
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives
Wen-Ting
Wu
a,
Liming
Zhang
*b and
Shu-Li
You
*a
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn
bDepartment of Chemistry & Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: zhang@chem.ucsb.edu
First published on 22nd January 2016
Abstract
Phenols are widely used as starting materials in both industrial and academic society. Dearomatization reactions of phenols provide an efficient way to construct highly functionalized cyclohexadienones. The main challenge to make them asymmetric by catalytic methods is to control the selectivity while overcoming the loss of aromaticity. In this tutorial review, an up to date summary of recent progress in CADA reactions of phenol and aniline derivatives is presented.
 Wen-Ting Wu | Wen-Ting Wu was born in 1989 in Suzhou, Jiangsu province, China, and received her BS degree from Soochow University in 2012. She then joined Shanghai Institute of Organic Chemistry (SIOC) for her PhD degree under the supervision of Prof. Shu-Li You and Prof. Liming Zhang. Her research focused on the gold-catalyzed dearomatization reactions. |
 Liming Zhang | Liming Zhang was born in Pingxiang, China, in 1972. He received his B.S. degree in chemistry from Nanchang University in 1993, his first Master's degree in organometallic chemistry with Professor Zhengzhi Zhang from Nankai University in 1996, and his second Master's degree in organic chemistry with Professor Michael P. Cava from the University of Alabama in 1998. He obtained his Ph.D. degree with Professor Masato Koreeda from the medicinal chemistry program at the University of Michigan in 2003 and then carried out a post-doctoral study with Professor Sergey A. Kozmin at the University of Chicago. He started his independent academic career as Assistant Professor at the Department of Chemistry, University of Nevada, Reno, in July of 2005 and continued at the University of California Santa Barbara in July of 2009. He is currently a Professor of Organic Chemistry. His research interests include late transition metal-catalyzed reactions, natural product synthesis and medicinal chemistry. |
 Shu-Li You | Dr Shu-Li You received his BSc in chemistry from Nankai University in 1996. He obtained his PhD from Shanghai Institute of Organic Chemistry (SIOC) in 2001 under the supervision of Prof. Li-Xin Dai before doing postdoctoral studies with Prof. Jeffery W. Kelly at The Scripps Research Institute. From 2004, he worked at the Genomics Institute of the Novartis Research Foundation as a Principal Investigator before returning to SIOC in 2006. His research interests include asymmetric catalysis, synthetic methodology, natural product synthesis, as well as medicinal chemistry. |
Key learning points(1) Overview of current developments in the catalytic asymmetric dearomatization reactions of phenol and aniline derivatives.(2) Main challenges confronted in this field: control of chemo-, regio- and enantioselectivities while overcoming the high energy barrier during the dearomatization process. (3) General strategies employed in this area: oxidative and non-oxidative dearomatization reactions of phenol and aniline derivatives. |
1. Introduction
Phenol and its derivatives are widespread in nature such as natural products, bioactive molecules and lignin, the latter of which is a significant component in the support tissue of plants (Fig. 1). They often serve as a class of readily available chemical feedstocks and are mainly obtained via the Hock process starting from cumene. On the other hand, the intriguing chemical properties of phenol and its derivatives1 have captured enormous attention of chemists from both industry and academia for centuries. As a class of electron-rich arenes containing a hydroxyl group bound directly to the aromatic ring, phenols are sensitive towards oxidation, which is also the reason why phenols are suitable radical scavengers and often employed as oxidation inhibitors. Meanwhile, there exists an intrinsic tautomeric keto–enol equilibrium in phenol and its derivatives, and the enol form is more stable than its keto tautomer due to the formation of the aromatic system.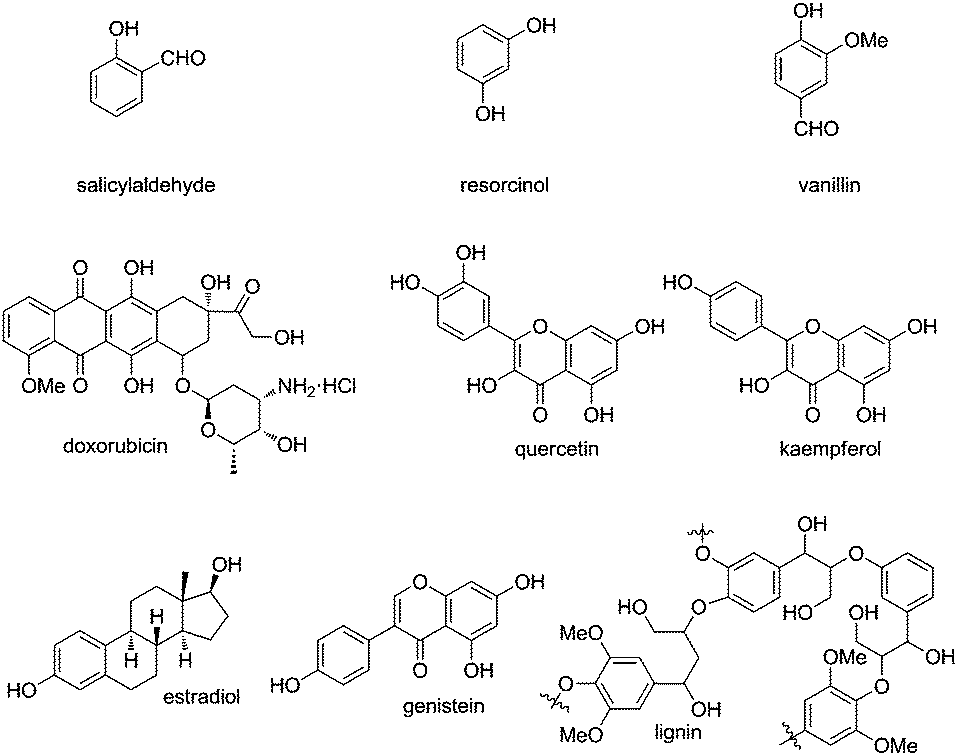 | ||
| Fig. 1 Natural products and bioactive molecules containing phenol motifs. | ||
In addition, dearomatization reactions of phenol and its derivatives have been studied intensively for a long time since they provide an efficient way to construct highly functionalized cyclohexadienones, which often appear in diverse biologically active natural products and pharmaceuticals (Fig. 2). However, how to make them asymmetric by catalytic methods has been daunting chemists for dozens of decades. In the past decade, considerable progress has been achieved in catalytic asymmetric dearomatization (CADA) reactions, which are attractive synthetic strategies to transform aromatic compounds to three-dimensional molecules containing quaternary carbon centers, and spiro or bridged backbones in many cases.2 The main challenge in the CADA reactions of phenol and aniline derivatives is to control the reaction selectivity including chemo-, regio-, and enantioselectivities while overcoming the loss of aromaticity.
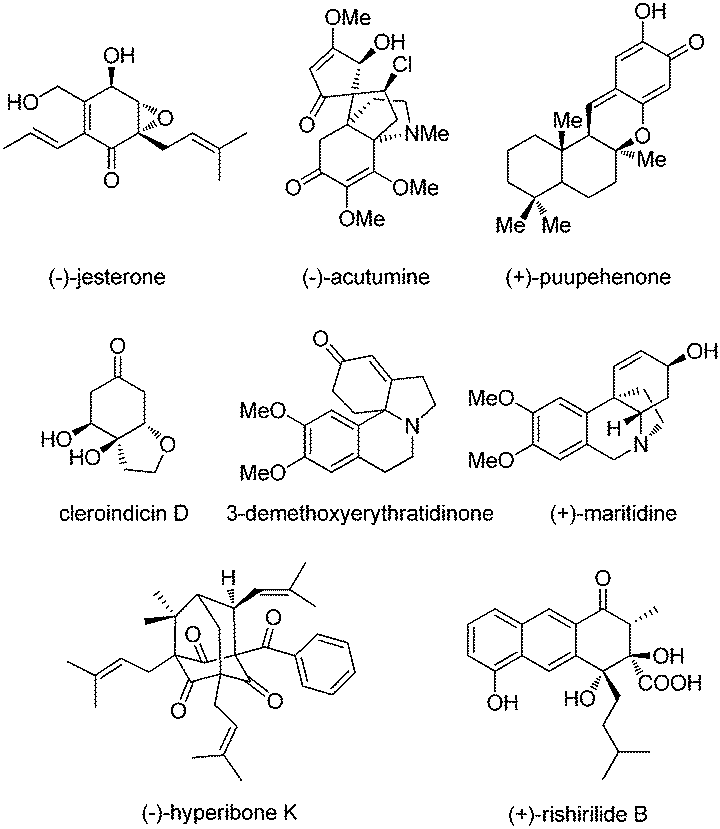 | ||
| Fig. 2 Natural products and bioactive molecules containing cyclohexadienone backbones. | ||
Thanks to the development of asymmetric catalysis in general, breakthroughs have recently been achieved in CADA reactions with excellent selectivities. In this tutorial review, these breakthroughs will be discussed under two categories, i.e., oxidative and non-oxidative CADA reactions of phenol and aniline derivatives.
2. CADA reactions of phenol and its derivatives under oxidative conditions
Phenols are electron-rich aromatic rings and are readily oxidized through the loss of one or two electrons. Historically, oxidative dearomatization reactions of phenol and its derivatives have been investigated intensely. Due to the commercial availability of oxidants, such as PhI(OAc)2 and 2,3-dichloro-5,6-dicyano-p-quinone (DDQ), and the development of efficient catalytic systems, oxidative dearomatization reactions of phenols have become a conventional methodology in organic synthesis and particularly in natural product synthesis.2.1 Chiral hypervalent iodine involved reactions
Since the first organic hypervalent iodine was prepared by Willgerodt in 1886, hypervalent iodine reagents have evolved into commonly used oxidants in organic synthesis and regarded as of low toxicity, in contrast to many metal oxidants.3 Generally, it was postulated that there exist two types of key intermediates in the hypervalent iodine mediated oxidative dearomatization reactions of phenols: one is phenoxy-λ3-iodane species A and another is discrete phenoxenium ion B (Scheme 1).4 With the utilization of chiral hypervalent iodine reagents, the asymmetric reactions could be achieved via the associative intermediate A.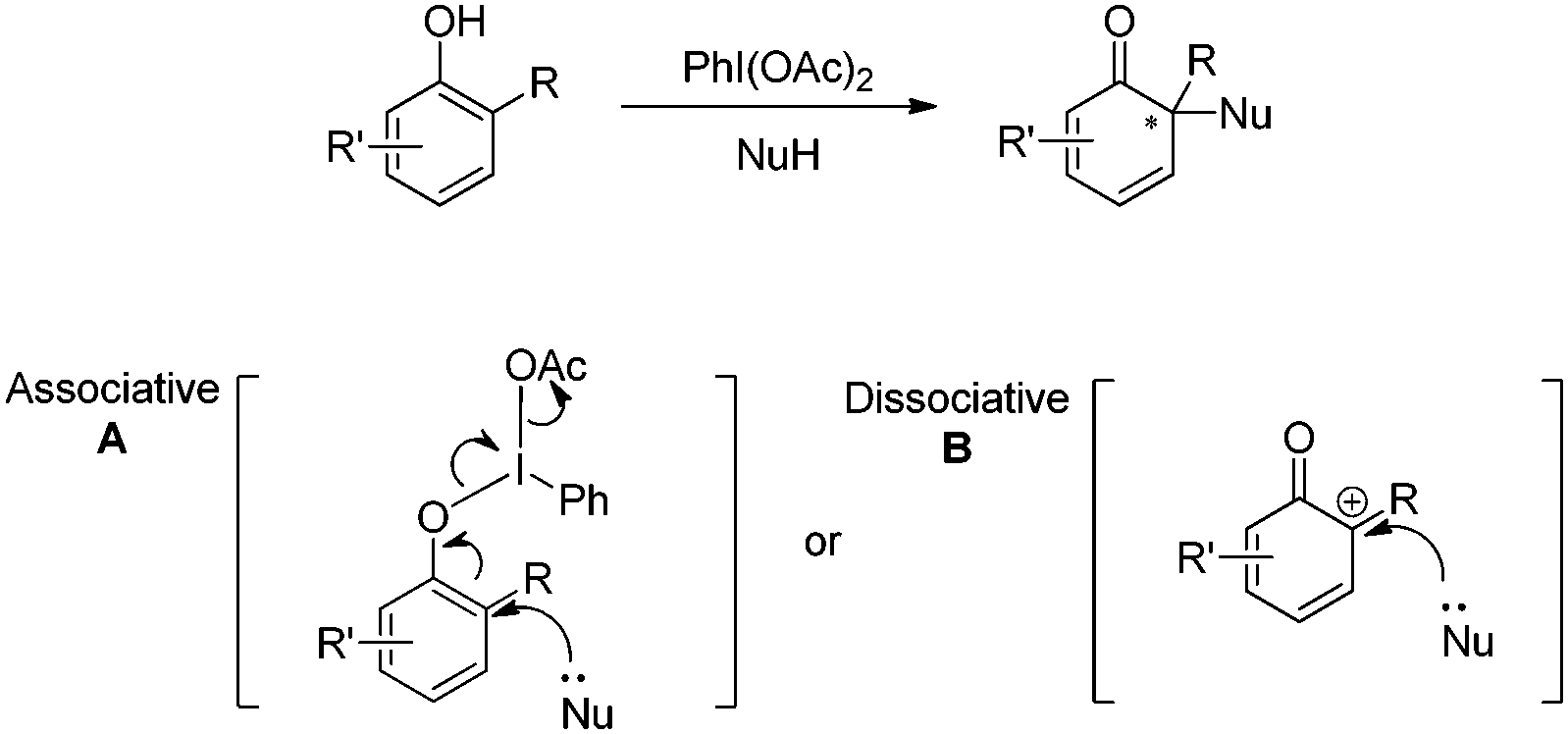 | ||
| Scheme 1 Two postulated intermediates of hypervalent iodine mediated oxidative dearomatization reactions of phenols. | ||
However, the development of chiral hypervalent iodines as reagents or even catalysts with high efficiency and satisfactory enantioselectivity has been a challenging task for many years. Breakthroughs were achieved in the past decade attributed to the considerable efforts from the groups of Kita, Ishihara, Quideau, Ibrahim and Gong, respectively. The commonly used strategy to realize the enantioselective oxidative reactions of phenol and its derivatives is the combination of catalytic chiral iodides as pre-catalysts and m-CPBA as the stoichiometric co-oxidant.
In 2008, Kita and coworkers5 reported a new chiral hypervalent iodine(III) reagent (R)-C1a bearing a rigid spirobiindane backbone to achieve the first enantioselective oxidative dearomatization reaction of 1-naphthol derivatives 1 to construct a chiral ortho-spirolactone structure 2 in 66–86% yields and 78–86% ee (enantiomeric excess) (Scheme 2, eqn (1)). They also found that the combination of m-chloroperoxybenzoic acid (m-CPBA) and 15 mol% of the corresponding chiral iodoarene (R)-C1b could generate (R)-C1ain situ and achieve comparative results (Scheme 2, eqn (2)). Moreover, the chiral iodoarene (R)-C1b can be easily recovered by column chromatography, which renders this new asymmetric oxidative dearomatization method practical. Later on, Kita and coworkers6 modified the rigid chiral spirobiindane iodoarene catalysts by ortho-functionalization in 2013 and discovered (R)-C1c as the optimal catalyst, which has ethyl groups ortho to the iodine atoms and hence provides extensive equatorial surroundings around them (Scheme 2, eqn (3)). Thus, an excellent level of asymmetric induction is realized in the catalytic oxidation of naphthol 1a. A plausible transition-state model was established based on the ortho-substituent effect and the results of X-ray analysis, where the tethered carboxylic acid moiety attacks the re-face of the enol moiety of 1a to afford 2a with the R configuration.
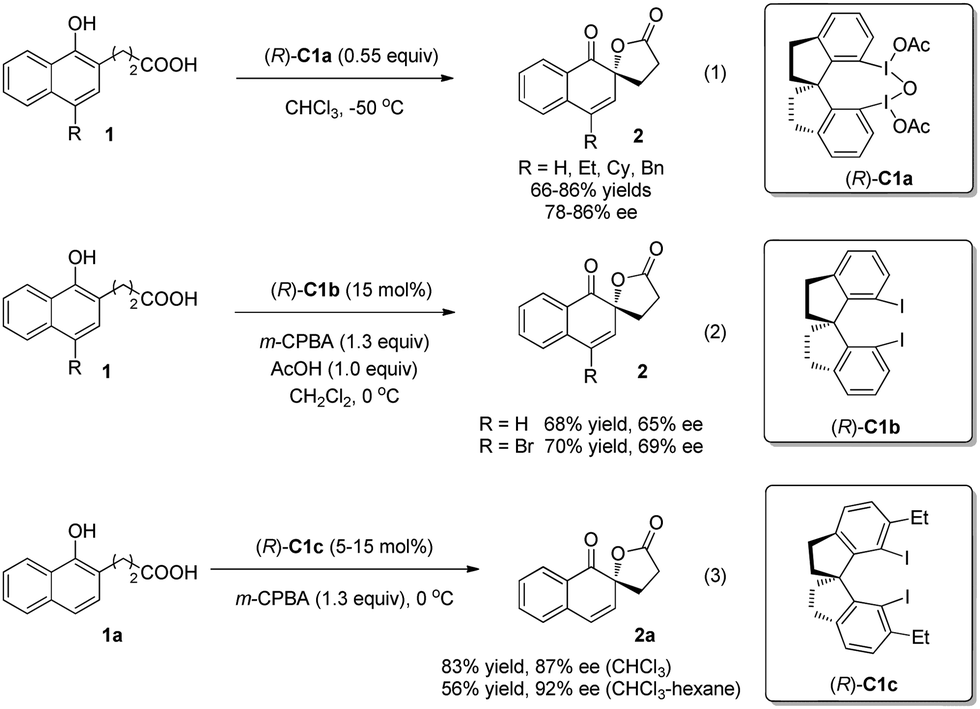 | ||
| Scheme 2 Enantioselective intramolecular oxidative dearomatization reaction of naphthols reported by Kita. | ||
Different from Kita's rigid chiral iodoarene (R)-C1b, Ishihara and coworkers7 designed and developed a series of conformationally flexible C2-symmetric chiral iodoarenes, represented by C2, which turned out to be an effective type of precatalysts for the enantioselective Kita oxidative spirolactonization reaction. Three units contained in the C2-symmetric chiral iodoarenes – an iodoaryl moiety, chiral linkers, and subfunctional groups – work cooperatively based on hydrogen-bonding. Under analogous conditions, the desired dearomatized products 2 could be obtained in up to 94% yield and 92% ee (Scheme 3).
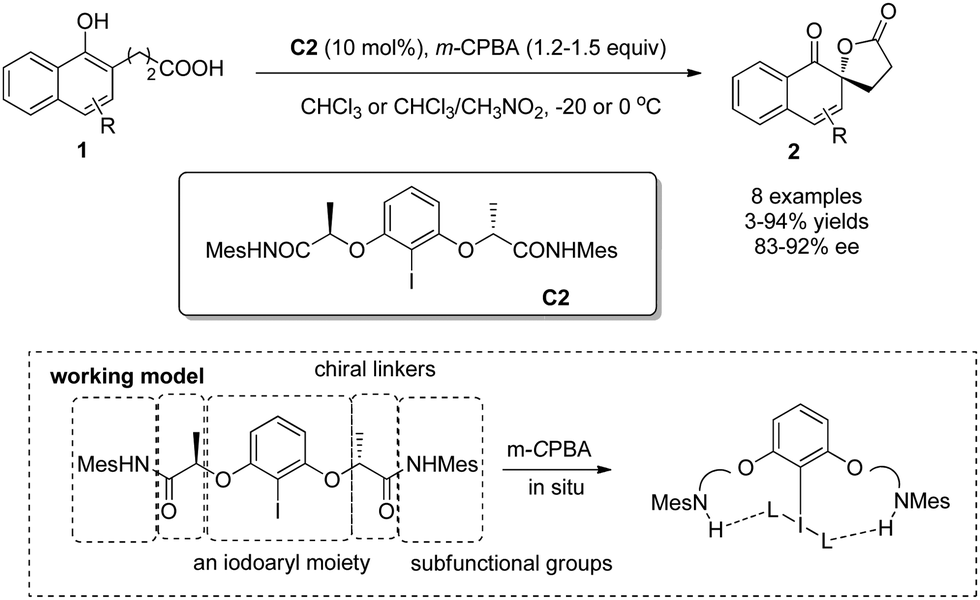 | ||
| Scheme 3 Enantioselective intramolecular oxidative dearomatization reaction of naphthols reported by Ishihara. | ||
In 2013, with the further modified C2-symmetric chiral iodoarene C3, Ishihara and coworkers8 expanded the substrate scope, previously limited to 1-naphthol derivatives,5–7 to phenols, and realized their highly enantioselective oxidative dearomatization reactions. Under optimized conditions, both electron-rich and electron-deficient phenols 3 were oxidized into various cyclohexadienones 4 in excellent enantiomeric excess. For the reactive cyclohexadienone products, Diels–Alder reactions were carried out in one-pot with different dienophiles to deliver the corresponding bridged products 5 as single diastereomers in up to 99% ee. Control experiments implied that the addition of protic polar MeOH or HFIP is essential for achieving satisfactory yields and enantioselectivity and it was proposed by the authors that the alcohol additives play the role of ligands of iodine(III) species (Scheme 4).
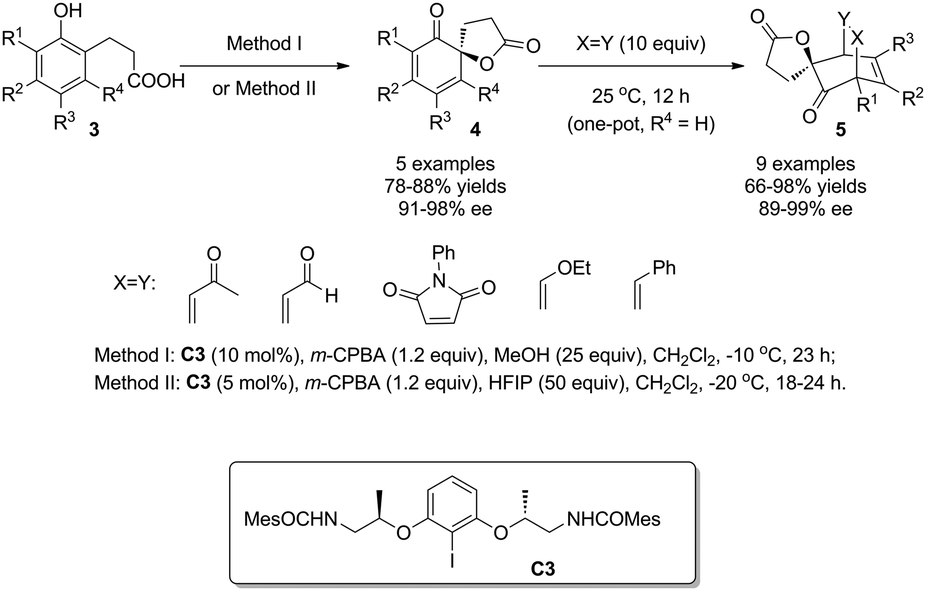 | ||
| Scheme 4 Enantioselective intramolecular oxidative dearomatization reaction of phenols reported by Ishihara. | ||
Recently, Ibrahim and coworkers9 reported another chiral iodoarene C4 based on a totally different backbone. Bearing a rigid and congested all-carbon anti-dimethanoanthracene framework, the C2-symmetric C4, in the presence of m-CPBA, can catalyze the Kita oxidative spirolactonization reaction in moderate yields and enantioselectivity (Scheme 5).
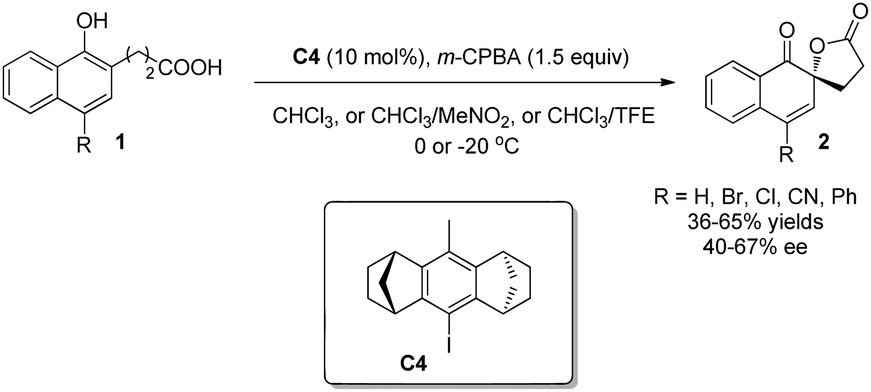 | ||
| Scheme 5 Enantioselective intramolecular oxidative dearomatization reaction of naphthols reported by Ibrahim. | ||
Previously, intramolecular nucleophiles were limited to carboxylic acids. As a result, the oxidative dearomatization reactions could only deliver spirolactone products. In 2015, Gong and coworkers10 expanded the nucleophiles to include appropriately tethered electron-rich arenes in the form of anilides. Hence, with C2 as precatalyst and m-CPBA as the stoichiometric oxidant, various chiral spirooxindoles 7 possessing all-carbon spiro-stereogenic centers can be obtained in satisfactory yields and enantioselectivity (42–80% yields and 80–92% ee). They also investigated the effect of alcohol and water as additives and discovered that the addition of both TFE (2,2,2-trifluoroethanol) and H2O is beneficial for both yield and enantioselectivity due to the facilitation of the associative pathway (Scheme 6).
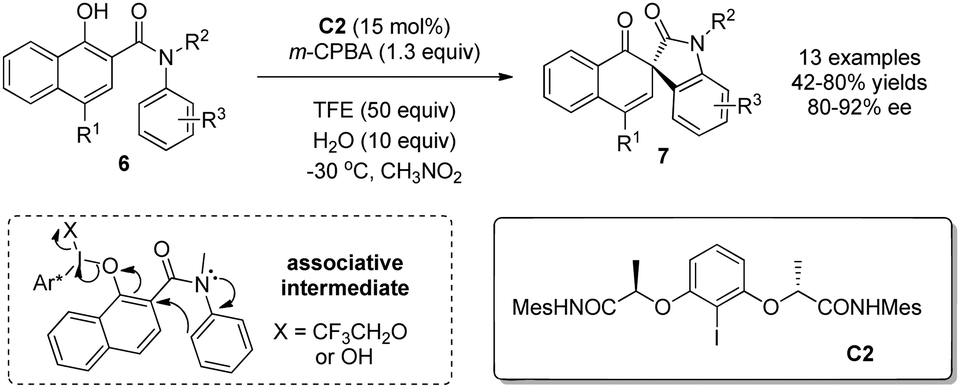 | ||
| Scheme 6 Enantioselective synthesis of spirooxindoles reported by Gong. | ||
Besides intramolecular oxidative dearomatization reactions, asymmetric intermolecular dearomatization reactions have also been realized with chiral iodoarenes. In 2009, Quideau and coworkers11 reported asymmetric dearomatization reactions of 2-methyl-1-naphthol (Scheme 7). When excess chiral iodoarene C5 (2 equiv.) and m-CPBA (1 equiv.) are used, the ortho-hydroxylative phenol dearomatization reaction is achieved with a moderate level of asymmetric induction (50% ee). On the other hand, the use of a catalytic amount of C5 (0.1 equiv.) and excess m-CPBA (2.5 equiv.) led to the double oxidized product 9 with decreased enantioselectivity (29% ee). Experimental observations support that a chiral iodine(V) species was generated in situ from the iodoarene and m-CPBA. With further modified chiral bis(λ5-iodane) C5′, dearomatized product 8 can be obtained in 73% ee.12
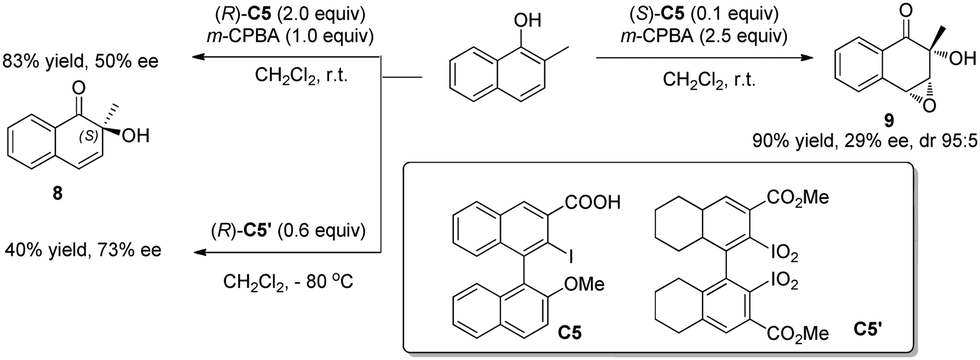 | ||
| Scheme 7 Enantioselective intermolecular oxidative dearomatization reaction of naphthols reported by Quideau. | ||
Despite the difficulty, intermolecular oxidative dearomatization reactions of phenols were achieved by Harned and coworkers in 2013.13 On the basis of computational molecular modeling of the transient associative intermediate of type A (Scheme 1), a new chiral aryl iodide catalyst C6 derived from 8-iodotetralone and tartaric acid was designed and applied in an intermolecular oxidative dearomatization reaction of phenols 10. The para-quinols 11 were formed with low to moderate enantioselectivity. In comparison, the intramolecular dearomatization reaction of para-substituted phenol 12 afforded the spirocycle 13 without notable improvement in asymmetric induction (Scheme 8).
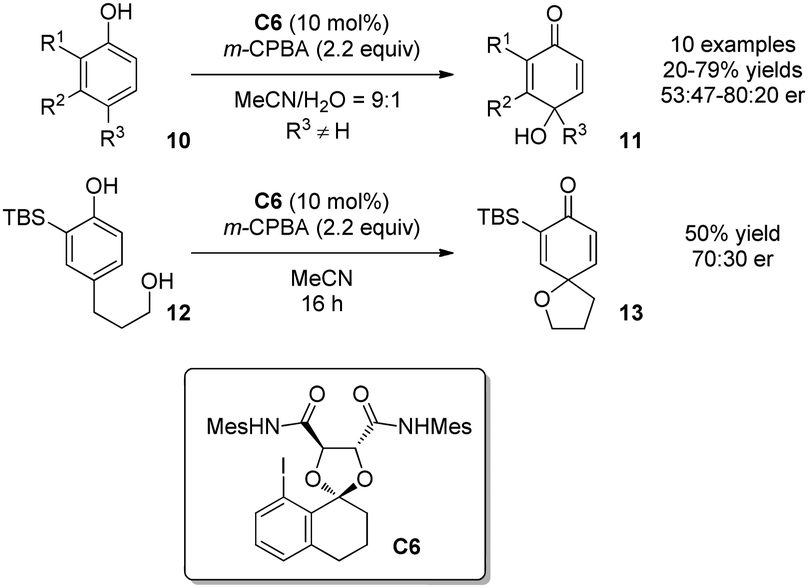 | ||
| Scheme 8 Enantioselective oxidative dearomatization reaction of phenols reported by Harned. | ||
2.2 SET oxidation reactions
In the previous section, phenol derivatives are oxidized by chiral hypervalent iodines in two-electron processes via the attack of either the associative intermediate A or the dissociative intermediate B by nucleophiles. In addition, these electron-rich arenes can also undergo oxidations in single electron transfer processes (SET) involving radical intermediates.Oguma and Katsuki disclosed that the Fe(salan) complex C7 can catalyze the aerobic intermolecular oxidative dearomatization reaction of 1,3-disubstituted 2-naphthols 14 with nitroalkanes14 or phenols 1615 as external nucleophiles, affording cyclic enones 15 and 17 in excellent yields and enantioselectivity, respectively. It was proposed that once 2-naphthols 14 were coordinated by C7, radical cation species C would be generated from SET oxidation by O2 in the air. The radical cation species C could be trapped by nitroalkane to deliver the corresponding dearomatized products 15 bearing an all-carbon quaternary stereocenter. On the other hand, the radical cation species C could be further transformed into a strong Michael acceptor by γ-H abstraction and the subsequent Michael addition with phenols 16 took place to yield useful spirocyclic (2H)-dihydrobenzofurans 17 (Scheme 9).
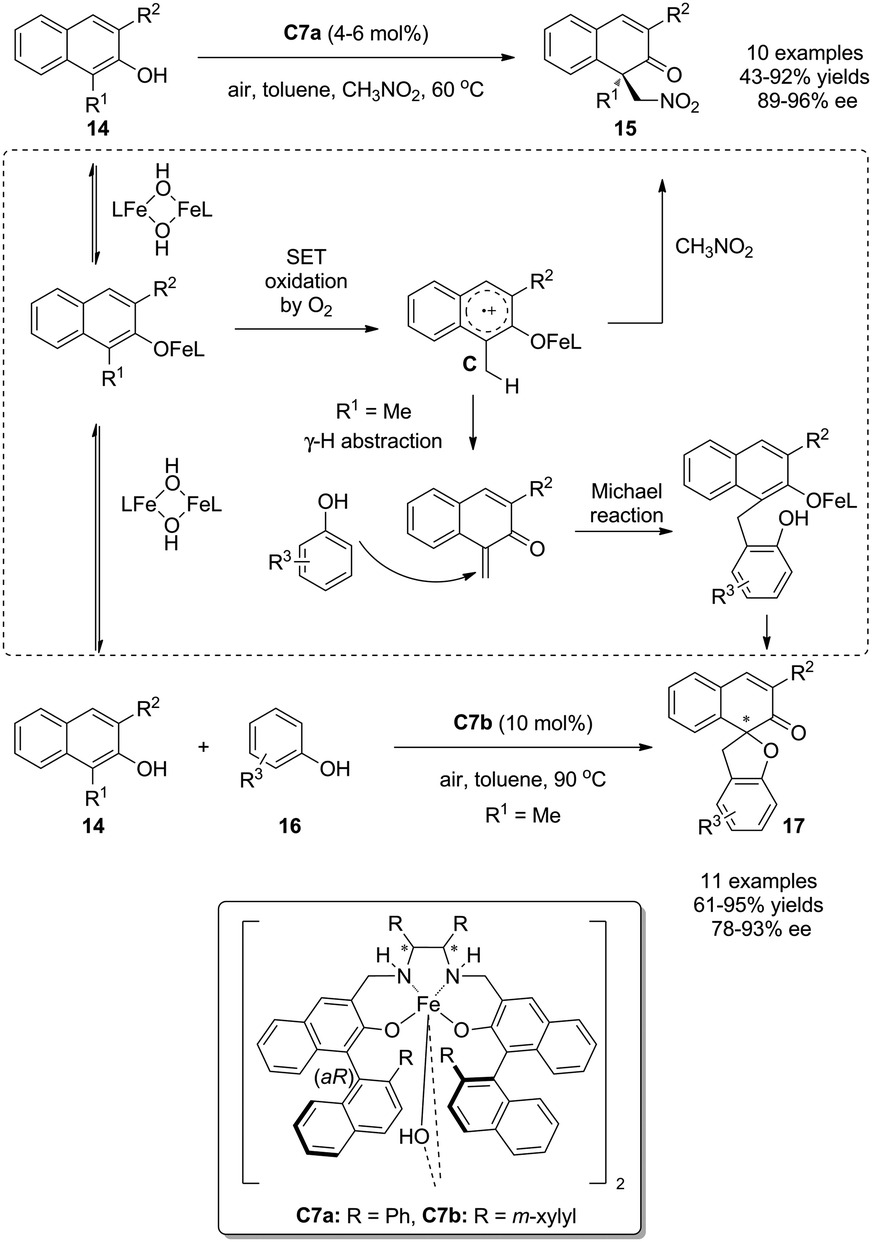 | ||
| Scheme 9 Fe(salan) complex-catalyzed aerobic asymmetric dearomatization reactions of naphthols reported by Katsuki. | ||
3. CADA reactions of phenol and aniline derivatives under non-oxidative conditions
Phenols are electron-rich aromatic compounds and hence display considerable nucleophilicity (Fig. 3). There are several nucleophilic sites on the phenol ring, O, C2 and C4, and only the reactions involving the carbon nucleophilic sites can be employed in dearomatization reactions. Moreover, the selectivity over these nucleophilic sites can be challenging. As such, substrates are often delicately designed to achieve desired chemo-, regio- and enantioselectivities.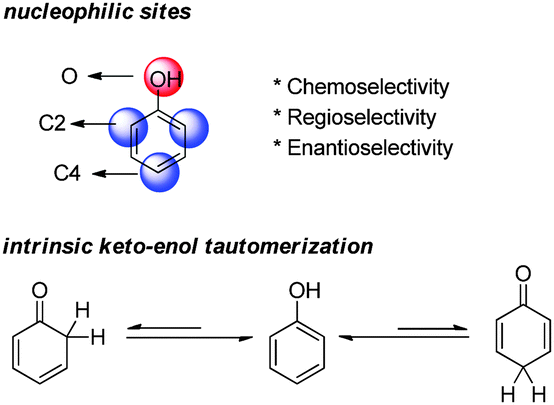 | ||
| Fig. 3 Nucleophilic sites on the phenol ring and the intrinsic keto–enol tautomerization of phenol. | ||
Due to the aromaticity, the enol form of phenol is much more stable than its keto tautomers, which are dearomatized. If some substrate intrinsically prefers the keto tautomers, its dearomatization reaction can be relatively facile. The most common factors that contribute to the keto forms are (1) additional hydroxy groups on the benzene ring; (2) additional arenes annulated to the phenolic benzene ring; (3) the formation of phenolate; (4) bulky groups in the ortho-positions of phenol; and (5) electron-withdrawing substituents in the ortho- and para-positions of phenol.
Besides phenols, naphthols have frequently been utilized as model substrates due to their relatively weak aromaticity. In addition, anilines and aminonaphthalenes are also electron-rich aromatic compounds and can undergo similar dearomatization reactions.
In general, dearomatization reactions of phenols can be regarded as functionalization of enols, and selected examples below are classified according to the types of functionalization.
3.1 Alkylative dearomatization reactions
ortho-Substituted phenols easily undergo alkylation with carbon-based electrophiles, and racemic protocols have been extensively documented and broadly applied in the synthesis. However, only a few catalytic asymmetric cases have been reported.In 2007, Porco and coworkers16 succeeded in the construction of the bicyclo[3.3.1]nonane framework via alkylative dearomatization annulation during the synthesis of polyprenylated phloroglucinol natural products. In 2010, they realized the asymmetric variant17 by utilizing a chiral phase-transfer catalyst derived from cinchona alkaloids. Hence, with C8 as the chiral phase-transfer catalyst and 5 equivalents of CsOH·H2O as the base, the previously reported cascade, i.e., Michael addition–elimination–Michael addition–aldol reaction sequence, occurs smoothly with clusiaphenone B as substrate in a nonracemic manner, affording adamantane 18 in 71% yield and 90% ee. The product was further transformed into (−)-hyperibone K, which permits the assignment of its absolute configuration (Scheme 10).
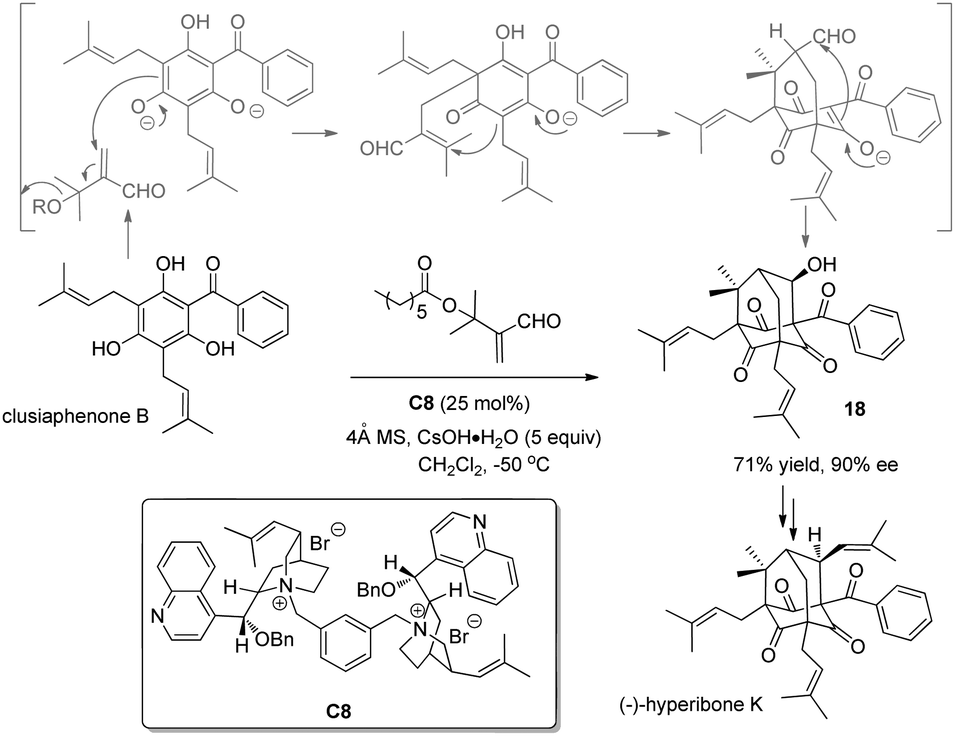 | ||
| Scheme 10 Enantioselective alkylative dearomatization reaction of clusiaphenone B reported by Porco. | ||
Using aziridines 19 as alkylative reagents and the catalyst generated from nBu2Mg and a newly designed Box–OH ligand L1, Wang and coworkers realized in 2015 asymmetric dearomatization reactions of β-naphthol derivatives 14 in excellent yields and enantioselectivity.18 Their investigations on the possible activation mode indicate that the in situ generated magnesium catalyst interacts with both β-naphthol 14a and N-(2-picolinoyl)-meso-aziridine 19a to form a relatively rigid chiral environment. Interestingly, a positive nonlinear effect was also observed in the reaction (Scheme 11).
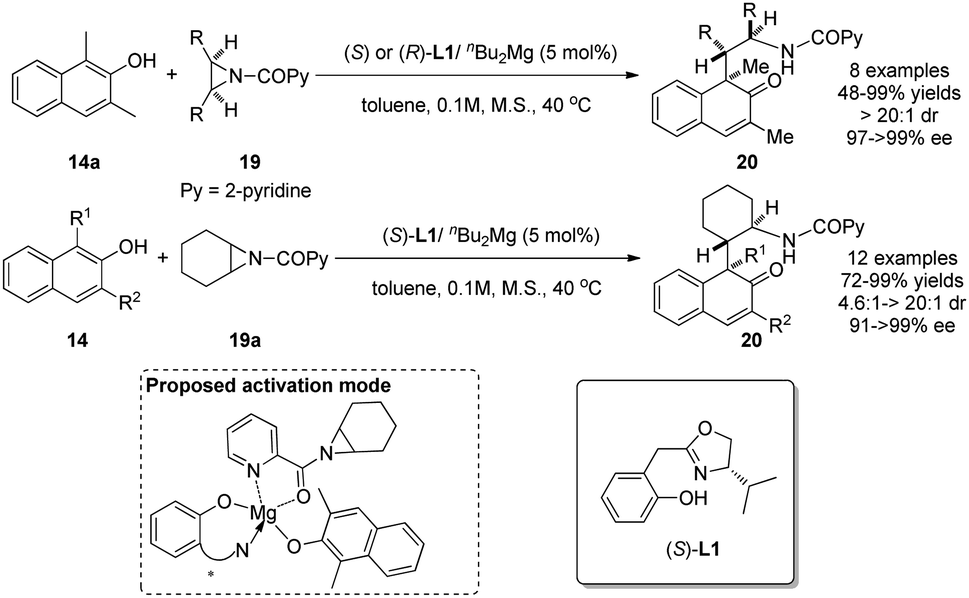 | ||
| Scheme 11 Enantioselective alkylative dearomatization reaction of β-naphthol derivatives reported by Wang. | ||
In the same year, You and coworkers19 employed chiral thiourea C9 as catalyst to achieve intermolecular asymmetric dearomative Michael additions of β-naphthols 14′ to nitroethylene. Functionalized β-naphthalenones 21 bearing an all-carbon quaternary stereogenic center are formed in good yields with excellent enantio control. To demonstrate the synthetic utility of this chemistry, enantioenriched 21 is transformed into aminotetralin 21aa and carbamate 21bb, the latter of which serves as a key intermediate in the synthesis of the common propellane core structure of the hasubanan alkaloids. A plausible working model was also postulated, where the hydrogen bonds between thiourea C9 and nitroethylene and the hydroxy group of β-naphthol provide a highly ordered transition state, which enables the high level of enantioselective induction (Scheme 12).
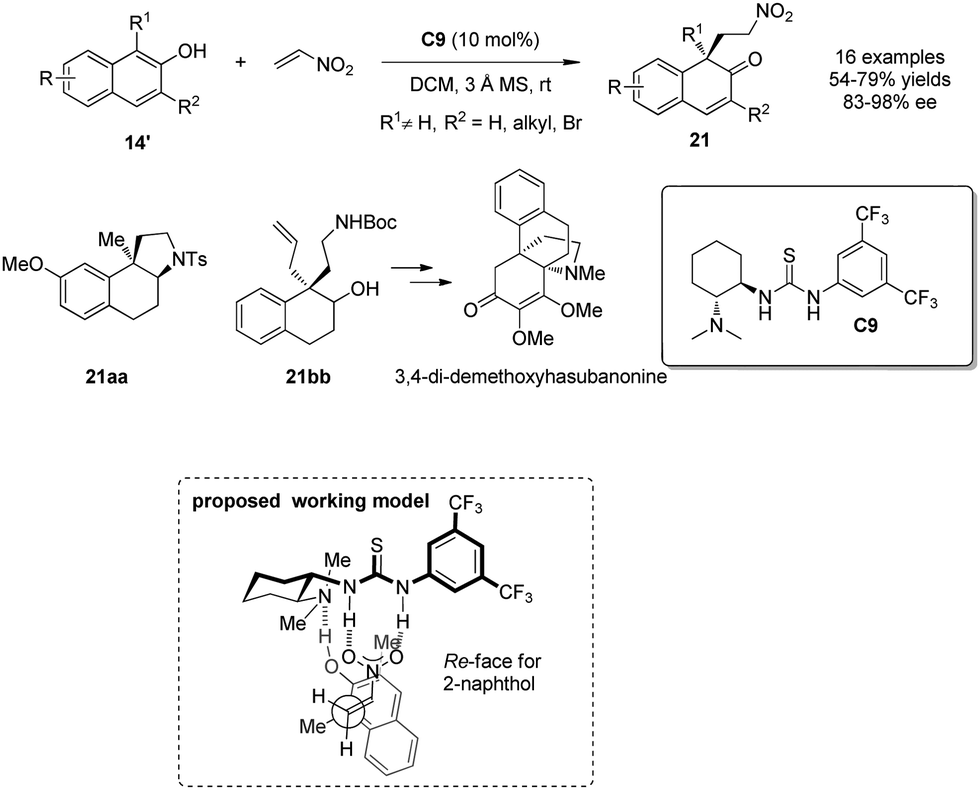 | ||
| Scheme 12 Enantioselective alkylative dearomatization reaction of β-naphthol derivatives reported by You. | ||
3.2 Allylative dearomatization reactions
Asymmetric allylic alkylation (AAA) reactions have become one of the most reliable and versatile methods for the formation of C–C bonds with desirable enantioselectivity, and a wide range of nucleophiles including phenol and its derivatives can be employed. However, O-allylation of phenol substrates dominates over C-allylation, which has only been realized in a handful of cases and rarely displays good chemo-, stereo- and enantioselectivities.In 2010, Hamada and coworkers20 reported one successful study on Pd-catalyzed enantioselective intramolecular allylic dearomatization reaction of para-substituted phenol 22a. With L2 as the chiral ligand, the spirocyclohexadienone 23a was formed in 80% yield, 9.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 89% ee (Scheme 13).
:1 dr and 89% ee (Scheme 13).
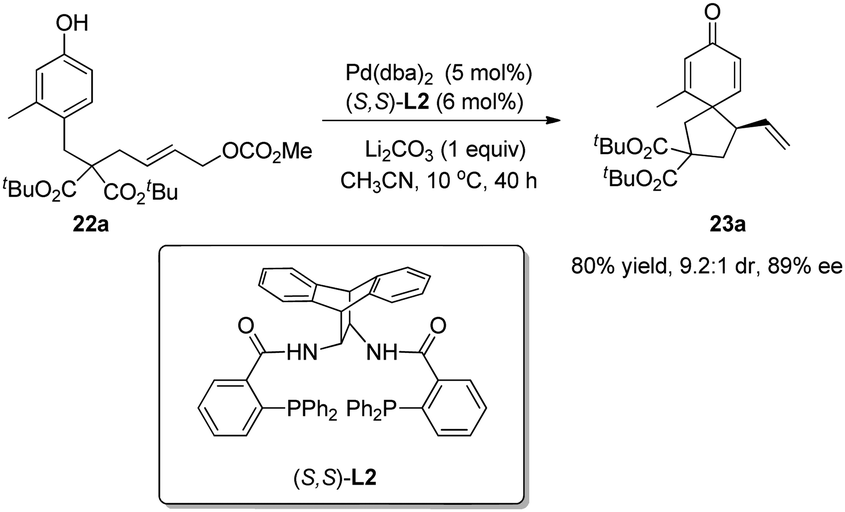 | ||
| Scheme 13 Pd-catalyzed asymmetric intramolecular allylic dearomatization reaction of phenols reported by Hamada. | ||
In 2011, You and coworkers21 accomplished an Ir-catalyzed intramolecular asymmetric allylic dearomatization reaction of phenols. In the presence of a catalytic amount of [Ir(cod)Cl]2, the chiral phosphoramidite ligand L3 and 2 equivalents of Li2CO3, a series of para-substituted phenols 22 were converted into various 5- or 6-membered spirocyclohexadienone derivatives 23 in excellent yields and enantioselectivity (up to 97% ee) (Scheme 14).
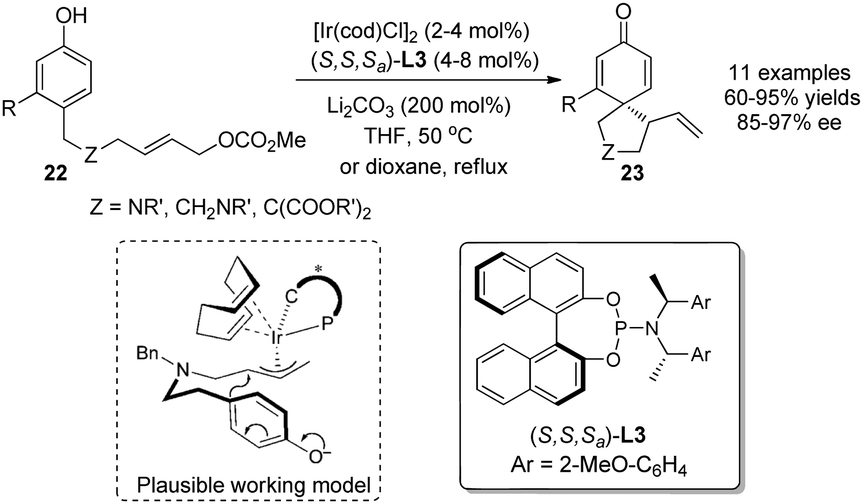 | ||
| Scheme 14 Ir-catalyzed asymmetric intramolecular allylic alkylation dearomatization reaction of phenols reported by You. | ||
The two cases above both have the nucleophilic moiety substituted on the phenol para-position to avoid the potential competition of alkylations at the phenolic oxygen and/or at the ortho position. When it comes to the intermolecular scenario, it can be a great challenge to address all three issues: chemo-, regio-, and enantioselectivities. In 2013, You and coworkers22 realized a palladium-catalyzed intermolecular asymmetric allylic dearomatization reaction of naphthol derivatives 14. Hence, with various substituted allylic carbonates 24 as the allylating reagents, an array of β-naphthalenones 25 with an all-carbon quaternary chiral center could be constructed in good to excellent yields, and with excellent chemo- and enantioselectivities. This intermolecular example establishes a promising precedent for future development in this area (Scheme 15).
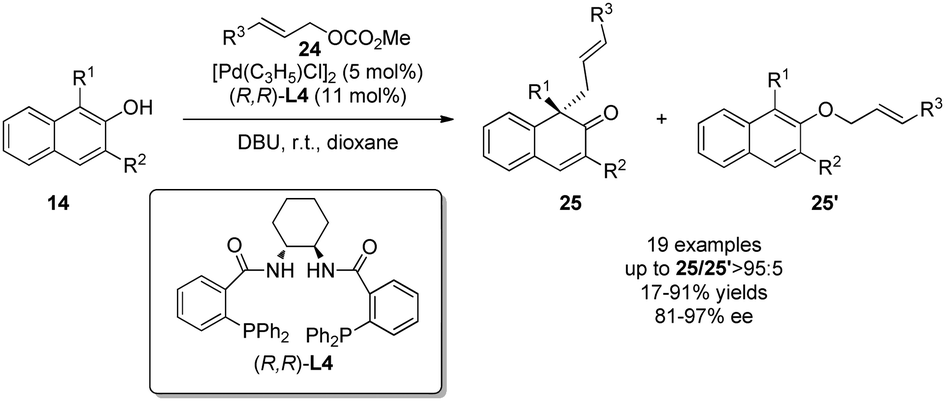 | ||
| Scheme 15 Pd-catalyzed asymmetric intermolecular allylic dearomatization reaction of naphthols reported by You. | ||
3.3 Arylative dearomatization reactions
Cross-coupling reactions are powerful tools in organic synthesis. Especially, Pd-catalyzed asymmetric cross-coupling reactions have been investigated intensively, and ground-breaking discoveries have been realized quite recently owing to the development of various chiral ligands. In general, phenols are employed as intramolecular nucleophiles to attack electrophilic Pd intermediates en route to multi-functionalized dearomatized products.An early study in this area was reported by Buchwald in 2009, where anilines instead of phenols undergo asymmetric dearomatization reaction.23 In the reaction, LiOtBu was used to promote deprotonation of the aniline NH, which in turn increases the nucleophilicity of the naphthalene C2-position and hence facilitates its attack at the Pd(II) center. With KenPhos L5 as the chiral ligand, various benzocarbazole derivatives 27 were obtained in good to excellent yields and with mostly outstanding enantioselectivity (Scheme 16).
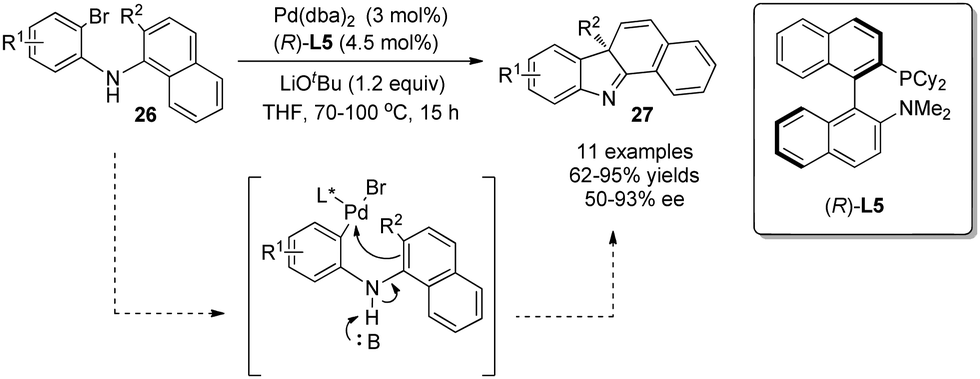 | ||
| Scheme 16 Pd-catalyzed asymmetric intramolecular arylative dearomatization reaction of aminonaphthalenes reported by Buchwald. | ||
In 2011, Buchwald and coworkers24 broadened the substrate scope to include phenols. On the basis of racemic Pd-catalyzed arylative dearomatization reaction of phenols 28, the asymmetric version was carried out with (S,S)-L6 as ligand, and the spirocyclohexadienone products 29 were isolated in good to excellent yields and enantioselectivity (up to 91% ee) (Scheme 17).
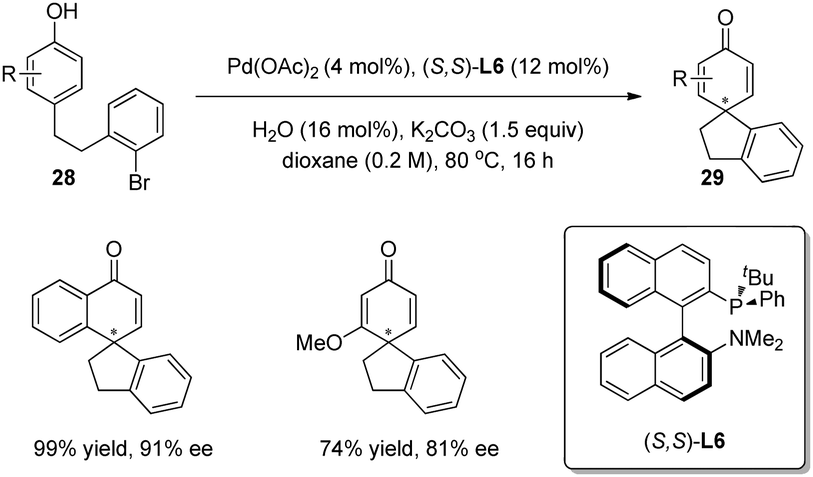 | ||
| Scheme 17 Pd-catalyzed asymmetric intramolecular arylative dearomatization reaction of phenols reported by Buchwald. | ||
The above two studies demonstrated that Pd-catalyzed cross-coupling reactions can offer versatile strategies for the CADA reaction of phenol derivatives. In 2014, You and coworkers25 reported another application of this strategy. With a broad range of 5-hydroxyl indolines 30 as substrate, Pd-catalyzed intramolecular arylative dearomatization reactions delivered various sterically congested tetracyclic spiroamines 31 in excellent yields. One of the products was converted to 3-demethoxyerythratidinone upon selective hydrogenation, thereby offering an efficient and straightforward synthetic route to this natural product (Scheme 18, eqn (1)). Their preliminary studies suggested that asymmetric catalysis is also feasible (Scheme 18, eqn (2)).
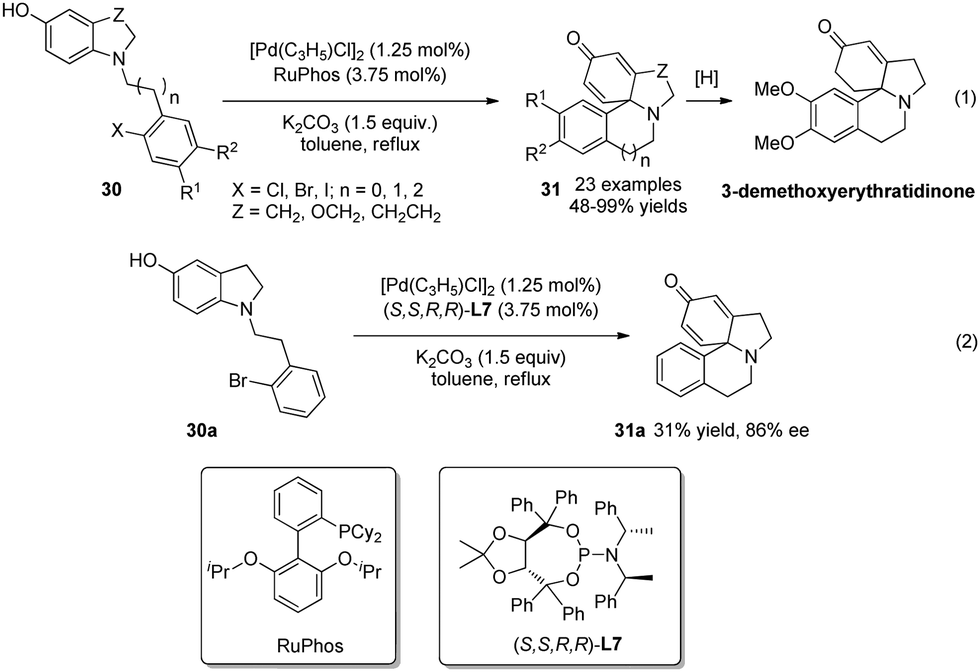 | ||
| Scheme 18 Pd-catalyzed asymmetric intramolecular arylative dearomatization reaction of 5-hydroxyl indolines reported by You. | ||
In the studies discussed so far, excellent asymmetric inductions were achieved only in limited cases. In 2015, Tang and coworkers26 developed a P-chiral biaryl monophosphine ligand L8 and applied it in a novel enantioselective palladium-catalyzed dearomative cyclization reaction. A range of chiral phenanthrenone derivatives 33 were obtained in excellent enantiomeric excess. This efficient method provides a facile way to synthesize terpenes and steroids, which is demonstrated in the syntheses of chiral kaurene intermediate 33a, boldenone skeleton 33b, and antimicrobial diterpene totaradiol 33cc (Scheme 19).
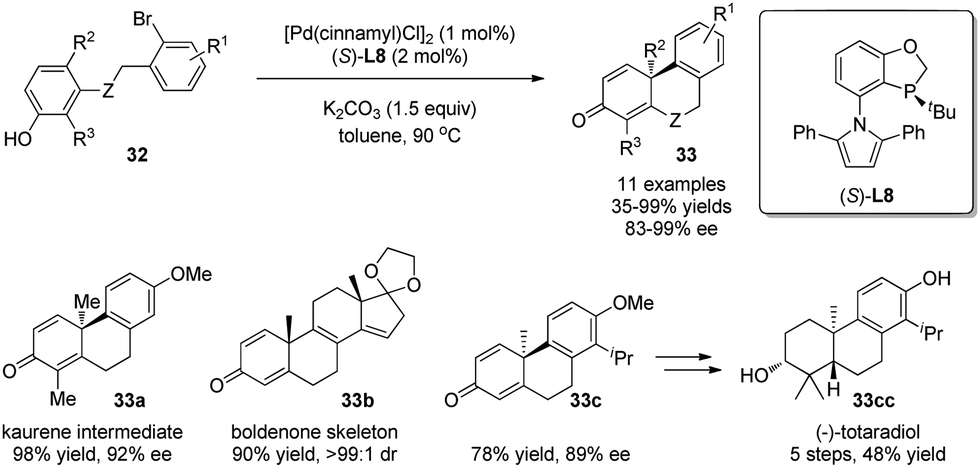 | ||
| Scheme 19 Pd-catalyzed asymmetric intramolecular arylative dearomatization reaction of phenols reported by Tang. | ||
3.4 Alkenylative dearomatization reactions
The electrophilic Pd(II) intermediates mentioned above can undergo insertion reactions with alkynes prior to dearomatization, which can be regarded as alkenylative dearomatization reactions of phenols. In 2015, Luan and coworkers27 realized a Pd-catalyzed enantioselective annulation of phenol derivatives 34 with alkynes 35, affording the spirocycle 36 with an all-carbon quaternary stereogenic center in superb yields and excellent enantioselectivity (up to 97% ee). Interestingly, the reaction achieves an axial-to-central chirality transfer, where the spirocyclic molecules 36 of central chirality are formed from dynamic kinetic resolutions of racemic axial chiral biaryls 34 (Scheme 20).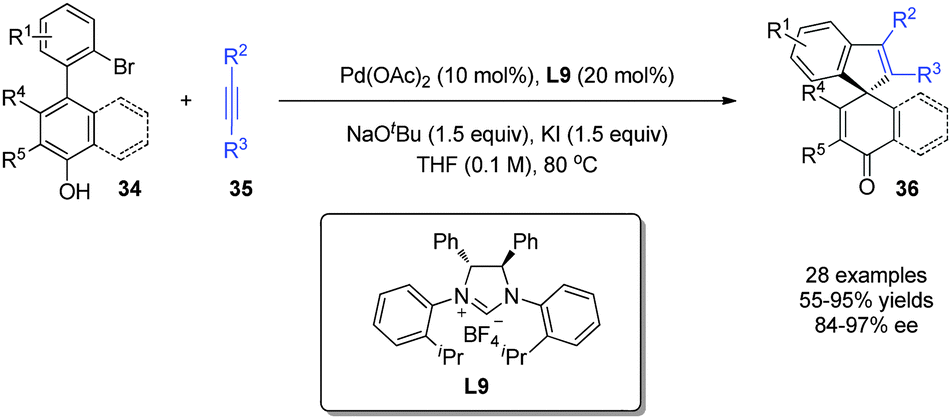 | ||
| Scheme 20 Pd-catalyzed asymmetric intramolecular dearomatization reaction of phenols reported by Luan. | ||
Besides Pd, Rh can also catalyze insertion reactions of alkynes. Almost at the same time, You and coworkers28 succeeded in a Rh-catalyzed asymmetric alkenylative dearomatization reaction of 1-aryl-2-naphthols 37via C(sp2)–H functionalization/alkyne insertion/annulation reaction. In the presence of a chiral Cp/Rh catalyst C10 and Cu(OAc)2 and air as the oxidants, the reaction was initiated by a hydroxyl group-directed C(sp2)–H functionalization by a Rh complex, followed by alkyne insertion and dearomative reductive elimination to afford highly enantioenriched spirocyclic enones 38 bearing an all-carbon quaternary stereogenic center in mostly good yields (Scheme 21).
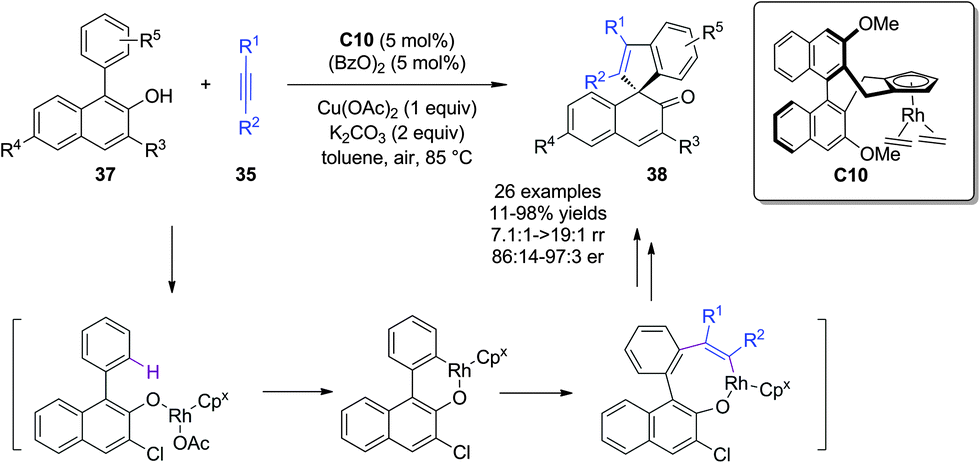 | ||
| Scheme 21 Rh-catalyzed asymmetric alkenylative dearomatization reaction of naphthols reported by You. | ||
It is well-known that cationic gold(I) can activate alkynes toward attack by external nucleophiles, resulting eventually in the trans addition of H–Nu across the C–C triple bond. With phenols and anilines as nucleophiles, this fundamental gold catalysis can lead to alkenylative dearomatization reactions of these electron-rich arenes. In 2015, Tanaka and coworkers29 realized the conversion of 1-aminonaphthalene derivatives 39 to two different products 40 and 40′, the ratio of which depends on the R′ group, in good to excellent yields and ee (Scheme 22, eqn (1)). In addition, the substrates with electron-rich heteroarenes appended to the ynamide (Scheme 22, eqn (2)) or displaying altered arrangement of the functional groups (Scheme 22, eqn (3)) can also undergo similar dearomatization reaction.30
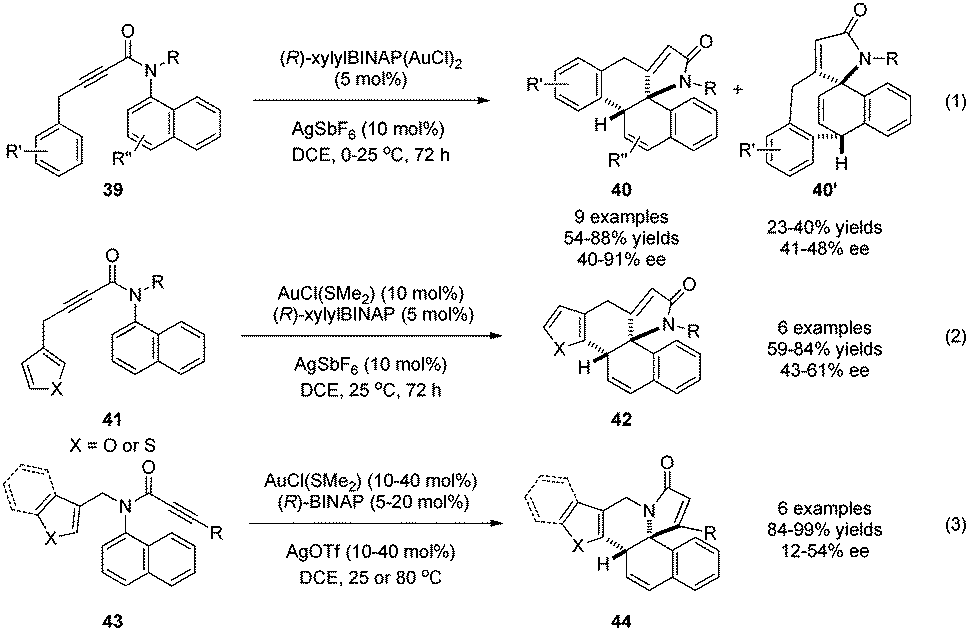 | ||
| Scheme 22 Gold-catalyzed asymmetric alkenylation dearomatization reaction of aminonaphthalenes reported by Tanaka. | ||
In addition to the intramolecular enantioselective alkenylative dearomatization reactions of phenols, an intermolecular Mg-catalyzed conjugate addition of β-naphthols 14 to ynones 45 was recently realized by Wang and coworkers.31 Like the reaction discussed in Scheme 11,15 the chiral magnesium catalyst generated in situ by reaction of nBu2Mg with L10 is proposed to interact with both substrates in the transition state. Notably, good to excellent Z-selectivity and enantioselectivity were obtained for a wide range of substrates (Scheme 23).
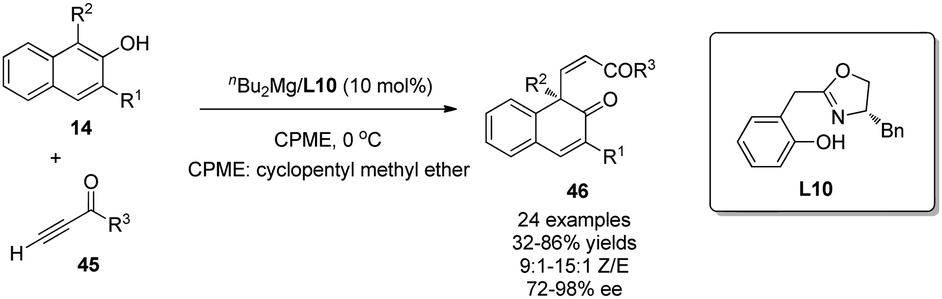 | ||
| Scheme 23 Mg-catalyzed asymmetric alkenylative dearomatization reaction of naphthols reported by Wang. | ||
3.5 Halogenative dearomatization reactions
The aforementioned alkylative, allylative, arylative and alkenylative dearomatization reactions form C–C bonds and are generally promoted or catalyzed by metal complexes. When it comes to asymmetric C–X (X![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) F, Cl) bond formation in dearomatization reactions, organocatalysts have exhibited outstanding performance.
F, Cl) bond formation in dearomatization reactions, organocatalysts have exhibited outstanding performance.
In 2013, Toste and coworkers32 realized a fluorinative dearomatization reaction of phenols 47 by using a BINOL-derived phosphate [e.g., (S)-C11] as the chiral anion phase-transfer catalyst (PTC). Selectfluor acts as the electrophilic fluorine source and reacts with 47 to yield various cyclohexadienones 48 bearing a chiral quaternary F-containing stereocenter in generally satisfactory yields and with excellent asymmetric induction. In addition, para-fluorination is achieved with the substrate 49 lacking ortho-substituents in good ee. Interestingly, products of type 52 readily undergo [4+2] dimerization, leading to single diastereomers 52′. A working model, as shown in Scheme 24, is proposed to rationalize the asymmetric induction.
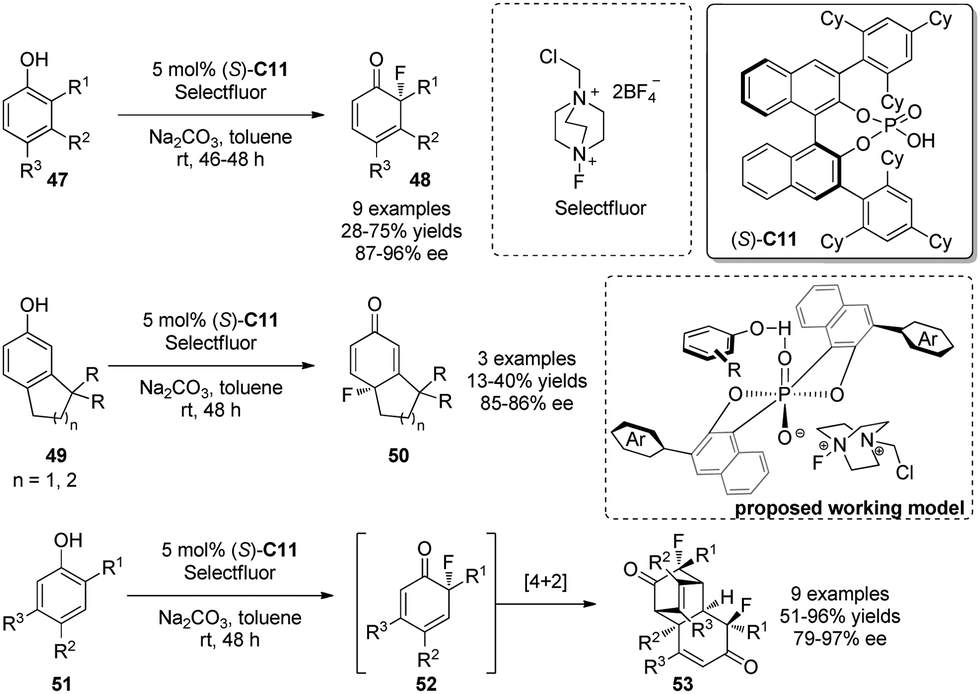 | ||
| Scheme 24 PTC-catalyzed asymmetric flourinative dearomatization reaction of phenols reported by Toste. | ||
Subsequently, a homogeneous organocatalytic asymmetric chlorinative dearomatization reaction of naphthols 54 was realized by You and coworkers in 2015.33 Using (DHQD)2PHAL derived from cinchonine as catalyst and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) as the electrophilic chlorine source, the operationally simple reaction provides facile access to chiral chlorinated naphthalenones 55 in excellent yields and ee under mild conditions. In addition, it tolerates a broad range of functional groups. The postulated mechanism entails that the phthalazine nitrogen in the catalyst interacts with the hydroxyl group of naphthol via a hydrogen bond and the tertiary amine nitrogen in quinuclidine acts as a Lewis base to deliver Cl+. Notably, intermolecular dearomative chlorination of 1-naphthol derivative 56 was also realized with high enantioselectivity (Scheme 25).
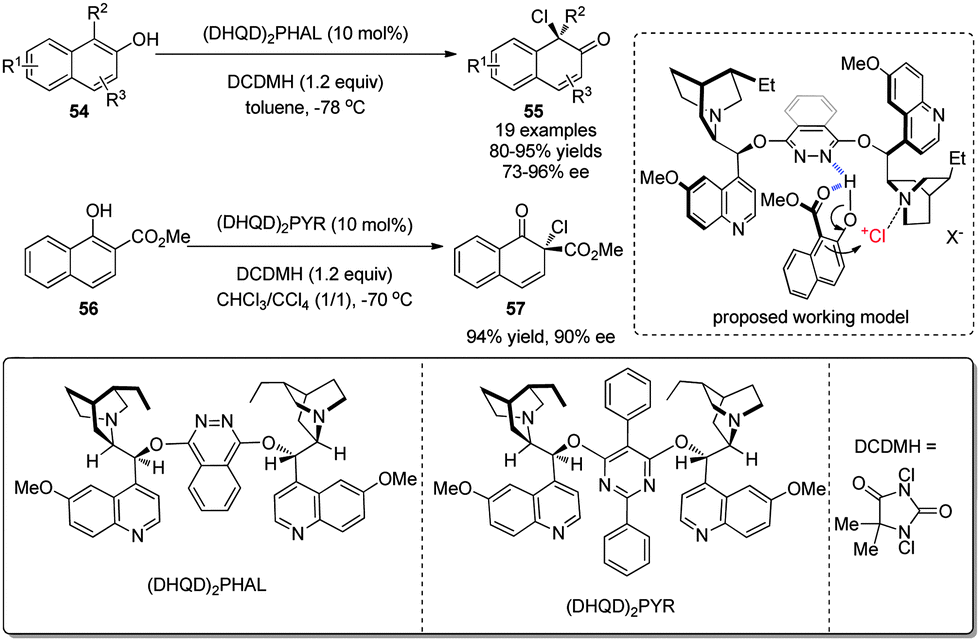 | ||
| Scheme 25 Organocatalytic asymmetric chlorinative dearomatization reaction of naphthols reported by You. | ||
3.6 Aminative dearomatization reactions
CADA reactions of phenol derivatives can also be achieved via electrophilic amination reactions. Recently, both Brønsted and Lewis acids have been employed as catalysts to activate azodicarboxylates 58 toward nucleophilic attack by naphthols 14. This dearomatization reaction can furnish aminated-naphthalenones 59 featuring a tertiary amine moiety in mostly excellent yields and ee (Scheme 26).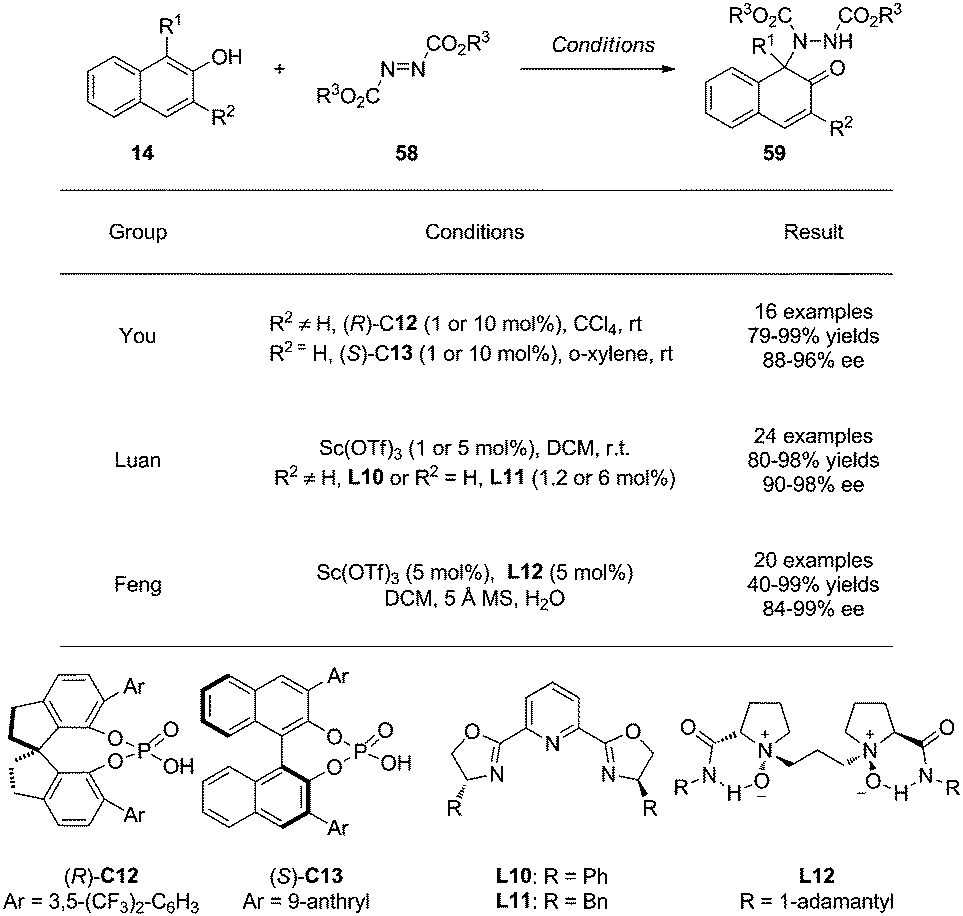 | ||
| Scheme 26 Asymmetric dearomatization reaction of naphthols via amination reactions reported by You, Luan and Feng. | ||
In 2015, You and coworkers34 realized this type of reactions by using chiral phosphoric acid (CPA) as catalyst. Notably, the catalyst loading can be as low as 0.1 mol% without affecting the yields and enantioselectivity. Intriguingly, C13 was found to be the optimal catalyst when the substituent on the naphthol 3-position is H.
Besides chiral Brønsted acids, the chiral Sc(III)/pybox complexes derived from L10 and L11 were employed by Luan and coworkers35 in the same year as effective catalysts for the same aminative dearomatization reactions, affording 59 in excellent yields and ee.
Moreover, the combination of Sc(OTf)3 and the N,N′-di-oxide L12, developed by Feng's group, is also highly effective in achieving the aminative dearomatization reaction in good to superb yields and with excellent enantioselectivity.36
3.7 Rearrangement dearomatization reactions
Inspired by the dearomatizing [3,3]-sigmatropic diaza-Cope rearrangement in the Fischer indole synthesis, List and coworkers recently developed an asymmetric version of this process by using chiral Brønsted acid (R)-C14.37 This mild reaction converts aryl hydrazines 60 and ketones 61 to useful 1,4-diketones 62 possessing an all-carbon quaternary stereocenter with good to excellent enantioselectivity. It was proposed that the chiral Brønsted acid promotes the formation of the ene hydrazine intermediate D and facilitates its subsequent rearrangement to diimine E, which then undergoes hydrolysis instead of indole formation due to the nascent quaternary carbon center (Scheme 27).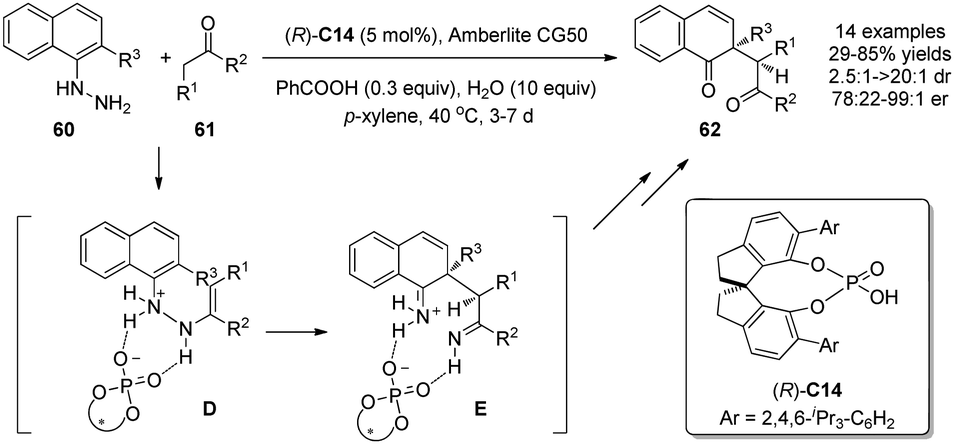 | ||
| Scheme 27 Asymmetric dearomatization reaction of naphthols via aryl hydrazine rearrangement reaction reported by List. | ||
4. Conclusions
In conclusion, two types of CADA reactions of phenol and aniline derivatives are described in this tutorial review. In the oxidative dearomatization reactions, the phenol moiety is oxidized, and the thus-generated intermediate reacts with nucleophiles; on the other hand, phenol itself is electron-rich and can react with electrophiles in the non-oxidative dearomatization reactions. The recent progress discussed in this tutorial reveals that a variety of strategies can be employed to achieve CADA reactions of phenol and its derivatives. In general, naphthols are more susceptible to dearomatization reactions than phenols, and the addition of base or additional interaction between chiral catalysts and the hydroxyl group of phenols is beneficial to both yield and enantioselectivity.This progress also illustrates that the CADA reactions of readily available phenol derivatives provide unconventional strategies for streamlining synthesis of complex molecules and natural products of high value. It is expected that the strategies successfully employed in these CADA reactions would be soon applied to other aromatic systems, thereby enabling CADA reaction as a general approach to the manipulation of aromatic compounds.
Acknowledgements
This research was supported by the CAS/SAFEA International Partnership Program for Creative Research Teams, the National Basic Research Program of China (973 Program 2015CB856600) and NSFC (21332009, 21421091). LZ acknowledges the support by NSF of USA (CHE-1301343).Notes and references
- Z. Rappoport, The Chemistry of Phenols, John Wiley & Sons Ltd, 2003 Search PubMed.
- C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed.
- V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, UK, 2014 Search PubMed.
- A. M. Harned, Tetrahedron Lett., 2014, 55, 4681–4689 CrossRef CAS PubMed.
- T. Dohi, A. Maruyama, N. Takenage, K. Senami, Y. Minamitsuji, H. Fujioka, S. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787–3790 CrossRef CAS PubMed.
- T. Dohi, N. Takenaga, T. Nakae, Y. Toyoda, M. Yamasaki, M. Shiro, H. Fujioka, A. Maruyama and Y. Kita, J. Am. Chem. Soc., 2013, 135, 4558–4566 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175–2177 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215–9218 CrossRef CAS PubMed.
- S. J. Murray and H. Ibrahim, Chem. Commun., 2015, 51, 2376–2379 RSC.
- D.-Y. Zhang, L. Xu, H. Wu and L.-Z. Gong, Chem. – Eur. J., 2015, 21, 10314–10317 CrossRef CAS PubMed.
- S. Quideau, G. Lyvinec, M. Marguerit, K. Bathany, A. Ozanne-Beaudenon, T. Buffeteau, D. Cavagnat and A. Chénedé, Angew. Chem., Int. Ed., 2009, 48, 4605–4609 CrossRef CAS PubMed.
- C. Bosset, R. Coffinier, P. A. Peixoto, M. El Assal, K. Miqueu, J. M. Sotiropoulos, L. Pouységu and S. Quideau, Angew. Chem., Int. Ed., 2014, 53, 9860–9864 CrossRef CAS PubMed.
- K. A. Volp and A. M. Harned, Chem. Commun., 2013, 49, 3001–3003 RSC.
- T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2012, 134, 20017–20020 CrossRef CAS PubMed.
- T. Oguma and T. Katsuki, Chem. Commun., 2014, 50, 5053–5056 RSC.
- J. Qi and J. A. Porco, Jr., J. Am. Chem. Soc., 2007, 129, 12682–12683 CrossRef CAS PubMed.
- J. Qi, A. B. Beeler, Q. Zhang and J. A. Porco, Jr., J. Am. Chem. Soc., 2010, 132, 13642–13644 CrossRef CAS PubMed.
- D. Yang, L. Wang, F. Han, D. Li, D. Zhao and R. Wang, Angew. Chem., Int. Ed., 2015, 54, 2185–2189 CrossRef CAS PubMed.
- S.-G. Wang, X.-J. Liu, Q.-C. Zhao, C. Zheng, S.-B. Wang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14929–14932 CrossRef CAS PubMed.
- T. Nemoto, Y. Ishige, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020–5023 CrossRef CAS PubMed.
- Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455–4458 CrossRef CAS PubMed.
- C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2013, 52, 10056–10059 CrossRef CAS PubMed.
- J. Garcia-Fortanet, F. Kessler and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 6676–6677 CrossRef CAS PubMed.
- S. Rousseaux, J. Garcia-Fortanet, M. A. D. A. Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282–9285 CrossRef CAS PubMed.
- R.-Q. Xu, Q. Gu, W.-T. Wu, Z.-A. Zhao and S.-L. You, J. Am. Chem. Soc., 2014, 136, 15469–15472 CrossRef CAS PubMed.
- K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033–3037 CrossRef CAS PubMed.
- L. Yang, H. Zheng, L. Luo, J. Nan, J. Liu, Y. Wang and X. Luan, J. Am. Chem. Soc., 2015, 137, 4876–4879 CrossRef CAS PubMed.
- J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880–4883 CrossRef CAS PubMed.
- J. Oka, R. Okamoto, K. Noguchi and K. Tanaka, Org. Lett., 2015, 17, 676–679 CrossRef CAS PubMed.
- T. Baba, J. Oka, K. Noguchi and K. Tanaka, Eur. J. Org. Chem., 2015, 4374–4382 CrossRef CAS.
- D. Yang, L. Wang, M. Kai, D. Li, X. Yao and R. Wang, Angew. Chem., Int. Ed., 2015, 54, 9523–9527 CrossRef CAS PubMed.
- R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed.
- Q. Yin, S.-G. Wang, X.-W. Liang, D.-W. Gao, J. Zheng and S.-L. You, Chem. Sci., 2015, 6, 4179–4183 RSC.
- S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647–650 CAS.
- J. Nan, J. Liu, H. Zheng, Z. Zuo, L. Hou, H. Hu, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 2356–2360 CrossRef CAS PubMed.
- X. Lian, L. Lin, G. Wang, X. Liu and X. Feng, Chem. – Eur. J., 2015, 21, 17453–17458 CrossRef CAS PubMed.
- S. Huang, L. Kötzner, C. K. De and B. List, J. Am. Chem. Soc., 2015, 137, 3446–3449 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |