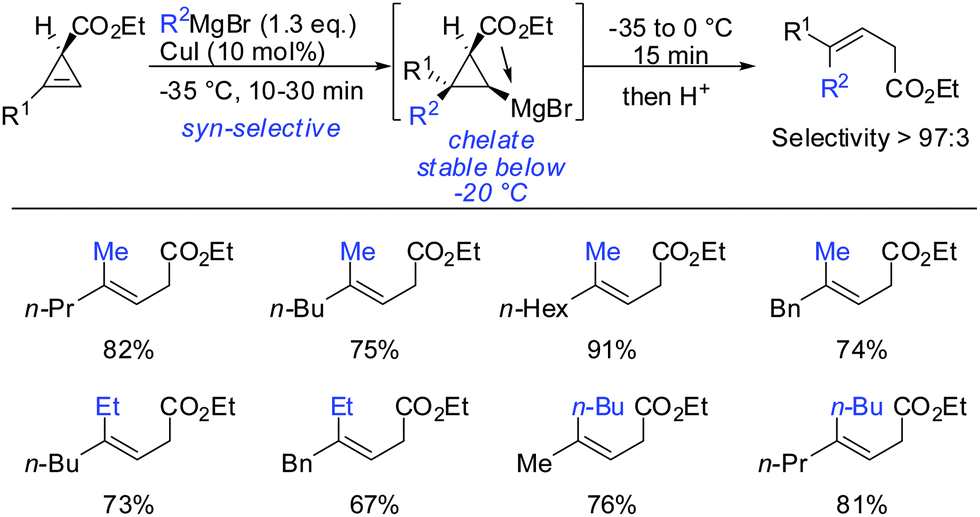DOI:
10.1039/C5CS00897B
(Review Article)
Chem. Soc. Rev., 2016,
45, 4552-4566
Copper mediated carbometalation reactions†
Received
7th December 2015
First published on 25th January 2016
Abstract
Since the first discovery of carbocupration of alkynes in the 1970s a tremendous amount of research has been carried out in this field. The exceptionally high selectivities obtained attribute to the great synthetic value of carbocupration reactions. This tutorial review will present the most important features of carbocupration of alkynes and highlight the most relevant reviews. Then a comprehensive review of copper mediated carbometalation of cyclopropenes will follow. The latter method has received much attention over the last decade as it allows the highly selective construction of poly-substituted cyclopropanes which can be transformed into acyclic derivatives bearing one or multiple tertiary or quaternary carbon stereocenters.

D. S. Müller
| Daniel S. Müller received his diploma in chemistry in 2008 under the guidance of Prof. Bernhard Breit at the University of Freiburg, Germany. In 2012, he earned his PhD in chemistry from the University of Geneva under the directions of Prof. Alexandre Alexakis. As a SNSF (Swiss National Science Foundation) fellow, Daniel conducted postdoctoral studies in the area of total synthesis with Prof. Larry Overman at the University of California, Irvine. In 2014, Daniel joined Prof. Ilan Marek's group at the Technion, where he is currently working on carbometalation reactions and asymmetric catalysis. |

I. Marek
| Ilan Marek, FRSC was born in Haifa (Israel), educated in France, and received his PhD thesis in 1988 from the University Pierre et Marie Curie, Paris, (France) under the guidance of Professor J. F. Normant and Prof. Alexandre Alexakis. In 1989, he was a postdoctoral fellow in Louvain-la-Neuve (Belgium) with Professor L. Ghosez and obtained a research position at the CNRS in France in 1990. After obtaining his Habilitation in Organic Chemistry, he moved to the Technion-Israel Institute of Technology at the end of 1997 where he currently holds a full Professor position. Since 2005, he holds the Sir Michael and Lady Sobell Academic Chair. |
1. Introduction
In the last 50 years an enormous number of stereoselective reactions have appeared. Literally thousands of chiral catalysts, auxiliaries and reagents were invented and are used today with great success. In this context, the stereoselective synthesis of double bonds, which initially received little attention since the historical Wittig- or Julia olefination reactions, became a vibrant field of research.1 The most recent contributions of stereoselective cross-metathesis and transition metal catalysed cross coupling reactions are noteworthy with the latter being dependent isomerically on pure and configurationally stable alkenyl coupling partners.2–4 With the advent of copper mediated carbometalation reactions of alkynes, it soon became obvious that this reaction constitutes one of the best approaches to generate polysubstituted olefins with a defined substitution pattern. In particular, tri- and tetra-substituted alkenes can be prepared with very high stereochemical purity.5 More recent research focuses on translating this exceptional selectivity from the two dimensional space (E/Z-selectivity) into the three dimensional space (creation of stereocenters) by replacing the alkyne substrates with alkenes.6 The copper mediated carbometalation reaction of strained alkenes provides stereoselective access to cyclic- and acyclic stereocenters, which are challenging to synthesize using traditional methods. Given many reviews on carbometalation of alkynes, this review will only provide a short introduction on carbocupration of alkynes. The main intention in the short overview below is to guide the readers to the most relevant reviews with a special emphasis on the basic principles governing the copper-mediated carbometalation reactions. Then a comprehensive review of copper mediated carbometalation of strained alkenes, mainly cyclopropenes, will follow. The nucleophilic species is shown in blue whereas the electrophile is given in red (abbreviated E+). If the electrophile is a proton, the hydrogen atom in the product is not shown for the sake of clarity.
2. Short overview of the carbocupration reactions of alkynes
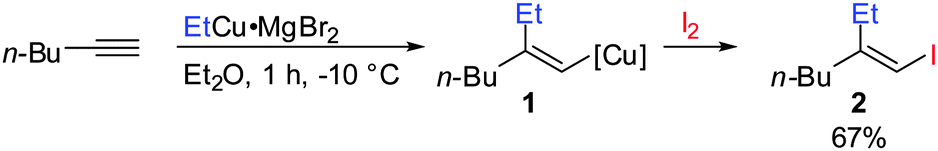
Gilman cuprates (R2CuLi), formed by the reaction of two equivalents of organolithium reagent with one equivalent of organocopper salt, react with monosubstituted alkynes through deprotonation in tetrahydrofuran.7 In 1971 Normant and Bourgain found that a less basic ethyl copper reagent, prepared from an equimolar amount of EtMgBr and CuBr, reacted with n-hexyne in low-polar solvents such as diethyl ether by regio- and stereospecific syn-addition to give, after treatment with iodine, pure (E)-iodoolefin 2 (Scheme 1).7 Throughout this review, the [Cu] notation will be used to indicate a copper metal bearing ligands.
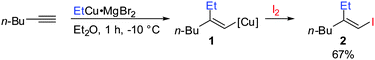 |
| | Scheme 1 First carbocupration of non-functionalized alkynes by Normant and Bourgain. | |
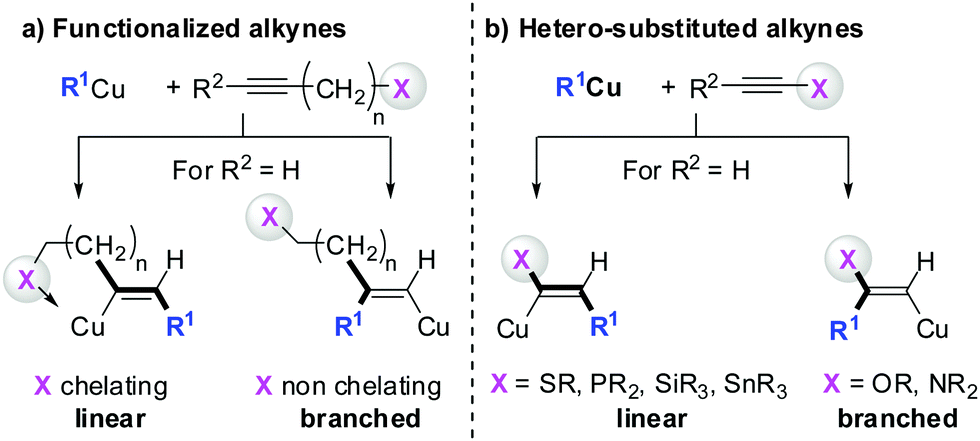
Three key features of this reaction are remarkable: (a) carbometalation is faster than deprotonation; (b) the excellent regio- and cis-stereoselectivity; and (c) synthetic usefulness of the in situ generated copper reagent 1 which can be trapped by a large variety of electrophiles including transmetalation reactions with various metals to catalyse subsequent cross-coupling reactions.8 Importantly many factors such as the presence of salts, nature of solvents, temperature as well as steric effects of the organometallic species and alkyne can greatly influence the outcome of the reaction. It is noteworthy that the most simple alkyne, acetylene, does not show deprotonation with Gilman cuprates but carbocupration reactions. The carbometalation of acetylene was intensively investigated and its usefulness culminated by the total synthesis of many natural products containing Z-alkenes. Comprehensive reviews by Alexakis, Normant and Lipshutz as well as a more recent book chapter by Chemla and Ferreira discuss in detail the diverse effects of carbocupration reactions with various types of alkynes.8–10 Yorimitsu also reviewed the transition-metal catalysed carbomagnesiation and carbozincation of alkynes.11 The directional effect of heteroatoms attached to the alkyne on the carbometalation reaction is particularly interesting and therefore has stimulated much research efforts in the last few decades. While dialkylsubstituted alkynes typically react inefficiently with organocopper reagents, mono- or disubstituted alkynes bearing heteroatoms or functional groups may react smoothly. Alkynes substituted with a directing (or alternatively blocking group) X in general afford the linear (or the branched product). Branched and linear refer to the hydrolysed products where R2 = H. Bold bonds were used to emphasize the linear or branched shape of the alkenes. Hetero-substituted alkynes lead to the linear or branched product depending on the nature of X. Both cases shown in Scheme 2 were covered by several excellent reviews.8–10,12–15
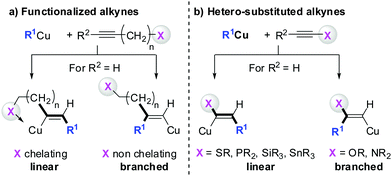 |
| | Scheme 2 Functional groups or heteroatoms direct carbocupration reactions. | |
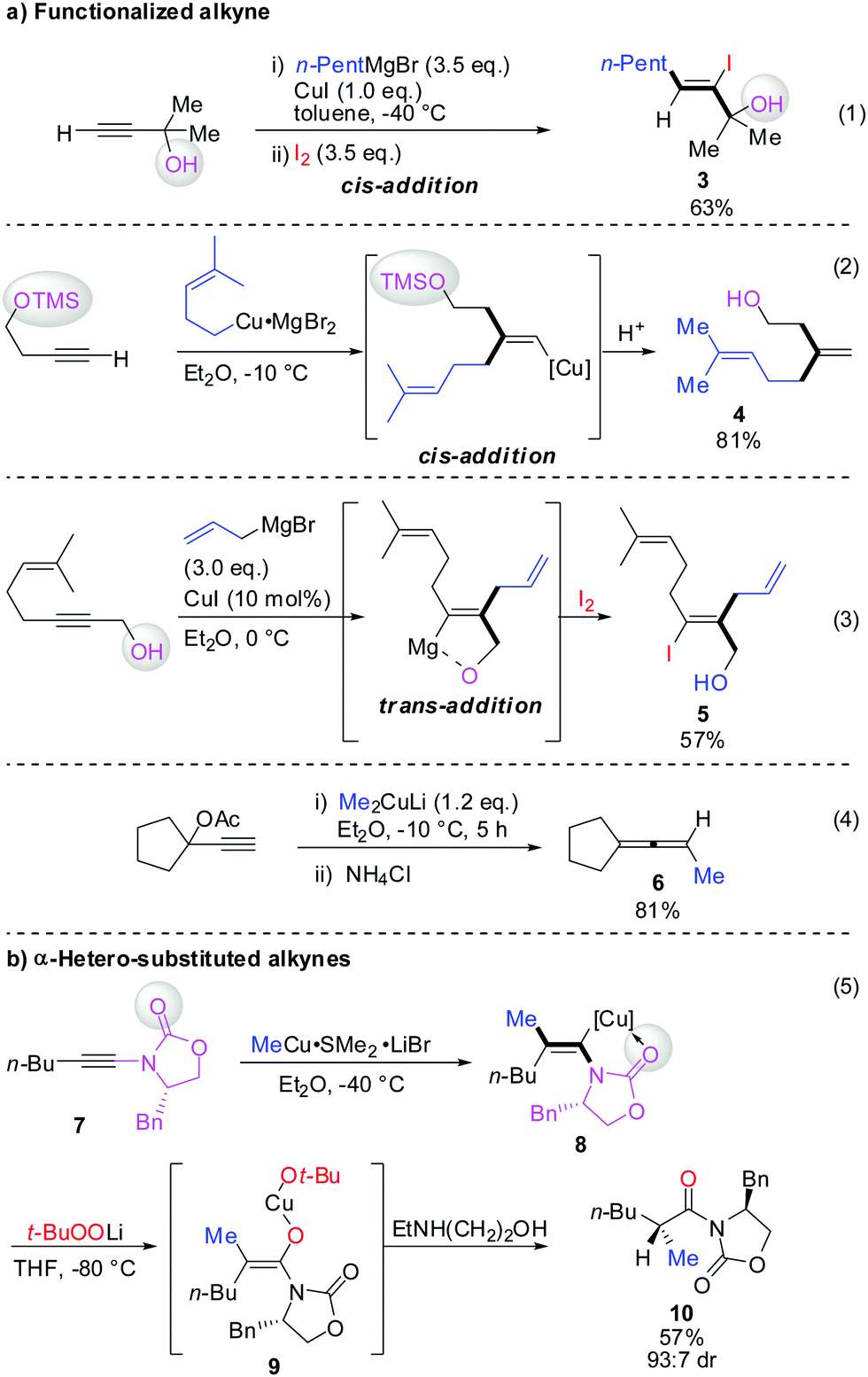
Scheme 3 illustrates several examples for the directing effect of functionalized alkynes (a) or α-hetero-substituted alkynes (b). The first example shows the effect of a chelating tertiary metal alcoholate to direct the copper into the α-position to afford the linear product 3 (eqn (1), Scheme 3).16 When the alcohol is slightly further away and protected by a TMS group, it acts as a blocking group providing the normal branched product 4 (eqn (2)).17 Eqn (3) shows the exceptional but highly important case of primary propargylic alcohol which undergoes unexpected trans allyl-addition to provide the branched product 5 (eqn (3)).18 However, when the propargylic alcohol is transformed into a leaving group (OLG, e.g. OAc) then propargylic displacement is usually obtained providing the corresponding allene 6 (eqn (4)).19 It is noteworthy that carbocupration reactions with methylcopper or cuprate reagents usually suffer from low efficiency.8 Negishi was able to solve this important problem by developing a well-known zirconium-catalysed carboalumination reaction.20 Eqn (5) shows that the directing effect of a functional group outweighs the effect of heteroatom substitution. The carbonyl group of oxazolidinone 7 directs the copper, in a low polar solvent, into the α-position and overwrites the tendency of the nitrogen substituent to direct the substituent into the α-position. The highly regioselective reaction affords chiral alkenyl copper reagent 8 as a reactive intermediate which was selectively oxidized with t-BuOOLi into the β,β′-stereodefined trisubstituted copper-enolate 9 as a single isomer. When treated with a proton source, imide 10 was obtained with high diastereoselectivity.21
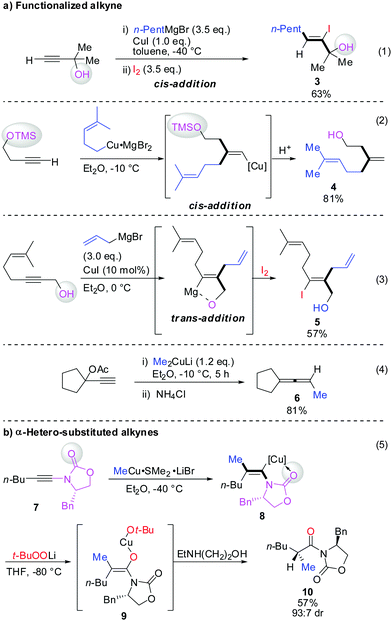 |
| | Scheme 3 Carbocupration with functionalized alkynes. | |
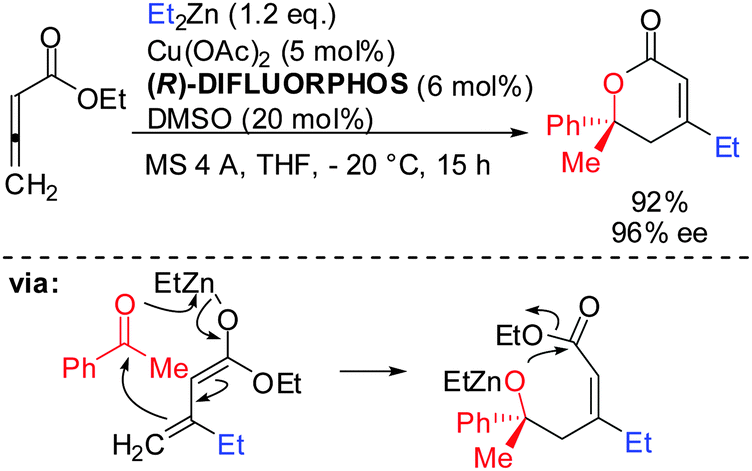
Allenes, which are less reactive than alkynes, can also undergo carbocupration reactions. The presence of three possible centers where carbometalation may proceed makes the regioselective carbocupration much more difficult. A recent enantioselective example with a strongly biased allene is shown in Scheme 4.22
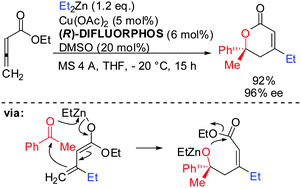 |
| | Scheme 4 Carbocupration of biased allene. | |
Mechanistic studies by Nakamura concerning the carbometalation of acetylenic carbonyl derivatives showed that in this case one cannot distinguish anymore between 1,4-addition reaction and carbometalation reaction because both reaction pathways proceed via the same intermediate.23 Based on the pioneering work of Iwai and Konotsune for the addition of organocopper reagents to acetylenic acids,24 several research groups investigated the E/Z selectivity of these reactions in more detail.25–27 It was shown that at low temperatures the cis-addition product could be obtained with very high selectivity (Scheme 5).27
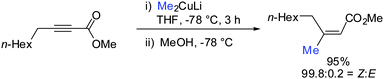 |
| | Scheme 5 Ambiguity: 1,4-addition or carbocupration. | |
The examples discussed above demonstrate how diverse carbocuprations with alkynes or allenes can be. The details matter and slight changes in the substrates, the presence of functional groups and the choice of nucleophile can greatly affect the reaction outcome. The subtle interplay of the reaction parameters can be exploited to access many diverse products from very similar starting materials.
3. Carbocupration reactions of cyclopropenes
3.1 Introduction
Before introducing the topic of copper mediated carbometalation of cyclopropenes it should be noted that several excellent reviews have been published concerning the synthesis and reactivity of cyclopropenes and alkylidenecyclopropanes.28–41 In particular recent reviews of Nakamura, Rubin and Gevorgyan, Fox, Marek cover the carbometalation reactions of cyclopropenes.6,42–46 Several reviews have appeared concerning the strain energy and geometry of cyclopropenes.47,48
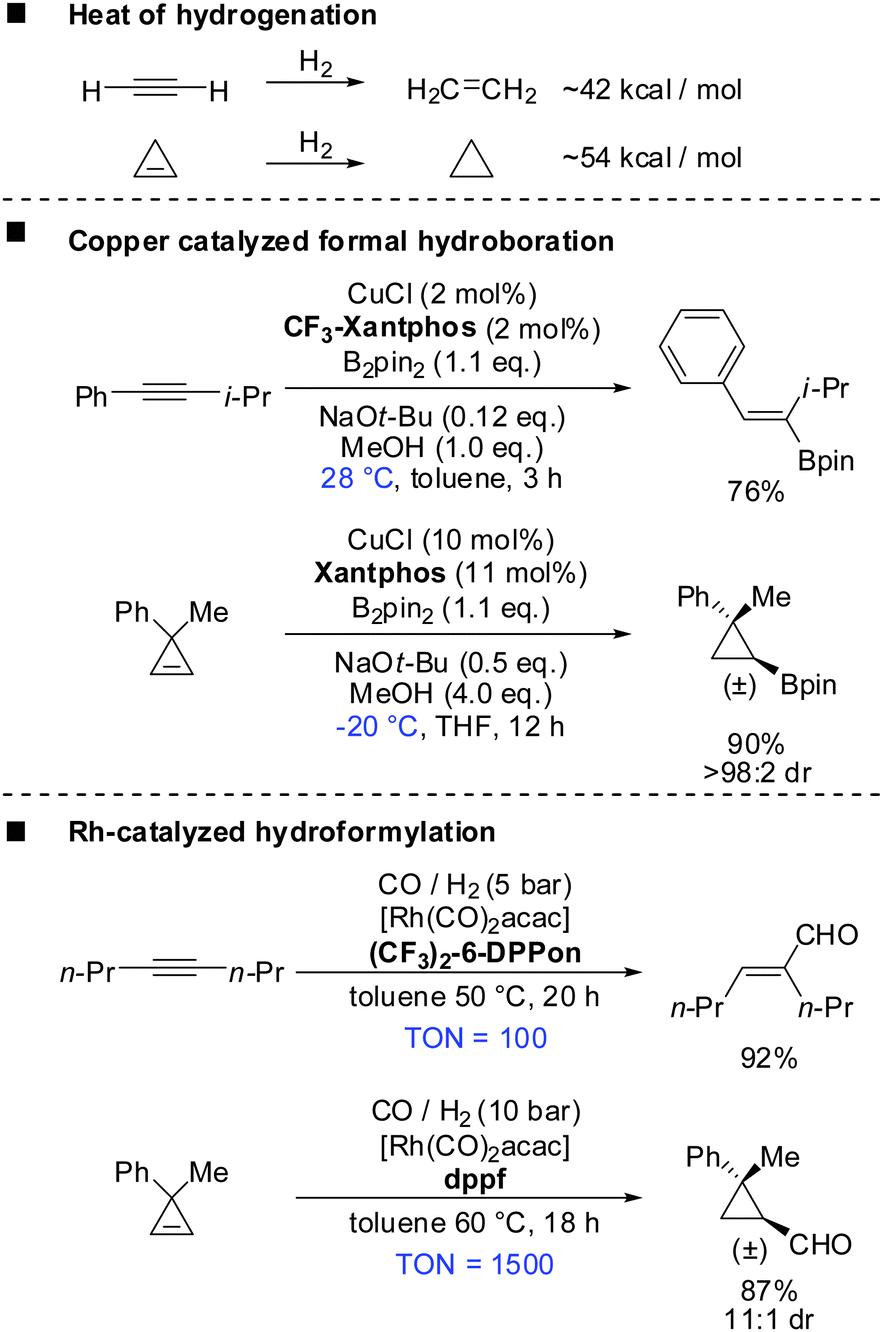
Cyclopropenes show similar reactivity compared to alkynes. For example the heat of hydrogenation for the conversion of cyclopropene to cyclopropane is ∼54 kcal mol−1 and is considerably larger than that for the conversion of acetylene to ethylene (∼42 kcal mol−1) (Scheme 6).49 The true nature of the strain in these compounds is very complex as theoretical studies by Wiberg, Borden, Kass and Bach have shown.50–54 However, this simplified comparison suggests the following two principles: (a) reactions which are possible with alkynes should also be applicable to cyclopropenes. (b) Reactions with cyclopropenes, if carried out under identical conditions, should require less harsh conditions compared to alkynes due to the release of additional 12 kcal mol−1 of strain. A closer look into the literature reveals that these two principles apply in many cases. Scheme 6 describes the copper catalysed formal hydroboration and the rhodium catalysed hydroformylation reaction which were carried out on alkynes and cyclopropenes under very similar conditions.55–58 Even though reaction conditions are not identical, both reactions suggest that the reaction with cyclopropenes can be carried out at lower temperatures, lower catalyst loading or reduced reaction times. In both cases the reaction with cyclopropenes creates a new stereogenic center. Tortosa and Rubin were able to find chiral copper or rhodium complexes which afforded the corresponding enantioenriched cyclopropanes.56,58
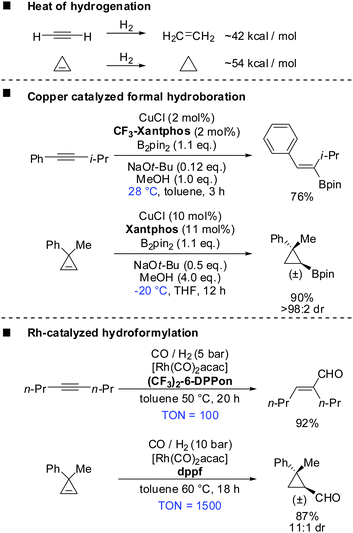 |
| | Scheme 6 Comparison: alkynes and cyclopropenes. | |
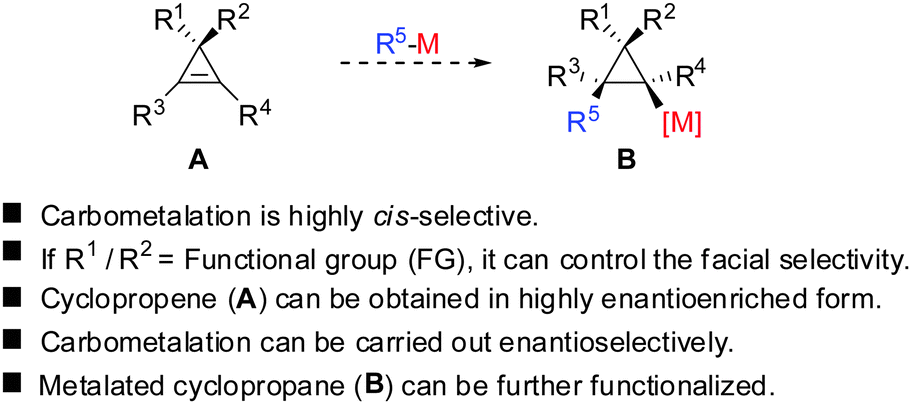
Given the similarity of cyclopropenes and alkynes, carbocupration of cyclopropenes should be a feasible reaction. Furthermore, in analogy to alkynes a functional group incorporated in the cyclopropenyl scaffold should be able to direct the carbometalation reaction selectively from one face. The obtained metalated cyclopropane would be a useful reagent, which could undergo many subsequent reactions: trapping with electrophiles, copper- and palladium catalysed carbon–carbon bond forming reactions, oxidation and many others (Scheme 7).
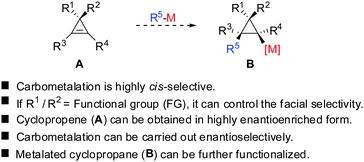 |
| | Scheme 7 Carbocupration of cyclopropenes. | |
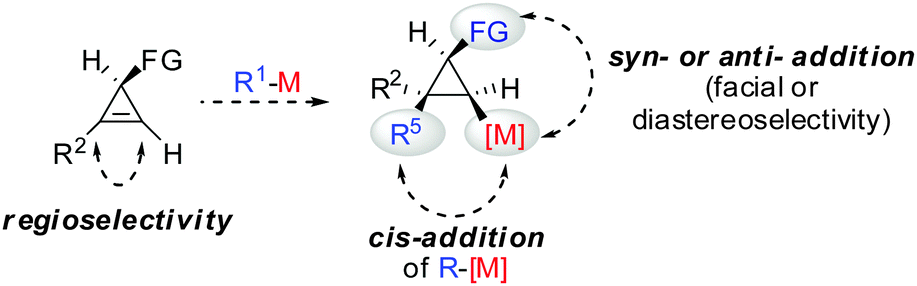
All these assumptions based on the analogy with alkynes have shown to be true and the herein described overview of copper mediated carbometalation of cyclopropenes will illustrate very well the similarity of cyclopropenes and alkynes. Nevertheless, it is important to stress that this comparison cannot always be applied. Cyclopropenes, due to their highly strained character, are more prone to decomposition and side reactions as compared to their alkyne counterparts, especially when exposed to strong Lewis acids. The following tutorial review first discusses the addition of stoichiometric amounts of organocopper reagents to cyclopropenes. Next, the copper catalysed carbomagnesiation and carbozincation reaction will be described. Finally, carbometalation reactions followed by elimination reactions, oxidation and cyclopropane ring opening will be presented. The review will not cover carbometalation which does not involve copper in stoichiometric or catalytic amounts. Copper mediated addition reactions with non-carbon nucleophiles are also not included in this comprehensive account. These types of addition reactions with cyclopropenes are reviewed elsewhere.28–41 It is important to mention that when describing syn- and anti-addition to cyclopropenes, we refer to the facial selectivity as regards to a functional group (or any group) present in the cyclopropenyl substrate. The words diastereo-selectivity and facial-selectivity also refer to the syn- or anti-selectivity. In all the described examples the addition of the metal–carbon bond across the cyclopropene double bond proceeds in an exclusive cis-fashion and will therefore not be mentioned anymore. The regioselectivity of the addition reactions is explained in Scheme 9 and refers to the way the carbon–metal bond is added across the double bond. All the different types of selectivities are depicted in Fig. 1.
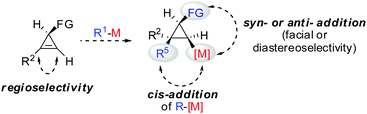 |
| | Fig. 1 Different types of selectivity. | |
3.2 Carbocupration reactions with organocuprate or organocopper reagents
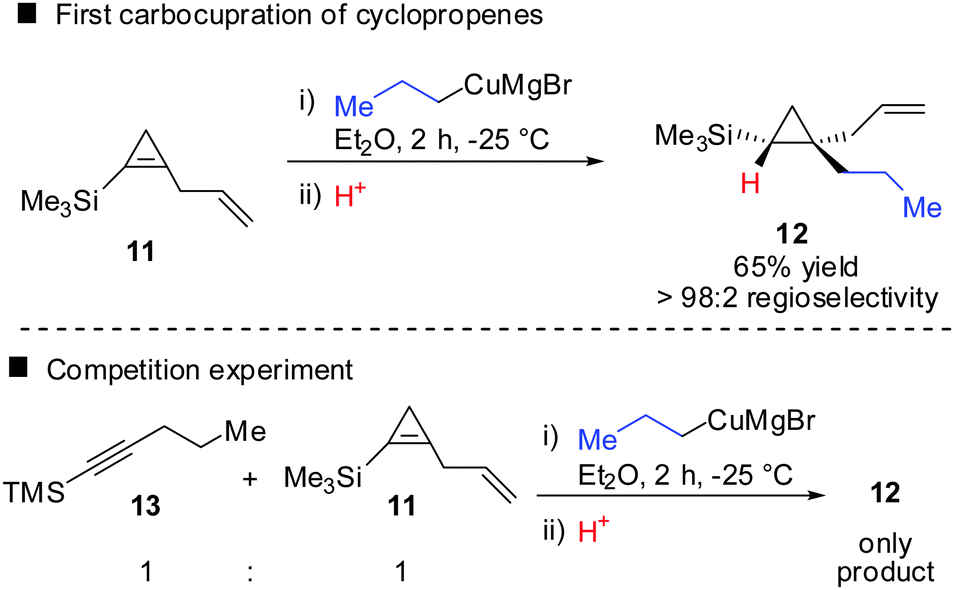
In 1985, Negishi reported the first carbocupration of cyclopropenes (Scheme 8).49 As previously discussed for the reactions displayed in Scheme 6, he noticed that carbometalation of cyclopropenes proceeds indeed faster compared to the corresponding alkynes. In a competition experiment of alkyne 13 and cyclopropene 11 only cyclopropane 12 and unreacted alkyne 13 were obtained. This implies that cyclopropene 11 reacted much faster compared to alkyne 13. The high regioselectivity can be explained by the α-effect of silicon, stabilizing the intermediate tertiary copper reagent α to the TMS-group.
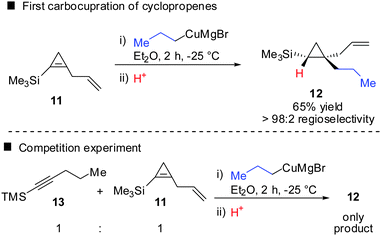 |
| | Scheme 8 First reported carbocupration reaction of cyclopropene by Negishi. | |
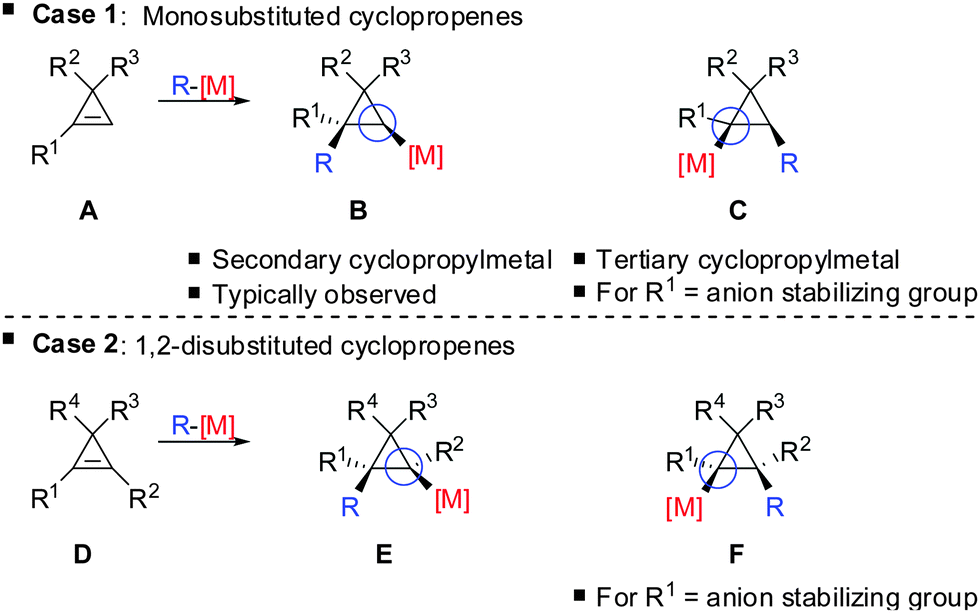
The preference for the most stabilized cyclopropyl metal reagent is a general rule that dictates the regioselectivity of the carbometalation reactions of cyclopropenes. Two cases have to be distinguished: (1) monosubstituted cyclopropene (A, Scheme 9) leads to product B exclusively due to the greater stability of the secondary compared to the tertiary organometallic reagent C. However, C may also be observed in some cases when R1 is an anion stabilizing group (e.g. phenyl as shown in Scheme 21). (2) For disubstituted cyclopropenes of type D mixtures of cyclopropyl metal compounds E and F are observed. However, if substituent R1 is an anion stabilizing group (e.g. TMS, Scheme 8) then product F becomes major or even an exclusive product of the reaction.
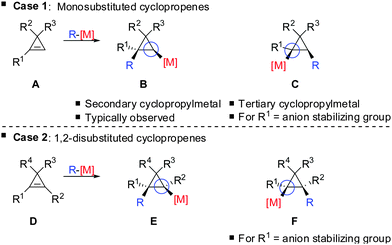 |
| | Scheme 9 Regioselectivity of carbocupration reactions. | |
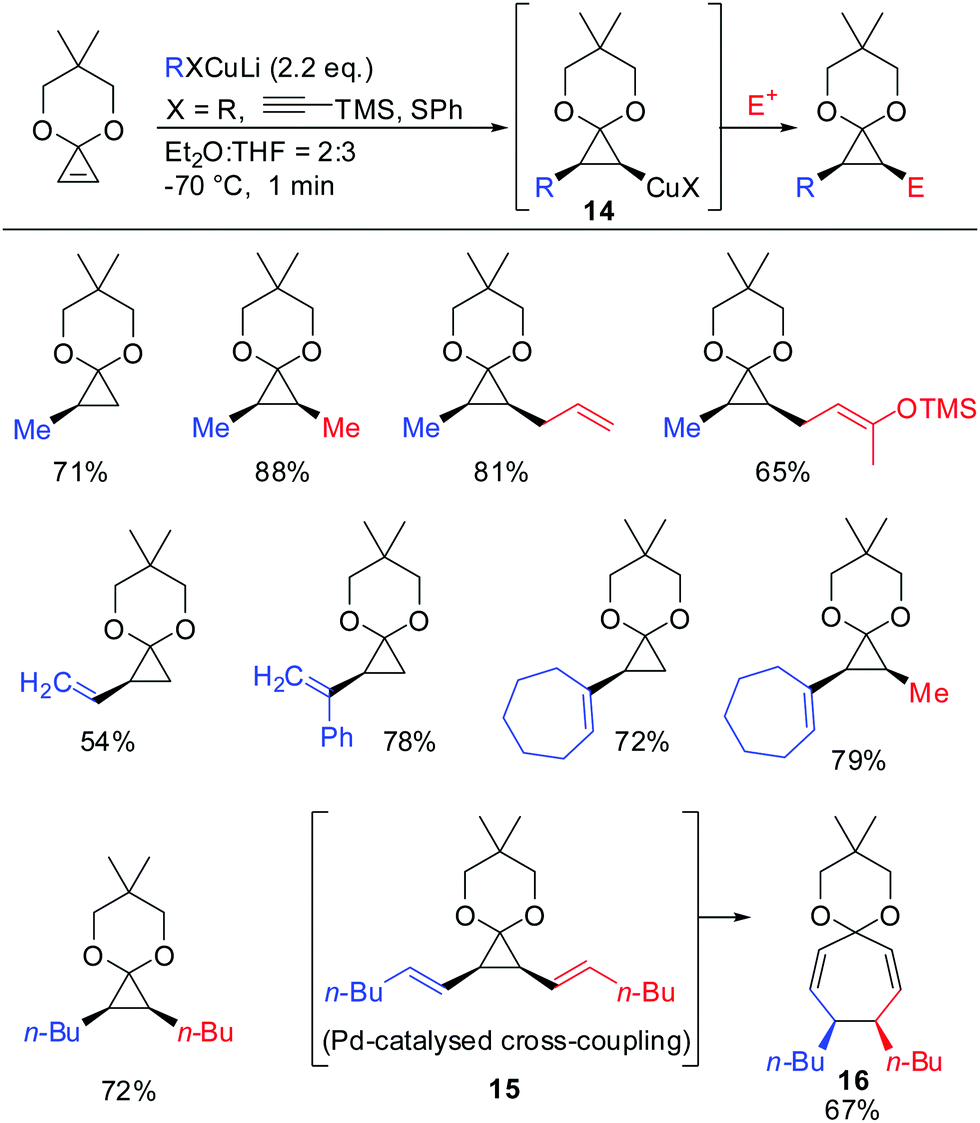
In 1988 Nakamura and co-workers examined for the first time the carbocupration of cyclopropene ketals.59 They found that cyclopropene ketals, when treated with an organocuprate or hetereocuprate, efficiently underwent a carbocupration reaction to afford cyclopropyl copper species 14 which was reported to be stable at temperature lower than −50 °C. Subsequent reactions with electrophiles, or palladium catalysed cross-coupling reactions gave the corresponding cyclopropane products in good yields. Palladium catalysed cross coupling reaction with (E)-1-iodohex-1-ene afforded 15, which immediately underwent divinylcyclopropane–cycloheptadiene rearrangement to afford cycloheptadienone ketal 16 in 67% yield. In general, the cyclopropyl anion is configurationally stable unless the anion is substituted by an electron withdrawing group (Scheme 10).
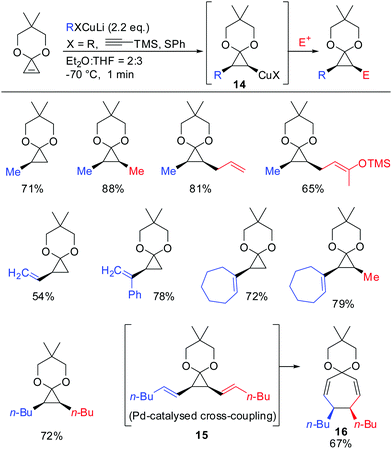 |
| | Scheme 10 Carbocuprations with cyclopropene ketals. | |
Two years later, in 1990, Nakamura reported the stereo- and regioselective carbocupration of cyclopropenes (Scheme 11).60 Protection of the cyclopropenone with a chiral ketal moiety transmitted successfully the stereoinformation during the carbocupration reaction. Interestingly, 1-substituted cyclopropene afforded the opposite diastereomer compared to the unsubstituted cyclopropene. In the case of monosubstituted cyclopropenes the reaction is also highly regio- and stereoselective: the more stable secondary cyclopropyl reagent is obtained exclusively (see Scheme 9 for explanation). The extension of the nucleophile scope to vinylic cyanocuprates and Normant cuprates (R2CuMgBr) is noteworthy.
 |
| | Scheme 11 Carbocuprations with enantioenriched cyclopropene ketals. | |
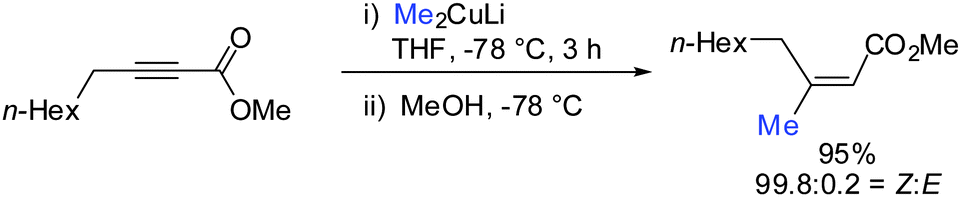
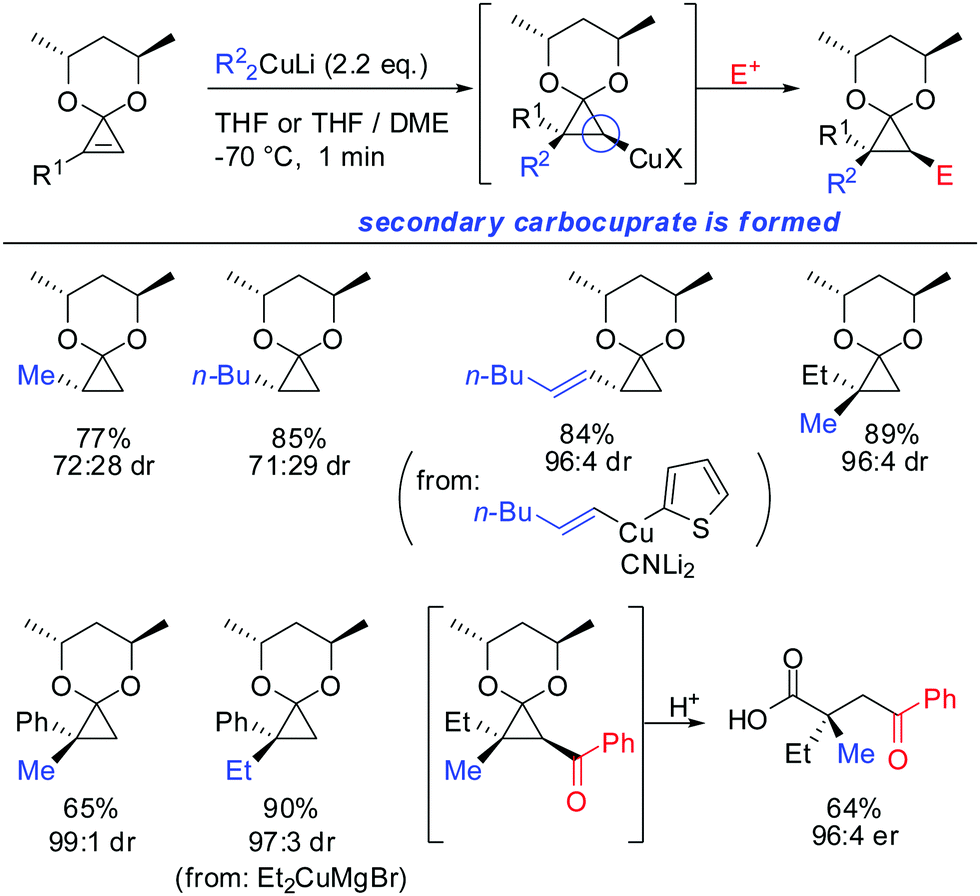
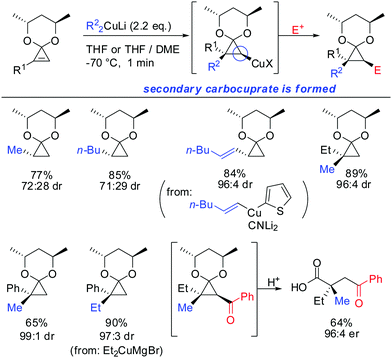
In 2007, we reported that 1-hydroxymethyl substituted cyclopropenes have the ability of direct addition of Gilman type cuprates (R2CuLi) with high facial control.61 The high selectivity is rather surprising as the Gilman cuprates (Scheme 12) provide the opposite diastereoisomer than the Normant type cuprates (R2MgX), formed from the Grignard reagent in the presence of catalytic amounts of copper, (Scheme 23).61 Although the origin of this diastereodivergency is still unknown, it is interesting to note that both diastereoisomers can be obtained at will and in a good ratio by simply changing the nature of the organometallic species involved in the carbometalation reaction.
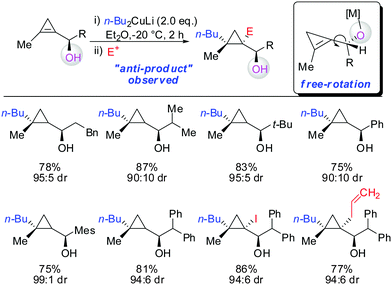 |
| | Scheme 12 Cyclopropanes obtained by addition of Gilman type cuprates. | |
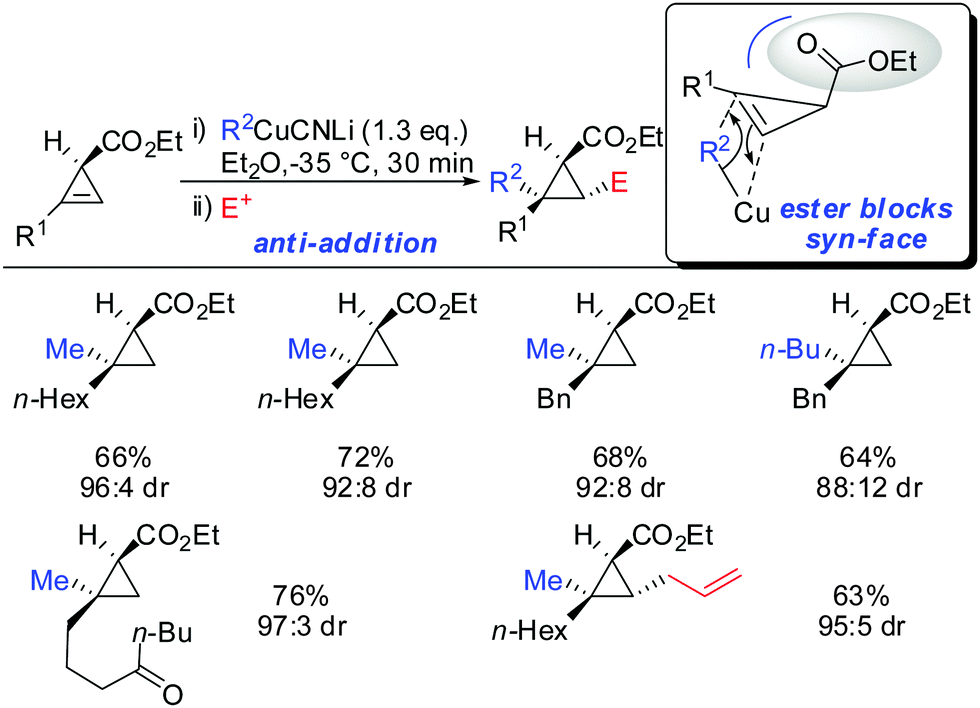
When treating cyclopropene esters with lower order cyanocuprates R2CuCNLi an unusual anti-addition was observed (Scheme 13).62 It was hypothesized that chelation does not take place, probably due to the low availability of free lithium cations in lower order cyanocuprates R2CuCNLi. Hence, no efficient chelation takes place and the ester group instead of directing the cuprate to the syn-face of the cyclopropene blocks this face and the cuprate adds from the less-hindered face, anti to the ester group.
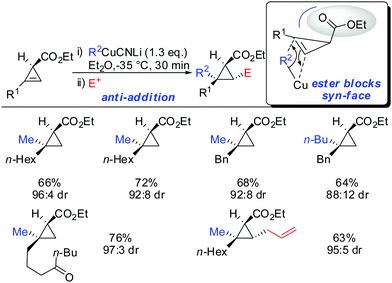 |
| | Scheme 13
anti-Addition with R2CuCNLi. | |
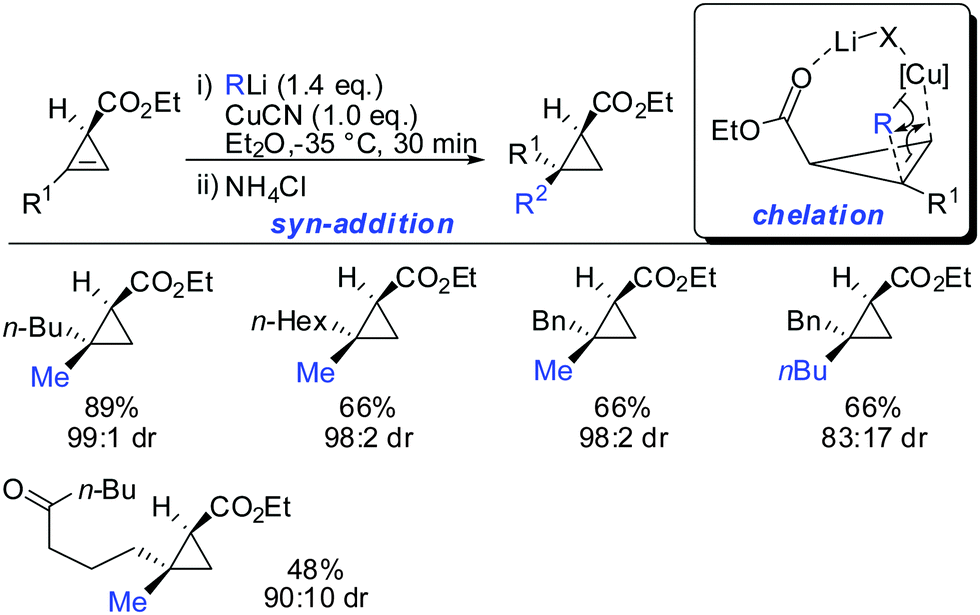
In this special case the reaction was very sensitive to stoichiometry. The occurrence of small amounts of free alkyllithium in the presence of RCuCNLi was able to reverse the facial selectivity back to the expected syn-selectivity which is probably due to the better availability of lithium ions for chelation (Scheme 14).63
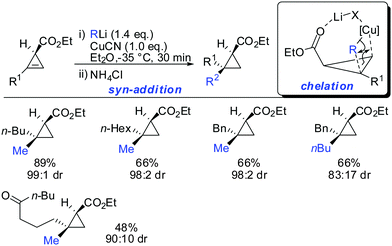 |
| | Scheme 14
syn-Addition when excess of RLi was used. | |
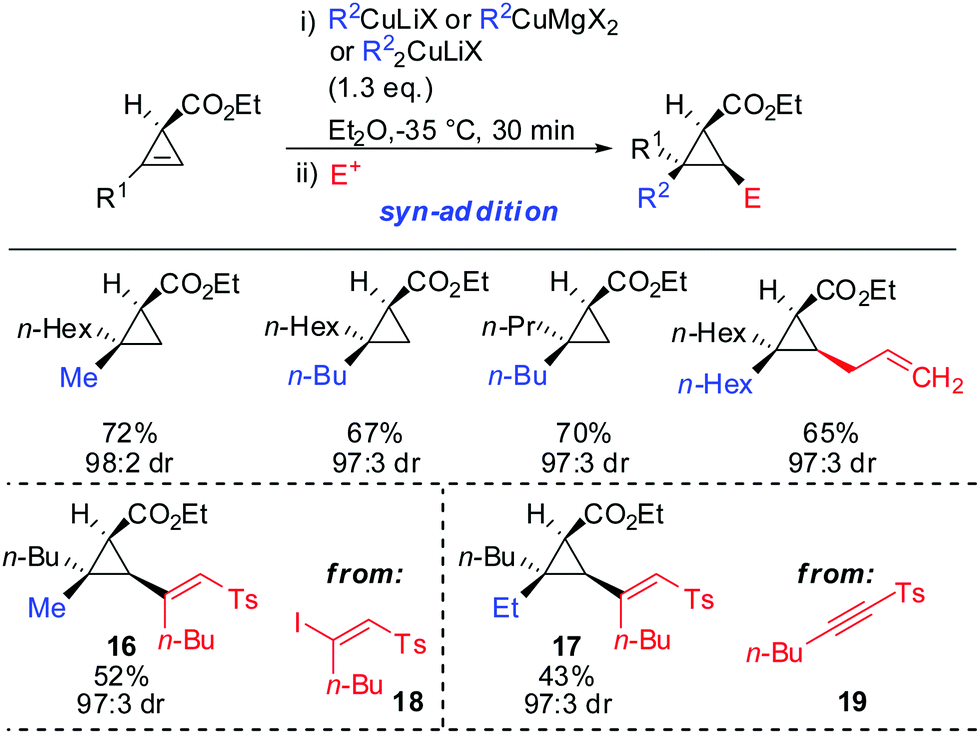
syn-Selectivity was also observed for the addition of organocopper reagents (RCuLiX or RCuMgX2) and classical Gilman cuprate (Me2CuLi). The depicted vinyl sulfones 16 and 17 were obtained from the corresponding alkenyl iodide 18via an addition–elimination sequence or from the alkyne 19via addition-hydrolysis (Scheme 15).63
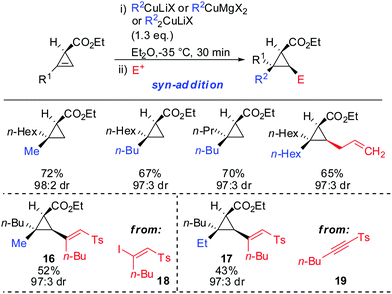 |
| | Scheme 15
syn-Addition with copper reagents and Gilman cuprates. | |
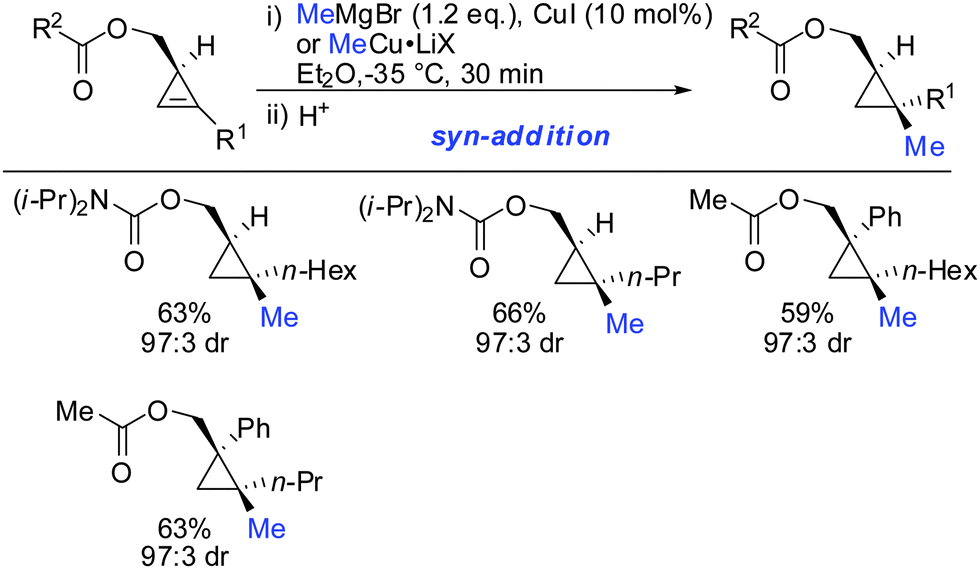
Furthermore, the substrate scope was extended to homologous esters and amides (Scheme 16). Despite the greater flexibility of the directing group, very high diastereoselectivity was observed with organocopper reagents. The copper-catalysed carbomagnesiation worked equally well.
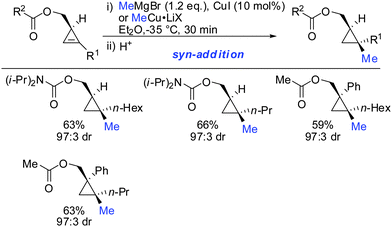 |
| | Scheme 16
syn-Addition with ester- and amide-homologues. | |
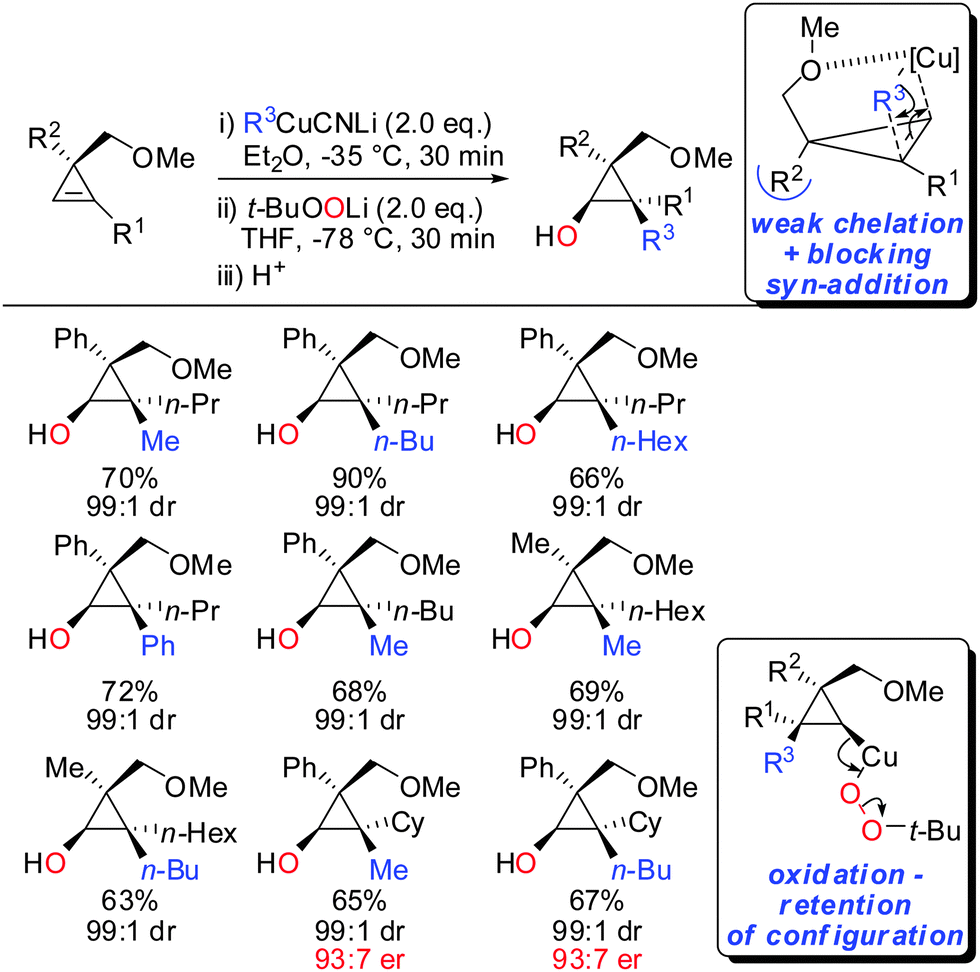
In 2015, it was shown that lower cyano cuprates can also undergo syn-addition with cyclopropenes bearing a weak ether-coordinating group in the 3-position (Scheme 17).64 The dramatic influence of an additional substituent at the 3-position on the facial selectivity was also reported by Fox in the context of iron-catalysed carbomagnesiation of cyclopropenes.65 This reversal of selectivity compared to the previous reaction depicted in Scheme 13 could be rationalized by the additional substituent at the 3-position of the cyclopropene. This additional substituent exerts a strong steric effect and now the weak coordination of the ether group is sufficient enough to direct the incoming organometallic reagent. The cyclopropyl copper intermediate was in situ oxidized with retention of configuration with t-BuOOLi. It should be noted that oxygen cannot be used for this type of oxidation because organocopper reagents dimerize when exposed to oxygen.8t-BuOOLi was found by Whitesides and Boche to be an excellent oxidizing reagent for organolithium and copper reagents.66,67 After aqueous work-up highly-substituted cyclopropanols were obtained in good yields and excellent diastereoselectivities. The starting material is easily obtained in enantioenriched form by enantioselective cyclopropenation of alkynes with methyl aryldiazoacetates in the presence of a chiral rhodium catalyst.68 Two examples were prepared in their enantioenriched form (Scheme 17).
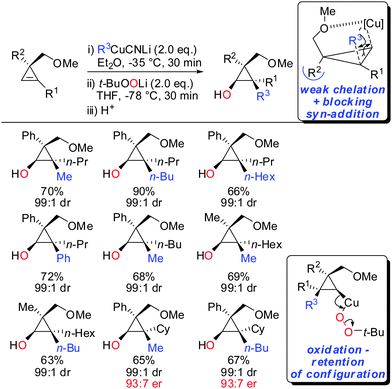 |
| | Scheme 17 Carbocupration–oxidation sequence. | |
3.3 Copper catalysed carbomagnesiation
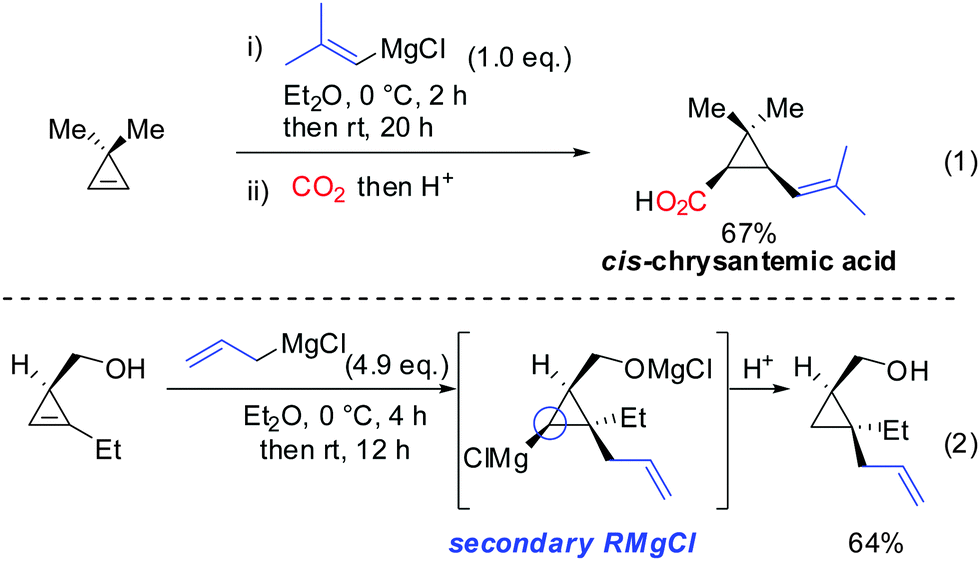
Interestingly, early studies by Lukina, Rudavshevskaya and Nesmeyanova showed that cyclopropenes react with alkyl Grignard reagents to give mainly the carbometalated product.69 Alkenyl- and allylmagnesium bromides also react cleanly with cyclopropenes to afford the desired products in moderate to good yields.70 Lehmkuhl utilized the potential of this chemistry for a one-pot synthesis of cis-chrysanthemic acid (eqn (1), Scheme 18).70 The depicted example illustrates the increased reactivity of cyclopropenes compared to disubstituted alkynes which under similar conditions do not undergo carbomagnesiation with Grignard reagents.8 Richey found that, in analogy to propargylic alcohols, the alcohol function of 3-(hydroxymethyl)cyclopropenes was able to direct the carbomagnesiation of allyl Grignard reagents with high diastereoselectivity (eqn (2), Scheme 18).71 The new carbon substituent adds to the more substituted site of the cyclopropene to avoid the generation of the less-stabilized tertiary organomagnesium reagent. Therefore, the overall process is completely regio- and stereoselective.
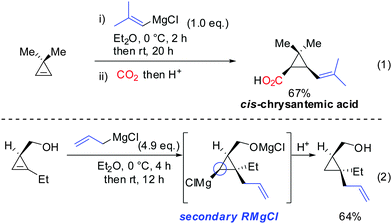 |
| | Scheme 18 Non-catalysed additions of Grignard reagents to cyclopropenes. Eqn (2): Stereochemistry and Mg-alcoholate. | |
Other Grignard reagents employed by Richey such as alkyl-, benzyl- and aryl magnesium halides failed to react.71 With the desire to expand the scope of potential nucleophiles, Fox in 2002 reported the replacement of the hydroxyl group by a methoxymethyl acetal (MOM)-group which is well known for its strong chelating properties. After the optimization of the reaction conditions he found that the replacement of ethereal solvents by pentane afforded the addition of copper catalysed alkyl- and vinyl-magnesium halide with high diastereoselectivity and good yield (Scheme 19).72
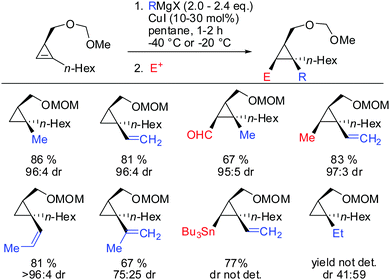 |
| | Scheme 19 Copper-catalysed carbomagnesiation of cyclopropenyl methyl methoxymethyl acetal. | |
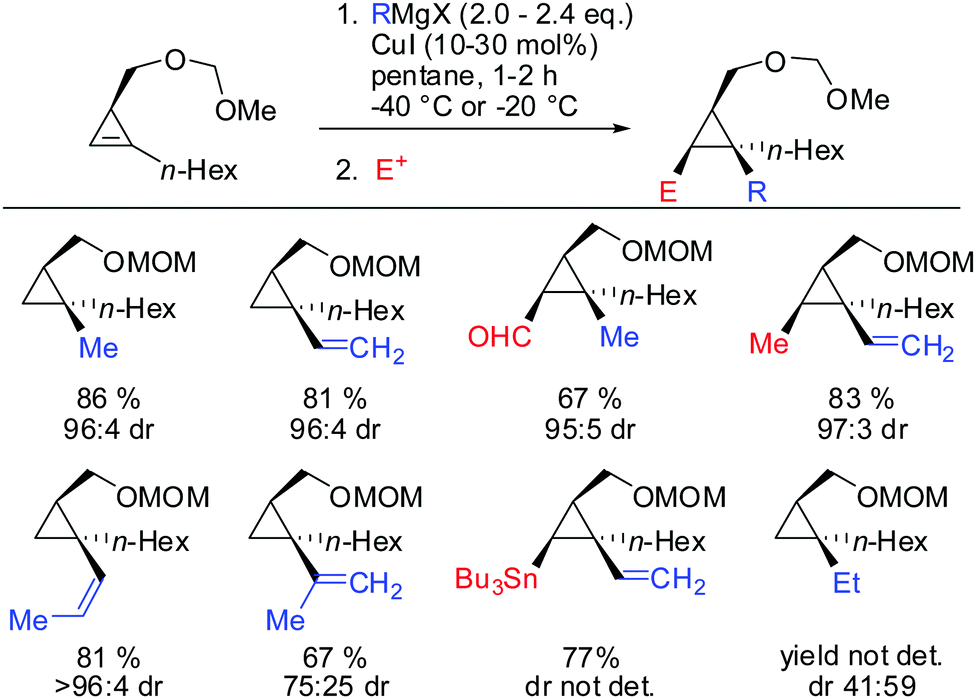
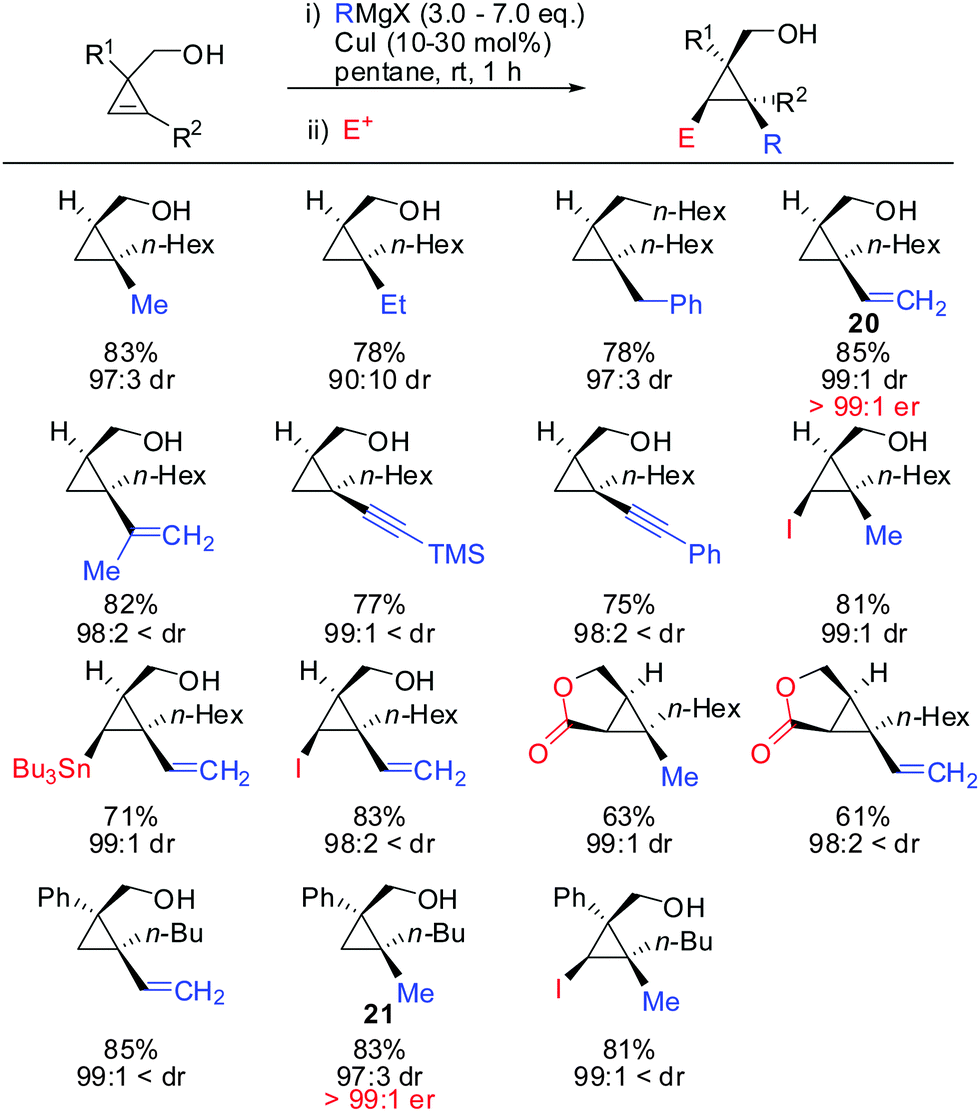
Under these conditions, many nucleophiles could be added to 3-(hydroxymethyl)cyclopropene. Previously unsuccessful alkyl-, alkenyl- and even alkynyl Grignard reagents underwent carbometalation in good yields and excellent facial selectivity (Scheme 20).72 Fox and co-workers also developed a cost-efficient synthesis method providing enantiomerically pure cyclopropene substrates on gram scale.73 After the copper-catalysed carbomagnesiation reaction the corresponding cyclopropanes were obtained with high facial selectivity and complete preservation of enantiomeric purity. Two examples were provided in their study (20 and 21, Scheme 20).
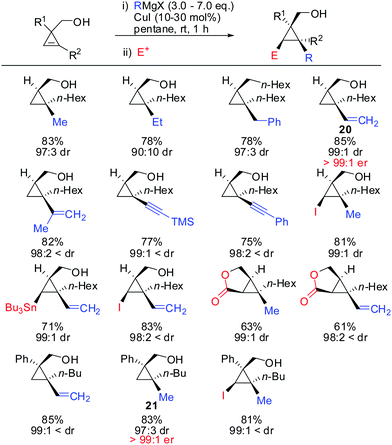 |
| | Scheme 20 High diastereoselectivities with CH2OH directing group in 3-position of cyclopropene. | |
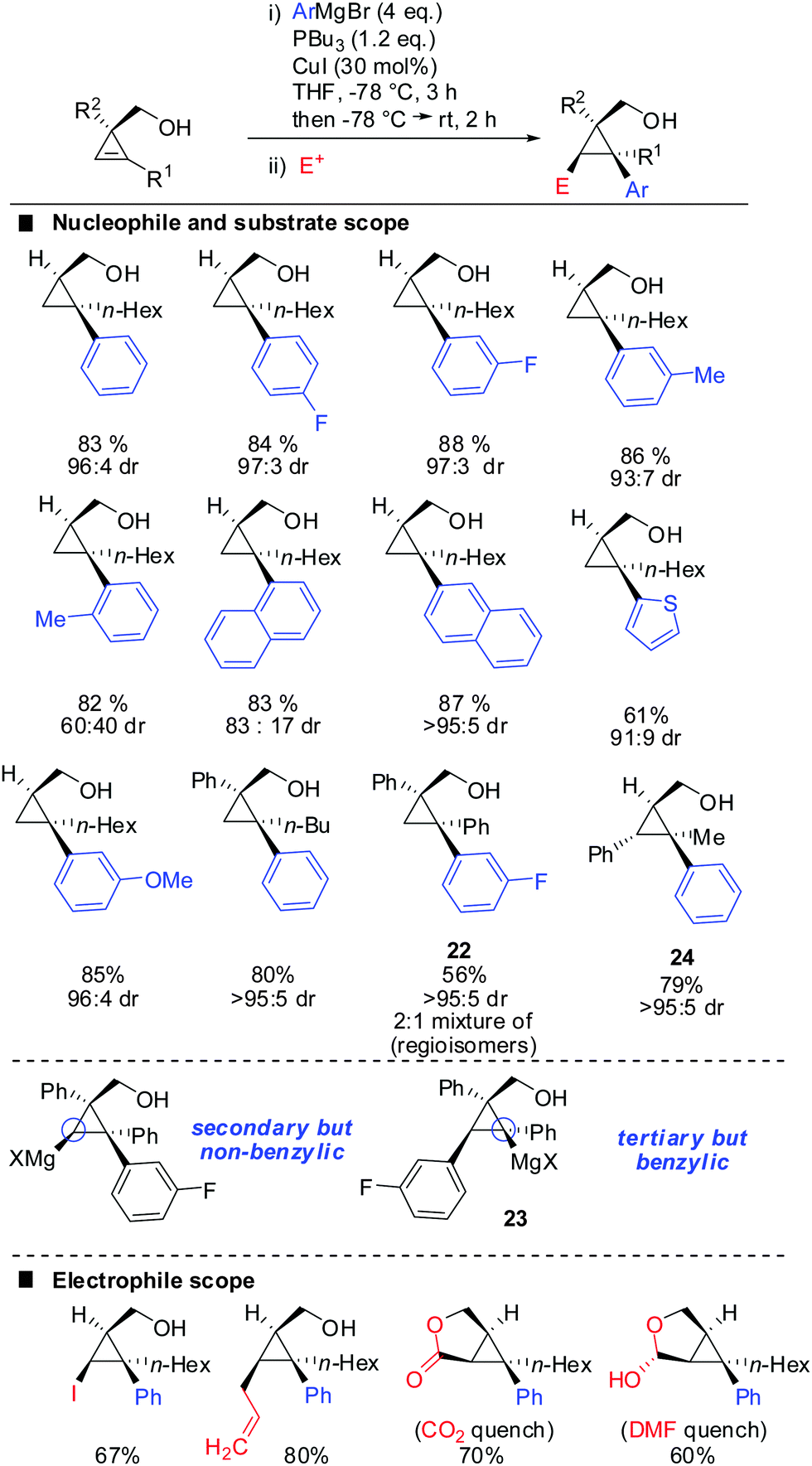
One of the last challenging issues that remained was the introduction of an aryl nucleophile. Standard conditions shown in Scheme 20 afforded less than 1% of product when PhMgBr was used as a nucleophile. After much experimentation Fox and co-workers found that super stoichiometric amounts of electron rich phosphine ligand P(n-Bu)3 allowed for the efficient copper-catalysed carbomagnesiation with aryl Grignard reagents (Scheme 21).74 High syn-selectivity was observed in all cases, especially for 3,3′-disubstituted cyclopropenes providing the corresponding products with excellent selectivity. The facial selectivity only dropped when sterically encumbered aryl-nucleophiles were used. Reaction with ortho-tolylmagnesium bromide gave a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 mixture of syn- and anti-diastereomers, whereas phenylmagnesium bromide and meta-tolylmagnesium chloride provided the syn-diastereomer with high selectivity. As explained in Scheme 9 the regioselectivity is determined by the stability of the intermediate cyclopropylmetal species. In general, the organic substituent is added to the more substituted carbon. Only in the case of phenyl-substituted cyclopropenes the regioselectivity may be significantly lower. Following the guidelines provided in Scheme 9 one can easily rationalize the eroded regioselectivity for product 22 as 23 benefits from benzylic stabilization. The low selectivity suggests that a tertiary benzylic carbanion is almost as stable as a secondary carbanion. For the formation of product 24 the situation is rather simple as both regioisomers of addition are tertiary cyclopropylmagnesium products but only one of them benefits from benzylic stabilization. Hence, only product 24 is formed.
:40 mixture of syn- and anti-diastereomers, whereas phenylmagnesium bromide and meta-tolylmagnesium chloride provided the syn-diastereomer with high selectivity. As explained in Scheme 9 the regioselectivity is determined by the stability of the intermediate cyclopropylmetal species. In general, the organic substituent is added to the more substituted carbon. Only in the case of phenyl-substituted cyclopropenes the regioselectivity may be significantly lower. Following the guidelines provided in Scheme 9 one can easily rationalize the eroded regioselectivity for product 22 as 23 benefits from benzylic stabilization. The low selectivity suggests that a tertiary benzylic carbanion is almost as stable as a secondary carbanion. For the formation of product 24 the situation is rather simple as both regioisomers of addition are tertiary cyclopropylmagnesium products but only one of them benefits from benzylic stabilization. Hence, only product 24 is formed.
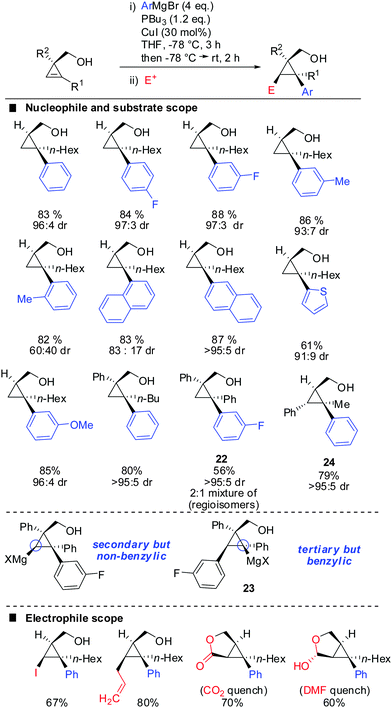 |
| | Scheme 21 Extension of scope: aryl nucleophiles. | |
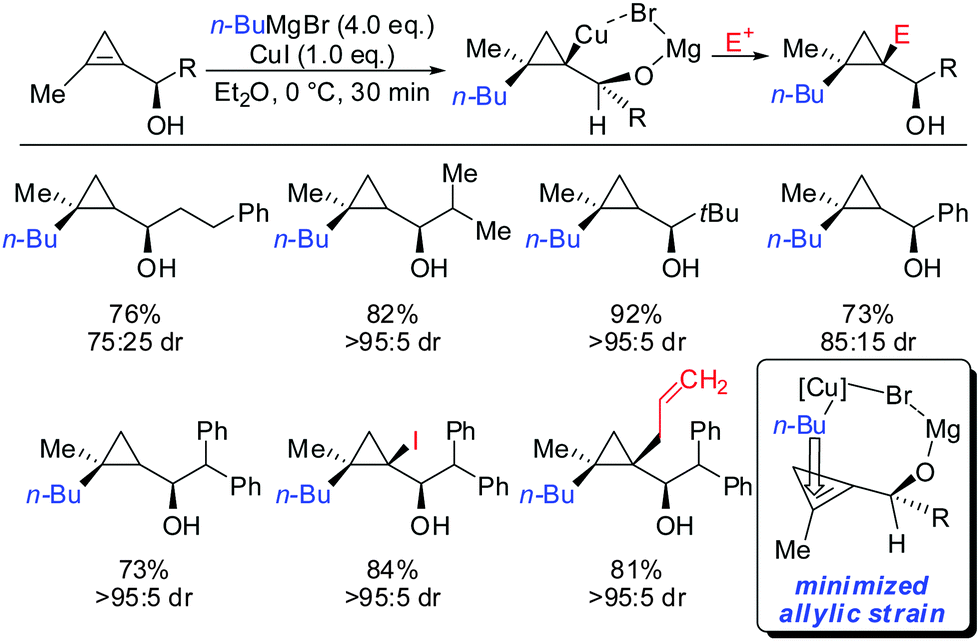
As already described in Scheme 12, a secondary alcohol in the 1-position of the cyclopropene can lead to facial differentiation. The organocuprate reagent generated from n-BuMgBr and copper iodide (Normant cuprates) with high selectivity was added (Scheme 22).61 Only when the secondary alcohol was substituted with alkyl and aryl-substituents the diastereoselectivity was moderate. The facial selectivity can be rationalized by considering the most stable conformer (minimization of allylic strain).
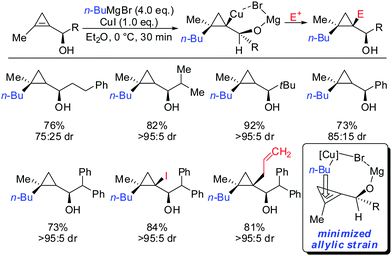 |
| | Scheme 22 High diastereoselectivities with allylic secondary alcohol functionality as directing group. | |
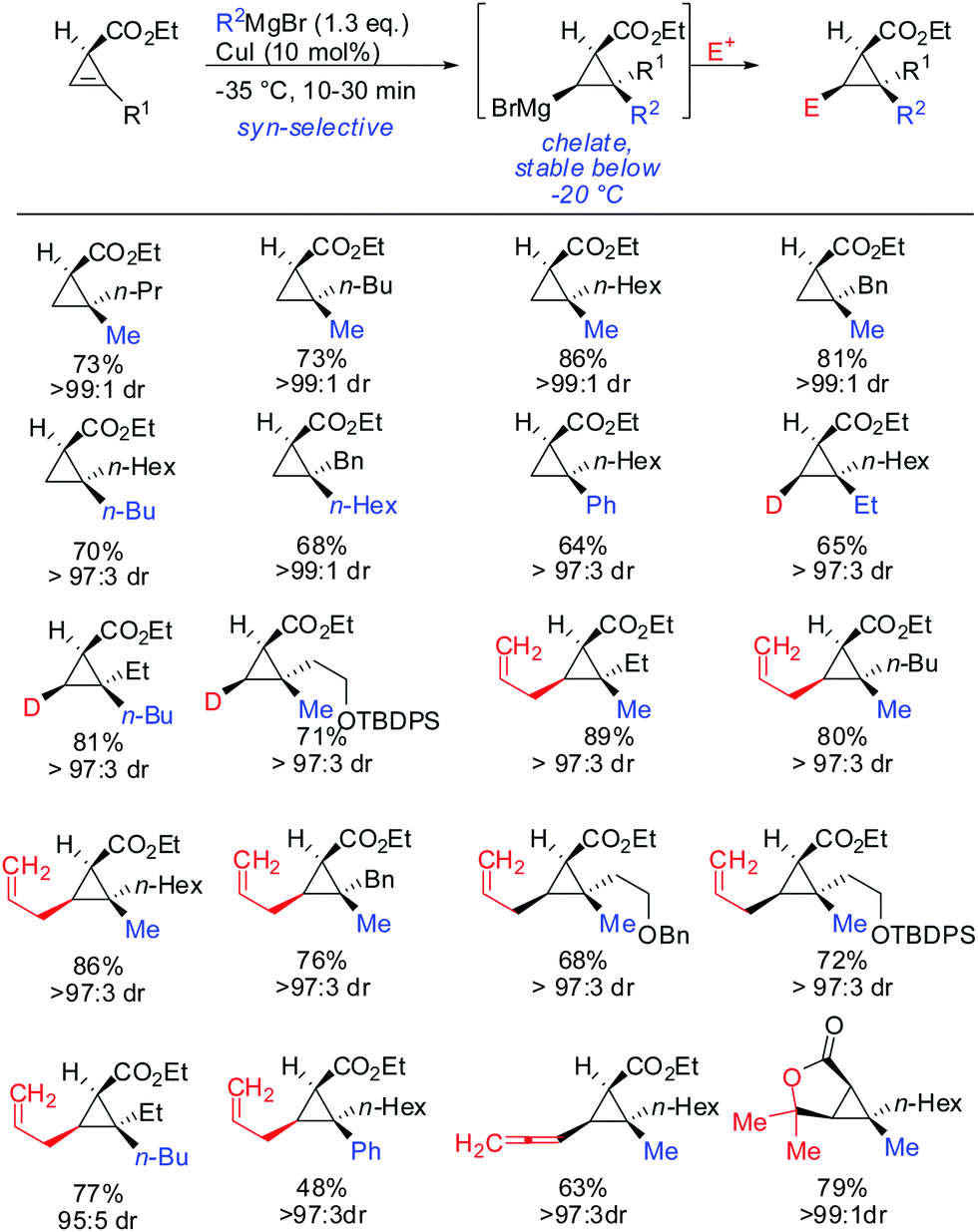
The copper-catalysed carbomagnesiation of cyclopropenyl esters was also investigated. Methyl magnesium bromide in the presence of catalytic amounts of copper iodide gave the desired products in excellent syn-selectivity (Scheme 23).62 Interestingly, even in a more coordinating solvent such as THF, the syn-chelated adducts are formed with the same diastereomeric ratios. Different copper salts such as CuBr and CuCN could be used with equal efficiency. The ester moiety was not attacked by the Grignard reagents at low temperatures. In contrast, lower order cyanocuprates RCuCNLi afforded the anti-product with high selectivity (>90:10; Scheme 13).62 It is important to note that the Cu-catalysed carbomagnesiation of cyclopropenyl ester has to be carried out at temperature below −20 °C. If the reaction is warmed up to 0 °C, a stereospecific fragmentation reaction occurs (Scheme 36).63
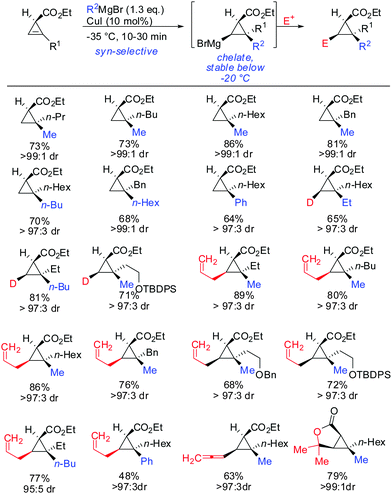 |
| | Scheme 23
syn-Selective copper-catalysed carbomagnesiation reactions of cyclopropene esters. | |
In 2015, Endo showed that catalytic amounts of copper salt also allowed high yielding reactions with unfunctionalized cyclopropenes (Scheme 24).75 A great number of copper catalysed carbomagnesiation reactions of aryl- and alkyl-3,3′-disubstituted cyclopropenes were reported. The copper-catalysed carbomagnesiation with various Grignard reagents resulted in high yields of the corresponding cyclopropanes. Only the addition of encumbered Grignard reagents such as t-BuMgBr and isopropenyl Grignard reagent did not afford the desired products. Unsymmetrical cyclopropenes resulted in a mixture of unassigned diastereomers 25 and 26.
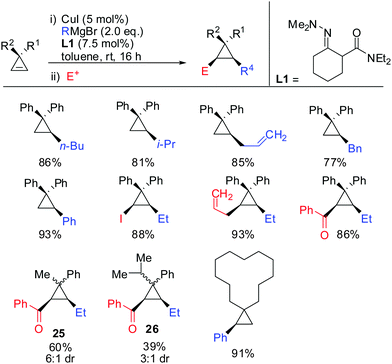 |
| | Scheme 24 Copper-catalysed carbomagnesiation reaction of unfunctionalized cyclopropenes. | |
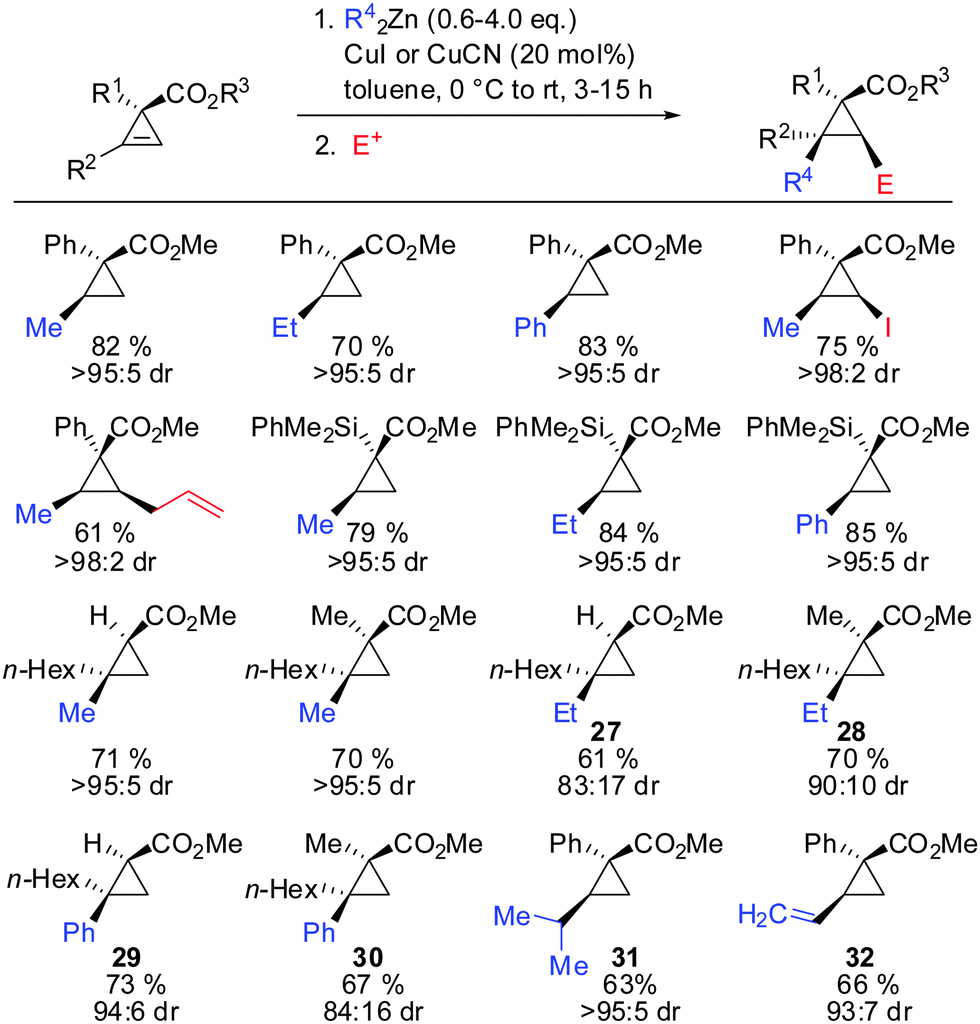
3.4 Copper catalysed carbozincation
In 2009, Fox showed that the addition of dialkyl- and diaryl-zinc reagents to cyclopropenyl esters was more efficient in the presence of catalytic amounts of copper. The use of toluene as a solvent was crucial to afford high facial selectivity (Scheme 25).76 All products were obtained in good to excellent diastereoselectivity. Fox and co-workers were also able to generate non-commercial diorganozinc reagents from Grignard reagents and ZnCl2 which successfully afforded the corresponding products 31 and 32. If cyclopropane is substituted with an alkyl substituent at the 3-position (R1 ≠ H) improved diastereoselectivity was found (compare diastereoselectivity of 27 and 28). Surprisingly, a decrease of diastereoselectivity was found for the addition of Ph2Zn leading to compound 29 when substituted at the 3-position (30).
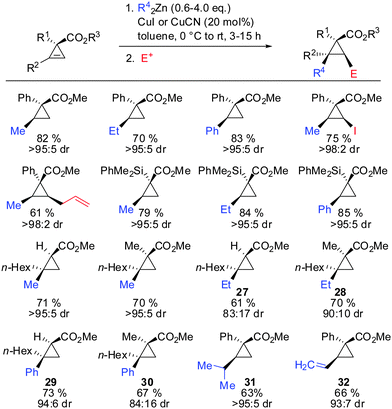 |
| | Scheme 25 First copper-catalysed carbozincation reaction of cyclopropenes. | |
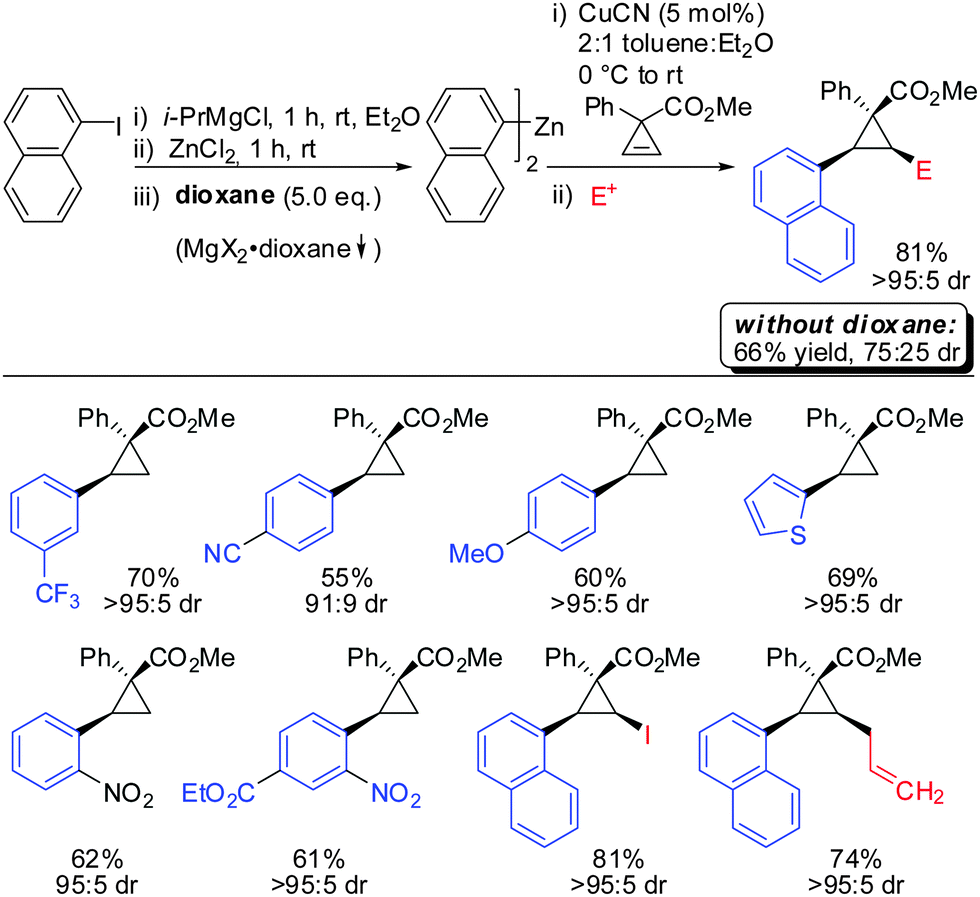
In 2012, Fox reported an extension of his previous work. Several diarylzinc reagents were prepared in situ from the corresponding aryl iodides by a iodine–magnesium–zinc exchange (Scheme 26).77 An efficient protocol was developed which includes the removal of magnesium salts by dioxane, which was shown to be crucial for the diastereoselectivity of the reaction. Importantly, a wide range of functionality (nitrile, ester, nitro, alkoxy, CF3) was tolerated. When nitro groups were present, the iodine–magnesium exchange had to be done with PhMgBr instead of the “turbo Grignard (i-PrMgBr)”.78
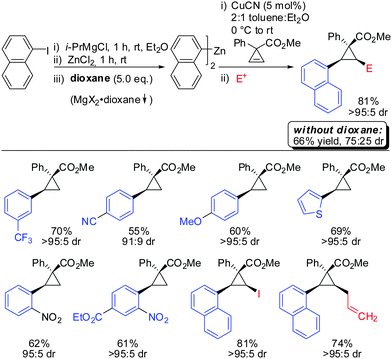 |
| | Scheme 26 Expanding the nucleophile scope: in situ generated diarylzinc reagents. | |
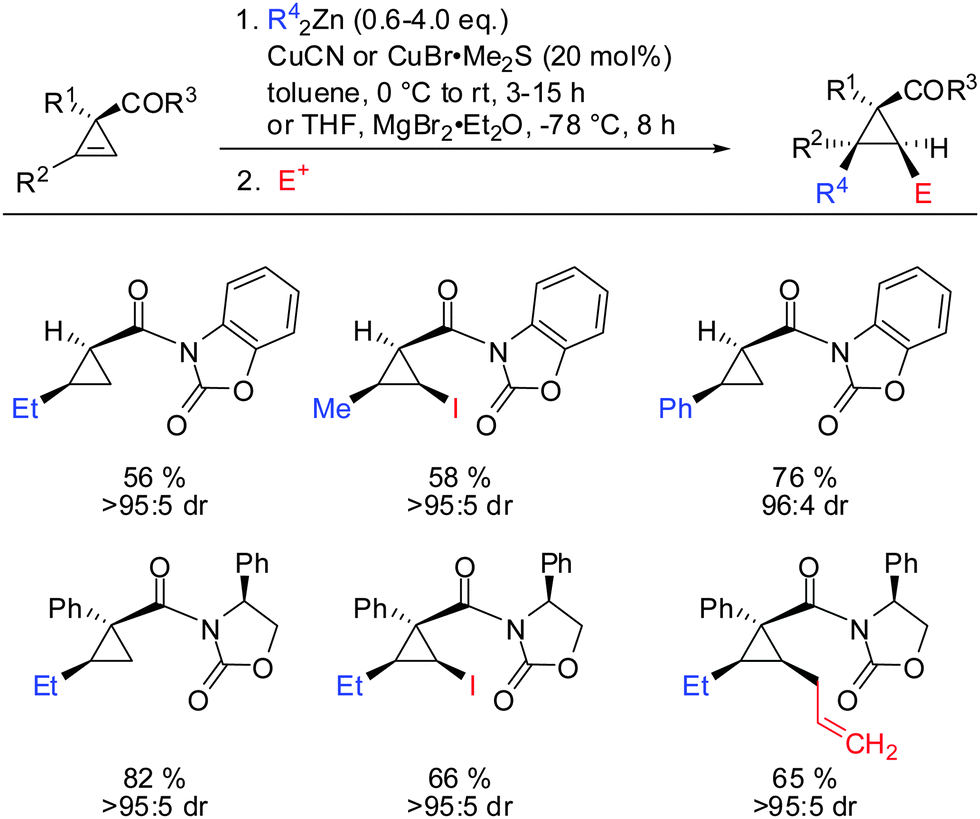
Related cyclopropenes such as chiral and non-chiral acyloxazolidinones were also shown to direct the carbozincation with high facial and enantioselectivity (Scheme 27).76
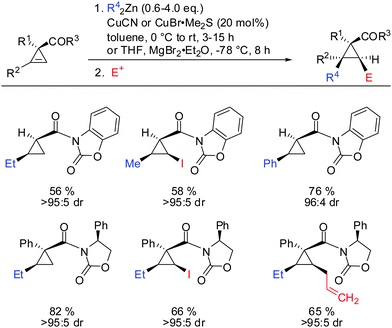 |
| | Scheme 27 Acyloxazolidinones: highly efficient directing groups. | |
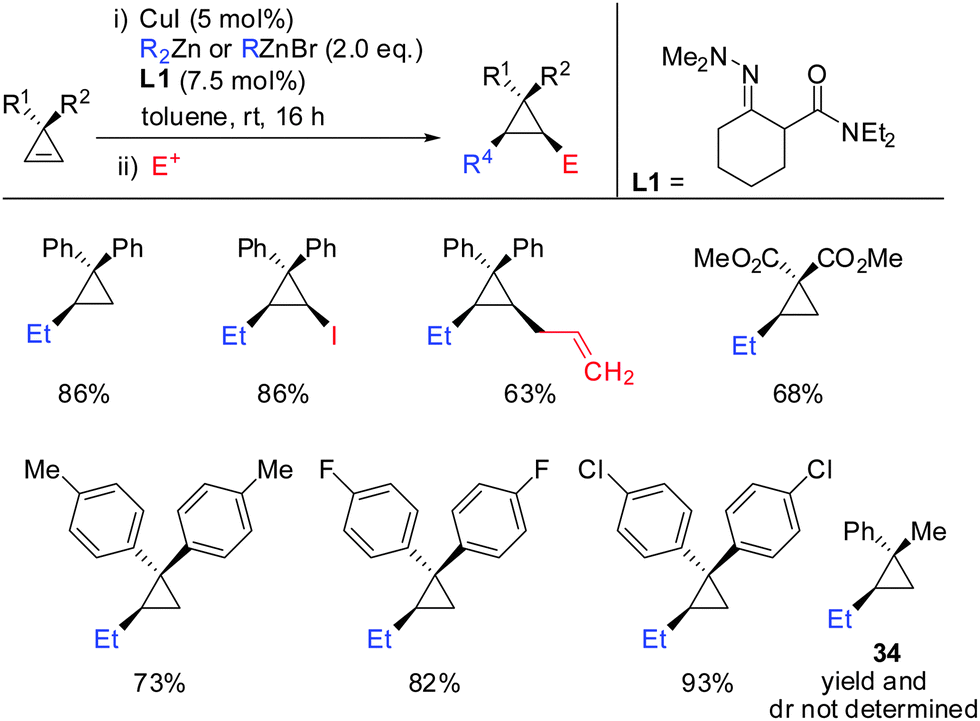
Very recently, in 2015, Endo showed that the addition of hydrazone ligand L1 greatly improved the yield of the copper catalysed carbozincation of unfunctionalized cyclopropenes (Scheme 28).75 The obtained cyclopropylzinc reagents were usually hydrolysed but also functionalization with iodine or allylbromide was reported. Compound 34 was observed as a single spot by TLC but could not be isolated as a pure product.
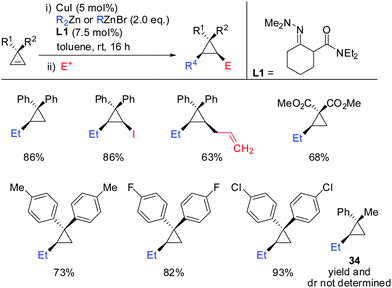 |
| | Scheme 28 Copper-catalysed carbozincation reaction of non-functionalized cyclopropenes. | |
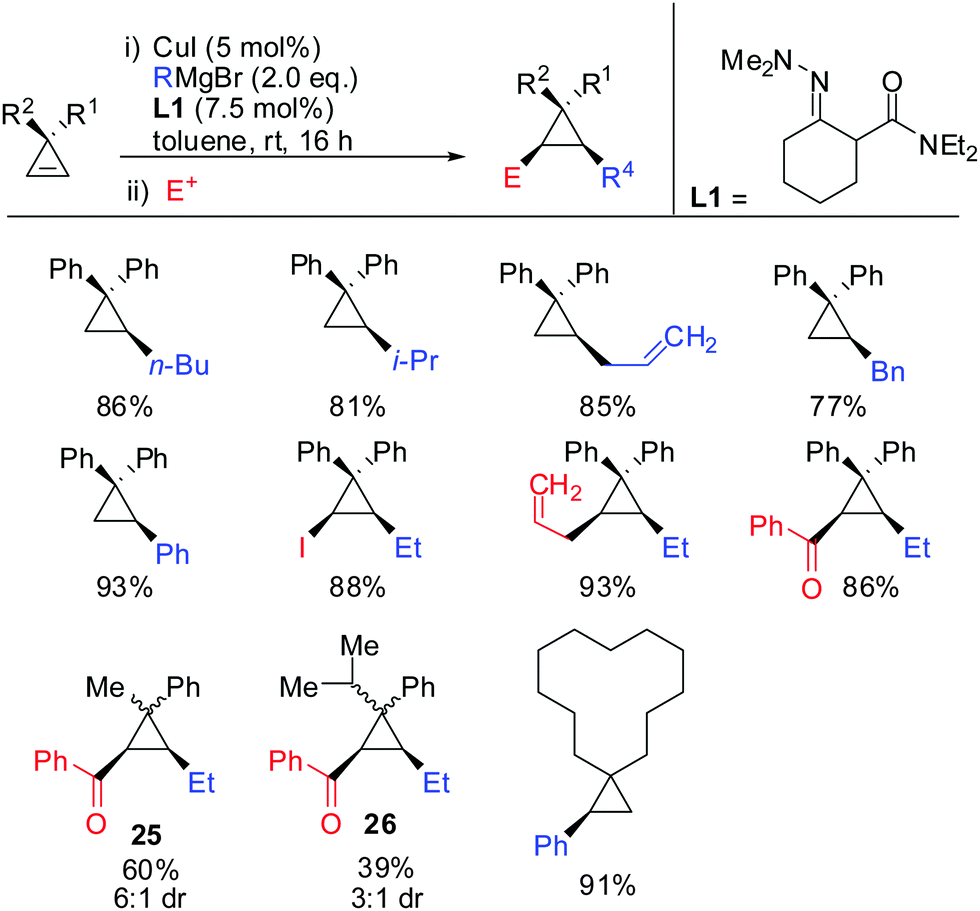
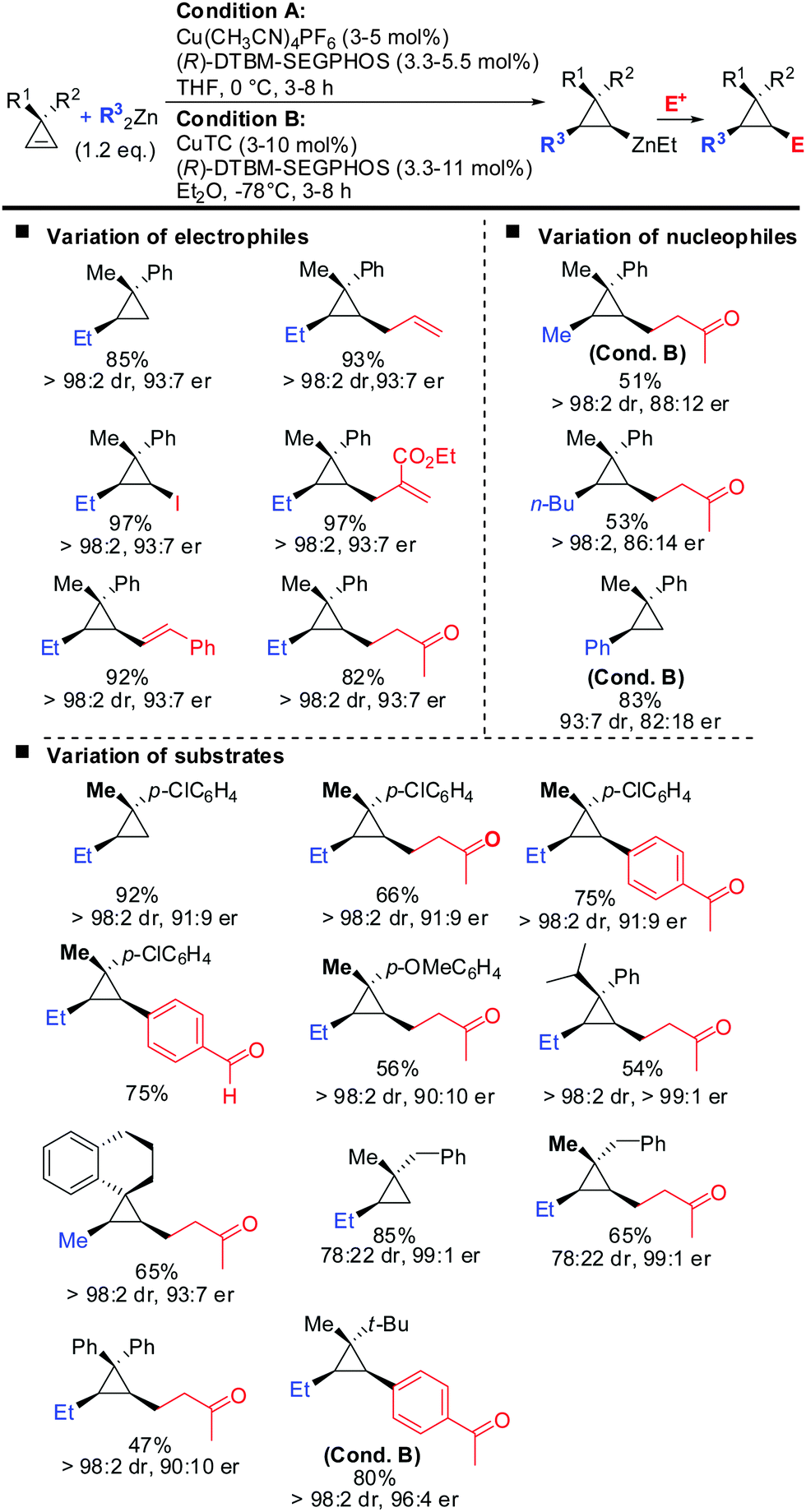
Very recently, the authors reported the first asymmetric copper catalysed carbozincation of cyclopropenes (Scheme 29).79 Following the pioneering work by Nakamura and Lautens they were able to identify two chiral copper catalysts which efficiently furnished highly enantioenriched polysubstituted cyclopropanes.80,81 Several features of the method are noteworthy: (1) the combination of highly encumbered phosphine ligand (R)-DTBM-SEGPhos with Cu(CH3CN)4PF6 or CuTC (copper(I) thiophene-2-carboxylate) completely suppressed dimeric side products which appeared otherwise in significant amounts (typically 15–20%). (2) The developed method provided the products with excellent diastereoselectivity (in general >95:5). The carbozincation is directed by the substituents in the 3-position and occurs on the less hindered face of the cyclopropene. In the absence of the ligand low diastereoselectivities were observed. Hence, the developed method also provides an efficient and highly diastereoselective route to racemic cyclopropanes. Only in the case where the two substituents in the 3-position lack steric bias, the diastereoselectivity decreases. (3) The enantiomeric ratio of the obtained products is in general high (>90:10) and in some cases even excellent (>99:1). It is noteworthy that diaryl- and dialkylsubstituted cyclopropenes are also obtained in high optical purity. (4) The in situ generated cyclopropylzinc reagent is configurationally stable and can be further functionalized under mild conditions with a great variety of electrophiles and coupling partners. Importantly palladium-catalysed cross coupling reactions with commercially available alkenyl- and aryl bromides allows for the rapid synthesis of a library of cyclopropanes from a single cyclopropylzinc precursor.
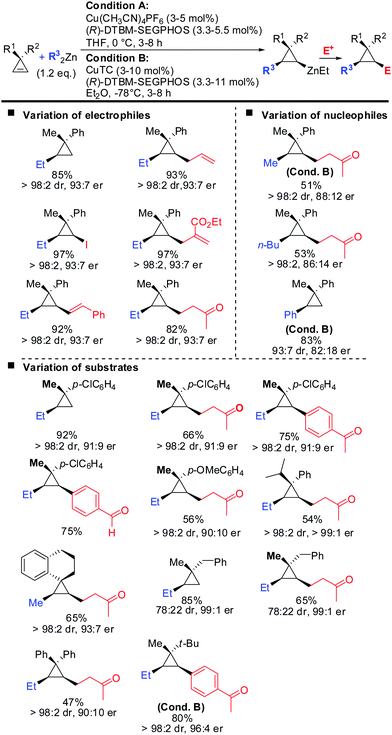 |
| | Scheme 29 Copper-catalysed asymmetric carbozincation of unfunctionalized cyclopropenes; unless otherwise noted conditions A were applied. | |
3.5 Carbocupration followed by elimination
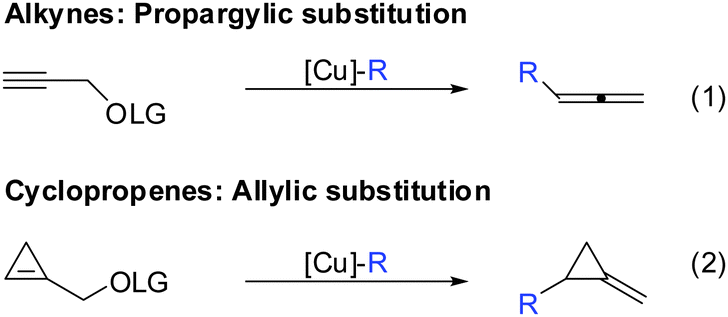
If one takes into consideration the similar chemistry of cyclopropenes and alkynes, the propargylic displacement shown in Scheme 3 (eqn (4)) should also apply to cyclopropenes (Scheme 30).
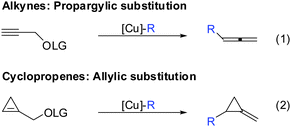 |
| | Scheme 30 Propargylic displacements – analogy for cyclopropenes. | |
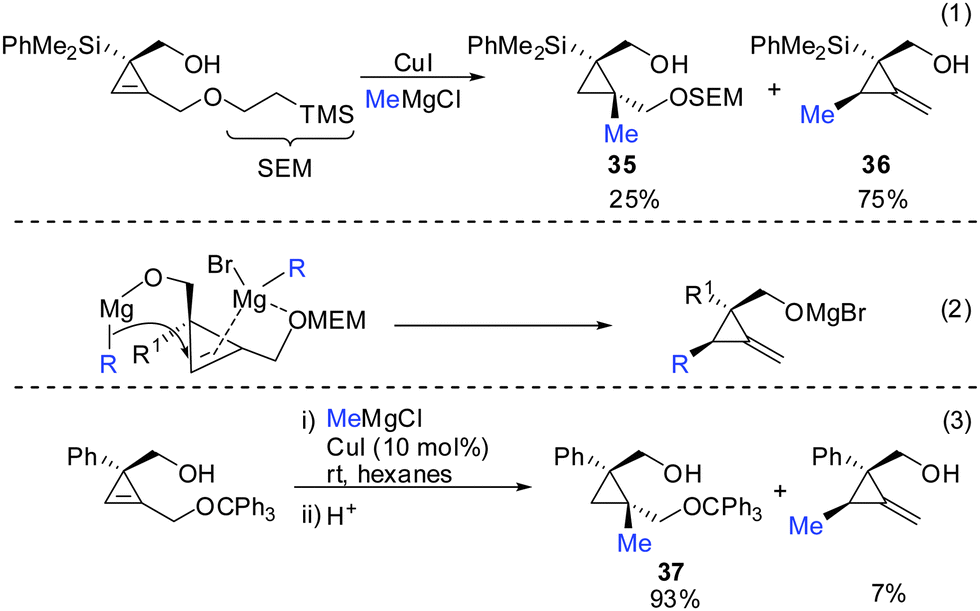
Indeed, in 2006 Fox and Marek reported simultaneously the diastereoselective synthesis of methylenecyclopropane (MCP) and alkylidenecyclopropane derivatives (ACP) respectively from cyclopropenes.82,83 Fox showed that cyclopropenes substituted with suitable leaving groups such as methyl, MEM- and SEM ethers gave a mixture of addition (35) and addition–elimination (36) products (Scheme 31).82 Further investigations showed that the presence of copper salts was not necessary to induce the elimination reaction. Changing the Grignard counter ion from chloride to bromide resulted in the exclusive formation of the addition–elimination product 36. The change in regioselectivity to form the less substituted product contrasted all prior studies on the copper-catalysed carbomagnesiation reaction of 1-alkylcyclopropenes, where the normal course of reactivity is for the nucleophile to add to the more substituted carbon-center of the double bond (Scheme 20). Fox proposed the simultaneous chelation of magnesium by the hydroxymethyl and the OMEM group (eqn (2)). Then, similar to propargylic substitution the organic substituent is delivered while the leaving group is expulsed. A large excess of the respective Grignard reagent is required for efficient reactions (eqn (5) and (6)). Poorly coordinating groups such as trityl ethers afforded mainly the addition product 37 (eqn (3)). Noteworthy is that the cyclopropene precursors are readily available in the enantioenriched form and therefore this route can provide access to the enantiomerically enriched MCP.
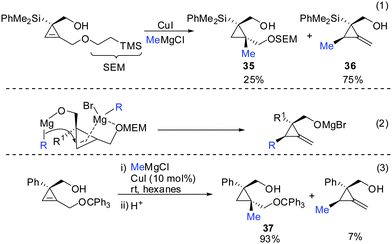 |
| | Scheme 31 Addition–elimination reaction to provide MCP. | |
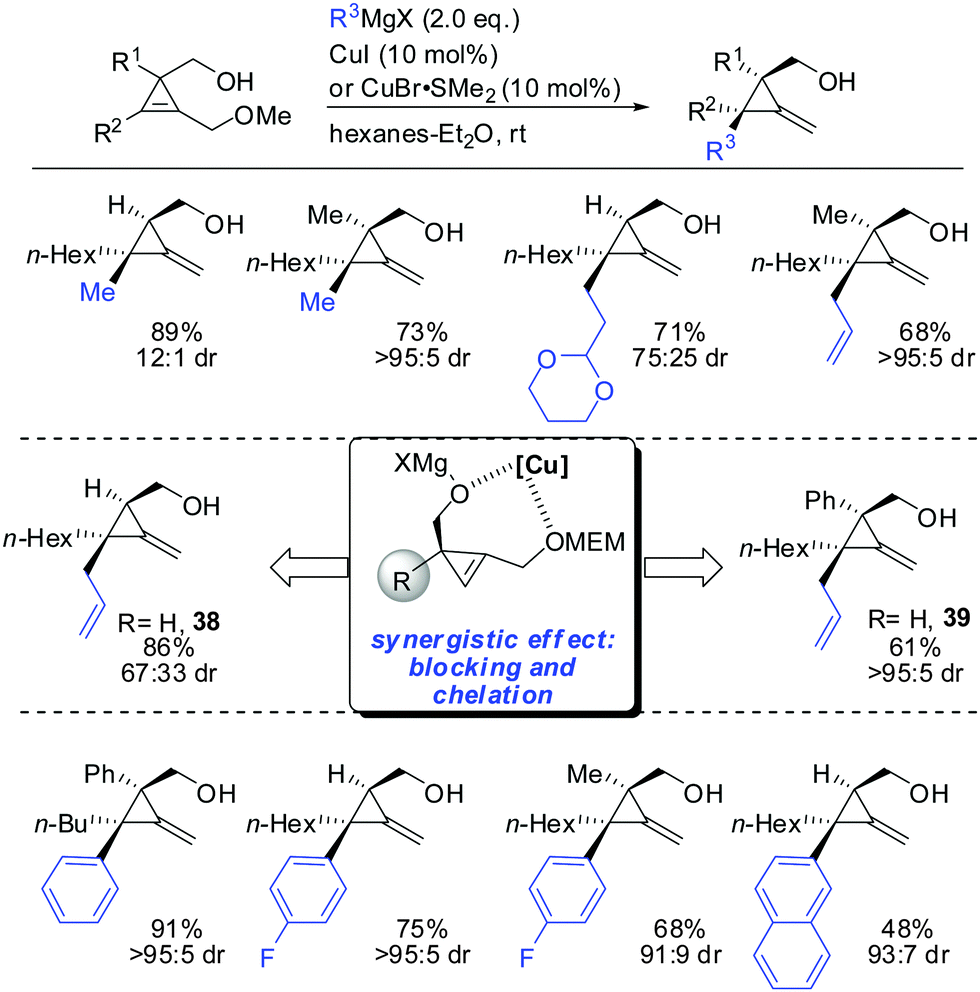
In 2013, Fox described the synthesis of highly substituted MCP from 1,2-disubstituted 3-(hydroxymethyl) cyclopropenes bearing a methoxy substituent as the leaving group (Scheme 32).84 Copper-catalysed displacement reactions with Grignard reagents proceeded in good yield and high diastereoselectivity. When substituent R1 was introduced the diastereoselectivity increased dramatically (38 and 39). Interestingly, iron catalysis furnished the anti-addition product whereas copper-catalysis afforded the syn-addition product.84
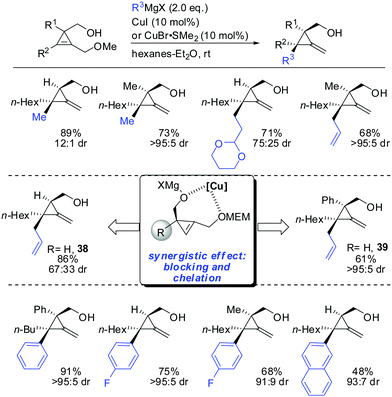 |
| | Scheme 32 Synthesis of various MCP. | |
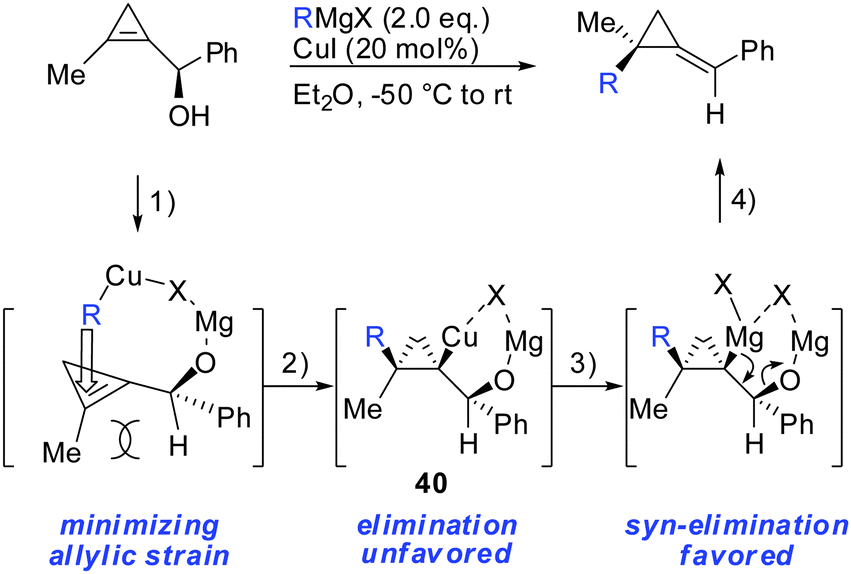
In parallel, it has been shown that when 1-(1-hydroxyalkyl)cycloprop-1-ene was treated with copper salt and alkyl Grignard reagents, alkylidenecyclopropanes were obtained in good yield, high diastereoselectivity and E/Z selectivity (Scheme 34).83 Without the addition of copper salt, the reaction did not proceed. The absolute configuration of the starting cyclopropenylcarbinols and the final alkylidenecyclopropanes imply an overall syn SN2′ displacement of the alcohol moiety. The reaction proceeds by a four step procedure (Scheme 33): (1) deprotonation of the allylic alcohol by the Grignard reagent and subsequent chelation of the organocopper reagent. (2) Copper-catalysed carbomagnesiation reaction of the most stable conformer. (3) Transmetalation from copper to magnesium (4) subsequent β-elimination. The transmetalation of copper to magnesium (step 3) is crucial for efficient elimination reactions. If copper is used stoichiometrically, the reaction stalls at intermediate 40 and can be trapped by electrophiles (Scheme 22). This is in accordance with the previous reports which show that the carbon–copper bond is less prone to β-elimination compared to the carbon–magnesium bond.85,86
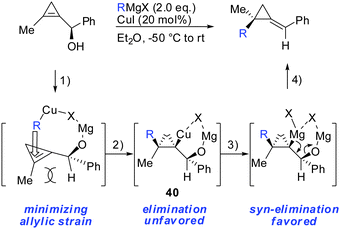 |
| | Scheme 33 Carbomagnesiation–elimination sequence. | |
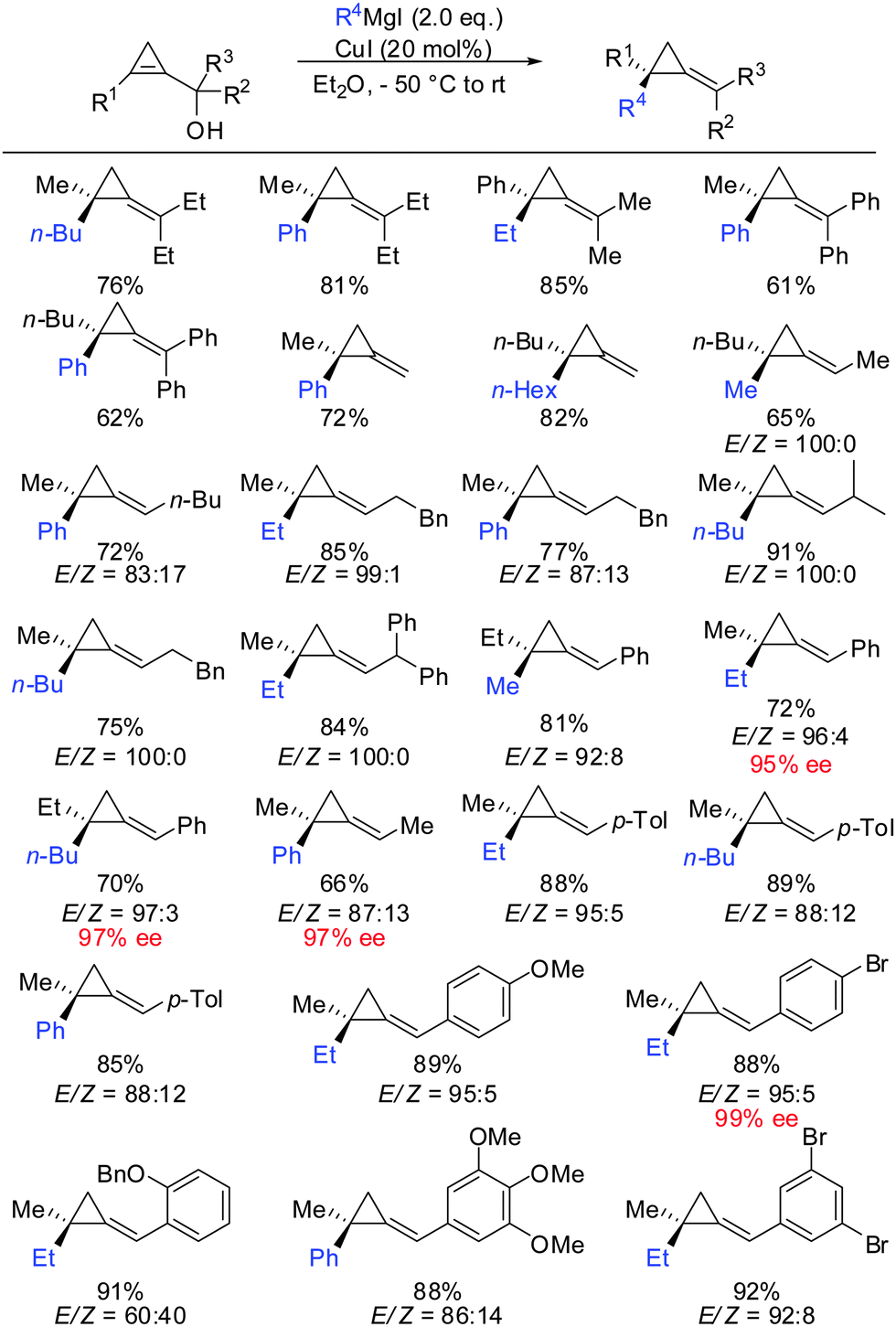
It should be noted that highly enantioenriched cyclopropenes can easily be obtained by Sharpless kinetic resolution or enzyme-mediated acylation of the racemic starting material. In a full account the generality of this method was demonstrated by providing a great number of examples (Scheme 34).87 It is important to note that the counter ion of the Grignard reagent is not an innocent spectator in such displacement reactions.88 In some cases, the stereochemistry of β-elimination reactions (syn versus anti) significantly improved when changing from alkyl magnesium bromides to iodides. In general arylmagnesium halides reacted with lower selectivities compared to alkylmagnesium halides.
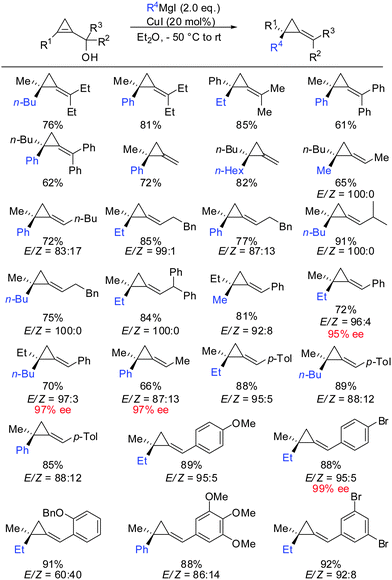 |
| | Scheme 34 Extended scope for the formation of ACP. | |
3.6 Carbocupration followed by ring opening
The similar reactivity of cyclopropene and alkyne is especially striking when cyclopropane ring-opening reactions are involved. A vinylmetal reagent generated from the carbometalation of an alkyne is well known to react with a great variety of electrophiles (eqn (1), Scheme 35). In analogy, cyclopropyl metals can be trapped with electrophiles to provide cyclopropane products. However, cyclopropanes substituted by an electron withdrawing group (EWG) possess an “internal electrophile” and can undergo ring-opening to provide acyclic products which resemble very much the products obtained via alkyne-chemistry (eqn (2)).89 The same holds true for the oxidized metal species. Alkyne chemistry and cyclopropene chemistry provide aldehydes with an alpha all-carbon quaternary stereocenter (eqn (3) and (4)).90 Obviously, if the electron-withdrawing group is replaced by a suitable leaving group, elimination occurs and an alkene is obtained (eqn (5)).
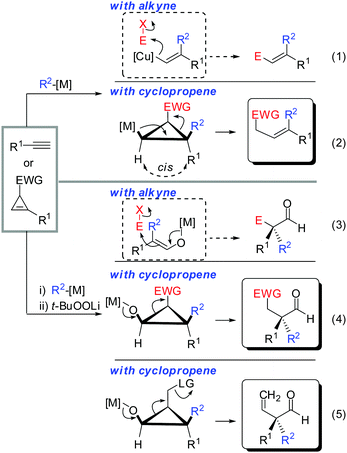 |
| | Scheme 35 Cyclopropane ring-opening reactions. | |
We could demonstrate the feasibility of all three pathways. First, the ring opening of cyclopropyl-magnesium species containing an ester group as an electron withdrawing group was observed upon warming the reaction to 0 °C (Scheme 36).63 The formation of the alkene is highly selective due to the preorganized position of the substituents in the cyclopropane scaffold (eqn (2), Scheme 35).
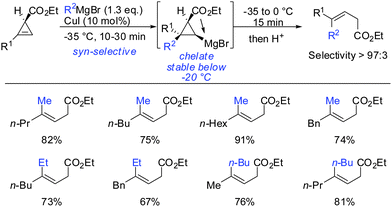 |
| | Scheme 36 Thermally induced ring-opening reaction. | |
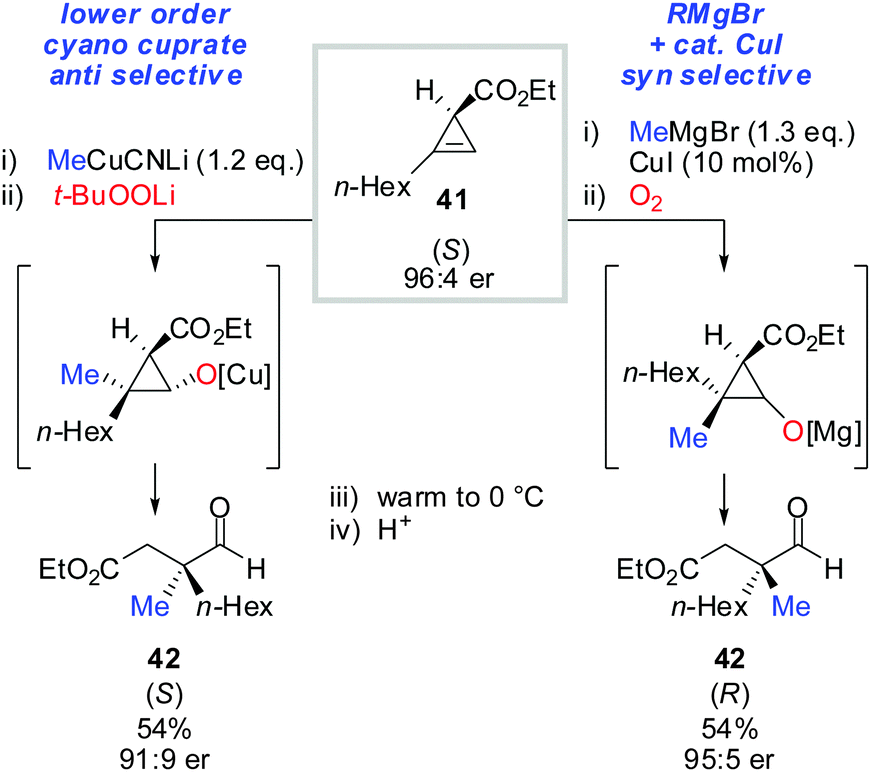
Next, the oxidation-ring opening sequence was shown to efficiently provide the corresponding acyclic aldehydes (Scheme 37).63 The diastereodivergent carbometalation reaction of cyclopropenyl esters (Schemes 13–15) was cleverly exploited to access both enantiomers of the corresponding aldehyde (S)-42 and (R)-42 starting from the same cyclopropenyl ester 41 (Scheme 37). It is noteworthy that in the case of copper-catalysed carbomagnesiation, molecular oxygen provided clean oxidation without leading to dimerized products which are typically observed with organocopper reagents.
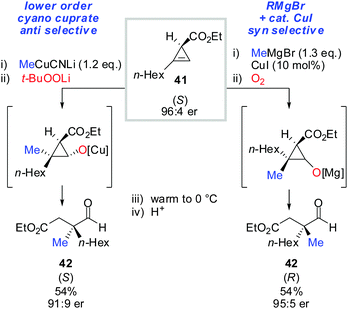 |
| | Scheme 37 Diastereodivergent carbometalation reaction allows for the synthesis of both enantiomers of aldehydes 42. | |
Finally, the addition–oxidation–elimination sequence described in eqn (5), Scheme 35 was realized (Scheme 38).64 Acetate was found to be the best leaving group. As previously discussed for Scheme 17, substituent R2 is necessary to block one face of the cyclopropene allowing the high observed diastereoselectivity. Two aldehydes were prepared in their enantioenriched form starting from enantioenriched cyclopropenes. It has been noticed that the corresponding aldehydes are prone to decomposition. Very likely oxidation of the aldehyde into the corresponding acid occurred. It is well known that aldehydes with a high degree of substitution are prone to aerobic oxidation.91
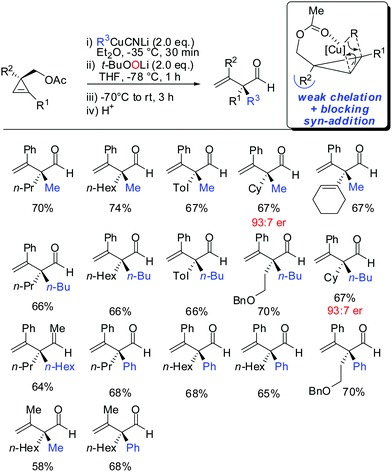 |
| | Scheme 38 Carbometalation–oxidation–elimination sequence. | |
4. Conclusions
Copper mediated carbometalation remains one of the best methods to construct carbon–carbon bonds with high selectivity. The product, itself an organometallic reagent, can enter a manifold of subsequent reactions such as electrophile trapping but also oxidation and carbon–carbon bond cleavage. Most of the past research focused on the highly selective construction of E/Z alkenes. Nowadays the high syn-selectivity and the possibility of employing substrate controlled carbometalation have opened new possibilities for the highly stereoselective construction of tertiary and quaternary stereocenters. Finally, the first highly enantioselective copper-catalysed carbometalation was shown which is the most recent way to synthesize non-functionalized enantioenriched cyclopropanes.
Acknowledgements
We are grateful for financial support from the Israel Science Foundation administrated by the Israel Academy of Sciences and Humanities (140/12) and the European Research Council under the Seventh Framework Program of the European Community (ERC grant agreement no. 338912).
Notes and references
-
Topics in Current Chemistry: Stereoselective Alkene Synthesis, ed. J. B. Wang, Springer, Heidelberg, 2012 Search PubMed
 .
.
- For a recent review see: M. B. Herbert and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 5018 CrossRef CAS PubMed and references cited therein.
- For an automated iterative assembly of alkene building blocks see: J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297 CrossRef CAS PubMed .
- For synthesis of polysubstituted alkenes using cross-coupling reactions with polyborylalkenes see: L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC .
- A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed .
-
D. Didier and I. Marek, in Carbometallation Reactions in Copper-Catalyzed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, ch. 10, p. 267 ff DOI:10.1002/9783527664573 .
- J. F. Normant and M. Bourgain, Tetrahedron Lett., 1971, 27, 2583 CrossRef .
- J. F. Normant and A. Alexakis, Synthesis, 1981, 841 CrossRef CAS .
- B. H. Lipshutz and S. Sengupta, Org. React., 1992, 41, 135 CrossRef CAS .
-
F. Chemla and F. Ferreira, in The Chemistry of Organocopper Compounds, ed. Z. Rappoport and I. Marek, Patai series, John Wiley & Sons, Chichester, England, 2009, p. 527 ff Search PubMed .
- K. Murakami and H. Yorimitsu, Beilstein J. Org. Chem., 2013, 9, 278 CrossRef CAS PubMed .
- S. Casson and P. Kocienski, Contemp. Org. Synth., 1995, 19, 19 RSC .
- A. G. Fallis and P. Forgione, Tetrahedron, 2001, 57, 5899 CrossRef CAS .
- A. Basheer and I. Marek, Beilstein J. Org. Chem., 2010, 6, 77 Search PubMed .
- Y. Minko, M. Pasco, H. Chechik and I. Marek, Beilstein J. Org. Chem., 2013, 9, 526 CrossRef CAS PubMed .
- S. Ma and Z. Lu, Adv. Synth. Catal., 2006, 348, 1894 CrossRef CAS .
- A. Alexakis, J. F. Normant and J. Villieras, J. Organomet. Chem., 1975, 96, 471 CrossRef CAS .
- L. Anastasia, Y. R. Dumond and E. Negishi, Eur. J. Org. Chem., 2001, 3039 CrossRef CAS .
- P. Rona and P. Crabbe, J. Am. Chem. Soc., 1968, 90, 4733 CrossRef CAS .
- D. E. Van Horn and E. Negishi, J. Am. Chem. Soc., 1977, 100, 2252 CrossRef .
- Y. Minko, M. Pasco, L. Lercher, M. Botoshansky and I. Marek, Nature, 2012, 490, 522 CrossRef CAS PubMed .
- K. Oisaki, D. Zhao, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 7439 CrossRef CAS PubMed .
- N. Yoshikai and E. Nakamura, Chem. Rev., 2012, 112, 2339 CrossRef CAS PubMed .
- I. Iwai and T. Konotsune, Yakugaku Zasshi, 1962, 82, 106 Search PubMed .
- J. Klein and R. M. Turckel, J. Am. Chem. Soc., 1969, 91, 6186 CrossRef CAS .
- J. B. Siddal, M. F. Biskup and J. H. Fried, J. Am. Chem. Soc., 1969, 91, 1852 Search PubMed .
- J. A. Katzenellenbogen and E. J. Corey, J. Am. Chem. Soc., 1969, 91, 1851 CrossRef .
- M. L. Deem, Synthesis, 1972, 675 CrossRef CAS .
- V. R. Kartashov, E. V. Skorobogatova and N. S. Zefirov, Russ. Chem. Rev., 1993, 935 CrossRef .
- A. Goti, F. M. Cordero and A. Brandi, Top. Curr. Chem., 1996, 178 Search PubMed .
- A. Brandi and A. Goti, Chem. Rev., 1998, 98, 589 CrossRef CAS PubMed .
- M. S. Baird, Chem. Rev., 2003, 103, 1271 CrossRef CAS PubMed .
- A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed .
- M. Shi, L. Shao, J. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed .
- H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS .
- G. Audrana and H. Pellisier, Adv. Synth. Catal., 2010, 352, 575 CrossRef .
- F. Miege, C. Meyer and J. Cossy, Beilstein J. Org. Chem., 2011, 7, 717 CrossRef CAS PubMed .
- Z. Zhu, Y. Weib and M. Shi, Chem. Soc. Rev., 2011, 40, 5534 RSC .
- L. H. Phun, J. Aponte-Guzman and S. France, Synlett, 2012, 2723 CAS .
- R. M. Wilson and R. E. Taylor, Angew. Chem., Int. Ed., 2013, 52, 4078 CrossRef PubMed .
- A. Archambeau, F. Miege, C. Meyer and J. Cossy, Acc. Chem. Res., 2015, 48, 1021 CrossRef CAS PubMed .
- M. Nakamura, H. Isobe and E. Nakamura, Chem. Rev., 2003, 103, 1295 CrossRef CAS PubMed .
- J. M. Fox and N. Yan, Curr. Org. Chem., 2005, 9, 719 CrossRef CAS .
- M. Rubin, M. Rubina and V. Gevorgyan, Synthesis, 2006, 1221 CAS .
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed .
- I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed .
- F. Allen, Tetrahedron, 1982, 38, 645 CrossRef CAS .
- P. Rademacher, Chem. Rev., 2003, 103, 933 CrossRef CAS PubMed .
- A. T. Stoll and E. Negishi, Tetrahedron Lett., 1985, 26, 5671 CrossRef CAS .
- K. G. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312 CrossRef .
- W. T. G. Johnson and W. T. Borden, J. Am. Chem. Soc., 1997, 119, 312 Search PubMed .
- R. D. Bach and O. Dmitrenko, J. Am. Chem. Soc., 2004, 126, 4444 CrossRef CAS PubMed .
- A. Fattahi, R. E. McCarthy, M. R. K. Ahmad and S. R. Kass, J. Am. Chem. Soc., 2003, 125, 11746 CrossRef CAS PubMed .
- A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213 CrossRef CAS PubMed .
- K. Semba, T. T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2012, 18, 4179 CrossRef CAS PubMed .
- A. Parra, L. Amenós, M. Guisán-Ceinos, A. López, J. L. Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833 CrossRef CAS PubMed .
- V. Agabekov, W. Seiche and B. Breit, Chem. Sci., 2013, 4, 2418 RSC .
- W. M. Sherrill and M. Rubin, J. Am. Chem. Soc., 2008, 130, 13804 CrossRef CAS PubMed .
- E. Nakamura, M. Isaka and S. Matsuzawa, J. Am. Chem. Soc., 1988, 110, 1297 CrossRef CAS .
- M. Isaka and E. Nakamura, J. Am. Chem. Soc., 1990, 112, 7428 CrossRef CAS .
- S. Simaan and I. Marek, Org. Lett., 2007, 9, 2569 CrossRef CAS PubMed .
- P. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333 CrossRef CAS PubMed .
- D. Didier, P. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem. – Eur. J., 2014, 20, 1038 CrossRef CAS PubMed .
- M. Simaan, P. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345 CrossRef CAS PubMed .
- X. Xie and J. M. Fox, Synthesis, 2013, 1807 CAS .
- G. M. Whitesides, J. C. San Filippo, C. P. Casey and E. J. Panek, J. Am. Chem. Soc., 1967, 80, 5302 CrossRef .
- M. Möller, M. Husemann and G. Boche, J. Organomet. Chem., 2001, 624, 47 CrossRef .
- H. M. L. Davies and G. H. Lee, Org. Lett., 2004, 6, 1233 CrossRef CAS PubMed .
- M. Y. Lukina, T. Y. Rudavshevskaya and O. A. Nesmeyanova, Dokl. Akad. Nauk SSSR, 1970, 190, 133 Search PubMed .
- H. Lehmkuhl and K. Mehler, Liebigs Ann. Chem., 1978, 1841 CrossRef CAS .
- H. G. Richey and R. M. Bension, J. Org. Chem., 1980, 45, 5036 CrossRef CAS .
- L. Liao and J. M. Fox, J. Am. Chem. Soc., 2002, 124, 14322 CrossRef CAS PubMed .
- L. Liao, F. Zhang, N. Yan, J. A. Golen and J. M. Fox, Tetrahedron, 2004, 60, 1803 CrossRef CAS .
- N. Yan, X. Liu and J. M. Fox, J. Org. Chem., 2008, 73, 563 CrossRef CAS PubMed .
- T. Nakano, K. Endo and Y. Ukaji, Synlett, 2015, 671 CAS .
- V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382 CrossRef CAS PubMed .
- V. Tarwade, R. Selvaraj and J. M. Fox, J. Org. Chem., 2012, 77, 9900 CrossRef CAS PubMed .
- R. L. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884 RSC .
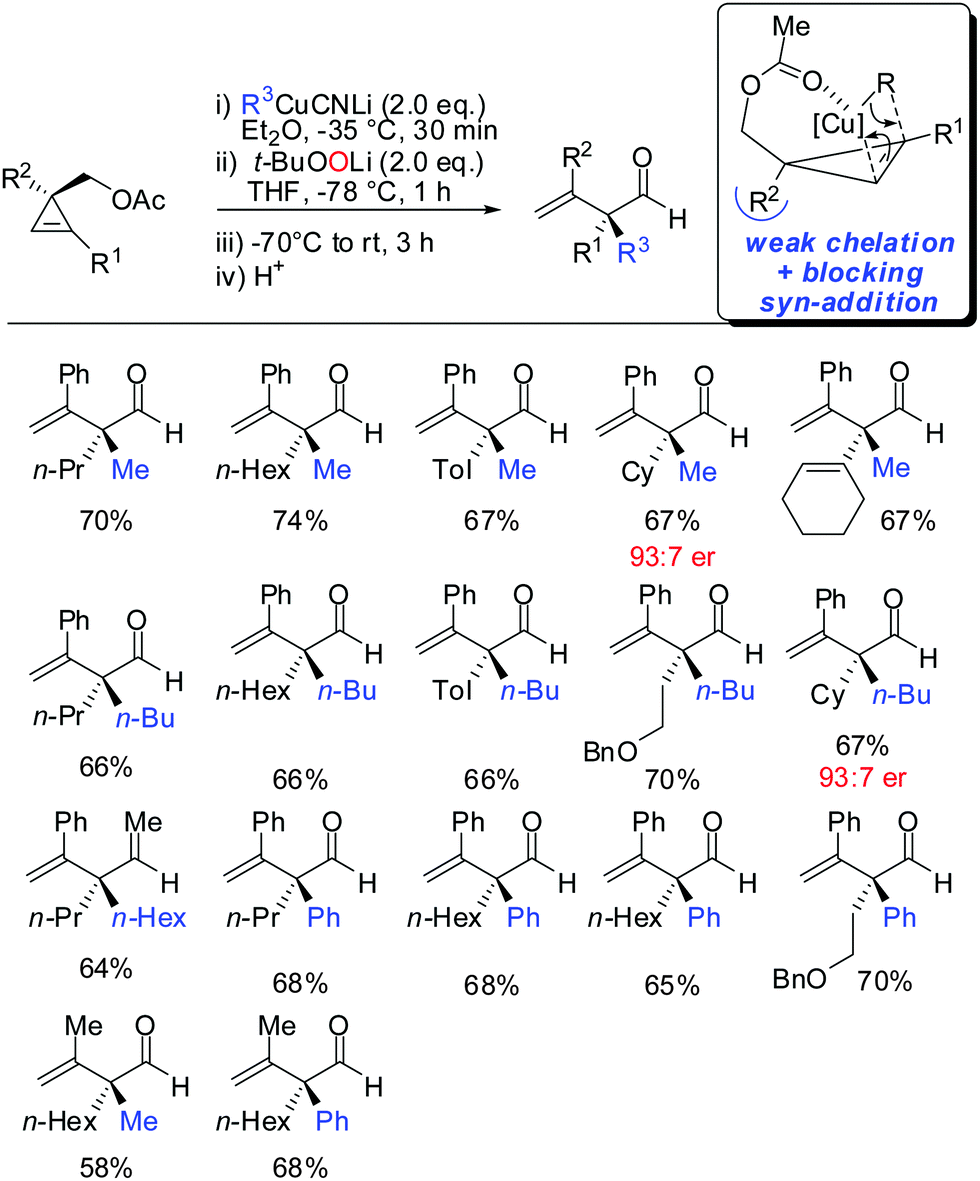
- D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414 CrossRef PubMed .
- M. Nakamura, E. Hirai and E. Nakamura, J. Am. Chem. Soc., 2000, 122, 978 CrossRef CAS .
- K. Kramer, P. Leong and M. Lautens, Org. Lett., 2011, 13, 819 CrossRef CAS PubMed .
- Z. Yang, X. Xie and J. M. Fox, Angew. Chem., Int. Ed., 2006, 45, 3960 CrossRef CAS PubMed .
- S. Simaan, A. Masarwa, P. Bertus and I. Marek, Angew. Chem., Int. Ed., 2006, 45, 3963 CrossRef CAS PubMed .
- X. Xie and J. M. Fox, Synthesis, 2013, 1807 CAS .
- A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, Tetrahedron, 1991, 47, 1677 CrossRef CAS .
- I. Marek, A. Alexakis, P. Mangeney and J. F. Normant, Bull. Soc. Chim. Fr., 1992, 129, 171 CAS .
- S. Simaan, A. Masarwa, E. Zohar, A. Stanger, P. Bertus and I. Marek, Chem. – Eur. J., 2009, 15, 8449 CrossRef CAS PubMed .
- A. Alexakis, I. Marek, P. Mangeney and J. F. Normant, J. Am. Chem. Soc., 1990, 112, 8042 CrossRef CAS .
- T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed .
- D. Zhang and J. M. Ready, Org. Lett., 2005, 7, 5681 CrossRef CAS PubMed .
- L. Vanoye, A. Favre-Reguillon, A. Aloui, R. Philippea and C. de Bellefon, RSC Adv., 2013, 3, 18931 RSC .
Footnote |
| † Dedicated to Prof. Jean F. Normant for his 80th birthday. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence