
- F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- R. Ramage, J. Green and O. M. Ogunjobi, Tetrahedron Lett., 1989, 30, 2149–2152 CrossRef CAS.
- R. Ramage, J. Green, T. W. Muir, O. M. Ogunjobi, S. Love and K. Shaw, Biochem. J., 1994, 299, 151–158 CrossRef CAS PubMed.
- D. Bang, G. I. Makhatadze, V. Tereshko, A. A. Kossiakoff and S. B. Kent, Angew. Chem., Int. Ed., 2005, 44, 3852–3856 CrossRef CAS PubMed.
- L. A. Erlich, K. S. A. Kumar, M. Haj-Yahya, P. E. Dawson and A. Brik, Org. Biomol. Chem., 2010, 8, 2392–2396 CAS.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS PubMed.
- S. N. Bavikar, L. Spasser, M. Haj-Yahya, S. V. Karthikeyan, T. Moyal, K. S. Ajish Kumar and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 758–763 CrossRef CAS PubMed.
- R. Ekkebus, D. Flierman, P. P. Geurink and H. Ovaa, Curr. Opin. Chem. Biol., 2014, 23, 63–70 CrossRef CAS PubMed.
- H. Ovaa, Nat. Rev. Cancer, 2007, 7, 613–620 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- C. M. Pickart and I. A. Rose, J. Biol. Chem., 1986, 261, 10210–10217 CAS.
- A. Hershko and I. A. Rose, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1829–1833 CrossRef CAS.
- Y. A. Lam, W. Xu, G. N. DeMartino and R. E. Cohen, Nature, 1997, 385, 737–740 CrossRef CAS PubMed.
- C. Schlieker, W. A. Weihofen, E. Frijns, L. M. Kattenhorn, R. Gaudet and H. L. Ploegh, Mol. Cell, 2007, 25, 677–687 CrossRef CAS PubMed.
- A. Borodovsky, H. Ovaa, W. J. N. Meester, E. S. Venanzi, M. S. Bogyo, B. G. Hekking, H. L. Ploegh, B. M. Kessler and H. S. Overkleeft, ChemBioChem, 2005, 6, 287–291 CrossRef CAS PubMed.
- K. Routenberg Love, R. K. Pandya, E. Spooner and H. L. Ploegh, ACS Chem. Biol., 2009, 4, 275–287 CrossRef CAS PubMed.
- H. Ovaa, B. M. Kessler, U. Rolen, P. J. Galardy, H. L. Ploegh and M. G. Masucci, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2253–2258 CrossRef CAS.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. R. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- S. Sommer, N. D. Weikart, U. Linne and H. D. Mootz, Bioorg. Med. Chem., 2013, 21, 2511–2517 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- A. de Jong, R. Merkx, I. Berlin, B. Rodenko, R. H. M. Wijdeven, D. El Atmioui, Z. Yalcin, C. N. Robson, J. J. Neefjes and H. Ovaa, ChemBioChem, 2012, 13, 2251–2258 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, J.-P. Pellois and T. W. Muir, Angew. Chem., Int. Ed., 2007, 46, 2814–2818 CrossRef CAS PubMed.
- C. E. Weller, W. Huang and C. Chatterjee, ChemBioChem, 2014, 15, 1263–1267 CrossRef CAS PubMed.
- K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS PubMed.
- R. Yang, K. K. Pasunooti, F. Li, X.-W. Liu and C.-F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS PubMed.
- M. Haj-Yahya, K. S. Ajish Kumar, L. A. Erlich and A. Brik, Biopolymers, 2010, 94, 504–510 CrossRef CAS PubMed.
- K. S. A. Kumar, L. Spasser, S. Ohayon, L. A. Erlich and A. Brik, Bioconjugate Chem., 2011, 22, 137–143 CrossRef CAS PubMed.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y.-c. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS PubMed.
- K. S. A. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2010, 49, 9126–9131 CrossRef CAS PubMed.
- S. Eger, M. Scheffner, A. Marx and M. Rubini, J. Am. Chem. Soc., 2010, 132, 16337–16339 CrossRef CAS PubMed.
- L. Yin, B. Krantz, N. S. Russell, S. Deshpande and K. D. Wilkinson, Biochemistry, 2000, 39, 10001–10010 CrossRef CAS PubMed.
- C. Castaneda, J. Liu, A. Chaturvedi, U. Nowicka, T. A. Cropp and D. Fushman, J. Am. Chem. Soc., 2011, 133, 17855–17868 CrossRef CAS PubMed.
- D. Komander, C. J. Lord, H. Scheel, S. Swift, K. Hofmann, A. Ashworth and D. Barford, Mol. Cell, 2008, 29, 451–464 CrossRef CAS PubMed.
- M. Hu, P. Li, L. Song, P. D. Jeffrey, T. A. Chenova, K. D. Wilkinson, R. E. Cohen and Y. Shi, EMBO J., 2005, 24, 3747–3756 CrossRef CAS PubMed.
- F. Koyano, K. Okatsu, H. Kosako, Y. Tamura, E. Go, M. Kimura, Y. Kimura, H. Tsuchiya, H. Yoshihara, T. Hirokawa, T. Endo, E. A. Fon, J.-F. Trempe, Y. Saeki, K. Tanaka and N. Matsuda, Nature, 2014, 510, 162–166 CAS.
- L. A. Kane, M. Lazarou, A. I. Fogel, Y. Li, K. Yamano, S. A. Sarraf, S. Banerjee and R. J. Youle, J. Cell Biol., 2014, 205, 143–153 CrossRef CAS PubMed.
- A. Kazlauskaite, C. Kondapalli, R. Gourlay, D. G. Campbell, M. S. Ritorto, K. Hofmann, D. R. Alessi, A. Knebel, M. Trost and M. M. K. Muqit, Biochem. J., 2014, 460, 127–139 CrossRef CAS PubMed.
- A. Ordureau, S. A. Sarraf, D. M. Duda, J.-M. Heo, M. P. Jedrychowski, V. O. Sviderskiy, J. L. Olszewski, J. T. Koerber, T. Xie, S. A. Beausoleil, J. A. Wells, S. P. Gygi, B. A. Schulman and J. W. Harper, Mol. Cell, 2014, 56, 360–375 CrossRef CAS PubMed.
- T. Wauer, K. N. Swatek, J. L. Wagstaff, C. Gladkova, J. N. Pruneda, M. A. Michel, M. Gersch, C. M. Johnson, S. M. V. Freund and D. Komander, EMBO J., 2015, 34, 307–325 CrossRef CAS PubMed.
- S. Bondalapati, W. Mansour, M. A. Nakasone, S. K. Maity, M. H. Glickman and A. Brik, Chem. – Eur. J., 2015, 21, 7360–7364 CrossRef CAS PubMed.
- L. Herhaus and I. Dikic, EMBO Rep., 2015, 16, 1071–1083 CrossRef CAS PubMed.
- N. S. Russell and K. D. Wilkinson, Biochemistry, 2004, 43, 4844–4854 CrossRef CAS PubMed.
- I. Dikic, S. Wakatsuki and K. J. Walters, Nat. Rev. Mol. Cell Biol., 2009, 10, 659–671 CrossRef CAS PubMed.
- Y. Ye, G. Blaser, M. H. Horrocks, M. J. Ruedas-Rama, S. Ibrahim, A. A. Zhukov, A. Orte, D. Klenerman, S. E. Jackson and D. Komander, Nature, 2012, 492, 266–270 CrossRef CAS PubMed.
- A. B. Datta, G. L. Hura and C. Wolberger, J. Mol. Biol., 2009, 392, 1117–1124 CrossRef CAS PubMed.
- N. Haj-Yahya, M. Haj-Yahya, C. A. Castaneda, L. Spasser, H. P. Hemantha, M. Jbara, M. Penner, A. Ciechanover, D. Fushman and A. Brik, Angew. Chem., Int. Ed., 2013, 52, 11149–11153 CrossRef CAS PubMed.
- M. Haj-Yahya, N. Eltarteer, S. Ohayon, E. Shema, E. Kotler, M. Oren and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 11535–11539 CrossRef CAS PubMed.
- H. Pham Grace, S. J. B. Rana Ambar, E. N. Korkmaz, H. Trang Vivian, Q. Cui and R. Strieter Eric, Protein Sci., 2016, 25, 456–471 CrossRef CAS PubMed.
- J. Lopez-Mosqueda and I. Dikic, Cell, 2014, 157, 767–769 CrossRef CAS PubMed.
- E. K. Dixon, C. A. Castaneda, T. R. Kashyap, Y. Wang and D. Fushman, Bioorg. Med. Chem., 2013, 21, 3421–3429 CrossRef CAS PubMed.
- J. F. McGouran, S. R. Gaertner, M. Altun, H. B. Kramer and B. M. Kessler, Chem. Biol., 2013, 20, 1447–1455 CrossRef CAS PubMed.
- G. Li, Q. Liang, P. Gong, A. H. Tencer and Z. Zhuang, Chem. Commun., 2014, 50, 216–218 RSC.
- N. Haj-Yahya, H. P. Hemantha, R. Meledin, S. Bondalapati, M. Seenaiah and A. Brik, Org. Lett., 2014, 16, 540–543 CrossRef CAS PubMed.
- M. P. C. Mulder, F. El Oualid, J. ter Beek and H. Ovaa, ChemBioChem, 2014, 15, 946–949 CrossRef CAS PubMed.
- J. B. Schaefer and D. O. Morgan, J. Biol. Chem., 2011, 286, 45186–45196 CrossRef CAS PubMed.
- N. Shabek, Y. Herman-Bachinsky, S. Buchsbaum, O. Lewinson, M. Haj-Yahya, M. Hejjaoui, H. A. Lashuel, T. Sommer, A. Brik and A. Ciechanover, Mol. Cell, 2012, 48, 87–97 CrossRef CAS PubMed.
- M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar, S. V. Karthikeyan, A. Ciechanover, H. A. Lashuel and A. Brik, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17726–17731 CrossRef CAS PubMed.
- R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453, 812–816 CrossRef CAS PubMed.
- M. Hejjaoui, M. Haj-Yahya, K. S. A. Kumar, A. Brik and H. A. Lashuel, Angew. Chem., Int. Ed., 2011, 50, 405–409 CrossRef CAS PubMed.
- J. Chen, Y. Ai, J. Wang, L. Haracska and Z. Zhuang, Nat. Chem. Biol., 2010, 6, 270–272 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS PubMed.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- X. Bi, R. Yang, X. Feng, D. Rhodes and C.-F. Liu, Org. Biomol. Chem., 2016, 14, 835–839 CAS.
- K. Yang, P. Gong, P. Gokhale and Z. Zhuang, ACS Chem. Biol., 2014, 9, 1685–1691 CrossRef CAS PubMed.
- M. Morgan, M. Haj-Yahya, A. E. Ringel, P. Bandi, A. Brik and C. Wolberger, Science, 2016, 351, 725–728 CrossRef CAS PubMed.
- J. S. Thrower, L. Hoffman, M. Rechsteiner and C. M. Pickart, EMBO J., 2000, 19, 94–102 CrossRef CAS PubMed.
- L. C. Dang, F. D. Melandri and R. L. Stein, Biochemistry, 1998, 37, 1868–1879 CrossRef CAS PubMed.
- A. Tirat, A. Schilb, V. Riou, L. Leder, B. Gerhartz, J. Zimmermann, S. Worpenberg, U. Eidhoff, F. Freuler, T. Stettler, L. Mayr, J. Ottl, B. Leuenberger and I. Filipuzzi, Anal. Biochem., 2005, 343, 244–255 CrossRef CAS PubMed.
- P. P. Geurink, F. El Oualid, A. Jonker, D. S. Hameed and H. Ovaa, ChemBioChem, 2012, 13, 293–297 CrossRef CAS PubMed.
- R. A. Horton, E. A. Strachan, K. W. Vogel and S. M. Riddle, Anal. Biochem., 2007, 360, 138–143 CrossRef CAS PubMed.
- B. Nicholson, C. A. Leach, S. J. Goldenberg, D. M. Francis, M. P. Kodrasov, X. Tian, J. Shanks, D. E. Sterner, A. Bernal, M. R. Mattern, K. D. Wilkinson and T. R. Butt, Protein Sci., 2008, 17, 1035–1043 CrossRef CAS PubMed.
- D. Chauhan, Z. Tian, B. Nicholson, K. G. S. Kumar, B. Zhou, R. Carrasco, J. L. McDermott, C. A. Leach, M. Fulcinniti, M. P. Kodrasov, J. Weinstock, W. D. Kingsbury, T. Hideshima, P. K. Shah, S. Minvielle, M. Altun, B. M. Kessler, R. Orlowski, P. Richardson, N. Munshi and K. C. Anderson, Cancer Cell, 2012, 22, 345–358 CrossRef CAS PubMed.
- J. Weinstock, J. Wu, P. Cao, W. D. Kingsbury, J. L. McDermott, M. P. Kodrasov, D. M. McKelvey, K. G. Suresh Kumar, S. J. Goldenberg, M. R. Mattern and B. Nicholson, ACS Med. Chem. Lett., 2012, 3, 789–792 CrossRef CAS PubMed.
- U. Hassiepen, U. Eidhoff, G. Meder, J.-F. Bulber, A. Hein, U. Bodendorf, E. Lorthiois and B. Martoglio, Anal. Biochem., 2007, 371, 201–207 CrossRef CAS PubMed.
- J. Chen, T. S. Dexheimer, Y. Ai, Q. Liang, M. A. Villamil, J. Inglese, D. J. Maloney, A. Jadhav, A. Simeonov and Z. Zhuang, Chem. Biol., 2011, 18, 1390–1400 CrossRef CAS PubMed.
- Q. Liang, T. S. Dexheimer, P. Zhang, A. S. Rosenthal, M. A. Villamil, C. You, Q. Zhang, J. Chen, C. A. Ott, H. Sun, D. K. Luci, B. Yuan, A. Simeonov, A. Jadhav, H. Xiao, Y. Wang, D. J. Maloney and Z. Zhuang, Nat. Chem. Biol., 2014, 10, 298–304 CrossRef CAS PubMed.
- S. J. Orcutt, J. Wu, M. J. Eddins, C. A. Leach and J. E. Strickler, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 2079–2086 CrossRef CAS PubMed.
- S. Ohayon, L. Spasser, A. Aharoni and A. Brik, J. Am. Chem. Soc., 2012, 134, 3281–3289 CrossRef CAS PubMed.
- S. Ohayon, M. Refua and A. Brik, Org. Biomol. Chem., 2015, 13, 8182–8186 CAS.
- S. K. Maity, S. Ohayon and A. Brik, Org. Lett., 2015, 17, 3438–3441 CrossRef CAS PubMed.
- S. J. Goldenberg, J. L. McDermott, T. R. Butt, M. R. Mattern and B. Nicholson, Biochem. Soc. Trans., 2008, 36, 828–832 CrossRef CAS PubMed.
- C. Ndubaku and V. Tsui, J. Med. Chem., 2015, 58, 1581–1595 CrossRef CAS PubMed.
- W. Zhang and S. S. Sidhu, FEBS Lett., 2014, 588, 356–367 CrossRef CAS PubMed.
- M. Kemp, Prog. Med. Chem., 2016, 55, 149–192 Search PubMed.
- F. Colland, Biochem. Soc. Trans., 2010, 38, 137–143 CrossRef CAS PubMed.
- Y. Liu, H. A. Lashuel, S. Choi, X. Xing, A. Case, J. Ni, L.-A. Yeh, G. D. Cuny, R. L. Stein and P. T. Lansbury, Chem. Biol., 2003, 10, 837–846 CrossRef CAS PubMed.
- T. S. Dexheimer, A. S. Rosenthal, D. K. Luci, Q. Liang, M. A. Villamil, J. Chen, H. Sun, E. H. Kerns, A. Simeonov, A. Jadhav, Z. Zhuang and D. J. Maloney, J. Med. Chem., 2014, 57, 8099–8110 CrossRef CAS PubMed.
- S. A. Williams, H. L. Maecker, D. M. French, J.-F. Liu, A. Gregg, L. B. Silverstein, T. C. Cao, R. A. D. Carano and V. M. Dixit, Cell, 2011, 146, 918–930 CrossRef CAS PubMed.
- H. Mistry, G. Hsieh, S. J. Buhrlage, M. Huang, E. Park, G. D. Cuny, I. Galinsky, R. M. Stone, N. S. Gray, A. D. D'Andrea and K. Parmar, Mol. Cancer Ther., 2013, 12, 2651–2662 CrossRef CAS PubMed.
- J. L. Parsons, I. I. Dianova, S. V. Khoronenkova, M. J. Edelmann, B. M. Kessler and G. L. Dianov, Mol. Cell, 2011, 41, 609–615 CrossRef CAS PubMed.
- F. Colland, E. Formstecher, X. Jacq, C. Reverdy, C. Planquette, S. Conrath, V. Trouplin, J. Bianchi, V. N. Aushev, J. Camonis, A. Calabrese, C. Borg-Capra, W. Sippl, V. Collura, G. Boissy, J.-C. Rain, P. Guedat, R. Delansorne and L. Daviet, Mol. Cancer Ther., 2009, 8, 2286–2295 CrossRef CAS PubMed.
- C. Reverdy, S. Conrath, R. Lopez, C. Planquette, C. Atmanene, V. Collura, J. Harpon, V. Battaglia, V. Vivat, W. Sippl and F. Colland, Chem. Biol., 2012, 19, 467–477 CrossRef CAS PubMed.
- X. Tian, N. S. Isamiddinova, R. J. Peroutka, S. J. Goldenberg, M. R. Mattern, B. Nicholson and C. Leach, Assay Drug Dev. Technol., 2010, 9, 165–173 CrossRef PubMed.
- M. Altun, H. B. Kramer, L. I. Willems, J. L. McDermott, C. A. Leach, S. J. Goldenberg, K. G. Kumar, R. Konietzny, R. Fischer, E. Kogan, M. M. Mackeen, J. McGouran, S. V. Khoronenkova, J. L. Parsons, G. L. Dianov, B. Nicholson and B. M. Kessler, Chem. Biol., 2011, 18, 1401–1412 CrossRef CAS PubMed.
- B.-H. Lee, M. J. Lee, S. Park, D.-C. Oh, S. Elsasser, P.-C. Chen, C. Gartner, N. Dimova, J. Hanna, S. P. Gygi, S. M. Wilson, R. W. King and D. Finley, Nature, 2010, 467, 179–184 CrossRef CAS PubMed.
- B. Bingol, J. S. Tea, L. Phu, M. Reichelt, C. E. Bakalarski, Q. Song, O. Foreman, D. S. Kirkpatrick and M. Sheng, Nature, 2014, 510, 370–375 CAS.
- W. Yue, Z. Chen, H. Liu, C. Yan, M. Chen, D. Feng, C. Yan, H. Wu, L. Du, Y. Wang, J. Liu, X. Huang, L. Xia, L. Liu, X. Wang, H. Jin, J. Wang, Z. Song, X. Hao and Q. Chen, Cell Res., 2014, 24, 482–496 CrossRef CAS PubMed.
- X. M. Cotto-Rios, M. Bekes, J. Chapman, B. Ueberheide and T. T. Huang, Cell Rep., 2012, 2, 1475–1484 CrossRef CAS PubMed.
- Y. Kulathu, J. Garcia Francisco, E. T. Mevissen Tycho, M. Busch, N. Arnaudo, S. Carroll Kate, D. Barford and D. Komander, Nat. Commun., 2013, 4, 1569 CrossRef PubMed.
- J.-G. Lee, K. Baek, N. Soetandyo and Y. Ye, Nat. Commun., 2013, 4, 1568 CrossRef PubMed.
- M. Lo Conte and K. S. Carroll, J. Biol. Chem., 2013, 288, 26480–26488 CrossRef CAS PubMed.
- S. E. Leonard, K. G. Reddie and K. S. Carroll, ACS Chem. Biol., 2009, 4, 783–799 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, ACS Chem. Biol., 2010, 5, 47–62 CrossRef CAS PubMed.
- S. Ohayon, M. Refua, A. Hendler, A. Aharoni and A. Brik, Angew. Chem., Int. Ed., 2015, 54, 599–603 CAS.
- R. E. Savage, A. N. Tyler, X.-S. Miao and T. C. K. Chan, Drug Metab. Dispos., 2008, 36, 753–758 CrossRef CAS PubMed.
- Y. Zhang, L. Zhou, L. Rouge, A. H. Phillips, C. Lam, P. Liu, W. Sandoval, E. Helgason, J. M. Murray, I. E. Wertz and J. E. Corn, Nat. Chem. Biol., 2013, 9, 51–58 CrossRef CAS PubMed.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A.-M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P.-K. Chan, Y.-C. Juang, M.-C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, Science, 2013, 339, 590–595 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali and A. Brik, Chem. Rec., 2016, 16, 509–519 CrossRef CAS PubMed.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef CAS PubMed.
- M. Hochstrasser, Nature, 2009, 458, 422–429 CrossRef CAS PubMed.
- C. M. Guzzo, C. E. Berndsen, J. Zhu, V. Gupta, A. Datta, R. A. Greenberg, C. Wolberger and M. J. Matunis, Sci. Signaling, 2012, 5, ra88 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |