
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vacuo interfacial tetrapyrrole metallation
Katharina
Diller
 *ab,
Anthoula C.
Papageorgiou
a,
Florian
Klappenberger
a,
Francesco
Allegretti
a,
Johannes V.
Barth
a and
Willi
Auwärter
*a
*ab,
Anthoula C.
Papageorgiou
a,
Florian
Klappenberger
a,
Francesco
Allegretti
a,
Johannes V.
Barth
a and
Willi
Auwärter
*a
aPhysik-Department E20, Technische Universität München (TUM), James-Franck-Str. 1, 85748 Garching, Germany. E-mail: wau@tum.de
bInstitute of Condensed Matter Physics (ICMP), École Polytechnique Fédérale de Lausanne (EPFL), Station 3, CH-1015 Lausanne, Switzerland. E-mail: katharina.diller@epfl.ch
First published on 19th January 2016
Abstract
The metallation of tetrapyrroles at well-defined surfaces under ultra-high vacuum conditions represents an unconventional synthesis approach to achieve tetrapyrrole-based metal–organic complexes and architectures. Different protocols, pioneered over the last decade, and now widely applied in several fields, provide an elegant route to metallo-tetrapyrrole systems often elusive to conventional procedures and give access and exquisite insight into on-surface tetrapyrrole chemistry. As highlighted by the functionality of metallo-porphyrins in biological or other environments and by the eminent role of metallo-phthalocyanines in synthetic materials, the control on the metal centres incorporated into the macrocycle is of utmost importance to achieve tailored properties in tetrapyrrole-based nanosystems. In the on-surface scenario, precise metallation pathways were developed, including reactions of tetrapyrroles with metals supplied by physical vapour deposition, chemical vapour deposition or the tip of a scanning tunnelling microscope, and self-metallation by atoms of an underlying support. Herein, we provide a comprehensive overview of in vacuo tetrapyrrole metallation, addressing two-dimensional as well as three-dimensional systems. Furthermore, we comparatively assess the available library of on-surface metallation protocols and elaborate on the state-of-the-art methodology.
 Katharina Diller | Katharina Diller received her PhD from the Technical University of Munich in 2013, where she worked at the “Chair of Molecular Nanoscience and Chemical Physics of Interfaces” with Prof. Johannes Barth. After a postdoctoral fellowship in Theoretical Chemistry with Prof. Karsten Reuter, she moved to Lausanne in 2014 to work with Prof. Harald Brune at the Ecole polytechnique fédérale de Lausanne, where she currently is an EPFL/Marie Curie postdoctoral fellow. In her research she combines synchrotron radiation experiments with density functional theory simulations to study how the interaction with surfaces influences the structural, electronic, and magnetic properties of (metal–)organic molecules. |
 Anthoula C. Papageorgiou | Anthoula Papageorgiou studied physics and materials science in London, Cambridge and Thessaloniki. She obtained a chemistry PhD in 2007 from University College London under the supervision of Prof. Geoff Thornton (Nanoscience Group). She subsequently took a research appointment mentored by Prof. Richard Lambert (Surface Science and Catalysis Group) in the University of Cambridge. In 2010 she moved to the Technical University of Munich to work with Prof. Johannes Barth (Chair of Molecular Nanoscience and Chemical Physics of Interfaces) as a postdoctoral fellow. Since 2014 she pursues a habilitation in physics. She is particularly interested in the chemical changes of small molecules on surfaces. |
 Florian Klappenberger | Florian Klappenberger obtained his PhD in 2004 from the University of Regensburg, for investigating nonlinear electron transport in semiconductor diodes. After a post-doctoral stay at the Ecole Polytechnique Fédérale de Lausanne, he joined the chair E20 at the Technical University of Munich in 2007 where he brought the STM + XS (scanning tunneling microscopy plus X-ray spectroscopy) approach to perfection. Since his habilitation in late 2012, he has been the leader of the Klappenberger Group focusing on the atom-precise fabrication of functional nanoarchitectures with supramolecular and surface-templating mechanisms. |
 Francesco Allegretti | Francesco Allegretti received his PhD from the University of Modena and Reggio Emilia (Italy) in 2003. In the following years he held research posts at the University of Warwick (United Kingdom) and the Karl-Franzens University of Graz (Austria). In 2010 he joined the “Chair of Molecular Nanoscience and Chemical Physics of Interfaces” at the Technical University of Munich as a senior scientist. His research activity is focused on the study of the structural, electronic and chemical properties of functional hybrid interfaces based on organic and oxide nanostructures, particularly utilising synchrotron radiation techniques. |
 Johannes V. Barth | Johannes V. Barth is a Chair in Experimental Physics and Dean at the Physics Department, Technical University of Munich. His present research interests are mainly in molecular nanoscience and surface chemical physics. He holds a physics degree from Ludwig-Maximilians-Universität München and conducted his PhD studies in Physical Chemistry at the Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin. He was a postdoctoral fellow at IBM Almaden Research Center and Fritz-Haber-Institut, Team Leader, Docent and Visiting Professor at Ecole Polytechnique Fédérale de Lausanne, and nominated a Canada Research Chair at The University of British Columbia, Vancouver, prior to the appointment at Technical University of Munich in 2007. |
 Willi Auwärter | Willi Auwärter completed his PhD at the University of Zurich in 2003. Subsequently he worked as a postdoctoral fellow at the University of British Columbia, Vancouver, and at the Ecole Polytechnique Fédérale de Lausanne. In 2007, he joined the Technical University of Munich where he was a fellow of the TUM Institute for Advanced Study. He presently holds an ERC consolidator grant and a Heisenberg professorship position. His current research interest focuses on the creation of nanoscale model systems on atomically tailored surfaces, enabling the study and control of single-molecule processes as well as the self-assembly of supramolecular structures. |
1 Introduction
The synthesis, constitution and coordination chemistry of tetrapyrrole complexes has fascinated and challenged scientists since the 19th century, where among other pioneering achievements the denomination and exploration of key compounds such as chlorophyll1 or haemoglobin2–4 emerged and the cytochromes have been discovered.5 Notably the pioneering work of R. Willstätter6 and H. Fischer7–9 on chlorophyll and haem, rewarded with the Nobel Prize in Chemistry in 1915 and 1930, respectively, unveiled the nature of metalloporphyrins and related compounds. Their crucial role in manifold life processes, such as transport of respiratory gases, metabolic catalytic conversion or light-harvesting is undisputed. While metalloporphyrins originally appeared in nature, later studies striving for novel materials and applications strongly rely on synthetic tetrapyrrolic compounds, including the phthalocyanine family.10,11Fig. 1 introduces their basic features as well as the corresponding nomenclature. The large variety of possible substituents and metal centres determines the system's topological, chemical, optical, electronic, and magnetic properties and hence their overall functionality in different fields like photochemistry, photovoltaics, medicine, sensing, catalysis or molecular electronics.12–17 In this respect, the interaction of tetrapyrroles with surfaces plays an important role, and their adsorption and organisation at interfaces have been extensively studied under well-defined ultra-high-vacuum (UHV) conditions and in solution.18–25 However, the bulk of these investigations is limited to the deposition of pre-synthesized molecules. The so-called in vacuo metallation approach, i.e., unconventional synthesis protocols that provide an elegant route to create advanced tetrapyrrole-based metal–organic structures on surfaces, only emerged rather recently and yields unprecedented insights into interfacial tetrapyrrole chemistry.
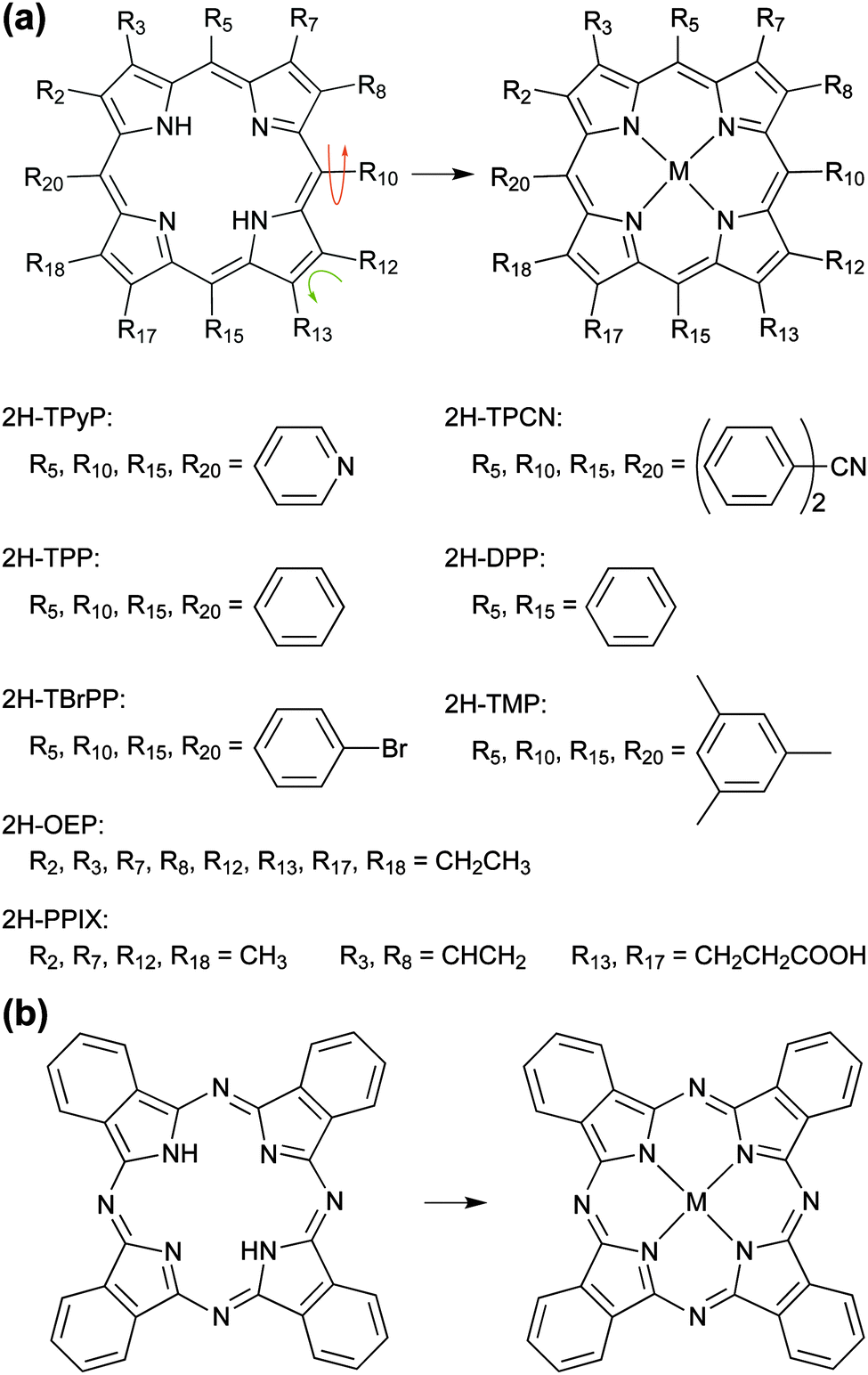 | ||
| Fig. 1 Metallation of tetrapyrrole compounds. The basic building blocks are (a) the free-base porphine (2H-P, top left, with all Ri = H) and (b) the free-base phthalocyanine (2H-Pc, left). Tailored complexes can then be built by attaching substituents and/or inserting a metal ion (M) in the centre of the tetrapyrrole macrocycle. Depending on the chosen substituent the molecules can exhibit a great conformational variety (indicated by arrows). A survey of compounds discussed in this review (cf.Tables 1–3) is given in (a). If not noted otherwise, Ri = H. | ||
The macrocycle metallation is a decisive step in the formation of metallo-tetrapyrroles and was extensively studied.26–31 Most synthesis protocols initially afford a free-base species32–34 that is subsequently metallated. Whereas for biological systems enzymatic pathways prevail, in solution typically dissolved metal salts are employed. There is evidence both from experiment and theory that a multi-step reaction sequence exists, whereby appreciable deformations of the macrocycle interfere.35,36
The formation of porphyrins and metallation events occur at silicate surfaces of montmorillonite under simulated geochemical conditions and in the presence of hydrated metal ions, which findings are of interest in relation to the question of prebiotic porphyrin evolution.37–39 Furthermore, indications for interfacial metallation of free-base tetrapyrroles were recognized as spurious effects without the reaction being targeted or even adequately identified. For example, evidence of porphyrin metallation by atoms from the support – a process later termed as self-metallation – was provided by surface-enhanced resonant Raman studies based on porphyrin coated silver electrodes.40 Also demetallation41 and the influence of the substrate structure on porphyrin metallation was explored for silver supports.42 Such metallation or demetallation reactions are not restricted to silver surfaces, but are also reported for more complex supports as sodium hectorate43 or metal oxides grown via atomic layer deposition and thus play a considerable role in chemistry, organic photovoltaics, nanoscience and materials engineering.44,45
Beyond metallation reactions, demetallation and transmetallation processes are well documented in solution and utilized to rank metallotetrapyrroles regarding their stability.46 Interestingly, the exploration of analogous procedures on surfaces is just in its infancy.
The first stringent, molecular-level evidence for direct and controlled in vacuo metallation events in a tetrapyrrole adlayer induced by a beam of transition metal atoms was a scanning tunnelling microscopy (STM) and spectroscopy (STS) study presented in 2005 at the European Conference on Surface Science (ECOSS 23) and later published in ref. 47. This serendipitous finding appeared in an attempt to generate metal–organic coordination networks using a free-base TPyP layer on a Ag(111) support, where a site-specific uptake of Fe atoms in the tetradentate ligands took place readily and efficiently at room temperature (RT).47 The communication of these unexpected phenomena rapidly spurred further investigations, and the reaction pathway was confirmed by space-averaging X-ray photoelectron spectroscopy (XPS) studies demonstrating Co metallation in a TPP film.48 Ever since, a rising number of investigations has dealt with developing metallation recipes, analysing the occurring chemical reactions and defining the educts in terms of their chemical, electronic or magnetic properties. Reflecting the large variety of tetrapyrrole species and atomically defined supports, a manifold of systems and in situ metallation routes have been reported by different research groups over the last few years.25
The in vacuo tetrapyrrole metallation can provide complexes and nanostructures unachievable by conventional synthesis protocols, but nevertheless shares some common aspects with established solution-based synthesis procedures33 and even biosynthetic pathways to metalloporphyrins.50 Specifically, while a complicated sequence of reactions is involved in the latter,36 the key steps include the deprotonation of the free-base tetrapyrrole core, supply of the metal ion including the removal of ligands, coordination of the metal ion with the nitrogens of the tetrapyrrole core and completion of the coordination sphere.36,50 Hereby, a so-called sitting atop complex might play a relevant role as an intermediate where the metal centre already forms a coordination bond to the macrocycle, while the central hydrogens are not yet released.30,32,51 In addition, non-planar deformations of the tetrapyrrole ring influence the metallation reaction.30,36,52,53 All these aspects and steps of metallotetrapyrrole formation are also reflected in a solvent-free on-surface scenario, however the role of the support introduces distinct differences providing a unique platform for tetrapyrrole chemistry in reduced dimensions. For example, the surface readily induces deformations of porphyrins and might catalyse the metallation reaction mimicking the role of proteins in biosynthesis.54
The emphasis of the present review is placed on on-surface synthesis approaches for metallotetrapyrrole model systems in UHV and on the resulting complexes and nanostructures. It does not cover processes in solution (as e.g. described in ref. 55). The review is organized as follows: after introducing the methodology generally applied to study on-surface metallation in Section 2, Section 3 addresses different metallation scenarios reviewing the most important studies and systems. As highlighted in Fig. 2, which serves as an outline of this section, we discriminate four on-surface metallation methods, namely physical vapour deposition, self-metallation, alternative routes, and the metallation of three-dimensional structures. We close by providing a comparative assessment of the metallation strategies, a summary of the key findings and a perspective on future trends in Section 4.
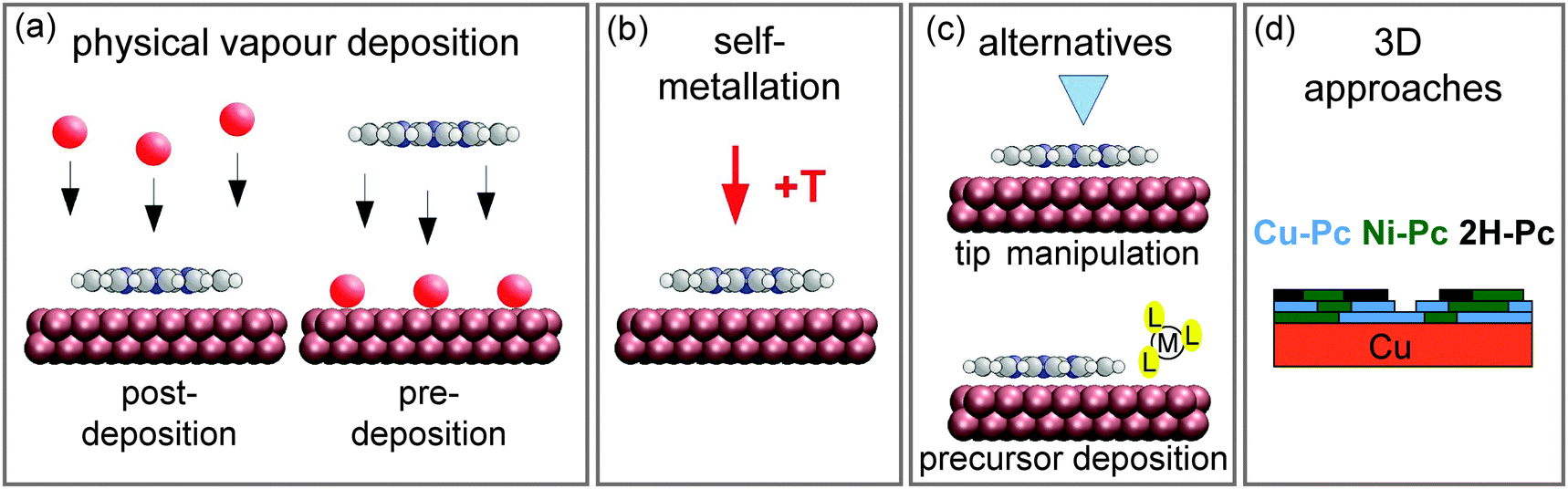 | ||
| Fig. 2 Schematics summarising different on-surface metallation methods discussed in this review. (a) Metallation by physical vapour deposition addressed in Section 3.1. (b) The self-metallation of the molecule–substrate system where surface atoms are inserted into the macrocycle (Section 3.2). (c) Alternative metallation methods discussed in Section 3.3: metallation by tip manipulation and co-adsorption of a metal-containing precursor molecule. (d) The metallation of three-dimensional structures summarised in Section 3.4. (Adapted with permission from ref. 49. Copyright 2014 American Chemical Society). | ||
2 Methodology
This section introduces the principal techniques that are used for the investigation of on-surface metallation. It provides some basics of the modelling and the experimental procedures used for surface preparation and analysis.2.1 Experimental
Systems discussed in this review were prepared and analysed under UHV conditions, with typical pressures in the 10−9 to 10−11 mbar regime. Metal substrates are usually conditioned by repeated cycles of sputtering (mainly with Ar+ and Ne+ ions) and annealing, to create atomically clean and well-ordered surfaces. Tetrapyrrole molecules are deposited by supersonic molecular-beam deposition (SuMBD, in ref. 56) or organic molecular beam epitaxy (OMBE) on the pristine surfaces. Metal atoms are deposited by either physical vapour deposition (PVD, Fig. 2a) via electron beam or resistive heating or by chemical vapour deposition of suitable precursor molecules (Fig. 2c, lower panel). The samples are then characterised in situ using one or a combination of the techniques described below.2.2 Photoelectron spectroscopy (PES)
One of the most straightforward techniques to monitor the metallation of tetrapyrroles is X-ray photoelectron spectroscopy (XPS) of the N 1s core-level region. The basic principle of XPS is illustrated in Fig. 3a: the sample is irradiated with a monochromatic X-ray beam of fixed photon energy, which causes the emission of photoelectrons by the photoelectric effect, with the kinetic energy, Ekin, of the photoelectrons depending on the binding energy, Eb, of the initial core level. For a typical XPS experiment the number of emitted electrons is recorded as a function of Ekin in the vicinity of the core level of interest by using a hemispherical electron energy analyser. The binding energy Eb of the electrons can then be determined by the formula Eb = ℏω − Ekin − Φa whereby Ekin is given with respect to the vacuum level of the analyser, ℏω denotes the photon energy, and Φa the work function of the analyser. In general, the binding energy scale is calibrated assuming Eb = 0 at the Fermi level of the sample. XPS is mainly used to probe the core levels of the elements present in a material: each distinct energy level gives rise to a distinct photoemission line in the spectrum, at a binding energy characteristic of the elemental species. These lines are peaks of finite width due to the intrinsic lifetime of the core hole created upon photoemission, and they are broadened by the energy resolution of the experimental set-up mainly given by the spectral width of the exciting photon beam and the energy resolution of the analyser. XPS is not only element specific, but also discerns chemical states: for the core level of a given element the exact binding energy position depends sensitively on changes in the bonding configuration, i.e., the chemical environment.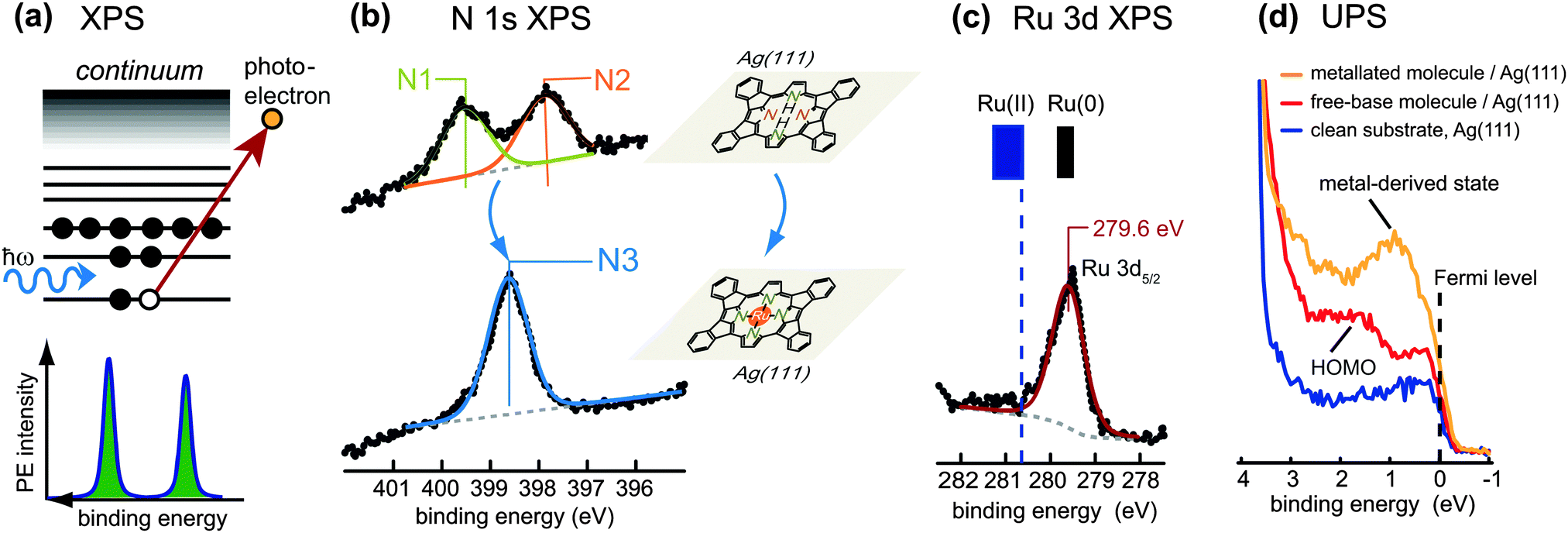 | ||
| Fig. 3 Photoelectron spectroscopy (PES) used to monitor the metallation of tetrapyrrole molecules. (a) Schematic view of the photoemission process in X-ray photoelectron spectroscopy (XPS). Upon absorption of a photon, a core-level electron is excited into a state in the continuum above the vacuum level and ejected. Correspondingly, a sharp line appears in the PE spectrum at a binding energy characteristic of the atom from which the photoelectron was emitted. (b) Typical signatures of tetrapyrrole compounds in the N 1s XPS region: free-base tetrapyrrole molecules (top; here: planar 2H-TPP on Ag(111) display two peaks corresponding to the two inequivalent nitrogen species in the macrocycle, whereas after metallation (bottom; here: planar Ru-TPP on Ag(111)) a single peak is observed. (c) XPS from a core level of the metal centre: after metallation a distinct peak is observed, whose binding energy bears information on the oxidation state of the metal. In the specific case shown, where planar 2H-TPP molecules adsorbed on Ag(111) were metallated with ruthenium (see panel (b)), the oxidation state of the Ru centres is close to Ru(0). (d) Ultraviolet photoelectron spectroscopy (UPS) provides information on the valence states close to the Fermi level of the substrate. In the example here, a Ru-derived state develops just below the Fermi level upon metallation of the free-base molecules. (b–d) Adapted with permission from ref. 57. Copyright 2013 American Chemical Society. | ||
A typical N 1s XP spectrum from free-base tetrapyrrole molecules (e.g., porphyrins) shows two components arising from the two inequivalent nitrogen atoms of the macrocycle (see Fig. 3b, top, ref. 57). They can be identified easily even in set-ups with limited energy resolution, as the chemical shift results in a peak splitting of approximately 2 eV.58 The peak at lower binding energy (usually located close to 398 eV) originates from the iminic nitrogen, the one at higher binding energy (located at about 400 eV) from the pyrrolic nitrogen. For porphyrins the intensity ratio of the two components should be close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while for phthalocyanines a 3:1 ratio is expected due to different stoichiometry of the respective nitrogen types. However, the interaction with the surface can affect the peak splitting and the relative intensities (see Section 3.2.2). After metallation of the free-base species the chemical environment of the nitrogen atoms becomes more similar, and a single peak appears in the N 1s XP spectrum (Fig. 3b, bottom). For porphyrins this peak corresponds to metallated nitrogen species. For phthalocyanines it also overlaps with contributions of the unreacted outer nitrogen atoms. The binding energy of the metallated nitrogen species typically falls in the region between the two components of the free-base compound (at approximately 398.6–398.9 eV, e.g. in ref. 48 and 59–61), but in some cases it can shift downwards due to the interaction with the substrate.
:1, while for phthalocyanines a 3:1 ratio is expected due to different stoichiometry of the respective nitrogen types. However, the interaction with the surface can affect the peak splitting and the relative intensities (see Section 3.2.2). After metallation of the free-base species the chemical environment of the nitrogen atoms becomes more similar, and a single peak appears in the N 1s XP spectrum (Fig. 3b, bottom). For porphyrins this peak corresponds to metallated nitrogen species. For phthalocyanines it also overlaps with contributions of the unreacted outer nitrogen atoms. The binding energy of the metallated nitrogen species typically falls in the region between the two components of the free-base compound (at approximately 398.6–398.9 eV, e.g. in ref. 48 and 59–61), but in some cases it can shift downwards due to the interaction with the substrate.
XPS can also be exploited to obtain information concerning the chemical state of the metal centres. This is more suited for metallation with metal centres different to the substrate atoms (Sections 3.1, 3.3, 3.4), whereas for the self-metallation (Section 3.2) it is challenging to separate the weak signal of the central metal ion from the dominant contribution of the substrate atoms. Fig. 3c shows the Ru 3d region after metallation of planar, cyclodehydrogenated, 2H-TPP porphyrins, adsorbed on Ag(111), with ruthenium.57 The Ru 3d5/2 binding energy position shows that after metallation the actual oxidation state of Ru is close to Ru(0), in contrast with the formal Ru(II) charge state of a Ru ion in a porphyrin macrocycle. Note that the metal atoms in metallated porphyrins and phthalocyanines are in a formal oxidation state of +2 in the absence of other axial ligands. Situations as that depicted in Fig. 3c are typically rationalized in terms of charge transfer from the substrate to the molecules. Therefore, XPS from the core levels of the metal centres may allow to draw valuable conclusions about the interaction of the metallated molecules with the underlying surface (cf. Section 3.1.2).
While XPS is a variant of photoelectron spectroscopy (PES) that probes the core levels of a sample, ultraviolet photoelectron spectroscopy (UPS) is principally used to study the density of occupied states in the valence region. In this case ultraviolet photons are used, which result in a strong enhancement of the photoionization cross-sections for valence band states and occupied molecular orbitals close to the Fermi level in comparison to X-ray excitation. The interpretation of valence band spectra is generally more difficult, as the electronic states are broader and the background is often more complicated. Moreover, the signatures of free-base and metallated molecules are not as characteristic as those in XPS, therefore UPS is not well suited to study the metallation itself. Nonetheless, the technique can be used to characterise the valence region of the molecular entities before and after the reaction.59,62,63Fig. 3d depicts an exemplary case where the highest occupied molecular orbital (HOMO) of Ag(111)-supported free-base porphyrins is clearly visible at a binding energy of about 1.5 eV (red spectrum). After metallation, however, the electronic structure is strongly affected and a metal-derived state appears just below the Fermi level (orange spectrum).57
2.3 Scanning tunnelling microscopy (STM)
One of the most widely used surface science techniques is scanning tunnelling microscopy (STM) providing real space, single molecule information. It can thus give valuable insight into the spatial distribution of free-base and metallated species in not fully metallated samples, the self-assembly properties before and after the metallation reaction, and (to some extent) the conformation of the adsorbed (metal–)organic compounds. STM exploits the probability for an electron to tunnel through a potential barrier, the tunnelling current depending exponentially on the distance that the electron must travel through the vacuum barrier. Accordingly, if an atomically sharp (metallic) tip is brought close enough to the surface of a (semi)conducting material, a tunnelling current (I) flows between the tip and the sample when a bias voltage (V) is applied (Fig. 4a). Notably, the tunnelling current depends not only on the tip–surface distance but also on the electronic structure of both the tip and the surface. Therefore, if the tip is scanned laterally across the surface (e.g., at constant tunnelling current as in Fig. 4a), insight into the surface topography convoluted with the electronic structure can be gained. | ||
| Fig. 4 (a) Principle of scanning tunnelling microscopy (STM) in constant current mode operation. A metallic tip is scanned laterally across the surface of an adsorbate-covered sample and is brought closer or further from the surface to maintain a constant tunnelling current. As a result, one obtains a measurement of the tip displacement normal to the surface as a function of the lateral position, i.e. a two-dimensional map of the sample topography convoluted with the electronic structure at the probed bias. (b) STM image of planar (cyclodehydrogenated) 2H-TPP and Ru-TPP molecules coexisting on a Ag(111) surface. While the free-base species exhibit a central depression, the metal centre of the metallated species is clearly imaged as a protrusion. The overview and magnified images were all recorded under the same conditions: I = 0.2 nA, V = −0.5 V, 7 K. Adapted with permission from ref. 57. Copyright 2013 American Chemical Society. (c) STS measurements: dI/dV curves measured as a function of the sample bias V. The ordinate (dI/dV) is approximately proportional to the density of states of the surface, both occupied (negative bias) and unoccupied (positive bias) states are probed. | ||
In the case of adsorbed (metal–)organic compounds (such as porphyrins) submolecular resolution is typically attained, as exemplified in Fig. 4b. Here, planar free-base porphyrins are imaged with a depression in their centre, whereas after metallation with ruthenium they exhibit a clear protrusion, signalling the presence of the metal ion inside the macrocycle.57 It should be noted that Fig. 4b illustrates a particularly favourable case, where the increased contrast for the metal centres arises from the tunnelling of electrons from the occupied metal-derived state highlighted in Fig. 3d. In general, the apparent height and contrast in the STM images may depend strongly on the bias voltage (in magnitude and sign) applied (cf. Section 3.1), and therefore the interpretation of the topographic images before and after metallation is not always straightforward.
STM is often combined with scanning tunnelling spectroscopy (STS). In this technique, the tip is immobilised at a position of interest and a dI/dV curve is recorded as a function of the bias voltage V, as sketched in Fig. 4c. Within some approximations,65 the tunnelling conductance dI/dV at a given bias voltage can be considered as being proportional to the local density of states of the sample. Accordingly, STS measurements provide information on the electronic structure of the surface at the probed position. Importantly, the sign of V can be changed from positive to negative, hence both unoccupied and occupied states of the surface can be probed. For example, in Fig. 4c the lowest unoccupied molecular orbital (LUMO) of free-base molecules results in a clear spectroscopic feature at positive bias (in this case, tunnelling from the tip to the sample occurs); for metallated molecules, a metal-derived state just below the Fermi level is observed, analogous to that observed in UPS (Fig. 3d). Finally, if dI/dV data are recorded at a bias voltage of interest with the tip being laterally scanned across the area occupied by the molecules, a map of the spatial distribution of, e.g., the frontier orbitals of the molecule (HOMO or LUMO) can be obtained.66
2.4 Near edge X-ray absorption fine structure (NEXAFS) spectroscopy
While XPS and UPS can be exploited to study the occupied density of states in a material, near edge X-ray absorption fine structure (NEXAFS) is a spectroscopic tool which provides complementary information on the unoccupied states. Soft X-ray radiation is typically used to probe the absorption K-edge of low atomic number elements such as C and N. If the photon energy is not sufficient to eject an electron from the sample into the vacuum, but instead corresponds to the transition energy between a core level (e.g., the 1s level) and an unoccupied level of a molecule (e.g., the LUMO), the core electrons can be resonantly excited into an unoccupied bound molecular orbital (Fig. 5a). These transitions give rise to characteristic resonances close to the absorption edge (the so-called “fine structure”), as shown schematically in the bottom panel of Fig. 5a. Note that in the NEXAFS measurements the photon energy is scanned across the absorption edge of the chosen element while the corresponding absorption is measured; therefore the use of tunable synchrotron radiation is required. In this way, it is possible to identify transition energies and to gain information on the unoccupied electronic states of the sample. Similar to XPS, NEXAFS is elemental-sensitive; moreover, the absorption fine structure is strongly dependent on the chemical and structural environment of the atoms, and different organic molecules generally exhibit very different NEXAFS signatures. As a consequence, the interpretation of NEXAFS spectra can be challenging. In fact, while in XPS each chemical species produces one single peak, the NEXAFS spectrum of a single chemical species corresponds to a whole set of transitions with varying intensities. It follows that for complex molecules the assignment of the individual spectral features in NEXAFS may require considerable effort.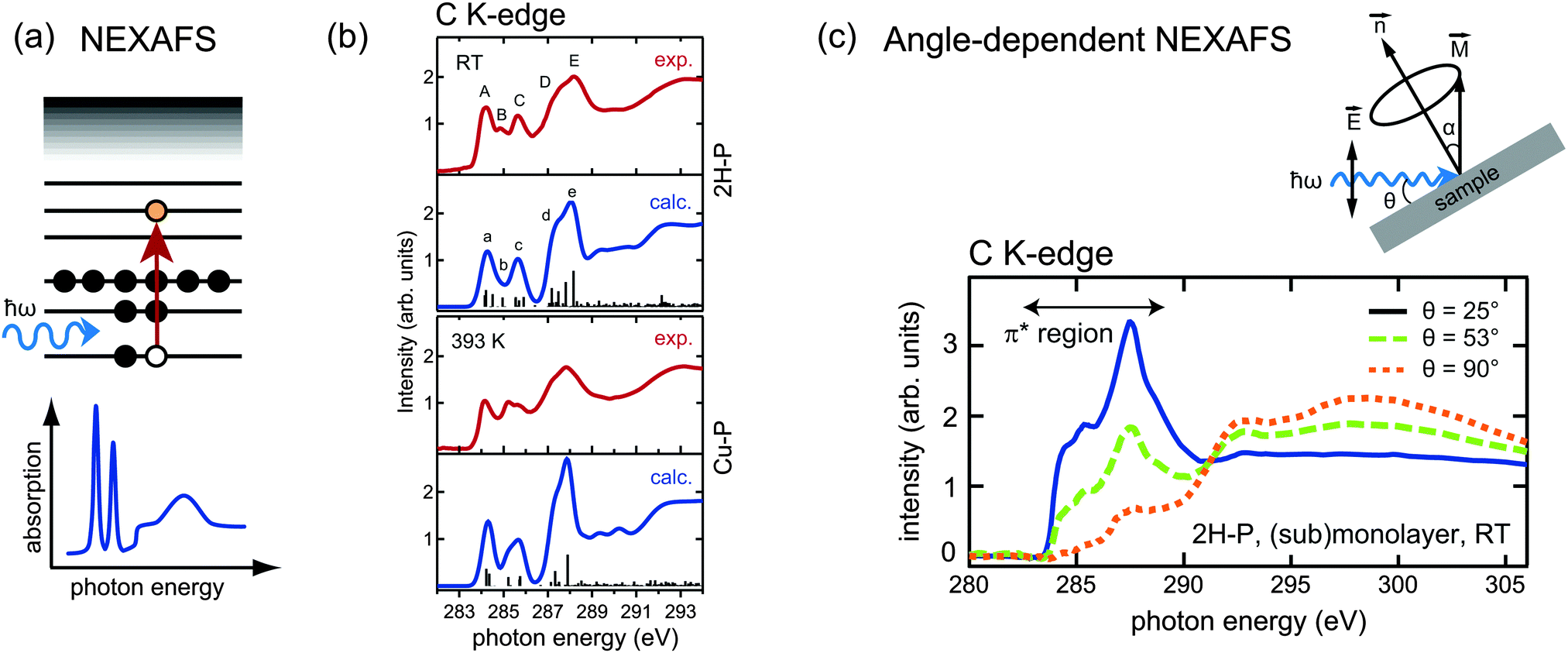 | ||
Fig. 5 (a) Schematic energy diagram of a resonant absorption process in near edge X-ray absorption fine structure (NEXAFS). The absorption of a photon with energy matching exactly the transition energy leads to the excitation of a core electron into an unoccupied orbital. The “fine structure” at the absorption edge, sketched in the bottom panel, consists of several such resonances. (b) Comparison of simulation and experiment of the C K-edge of 2H-P (top panels) and Cu-P (lower panels): the metallation is reflected in the quenching of peak B, which is attributed to carbon atoms bonded to the iminic nitrogen species. Adapted from ref. 64. (c) Principle of angle-dependent NEXAFS. The absorption spectra are recorded for different angles θ between the incident radiation and the sample surface. The intensity is maximized when the electric field ![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) is collinear with the direction of the excited molecular orbital is collinear with the direction of the excited molecular orbital ![[M with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004d_20d1.gif) . The main panel displays the angle-dependent NEXAFS spectra at the C K-edge of 2H-P adsorbed on Cu(111). As the π* orbitals of 2H-P can be represented by vectors perpendicular to the macrocycle, the quenching of the π*-resonances at θ = 90° is indicative of a flat orientation of the adsorbed molecules. (b and c) Adapted with permission from ref. 64. Copyright 2013, AIP Publishing LLC. . The main panel displays the angle-dependent NEXAFS spectra at the C K-edge of 2H-P adsorbed on Cu(111). As the π* orbitals of 2H-P can be represented by vectors perpendicular to the macrocycle, the quenching of the π*-resonances at θ = 90° is indicative of a flat orientation of the adsorbed molecules. (b and c) Adapted with permission from ref. 64. Copyright 2013, AIP Publishing LLC. | ||
For the analysis of on-surface metallation reactions NEXAFS is therefore less widely used than XPS or STM, but still a significant number of studies exist, mainly in combination with those techniques. From the point of view of the metallation, the most informative region is again the nitrogen region, due to the low number of inequivalent species. However, for monolayer and submonolayer coverages, the signal at the N K-edge can be very weak, and commonly measurements at the C K-edge are also performed. Due to the degree of complexity of the spectra, these experiments are often accompanied by DFT calculations, which simulate the curves for different species and thus allow a detailed peak assignment. Fig. 5b shows an example of such simulations. It compares the C K-edge spectra of free-base porphine, 2H-P, before and after the self-metallation to Cu-P (cf. Section 3.2) on Cu(111).64 This is one of the rare examples where the metallation can be followed at the carbon edge, because in general (i) the change in the carbon atoms upon metallation is small, as they are not directly bonded to the metal, and (ii) the large number of carbon atoms with a very similar chemical environment makes it very hard to observe these moderate changes. As 2H-P is the smallest porphyrin, in this case it is possible to identify the changes. The most prominent modification of the 2H-P spectrum following annealing to 393 K affects the spectral feature labelled B in the top panel of Fig. 5b. The DFT analysis shows that this feature exclusively originates from the carbon atoms bonded to the iminic nitrogen of the porphine macrocycle, whereby it is suppressed upon metallation.64
It is worth noting that the absorption of soft X-rays can be described within the dipole transition approximation. This implies that the NEXAFS intensities crucially depend on the angle between the polarization vector of the light and the direction of the final state molecular orbitals, being maximal for collinear orientation. If linearly polarized synchrotron radiation is utilized and the angle between the photon direction and the sample surface is varied during the measurements, the angular dependence of the NEXAFS spectra can be used to determine the adsorption geometry of adsorbed molecules. For example, in Fig. 5c the strong dichroism at the C K-edge of 2H-P on Cu(111) indicates a flat orientation of the molecule (the π*-resonances are strongly suppressed when the electric field is parallel to the surface).64 This structural sensitivity of angle-dependent NEXAFS also enables to detect changes in the molecular conformation upon incorporation of a metal centre inside the macrocycle.60
2.5 Temperature programmed desorption (TPD)
The methods discussed so far analyse the free-base and metallated tetrapyrroles constituting reactants and products. There is, however, another product of metallation, namely the hydrogen released during the metallation process. An effective means to monitor the release is provided by temperature programmed desorption (TPD) experiments, where the sample is positioned in front of a mass spectrometer and heated with a constant rate (Fig. 6). The mass spectrometer detects desorption products as a function of the sample temperature, hence their desorption temperatures are precisely determined. In the study of metallation reactions this can be exploited in different ways. Firstly, the onset of metallation can be determined much more precisely by monitoring the release of molecular hydrogen as compared to, e.g., STM, where the experimental set-up does not allow to apply a constant heating ramp during the measurements. Moreover, the isotopic labelling at selected sites can provide further insight into the mechanism of the metallation reaction. For example, in Fig. 6 the TPD spectra from a checkerboard-like67 layer of deuterated 2D-TPP molecules on Cu(111) are reported; in ref. 68 the peak at 450 K was assigned to the metallation with Cu atoms and thus marks the onset of the reaction, whereas the second peak centred just below 550 K and the broader structure above 700 K were attributed to partial and complete dehydrogenation reactions in the periphery of the molecules, respectively. Interestingly, as the deuteration was found to be limited to the two pyrrolic nitrogen atoms of the macrocycle, the small D2 signal upon metallation and the observed desorption of HD and D2 during dehydrogenation of the periphery of the molecules can be taken as an indication of scrambling of deuterium on the surface during metallation. On this basis a reaction model for the metallation was proposed, according to which the two pyrrolic hydrogen atoms do not directly recombine into molecular hydrogen above a partially inserted metal centre (as predicted for the gas phase reaction, see ref. 69), but instead spill over to the copper surface as atomic hydrogen. Furthermore, in some cases the desorption of the tetrapyrrole species themselves is monitored by TPD,49 thus providing information on their metallation state.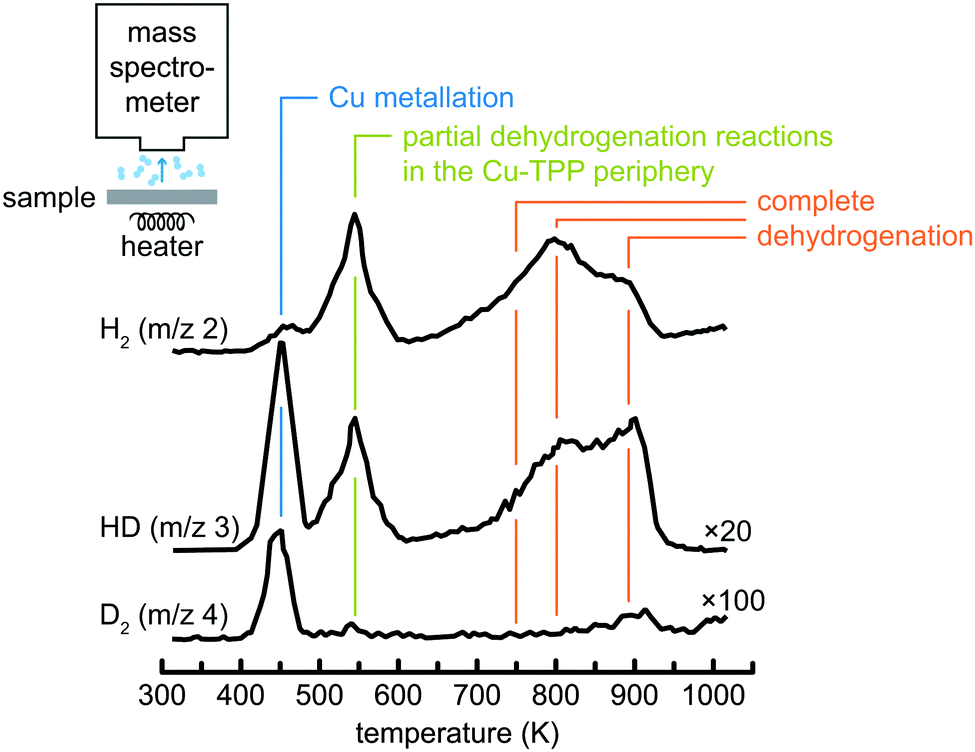 | ||
| Fig. 6 TPD spectra of deuterated tetraphenylporphyrins (2D-TPP) on a Cu(111) surface. The peak at approximately 450 K is assigned to metallation with Cu atoms. At higher temperatures dehydrogenation reactions in the periphery of the molecule start to occur. Note the multiplication factor for the signals of different masses. Adapted with permission from ref. 68. Copyright 2014 American Chemical Society. | ||
2.6 Density functional theory (DFT)
This section will only briefly outline how density functional theory (DFT) is used to analyse and understand metallation reactions. There are different ways in which DFT can aid the interpretation of experimental data. An important aspect is the simulation of spectra for comparison with the experimental results, as already shown in Section 2.4. Ab initio simulations can either be used to analyse spectral lines whose interpretation would be difficult without theoretical support, or to test hypotheses such as the formation of specific metalloporphyrins by simulation of spectra of the corresponding compound. An example for the first scenario is the work of Schmidt et al., who used time-dependent DFT to interpret the NEXAFS spectra of 2H-TPP, Co-TPP, and Zn-TPP.70 The second approach was followed, for instance, to show that the changes observed in the XP and NEXAFS spectra of 2H-TPP60 and 2H-P64 layers on Cu(111) after thermal treatment were indeed related to the formation of the corresponding copper complex. While these and other examples71–75 do not explicitly take into account the metallation reaction itself, the simulation of the experimental signatures enables to assign and rationalize the experimentally observed spectral features and is therefore a very valuable tool. Moreover, an indirect way of verifying conclusions drawn from the experiments is the computation of molecular properties, such as (adsorption) geometries and energetics,73,76–81 or the charge distribution and electronic structure.73,77,80–84Conversely, only few investigations have been dedicated to the metallation mechanism. To limit the computational effort, large tetrapyrrole molecules are often replaced by the smallest porphyrin, porphine. This was the case, for example, in the study of Shen et al.36 who studied the metallation of porphine in solution, or in the work of Shubina et al., who identify possible intermediate steps and were the first to compute energy diagrams for the gas phase metallation of free-base porphine with different bare metal atoms.69,85 The derived activation barrier for metallation with copper (1.04–1.60 eV) was recently verified by an experiment of Ditze et al., who determined the barrier for the self-metallation of 2H-TPP/Cu(111) to be 1.48 eV (Section 3.2.1).86 Dyer et al. used DFT to show that there is a weak attractive interaction of free-base porphine with copper adatoms on a Cu(110) surface, which is an important argument for the involvement of adatoms in self-metallation (Section 3.2.3).80 An analogous study was performed by Goldoni et al.: they showed that the insertion of a nickel adatom during the self-metallation of 2H-TPP/Ni(111) is preferred to the creation of a surface vacancy.76 Very recently, Smykalla et al. studied the reaction path for self-metallation of the free-base phthalocyanine on a Ag(110) surface87 (cf. Section 3.2).
3 On-surface metallation protocols
3.1 Metallation via physical vapour deposition (PVD)
In this case the on-surface metallation of tetrapyrrole molecules follows a generic scheme: both free-base molecules and metal atoms are deposited on a supporting substrate, and the temperature-dependent metallation is monitored by techniques described in Section 2. In most cases, relatively inert supports such as the Ag(111) or Au(111) surfaces are considered for this approach, as the stronger molecule–substrate and metal–substrate interactions on more reactive surfaces (e.g., Cu(111), Ni(111) or Fe(110)) may introduce complications and lead to competing phenomena (e.g., self-metallation, changes in the molecular conformation or alloying of the metal) even at ambient temperatures. Depending on the supplied amount of metal atoms only single molecules are metallated (Fig. 7a, bright, in panel A, or dark spots, in panel B, represent Fe-TPyP, grey areas are unreacted 2H-TPyP, ref. 47) or the sample is fully reacted (Fig. 7b, evolution from two peaks, 2H-TPP, to a single peak, Co-TPP, upon incremental deposition of cobalt, ref. 48). As introduced in Section 2.3, bias-dependent STM imaging and STS yield fingerprints of the chemical nature of the metal centre and allow one, e.g., to discriminate Co-TPP and Fe-TPP species at the single molecule level (Fig. 7a and c).88 The molecule–substrate–metal combinations explored so far are listed in Table 1, together with the reaction temperatures and the techniques used to investigate the respective system. It should be pointed out that while the vast majority of reports focus on tetrapyrrole metallation on noble metal surfaces, the generic metallation scheme can also be applied on ultra thin films including Pb overlayers or boron-nitride sheets (Fig. 7e). In the following subsection we will outline the reaction pathway as derived from DFT for a free porphyrin (Fig. 7d, for the reaction from 2H-P to Zn-P, ref. 69) and examine the metallation temperatures in the context of the choice of metal and molecule, as well as of the preparation conditions. Section 3.1.2 focuses on the characteristics of the metal atoms after the metallation process, i.e., the oxidation state of the inserted atom and the identification of potential excess metal and its properties. | ||
| Fig. 7 On-surface metallation of free-base molecules via PVD. (a) STM study of the metallation of 2H-TPyP on a Ag(111) surface with iron atoms at RT. The three top images show a closed 2H-TPyP island with a few Fe-TPyP molecules, appearing as bright (left) or dark (middle) species. The appearance depends on the applied bias voltage, which can be explained by the different electronic structure probed by STS (bottom panels). The occupied electronic states are characteristic for the metal centre and the LUMO of the Fe species is shifted to higher energies, inducing the bias-dependent contrast. For high voltages the apparent height of both molecules appears to be nearly identical (right) (adapted with permission from ref. 47. Copyright Wiley-VCH Verlag GmbH & Co. KGaA). (b) XP spectra for the metallation of 2H-TPP/Ag(111) with Co atoms. Upon incremental Co deposition the two distinct peaks corresponding to the two inequivalent nitrogen species in 2H-TPP (top panel) are reduced, and the metallated species is formed until the whole sample is metallated (panel D). The signature is the same as that for a predeposited Co-TPP (panel E) (adapted with permission from ref. 48). (c) A bimetallic TPP array featuring Co-TPP and in situ metallated Fe-TPP. Bias-dependent imaging yields a fingerprint of the different metal centres (Co versus Fe, compare (a)) (reprinted with permission from ref. 88. Copyright 2009 American Chemical Society). (d) Energy level diagram and some of the bond lengths (in Å) simulated with DFT using two different parameter sets for the insertion of Zn into a free-base porphyrin (reprinted with permission from ref. 69. Copyright 2007 American Chemical Society). (e) Cobalt metallation of TPCN on an sp2 bonded boron-nitride monolayer grown on Cu(111). STS corroborates the successful metallation on this insulating spacer layer and evidences an electronic signature of the Co-porphyrin distinctly different from adsorption on a metal (reprinted with permission from ref. 89. Copyright 2015 American Chemical Society). | ||
| Molecule | Substrate | Centre | Temp. | Comment | Technique(s) | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | 2H-TPyP | Ag(111) | Fe | RT | — | STM, STS, calc. | 47 and 71 |
| 2 | 2H-TPyP | Au(111) | Cu | 450 K | — | STM, XPS, DFT | 85 |
| 3 | 2H-TPyP | Au(111) | Fe | RT | — | STM, STS | 90 |
| 4 | 2H-TPyP | Au(111) | Co | RT | — | STM | 91 |
| 5 | 2H-TPyP | Au(111) | Cr | RT | — | STM | 91 |
| 6 | 2H-TPP | Ag(111) | Co | RT | — | XPS, VB, NEXAFS, STM, DFT | 48, 59 and 69 |
| 7 | 2H-TPP | Ag(111) | Fe | RT | Mol. + metal | STM, XPS, VB, NEXAFS | 59, 62 and 92 |
| 8 | 2H-TPP | Ag(111) | Fe | 550 K | Metal + mol. | XPS | 92 |
| 9 | 2H-TPP | Ag(111) | Zn | 550 K (XPS) | Order irrelevant | STM, XPS, DFT | 69 and 93 |
| 10 | 2D-TPP | Ag(111) | Zn | 510 K (TPD) | Order irrelevant | TPD, DFT | 69 |
| 11 | 2H-TPP | Ag(111) | Ce | 550 K | Double decker | STM | 94 |
| 12 | 2H-TPP | Ag(111) | Ce | RT | — | STM | 66 |
| 13 | 2H-TPP | Ag(111) | Mn | RT | — | XPS | 95 |
| 14 | 2H-TPP | Ag(111) | Rh | RT | — | XPS | 95 |
| 15 | 2H-TPP | Ag(111) | Ti | 500 K | — | XPS | 96, SI |
| 16 | 2H-TPP | Ag(111) | Gd | 550 K | Double decker | STM | 97 |
| 17 | 2H-TPP | Ag(100) | Fe | RT | — | XPS | 98 |
| 18 | 2H-TPP | Au(111) | Ni | RT | Mol. + metal | XPS, NEXAFS | 99 |
| 19 | 2H-TPP | Au(111) | Ni | 512 K | Metal + mol. | XPS, NEXAFS | 99 |
| 20 | 2H-TPP | TiO2(110) | Ni | RT | Mol. + metal | XPS, STM | 100 |
| 21 | 2H-TPP | TiO2(110) | Ni | 550 K | Metal + mol. | XPS, STM | 100 |
| 22 | 2H-TPP | SiO2/Si(100) | Er | RT | Co-deposition | XPS, STM | 56 |
| 23 | 2H-OEP | Ag(111) | Fe | RT | — | VB, XPS, NEXAFS | 62 |
| 24 | 2H-OEP | Cu(111) | Ni | RT | Order irrelevant | STM | 101 |
| 25 | 2H-Pc | Ag(111) | Fe | RT | — | STM, XPS | 102 |
| 26 | 2H-Pc | Ag(111) | V | 450 K | — | XPS, NEXAFS | 103 |
| 27 | 2H-TMP | Cu(100) | Fe | RT | — | STM, VB | 104 |
| 28 | 2H-TPCN | Ag(111) | Co | RT | — | STM | 105 |
| 29 | 2H-DPP | Au(111) | Cu | 363 K | — | STM | 106 |
| Films: | |||||||
| a | 2H-TPCN | BN/Cu(111) | Co | RT | — | STM, STS | 89 |
| b | 2H-Pc | Pb/Si(111) | Fe | RT | — | STM | 107 |
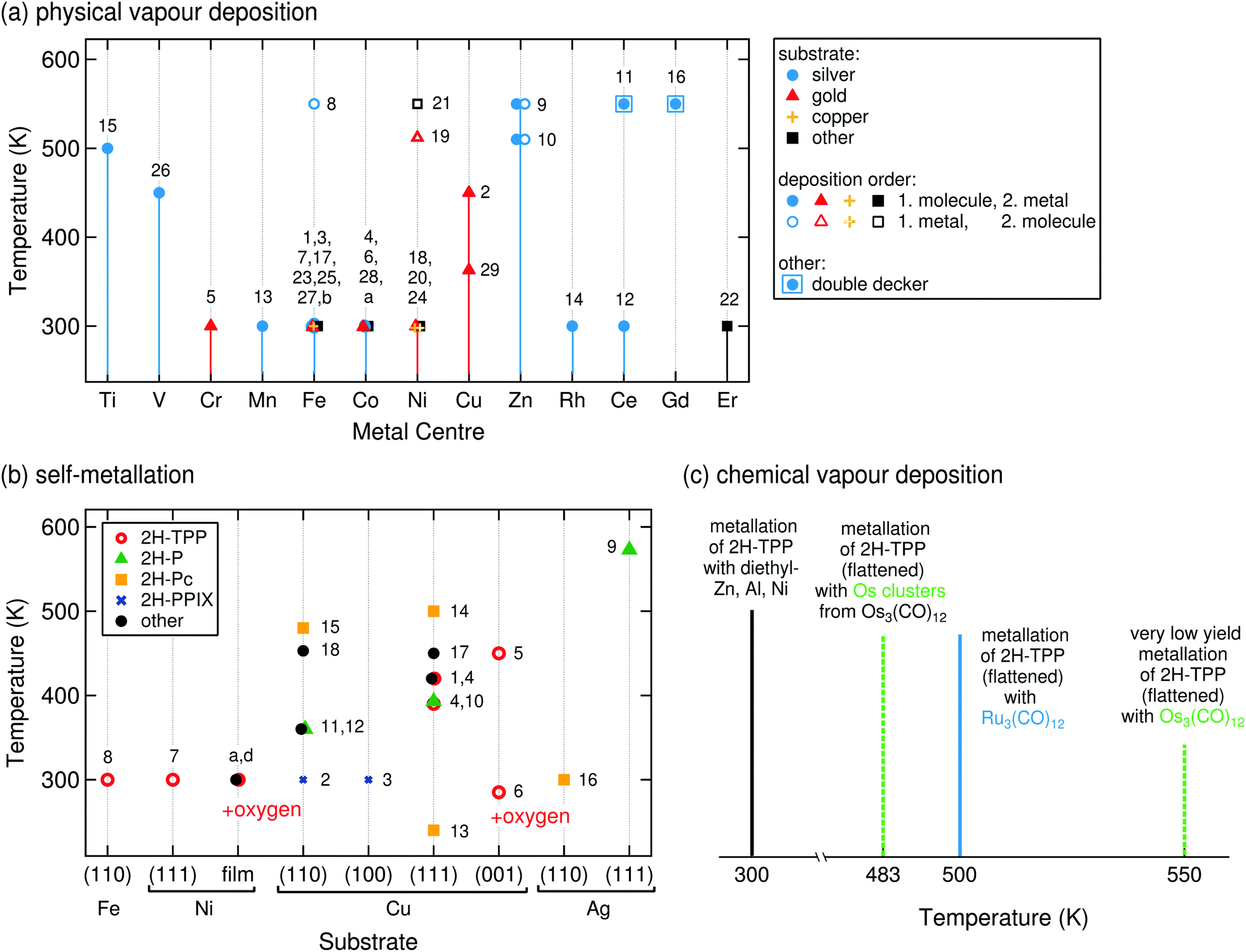 | ||
| Fig. 8 On-surface metallation temperatures. The numbers aside the symbols correspond to the systems of Tables 1 and 2, each data point might correspond to several references. (a) Reaction temperatures for PVD metallation with different elements. Measurements on a silver substrate are indicated with blue circles, on gold with red triangles, and on copper with orange crosses. As predicted, Zn and Cu metallation processes require elevated temperatures, regardless of molecule/atom deposition order. When molecules are dosed first and then exposed to a beam of metal atoms the reactions occur at RT (full symbols), when the order is reversed sometimes elevated temperatures are required (empty symbols, Fe, Ni), indicating an additional rate limiting step such as the dissolution of metal clusters.92,99 Systems 11 and 16 (square) are special cases, as the value refers to the formation of a ML of double deckers (Section 3.4.2). “Room temperature” was entered as 300 K. (b) Reaction temperatures for self-metallation on different substrates. A whole range of temperatures can be found for copper, depending strongly on the choice of the specific molecule–substrate system. (c) For CVD an additional potential rate-limiting process has to be considered, namely the decomposition of the precursor complex. Ru3(CO)12 is thermally less stable than Os3(CO)12,108 a high metallation yield with Ru can thus already be obtained at 500 K,109 while for Os even higher temperatures only yield a small amount of Os-TPP molecules.110 In contrast, the reaction with pre-decomposed Os3(CO)12 (i.e., Os clusters) proceeds at lower temperatures with a much higher yield. | ||
Role of the inserted metal. In a first approximation one can neglect the surface and only look at the metallation in gas phase, simulated using density functional theory at a hybrid functional (B3LYP) level by Shubina et al. in ref. 69 and by Shen and Ryde in ref. 36. The latter studied in detail the metallation of free-base porphine with the solvated metal ions Mg2+ or Fe2+, i.e., with Mg(H2O)6 and Fe(H2O)6. In both cases the highest activation barrier (which is lower for Fe than for Mg) is required for the formation of the sitting-atop-complex, where the metal centre already forms a coordination bond to the macrocycle, while the central hydrogen atoms are not yet released. During this process, the porphyrin gets deformed considerably; only by the final deprotonation this strain is released.36 For the metallation with bare metal atoms Shubina et al. calculated possible intermediate steps and the corresponding energy levels, so that energy level diagrams are available for the metallation of free-base porphine with Fe, Co, Ni, Cu, and Zn.69 The energy barrier for the last metal is the highest (the reaction the slowest), allowing to access the intermediate steps experimentally. Therefore, the metallation with Zn will be discussed in the following as the exemplary case. The pathway proposed in ref. 69 (reproduced in Fig. 7d) predicts an intermediate metal–organic complex where the hydrogen atoms are still attached (sitting-atop complex, cf. also discussion of ref. 36 above). The limiting step is the transfer of one hydrogen atom to the Zn atom with a barrier of 32.6–34.8 kcal mol−1 (corresponding to 1.4–1.5 eV, depending on the basis set). This hydrogen atom can then approach the other, form H2, and subsequently split off from the Zn centre. This pathway was verified by XPS and TPD measurements of 2H-TPP (XPS) and isotopically labelled 2D-TPP (TPD) molecules, respectively, adsorbed on Ag(111) and metallated with Zn atoms. The authors observed an intermediate state in the Zn 2p3/2 XP spectra and a reaction temperature of 510 K (corresponding to approximately 32 kcal mol−1), in good agreement with the theoretical predictions. In the XPS experiments 550 K are typically sufficient to metallate all free-base molecules of the sample.69,93
Of the elements tested in ref. 69 only Zn and Cu exhibit a global reaction barrier, whereas the reaction for Fe, Co, and Ni proceeds much faster. It should also be noted that there is another difference between the two groups: the metallation reactions for Cu and Zn take place on a single potential energy surface, while two possible pathways (via a low- and a high-spin transition state) are involved for Fe, Co, and Ni.69 Taking into account (for the moment) only the full symbols in Fig. 8a, which represent experiments where pre-deposited molecules were metallated with on-top evaporated metal atoms, the known experimental data fit these predictions very well. Metallation with Fe, Co, and Ni already proceeds at room temperature, while the metallation with Zn and Cu requires elevated temperatures. Incidentally, we note that this is also the case for the self-metallation, which occurs readily on Ni(111) and Fe(110) surfaces at room temperature (cf. discussion in Section 3.2 and Fig. 8b), but is more complicated for the incorporation of copper atoms.
Role of the molecular species. The results described in the previous section do not seem to depend on the employed free-base molecules. Of course, one has to handle with care systems where only one data point (e.g., one type of molecular species) in Fig. 8a is known, but especially for the well-studied metallation with iron and nickel the data are consistent. This can be explained by the similar adsorption environment: most of the studies presented here were conducted using silver or gold substrates, which do not induce very pronounced changes in the molecular conformation. On these surfaces the molecules only exhibit a (compared to molecules adsorbed onto more reactive substrates) small macrocycle deformation (see for example the case of 2H-TPyP/Ag(111),111 2H-P/Ag(111),112 2H-Pc/Ag(110),113 and ref. 18 for a general overview) of comparable magnitude. The interaction with the surface only leads to moderate (if any) electron transfer to the molecule,77 and diffusion parameters are considered as comparable. Thus, for the so far discussed metallation by exposing free-base tetrapyrrole molecules to a beam of metal atoms, the reaction barriers for the pathways calculated in ref. 69 seem to be applicable. As will be discussed in Sections 3.2.1 and Section 4 this is not generally the case for all metallation protocols.
Role of the deposition order. Experiments have been performed with reversed deposition sequence, i.e., molecules are added after depositing metal atoms. This was explored for the metallation of 2H-TPP with Fe, Ni, and Zn, and for 2H-OEP with Ni.92,93,99,101 As could be expected from the considerations above, the deposition order is irrelevant for the metallation with Zn,93 as high temperatures are required to overcome the reaction barrier.
The metallation of 2H-TPP/Ag(111) with iron atoms which was described by Buchner et al. in ref. 92 offers more insight. For pre-deposited molecules the metallation already proceeds at room temperature, as predicted by DFT. But if the metal is dosed first, elevated temperatures (550 K in their experiment, open circle in Fig. 8a) are required for the same process. In this case the rate limiting step is the formation of an equilibrium of diffusing Fe atoms and the formation of Fe clusters at the step edges (as observed by STM).92 This seems plausible, as in the same publication a similar effect is observed for the metallation of 2H-TPP multilayers: even an excess of iron does not lead to a metallation of all layers, which can be explained by the formation of iron clusters in the film. Depositing the molecules first should lead to the decoration of the step edges with molecules and thus possibly hinder the diffusion of Fe atoms. Another tentative explanation presented by Buchner et al. is that for the insertion of the iron atom inside the macrocycle an additional bond between the atom and the substrate has to be broken, which is easier if the atom additionally brings kinetic energy through deposition.92 The same observation was made for 2H-TPP/TiO2(110) (Fig. 8a: black square at RT and open square at 550 K100) and for 2H-TPP/Au(111) metallated with Ni by Chen et al.,99 who showed that pre-deposited molecules react at room temperature while dosing molecules on top of pre-deposited nickel requires higher temperatures (512 K, open triangle in Fig. 8a). In both cases the difference was attributed to the formation of nickel clusters in the absence of molecules, which need to be activated before they act as a source of adatoms available for incorporation.99,100
It is worth noting that the metallation with pre-deposited metal does not always require elevated temperatures. A case study was performed for 2H-OEP/Cu(111), where the metallation with Ni at room temperature was achieved for both pre-deposited molecules and molecules adsorbed on nickel islands which were grown on the copper substrate.101 However, in the same work 2H-TPP molecules were deposited on the nickel islands, and it was shown that they did not metallate at room temperature but instead remained as free-base species in a saddle-shape conformation. The latter is characterised by two opposite pyrrole rings oriented with their nitrogen atoms towards the surface, while the other two pyrrole rings are pointing upwards (“saddle shape”, cf. also Section 4). A very similar result was also found for 2H-TPP on a Ni film on Cu(001), deposited at room temperature (see also the overview in Table 2).114 These findings imply that, in contrast to the metallation with pre-deposited molecules described above, for pre-deposited metal the specific identity of the molecules (and with it the details of the molecule–molecule, molecule–substrate, and molecule–atom interactions) does play an important role. Similar tendencies are observed in the case of self-metallation described in Section 3.2, where the molecule is metallated by substrate atoms and the reaction temperatures also depend on the chosen molecule. Indeed, the metallation of free-base tetrapyrrole molecules on pre-deposited islands may be regarded as an intermediate case between metallation by PVD and self-metallation.
| System | Molecule | Substrate | Temperature | Technique(s) | Ref. |
|---|---|---|---|---|---|
| 1 | 2H-TBrPP | Cu(111) | 420 K | STM, XPS, NEXAFS | 127 |
| 2 | 2H-PPIX | Cu(110) | RT | STM, XPS | 128 and 129 |
| 3 | 2H-PPIX | Cu(100) | RT | STM, XPS | 128 |
| 4 | 2H-TPP | Cu(111) | 39086–420 K60 | XPS, NEXAFS, DFT, STM, XSW, TPD | 60, 63, 86 and 130–132 |
| 5 | 2H-TPP | Cu(001) | 450 K | STM, XPS | 114 |
| 6 | 2H-TPP | O/Cu(001) | 285 K | STM, XPS | 114 |
| 7 | 2H-TPP | Ni(111) | RT | XPS, NEXAFS, calc. | 76 |
| 8 | 2H-TPP | Fe(110) | RT | XPS, NEXAFS, calc. | 76 |
| — | 2H-TPP | O/Pd(100) | 430–480 K | — | 133 |
| 9* | 2H-P | Ag(111) | 573 K | STM | 134 |
| 10 | 2H-P | Cu(111) | 393 K | STM, XPS, NEXAFS, DFT | 64 |
| 11 | 2H-P | Cu(110) | 360–410 K | STM, DFT, TPD | 135 |
| 12 | 2H-DPP | Cu(110) | 360–410 K | STM, DFT, TPD | 135 |
| 13 | 2H-Pc | Cu(111) | 240 K (submonolayer) | XPS, TPD | 49 |
| 14 | 2H-Pc | Cu(111) | 500 K (multilayer) | XPS, TPD | 49 |
| 15 | 2H-Pc | Cu(110) | 480 K | XPS, NEXAFS, STM | 136 |
| 16 | Dehydr. 2H-Pc | Ag(110) | RT | XPS, STM | 87 |
| 17* | 2H-TPyP | Cu(111) | 450 K | STM, XPS, NEXAFS | 137 |
| 18 | 2H-TMP | Cu(110) | 453 K | LDI-TOF | 138 |
| Films: | |||||
| a | 2H-OEP | Ni/Cu(111) | RT | STM | 101 |
| b | 2H-TPP | Ni/Cu(111) | No metall. at RT | STM | 101 |
| c | 2H-TPP | Ni/Cu(001) | No metall. at RT | XPS | 114, SI |
| d | 2H-TPP | O/Ni/Cu(001) | RT | XPS | 114, SI |
Chemical transformation of the molecular periphery. Even before the actual metallation experiment is performed, the molecular layers are often annealed to produce a well-defined single layer (monolayer) upon desorption of multilayer components.59,72,92,93,115–117 Depending on the chosen free-base compound, the temperature of this annealing step ranges from 510115,116 to 550 K.92,116 As discussed above, the subsequent metallation likewise sometimes requires temperatures up to 550 K. In view of the intriguing and multi-faceted covalent chemistry taking place under similar conditions,118–121 the question arises whether the tetrapyrrolic molecules (free-base or not) are actually stable against such thermal treatment. Indeed, XPS studies indicated that Co-TPP and Co-TTBPP monolayers, formed after annealing to 525 K, start to decompose between 600 and 700 K.48,117 For 2H-TPP/Au(111) the respective values are 581 K and 631 K, for Ni-TPP/Au(111) 520 K and 570 K.99 For the prototypical system 2H-TPP/Ag(111), Di Santo et al. studied the effect of annealing by angle-dependent NEXAFS, electron energy loss spectroscopy, STM, TPD, and DFT.74 They found that annealing to 550 K induces a cyclodehydrogenation reaction which causes the rotation and fusing of the phenyl rings, such that the transformed compound lies flat on the surface. Several dehydrogenated products are possible, the by far most frequently occurring one122 being shown in Fig. 3b.57 The cyclodehydrogenation reaction is accompanied by a shift to lower binding energies of the N and C 1s XPS peaks,57 which was interpreted as start of decomposition in ref. 99 and 117. Recent TPD measurements indicate that the reaction onset is even lower, and already for 500 K flattened layers can be obtained.122 Therefore, we note that the on-surface metallation of tetrapyrrole compounds is limited by the chemical stability of the employed species and accordingly this approach might not be applicable for specific molecule/substrate/metal combinations. On the other hand, when the comparison is possible, the on-surface metallation was found to be hardly affected by the conformation of the free-base porphyrin. Metallation using Fe59 and Co123 was attained at the same temperatures for the planar as for the pristine TPP species. This may not be surprising in the light of the previous remarks on the role of the molecular species for post-deposition of metal atoms. Moreover, metallation of the inherently planar phthalocyanines very much follows the trends of the porphyrins.
To answer this question the XP spectra from the core level of the metal can be informative, as they can give quantitative insight about the different metal species and their chemical environment. Unreacted metal atoms are observed at binding energies corresponding to metallic species, i.e., with formal oxidation state M(0). For metallated porphyrin and phthalocyanine molecules the metal is expected to be in a formal +2 state, i.e., the XP signals should be shifted to higher binding energies. For the metallation with Zn this is indeed the case,69,93 as well for the metallation with Ni99 and Cu.63,85 In these cases, therefore, XPS is able to differentiate between unreacted metal atoms and those incorporated into the molecules. For metallotetrapyrroles containing Fe, Co, and Ru on silver and/or gold surfaces, however, this is not the case. After metallation to Co-TPP,48 Fe-Pc,92,102 Ru-TPP and Ru-P57,109 the Co, Fe, and Ru centres retain their (0) state (see e.g.Fig. 3c). This is not a side-effect of the on-surface metallation, but was also reported for the adsorption of prefabricated cobalt, iron and ruthenium porphyrins and phthalocyanines, whose (II) state in the multilayer shifts to a (0) position in the first layer in contact with the respective silver and gold substrates.82,96,102,109,117,124–126 As anticipated in Section 2.2, a widely invoked rationale for this effect is that through the interaction with the substrate (presumably through the dz2 orbitals82) a charge transfer is induced reducing the oxidation state of the metal. However, it should be noted that to verify the occurrence of charge transfer, XPS is not the most appropriate tool. Spectroscopic signatures in UPS (occupied states), NEXAFS (unoccupied states), and STS (both) may then be desirable, in order to assess the population (depopulation) of unoccupied (occupied) molecular orbitals. Thus, XPS can only directly detect excess of Zn, Ni, and Cu, where the core-level lines of inserted metal atoms can be distinguished from those of unreacted atoms. For the reaction of 2H-TPP/Au(111) with Ni, the presence of two peaks in the Ni 2p3/2 spectrum clearly indicates that after annealing to 510 K and full metallation of the porphyrins (as evidenced by the N 1s XPS), unreacted nickel atoms remained on the surface.99 In contrast, for the reactions of 2H-TPP/Ag(111) with Zn and for 2H-TPyP/Au(111) with Cu the M(0) component vanishes after the annealing required to induce the reaction with Zn and Cu (see previous section). This was explained by dissolution of the excess metal into the substrate.85,93
Summary. Tetrapyrrole molecules can be metallated by evaporating molecules and metal atoms on a suitable substrate. The reaction is likely to proceed via the formation of an intermediate metal–organic complex before the hydrogen atoms are finally split off and desorb as H2. The desorption of molecular hydrogen can be observed with TPD to determine the reaction temperature, whereas the changes in the molecule are typically monitored using STM, XPS, and/or NEXAFS. DFT predicts activation barriers for the metallation with Zn and Cu, and consistently the metallation itself (and not, e.g., diffusion processes) is identified as the rate limiting step in the corresponding experiments where the metallation requires elevated temperatures (>510–550 K). For other systems, e.g., the metallation of porphyrins with Ni and Fe, where no barrier is predicted, the deposition order can play an important role for the necessary reaction temperature, as the metallation process competes with the morphology and pertaining physical properties of the surface metal reservoirs, such as island or cluster formation or the diffusion of single atoms. For pre-deposited molecules and subsequent addition of metal atoms, the energy barrier of the metallation itself seems to be the dominant factor determining the metallation temperature, explaining why the substituents in the molecules and the details of the molecular conformation hardly seem to play any role. In contrast, for the metallation with pre-deposited atoms the interaction with the metal islands and the substrate appears to be crucial, so that for different molecules on the same kind of metal islands different behaviours are observed.
3.2 Self-metallation
Contrasting the PVD metallation described in the previous section, the self-metallation occurs without additional metal deposition. The term “self-” refers to the fact that the molecule–substrate system itself provides all reactants required for the metallation as the metal centre-to-be is contributed by the substrate. Similar to the studies discussed in the previous section, typical self-metallation experiments consist of in vacuo deposition of the tetrapyrrole molecules on a metal support (commonly a single crystal) followed by a thermally induced metallation reaction, thus offering simple means to inspect metallation-induced changes of the system’s chemical, conformational, and electronic properties.In favourable cases the insertion of a metal atom can be directly visualised by STM (e.g., Fig. 9b of 2H-P/Cu(111)64). For all systems the XPS and NEXAFS spectra clearly allow to differentiate between the free-base and the metallated species (e.g., Fig. 9a of 2H-TBrPP/Cu(111)127), and TPD gives evidence of the release of hydrogen during the metallation process. For example, TPD studies using isotopically labelled 2D-TPP showed that the mechanism of metallation involves D or H spillover to the Cu surface and not recombination to molecular D2 above the inserted metal centre, as seen in the case of Zn metallation in the previous section (see also Fig. 6).68,131 In contrast to the metallation by co-deposition, where the substrate and incorporated metal ions usually do not belong to the same element, it is hardly feasible to measure the charge state of the central metal ion with spectroscopy, because its signal would be obscured by the signal of the substrate at typical coverages.
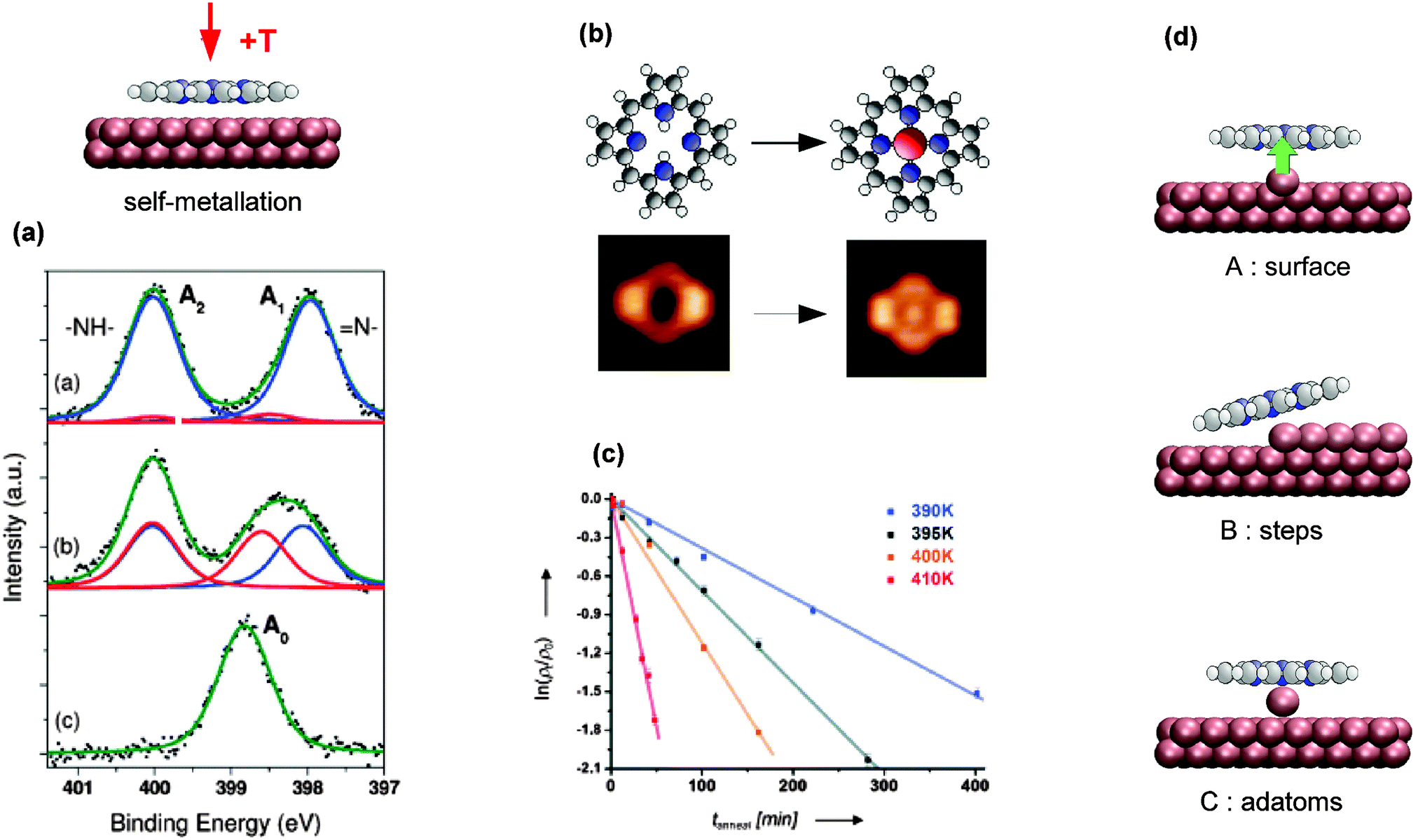 | ||
Fig. 9 Self-metallation of tetrapyrrole molecules on metal surfaces. As discussed in Section 2 the metallation can be followed for example by (a) XPS or (b) STM. (a) XP spectra of 2H-TBrPP/Cu(111) corresponding to a multilayer (top), a bilayer (middle) and a bilayer annealed to 420 K (bottom). The middle panel indicates an intermediate state, where some of the molecules are still intact but strongly bound to the copper substrate via the iminic (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) nitrogen atoms of the porphyrin (ref. 127). (b) STM images showing the self-metallation of 2H-P/Cu(111) upon annealing to 423 K (reproduced with permission from ref. 64. Copyright 2013, AIP Publishing LLC). (c) By counting metallated vs. non-metallated porphyrin species in STM images after different annealing times and temperatures it is possible to determine the activation barrier for the self-metallation of 2HTPP/Cu(111). (Adapted from ref. 86. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.) (d) Schematics illustrating different scenarios under discussion for the origin of the incorporated substrate atom (A: direct extraction from the surface, B: metallation at step edges, C: involvement of adatoms). N–) nitrogen atoms of the porphyrin (ref. 127). (b) STM images showing the self-metallation of 2H-P/Cu(111) upon annealing to 423 K (reproduced with permission from ref. 64. Copyright 2013, AIP Publishing LLC). (c) By counting metallated vs. non-metallated porphyrin species in STM images after different annealing times and temperatures it is possible to determine the activation barrier for the self-metallation of 2HTPP/Cu(111). (Adapted from ref. 86. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission.) (d) Schematics illustrating different scenarios under discussion for the origin of the incorporated substrate atom (A: direct extraction from the surface, B: metallation at step edges, C: involvement of adatoms). | ||
An advantage of self-metallation over other metallation methods is that no additional metal evaporation is required, which simplifies the experimental procedures. On the downside, currently only a rather small number of self-metallating systems is known (cf.Table 2 and discussion below). An exact knowledge of self-metallating processes is indispensable for the interpretation of tetrapyrrole experiments, as already for preparations at room temperature free-base molecules might self-metallate and thus not be present in their original state any more (Section 3.2.1). The exact origin of the inserted metal atom is still under discussion, and different scenarios will be outlined in Section 3.2.3.
Table 2 lists the self-metallating tetrapyrrole systems known up to date. The majority of examined systems employed porphyrins adsorbed onto copper surfaces, which allows a direct comparison to determine the role of different surface facets and the influence of the molecules' substituents on the activation temperature. Many studies, involving the whole range of available techniques, were dedicated to the 2H-TPP/Cu(111) system, which thus can serve as a model system to understand self-metallation.
A further extension of this scenario is the transmetallation reported in ref. 139 by Doyle et al. (not listed in Table 2). For two different nickel porphyrin compounds (NiDBrDPP and NiTBrPP) adsorbed onto a Cu(111) surface, the authors found that after annealing to 430 K nickel was removed from the porphyrin core and replaced by a substrate copper atom. This is well in line with the results from experiments in solution, where transmetallation reactions are used to test the relative stability of different metalloporphyrins.140
For different surface facets the employed molecules differ, such that a direct comparison is not possible. Interestingly, self-metallation can be promoted by introducing an oxygen reconstruction which reduced the metallation temperature for 2H-TPP/Cu(001) from 450 K to 285 K.114 A possible explanation for the reduced activation barrier is the formation of an additional intermediate state including oxygen–copper bonds.114
We can assess the influence of the chosen free-base tetrapyrrole. On Cu(111) three different compounds can be compared: 2H-P,64 2H-TPP,60,63,86 and 2H-TBrPP,127 of which 2H-P is the smallest and most planar, while the other two feature four substituents each. 2H-P requires apparently slightly lower temperatures for metallation (onset at 373 K for 2H-P64vs. 390 K for 2H-TPP86): this can be rationalized in terms of a lower height of the macrocycle above the surface, which increases the surface interaction. While the deformation of the substituted molecules can facilitate the formation of specific bonds to the surface,60,127 it is plausible that the interaction is stronger for 2H-P. The same trend can be observed for the Cu(100) facet, where the rather flat 2H-PPIX metallates already at room temperature,128 while metallation was ruled out by Écija et al. for the more bulky 2H-TMP.104 However, the assumption that metallation of the flat 2H-P always proceeds at lower temperatures cannot be extrapolated to the Cu(110) surface, where 2H-P requires slightly higher reaction temperatures (onset at 360 K135) than the 2H-PPIX (metallation at room temperature128). It should be noted, though, that for the latter a complete metallation of the film was not achieved. From the experimental point of view, one problem are the uncertainties introduced by the different methods to determine the metallation onset, another the accuracy of the actual temperature readings.
A more quantitative approach was shown in ref. 86 by Ditze et al., who were the first to determine activation barriers and rate constants for a self-metallation reaction. For 2H-TPP/Cu(111) the authors exploited the fact that through a strong interaction with the substrate 2H-TPP is not mobile and can hence be imaged easily with STM, while Cu-TPP molecules are mobile at room temperature and are not resolved. By counting the visible 2H-TPP molecules it was hence possible to perform a quantitative measurement of the metallation rate. The corresponding density and Arrhenius plots of the rate constant for the range 390–410 K are reproduced in Fig. 9c. For the rate-limiting step the determined value of (1.48 ± 0.12) eV in ref. 86 is well in line with the gas phase values predicted by DFT for the insertion of a copper atom into the model system 2H-P treated in ref. 69 and 85 (24–37 kcal mol−1, corresponding to 1.04–1.60 eV). Assuming that the good agreement is not based on an error cancellation effect, this would suggest that the rate limiting step is given by the insertion of the copper ion into the macrocycle. The extraction of the atom from the copper substrate may therefore not be the dominating effect, and the experimental value of (1.48 ± 0.12) eV can thus be seen as an upper limit for the extraction process (a discussion of the origin of the substrate metal atom follows below). One point that has to be kept in mind is that (at least for 2H-TPP/Cu(111)) the self-metallation rate strongly depends on the surface coverage as this can affect the distance of the macrocycle from the surface. Röckert et al. found a substantial rate enhancement130 when reaching the checkerboard structure67,146 at higher coverages. Also the onset of cyclodehydrogenation (cf. Section 3.1.1) depends on the coverage, as shown by TPD measurements of the same authors.131
The same effect was reported by several groups for other porphyrins adsorbed on Cu(111), even though the details vary slightly. For 2H-TPP/Cu(111), Buchner et al. observed a reduction in the XPS peak splitting from 2.0 eV in the multilayer to 1.5 eV in the monolayer, although in this case both peaks shifted: the pyrrolic peaks downwards, the iminic peak upwards.147 For the same system we reported a reduction from 2.0 to 1.6 eV, with a stronger downward shift of the pyrrolic contribution, and hardly any change in the iminic nitrogen peak.60 Similar trends exist for 2H-P/Cu(111), where both peaks moved towards each other in the monolayer.64 The small differences can be attributed to different ways of calibrating the energy scale (in ref. 64 and 127 bilayers were fitted, in ref. 60 and 147 multi- and monolayers were measured separately) and/or of fitting the single components. These changes were unanimously attributed to a strong interaction of the porphyrin with the surface, facilitated either by an already planar adsorption conformation (2H-P64) or by a substantial deformation of the macrocycle with the iminic nitrogen atoms pointing towards the substrate (2H-TBrPP127 and 2H-TPP60). In some cases an additional electron transfer from the copper substrate to the molecule was observed.60,64,77 However, only in ref. 127 the intermediate step is observed as a clear feature in the N K-edge NEXAFS data of a 2H-TBrPP bilayer, while it was not found for other systems. For 2H-P and 2H-TPP the submonolayer data are strongly influenced by hybridization and electron transfers60,64 such that hardly any clear features can be derived. This is not the case for 2H-TBrPP, possibly as a result of different adsorption conformations which slightly alter the interaction between the copper and the LUMO of the molecule. For the self-metallation on Fe(110) and Ni(111) a possible intermediate step was not studied, as experiments were performed at room temperature where the sample was already fully metallated.76
Unfortunately, there is no consensus in the literature on this possible intermediate step and its definition. In ref. 128, for example, González-Moreno et al. also use the term “intermediate situation”, referring to a mix between metallated and non-metallated molecules. In ref. 63, Xiao et al. mention an “intermediate state” including adatoms, and the adsorbate–substrate complex formed upon adsorption of 2H-TPP on Cu(111) is called a “precursor state”. Another utilized expression is “sitting-atop complex” (see also Introduction and Discussion in Section 4), which refers to an intact molecule interacting strongly with a metal atom.147 It should finally be noted that similar observations, i.e., the formation of a state bound via iminic nitrogen atoms to the metal surface, can be found for free-base meso-tetradodecylporphyrin adsorbed onto Au(111), where self-metallation was ruled out.144
The currently most favoured explanation is the involvement of adatoms.60,63,76,128 On Cu(111), for example, the adatom detachment from kinks requires only 0.76 eV,152 which is considerably lower than the 2 eV discussed above. Mass exchange with the terraces by adatom extraction sets in at 500 K and becomes the dominant mass transport mechanism at 600 K,152,153 leading to a surface adatom gas with a coverage of typically several percent of a monolayer.154 The presence of adsorbates might even lower the temperature required for the metallation,114 such that at the reaction temperatures reported here a sufficient amount of adatoms is present. The DFT calculations by D. Passerone and co-workers support the involvement of adatoms: their simulations for 2H-TPP/Ni(111) show that the metallation involving a surface atom is exothermic, while metallation creating a vacancy is energetically unfavourable.76
Summary. The self-metallation of tetrapyrrole molecules has been observed on a wide range of metal substrates in the form of single crystals or thin films. These include Ni, Fe, Cu and Ag, with metallation on Cu being the most extensively investigated. In general, the more reactive the metal substrate, the lower the activation energy required for metallation. An “intermediate” state can be inferred from the electronic shift of the nitrogen signals observed by XPS; the surface, e.g. of Cu, causes shifts in binding energy consistent with a strong interaction with the metal. For the case of 2H-TPP on Cu(111) the metallation energy was experimentally determined by Arrhenius plots based on STM data. A different mechanism was proposed for the self-metallation of 2H-Pc on a Ag surface, whereby the 2H-Pc converts to Pc prior to the formation of Ag-Pc. The origin of the inserted metal is at present ambiguous in most cases.
3.3 Alternative on-surface metallation routes
Both the PVD metallation and the self-metallation offer a wide range of possible systems that can be prepared in vacuo. However, not all metal–molecule combinations are easily accessible, and the development of other techniques is desirable. An overview over alternative approaches presented in this section is given in Table 3.| Molecule | Substrate | Centre | Comment | Technique(s) | Ref. |
|---|---|---|---|---|---|
| 2H-Pc | Ag(111) | Ag | Tip manipulation | STM, STS, calc. | 78 |
| 2H-P | Ag(111) | Ru | CVD | XPS, NEXAFS | 109 |
| 2H-TPP | Ag(111) | Ru | CVD | XPS, NEXAFS | 109 |
| 2H-TPP | Ag(111) | Os | CVD | STM | 110 |
| 2H-TPP | SiO2/p-doped Si(100) | Zn | CVD | XPS, DFT, Raman, AFM | 148 |
| 2H-TPP | Unknown | Al | CVD | Unknown | Mentioned in ref. 148 |
| 2H-TPP | Unknown | Ni | CVD | Unknown | Mentioned in ref. 148 |
| 2H-TPP (flattened) | Ag(111) | Ru | CVD | STM, XPS, NEXAFS | 57 |
| 2H-TPP (flattened) | Ag(111) | Os | CVD | STM | 110 |
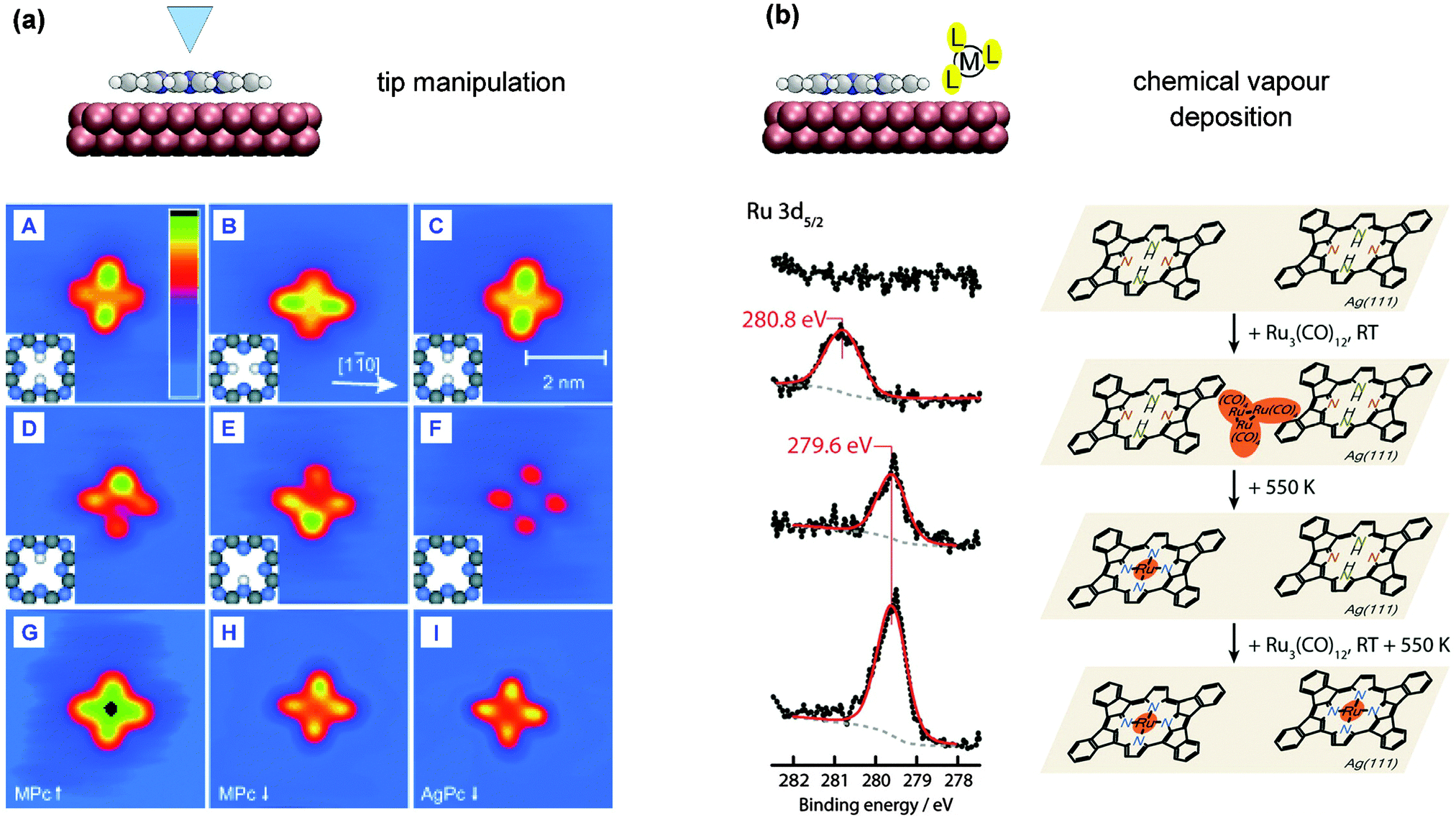 | ||
| Fig. 10 Alternative tetrapyrrole metallation routes. (a) Tip manipulation of 2H-Pc/Ag(111): the inner hydrogen atoms are removed sequentially, followed by insertion of a silver atom with an STM tip (adapted with permission from ref. 78. Copyright Wiley-VCH Verlag GmbH & Co. KGaA). (b) Codeposition of a precursor molecule: a cyclodehydrogenated 2H-TPP molecule on Ag(111) is metallated after exposure to Ru3(CO)12 at RT followed by annealing to 550 K (adapted with permission from ref. 57. Copyright 2013 American Chemical Society). | ||
It should be noted that the manipulation of tetrapyrrole molecules with an STM tip does not automatically lead to an incorporation of adatoms. Mielke et al. showed for example that 2H-TPP adsorbed onto Au(111) remains intact, even when residing on top of gold adatoms.142
More recently the corresponding Os precursor complex, Os3(CO)12, was employed to metallate 2H-TPP and its planar high temperature derivatives on Ag(111) as investigated by STM.110 In contrast to the case of the Ru precursor, it was found that the more stable Os3(CO)12108 did not efficiently metallate the porphyrin species when post deposited on a surface of the high temperature planar derivatives of 2H-TPP, but rather promoted the porphyrin polymerisation by new C–C bond formations. By inversion of the dosing sequence of metal precursor and porphyrins, the decomposition of the metal precursor to Os clusters on Ag(111) was achieved by annealing to 623 K. Subsequent deposition of 2H-TPP and annealing to 453 K showed a partially metallated layer of pristine and partially flattened 2H-TPP. Annealing to 483 K was required to dissolve the surface Os clusters and achieve a high degree of porphyrin metallation by Os (as illustrated in Fig. 8c). By this annealing temperature, due to the presence of Os, all porphyrins had been converted to planar TPPs.
A similar approach was recently demonstrated by Kim et al., who used diethylzinc to metallate 2H-TPP layers on a silicon dioxide support to achieve multilayer films of Zn-TPP.148 In contrast to the examples presented above, the dosage of the precursor complex takes place at relatively high pressures (2.8 × 10−1 Torr), which allows the chemisorption of the diethylzinc molecules onto the porphyrin films. This example, as well as the metallation of 2H-TPP by diethylzinc during atomic layer deposition presented in ref. 44 might be regarded as cases bordering to experiments conducted under near-ambient conditions.
Summary. Emerging metallation techniques include atomic manipulation and CVD of appropriate metal precursors. The former route was demonstrated for 2H-Pc on Ag(111). It provides a model study, where single molecules can be addressed and even unfavourable metallations can be promoted so as to investigate the electronic properties of these metalloporphyrins at interfaces. With CVD, efficient Ru and Os metallation of the planar derivatives of 2H-TPP was shown on Ag(111) by using trimetal dodecacarbonyl precursors. The formation of Ru-TPP, Os-TPP and Ru-P via CVD was also observed. Here, the thermal stability of the precursor was suggested to further affect the metallation process.
3.4 Tackling the third dimension
For the PVD approach, achieving full metallation of a multilayer can be a challenge. This became clear with the system 2H-TPP and Fe92 or Rh:95 The metallation of 2H-TPP on Ag(111) with Fe atoms is complete for a monolayer, but proceeds only partially for a thin film of approximately 20 layers. The Fe region XPS shows an excess of neutral iron in the sample, the authors thus concluded that the formation of iron clusters inside the multilayers prevents complete metallation of the sample.92 In contrast, the same experiment for 2H-OEP on Ag(111) in ref. 62 and 2H-TPP on Ag(111) in ref. 59 led to a (nearly) complete metallation of the free-base molecules with Fe. This might result from the smaller film thickness in the latter experiments (approximately four layers), which prevents the formation of iron clusters before the full metallation of the sample succeeds. To prepare thicker, fully metallated samples, it is possible to repeat the procedure adopted for a single layer until the desired thickness is reached (shown for 10 layers of vanadium phthalocyanine in ref. 103).
Very recently Kim et al. demonstrated the formation of Zn-TPP layers in the 100 nm regime by repeated cycles (up to 300) of CVD metallation of 2H-TPP with diethylzinc precursors.148 By sequential creation of Zn-TPP layers with controllable thickness and dosing of pristine 2H-TPP molecules they could even embed the metalloporphyrin layers in a free-base porphyrin film.
Surprisingly, also self-metallation of more than one layer was occasionally observed. Moderate annealing to 420 K of a 2H-TPP multilayer (approximately three layers) adsorbed onto a Cu(111) surface resulted in a near complete metallation of the film,60 as evidenced by a single peak in the nitrogen XPS region (Fig. 11a, second spectrum from the bottom). Further annealing to 490 K caused the desorption of excess molecules, such that only one monolayer of Cu-TPP remained on the surface (Fig. 11a, bottom spectrum). Also for 2H-P/Cu(111) a metallated second layer was observed.64 This has the big advantage that the effect of the surface on the metallated species can be studied directly by comparison to the metallated multilayer. The N 1s energy of the Cu–N species in Cu-TPP is typical for metalloporphyrins (located between the iminic and the pyrrolic nitrogen peak, Fig. 11a, second spectrum from the bottom), in the first layer this peak shifts to lower binding energies due to screening of the surface (Fig. 11a, bottom spectrum).
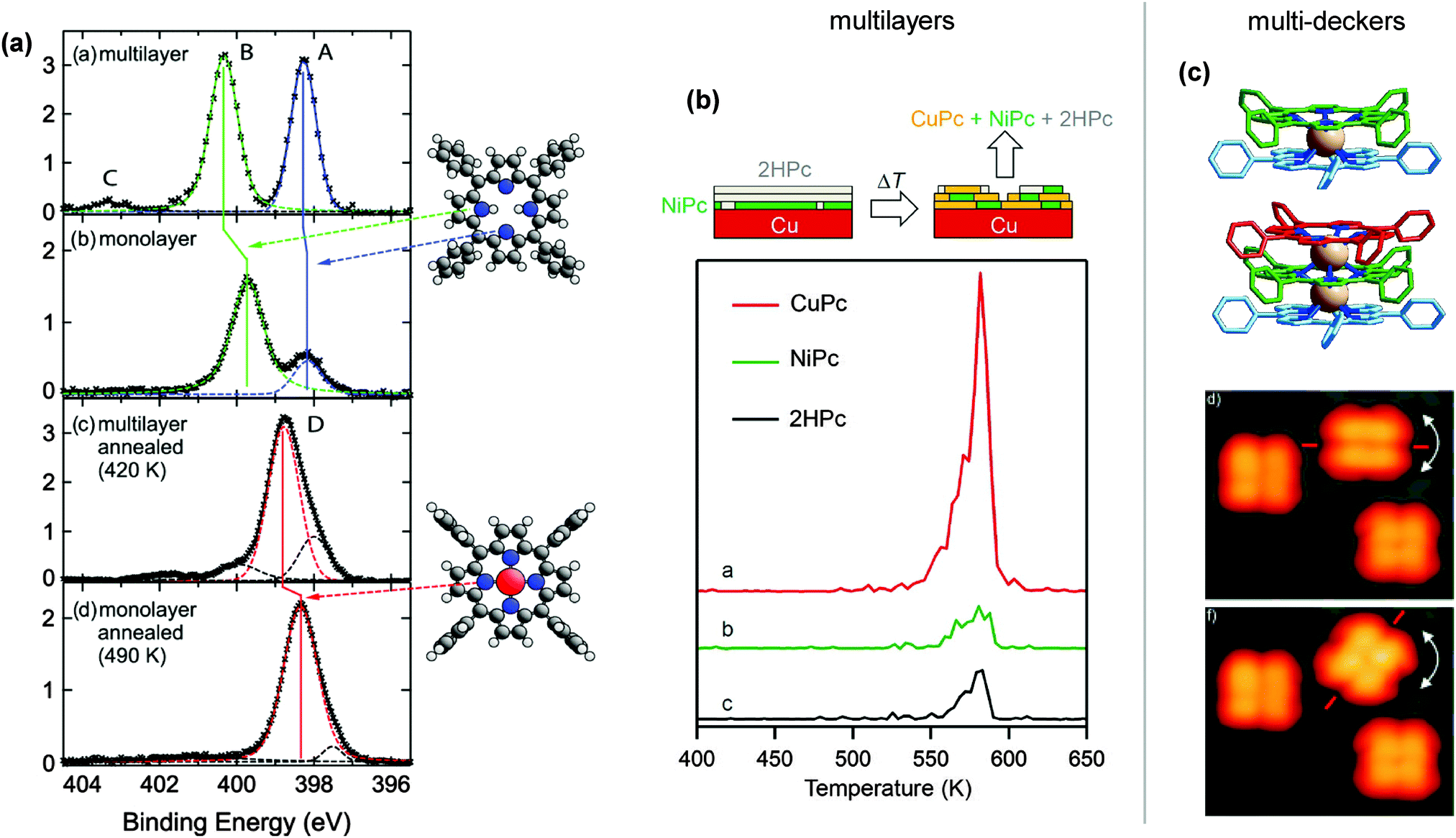 | ||
| Fig. 11 Three-dimensional structures created by on-surface metallation. (a) N 1s XP spectra illustrating the self-metallation of mono- and multilayers of 2H-TPP on a Cu(111) surface as indicated by a single peak in the nitrogen region (reproduced with permission from ref. 60. Copyright 2012, AIP Publishing LLC). (b) To understand the transport mechanisms involved in the self-metallation of 2H-Pc multilayers on Cu(111) TPD measurements were carried out. They showed that a buffer layer of NiPc placed between the 2H-Pc and the copper did not prevent metallation and that molecular exchange took place in the layers (adapted with permission from ref. 49. Copyright 2014 American Chemical Society). (c) Ball-and-stick models of double and triple decker porphyrins (Ce(TPP)2 and Ce2(TPP)3). STM images of the rotation of the top of a single Ce2(TPP)3 complex which is embedded in an island of Ce(TPP)2 molecules (adapted with permission from ref. 94. Copyright Wiley-VCH Verlag GmbH & Co. KGaA). | ||
These results raise questions concerning the transport mechanism involved in the self-metallation of thin films thicker than one monolayer. Three scenarios seem plausible: (a) the metallation only takes place directly at the molecule–substrate interface. Diffusion of the metallated porphyrins within the film then leads to several layers of metalloporphyrins. (b) The molecules are not mobile and metallation occurs by transferring the metal atom from the macrocycle to the macrocycle between the layers. (c) Substrate atoms diffuse into the molecular film,160 similar to the metallation with evaporated metal atoms discussed in the previous paragraph. For the self-metallation of 2H-Pc multilayers on Cu(111),49 Chen et al. could show that the additional evaporation of a NiPc layer between 2H-Pc and the copper substrate did not prevent metallation. Employing TPD they proved that there is an exchange between 2H-Pc, Ni-Pc and Cu-Pc molecules (Fig. 11b).49 The authors concluded that the metallation occurs at the molecule–metal interface, followed by a migration of the formed Cu-Pc to higher layers. In comparison, the diffusion of single copper atoms is deemed energetically unlikely.49
Summary. In vacuo metallation was demonstrated for multilayer structures by both PVD and self-metallation. In particular PVD of Fe and Rh metallated multilayers of various porphyrins on the Ag(111) surface was successfully performed. For 2H-TPP/Fe-TPP, the additional formation of Fe clusters in the multilayer was observed. By self-metallation both porphyrin and Pc multilayers were shown to metallate on Cu(111). Molecular transport across the multilayer is proposed to facilitate the multilayer metallation with substrate atoms. Finally, the formation of rare earth porphyrin multideckers by PVD can be regarded as a further achievement of the metallation of multilayers, extending metal–organic architectures in the third dimension.
4 Comparative assessment – conclusions
This section will compare the protocols described in the previous sections. To this end, we will first discuss the effect the insertion of the metal atom has on the conformation, electronic and self-assembly properties of the tetrapyrroles, followed by a summary of different processes involved in the on-surface metallation. This will allow to rationalise the trends in Fig. 8 and the (seeming) exceptions. Similarities and differences for different protocols are discussed afterwards, as well as their advantages and disadvantages.Effects of metallation
First, we shall briefly review the differences between adsorbed free-base and metallotetrapyrrole molecules, focusing on the conformation, the interaction with the surface, the self-assembly, and the electronic and magnetic properties. These properties are of course interconnected and might strongly depend on various substituents, as discussed in a variety of reviews (e.g., ref. 18–20). Here, we only address some trends focusing on the most studied on-surface metallation systems described in Sections 3.1 and 3.2, 2H-P and 2H-TPP adsorbed on Ag(111) and Cu(111) (cf.Tables 1 and 2).2H-P is planar in the gas phase and also largely retains its geometry when adsorbed onto Ag(111) and Cu(111) surfaces, featuring a maximal deviation from planarity of 15° on the latter substrate.64,112 After metallation of 2H-P to Cu-P the macrocycle retains its flat conformation.64 Such behaviour is not generally the case for the substituted porphyrins. The phenyl substituted porphyrin 2H-TPP typically adsorbs in the so-called “saddle shape” conformation,169 where two pyrrole groups of the macrocycle point towards the substrate and the other two point upwards (cf.Fig. 12c60). The saddle shape conformation is found both on Ag(111)77 and Cu(111),60,77 but the deformation is typically stronger on the latter, allowing an interaction between the nitrogen lone pairs and the substrate (cf. discussion in Section 3.2.2). The metallated TPP species generally exhibit a rather flat macrocycle with steeper upwards tilted phenyl rings,60,170 regardless of whether the free-base compound was the only mildly deformed species on Ag(111) or the strongly deformed one on Cu(111).
 | ||
| Fig. 12 Self-assembly of free-base and metallated tetraphenylporphyrins. (a) 2H-TPP adsorbed onto Ag(111) forms islands (left), this is not the case on Cu(111) (right), where the molecules remain far apart (adapted from ref. 77). (b) After self-metallation to Cu-TPP the molecules assemble into islands (adapted with permission from ref. 63. Copyright 2012 American Chemical Society). (c) This can be explained by a different surface anchoring and modified molecule–molecule interactions induced by the change in conformation: for 2H-TPP/Cu(111) the phenyl rings adsorb with an angle of 20° relative to the surface and the macrocycle is deformed. The latter becomes flat in the metalloporphyrin and the substituents stand more upright, allowing T-type interactions to adjacent Cu-TPP molecules.63,131 (Reproduced with permission from ref. 60. Copyright 2012, AIP Publishing LLC.) | ||
The different conformations have implications both on the electronic structure and the self-assembly properties. 2H-P on Ag(111) and Cu(111) adsorb as isolated species, and for both substrates the LUMO is partially filled upon adsorption (more on copper than on silver).64,112 After self-metallation on Cu(111) this is still the case: the Cu-P molecules remain isolated and partial charge transfer to the LUMO is observed.64
Also for 2H-TPP the charge rearrangement leads to a net electron transfer to the molecule, which is much stronger on Cu(111) than on silver.60,77 On Ag(111) 2H-TPP islands are found, while the molecules form no ordered assembly on Cu(111).77 After self-metallation to Cu-TPP the macrocycle becomes flatter, while the phenyl rings are standing much more upright (Fig. 12c). Through the modified conformation the macrocycle is lifted away from the surface, which is presumably the reason why (in contrast to the flat Cu-P) no charge transfer to the Cu-TPP was observed.60 The modified surface anchoring, as well as the T-type bonds that now can be established between the tilted phenyl rings of adjacent molecules lead to the formation of islands (Fig. 12a and b).63,131 These effects are not exclusively limited to the 2H-TPP/Cu(111) system. A very similar observation was reported for the self-metallation of 2H-TTBPP to Cu-TTBPP on Cu(111) in ref. 171. The metallation induces a substantial change in conformation, where the incorporated Cu atom is lifted away from the surface. Similar to the self-metallation of 2H-TPP to Cu-TPP this leads to a reduced interaction with the substrate and a slightly modified self-assembled structure.
The metallation process
The process that leads to the incorporation of a metal ion in tetrapyrrole compounds is the result of many complex, and potentially competing effects which cannot be separated easily. Only by taking into account the information obtained by different complementary experimental and theoretical techniques, as well as a comparison of the different on-surface metallation protocols described in Sections 3.1, 3.2, and 3.3 it is possible to develop a full picture. A special case are hereby the “films” and islands discussed in Sections 3.1 and 3.2, which constitute an intermediate situation between the metallation with single atoms provided by physical vapour deposition and the self-metallation. The metallation can be separated into three main processes: (1) the actual incorporation of the metal atom into the macrocycle, (2) the availability of the metal centre-to-be, and (3) the interaction of the tetrapyrrole molecule with the surface, other molecules, and the metal atoms (Fig. 13, left side). Depending on the employed on-surface metallation protocol, these three main processes can be broken down into the effects discussed in the following.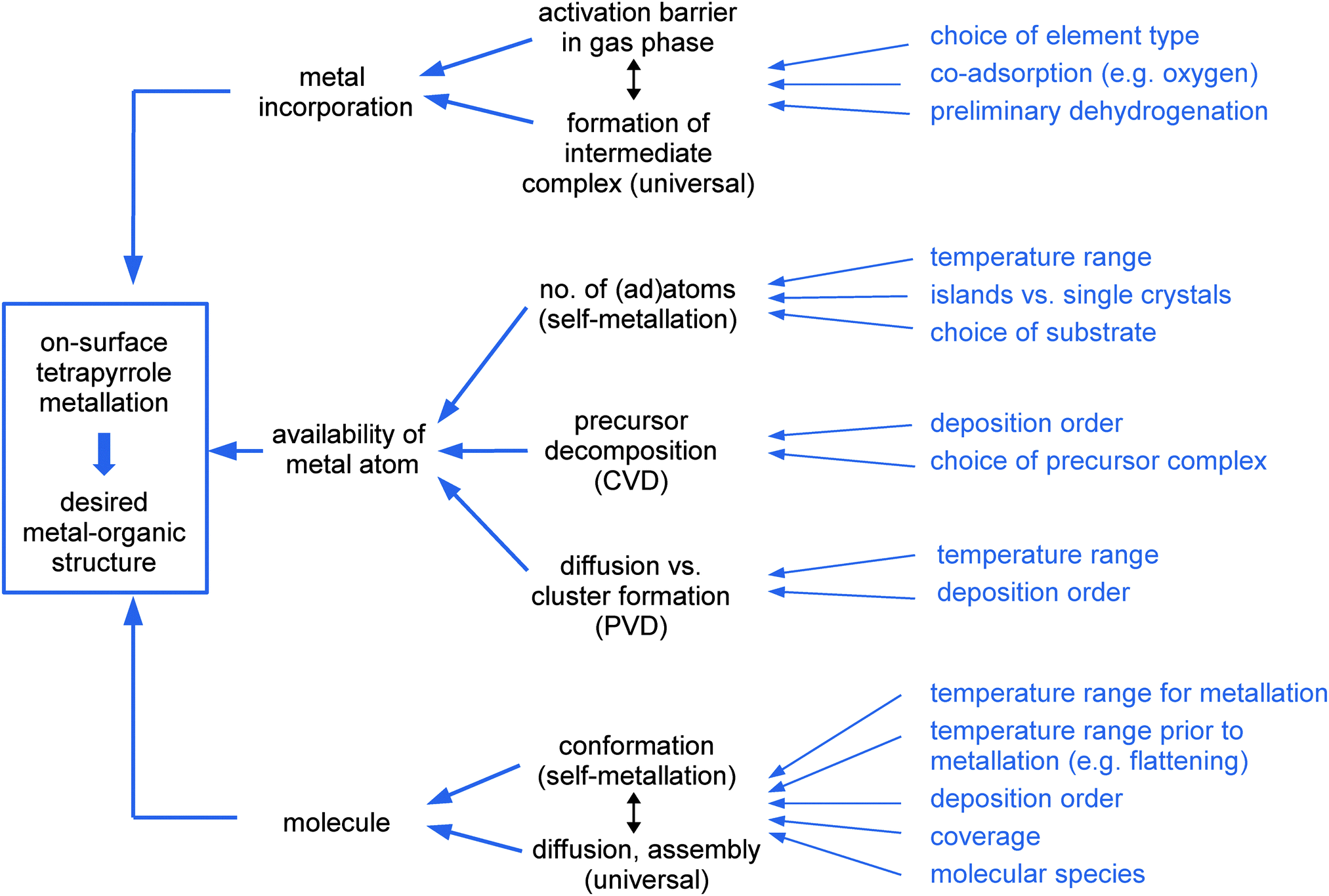 | ||
| Fig. 13 Schematics illustrating the large number of effects which contribute to the metallation process as a whole. In turn, each of these single processes (middle) can be influenced by careful choice of the molecule–metal–substrate system and the experimental conditions (right side, in blue). | ||
The crucial part is of course the actual metal incorporation. Density functional theory of metallation in the gas phase can predict possible intermediate states and the corresponding reaction barriers which determine whether the metallation proceeds at all and at which temperature. The most commonly accepted model for the reaction path via an intermediate state is a macrocycle which is not yet deprotonated, but already bound to the metal atom, followed by a transfer of the hydrogen atoms to the metal, which are split off in the next step. Reaction barriers of this process depend mainly on the elemental identity of the incorporated metal ion, regardless of the employed metallation protocol. Both for the metallation with PVD and the self-metallation, the incorporation of Fe and Ni atoms typically proceeds at room temperature, while for Cu and Zn elevated temperatures are required (Fig. 8a, full markers, and Fig. 8b). For even less reactive elements either no metallation (for Au) is observed or high temperatures are required (573 K for the self-metallation of Ag). This is in agreement with the predictions from gas-phase DFT,69i.e., the incorporation of the metal atom constitutes the rate-limiting step for these systems, independent of the employed method. At first glance, this trend seems to be contradicted by several systems presented in Fig. 8: for example, for PVD metallation with pre-deposited Ni and Fe elevated temperatures were required (Fig. 8a, empty markers, systems 8, 19, and 21), and no self-metallation could be observed for 2H-TPP on Ni/Cu(001) or Ni/Cu(111) (Table 2, systems b and c). These examples give a first indication on the importance of deposition order and will be discussed below. In contrast, sometimes the necessary temperature is lower than what could be expected from the calculated gas phase barriers. This is the case for the self-metallation of 2H-Pc with Ag on Ag(110)87 which proceeds already at room temperature, as well as that for 2H-TPP with Cu on O/Cu(001).114 For the aforementioned metallation with Ni on Ni/Cu(001) the co-adsorption of oxygen lowers the reaction barrier such that room temperature metallation can be observed. In all these cases the authors ascribed the changes to the formation of modified intermediate complexes with lower activation barriers, through dehydrogenation of the 2H-Pc in the first case, and a participation of the co-adsorbed oxygen in the latter cases. For the CVD metallation with Zn, which also takes place already at room temperature we suggest a similar mechanism. For applications the metal incorporation process can thus be tuned by choice of the suitable metal type, by an exploration of possible co-adsorbates, and by a modification of the molecules' macrocycle prior to metallation.
As could be seen from the examples described in the previous paragraph, the incorporation of the metal atom inside the macrocycle is not the only possible rate-limiting step in the metallation process. The intermediate complex is formed as soon as metal is available and can interact with the tetrapyrrole species. This has different implications for the various metallation protocols. For the sublimation of metal atoms by PVD or CVD the deposited amount can be steered by the evaporation parameters (i.e., time and rate), e.g., to achieve only partially metallated layers. However, even when enough metal is provided, in some cases full metallation could only be achieved at elevated temperatures or not at all. Examples are the aforementioned metallation experiments with Ni and Fe, which typically readily occur at room temperature. If however, the deposition order is reversed (i.e., the metal is deposited before the free-base molecules) temperatures of up to 550 K are required for the reaction (Fig. 8a, cf. empty vs. full markers). Multilayers of 2H-TPP with post-deposited metal cannot be fully metallated in one step. Both kinds of effects can be attributed to the formation of metal clusters, reducing the availability of single atoms. While there might be a nominal excess of metal on the surface, the atoms cannot reach the macrocycle to be incorporated. However, it cannot be generally concluded that a pre-deposition of the molecules is in all cases advantageous. For the metallation of 2H-TPP with Os on Ag(111) (Fig. 8c) a pre-deposition of the free-base species does not lead to a high yield of metallated porphyrins: the decomposition temperature for the Os3(CO)12 precursor complex lies above the temperature that would induce a polymerisation of the molecules. If however, the precursor is deposited first and annealed to yield Os clusters on Ag(111), followed by the deposition of the porphyrins and an annealing step to 483 K, a high metallation yield of desired Os-porphyrin species could be obtained. For the self-metallation the situation is more complex, as the origin of the incorporated metal atoms is not yet unanimously clear. While the temperature certainly plays an important role it is not evident whether a higher temperature allows the direct extraction of an atom from the surface, or only leads to an increased number of adatoms detached from step edges. Most of the studies published so far seem to favour an explanation including adatoms, which is supported by accompanying density functional theory calculations. If adatoms are the crucial factors, this might explain why 2H-TPP readily self-metallates at room temperature on a Ni(111) single crystal, but not on a nickel film deposited on Cu(111) (Table 2, systems 7 and b).
The last contributing main factor is the availability of the tetrapyrrole species in a suitable configuration. Tetrapyrrole species and their conformations hardly affect the metallation reaction using PVD or CVD, however they clearly influence their ability to self-metallate (Fig. 8b). A possible explanation is that different conformations cause the formation of different intermediate complexes, which (as detailed above) may have substantial influence on the activation barriers involved in the reaction. This might also explain why the coverage (and therewith the associated changes in molecular arrangement) was found to play a crucial role for the rate of the self-metallation of 2H-TPP on Cu(111).
Advantages and disadvantages of different protocols
In addition to different possibilities to tune the metallation process discussed in the previous paragraphs, the different on-surface metallation protocols all have their own intrinsic limitations and advantages. As can be seen from Fig. 8 the metallation through PVD offers the largest range of possible metal centres and the additional advantage of being able to define the deposition order. It is thus possible to obtain metal–organic structures with desired properties, by selecting metals with for example catalytic (Ti), magnetic (Co, Fe, Cr), or non-magnetic (Cu) properties, as well as rare earths with a large radius (Ce, Gd) to create sandwich complexes. However, there might be limitations in the choice of metal due to high sublimation temperatures and reactivities, which might prevent easy preparation of all targeted structures. Another problem is the presence of excess metal after the reaction if the metal deposition is not tuned carefully.Both these disadvantages can be overcome by using metallation through CVD of precursor complexes. The ligands in the complexes protect the chemical environment of reactive metals; in the examples presented here the molecules can typically be dosed easily at ambient temperatures. For the metallation with Ru–dodecacarbonyl complexes a self-limiting protocol has been found, i.e., only reacted species without undesired side-products are left on the surface after the reaction. The metallation with diethylzinc even allows to create films of defined thickness, which might play an important role for the fabrication of devices such as organic transistors. The application of metallation through CVD is intrinsically limited by the availability of suitable precursor molecules and currently only few examples are known. Another drawback is the possibly high thermal stability of the precursor complexes, which (while advantageous for the sublimation) may require high annealing temperatures for the scission of the ligands.
Using an STM tip to directly metallate tetrapyrrole macrocycles can be an alternative route to create systems which could not be achieved otherwise. However, as only single, selected molecules can be metallated in this way, the approach is not suitable for the metallation of molecular films, thus excluding technological applications.
The method with the lowest experimental complexity is certainly the self-metallation which does not require any deposition of reactants apart from the free-base tetrapyrrole species. Comparable to the metallation through CVD of precursor complexes, no unwanted side products are left on the substrate after the reaction. Self-metallation cannot be used to create all structures, as the central metal atom will be of the same element type as the substrate, and the range of suitable substrates is small. However, as discussed above, first results showing that co-adsorption and dehydrogenation enable self-metallation on hitherto unreactive substrates might play an important role for further exploration of suitable systems.
Future trends
We anticipate a considerable effect of in vacuo metallation procedures on the engineering of advanced metallo-supramolecular architectures and hybrid systems. Specifically, the fabrication of sandwich compounds with prospects for applications is not fully exploited to date, but bears great promise as the sublimation of intact ex situ synthesized multi-deckers and molecular magnets is usually prevented by their limited thermal stability. The sequential metallation of pre-fabricated multi-component free-base tetrapyrrole assemblies can provide intricate arrays of coordinatively unsaturated metal centres at interfaces. Importantly, supports are not restricted to low-index metal single crystal facets, but can include vicinal surfaces, metal films, nano-structured templates or two-dimensional materials. The possibility to utilise most elements throughout the periodic table will allow one to engineer distinct electronic and magnetic properties of the resulting architectures. In this respect, constraints introduced by reduced macrocycle dimensions in tailored species such as corroles might be beneficial to stabilize specific oxidation states of the metal centres at interfaces. Thus, interfacial in vacuo metallation provides prospects for functionalities, e.g., in heterogeneous catalysis, molecular spintronics, single molecule rotors/machinery, quantum information or light-harvesting and photonic devices.Acknowledgements
We are grateful for support from the European Research Council Advanced Grant MolArt (no. 247299), the Munich Centre for Advanced Photonics (MAP) and the TUM Institute for Advanced Study funded by the German Research Foundation (DFG) via the German Excellence Initiative. W. A. acknowledges funding by the DFG via a Heisenberg professorship and by the European Research Council Consolidator Grant NanoSurfs (no. 615233). K. D. acknowledges support from the ‘EPFL Fellows’ fellowship programme co-funded by Marie Curie, FP7 Grant agreement no. 291771. We thank all team members and project partners co-authoring cited joint publications.Notes and references
- P. Pelletier and J. Caventou, Ann. Chim. Phys., 1818, 9, 194–196 Search PubMed.
- F. Hoppe-Seyler, Virchows Arch. Pathol. Anat. Physiol., 1864, 29, 597–600 CrossRef.
- F. Hoppe-Seyler, Z. Anal. Chem., 1864, 3, 432–439 CrossRef.
- F. Hoppe-Seyler, Virchows Arch. Pathol. Anat. Physiol., 1864, 29, 233–235 CrossRef.
- C. A. McMunn, J. Physiol., 1884, 5, XXIV–XXVI Search PubMed.
- R. Willstätter and A. Stoll, Untersuchungen über Chlorophyll, Springer, 1913 Search PubMed.
- H. Fischer and H. Orth, Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1934 Search PubMed.
- H. Fischer and H. Orth, Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1937 Search PubMed.
- H. Fischer and A. Stern, Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1940 Search PubMed.
- A. Braun and J. Tcherniac, Ber. Dtsch. Chem. Ges., 1907, 40, 2709–2714 CrossRef CAS.
- H. de Diesbach and E. von der Weid, Helv. Chim. Acta, 1927, 10, 886–888 CrossRef CAS.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710–713 CrossRef CAS PubMed.
- W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- S. Mohnani and D. Bonifazi, Coord. Chem. Rev., 2010, 254, 2342–2362 CrossRef CAS.
- J. Otsuki, Coord. Chem. Rev., 2010, 254, 2311–2341 CrossRef CAS.
- S. Yoshimoto and K. Itaya, J. Porphyrins Phthalocyanines, 2007, 11, 313–333 CrossRef CAS.
- J. Gottfried and H. Marbach, Z. Phys. Chem., 2009, 223, 53–74 CrossRef CAS.
- Y. Wang, K. Wu, J. Kröger and R. Berndt, AIP Adv., 2012, 2, 041402 CrossRef.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS PubMed.
- E. B. Fleischer and J. Wang, J. Am. Chem. Soc., 1960, 82, 3498–3502 CrossRef CAS.
- B. Berezin and O. Koirman, Russ. Chem. Rev., 1973, 42, 922–938 CrossRef.
- S. Baum and R. Plane, J. Am. Chem. Soc., 1966, 88, 910–913 CrossRef CAS.
- A. Adler, F. Longo and W. Shergalis, J. Am. Chem. Soc., 1964, 86, 3145–3149 CrossRef CAS.
- D. K. Lavallee, Coord. Chem. Rev., 1985, 61, 55–96 CrossRef CAS.
- Z.-C. Sun, Y.-B. She, Y. Zhou, X.-F. Song and K. Li, Molecules, 2011, 16, 2960–2970 CrossRef CAS PubMed.
- E. B. Fleischer, E. I. Choi, P. Hambright and A. Stone, Inorg. Chem., 1964, 3, 1284–1287 CrossRef CAS.
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, J. Inorg. Nucl. Chem., 1970, 32, 2443–2445 CrossRef CAS.
- R. P. Evstigneeva, Pure Appl. Chem., 1981, 53, 1129–1140 CrossRef CAS.
- S. Funahashi, Y. Inada and M. Inamo, Anal. Sci., 2001, 17, 917–927 CrossRef CAS PubMed.
- Y. Shen and U. Ryde, Chem. – Eur. J., 2005, 11, 1549–1564 CrossRef CAS PubMed.
- S. S. Cady and T. J. Pinnavaia, Inorg. Chem., 1978, 17, 1501–1507 CrossRef CAS.
- G. W. Hodgson, Nature, 1967, 216, 29–32 CrossRef CAS PubMed.
- G. W. Hodgson, B. Hitchon, K. Taguchi, B. L. Baker and E. Peake, Geochim. Cosmochim. Acta, 1968, 32, 737–772 CrossRef CAS.
- T. M. Cotton, S. G. Schultz and R. P. Van Duyne, J. Am. Chem. Soc., 1982, 104, 6528–6532 CrossRef CAS.
- A. H. R. Al-Obaidi, S. J. Rigby, S. E. J. Bell and J. J. McGarvey, J. Phys. Chem., 1992, 96, 10960–10963 CrossRef CAS.
- M. Prochazka, J. Stepanek, P.-Y. Turpin and J. Bok, J. Phys. Chem. B, 2002, 106, 1543–1549 CrossRef CAS.
- S. Abdo, M. I. Cruz and J. J. Fripiat, Clays Clay Miner., 1980, 28, 125–129 CAS.
- L. Zhang, A. Patil, L. Li, A. Schierhorn, S. Mann, U. Gösele and M. Knez, Angew. Chem., 2009, 121, 5082–5085 CrossRef.
- R. González-Moreno, P. L. Cook, I. Zegkinoglou, X. Liu, P. S. Johnson, W. Yang, R. E. Ruther, R. J. Hamers, R. Tena-Zaera, F. J. Himpsel, J. E. Ortega and C. Rogero, J. Phys. Chem. C, 2011, 115, 18195–18201 Search PubMed.
- T. Mashiko and D. Dolphin, Porphyrins, Hydroporphyrins, Azaporphyrins, Phthalocyanins, Corroles, Corrins and Related Macrocycles, Pergamon Press, 1987, vol. 2 Search PubMed.
- W. Auwärter, A. Weber-Bargioni, S. Brink, A. Riemann, A. Schiffrin, M. Ruben and J. V. Barth, ChemPhysChem, 2007, 8, 250–254 CrossRef PubMed.
- J. M. Gottfried, K. Flechtner, A. Kretschmann, T. Lukasczyk and H.-P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644–5645 CrossRef CAS PubMed.
- M. Chen, M. Röckert, J. Xiao, H.-J. Drescher, H.-P. Steinrück, O. Lytken and J. M. Gottfried, J. Phys. Chem. C, 2014, 118, 8501–8507 CAS.
- C. J. Walker and R. D. Willows, Biochem. J., 1997, 327, 321–333 CrossRef CAS PubMed.
- G. De Luca, A. Romeo, L. M. Scolaro, G. Ricciardi and A. Rosa, Inorg. Chem., 2009, 48, 8493–8507 CrossRef CAS PubMed.
- R. Khosropour and P. Hambright, J. Chem. Soc., Chem. Commun., 1972, 13–14 RSC.
- R. Franco, S. Al-Karadaghi and G. C. Ferreira, J. Porphyrins Phthalocyanines, 2011, 15, 357–363 CrossRef CAS PubMed.
- M. M. Conn, J. R. Prudent and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 7012–7013 CrossRef CAS.
- J. Schneider, F. Kollhoff, J. Bernardi, A. Kaftan, J. Libuda, T. Berger, M. Laurin and O. Diwald, ACS Appl. Mater. Interfaces, 2015, 7, 22962–22969 CAS.
- M. Nardi, R. Verucchi, R. Tubino and S. Iannotta, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125404 CrossRef.
- A. C. Papageorgiou, S. Fischer, S. C. Oh, O. Sağlam, J. Reichert, A. Wiengarten, K. Seufert, S. Vijayaraghavan, D. Écija, W. Auwärter, F. Allegretti, R. G. Acres, K. C. Prince, K. Diller, F. Klappenberger and J. V. Barth, ACS Nano, 2013, 7, 4520–4526 CrossRef CAS PubMed.
- J. P. Macquet, M. M. Millard and T. Theophanides, J. Am. Chem. Soc., 1978, 100, 4741–4746 CrossRef CAS.
- G. Di Santo, C. Castellarin-Cudia, M. Fanetti, B. Taleatu, P. Borghetti, L. Sangaletti, L. Floreano, E. Magnano, F. Bondino and A. Goldoni, J. Phys. Chem. C, 2011, 115, 4155–4162 CAS.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- Y. Niwa, H. Kobayashi and T. Tsuchiya, J. Chem. Phys., 1974, 60, 799–807 CrossRef CAS.
- P. Borghetti, G. D. Santo, C. Castellarin-Cudia, M. Fanetti, L. Sangaletti, E. Magnano, F. Bondino and A. Goldoni, J. Chem. Phys., 2013, 138, 144702 CrossRef PubMed.
- J. Xiao, S. Ditze, M. Chen, F. Buchner, M. Stark, M. Drost, H.-P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2012, 116, 12275–12282 CAS.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, A. Wiengarten, S. Joshi, K. Seufert, D. Écija, W. Auwärter and J. V. Barth, J. Chem. Phys., 2013, 138, 154710 CrossRef CAS PubMed.
- W. Krenner, D. Kühne, F. Klappenberger and J. V. Barth, Sci. Rep., 2013, 3, 1454 Search PubMed.
- A. Weber-Bargioni, J. Reichert, A. P. Seitsonen, W. Auwärter, A. Schiffrin and J. V. Barth, J. Phys. Chem. C, 2008, 112, 3453–3455 CAS.
- The checkerboard structure was observed for 2H-TPP/Cu(111), where at coverages between 0.02 and 0.03 2H-TPP molecules per substrate atom half of the molecules adsorb in direct contact with the surface. The other half are elevated and bridge the gaps between the molecules of the first layer.
- M. Röckert, M. Franke, Q. Tariq, D. Lungerich, N. Jux, M. Stark, A. Kaftan, S. Ditze, H. Marbach, M. Laurin, J. Libuda, H.-P. Steinrück and O. Lytken, J. Phys. Chem. C, 2014, 118, 26729–26736 Search PubMed.
- T. E. Shubina, H. Marbach, K. Flechtner, A. Kretschmann, N. Jux, F. Buchner, H. Steinrück, T. Clark and J. M. Gottfried, J. Am. Chem. Soc., 2007, 129, 9476–9483 CrossRef CAS PubMed.
- N. Schmidt, R. Fink and W. Hieringer, J. Chem. Phys., 2010, 133, 054703 CrossRef PubMed.
- L. Zotti, G. Teobaldi, W. Hofer, W. Auwärter, A. Weber-Bargioni and J. Barth, Surf. Sci., 2007, 601, 2409–2414 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Klappenberger, J. Reichert, A. Weber-Bargioni, A. Verdini, D. Cvetko, M. Dell'Angela, L. Floreano, A. Cossaro, G. Bavdek, A. Morgante, A. P. Seitsonen and J. V. Barth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245403 CrossRef.
- K. Boukari, P. Sonnet and E. Duverger, ChemPhysChem, 2012, 13, 3945–3951 CrossRef CAS PubMed.
- G. Di Santo, S. Blankenburg, C. Castellarin-Cudia, M. Fanetti, P. Borghetti, L. Sangaletti, L. Floreano, A. Verdini, E. Magnano, F. Bondino, C. A. Pignedoli, M.-T. Nguyen, R. Gaspari, D. Passerone and A. Goldoni, Chem. – Eur. J., 2011, 17, 14354–14359 CrossRef CAS PubMed.
- S. Müllegger, W. Schöfberger, M. Rashidi, T. Lengauer, F. Klappenberger, K. Diller, K. Kara, J. V. Barth, E. Rauls, W. G. Schmidt and R. Koch, ACS Nano, 2011, 5, 6480–6486 CrossRef PubMed.
- A. Goldoni, C. A. Pignedoli, G. Di Santo, C. Castellarin-Cudia, E. Magnano, F. Bondino, A. Verdini and D. Passerone, ACS Nano, 2012, 6, 10800–10807 CrossRef CAS PubMed.
- G. Rojas, S. Simpson, X. Chen, D. A. Kunkel, J. Nitz, J. Xiao, P. A. Dowben, E. Zurek and A. Enders, Phys. Chem. Chem. Phys., 2012, 14, 4971–4976 RSC.
- A. Sperl, J. Kröger and R. Berndt, Angew. Chem., Int. Ed., 2011, 123, 5406–5409 CrossRef.
- F. Hanke, S. Haq, R. Raval and M. Persson, ACS Nano, 2011, 5, 9093–9103 CrossRef CAS PubMed.
- M. S. Dyer, A. Robin, S. Haq, R. Raval, M. Persson and J. Klimeš, ACS Nano, 2011, 5, 1831–1838 CrossRef CAS PubMed.
- K. Leung, S. B. Rempe, P. A. Schultz, E. M. Sproviero, V. S. Batista, M. E. Chandross and C. J. Medforth, J. Am. Chem. Soc., 2006, 128, 3659–3668 CrossRef CAS PubMed.
- M. Fanetti, A. Calzolari, P. Vilmercati, C. Castellarin-Cudia, P. Borghetti, G. Di Santo, L. Floreano, A. Verdini, A. Cossaro, I. Vobornik, E. Annese, F. Bondino, S. Fabris and A. Goldoni, J. Phys. Chem. C, 2011, 115, 11560–11568 CAS.
- J. M. García-Lastra, P. L. Cook, F. J. Himpsel and A. Rubio, J. Chem. Phys., 2010, 133, 151103 CrossRef PubMed.
- I. Reid, Y. Zhang, A. Demasi, A. Blueser, L. Piper, J. E. Downes, A. Matsuura, G. Hughes and K. E. Smith, Appl. Surf. Sci., 2009, 256, 720–725 CrossRef CAS.
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H.-P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 CrossRef CAS PubMed.
- S. Ditze, M. Stark, M. Drost, F. Buchner, H.-P. Steinrück and H. Marbach, Angew. Chem., Int. Ed., 2012, 51, 10898–10901 CrossRef CAS PubMed.
- L. Smykalla, P. Shukrynau, D. R. T. Zahn and M. Hietschold, J. Phys. Chem. C, 2015, 119, 17228–17234 CAS.
- F. Buchner, K.-G. Warnick, T. Wölfle, A. Görling, H.-P. Steinrück, W. Hieringer and H. Marbach, J. Phys. Chem. C, 2009, 113, 16450–16457 CAS.
- J. I. Urgel, M. Schwarz, M. Garnica, D. Stassen, D. Bonifazi, D. Écija, J. V. Barth and W. Auwärter, J. Am. Chem. Soc., 2015, 137, 2420–2423 CrossRef CAS PubMed.
- T. Lin, G. Kuang, W. Wang and N. Lin, ACS Nano, 2014, 8, 8310–8316 CrossRef CAS PubMed.
- K. Schouteden, T. Ivanova, Z. Li, V. Iancu, E. Janssens and C. Van Haesendonck, J. Phys. Chem. C, 2015, 6, 1048–1052 CAS.
- F. Buchner, K. Flechtner, Y. Bai, E. Zillner, I. Kellner, H.-P. Steinrück, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 15458–15465 CAS.
- A. Kretschmann, M.-M. Walz, K. Flechtner, H.-P. Steinrück and J. M. Gottfried, Chem. Commun., 2007, 568–570 RSC.
- D. Écija, W. Auwärter, S. Vijayaraghavan, K. Seufert, F. Bischoff, K. Tashiro and J. V. Barth, Angew. Chem., Int. Ed., 2011, 50, 3872–3877 CrossRef PubMed.
- M. Panighel, G. D. Santo, M. Caputo, C. Lal, B. Taleatu and A. Goldoni, J. Phys. Chem. Solids, 2013, 470, 012012 Search PubMed.
- D. A. Duncan, P. S. Deimel, A. Wiengarten, R. Han, R. G. Acres, W. Auwärter, P. Feulner, A. C. Papageorgiou, F. Allegretti and J. V. Barth, Chem. Commun., 2015, 51, 9483–9486 RSC.
- J. Rombach, S. Vijayaraghavan, W. Auwärter, D. Écija and J. V. Barth, in preparation.
- M. Röckert, M. Franke, Q. Tariq, H.-P. Steinrück and O. Lytken, Chem. Phys. Lett., 2015, 635, 60–62 CrossRef.
- M. Chen, X. Feng, L. Zhang, H. Ju, Q. Xu, J. Zhu, J. M. Gottfried, K. Ibrahim, H. Qian and J. Wang, J. Phys. Chem. C, 2010, 114, 9908–9916 CAS.
- C. Wang, Q. Fan, S. Hu, H. Ju, X. Feng, Y. Han, H. Pan, J. Zhu and J. M. Gottfried, Chem. Commun., 2014, 50, 8291–8294 RSC.
- S. Ditze, M. Röckert, F. Buchner, E. Zillner, M. Stark, H.-P. Steinrück and H. Marbach, Nanotechnology, 2013, 24, 115305 CrossRef PubMed.
- Y. Bai, F. Buchner, M. T. Wendahl, I. Kellner, A. Bayer, H.-P. Steinrück, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 6087–6092 CAS.
- K. Eguchi, T. Nakagawa, Y. Takagi and T. Yokoyama, J. Phys. Chem. C, 2015, 119, 9805–9815 CAS.
- D. Écija, M. Trelka, C. Urban, P. d. Mendoza, E. Mateo-Martí, C. Rogero, J. A. Martín-Gago, A. M. Echavarren, R. Otero, J. M. Gallego and R. Miranda, J. Phys. Chem. C, 2008, 112, 8988–8994 Search PubMed.
- J. I. Urgel, D. Écija, W. Auwärter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163–6167 CrossRef CAS PubMed.
- F. Xiang, C. Li, Z. Wang, X. Liu, D. Jiang, X. Leng, J. Ling and L. Wang, Surf. Sci., 2015, 633, 46–52 CrossRef CAS.
- C.-L. Song, Y.-L. Wang, Y.-X. Ning, J.-F. Jia, X. Chen, B. Sun, P. Zhang, Q.-K. Xue and X. Ma, J. Am. Chem. Soc., 2010, 132, 1456–1457 CrossRef CAS PubMed.
- D. Chandra, M. Garner and K. Lau, J. Phase Equilib. Diffus., 1999, 20, 565–572 CrossRef CAS.
- K. Diller, PhD thesis, TU München, 2013.
- O. Sağlam, G. Yetik, J. Reichert, J. V. Barth and A. C. Papageorgiou, Surf. Sci., 2015 DOI:10.1016/j.susc.2015.08.020 , accepted.
- W. Auwärter, A. Weber-Bargioni, A. Riemann, A. Schiffrin, O. Gröning, R. Fasel and J. V. Barth, J. Chem. Phys., 2006, 124, 194708 CrossRef PubMed.
- F. Bischoff, K. Seufert, W. Auwärter, S. Joshi, S. Vijaraghavan, D. Écija, K. Diller, A. C. Papageorgiou, S. Fischer, F. Allegretti, D. A. Duncan, F. Klappenberger, F. Blobner, R. Han and J. V. Barth, ACS Nano, 2013, 7, 3139–3149 CrossRef CAS PubMed.
- E. Rauls, W. Schmidt, T. Pertram and K. Wandelt, Surf. Sci., 2012, 606, 1120–1125 CrossRef CAS.
- J. Nowakowski, C. Wäckerlin, J. Girovsky, D. Siewert, T. A. Jung and N. Ballav, Chem. Commun., 2013, 49, 2347–2349 RSC.
- Y. Bai, F. Buchner, I. Kellner, M. Schmid, F. Vollnhals, H.-P. Steinrück, H. Marbach and J. M. Gottfried, New J. Phys., 2009, 11, 125004 CrossRef.
- Y. Bai, M. Sekita, M. Schmid, T. Bischof, H.-P. Steinrück and J. M. Gottfried, Phys. Chem. Chem. Phys., 2010, 12, 4336–4344 RSC.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H.-P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CAS.
- A. Gourdon, Angew. Chem., Int. Ed., 2008, 47, 6950–6953 CrossRef CAS PubMed.
- M. Lackinger and W. M. Heckl, J. Phys. D: Appl. Phys., 2011, 44, 464011 CrossRef.
- J. Björk and F. Hanke, Chem. – Eur. J., 2014, 20, 928–934 CrossRef PubMed.
- F. Klappenberger, Y.-Q. Zhang, J. Björk, S. Klyatskaya, M. Ruben and J. V. Barth, Acc. Chem. Res., 2015, 48, 2140–2150 CrossRef CAS PubMed.
- A. Wiengarten, J. A. Lloyd, K. Seufert, J. Reichert, W. Auwärter, R. Han, D. A. Duncan, F. Allegretti, S. Fischer, S. C. Oh, O. Sağlam, L. Jiang, S. Vijayaraghavan, D. Écija, A. C. Papageorgiou and J. V. Barth, Chem. – Eur. J., 2015, 21, 12285–12290 CrossRef CAS PubMed.
- G. Di Santo, C. Sfiligoj, C. Castellarin-Cudia, A. Verdini, A. Cossaro, A. Morgante, L. Floreano and A. Goldoni, Chem. – Eur. J., 2012, 18, 12619–12623 CrossRef CAS PubMed.
- K. Flechtner, PhD thesis, Friedrich-Alexander Universität Erlangen-Nürnberg, 2007.
- Y. Bai, PhD thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2010.
- M. Schmid, A. Kaftan, H.-P. Steinrück and J. M. Gottfried, Surf. Sci., 2012, 606, 945–949 CrossRef CAS.
- C. M. Doyle, S. A. Krasnikov, N. N. Sergeeva, A. B. Preobrajenski, N. A. Vinogradov, Y. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2011, 47, 12134–12136 RSC.
- R. González-Moreno, C. Sánchez-Sánchez, M. Trelka, R. Otero, A. Cossaro, A. Verdini, L. Floreano, M. Ruiz-Bermejo, A. García-Lekue, J. Martín-Gago and C. Rogero, J. Phys. Chem. C, 2011, 115, 6849–6854 Search PubMed.
- R. González-Moreno, A. Garcia-Lekue, A. Arnau, M. Trelka, J. M. Gallego, R. Otero, A. Verdini, C. Sánchez-Sánchez, P. L. de Andrés, J. A. Martín-Gago and C. Rogero, J. Phys. Chem. C, 2013, 117, 7661–7668 Search PubMed.
- M. Röckert, S. Ditze, M. Stark, J. Xiao, H.-P. Steinrück, H. Marbach and O. Lytken, J. Phys. Chem. C, 2014, 118, 1661–1667 Search PubMed.
- M. Röckert, M. Franke, Q. Tariq, S. Ditze, M. Stark, P. Uffinger, D. Wechsler, U. Singh, J. Xiao, H. Marbach, H.-P. Steinrück and O. Lytken, Chem. – Eur. J., 2014, 20, 8948–8953 CrossRef.
- C. Bürker, A. Franco-Canellas, K. Broch, T.-L. Lee, A. Gerlach and F. Schreiber, J. Phys. Chem. C, 2014, 118, 13659–13666 Search PubMed.
- A. Verdini, A. Goldoni and D. Passerone, private communication.
- A. Wiengarten, K. Seufert, W. Auwärter, D. Écija, K. Diller, F. Allegretti, F. Bischoff, S. Fischer, D. A. Duncan, A. C. Papageorgiou, F. Klappenberger, R. G. Acres, T. H. Ngo and J. V. Barth, J. Am. Chem. Soc., 2014, 136, 9346–9354 CrossRef CAS PubMed.
- S. Haq, F. Hanke, M. S. Dyer, M. Persson, P. Iavicoli, D. B. Amabilino and R. Raval, J. Am. Chem. Soc., 2011, 133, 12031–12039 CrossRef CAS PubMed.
- M. Abadía, R. González-Moreno, A. Sarasola, G. Otero-Irurueta, A. Verdini, L. Floreano, A. Garcia-Lekue and C. Rogero, J. Phys. Chem. C, 2014, 118, 29704–29712 Search PubMed.
- F. Klappenberger, A. Weber-Bargioni, W. Auwärter, M. Marschall, A. Schiffrin and J. V. Barth, J. Chem. Phys., 2008, 129, 214702 CrossRef CAS PubMed.
- M. In't Veld, P. Iavicoli, S. Haq, D. B. Amabilino and R. Raval, Chem. Commun., 2008, 1536–1538 RSC.
- C. M. Doyle, J. P. Cunniffe, S. A. Krasnikov, A. B. Preobrajenski, Z. Li, N. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2014, 50, 3447–3449 RSC.
- P. Hambright and P. B. Chock, J. Am. Chem. Soc., 1974, 96, 3123–3131 CrossRef CAS PubMed.
- I. Kröger, P. Bayersdorfer, B. Stadtmüller, C. Kleimann, G. Mercurio, F. Reinert and C. Kumpf, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195412 CrossRef.
- J. Mielke, F. Hanke, M. V. Peters, S. Hecht, M. Persson and L. Grill, J. Am. Chem. Soc., 2015, 137, 1844–1849 CrossRef CAS PubMed.
- G. Rojas, X. Chen, C. Bravo, J.-H. Kim, J.-S. Kim, J. Xiao, P. A. Dowben, Y. Gao, X. C. Zeng, W. Choe and A. Enders, J. Phys. Chem. C, 2010, 114, 9408–9415 CAS.
- N. Katsonis, J. Vicario, T. Kudernac, J. Visser, M. M. Pollard and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 15537–15541 CrossRef CAS PubMed.
- T. Yokoyama, S. Yokoyama, T. Kamikado and S. Mashiko, J. Chem. Phys., 2001, 115, 3814–3818 CrossRef CAS.
- M. Stark, S. Ditze, M. Drost, F. Buchner, H.-P. Steinrück and H. Marbach, Langmuir, 2013, 29, 4104–4110 CrossRef CAS PubMed.
- F. Buchner, J. Xiao, E. Zillner, M. Chen, M. Röckert, S. Ditze, M. Stark, H.-P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2011, 115, 24172–24177 CAS.
- S. J. Kim, S. Myung, W. Song, B. K. Min, S.-J. Hong, M. Chung, H. Kim, K.-J. Kong, J. Lim, T. M. Chung and K.-S. An, Chem. Mater., 2015, 27, 4546–4551 CrossRef CAS.
- H. Häkkinen and M. Manninen, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 1725–1742 CrossRef.
- G. C. Kallinteris, G. A. Evangelakis and N. I. Papanicolaou, Surf. Sci., 1996, 369, 185–198 CrossRef CAS.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, D. A. Duncan, R. J. Maurer, J. A. Lloyd, S. C. Oh, K. Reuter and J. V. Barth, J. Chem. Phys., 2014, 141, 144703 CrossRef PubMed.
- M. Giesen, Surf. Sci., 1999, 442, 543–549 CrossRef CAS.
- M. Giesen and G. Schulze Icking-Konert, Surf. Sci., 1998, 412–413, 645–656 CrossRef.
- J. Kolaczkiewicz, Surf. Sci., 1987, 183, 251–262 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Bischoff, D. Écija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41–46 CrossRef PubMed.
- K. Morgenstern, N. Lorente and K.-H. Rieder, Phys. Status Solidi B, 2013, 250, 1671–1751 CrossRef CAS.
- Q. Sun and W. Xu, ChemPhysChem, 2014, 15, 2657–2663 CrossRef CAS PubMed.
- A. Sperl, J. Kröger and R. Berndt, J. Am. Chem. Soc., 2011, 133, 11007–11009 CrossRef CAS PubMed.
- A. C. Papageorgiou, K. Diller, S. Fischer, F. Allegretti, F. Klappenberger, S. C. Oh, Ö. Sağlam, J. Reichert, A. Wiengarten, K. Seufert, W. Auwärter and J. V. Barth, in preparation.
- A. Gölzhäuser, S. Panov, M. Mast, A. Schertel, M. Grunze and C. Wöll, Surf. Sci., 1995, 334, 235–247 CrossRef.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. Mannini, F. Bertani, C. Tudisco, L. Malavolti, L. Poggini, K. Misztal, D. Menozzi, A. Motta, E. Otero and P. Ohresser, et al., Nat. Commun., 2014, 5, 4582 CAS.
- L. Vitali, S. Fabris, A. M. Conte, S. Brink, M. Ruben, S. Baroni and K. Kern, Nano Lett., 2008, 8, 3364–3368 CrossRef CAS PubMed.
- M. Urdampilleta, S. Klayatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2015, 9, 4458–4464 CrossRef CAS PubMed.
- T. Komeda, H. Isshiki, J. Liu, Y.-F. Zhang, N. Lorente, K. Katoh, B. K. Breedlove and M. Yamashita, Nat. Commun., 2011, 2, 217 CrossRef PubMed.
- H. Tanaka, T. Ikeda, M. Takeuchi, K. Sada, S. Shinkai and T. Kawai, ACS Nano, 2011, 5, 9575–9582 CrossRef CAS PubMed.
- J. Otsuki, Y. Komatsu, D. Kobayashi, M. Asakawa and K. Miyake, J. Am. Chem. Soc., 2010, 132, 6870–6871 CrossRef CAS PubMed.
- K. Seufert, PhD thesis, TU München, 2011.
- A. Rosa, G. Ricciardi and E. J. Baerends, J. Phys. Chem. A, 2006, 110, 5180–5190 CrossRef CAS PubMed.
- C. Castellarin-Cudia, P. Borghetti, G. Di Santo, M. Fanetti, R. Larciprete, C. Cepek, P. Vilmercati, L. Sangaletti, A. Verdini, A. Cossaro, L. Floreano, A. Morgante and A. Goldoni, ChemPhysChem, 2010, 11, 2248–2255 CrossRef CAS PubMed.
- M. Stark, S. Ditze, M. Lepper, L. Zhang, H. Schlott, F. Buchner, M. Röckert, M. Chen, O. Lytken, H.-P. Steinrück and H. Marbach, Chem. Commun., 2014, 50, 10225–10228 RSC.
| This journal is © The Royal Society of Chemistry 2016 |