
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalized hexagonal boron nitride nanomaterials: emerging properties and applications
Qunhong
Weng
*a,
Xuebin
Wang
a,
Xi
Wang
*b,
Yoshio
Bando
a and
Dmitri
Golberg
*a
aWorld Premier International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, Ibaraki, Japan. E-mail: Weng.Qunhong@nims.go.jp; wengqunhong@gmail.com; Golberg.Dmitri@nims.go.jp
bSchool of Science, Beijing Jiaotong University, Beijing, 100044, P. R. China. E-mail: xiwang@bjtu.edu.cn
First published on 13th May 2016
Abstract
Functionalization is an important way to breed new properties and applications for a material. This review presents an overview of the progresses in functionalized hexagonal boron nitride (h-BN) nanomaterials. It begins with an introduction of h-BN structural features, physical and chemical properties, followed by an emphasis on the developments of BN functionalization strategies and its emerging properties/applications, and ends with the research perspectives. Different functionalization methods, including physical and chemical routes, are comprehensively described toward fabrication of various BN derivatives, hetero- and porous structures, etc. Novel properties of functionalized BN materials, such as high water solubility, excellent biocompatibility, tunable surface affinities, good processibility, adjustable band gaps, etc., have guaranteed wide applications in biomedical, electronic, composite, environmental and “green” energy-related fields.
 Qunhong Weng | Qunhong Weng received his BS degree in Applied Chemistry from Huazhong Agricultural University, PR China, in 2007. In 2010, he obtained his MSc degree in Chemistry from Xiamen University while working on carbon clusters. After working as a research associate in XMU and Nanyang Technological University, Singapore, he joined Prof. Golberg's group at the National Institute for Materials Science (NIMS), Japan, and obtained his PhD degree in March of 2015. Since then, he has been a MANA postdoctoral researcher of Prof. Bando's group at NIMS. His research focuses on understanding layered material functionalization and their applications in clean energy storage and conversion technologies. |
 Xuebin Wang | Xuebin Wang received his BS and MS degrees from Nanjing University in China, and got his PhD degree from Waseda University in Japan in 2013. He worked as a Junior Researcher (2010–2013) and a Postdoctoral Researcher (2013–2014) at the National Institute for Materials Science (NIMS). He is working as an ICYS Researcher in the World Premier International Center for Materials Nanoarchitectonics (WPI-MANA) and NIMS since 2014. He has been pursuing the development of synthesis, novel properties and practical applications of low-dimensional functional materials. His research has recently focused on 3D-designed nanosheets, such as strutted-graphenes and BN nanosheets, and their applications in supercapacitors, polymeric composites etc. |
 Xi Wang | Xi Wang received his PhD degree in physical chemistry from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS). Then he joined the National Institute for Materials Science (NIMS) as a JSPS postdoctoral fellow within Prof. Bando's group and then as a Fellow of the International Center for Young Scientists (ICYS) researcher within NIMS. His research interests include the controlled synthesis and exploration of fundamental physical properties of inorganic functional nanomaterials, as well as their promising applications for energy storage and optoelectronics. |
 Yoshio Bando | Yoshio Bando received his PhD degree from Osaka University in 1975 and joined NIMS in the same year. From 1979 to 1981 he worked as a visiting researcher at Arizona State University. Currently, he is a Chief Operating Officer (COO) of WPI-MANA and a Fellow within NIMS. His current research concentrates on the synthesis and properties of inorganic nanostructures and their TEM characterizations. He has published more than 600 papers in international journals which were cited more than 25 |
 Dmitri Golberg | Dmitri Golberg obtained his PhD degree from Bardin Institute for Ferrous Metallurgy, Moscow, Russia, in 1990, and joined NIMS five years later. At present, he is a Group Leader and a Principal Investigator of WPI-MANA-NIMS, and a Professor of Tsukuba University. With more than 580 publications, over 25 |
1. Introduction
Boron nitrides (BN) are constructed from equal numbers of boron (B) and nitrogen (N) atoms. These materials were previously considered as only synthetic, but recently have also been discovered in nature.1 They have three crystalline forms that are isoelectronic to similarly structured carbon lattices: graphite-like hexagonal BN (h-BN), diamond-like cubic BN (c-BN), and wurtzite BN (w-BN). Among them, h-BN is the most stable BN phase under standard conditions. c-BN is well known for its superb hardness.2 Similarly to c-BN, the w-BN structure is also built from sp3-hybridized B and N atoms, but its neighbouring BN3 or NB3 tetrahedrons are stacked at a different angle.3 h-BN has a typical layered structure like graphite. Within a two-dimensional (2D) layer, alternating B and N atoms are linked with each other via strong B–N covalent bonds; whereas the 2D layers are held together by weak van der Waals forces. Unlike the case of graphite, the interlayer stacking pattern in the h-BN features its B atoms in every consecutive BN layer siting exactly above or below the N atoms in the adjacent layers. Such structural characteristics imply the polarity of B–N bonds, i.e. the partially ionic character of the covalent B–N bonds. Electron pairs in sp2-hybridized B–N σ bonds are more confined to the N atoms due to their higher electronegativity; and the lone pair of electrons in the N pz orbital is only partially delocalized with the B pz orbital, in contrast to the equally contributed and evenly distributed electrons along the C–C bonds of graphite layers.Due to a structural analogy with graphite, h-BN shares common properties with it, like anisotropy along and perpendicular to a basal plane, high mechanical strength and thermal conductivity, and good lubrication. On the other hand, the unique structural features of h-BN endow it many other important electrical, optical, and chemical characteristics. For example, the reduced electron-delocalization in the BN π bonds causes a large band gap and leads to the electrically insulating nature and colourless appearance of the material. These properties make h-BN very useful as insulating and thermally conductive fillers, deep ultraviolet light sources, dielectric layers, cosmetic products, microwave-transparent shields, etc.4 Furthermore, h-BN is highly thermally and chemically stable, and thus is also widely used for durable high-temperature crucibles, anti-oxidation lubricants, protective coatings, etc. in industry.5
It is expected that many novel properties can emerge from a material through its smart functionalization, either physical or chemical. This is also proven to be applicable in case of h-BN. Its properties can be tailored, and many brand-new features and applications can be created directly via such functionalization. However, the high chemical stability/inertness of h-BN hinders its modifications. This makes the corresponding attempts to functionalize BN structures, both physically and chemically, a challenging research topic. In this review, a comprehensive introduction of functionalized BN materials, from their synthesis, structural characterizations and properties toward real applications, is presented. We introduce a considerable number of recently developed functionalization strategies, which lead to non-classical properties and applications of functionalized BN nanomaterials. Finally, the general challenges and perspectives of this exciting area of research are highlighted and discussed.
2. Physical functionalization
Physical functionalization of h-BN nanomaterials mainly refers to their structural/morphological activations. Usually, the commercially available bulk h-BNs exhibit a laminar plate-like structure with a thickness varying from dozens to hundreds of nanometers, and a lateral size larger than a few micrometers. Certainly, their structures can be moulded into low-dimensional 0D particles/cages, 1D tubes/wires/ribbons, 2D sheets and 3D porous forms, as well as many other novel functional forms. We will selectively introduce and discuss different BN functional nanostructures in this section.2.1 Low-dimensional BN nanostructures
Low-dimensional BN materials refer to a class of BN nanostructures with at least one dimension constrained to the nanometer scale. This is also applicable to the family of functionalized BN materials. In the past decades, one witnessed a flourish of developments in the field of low-dimensional BN nanomaterials.21–29 Since these materials have already been reviewed in detail in numerous articles, like ref. 30 and 31 for 1D nanotubes, ref. 32 for 2D nanosheets, and ref. 4, 33 for all low-dimensional BN nanomaterials, we only present a general landscape highlighting the mechanical and thermal properties of these nanostructures.The mechanical properties of low-dimensional BN nanostructures are comparable to those of the bulk h-BN (Table 1). The measured elastic modulus of BNNTs along the axial direction ranges from 0.5 to 1.3 TPa,6–9 while the DFT calculated elastic modulus of BN monolayers was computed to be 0.72–0.95 TPa,13–18 both values are indeed close to the value for the bulk h-BN.20 It should be noted that to the best of our knowledge, to date, there have been no experimental data related to the elastic modulus and fracture strength of BN monolayers. Experimental measurements of CVD-grown BNNSs containing a few BN layers showed a decreased elastic modulus.15 These results confirm the ultrahigh strength and stiffness of the low-dimensional BN nanostructures and suggest their important applications for polymer, ceramic and light metal composite reinforcements.
| BN structures | Elastic modulus (TPa) | Fracture strength (GPa) | Thermal conductivity (W m−1 K−1) | Specific surface areas (m2 g−1) | |
|---|---|---|---|---|---|
| a All values for 2D BNNSs (monolayer) are taken from theoretical studies while the others are the experimental results. | |||||
| 1D | BNNTs | 0.5–1.36–9 | 339 | ∼180–30010 | 212–25411 |
| Bamboo-like BNNTs12 | 0.225 | 8 | — | — | |
| 2D | BNNSs (monolayer)a | 0.72–0.9513–18 | 70–21713–18 | >60019 | ∼2600 |
| Bulk | h-BN | 0.81120 | — | 39019 | <10 |
Unlike the carbon materials whose thermal conductivity is determined by free electrons and phonons, thermal conductivity in electrically-insulating BN materials is exclusively governed by phonons. It was revealed that the thermal conductivity of carbon materials increased along with a decrease of material dimensionality.34 Similarly, in BN materials, a rise in thermal conductivity under thinning BN to a monolayer was theoretically predicted, together with the stronger phonon–phonon scattering in the 2D layer.19 Thus, although being not as high as that of graphene, the thermal conductivity of BN 2D mono- and few-layered nanosheets is still extremely high. Some CVD-grown few-layered BN nanosheets were measured to have a lower thermal conductivity than that of bulk h-BN, probably due to the increased phonon scattering by polymer residues on the surfaces.35 Furthermore, higher thermal conductivity can be expected when the dimensionality of BN is further decreased to 1D BNNTs and BN nanoribbons; the value may rival that of their carbon counterparts.31,36
2.2 Porous structures
h-BN materials can be tailored into porous structures, which are useful for H2 storage, pollutant treatment, catalyst supports, drug delivery, etc. Like for the preparation of other porous materials, porous BNs could be obtained with and without involving templates during the synthesis: soft-/hard-templating and non-template syntheses were introduced. In regards to template directed synthesis, numerous templates, such as silica, activated carbons, graphene aerogels, zeolites, cationic surfactants, and block copolymers were employed as templates. The specific surface areas (SSAs) of these porous BNs ranged from 100 to 950 m2 g−1. Remarkable progress was made in the last three years based on the latter non-template strategy, the SSAs values reached up to 1900 m2 g−1.37,38 Theoretically modeled stable porous BN structures could reach a SSA up to 4800 m2 g−1,39,40 indicating that a large potential toward further textural properties improvements still remains.3. Chemical functionalization
Despite the structural similarity between h-BN and graphite, as well as between their nanostructures, functionalization of the BN materials has been far less well explored. Actually, functionalization of C-based materials can refer to many organic chemical reactions, while functionalization of BNs is not common with respect to well-established chemically active sites in organic chemistry. Although being very challenging, there have been several strategies successfully developed for BN chemical modifications.As a general rule, reaction of both h-BN and graphite on their basal plane sites always involves opening of their polarized or equally conjugated π bonds. Thus, the generated new bonds should always be present at an even number. When a functional group forms a single bond with B or N, a compensating group (no matter whether it is the same or not with the functional group) should also be attached to the unpaired N or B atom to balance the overall charge. Some other functional groups can form a bridging bond when a BN π bond is opened, similar to the case of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond epoxidation. The simplest functionalized BN structure model has usually two groups added to a neighboring B–N unit. However, both experimental and theoretical studies on the chemical modification of a conjugated carbon system have revealed that, when the charge and aromaticity are satisfied, the bonded functional groups are not limited to the neighbouring positions (they can be spatially separated).64–69 Such phenomena may also be applicable to the h-BN systems.
C bond epoxidation. The simplest functionalized BN structure model has usually two groups added to a neighboring B–N unit. However, both experimental and theoretical studies on the chemical modification of a conjugated carbon system have revealed that, when the charge and aromaticity are satisfied, the bonded functional groups are not limited to the neighbouring positions (they can be spatially separated).64–69 Such phenomena may also be applicable to the h-BN systems.
Since the B–N bonds in an h-BN structure have a partial ionicity, its B and N atoms are partially positively (electron deficient centers) and negatively (electron rich centers) charged, respectively. This property makes the B sites attackable by nucleophilic groups, while the N sites are reactive with electrophilic ones. To date, numerous functional groups, including hydroxyl (–OH, discussed in Section 3.1), amino (–NH2, Section 3.2), ether (–OR, Section 3.1), amine (–NHR, Section 3.2), arcyl (–COR, Section 3.4), alkyl (–R, Section 3.3), and halogen (–X, Section 3.4) groups, as well as heteroatoms (C and O, Section 3.5), have been experimentally introduced into BN skeletons through chemical functionalization, as shown in Fig. 1.
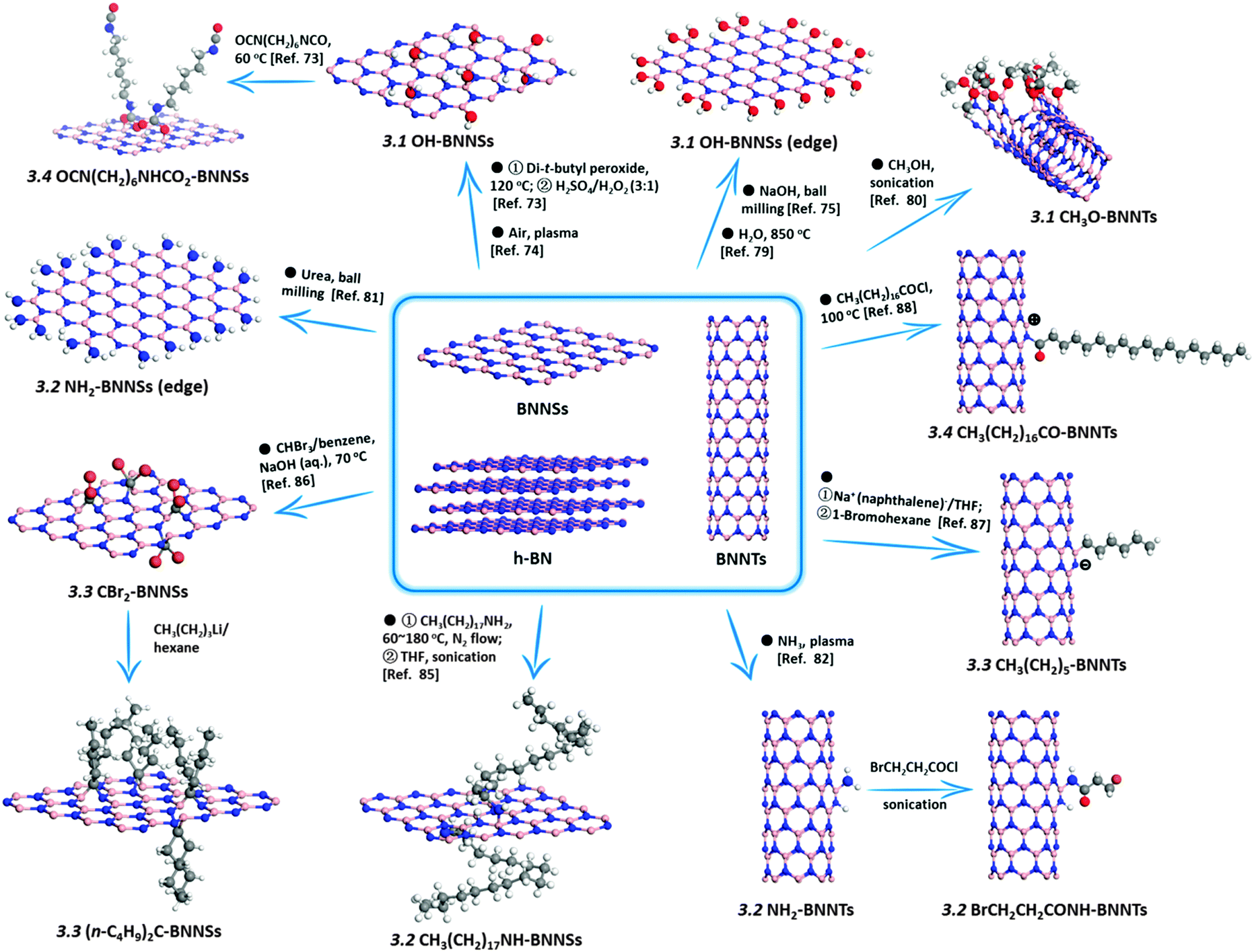 | ||
| Fig. 1 Summary of chemical functionalization strategies of h-BN bulk-/nanomaterials. A charge is denoted when the compensating functional group is unknown. | ||
3.1 Hydroxyl (–OH) and alkoxy (–OR) groups
When the BNNTs or BNNSs are used as the precursors, various methods become available to prepare the OH-BN derivatives, including plasma treatment, hydrothermal reactions with NaOH, NaOH-assistant ball-milling, and reactions with H2O at a high temperature, or using reagents that can generate ˙OH radicals, etc. For example, Zhi et al. reported the hydroxylation of BNNTs via chemical reactions between BNNTs with H2O2 that had been sealed in an autoclave, where H2O2 was dissociated to generate ˙OH radicals and reacted with BNNTs at 120 °C.71 The –OH groups were chemically bonded to the B sites of BNNTs in the final product, as verified by XPS and IR spectroscopy. The obtained OH-BNNTs contained ∼6 wt% oxygen and could form a stable aqueous solution at 0.25 mg mL−1 concentration. Later, a more simple method was found by Lin et al. for the preparation of the hydroxylated BNNSs through direct sonication of h-BN powders in water. The presence of monolayered and a-few-layered nanosheets was confirmed in the obtained dispersions (with an order of ∼0.05–0.1 mg mL−1). Sonication-assisted hydrolysis induced the exfoliation, cutting, and hydroxylation of BN layers.72 Coleman's group reported using an organic peroxide reagent (di-t-butyl peroxide) to firstly introduce alkoxy groups on BNNS surfaces. After further treatment with H2SO4/H2O2, the bonded t-butoxy groups were hydrolyzed to –OH groups.73 The estimated functionalization degree for the first step was ∼4 at%, whereas the resultant OH-BNNS could form a ∼0.1 mg mL−1 solution in water. More recently, our group has found an easier post-treatment method to prepare OH-BNNSs, i.e. exposing BNNSs to air plasma.74 This is because the air contains O2 and moisture that can also generate ˙OH radicals to react with BNNSs under plasma conditions. Since the ˙OH and ˙OR radicals are violent intermediates, the reactions between these reactants and B/N sites of BN surfaces are thought to proceed randomly. Both in-plane and edge sites may possibly be functionalized. The formed sp3 N–O bonds are considered to be not stable and would be degenerated or replaced by other groups.
Very different from the methods described above (always based on the post-reactions of BN bulk-/nanomaterials), the present authors have developed a new “reserved reaction” to obtain the OH-BN derivatives without using the corresponding BN precursors (Fig. 2).70 In this method, a hydroxyl-containing B precursor was used to replace the carbon component in an N-rich C3N4 solid at moderate temperatures. A portion of the –OH groups in the B precursors was successfully reserved in the obtained products, thus directly yielding the OH-BN derivatives without any post reactions. The prepared products featured the extra-high hydroxylation degrees, up to 30 at%, i.e. over 88% of B atoms had been hydroxylated, as was confirmed by XPS analysis. Their water colloid solutions, at a concentration of 2 mg mL−1, were found to be very stable and useful for drug loading and delivery applications. This strategy should also inspire the functionalization of other materials, such as graphene and carbon nanotubes (CNTs), because many C precursors also contain rich –OH, –NH2, and other groups, which could be reserved when the proper conditions are chosen during material growth/synthesis.
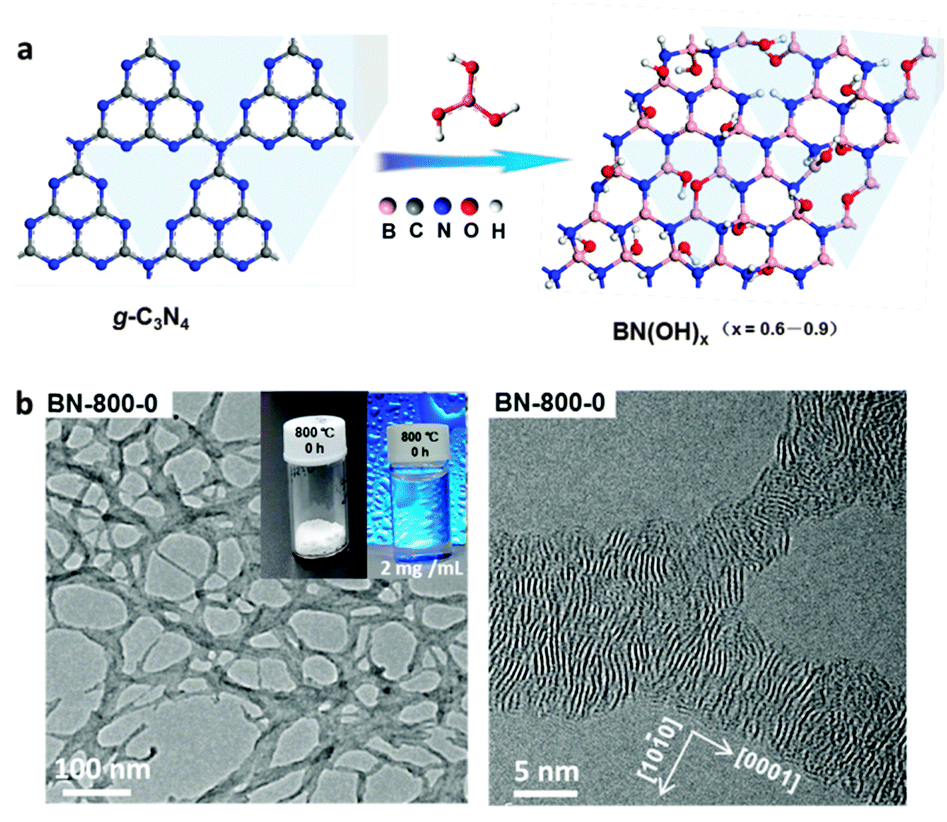 | ||
| Fig. 2 (a) Preparation of hydroxylated BNs through thermal substitution of carbon atoms in g-C3N4 using boric acid. (b) Low- (left) and high-resolution (right) TEM images of the precipitated hydroxylated BN materials extracted from an aqueous solution. The sample is prepared at 800 °C. The insets in (b) are the photographs of the as-prepared hydroxylated BN and its aqueous solution with a concentration of 2 mg mL−1. Reproduced from ref. 70 with permission. | ||
In recent years, remarkable progress has been made on edge hydroxylation of BNNSs. Using bulk h-BN powders as the precursor, Lee et al. have developed a hydroxide-assistant ball milling technique to simultaneously exfoliate and functionalize BN (Fig. 3a and b).75 The NaOH solution used in this method was suggested to cut and functionalize h-BN sheets via a reaction between the h-BN and OH− ions, while the high-speed balls provided the required shear force to exfoliate the materials. An increased oxygen content, up to 6.4 at%, for the OH-BNNS products indicated that, besides the edges, in-plane B sites had also been partly hydroxylated. The obtained OH-BNNSs were found to reduce the oxygen and water vapour permeability of a polymer matrix by 46% and 34%, respectively. Very recently, Xiao and Huang have reported that OH-BNNSs can be prepared via a steam treatment at high temperatures; this simultaneously realizes exfoliation and hydroxylation.79 Furthermore, they provided direct evidence for edge-hydroxylated BNNS characterizations using electron energy loss spectroscopy (EELS) maps (Fig. 4). The presence of oxygen at the edge positions and its absence within the in-plane locations was disclosed. In addition, as shown in Fig. 3c, the present authors have proposed the introduction of in-plane growth inhibitors to prepare edge exposed and –OH terminated BN porous sheets (BNPSs).76 The IR spectrum indicated the presence of sp2-hybridized B–O vibrations, which had arisen from the –OH groups that terminated at the BN(002) plane edges. The BNPSs were found to absorb visible light and showed a yellowish colour, indicating a narrowed band gap compared with h-BN. First-principle simulations have revealed that the band gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for the edge –OH terminated BN quantum dots are narrowed compared with those of –H terminated analogs and h-BN.77,78 Thus, both experimental and theoretical facts have revealed the feasibility of BNNS and BNNT band gaps modifications through edge-functionalization.
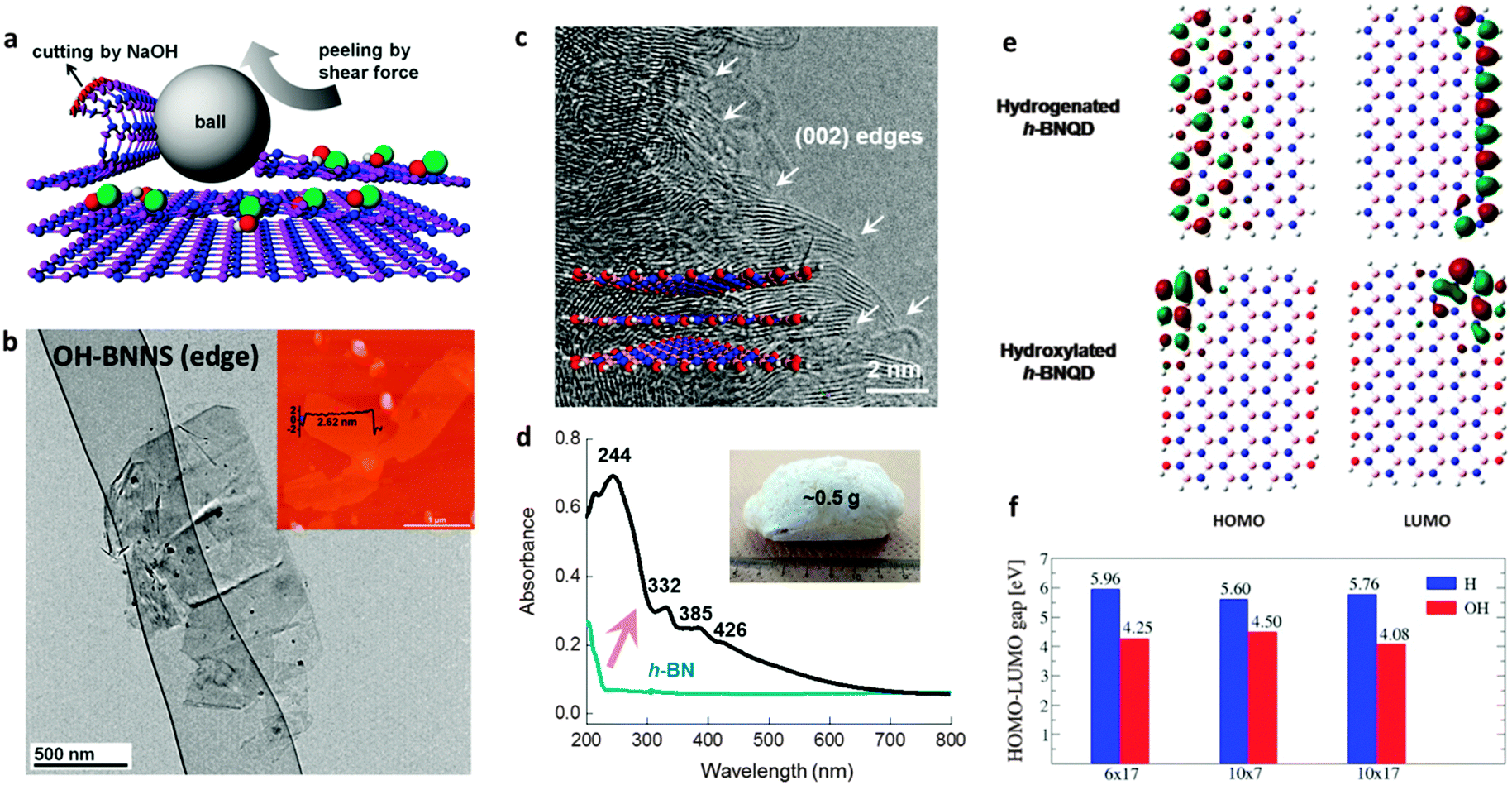 | ||
| Fig. 3 Preparation and band structures of edge-hydroxylated BN nanosheets (BNNSs). (a) Schematic illustration of the hydroxide-assisted ball milling method for h-BN exfoliation and edge functionalization. (b) TEM image of the prepared OH-BNNSs. Inset: AFM image of the material. (c) High-resolution TEM image and (d) UV-vis spectrum of basal edge exposed and –OH terminated BN porous sheets. Arrows in (c) show the edges of BN(002) plane interference fringes. Inset in (d) is the photo of a monolithic sample before mechanical crushing. (e) Comparison of calculated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) isosurfaces of edge hydrogenated (top) and hydroxylated (bottom) BN quantum dots (BNQDs). (f) HOMO–LUMO gaps of BNQDs with the edges terminated with –H and –OH groups. Simulations of (e and f) are performed at a B3LYP/6-31G** level of theory. (a and b) are reprinted from ref. 75; (c and d) are reprinted from ref. 76; (e and f) are reprinted from ref. 77 and 78 with permissions. | ||
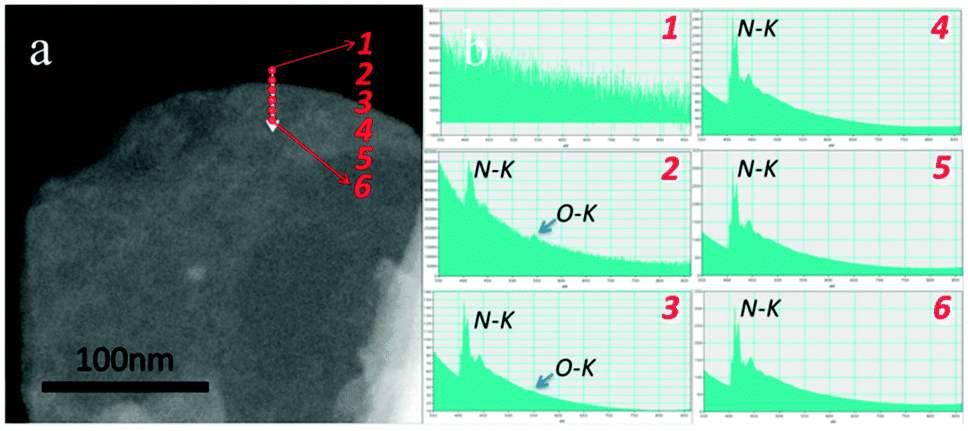 | ||
| Fig. 4 Electron energy loss spectroscopy (EELS) mapping of the edge area of OH-BNNSs. It confirms that the –OH groups are mainly located along the sheet edges (labeled as 2). There are no or very marginal O–K signals detected within basal regions of the sheet. Reprinted from ref. 79 with permission. | ||
3.2 Amino (–NH2) and amine (–NHR) groups
Just as with the –OH group, the electrophilic B centers of the BN surfaces can also be modified by –NH2 and –NHR groups. In 2007, using NH3 plasma, Zettl's group first reported that –NH2 groups could be attached to the BNNT surfaces covalently being accompanied by the amorphization of BNNT walls.82 Liao et al. found that BNNTs could be etched in ammonia solution under sonication. Entangled and freestanding BN nanoribbons were detected in the products due to –NH2 functionalization and unzipping.83 As shown in Fig. 5, Lei and Chen synthesized NH2-BNNSs via ball milling and using urea. The resultant NH2-BNNSs exhibited good solubility in water and formed colloidal solutions. Through different dehydration pathways, a light NH2-BNNS aerogel (complete dehydration via freeze drying) and a nearly transparent membrane (incomplete dehydration via natural drying in air) were obtained.81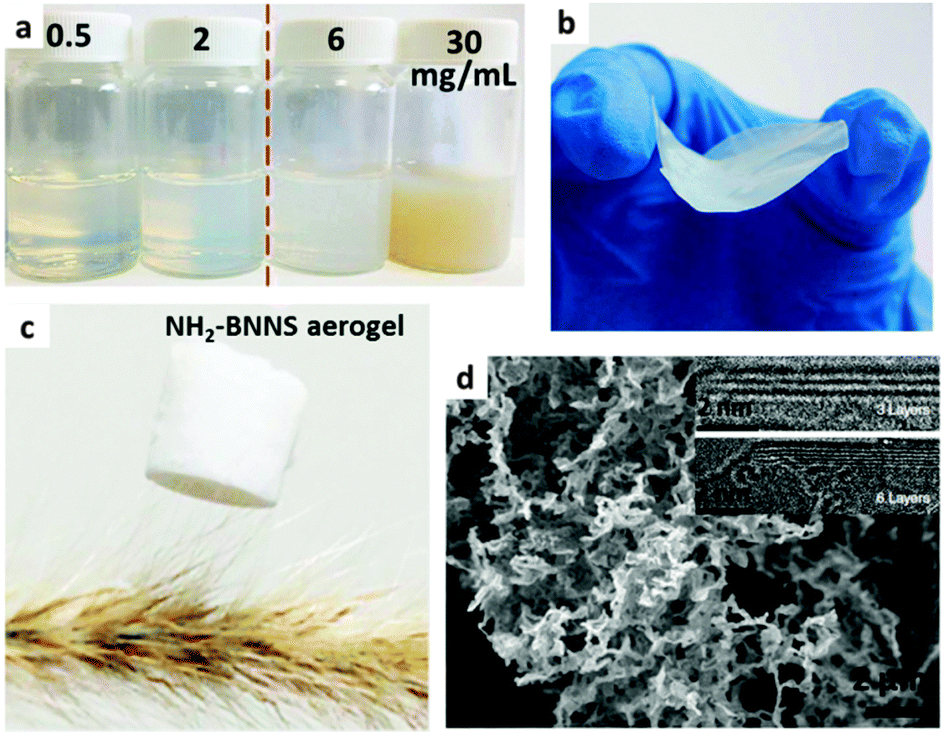 | ||
| Fig. 5 (a) Photographs of amino-functionalized BN nanosheets (NH2-BNNS) colloidal solutions with different concentrations. Photographs of a (b) prepared NH2-BNNS membrane and (c) NH2-BNNS aerogel with a low density (1.4 mg mL−1). (d) SEM image of the aerogel. Insets: High-resolution TEM images of the sheets with 3 and 6 layers, respectively. Reprinted from ref. 81 with permission. | ||
As discussed above, the positive charge of B atoms in BN structures enables them to interact with Lewis bases. Xie et al. treated the BNNTs with –NH2 terminated PEG chains, i.e. the first organic species used for BNNT functionalization, and observed that the yielded functionalized BNNTs could be introduced to and stabilized in aqueous solutions.84 After boosting the treatment temperature and time, Lin and coworkers found that such Lewis acid–base interactions between the –NH2 ends of PEG and octadecylamine take place, and the h-BN surfaces are effective for further exfoliations and functionalizations.85 Clear and transparent functionalized BNNS colloidal solutions in THF were obtained with a concentration of 0.5–1 mg mL−1.
3.3 Alkyl (–R) groups
Alkyl groups can link with BN by forming B–C–N bridged bonds or directly on B/N sites. In order to prepare the alkyl functionalized BNNSs, Sainsbury et al. designed a route to firstly insert dibromocarbenes into B–N bonds, followed by the butyl substitution of Br atoms to obtain the desired alkylated BNNSs, i.e. (n-C4H9)2C-BNNSs.86 The detailed spectroscopic characterizations of the intermediate and product confirmed the viability for BN novel surface-functionalization through such a carbene route. Another interesting method reported recently for BNNT alkylation has been based on an intermediate reduction step. BNNTs were firstly treated with sodium naphthalide solution to accept transferred electrons from the naphthalide ions. After the following reaction with 1-bromohexane, CH3(CH2)5-BNNTs were prepared (Fig. 1). The authors provided DFT calculations and IR spectra to prove that the introduced hexyl groups were bonded with B sites due to the extra injected electrons, which had to fill the empty p orbitals of B sites. This finally resulted in covalent bonding of hexyl groups with the unpaired electrons located at B sites.873.4 Other groups (–OCOR, –NHCOR, –COR, etc.)
Besides the functional groups discussed above, there have been also many reports on other functionalized BN nanostructures, including esterified (–OCOR), amidated (–NHCOR), acylated (–COR) ones, etc. These derivatives can be prepared directly from their BN precursors or via their hydroxylated (OH-BN) and aminated (NH2-BN) intermediates.The first covalent approach developed for BN nanostructure functionalization was based on the reaction between the electrophilic acyl group (–COR) and the amino-groups (defect sites) on BNNTs by refluxing.30,88 To date, there have been very few samples prepared in relation to this type of BN functionalization. The resultant modified BNNTs were dispersible in numerous organic solvents, such as chloroform, N,N-dimethylacetamide, THF, DMF, acetone, toluene, and ethanol. This property endows the functionalized BNs with good processability during fabrications of uniform and thermally/mechanically enhanced composite materials.
Starting from the as-synthesized OH-BN and NH2-BN, further derivatization reactions were conducted to construct more complex BN derivatives. –OH groups in the OH-BNNTs/OH-BNNSs could be esterified by perfluorobutyric acid (CF3CF2CF2COOH) or thioglycolic acid (HSCH2COOH), and could also be functionalized by isocyanate.71,73 Furthermore, in order to introduce polyhedral oligosilsesquioxane (POSS) structures onto the surfaces of BNNTs for dielectric modification purposes, Huang et al. designed a multi-step reaction.89 They used oxosilane to react with the –OH groups on BNNTs followed by coupling with POSS derivatives to achieve the goal. Such a reaction was also demonstrated to bond other oxosilane groups onto BNNT surfaces.90 With the presence of –NH2 groups in BNNTs, acylation of –NH2 groups became possible.82 These efforts to graft various groups onto BN nanostructures not only enrich the pool of BN chemical functionalization routes, but more importantly, provide tailored BN nanofillers for advanced composite applications, as will be discussed in Section 4.4 in more detail.
Hydrogen is the simplest group that can be used for BN structural modifications. Based on the first principle calculations, a fully hydrogenated BN nanosheet (H-BNNS) shows a narrowed band gap of 3.0–3.1 eV compared with the h-BN.91 Furthermore, the band gap of a half-hydrogenated BN nanosheet (on B sites) decreases to 2.24 eV, as predicted by Yang's group.92 Thus, this theoretically investigated BN structure should absorb the yellow-green light of the visible spectrum. The chemical adsorption energy of hydrogen is −0.31 eV per H atom, indicating that the proposed hydrogenation process is quite exothermic and thermodynamically favourable. And interestingly, the H+ reduction potential and H2O oxidation potential were calculated to locate within the band gap of this half-hydrogenated BN nanosheet, endowing it with promise for water photocatalytic splitting.92
Florine functionalization of BNNTs was achieved through one-step BNNT growth and simultaneous F functionalization using BF3 as the B and F source.93 The obtained F-BNNTs, with F concentration of ∼4 at%, were found to be semiconducting; the conductivity increases by 3 orders of magnitude compared with the pure BNNTs. First-principle simulations of fully fluorinated BN nanosheets revealed a comparable band gap (3.1–3.2 eV) to H-BNNSs, smaller than that of h-BN;91 this result was in accordance with the experimental results obtained for the BNNTs after F-functionalization.
Besides the functional groups discussed above, many other groups, such as –CH3, –CHO, –CN, etc. as well as their effects on the material band gaps/band structures, were also explored theoretically by Bhattacharya et al.91 The calculated band gaps of these chemically functionalized BN monolayers varied from 3.2 eV to 0.3 eV. Many functional groups, such as –H, –OH, –NH2, –CH3, etc., were predicted to generate a direct band gap in semiconductors. Furthermore, these chemically functionalized structures were suggested to be inherently stable taking into consideration their real vibrational modes based on phonon frequency calculations.91
3.5 Heteroatom doping (C and O)
Doping refers to the atomic substitution of B or N atoms by heteroatoms within a BN 2D in-plane structure without forming large domains made of the solo doping atoms. Introduction of special heteroatoms to the BN structures can bring some unique properties or significant changes of the pristine properties. Since pure h-BN is a wide band-gap semiconductor, one important research direction is to narrow its band gap while maintaining its original honeycomb-like structure, i.e. the opposite strategy to opening the band gap in graphene in order to meet practical semiconducting applications. In this field, however, it is difficult to find appropriate elements to be alloyed with BN within its 2D hexagonal monolayer. Many efforts, like doping BNNTs and BNNSs with carbon (C) and oxygen (O) atoms, were described; the band-gaps of the resultant doped BN products were proved to be tuneable both theoretically and experimentally. Such chemically doped BN materials can be prepared either using post-treatments or during the material growth.Within a BN monolayer, only a dynamic carbon substitution to a honeycomb-like BN structure under non-equilibrium conditions leads to the formation of C-doped BNNSs. In fact, in B–C–N materials, the thermodynamically stable situation is the phase separation, i.e., graphite and h-BN phases coexist.26,94 Carbon atoms in as-defined C-doped BN structures can be alloyed as C2 dopants and aromatic rings that are surrounded by BN lattices. At a low doping level, pairs of adjacent BN atoms in a 2D BN single-layer sheet are substituted by C2 atom pairs, as depicted in Fig. 6a–c. These C2 atom pairs can be isolated from each other, or connect together to form C6 aromatic rings.95 With an increase in C doping level, the C aromatic ring domains grow and start to merge. However, for a BC2N monolayer with a doping level of 50 at%, first-principle simulations have predicted the energy-favourable structure is composed of alternating and parallel BN or C zig-zag chains throughout the plane (structure II in Fig. 6d).96–98 Another stable isomer with every C atom bonded with one B, C and N atom is also considered to be stable due to the release of structural stress, i.e. the structure I shown in Fig. 6d. Experimentally, Loh's group prepared a BC2N monolayer with a right stoichiometry after exposure Ru(0001) facets to C2H4 and borazine vapours at controlled temperature.99 They used STM mapping to reveal the presence of predicted two stable BC2N isomers (Fig. 6d). After decoupling with the metal substrate, the measured average energy gap for the BC2N was around 2 eV (Fig. 6e), close to the predicted value.
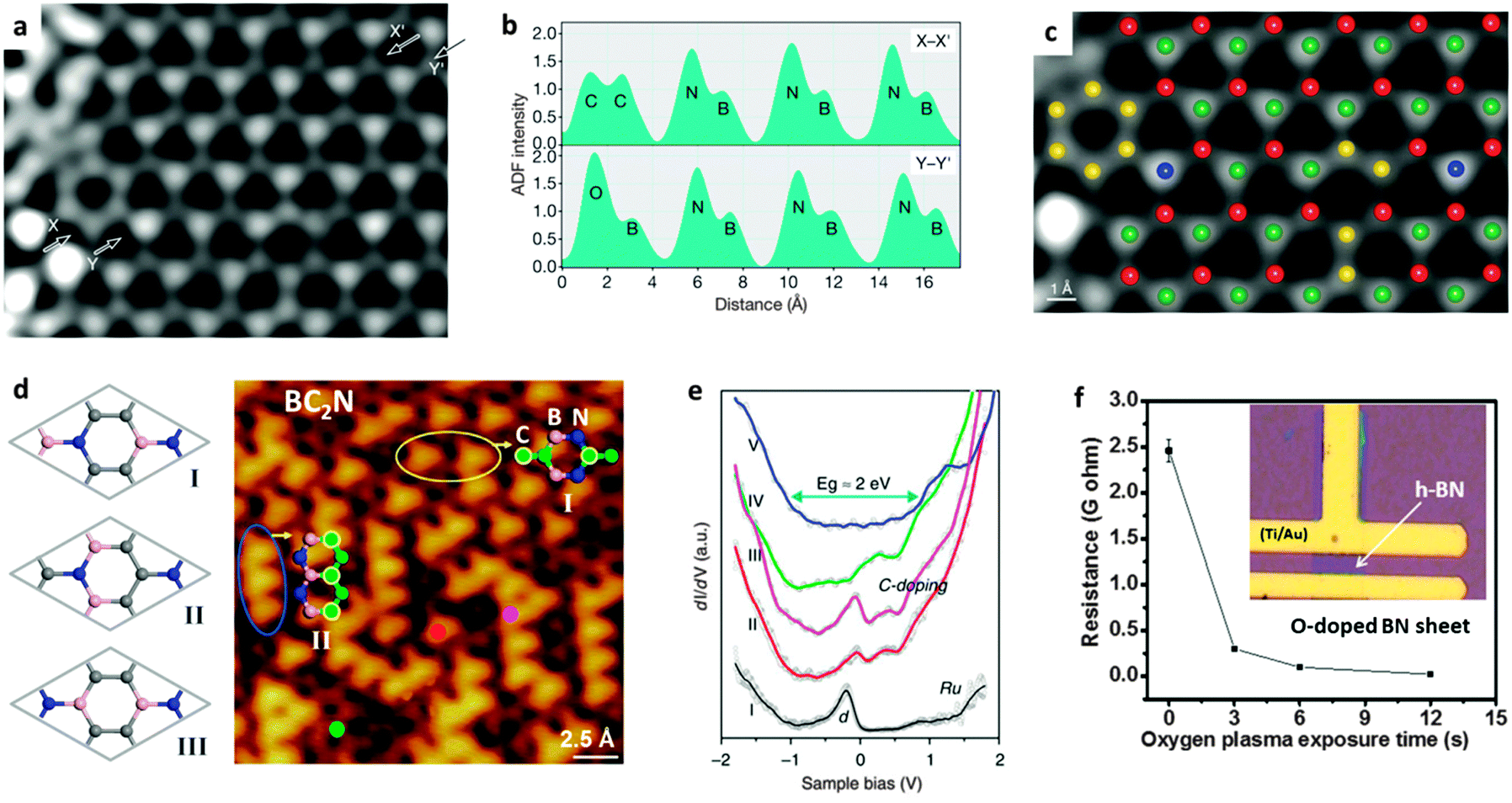 | ||
| Fig. 6 Carbon- and oxygen-doped BN nanosheets. (a) STEM annular dark-field (ADF) image of an h-BN monolayer after correction. (b) ADF intensity profiles along the X–X′ and Y–Y′ lines shown in image (a). (c) The resolved atomic structure of (a). Red, B; yellow, C; green, N; blue, O. (d) Three theoretically-predicted BC2N structures (left) and STM atomic image of the BC2N prepared on Ru(0001) surfaces reveal the two most stable BC2N isomers (right). (e) The dI/dV spectra of bare Ru(I), and the positions labeled in (d); decoupled BCN (V) shows an energy gap of ∼2 eV. (f) Electrical resistance of an h-BN film after exposure to O2 plasma over different times. Inset: The fabricated h-BN device for electrical resistance measurements. (a–c) are reprinted from ref. 95; (d–e) are reprinted from ref. 99; (f) is reprinted from ref. 100 with permissions. | ||
In addition, many other methods have also been developed and reported for the preparation of C-doped BNNSs or BNNTs.94,101–104 In this field, the major experimental obstacle is the lack of clear and efficient structure characterization technology. Clarifying a possible C-doped BN structure usually needs a comprehensive analysis combining the material chemical environment and its bond nature detections. Spectroscopy, like XPS, X-ray absorption near edge structure (XANES) spectroscopy, solid-state NMR, EELS and IR are useful tools that can give important information on whether the desired sp2-hybridized B–C and N–C bonds have been formed. Certainly, further development of state-of-the-art microscopic techniques, that are atom-resolved and element-distinguishable, such as annular dark-field imaging, aberration-corrected STEM, STM, AFM, etc. will provide ideal tools for solving the remaining challenges.
Doping BNNSs and BNNTs with O atoms has been investigated in theory and testified in experiment.100,105–107 Such doping takes place on N sites; O replacement of B atoms instead was predicted to be unstable due to the high structural deformation caused in theory.105 Interestingly, O doping will inject even electron numbers into the conjugated π bond, similar to the case of graphitic type N doping of graphene. This leads to the formation of narrow-band-gap BN nanostructures,105,108 and may also offer a new pathway for BN magnetic property manipulations,108 as well as new properties for electrochemical catalytic applications, like the oxygen reduction reaction (ORR), hydrogen evolution reaction (HER), etc. Recently, Teo and coworkers have treated a CVD-grown BN film with oxygen plasma, and found that the BN band gap decreased from 6 eV to 4.31 eV.100 And accordingly, the measured electrical conductivity of the BN film increased ∼100 times (Fig. 6f). Han et al. observed pronounced radiative transitions that were mainly located in UV and deep UV ranges in O-doped BNNTs while studying their cathodoluminescence (CL) spectroscopy.109 Later, based on first-principle calculations, Pan and Shi specified an O-doping configuration of BNNTs, i.e. via forming sp2-hybridized B3O6 fragments, that could properly explain the origin of radiative transition properties.110 Thus, CL and other luminescence spectroscopic techniques, can be applied to study the O-related defects in BN crystalline nanostructures.
3.6 BN heterostructures
2D materials, including h-BN, have attracted prime interest in microelectronics. A combination of various 2D structures with BN into stacked or in-plane built heterostructures can inherit the advantages/properties of both BN and hetero-components, and yield new functionalities, thus meeting new specific requirements for practical applications. BN and graphene domains can be merged within a single-atom-layer by covalent bonding or sandwiched in a layer-on-layer stacking fashion to realize the atomically thin circuitry. Such designs utilize the electrically insulating properties of BN and conductive properties of graphene, and arouse a wide interest of both the scientific and industrial communities. In addition, it has been shown that many other functional particles, domains and molecules, including metal oxide/chalcogenide/halide and metal nanoparticles, and MOF nanocrystals, can grow on the surfaces of BNNTs and BNNSs to form BN composites for a broad range of uses. Their interfaces could be engineered using either the weak van de Waals, π–π and charge–charge interactions or strong valence and ion bonding.Fig. 7a presents a general strategy to fabricate patterned planar graphene/h-BN heterojunctions. Firstly, BN or graphene is grown on a substrate surface with whole coverage by chemical vapour deposition (CVD), followed by patterned etching and epitaxial re-growth of the second component.111Fig. 7b and c illustrate SEM images of alternating strip-patterned and non-patterned graphene/h-BN planar heterojunctions, respectively. A focused ion beam (FIB) method allows one to generate thin lines as narrow as ∼100 nm. A STM image (Fig. 7d) reveals the seamless links between the lower graphene and upper BN domains; the height difference is much smaller than the interlayer spacing of graphene or BN (3.33–3.37 Å), indicating that the decent in-plane connection between graphene and BN domains has indeed been obtained. However, the electrical properties are rather abrupt across a one-dimensional interface.112 Without etching, Gong et al. found that a partial conversion of the graphene lattice to BN had happened after treatment with H3BO3–NH3 at a high temperature; this makes it possible to fabricate uniform graphene–BN planar heterojunctions.113 Besides the planar graphene–BN heterojunctions, a multiple-step CVD growth29,114 and one-step co-segregation growth,115 of layer-on-layer graphene–BN heterostructures have also been recently developed.
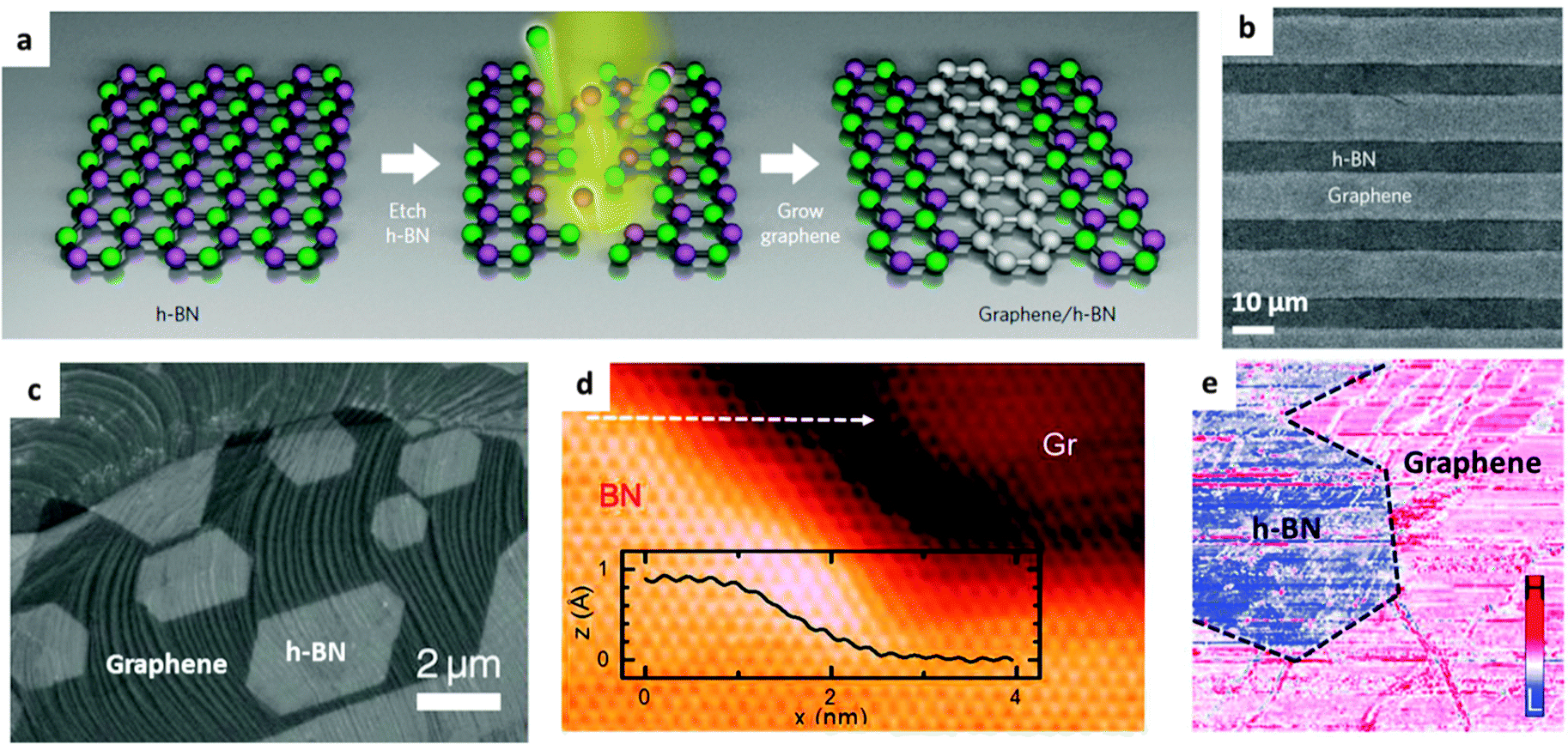 | ||
| Fig. 7 Fabrication and characterization of BN–graphene heterostructures. (a) Schematic illustration of the procedure for in-plane BN–graphene heterostructure fabrications. Briefly, CVD-grown h-BN sheets are firstly etched by argon ions to obtain the desired patterns, and then CVD growth of graphene on the etched regions is performed. (b) SEM image of strip-patterned BN–graphene heterostructures. (c) SEM image of graphene–BN monolayer heterostructure. The CVD-grown graphene was etched by H2 and then used for epitaxial growth of BN along the graphene edge. (d) STM image of a graphene–BN boundary. The inset is the height profile along the white dashed arrow. (e) The differential tunnelling conductance image at the same region in (d). (a and b) are reprinted from ref. 111; (c–e) are reprinted from ref. 112 with permissions. | ||
Due to the uneven ionic properties of a B–N bond, B and N atoms are slightly positively and negatively charged, respectively. This property makes BN materials interactive with both charge donors and acceptors. In an aqueous solution, BN surfaces are negatively charged (due to the tendency to adsorb more OH−), providing possibilities for adsorbing metal ions and growing uniform metal oxide/chalcogenide/halide, metal and MOF nanoparticles on the surfaces. These include TiO2,116–119 ZnO,120 SnO2,55 WO3,121 In2S3,122 AgBr,123 AgI,124 Fe3O4,125 Au,118 Ag,126,127 Pt,128 ZIF-8,129etc.130 Besides, a lot of other molecules/ions can also interact with BN nanomaterials through weak van de Waals, π–π, and charge–charge interactions, such as DNA, polymers, surfactants, small biomolecules, etc.31,131 Future advances in this field may bring revolutionary breakthroughs in electronic-, biomedical-, and energy-related researches.
4. Properties and applications
4.1 Water solubility and biological applications
The unique hollow structures of CNTs, and, later, the 2D structures of graphene oxide, have propelled great interests for these materials in biological applications, such as tumour labelling,133–136 sensing and targeting,136–138 drug/DNA/RNA delivery,136etc. However, toxicity related with the dose, time, and shape of these carbon-based nanomaterials has been confirmed through in vitro and in vivo investigations.139–142 BNNTs or other BN nanostructures, instead, seem to have a better biocompatibility and lower cytotoxicity than their C cousins, although more conclusive results are still required.143 In order to make the functionalized BN nanomaterials well qualified for biological applications, two main issues need to be addressed.The first challenge relates with the materials' water solubility/dispersibility. It is known that a material with poor solubility/suspension in physiological solutions is difficult to introduce into biological systems, while pure BN nanomaterials exhibit notable hydrophobicity when interacting with water or aqueous solutions.144–146 Over the last 10 years, many efforts were made to improve the solubility/suspension of BN materials via surface functionalization (as discussed in Section 3), interactions or wrapping by guest molecules. The phenomenon of improved water wettability on BNNS surfaces has been directly observed after hydroxylation through air plasma treatment (Fig. 8a).74 In recent years, remarkable progress has been made toward overcoming this severe problem. The present authors reported highly water-soluble BNs, which could be stabilized in water at a concentration as high as 2 mg mL−1. These materials were synthesized by an unconventional “reserved functionalization” method. The achieved concentration was more than 2 orders of magnitude higher, as compared with other reports, and also comparable with GO solutions.85,131 The formed colourless and transparent BN colloidal solutions were confirmed to be stable over at least a week. More recently, the NH2-BNNSs prepared by ball-milling method have been claimed to form a 30 mg mL−1 colloidal solution in water.81 These progressions indicate that the challenges related to BN materials' poor water solubility/dispersibility can be effectively addressed via chemical functionalization.
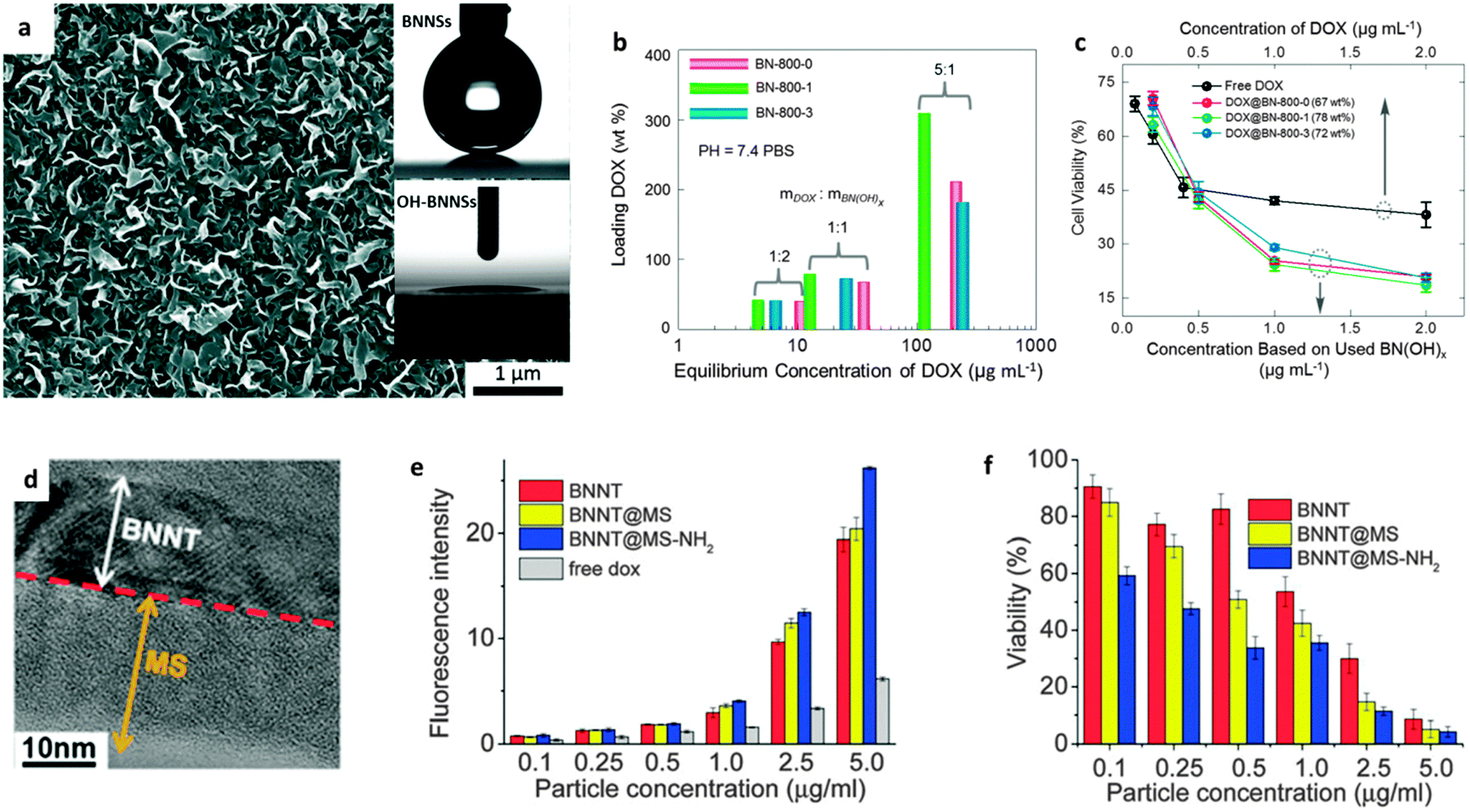 | ||
| Fig. 8 Hydrophility and drug delivery applications of functionalized BN nanomaterials. (a) SEM image of vertically-grown BNNS arrays on a Si/SiO2 substrate. The insets show contact angle measurements of BNNS and OH-BNNS surfaces, indicating that the hydrophobic BN surfaces become hydrophilic after hydroxylation. (b) Drug loading and (c) delivery performances of hydroxylated BNs prepared from the reaction between g-C3N4 and boric acid. The fewer anti-cancer drugs that loaded on the OH-BNs show a much better potency for reducing the cell viability. (d–f) Drug delivery of BNNTs coated with mesoporous silica (BNNT@MS) and the sample with the surfaces further grafted with –NH2 groups (BNNT@MS-NH2). (d) TEM image of BNNT@MS-NH2. (e) Fluorescence intensity of doxorubicin (DOX) uptake by LNcap prostate cancer cells after loading on BNNT, BNNT@MS and BNNT@MS–NH2. (f) Viability of LNcap prostate cancer cells after culturing with DOX-loaded BNNT, BNNT@MS and BNNT@MS-NH2. (a) is reprinted from ref. 74; (b and c) are reprinted from ref. 70; (d–f) are reprinted from ref. 132 with permissions. | ||
The second important issue is the biocompatibility problem of a material. The toxicity of carbon nanomaterials has been related to their aspect ratio, purity, concentration and dispersion reagents, also it is affected by the used cell/tissue/organ-types.147 As for BNNTs, there have been many toxicological results showing that they are nontoxic or have very low toxicity. Assaying using BNNTs with different purities and geometry, different dispersion reagents and cell types has proved this.148–152 As expected, the sphere-shaped BN nanoparticles with a low aspect ratio showed acceptable cytotoxicity.153 However, negative cytocompatibility was found when using an ultra-concentrated BNNT culture medium with the BNNT concentrations of 100 μg mL−1,154 and also long tubes (≥10 μm) in Horváth's report.155 As shown in Fig. 8b and c, the recently developed biocompatible OH-BN in our group could load ∼300 wt% of doxorubicin (DOX) drug, and exhibit much higher potency for reducing the viability of LNCaP cancer cells than free drugs.70 To improve the hydrophilicity and drug load capacity, Li et al. coated the shortened BNNTs with mesoporous SiO2, and found that the functionalized BNNTs could indeed adsorb more DOX drugs than the bare ones and show an improved cancer cell killing ability (Fig. 8d–f).132 These authors also fabricated BNNTs coated with europium doped sodium gadolinium fluoride (NaGdF4:Eu) for drug delivery, and found it to be advantageous for target directing and manipulating in an external magnetic field.156 In addition to drug delivery, more applications of BN nanomaterials are under exploration, e.g. using BNNTs for controlled differentiation of mesenchymal stem cells.157 Since the studies of BN nanomaterials for biological and biomedical applications have emerged only in the past few years, it is believed that the research depth and width should be developed in the near future, particularly with respect to functionalized nano-BNs.
4.2 Microelectronics
To date, two main types of applications have been targeted for BN materials in the microelectronics field. Firstly, while employing the electrically insulating property, BN nanosheets may be used as separators and barriers in highly conductive components, such as barriers utilized in field-effect tunnelling transistors and the atomically thin insulators to separate metallic channels. Secondly, benefiting from its surface smoothness, BN is also applied as a substrate for high-performance devices. h-BN surfaces are extremely smooth, free of charges and dangling bonds, and have excellent physical properties, such as superb in-plane mechanical strength, high chemical and thermal stability, and high thermal conductivity, etc. These properties make the h-BN a unique 2D material for the design and fabrication of high quality devices.Successful fabrication of hybrid structures allows one to build electrically isolated graphene devices with a monoatomic thickness, as shown in Fig. 9a and b. It is known that modern integrated circuitry presumes a precise spatial control of electrical properties of the used thin films, which in theory, could be thinned down to a monoatomic thickness. Levendorf et al. reported a versatile and scalable synthesis of basal junctions between electrically conductive graphene and insulating h-BN under precise spatial control.158 Conducting behaviour confined to the patterned graphene areas was observed; the h-BN segment showed no conductivity. Using electrostatic force microscopy, h-BN was also confirmed to be free of small conducting BN–C pockets that might form during the growth. This provides an important step toward developing atomically thin integrated circuits.
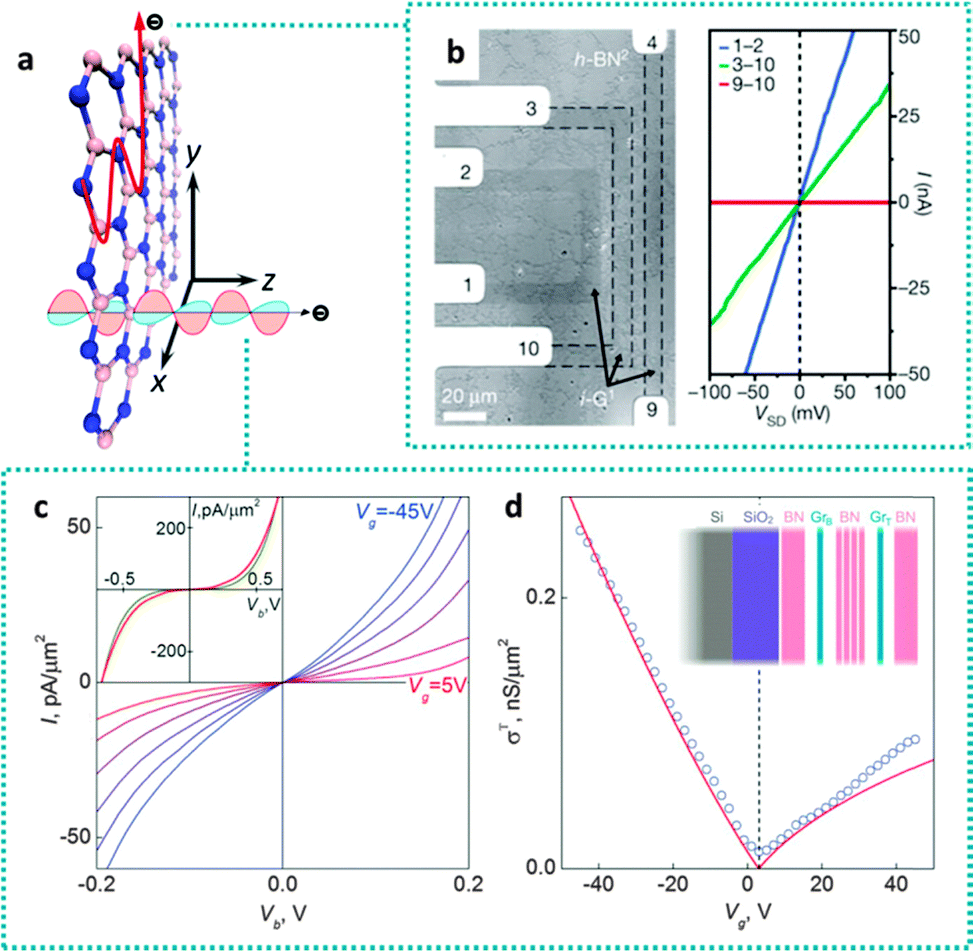 | ||
| Fig. 9 Electron transport behaviours along and through the BN basal plane directions. (a) Schematic illustration of electron conduction along the two directions in a BN nanosheet. (b) Optical image of a graphene–BN heterostructure device with 5 contacting electrodes (left). The right figure illustrates I–V curves for the indicated device. Graphene shows a conducting behaviour while h-BN is insulating along the basal plane direction. (c) I–V curves of a graphene–BN–graphene device with 6 ± 1 BN layers as the tunnel barrier. The inset compares the experimental (red curve) with theoretical (dark) simulation results at Vg = 5 V. (d) Zero-bias conductivity versus Vg. Experimental data (circles) are fitted with molding lines. The inset is the illustration of the graphene–BN–graphene device. (b) is reprinted from ref. 158; (c and d) are reprinted from ref. 159 with permissions. | ||
The zero-band-gap property of graphene is the main barrier for field-effect transistor (FET) applications because a low-power-dissipation OFF state is difficult to achieve under such a condition. Based on the quantum tunneling effect in a thin h-BN barrier, Britnell et al. fabricated a sandwich-like BN/graphene/BN (BN/G/BN) FET with high ON/OFF ratios, as depicted in Fig. 9c and d.159 The transit time for the electrons tunneling through a-few-layer BN barrier is expected to be only a few femtoseconds, thus faster than that for the conventional submicrometer-sized planar FETs. The field effect increases gradually up to gate voltages of ±50 V. The response performances (Fig. 9d) of a device containing 4-to-7-layer BNs show changing factors of ∼20 for negative and 6 for positive gate voltages, corresponding to a high ON/OFF ratio of >104.159 Other FET devices working on a similar principle have also been investigated while using various conducting materials (graphite, graphene, and gold) on the BN barrier sides. The tunneling current exponentially depends on the number of BN atomic layers; these function as a defect-free dielectric with a high breakdown field, offering a great potential to design tunneling devices and FETs with a high carrier density in the conducting channel.160
Mayorov et al. have observed that devices made from graphene encapsulated in h-BN exhibited a pronounced negative bend resistance and an anomalous Hall effect, as a consequence of room-temperature ballistic transport at a micrometer scale for a wide range of carrier concentrations.162 By aligning the crystallographic orientations of the two graphene layers in a G/BN/G heterostructure, Mishchenko et al. realized the resonant tunneling under both energy and momentum conservation. Such tunnel circuits can be operated in a THz regime.163 The electrically insulating BNNTs grown on graphene also enable its application as an effective digital switch. The STM characterizations of graphene–BNNT heterojunctions revealed a switching ratio as high as 105 at a turn-on voltage as low as 0.5 V. DFT calculations suggest that a mismatch of the density of states (DOS) is responsible for these novel switching behaviours.164
h-BN can provide an ultra-smooth and clean surface to improve device quality and its performance. Fig. 10a shows a device with a 2D graphene crystal encapsulated in h-BN, which was confirmed to be atomically flat by ADF-STEM and atomic force microscopy (AFM) characterizations (Fig. 10b).161 As reported by Wang et al., the resultant transport of the device reached room-temperature mobility over 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1. This realizes the large carrier density, and ballistic transport over distances >15 μm (only limited by the device size). Such a performance reflects the pristine electronic characteristics natural for graphene. It was also uncovered that there had been an extremely large local magnetoresistance of ∼2000% at 400 K and a non-local magnetoresistance of >90000% in a few-layer graphene/BN heterostructure.165 The local magnetoresistance is considered to arise from the large differential transport parameters (like carrier mobility) across the graphene layers in a normal magnetic field, whereas the non-local magnetoresistance is due to the magnetic field induced through the Ettingshausen–Nernst effect. Apart from graphene, other 2D materials can be envisioned for such applications. For example, single-layered WS2 shows a direct bandgap, which is attractive for optoelectronic and FET applications. However, the low mobility and environment-sensitive electronic characteristics limit its practical device applications. The remarkable improvement in the electrical characteristics for a single-layer WS2 FET was demonstrated once it had been placed onto h-BN.166 The structure showed a high mobility of 214 cm2 V−1 s−1 at room temperature, and 486 cm2 V−1 s−1 at 5 K, with the output current ON/OFF ratio of ∼107 at room temperature. A high field-effect mobility of ∼1350 cm2 V−1 s−1, ON/OFF ratios >105 at room temperature, together with a quantum oscillation phenomenon, have also been observed for a monolayer black phosphorus (BP) that is encapsulated into h-BN layers.167 The h-BN protects the BP layer from quick corrosion under ambient conditions. Such BN/WS2/BN or BN/BP/BN sandwich-like heterostructures offer a new way to develop high-quality and durable electronic devices.
000 cm2 V−1 s−1. This realizes the large carrier density, and ballistic transport over distances >15 μm (only limited by the device size). Such a performance reflects the pristine electronic characteristics natural for graphene. It was also uncovered that there had been an extremely large local magnetoresistance of ∼2000% at 400 K and a non-local magnetoresistance of >90000% in a few-layer graphene/BN heterostructure.165 The local magnetoresistance is considered to arise from the large differential transport parameters (like carrier mobility) across the graphene layers in a normal magnetic field, whereas the non-local magnetoresistance is due to the magnetic field induced through the Ettingshausen–Nernst effect. Apart from graphene, other 2D materials can be envisioned for such applications. For example, single-layered WS2 shows a direct bandgap, which is attractive for optoelectronic and FET applications. However, the low mobility and environment-sensitive electronic characteristics limit its practical device applications. The remarkable improvement in the electrical characteristics for a single-layer WS2 FET was demonstrated once it had been placed onto h-BN.166 The structure showed a high mobility of 214 cm2 V−1 s−1 at room temperature, and 486 cm2 V−1 s−1 at 5 K, with the output current ON/OFF ratio of ∼107 at room temperature. A high field-effect mobility of ∼1350 cm2 V−1 s−1, ON/OFF ratios >105 at room temperature, together with a quantum oscillation phenomenon, have also been observed for a monolayer black phosphorus (BP) that is encapsulated into h-BN layers.167 The h-BN protects the BP layer from quick corrosion under ambient conditions. Such BN/WS2/BN or BN/BP/BN sandwich-like heterostructures offer a new way to develop high-quality and durable electronic devices.
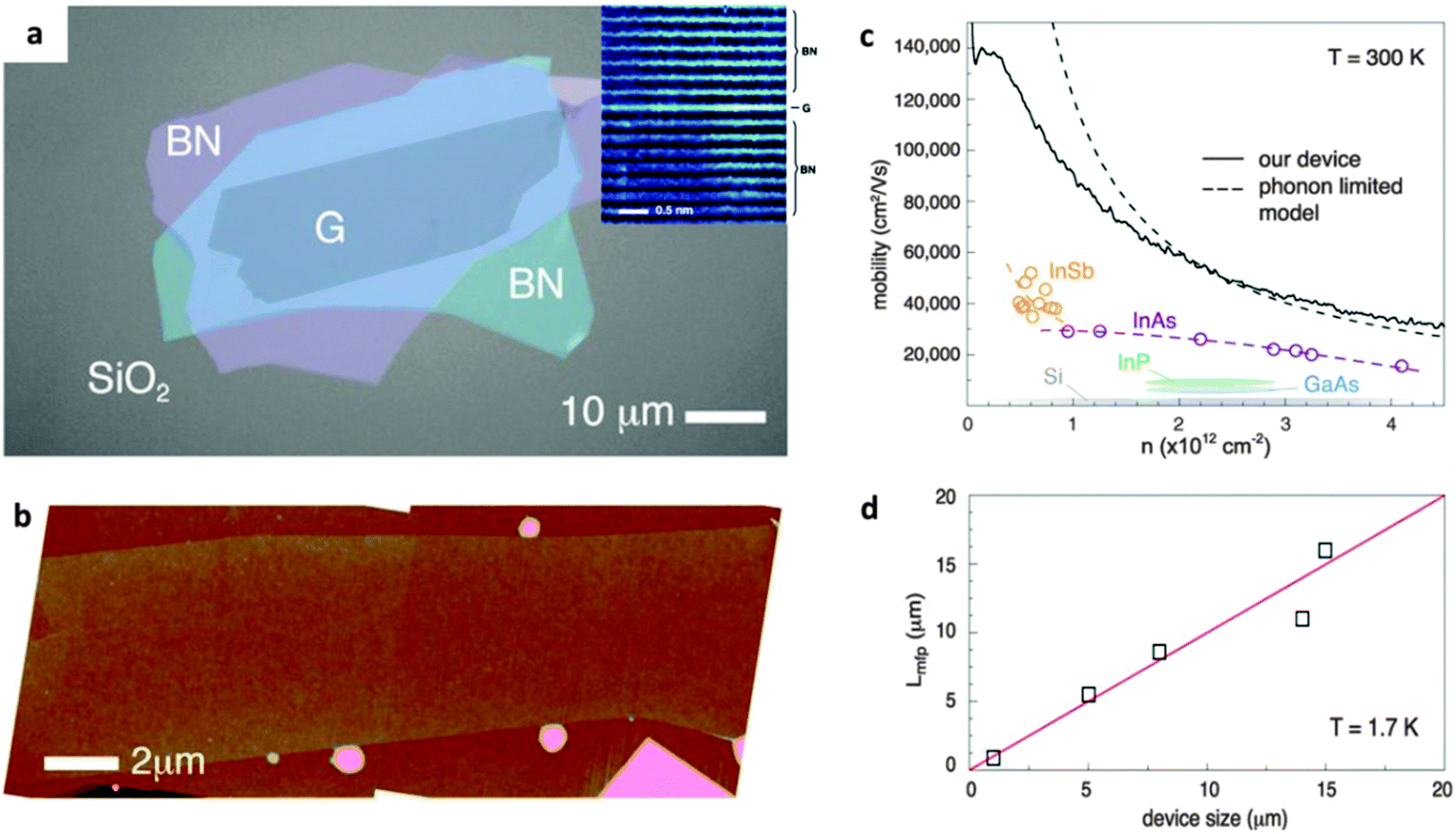 | ||
| Fig. 10 (a) Optical image of a hybrid BN/graphene/BN heterostructure. Inset: ADF-STEM image of the cross-section region of the heterostructure. (b) AFM image shows the flatness (free of wrinkles and bubbles) characteristics of the heterostructure. (c) Mobility versus density at room temperature of the device. The dashed curve depicts the theoretical mobility limit. (d) Lower bound mean free path of the devices of various sizes at T = 1.7 K. Reprinted from ref. 161 with permissions. | ||
4.3 Nanophotonics
In recent years, in-depth understanding of chemical and physical properties of h-BN and its heterostructures has boosted the development of BN-based photonic devices. Nanophotonics refers to the studies of confined lights in nanoscale dimensions (much smaller than a typical light wavelength) and their interactions with a nanomaterial.A mixture of polar dielectric materials at the atomic scale, such as alternating and complementary BN structures, can generate new nanophotonic devices resulting from the hybridized optical phonon behaviour of the components. These heterostructures could be used in nanophotonics ranging from mid-infrared to terahertz applications.168 h-BN was also reported to work as a multimode waveguide for hyperbolic phonon polariton propagations due to its unique dielectric property in the basal plane. Moreover, hybridization of surface plasmon polaritons in graphene with hyperbolic phonon polaritons in h-BN gives a waveguide tunability. Therefore, a graphene/BN heterostructure can be viewed as an electromagnetic metamaterial since its constituting elements alone can't exhibit the required properties.169 Ju et al. reported photo-induced doping in van der Waals G/BN heterostructures, which enables repeatable writing and erasing of the doping features under a visible light and preserves the very high mobility of G/BN. Microscopically coupled optical and electrical responses of G/BN heterostructures were also discovered.170 Likewise, through the advent of van der Waals heterostructures, Woessner et al. exploited near-field microscopy to detect plasmons propagating in a high-quality graphene within the BN/G/BN heterostructure. Unprecedentedly low plasmon damping combined with strong field confinement had been found, which was attributed to intrinsic thermal phonons in the graphene and dielectric losses in the h-BN (Fig. 11).171 Yang et al. successfully traced plasmons in h-BN with a lifetime of 1.6 picoseconds, which arise from the counterintuitive coupling of the transverse optical phonons in BN. This opens up the coupling mechanisms between plasmons and phonons in G/BN van der Waals heterostructures, and may provide a new way to tune the plasmon behaviour in plasmon–phonon polariton devices.172
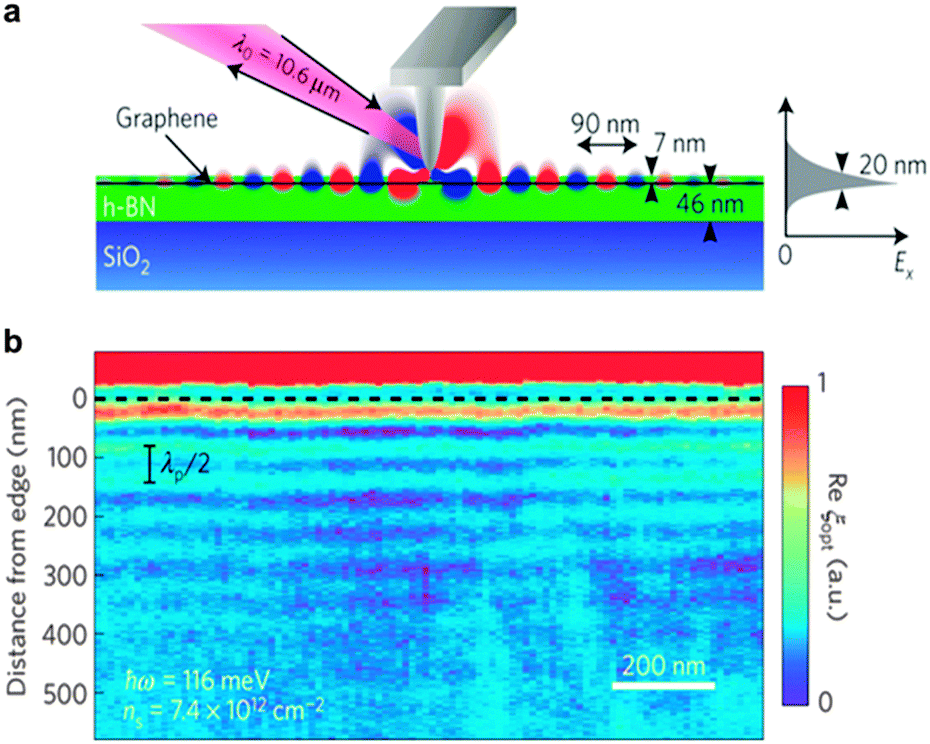 | ||
| Fig. 11 Plasmon propagating in a graphene/BN heterostructure. (a) Schematic of the plasmon imaging using a scanning near-field optical microscope, including a probe tip, excitation laser and detection apparatus. Plasmons are generated from the tip. The right panel shows the simulated field confinement in the out-of-plane plasmons along the x direction (Ex). (b) Recorded optical signals of the graphene edge positions (dashed line) at room temperature. The interference fringes are edge-reflected plasmons. Reprinted from ref. 171 with permissions. | ||
4.4 Mechanical and thermal properties and composite applications
| Ec = EmVm + EfVf | (1) |
| Vf = 1 − Vm |
 | (2) |
σf and σm are the shear stress of fillers and matrix, εf and εm are the strains of fillers and matrix. Thus, g reflects the interfacial shear modulus of the fillers and matrix. Considering that the applied load acts parallel to the composed phases, while the caused strain follows a series model, the empirical Halpin–Tsai equation can be deduced as follows:181,182
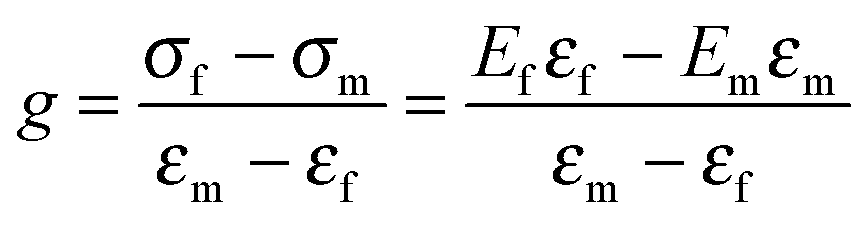
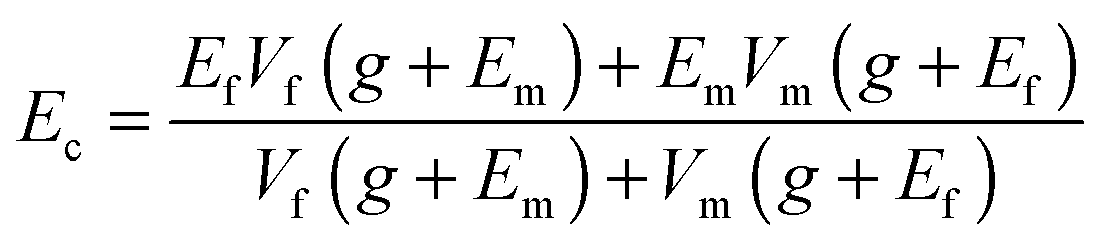 | (3) |
However, the critical problem faced by researchers during their mechanical reinforcement endeavours is how to efficiently transfer the loads to the nanofillers. Since BN fillers and the employed host matrices interact via weak van de Waals forces or π–π interactions, it is important to chemically modify the surfaces of BN nanofillers with appropriate functional groups to enhance such interactions and/or introduce other strong interactions.
As mentioned in the chemical functionalization section, BNNTs and BNNSs, that are functionalized with –OH, –OCOR, –COR, –R groups, have revealed a considerable reinforcement of polymer matrices compared with the effects of solo BN nanostructures. In 2006 Zhi et al. carried out a pioneering work utilizing BNNTs for polystyrene enhancement once the macroscopic fabrication of high-quality BNNTs had been realized within the same group.174 Afterward, the present authors' and Coleman's groups performed a considerable fraction of work in this field; various BN nanostructures and their functional derivatives were developed as advanced fillers for different polymers (Table 2). In this respect some conclusions can be drawn as follows: (1) both solo and functionalized BNNTs/BNNSs are effective for elastic modulus and tensile strength improvements for a variety of polymeric matrices, including PS, PVB, PMMA, PEVA, PVA, PU, PC, epoxy, PE, PNIPAM, etc.23,25,26,71,73,75,79,89,174,175,177,179 With an increase in the BN nanofiller fraction in a polymer, the elastic modulus continuously increases, while the tensile strength of the composites shows a more complicated trend; the effect depends on the BN filling fractions and the matrix types.25,179 Drawing of the fibrous BN composite can remarkably improve both its elastic modulus and tensile strength along the axial direction.176 (2) The chemically functionalized BNNTs/BNNSs are more effective for composite reinforcement, as compared to bare BNNTs/BNNSs. In fact, functionalization of BN nanostructures is able to modify the chemical and physical properties of the material surfaces, which improves the BN–polymer interfacial interactions and stimulates a more effective mechanical load transfer to BN structures. As shown in Fig. 12 and Table 2, Sainsbury et al. fabricated OH-BNNSs and OCN(CH2)6NHCO2-BNNSs to serve as nanofillers for PVA and PU, respectively. Their performances in composites were compared with those of unmodified BNNSs. The results show that adding 0.1 wt% OH-BNNSs can achieve a 186% increase in the elastic modulus of a composite compared with a 20% increase if using the same amount of standard BNNSs instead.73 Based on the Halpin–Tsai equation (eqn (3)), this result reveals near 70 times enhancement of the g values for the OH-BNNSs compared to the bare BNNSs in their PVA composites if the functionalized OH-BNNSs were assumed to have the same elastic modulus as the BNNSs. The advantages of functionalized BN nanostructures for the composite strength improvement are also remarkable, as compared with bare BN nanofillers.71,73 (3) For a specific polymer, a dedicated design of functionalized BN nanofillers, that can maximize the interactions with the polymeric host will optimize the composite mechanical reinforcements. Here, numerous factors need to be considered while designing new BN nanofillers, such as polymer structural features (linear or branched chains), hydrophilicity or lipophilicity, containing groups in the targeted polymers, etc. A single functionalization mode is not expected to be versatile and effective for all polymeric matrices.
| Polymeric substrates | BN fillers (fraction) | Elastic modulus | Tensile strength | Thermal conductivity | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| (GPa) | Reinforcement (%) | (MPa) | Reinforcement (%) | (W m−1 K−1) | Enhancement (fold) | |||
| Abbreviations: PS = polystyrene; PVB = poly(vinyl butyral); PMMA = poly(methyl methacrylate); PEVA = poly(ethylene vinyl alcohol); PC = polycarbonate; PVA = poly(vinyl alcohol), TPU = thermoplastic polyurethane; UHMWPE = ultrahigh molecular weight polyethylene; PU = polyurethane; PE = polyethylene; POSS = polyhedral oligosilsesquioxane; PNIPAM = poly(N-isopropylacrylamide). | ||||||||
| PS | BNNTs (1 wt%) | 2.67 | 21 | 29.6 | 22.3 | — | 174 | |
| PS | BNNTs (35 wt%) | — | — | 3.61 ± 0.21 | 19.1 | 175 | ||
| PVB | BNNTs (18 wt%) | — | — | 1.81 ± 0.08 | 6.5 | 175 | ||
| PMMA | BNNTs (24 wt%) | — | — | 3.16 ± 0.26 | 20.1 | 175 | ||
| PEVA | BNNTs (37 wt%) | — | — | 2.50 ± 0.05 | 13.7 | 175 | ||
| PMMA | BNNSs (0.3 wt%) | 2.13 | 22 | ∼42 | 11 | — | 23 | |
| PC | BNNSs (2 wt%) | ∼2.4 | ∼22 | ∼46 | ∼35 | — | 26 | |
| TPU | BNNSs (20 wt%) | ∼0.09 | ∼2150 | ∼21 | ∼0 | — | 25 | |
| UHMWPE (drawn treatment) | BNNSs (11 wt%) | 7.60 ± 2.3 | 152 | 605.59 ± 235.3 | −14.8 | 176 | ||
| Epoxy | BNNSs (40 wt%) | — | — | 6 | 14 | 177 | ||
| PVA | BNNSs (50 vol%) | 30 | — | 178 | ||||
| Nanofibrillated cellulose | BNNSs (50 wt%) | — | ∼50 | ∼−73 | 145.7 | 4162 | 179 | |
| Epoxy | POSS-BNNTs (30 wt%) | — | — | 2.77 | 13.6 | 89 | ||
| PC | BNNTs (1 wt%) | ∼2.5 | 13.6 | ∼48 | ∼5 | — | 71 | |
| PC | OH-BNNTs (1 wt%) | ∼2.8 | 31.8 | ∼49 | ∼25 | — | 71 | |
| PVB | BNNTs (1 wt%) | ∼2.6 | 25.0 | ∼45 | ∼7 | — | 71 | |
| PVB | OH-BNNTs (1 wt%) | ∼2.8 | 36.5 | ∼49 | ∼32 | — | 71 | |
| PVA | BNNSs (0.1 wt%) | 0.452 | 20 | 34 | 15 | — | 73 | |
| PVA | OH-BNNSs (0.1 wt%) | 1.08 | 186 | 49 | 66 | — | 73 | |
| PU | BNNSs (unknown) | 0.215 | 13 | 33.7 | 35 | — | 73 | |
| PU | OCN(CH2)6NHCO2-BNNSs (unknown) | 0.229 | 21 | 54.2 | 118 | — | 73 | |
| PE | OH-BNNSs (5 wt%) | 0.268 | 127 | 13.5 | 69 | — | 75 | |
| PNIPAM | OH-BNNSs (0.07 wt%) | — | — | ∼0.82 | 0.41 | 79 | ||
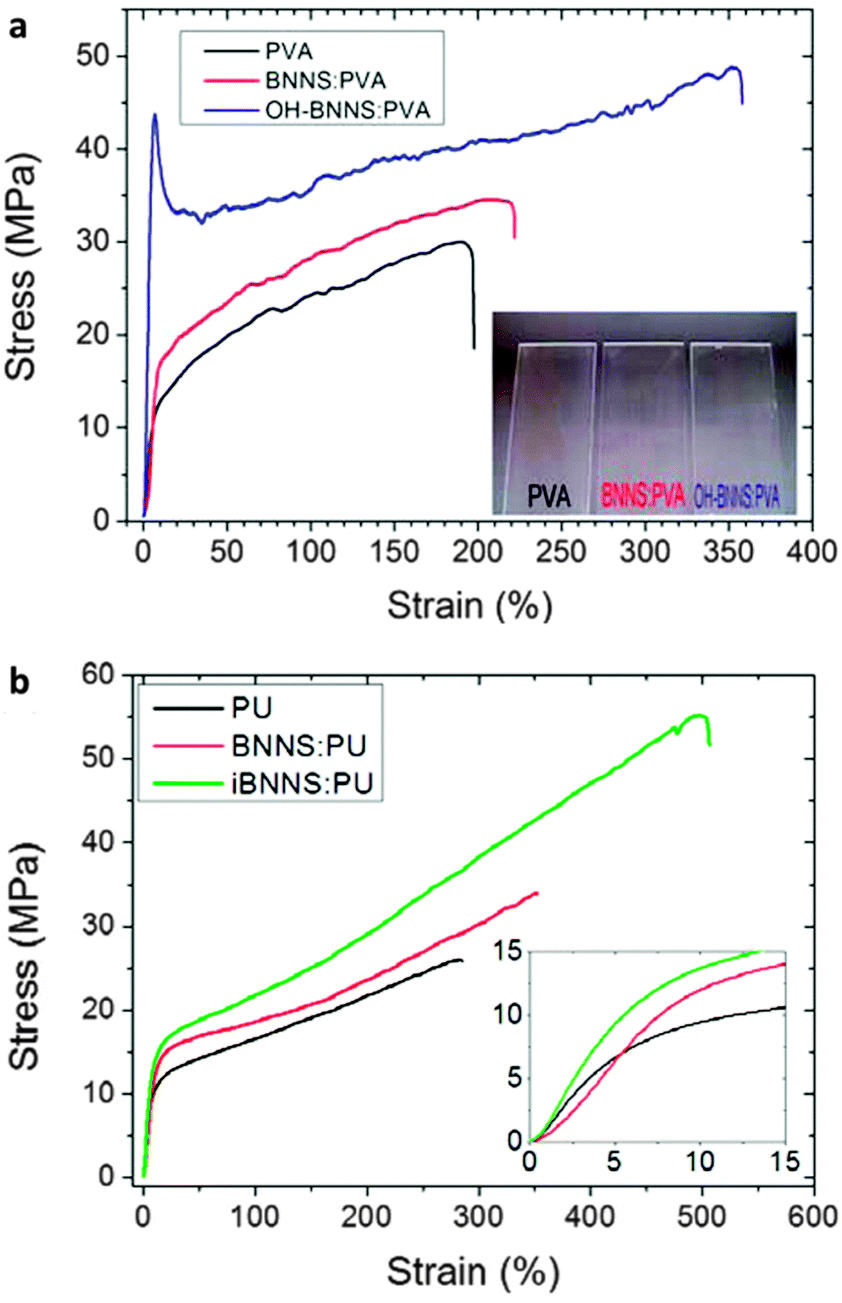 | ||
| Fig. 12 Mechanical reinforcement effects of functionalized BNNSs for polymers. (a) Typical stress–strain curves of polyvinyl alcohol (PVA), BNNSs/PVA and OH-BNNSs/PVA composites. Inset is the photograph of the as-prepared membrane samples on glass slides. (b) Typical stress–strain curves of polyurethane (PU), BNNSs/PU and iBNNSs/PU composites with the initial onset region inset. The iBNNSs is the isocyanate-functionalized-BNNSs: OCN(CH2)6NHCO2-BNNSs. Reprinted from ref. 73 with permission. | ||
Usually, a large fraction of BN fillers is necessary in order to let the polymer gain a clear improvement in thermal conductivity, much larger than the required fraction in the case of mechanical reinforcement. Under filling with 18–40 wt% BNNTs/BNNSs, over 10-fold improvements in thermal conductivity for different polymers was frequently achieved.175,177,183 It is noted that Zhu et al. fabricated a BNNSs/cellulose paper (Fig. 13a–c), and reported that the sample loaded with 50 wt% of BN phase shows a thermal conductivity of 145.7 W m−1 K−1 along the paper plane, which is over 4000 times improvement compared to bare cellulose and is also larger than that of the commercial Al alloy 135.179 Such exceptionally good performance may be governed by an improved ordering of BNNSs after filtering and mechanical pressing processes, and the possibility of measuring conductivity along the basal plane direction. And interestingly enough, the edge-hydroxylated BNNSs prepared via an h-BN reaction with steam at high temperatures were found to be useful as fillers in poly(N-isopropyl-acrylamide) (PNIPAM) hydrogel; the thermal conductivity of the hydrogel improved by 41% under incorporation of only 0.07 wt% of OH-BNNSs (Fig. 13d–f).79 This indicates that an effective thermal conductivity network forms via effective interactions between the hydrophilic –OH groups at the BNNS edges or through the hydrogen bonds with PNIPAM molecules. Other efforts, such as utilizing multiple BN nanostructures for thermal conductivity network construction, were also made.184 These results have proven the great advantages of functionalized BN nanomaterials for thermal conductivity enhancement of the composites.
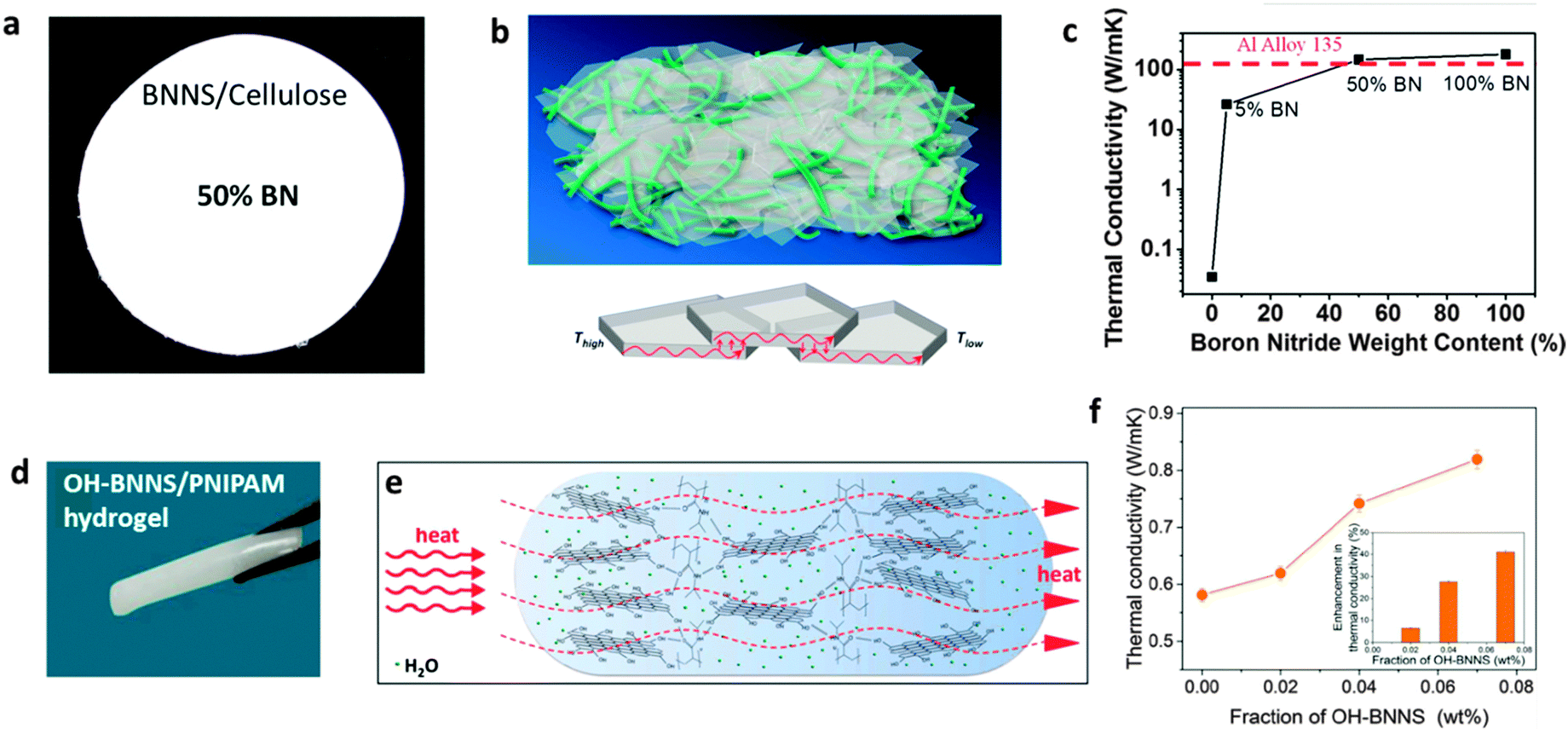 | ||
| Fig. 13 Thermal conductivity enhancement in BN-containing composites. (a) Photograph of a BNNS-loaded nanofibrillated cellulose composite with 50 wt% BN. (b) Schematic illustration of the composite and thermal conducting pathways along the plane direction. (c) Measured thermal conductivity along the BNNS/cellulose paper plane direction at different BN contents. (d) Photograph of the hydroxyl-functionalized BN nanosheets (OH-BNNS) filled poly(N-isopropylacrylamide) hydrogel (OH-BNNS/PNIPAM). (e) Schematic illustration of the enhanced thermal conductivity pathway in the OH-BNNS/PNIPAM hydrogel due to an effective construction of a thermal transferring network between BN and PNIPAM. (f) Thermal conductivity of the hydrogel with the increasing fraction of OH-BNNS fillers. The inset is the improvement displayed as a percentage. (a–c) are reprinted from ref. 179; (d–f) are reprinted from ref. 79 with permissions. | ||
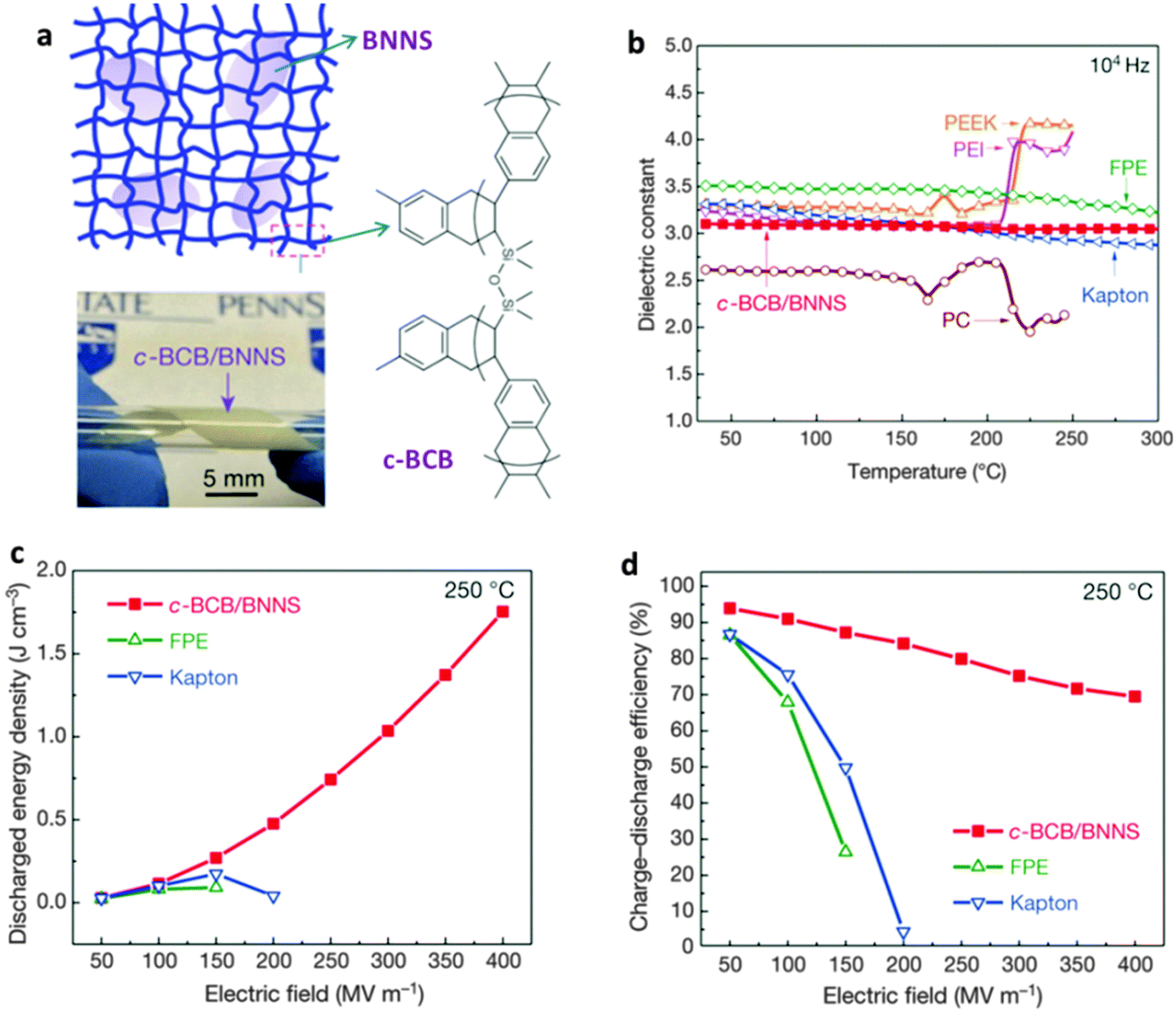 | ||
| Fig. 14 Dielectric-stabilized BN nanosheet (BNNS) composites for electrical energy storage applications. (a) Schematics and a photo of the crosslinked divinyltetramethyldisiloxane-bis(benzocyclobutene) and BNNS composite (c-BCB/BNNS). The molecular structure of c-BCB is shown on the right. (b) Thermal stability of dielectric constant for the composite compared with other polymers. PC = polycarbonate (Tg ≈ 150 °C), PEEK = poly(ether ketone) (Tg ≈ 150 °C), PEI = polyetherimide (Tg ≈ 217 °C), FPE = fluorine polyester (Tg ≈ 330 °C) and Kapton = polyimide (Tg ≈ 360 °C). (c) Discharged energy density and (d) charge–discharge efficiency of a c-BCB/BNNS composite (10 vol% BNNSs) measured at 250 °C compared with other polymeric dielectrics. Reprinted from ref. 185 with permission. | ||
Other applications of BN containing composites have also been studied. For example, additions of OH-BNNSs or BNNSs into a polymeric matrix can effectively enhance the barrier properties of the polymer for gas permeation.75,187 Kang et al. reported that a 2 wt% BNNT/polyimide composite exhibits an electroactive strain increase of ∼460% (piezoelectric and electrostrictive) compared to the polymeric host, implying bright prospects for future applications in energy harvesting and aerospace protection fields.188
4.5 Environmental and energy-related applications
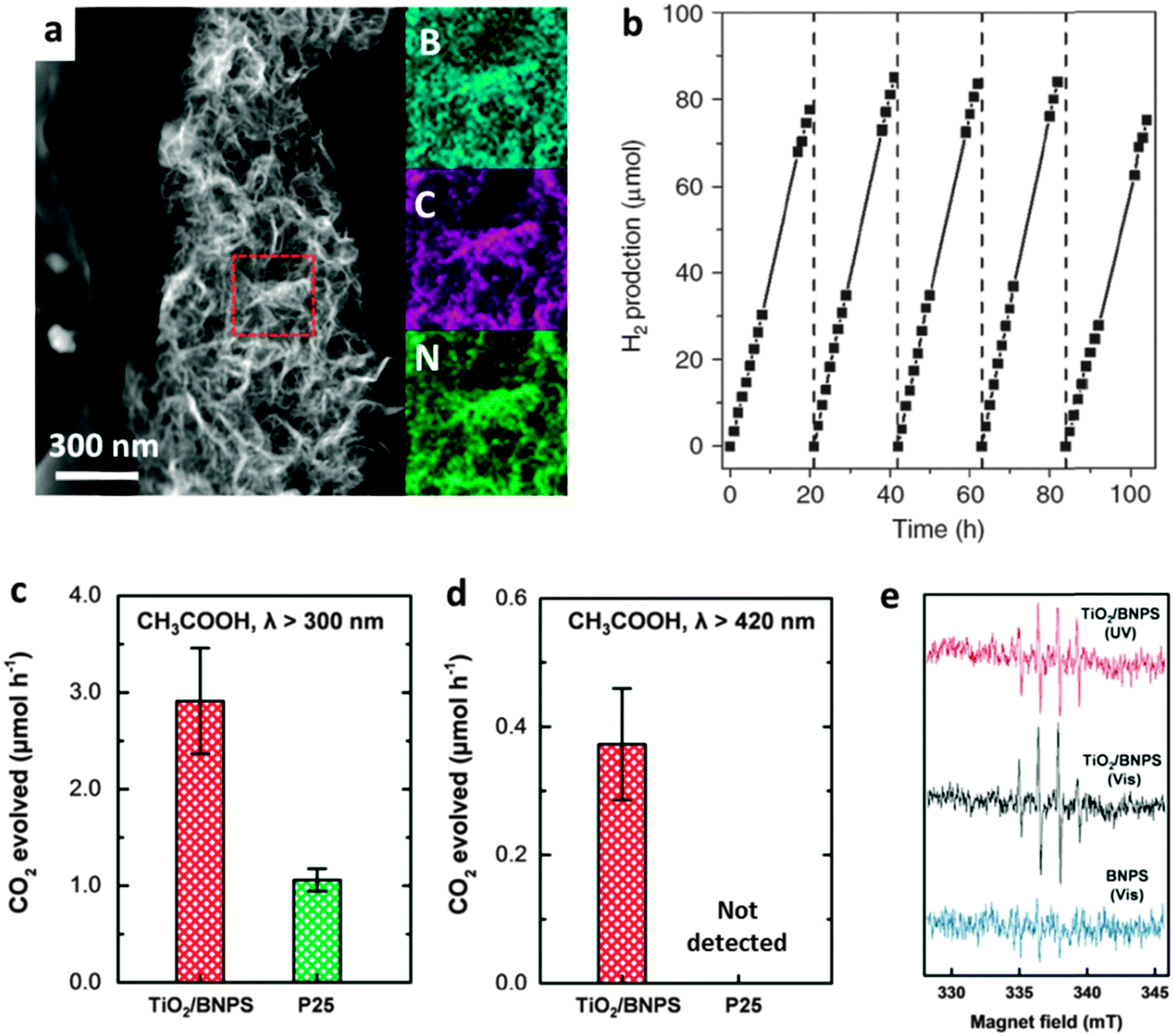 | ||
| Fig. 15 Photocatalytic applications of BN heterostructures. (a) Dark-field TEM image and the elemental maps of the selected area. (b) Photocatalytic production of H2 using BCN nanosheets under visible light irradiation (λ > 420 nm). (c and d) Photocatalytic degeneration of organic compounds (acetic acid) of edge –OH terminated BN porous sheets and the TiO2 composite (TiO2/BNPS) under different λ ranges. (e) Electron spin resonance spectra of the ˙OH radical spin adduct generated from TiO2/BNPS under UV (400 nm > λ > 300 nm) and visible light (λ > 420 nm) irradiations (top and middle, respectively), and the BNPS sample under visible light irradiation (bottom). (a and b) are reprinted from ref. 104; (c–e) are reprinted from ref. 76 with permissions. | ||
After functionalization with other metal oxides/sulfides/halides active particles, particularly, TiO2,116–119 ZnO,120 SnO2,55 and WO3,121 the photocatalytic performances of these BN heterostructures were improved remarkably for pollutant degenerations. This was due to the effective separation of the photo-generated holes and electrons. Besides carbon doping, our group has experimentally proven that the band gap of edge-hydroxylated BN porous sheets is narrowed compared with pure h-BN; the materials can be used as TiO2 visible light sensitizers for organic pollutants degeneration in water (Fig. 15c–e). Furthermore, we have demonstrated the existence of photo-generated ˙OH radicals in a TiO2/BN composite under visible-light irradiation using electron spin resonance (ESR) spectroscopy.76
4.6 Other applications
Many other applications of functionalized BN materials are also envisaged to have potential importance, although the respective studies are still in their infancy at the current stage. For example, proton transport through an atomic-thick BN monolayer was found to be unexpectedly high at room temperature, i.e. ∼2 orders of magnitude higher than through graphene and other 2D crystals. The H+ conductivity would be further boosted to >2 S cm−2 by decorating Pt nanoparticles on a BN monolayer surface. Such a value already meets the conductivity requirement for membranes used in currently operating hydrogen fuel cells (σ > 1 S cm−2).202 This may lead to a discovery of next-generation proton conduction membranes for advanced fuel cell applications. Besides, due to the structural diversity of BCN systems, great chances still exist to sieve high-performance catalysts for ORR, hydrogen evolution reactions (HER) and many other reactions, or develop BCN electrodes for electrochemical energy storage (supercapacitors and lithium/sodium ion batteries). Applications in photo-generated hole collectors in solar cells are also on the agenda. The emerging functional groups of chemically functionalized BN nanomaterials also provide good opportunities for the future interface engineering of BN heterostructures, which can chemically bond other components to the BN surfaces. This would be helpful for the development of efficient and durable nanosized catalysts or for tuning the band structures of a composite to improve photo-generated hole and electron separations. Future top-notch advances in these prospective fields would be timely and essential for a future eco-friendly and sustainable society.5. Summary and outlook
This article has presented an overview of the functionalized BN nanomaterials, from the developed chemical and physical functionalization methods to their emerging properties and applications. A variety of chemical groups/hetero-atom dopants/components, including hydroxyl (–OH), amino (–NH2), ether (–OR), amine (–NHR), arcyl (–COR), alkyl (–R), halogen (–X) groups, etc., have been introduced for the chemical modifications of BN surfaces. Other physical functionalization modes, such as preparations of BN low-dimensional and porous structures, have also been discussed. These efforts have generated new properties and functions of BN materials. These include but are not limited to: (1) water-soluble and biocompatible BN functional materials for biological and biomedical applications; (2) efficient load transfer using chemically functionalized BN nanofillers in polymeric composites; (3) construction of more effective thermal conducting networks toward creation of thermally conductive and electrically insulating composites; (4) BN–C heterostructures designed for ultimate atomically thin electrical circuitry and device applications; (5) band-gap-adjustable BNs and BN heterostructures used for photocatalytic H2 production and pollutant oxidation and electrochemical reaction catalysis.Despite the promising future of functionalized BN nanomaterials, the progress made in the related fields has still been rather modest, especially when compared with those achieved for carbon analogues. This situation is mainly explained by high chemical stability/inertness of BN materials that prevents them from immediate post-reactions/modifications. To date, various post-synthetic methods for BN functionalization have been established, however, the scope and efficiency of these strategies are still seldom satisfying. Alternatively, the recently proposed “reserved functionalization” route may provide a new way toward more efficient functionalization of BN and other materials.70 Many more advanced strategies need to be introduced to tackle this challenge.
In addition to the developments of synthetic methods, the design of analytical techniques that can precisely confirm the existence of functionalized BN nanostructures is also critical. For example, the cutting-edge high-resolution electron transmission microscopy is a powerful tool for resolving individual atoms spatially and chemically; this technology will certainly provide valuable information for BN functionalization studies, like precise determination of the functionalization sites on the BN surfaces, geometry and nature of the formed bonds, etc. However, this direction is still very challenging because the light B, C and N atoms typically show a close atomic size and analogous contrast under microscopic imaging, making them difficult to be resolved directly. In recent years, many important advances have been achieved in atomic and chemical determination of materials,95,99,203–205 such as the mentioned annular dark-field imaging technique, aberration-corrected transmission electron microscopy, scanning tunnelling microscopy, atomic force spectroscopy, etc. All these methods should become valuable instruments for uncovering material functionalization–structure–property relationships.
By taking into account the above-mentioned restrictions and challenges with respect to BN chemical and physical functionalization, precise characterization of the materials, and prospective applications, researchers should put more efforts into the development of more effective, scalable and economic BN functionalization strategies. This would pave the way toward exact property-oriented design of functionalized BN materials to meet the ever growing demands for future high-performance bio-, electronic, photoelectrical, and electrochemical devices for utilizing in energy, environmental and composite fields.
Acknowledgements
The authors thank Prof. Su-Yuan Xie in Xiamen University, and Prof. Chunyi Zhi in City University of Hong Kong for their valuable comments and suggestions on this article. Financial support from the International Center for Materials Nanoarchitectonics (MANA) of the National Institute for Materials Science (NIMS), Tsukuba, Japan, is acknowledged. This work was also partly supported by the Fundamental Research Funds for the Central Universities (2016RC008) and Thousand Youth Talents Plan.References
- L. F. Dobrzhinetskaya, R. Wirth, J. S. Yang, H. W. Green, I. D. Hutcheon, P. K. Weber and E. S. Grew, Am. Mineral., 2014, 99, 764–772 CrossRef CAS
.
- Y. J. Tian, B. Xu, D. L. Yu, Y. M. Ma, Y. B. Wang, Y. B. Jiang, W. T. Hu, C. C. Tang, Y. F. Gao, K. Luo, Z. S. Zhao, L. M. Wang, B. Wen, J. L. He and Z. Y. Liu, Nature, 2013, 493, 385–388 CrossRef CAS PubMed
- T. Soma, A. Sawaoka and S. Saito, Mater. Res. Bull., 1974, 9, 755–762 CrossRef CAS
- X. F. Jiang, Q. H. Weng, X. B. Wang, X. Li, J. Zhang, D. Golberg and Y. Bando, J. Mater. Sci. Technol., 2015, 31, 589–598 Search PubMed
- R. T. Paine and C. K. Narula, Chem. Rev., 1990, 90, 73–91 CrossRef CAS
- N. G. Chopra and A. Zettl, Solid State Commun., 1998, 105, 297–300 CrossRef CAS
- D. Golberg, P. M. F. J. Costa, O. Lourie, M. Mitome, X. D. Bai, K. Kurashima, C. Y. Zhi, C. C. Tang and Y. Bando, Nano Lett., 2007, 7, 2146–2151 CrossRef CAS
- V. Verma, V. K. Jindal and K. Dharamvir, Nanotechnology, 2007, 18, 435711 CrossRef
- X. L. Wei, M. S. Wang, Y. Bando and D. Golberg, Adv. Mater., 2010, 22, 4895–4899 CrossRef CAS PubMed
- C. Chang, A. Fennimore, A. Afanasiev, D. Okawa, T. Ikuno, H. Garcia, D. Li, A. Majumdar and A. Zettl, Phys. Rev. Lett., 2006, 97, 085901 CrossRef CAS PubMed
- C. C. Tang, Y. Bando, X. X. Ding, S. R. Qi and D. Golberg, J. Am. Chem. Soc., 2002, 124, 14550–14551 CrossRef CAS PubMed
- D.-M. Tang, C.-L. Ren, X. Wei, M.-S. Wang, C. Liu, Y. Bando and D. Golberg, ACS Nano, 2011, 5, 7362–7368 CrossRef CAS PubMed
- K. N. Kudin, G. E. Scuseria and B. I. Yakobson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 235406 CrossRef
- H. Sahin, S. Cahangirov, M. Topsakal, E. Bekaroglu, E. Akturk, R. T. Senger and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155453 CrossRef
- L. Song, L. J. Ci, H. Lu, P. B. Sorokin, C. H. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed
- M. Topsakal and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024107 CrossRef
- R. C. Andrew, R. E. Mapasha, A. M. Ukpong and N. Chetty, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125428 CrossRef
- R. Kumar, G. Rajasekaran and A. Parashar, Nanotechnology, 2016, 27, 085706 CrossRef PubMed
- L. Lindsay and D. A. Broido, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 155421 CrossRef
- A. Bosak, J. Serrano, M. Krisch, K. Watanabe, T. Taniguchi and H. Kanda, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041402 CrossRef
- N. G. Chopra, R. J. Luyken, K. Cherrey, V. H. Crespi, M. L. Cohen, S. G. Louie and A. Zettl, Science, 1995, 269, 966–967 CrossRef CAS PubMed
- H. B. Zeng, C. Y. Zhi, Z. H. Zhang, X. L. Wei, X. B. Wang, W. L. Guo, Y. Bando and D. Golberg, Nano Lett., 2010, 10, 5049–5055 CrossRef CAS PubMed
- C. Y. Zhi, Y. Bando, C. C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS
- A. Nag, K. Raidongia, K. P. S. S. Hembram, R. Datta, U. V. Waghmare and C. N. R. Rao, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS PubMed
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed
- X. B. Wang, C. Y. Zhi, L. Li, H. B. Zeng, C. Li, M. Mitome, D. Golberg and Y. Bando, Adv. Mater., 2011, 23, 4072–4076 CrossRef CAS PubMed
- D. Golberg, Nat. Nanotechnol., 2011, 6, 200–201 CrossRef CAS PubMed
- K. K. Kim, A. Hsu, X. T. Jia, S. M. Kim, Y. S. Shi, M. Hofmann, D. Nezich, J. F. Rodriguez-Nieva, M. Dresselhaus, T. Palacios and J. Kong, Nano Lett., 2012, 12, 161–166 CrossRef PubMed
- G. Y. Lu, T. R. Wu, Q. H. Yuan, H. S. Wang, H. M. Wang, F. Ding, X. M. Xie and M. H. Jiang, Nat. Commun., 2015, 6, 6160 CrossRef CAS PubMed
- D. Golberg, Y. Bando, C. C. Tang and C. Y. Zhi, Adv. Mater., 2007, 19, 2413–2432 CrossRef CAS
- C. Y. Zhi, Y. Bando, C. C. Tang and D. Golberg, Mater. Sci. Eng., R, 2010, 70, 92–111 CrossRef
- A. Pakdel, Y. Bando and D. Golberg, Chem. Soc. Rev., 2014, 43, 934–959 RSC
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. C. Tang and C. Y. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed
- I. Jo, M. T. Pettes, J. Kim, K. Watanabe, T. Taniguchi, Z. Yao and L. Shi, Nano Lett., 2013, 13, 550–554 CrossRef CAS PubMed
- Y. C. Chen, S. C. Lee, T. H. Liu and C. C. Chang, Int. J. Therm. Sci., 2015, 94, 72–78 CrossRef CAS
- Q. H. Weng, X. B. Wang, Y. Bando and D. Golberg, Adv. Energy Mater., 2014, 4, 1301525 Search PubMed
- P. Wu, W. Zhu, Y. Chao, J. Zhang, P. Zhang, H. Zhu, C. Li, Z. Chen, H. Li and S. Dai, Chem. Commun., 2016, 52, 144–147 RSC
- J. Dai, X. Wu, J. Yang and X. C. Zeng, J. Phys. Chem. Lett., 2013, 4, 3484–3488 CrossRef CAS
- J. Dai, X. Wu, J. Yang and X. C. Zeng, J. Phys. Chem. Lett., 2014, 5, 393–398 CrossRef CAS PubMed
- W. Q. Han, R. Brutchey, T. D. Tilley and A. Zettl, Nano Lett., 2004, 4, 173–176 CrossRef CAS
- P. Dibandjo, F. Chassagneux, L. Bois, C. Sigala and P. Miele, J. Mater. Chem., 2005, 15, 1917–1923 RSC
- A. Vinu, M. Terrones, D. Golberg, S. Hishita, K. Ariga and T. Mori, Chem. Mater., 2005, 17, 5887–5890 CrossRef CAS
- S. Schlienger, J. Alauzun, F. Michaux, L. Vidal, J. G. Alauzun, S. Ungureanu, N. Brun, S. Bernard, P. Miele, R. Backov and C. Sanchez, J. Mater. Chem., 2011, 21, 14025–14030 RSC
- S. Schlienger, J. Alauzun, F. Michaux, J. Parmentier, C. Gervais, F. Babonneau, S. Bernard, P. Miele and J. B. Parra, Chem. Mater., 2012, 24, 88–96 CrossRef CAS
- M. Rousseas, A. P. Goldstein, W. Mickelson, M. A. Worsley, L. Woo and A. Zettl, ACS Nano, 2013, 7, 8540–8546 CrossRef CAS PubMed
- Y. Song, B. Li, S. Yang, G. Ding, C. Zhang and X. Xie, Sci. Rep., 2015, 5, 10337 CrossRef CAS PubMed
- B. Rushton and R. Mokaya, J. Mater. Chem., 2008, 18, 235–241 RSC
- P. Dibandjo, L. Bois, F. Chassagneux and P. Miele, J. Eur. Ceram. Soc., 2007, 27, 313–317 CrossRef CAS
- S. Bernard and P. Miele, Mater. Today, 2014, 17, 443–450 CrossRef CAS
- P. R. L. Malenfant, J. Wan, S. T. Taylor and M. Manoharan, Nat. Nanotechnol., 2007, 2, 43–46 CrossRef CAS PubMed
- L. Wang, L. Xu, C. Sun and Y. Qian, J. Mater. Chem., 2009, 19, 1989–1994 RSC
- X. L. Meng, N. Lun, Y. Q. Qi, J. Q. Bi, Y. X. Qi, H. L. Zhu, F. D. Han, Y. J. Bai, L. W. Yin and R. H. Fan, Eur. J. Inorg. Chem., 2010, 3174–3178 CrossRef CAS
- X.-L. Meng, N. Lun, Y.-X. Qi, H.-L. Zhu, F.-D. Han, L.-W. Yin, R.-H. Fan, Y.-J. Bai and J.-Q. Bi, J. Solid State Chem., 2011, 184, 859–862 CrossRef CAS
- M. Wang, M. Li, L. Xu, L. Wang, Z. Ju, G. Li and Y. Qian, Catal. Sci. Technol., 2011, 1, 1159–1165 CAS
- C.-H. Sun, L.-Q. Xu, X.-J. Ma and Y.-T. Qian, Chin. J. Inorg. Chem., 2012, 28, 601–606 CAS
- X. B. Wang, A. Pakdel, J. Zhang, Q. H. Weng, T. Y. Zhai, C. Y. Zhi, D. Golberg and Y. Bando, Nanoscale Res. Lett., 2012, 7, 662 CrossRef PubMed
- Q. H. Weng, X. B. Wang, C. Y. Zhi, Y. Bando and D. Golberg, ACS Nano, 2013, 7, 1558–1565 CrossRef CAS PubMed
- J. Li, J. Lin, X. Xu, X. Zhang, Y. Xue, J. Mi, Z. Mo, Y. Fan, L. Hu, X. Yang, J. Zhang, F. Meng, S. Yuan and C. Tang, Nanotechnology, 2013, 24, 155603 CrossRef PubMed
- J. Li, Y. Huang, Z. Liu, J. Zhang, X. Liu, H. Luo, Y. Ma, X. Xu, Y. Lu, J. Lin, J. Zuo and C. Tang, J. Mater. Chem. A, 2015, 3, 8185–8193 CAS
- J. Lin, L. Xu, Y. Huang, J. Li, W. Wang, C. Feng, Z. Liu, X. Xu, J. Zou and C. Tang, RSC Adv., 2016, 6, 1253–1259 RSC
- W. W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4, 1777 CrossRef PubMed
- Y. Xue, P. Dai, X. Jiang, X. Wang, C. Zhang, D. Tang, Q. Weng, X. Wang, A. Pakdel, C. Tang, Y. Bando and D. Golberg, J. Mater. Chem. A, 2016, 4, 1469–1478 CAS
- S. Y. Xie, F. Gao, X. Lu, R. B. Huang, C. R. Wang, X. Zhang, M. L. Liu, S. L. Deng and L. S. Zheng, Science, 2004, 304, 699 CrossRef CAS PubMed
- X. Lu and Z. F. Chen, Chem. Rev., 2005, 105, 3643–3696 CrossRef CAS PubMed
- Y. Z. Tan, Z. J. Liao, Z. Z. Qian, R. T. Chen, X. Wu, H. Liang, X. Han, F. Zhu, S. J. Zhou, Z. P. Zheng, X. Lu, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Mater., 2008, 7, 790–794 CrossRef CAS PubMed
- Y. Z. Tan, S. Y. Xie, R. B. Huang and L. S. Zheng, Nat. Chem., 2009, 1, 450–460 CrossRef CAS PubMed
- Q. Weng, Q. He, T. Liu, H. Y. Huang, J. H. Chen, Z. Y. Gao, S. Y. Xie, X. Lu, R. B. Huang and L. S. Zheng, J. Am. Chem. Soc., 2010, 132, 15093–15095 CrossRef CAS PubMed
- I. V. Kuvychko, A. V. Streletskii, N. B. Shustova, K. Seppelt, T. Drewello, A. A. Popov, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2010, 132, 6443–6462 CrossRef CAS PubMed
- Q. H. Weng, B. J. Wang, X. B. Wang, N. Hanagata, X. Li, D. Q. Liu, X. Wang, X. F. Jiang, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 6123–6130 CrossRef CAS PubMed
- C. Y. Zhi, Y. Bando, T. Terao, C. C. Tang, H. Kuwahara and D. Golberg, Chem. – Asian J., 2009, 4, 1536–1540 CrossRef CAS PubMed
- Y. Lin, T. Williams, T.-B. Xu, W. Cao, H. Elsayed-Ali and J. Connell, J. Phys. Chem. C, 2011, 115, 2679–2685 CAS
- T. Sainsbury, A. Satti, P. May, Z. Wang, I. McGovern, Y. K. Gun'ko and J. Coleman, J. Am. Chem. Soc., 2012, 134, 18758–18771 CrossRef CAS PubMed
- A. Pakdel, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 10631–10639 CrossRef CAS PubMed
- D. Lee, B. Lee, K. H. Park, H. J. Ryu, S. Jeon and S. H. Hong, Nano Lett., 2015, 15, 1238–1244 CrossRef CAS PubMed
- Q. Weng, Y. Ide, X. Wang, X. Wang, C. Zhang, X. Jiang, Y. Xue, P. Dai, K. Komaguchi, Y. Bando and D. Golberg, Nano Energy, 2015, 16, 19–27 CrossRef CAS
- D. Krepel and O. Hod, J. Chem. Theory Comput., 2014, 10, 373–380 CrossRef CAS PubMed
- D. Krepel, L. Kalikhman-Razvozov and O. Hod, J. Phys. Chem. C, 2014, 118, 21110–21118 CAS
- F. Xiao, S. Naficy, G. Casillas, M. H. Khan, T. Katkus, L. Jiang, H. Liu, H. Li and Z. Huang, Adv. Mater., 2015, 27, 7196–7203 CrossRef CAS PubMed
- D. Kim, S. Nakajima, T. Sawada, M. Iwasaki, S. Kawauchi, C. Zhi, Y. Bando, D. Golberg and T. Serizawa, Chem. Commun., 2015, 51, 7104–7107 RSC
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS PubMed
- T. Ikuno, T. Sainsbury, D. Okawa, J. M. J. Fréchet and A. Zettl, Solid State Commun., 2007, 142, 643–646 CrossRef CAS
- Y. Liao, Z. Chen, J. W. Connell, C. C. Fay, C. Park, J.-W. Kim and Y. Lin, Adv. Funct. Mater., 2014, 24, 4497–4506 CrossRef CAS
- S. Y. Xie, W. Wang, K. A. Fernando, X. Wang, Y. Lin and Y. P. Sun, Chem. Commun., 2005, 3670–3672 RSC
- Y. Lin, T. V. Williams and J. W. Connell, J. Phys. Chem. Lett., 2010, 1, 277–283 CrossRef CAS
- T. Sainsbury, A. O'Neill, M. K. Passarelli, M. Seraffon, D. Gohil, S. Gnaniah, S. J. Spencer, A. Rae and J. N. Coleman, Chem. Mater., 2014, 26, 7039–7050 CrossRef CAS
- H. Shin, J. W. Guan, M. Z. Zgierski, K. S. Kim, C. T. Kingston and B. Simard, ACS Nano, 2015, 9, 12573–12582 CrossRef CAS PubMed
- C. Y. Zhi, Y. Bando, C. C. Tang, S. Honda, K. Sato, H. Kuwahara and D. Golberg, Angew. Chem., Int. Ed., 2005, 44, 7932–7935 CrossRef CAS PubMed
- X. Huang, C. Zhi, P. Jiang, D. Golberg, Y. Bando and T. Tanaka, Adv. Funct. Mater., 2013, 23, 1824–1831 CrossRef CAS
- G. Ciofani, G. G. Genchi, I. Liakos, A. Athanassiou, D. Dinucci, F. Chiellini and V. Mattoli, J. Colloid Interface Sci., 2012, 374, 308–314 CrossRef CAS PubMed
- A. Bhattacharya, S. Bhattacharya and G. P. Das, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 035415 CrossRef
- X. X. Li, J. Zhao and J. L. Yang, Sci. Rep., 2013, 3, 1858 Search PubMed
- C. C. Tang, Y. Bando, Y. Huang, S. L. Yue, C. Z. Gu, F. F. Xu and D. Golberg, J. Am. Chem. Soc., 2005, 127, 6552–6553 CrossRef CAS PubMed
- L. Song, Z. Liu, A. L. M. Reddy, N. T. Narayanan, J. Taha-Tijerina, J. Peng, G. H. Gao, J. Lou, R. Vajtai and P. M. Ajayan, Adv. Mater., 2012, 24, 4878–4895 CrossRef CAS PubMed
- O. L. Krivanek, M. F. Chisholm, V. Nicolosi, T. J. Pennycook, G. J. Corbin, N. Dellby, M. F. Murfitt, C. S. Own, Z. S. Szilagyi, M. P. Oxley, S. T. Pantelides and S. J. Pennycook, Nature, 2010, 464, 571–574 CrossRef CAS PubMed
- A. Y. Liu, R. M. Wentzcovitch and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 1760–1765 CrossRef CAS
- N. X. Qiu, C. H. Zhang and Y. Xue, ChemPhysChem, 2014, 15, 3015–3025 CrossRef CAS PubMed
- N. X. Qiu, Z. Y. Tian, Y. Guo, C. H. Zhang, Y. P. Luo and Y. Xue, Int. J. Hydrogen Energy, 2014, 39, 9307–9320 CrossRef CAS
- J. Lu, K. Zhang, X. F. Liu, H. Zhang, T. C. Sum, A. H. C. Neto and K. P. Loh, Nat. Commun., 2013, 4, 2681 Search PubMed
- R. S. Singh, R. Y. Tay, W. L. Chow, S. H. Tsang, G. Mallick and E. H. T. Teo, Appl. Phys. Lett., 2014, 104, 163101 CrossRef
- W. Q. Han, H. G. Yu and Z. X. Liu, Appl. Phys. Lett., 2011, 98, 203112 CrossRef
- T. W. Lin, C. Y. Su, X. Q. Zhang, W. J. Zhang, Y. H. Lee, C. W. Chu, H. Y. Lin, M. T. Chang, F. R. Chen and L. J. Li, Small, 2012, 8, 1384–1391 CrossRef CAS PubMed
- S. Y. Wang, L. P. Zhang, Z. H. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed
- L. D. A. Silva, S. C. Guerini, V. Lemos and J. M. Filho, IEEE Trans. Nanotechnol., 2006, 5, 517–522 CrossRef
- W. Lei, H. Zhang, Y. Wu, B. Zhang, D. Liu, S. Qin, Z. Liu, L. Liu, Y. Ma and Y. Chen, Nano Energy, 2014, 6, 219–224 CrossRef CAS
- R. S. Singh, AIP Adv., 2015, 5, 117150 CrossRef
- J. B. Wu and W. Y. Zhang, Solid State Commun., 2009, 149, 486–490 CrossRef CAS
- W. Q. Han, H. G. Yu, C. Zhi, J. Wang, Z. Liu, T. Sekiguchi and Y. Bando, Nano Lett., 2008, 8, 491–494 CrossRef CAS PubMed
- G. Y. Gou, B. C. Pan and L. Shi, J. Am. Chem. Soc., 2009, 131, 4839–4845 CrossRef CAS PubMed
- Z. Liu, L. Ma, G. Shi, W. Zhou, Y. Gong, S. Lei, X. Yang, J. Zhang, J. Yu, K. P. Hackenberg, A. Babakhani, J.-C. Idrobo, R. Vajtai, J. Lou and P. Ajayan, Nat. Nanotechnol., 2013, 8, 119–124 CrossRef CAS PubMed
- L. Liu, J. Park, D. A. Siegel, K. F. McCarty, K. W. Clark, W. Deng, L. Basile, J. C. Idrobo, A.-P. Li and G. Gu, Science, 2014, 343, 163–167 CrossRef CAS PubMed
- Y. Gong, G. Shi, Z. Zhang, W. Zhou, J. Jung, W. Gao, L. Ma, Y. Yang, S. Yang, G. You, R. Vajtai, Q. Xu, A. H. MacDonald, B. I. Yakobson, J. Lou, Z. Liu and P. M. Ajayan, Nat. Commun., 2014, 5, 3193 Search PubMed
- W. Yang, G. Chen, Z. Shi, C.-C. Liu, L. Zhang, G. Xie, M. Cheng, D. Wang, R. Yang and D. Shi, Nat. Mater., 2013, 12, 792–797 CrossRef CAS PubMed
- C. Zhang, S. Zhao, C. Jin, A. L. Koh, Y. Zhou, W. Xu, Q. Li, Q. Xiong, H. Peng and Z. Liu, Nat. Commun., 2015, 6, 6519 CrossRef CAS PubMed
- C. Tang, J. Li, Y. Bando, C. Zhi and D. Golberg, Chem. – Asian J., 2010, 5, 1220–1224 CrossRef CAS PubMed
- X. Fu, Y. Hu, Y. Yang, W. Liu and S. Chen, J. Hazard. Mater., 2013, 244, 102–110 CrossRef PubMed
- Y. Ide, F. Liu, J. Zhang, N. Kawamoto, K. Komaguchi, Y. Bando and D. Golberg, J. Mater. Chem. A, 2014, 2, 4150–4156 CAS
- D. Liu, W. Cui, J. Lin, Y. Xue, Y. Huang, J. Li, J. Zhang, Z. Liu and C. Tang, Catal. Commun., 2014, 57, 9–13 CrossRef CAS
- X. Fu, Y. Hu, T. Zhang and S. Chen, Appl. Surf. Sci., 2013, 280, 828–835 CrossRef CAS
- H. Xu, L. Liu, Y. Song, L. Huang, Y. Li, Z. Chen, Q. Zhang and H. Li, J. Hazard. Mater., 2016, 660, 48–54 CAS
- S. Meng, X. Ye, X. Ning, M. Xie, X. Fu and S. Chen, Appl. Catal., B, 2016, 182, 356–368 CrossRef CAS
- J. Chen, J. Zhu, Z. Da, H. Xu, J. Yan, H. Ji, H. Shu and H. Li, Appl. Surf. Sci., 2014, 313, 1–9 CrossRef CAS
- J. Choi, D. A. Reddy and T. K. Kim, Ceram. Int., 2015, 41, 13793–13803 CrossRef CAS
- Y. Huang, J. Lin, Y. Bando, C. Tang, C. Zhi, Y. Shi, E. Takayama-Muromachi and D. Golberg, J. Mater. Chem., 2010, 20, 1007–1011 RSC
- P. C. Dai, Y. M. Xue, X. B. Wang, Q. H. Weng, C. Zhang, X. F. Jiang, D. M. Tang, X. Wang, N. Kawamoto, Y. Ide, M. Mitome, D. Golberg and Y. Bando, Nanoscale, 2015, 7, 18992–18997 RSC
- F. Wang, X. Zeng, Y. Yao, R. Sun, J. Xu and C.-P. Wong, Sci. Rep., 2016, 6, 19394 CrossRef CAS PubMed
- A. Harley-Trochimczyk, T. Pham, J. Chang, E. Chen, M. A. Worsley, A. Zettl, W. Mickelson and R. Maboudian, Adv. Funct. Mater., 2016, 26, 433–439 CrossRef CAS
- R. Kumar, D. Raut, I. Ahmad, U. Ramamurty, T. K. Maji and C. Rao, Mater. Horiz., 2014, 1, 513–517 RSC
- Y. Song, H. Xu, C. Wang, J. Chen, J. Yan, Y. Xu, Y. Li, C. Liu, H. Li and Y. Lei, RSC Adv., 2014, 4, 56853–56862 RSC
- C. Y. Zhi, Y. Bando, C. C. Tang, R. G. Xie, T. Sekiguchi and D. Golberg, J. Am. Chem. Soc., 2005, 127, 15996–15997 CrossRef CAS PubMed
- X. Li, C. Zhi, N. Hanagata, M. Yamaguchi, Y. Bando and D. Golberg, Chem. Commun., 2013, 49, 7337–7339 RSC
- D. A. Heller, S. Baik, T. E. Eurell and M. S. Strano, Adv. Mater., 2005, 17, 2793–2799 CrossRef CAS
- A. De La Zerda, C. Zavaleta, S. Keren, S. Vaithilingam, S. Bodapati, Z. Liu, J. Levi, B. R. Smith, T.-J. Ma, O. Oralkan, Z. Cheng, X. Chen, H. Dai, B. T. Khuri-Yakub and S. S. Gambhir, Nat. Nanotechnol., 2008, 3, 557–562 CrossRef CAS PubMed
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed
- Z. Liu, S. Tabakman, K. Welsher and H. Dai, Nano Res., 2009, 2, 85–120 CrossRef CAS PubMed
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS PubMed
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed
- A. Nimmagadda, K. Thurston, M. U. Nollert and P. S. McFetridge, J. Biomed. Mater. Res., Part A, 2006, 76, 614–625 CrossRef PubMed
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6, 1 Search PubMed
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 111, 188–202 CrossRef CAS PubMed
- G. Ciofani, S. Danti, G. G. Genchi, B. Mazzolai and V. Mattoli, Small, 2013, 9, 1672–1685 CrossRef CAS PubMed
- C. H. Lee, J. Drelich and Y. K. Yap, Langmuir, 2009, 25, 4853–4860 CrossRef CAS PubMed
- J. Yu, L. Qin, Y. Hao, S. Kuang, X. Bai, Y.-M. Chong, W. Zhang and E. Wang, ACS Nano, 2010, 4, 414–422 CrossRef CAS PubMed
- A. Pakdel, C. Y. Zhi, Y. Bando, T. Nakayama and D. Golberg, ACS Nano, 2011, 5, 6507–6515 CrossRef CAS PubMed
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed
- X. Chen, P. Wu, M. Rousseas, D. Okawa, Z. Gartner, A. Zettl and C. R. Bertozzi, J. Am. Chem. Soc., 2009, 131, 890–891 CrossRef CAS PubMed
- G. Ciofani, L. Ricotti, S. Danti, S. Moscato, C. Nesti, D. D'Alessandro, D. Dinucci, F. Chiellini, A. Pietrabissa, M. Petrini and A. Menciassi, Int. J. Nanomed., 2010, 5, 285–298 CrossRef CAS
- G. Ciofani, S. Danti, D. D'Alessandro, S. Moscato and A. Menciassi, Biochem. Biophys. Res. Commun., 2010, 394, 405–411 CrossRef CAS PubMed
- T. H. Ferreira, P. Silva, R. Santos and E. Sousa, J. Biomater. Nanobiotechnol., 2011, 2, 426 CrossRef CAS
- A. Salvetti, L. Rossi, P. Iacopetti, X. Li, S. Nitti, T. Pellegrino, V. Mattoli, D. Golberg and G. Ciofani, Nanomedicine, 2015, 10, 1911–1922 CrossRef CAS PubMed
- I. V. Sukhorukova, I. Y. Zhitnyak, A. M. Kovalskii, A. T. Matveev, O. I. Lebedev, X. Li, N. A. Gloushankova, D. Golberg and D. V. Shtansky, ACS Appl. Mater. Interfaces, 2015, 7, 17217–17225 CAS
- G. Ciofani, S. Danti, D. D'Alessandro, L. Ricotti, S. Moscato, G. Bertoni, A. Falqui, S. Berrettini, M. Petrini, V. Mattoli and A. Menciassi, ACS Nano, 2010, 4, 6267–6277 CrossRef CAS PubMed
- L. Horvath, A. Magrez, D. Golberg, C. Zhi, Y. Bando, R. Smajda, E. Horvath, L. Forro and B. Schwaller, ACS Nano, 2011, 5, 3800–3810 CrossRef CAS PubMed
- X. Li, N. Hanagata, X. Wang, M. Yamaguchi, W. Yi, Y. Bando and D. Golberg, Chem. Commun., 2014, 50, 4371–4374 RSC
- X. Li, X. Wang, X. Jiang, M. Yamaguchi, A. Ito, Y. Bando and D. Golberg, J. Biomed. Mater. Res., Part B, 2015, 323–329 CAS
- M. P. Levendorf, C.-J. Kim, L. Brown, P. Y. Huang, R. W. Havener, D. A. Muller and J. Park, Nature, 2012, 488, 627–632 CrossRef CAS PubMed
- L. Britnell, R. Gorbachev, R. Jalil, B. Belle, F. Schedin, A. Mishchenko, T. Georgiou, M. Katsnelson, L. Eaves, S. V. Morozov, N. M. Peres, J. Leist, A. K. Geim, K. S. Novoselov and L. A. Ponomarenko, Science, 2012, 335, 947–950 CrossRef CAS PubMed
- L. Britnell, R. V. Gorbachev, R. Jalil, B. D. Belle, F. Schedin, M. I. Katsnelson, L. Eaves, S. V. Morozov, A. S. Mayorov, N. M. Peres, A. Neto, J. Leist, A. K. Geim, L. A. Ponomarenko and K. S. Novoselov, Nano Lett., 2012, 12, 1707–1710 CrossRef CAS PubMed
- L. Wang, I. Meric, P. Huang, Q. Gao, Y. Gao, H. Tran, T. Taniguchi, K. Watanabe, L. Campos, D. Muller, J. Guo, P. Kim, J. Hone, K. L. Shepard and C. R. Dean, Science, 2013, 342, 614–617 CrossRef CAS PubMed
- A. S. Mayorov, R. V. Gorbachev, S. V. Morozov, L. Britnell, R. Jalil, L. A. Ponomarenko, P. Blake, K. S. Novoselov, K. Watanabe, T. Taniguchi and A. K. Geim, Nano Lett., 2011, 11, 2396–2399 CrossRef CAS PubMed
- A. Mishchenko, J. Tu, Y. Cao, R. Gorbachev, J. Wallbank, M. Greenaway, V. Morozov, S. Morozov, M. J. Zhu, S. L. Wong, F. Withers, C. R. Woods, Y.-J. Kim, K. Watanabe, T. Taniguchi, E. E. Vdovin, O. Makarovsky, T. M. Fromhold, V. I. Fal'ko, A. K. Geim, L. Eaves and K. S. Novoselov, Nat. Nanotechnol., 2014, 9, 808–813 CrossRef CAS PubMed
- V. Parashar, C. P. Durand, B. Hao, R. G. Amorim, R. Pandey, B. Tiwari, D. Zhang, Y. Liu, A.-P. Li and Y. K. Yap, Sci. Rep., 2015, 5, 12238 CrossRef PubMed
- K. Gopinadhan, Y. J. Shin, R. Jalil, T. Venkatesan, A. K. Geim, A. H. C. Neto and H. Yang, Nat. Commun., 2015, 6, 8337 CrossRef CAS PubMed
- M. W. Iqbal, M. Z. Iqbal, M. F. Khan, M. A. Shehzad, Y. Seo, J. H. Park, C. Hwang and J. Eom, Sci. Rep., 2015, 5, 10699 CrossRef CAS PubMed
- X. Chen, Y. Wu, Z. Wu, Y. Han, S. Xu, L. Wang, W. Ye, T. Han, Y. He, Y. Cai and N. Wang, Nat. Commun., 2015, 6, 7315 CrossRef CAS PubMed
- J. D. Caldwell, I. Vurgaftman, J. G. Tischler, O. J. Glembocki, J. C. Owrutsky and T. L. Reinecke, Nat. Nanotechnol., 2016, 11, 9–15 CrossRef CAS PubMed
- S. Dai, Q. Ma, M. Liu, T. Andersen, Z. Fei, M. Goldflam, M. Wagner, K. Watanabe, T. Taniguchi, M. Thiemens, F. Keilmann, G. C. A. M. Janssen, S.-E. Zhu, P. Jarillo-Herrero, M. M. Fogler and D. N. Basov, Nat. Nanotechnol., 2015, 10, 682–686 CrossRef CAS PubMed
- L. Ju, J. Velasco Jr, E. Huang, S. Kahn, C. Nosiglia, H.-Z. Tsai, W. Yang, T. Taniguchi, K. Watanabe, Y. Zhang, G. Zhang, M. Crommie, A. Zettl and F. Wang, Nat. Nanotechnol., 2014, 9, 348–352 CrossRef CAS PubMed
- A. Woessner, M. B. Lundeberg, Y. Gao, A. Principi, P. Alonso-González, M. Carrega, K. Watanabe, T. Taniguchi, G. Vignale, M. Polini, J. Hone, R. Hillenbrand and F. Koppens, Nat. Mater., 2015, 14, 421–425 CrossRef CAS PubMed
- X. Yang, F. Zhai, H. Hu, D. Hu, R. Liu, S. Zhang, M. Sun, Z. Sun, J. Chen and Q. Dai, Adv. Mater., 2016, 28, 2931–2938 CrossRef CAS PubMed
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed
- C. Y. Zhi, Y. Bando, C. C. Tang, S. Honda, H. Kuwahara and D. Golberg, J. Mater. Res., 2006, 21, 2794–2800 CrossRef CAS
- C. Y. Zhi, Y. Bando, T. Terao, C. C. Tang, H. Kuwahara and D. Golberg, Adv. Funct. Mater., 2009, 19, 1857–1862 CrossRef CAS
- N. Tajaddod, K. Song, E. C. Green, Y. Zhang and M. L. Minus, Macromol. Mater. Eng., 2016, 301, 315–327 CrossRef CAS
- X. B. Wang, Q. H. Weng, X. Wang, X. Li, J. Zhang, F. Liu, X. F. Jiang, H. X. Guo, N. S. Xu, D. Golberg and Y. Bando, ACS Nano, 2014, 8, 9081–9088 CrossRef CAS PubMed
- W.-L. Song, P. Wang, L. Cao, A. Anderson, M. J. Meziani, A. J. Farr and Y.-P. Sun, Angew. Chem., Int. Ed., 2012, 51, 6498–6501 CrossRef CAS PubMed
- H. Zhu, Y. Li, Z. Fang, J. Xu, F. Cao, J. Wan, C. Preston, B. Yang and L. Hu, ACS Nano, 2014, 8, 3606–3613 CrossRef CAS PubMed
-
B. Harris, Engineering Composite Materials, The Institute of Metals, London, 1999 Search PubMed
- P. Liu and Z. Wan, J. Yangzhou Univ., Nat. Sci. Ed., 2007, 10, 21–23 Search PubMed
- H. Ku, H. Wang, N. Pattarachaiyakoop and M. Trada, Composites, Part B, 2011, 42, 856–873 CrossRef
- M. J. Meziani, W.-L. Song, P. Wang, F. Lu, Z. Hou, A. Anderson, H. Maimaiti and Y.-P. Sun, ChemPhysChem, 2015, 16, 1339–1346 CrossRef CAS PubMed
- C. Zhi, Y. Xu, Y. Bando and D. Golberg, ACS Nano, 2011, 5, 6571–6577 CrossRef CAS PubMed
- Q. Li, L. Chen, M. R. Gadinski, S. Zhang, G. Zhang, H. Li, A. Haque, L.-Q. Chen, T. Jackson and Q. Wang, Nature, 2015, 523, 576–579 CrossRef CAS PubMed
- Q. Li, G. Zhang, F. Liu, K. Han, M. R. Gadinski, C. Xiong and Q. Wang, Energy Environ. Sci., 2015, 8, 922–931 CAS
- S. Xie, O. M. Istrate, P. May, S. Barwich, A. P. Bell, U. Khan and J. N. Coleman, Nanoscale, 2015, 7, 4443–4450 RSC
- J. H. Kang, G. Sauti, C. Park, V. I. Yamakov, K. E. Wise, S. E. Lowther, C. C. Fay, S. A. Thibeault and R. G. Bryant, ACS Nano, 2015, 11942–11950 CrossRef CAS PubMed
- H. Fei, R. Ye, G. Ye, Y. Gong, Z. Peng, X. Fan, E. L. Samuel, P. M. Ajayan and J. M. Tour, ACS Nano, 2014, 8, 10837–10843 CrossRef CAS PubMed
- H. Jin, H. Huang, Y. He, X. Feng, S. Wang, L. Dai and J. Wang, J. Am. Chem. Soc., 2015, 137, 7588–7591 CrossRef CAS PubMed
- Y. Gong, H. Fei, X. Zou, W. Zhou, S. Yang, G. Ye, Z. Liu, Z. Peng, J. Lou, R. Vajtai, B. I. Yakobson, J. M. Tour and P. M. Ajayan, Chem. Mater., 2015, 27, 1181–1186 CrossRef CAS
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 125, 3192–3198 CrossRef
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201–1204 CrossRef CAS PubMed
- R. Z. Ma, Y. Bando, H. W. Zhu, T. Sato, C. L. Xu and D. H. Wu, J. Am. Chem. Soc., 2002, 124, 7672–7673 CrossRef CAS PubMed
- S.-H. Jhi and Y.-K. Kwon, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 245407 CrossRef
- D. Portehault, C. Giordano, C. Gervais, I. Senkovska, S. Kaskel, C. Sanchez and M. Antonietti, Adv. Funct. Mater., 2010, 20, 1827–1833 CrossRef CAS
- H. Zhang, C.-J. Tong, Y. Zhang, Y.-N. Zhang and L.-M. Liu, J. Mater. Chem. A, 2015, 3, 9632–9637 CAS
- A. Kinal and S. Sayhan, Int.
J. Hydrogen Energy, 2016, 41, 392–400 CrossRef CAS
- D. Liu, W. W. Lei, S. Qin and Y. Chen, Sci. Rep., 2014, 4, 4453 Search PubMed
- J. Li, P. Jin and C. Tang, RSC Adv., 2014, 4, 14815–14821 RSC
- J. Li, H. Jia, J. Lin, H. Luo, Z. Liu, X. Xu, Y. Huang, P. Jin, J. Zhang, S. Abbas and C. Tang, RSC Adv., 2015, 5, 71537–71543 RSC
- S. Hu, M. Lozada-Hidalgo, F. Wang, A. Mishchenko, F. Schedin, R. Nair, E. Hill, D. Boukhvalov, M. I. Katsnelson, R. A. W. Dryfe, I. V. Grigorieva, H. A. Wu and A. K. Geim, Nature, 2014, 516, 227–230 CrossRef CAS PubMed
- K. Suenaga and M. Koshino, Nature, 2010, 468, 1088–1090 CrossRef CAS PubMed
- J. Zhang, P. Chen, B. Yuan, W. Ji, Z. Cheng and X. Qiu, Science, 2013, 342, 611–614 CrossRef CAS PubMed
- D. G. de Oteyza, P. Gorman, Y.-C. Chen, S. Wickenburg, A. Riss, D. J. Mowbray, G. Etkin, Z. Pedramrazi, H.-Z. Tsai, A. Rubio, M. F. Crommie and F. R. Fischer, Science, 2013, 1434–1437 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |