
Organocatalytic enantioselective desymmetrisation
A.
Borissov†
,
T. Q.
Davies†
,
S. R.
Ellis†
,
T. A.
Fleming†
,
M. S. W.
Richardson†
and
D. J.
Dixon
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 27th June 2016
Abstract
Organocatalytic enantioselective desymmetrisation of achiral or meso compounds is a powerful strategy for the construction of enantiomerically enriched complex molecules, often with multiple stereocentres and in high selectivities. Recent years have seen increasing use of organocatalysts in desymmetrisation methodology, in contrast to traditional metal- or enzyme-catalysed reactions, with many impressive advances made in the current decade. This review will provide an overview of the field since 2010, with the aim of highlighting both the practical applications and elegance of enantioselective desymmetrisation to the wider synthetic community.
 A. Borissov | Arseni Borissov is a DPhil student in the Synthesis for Biology and Medicine CDT at the University of Oxford. Previously he has studied for MChem degree at the University of Edinburgh. He is currently working on a joint project with Prof. Martin Smith and Prof. Paul Beer. His work is focused on rational design of anion-binding molecules with the use of unconventional interactions for applications in molecular recognition and catalysis. |
 T. Q. Davies | Thomas Davies is a second year DPhil student in the Synthesis for Biology and Medicine CDT at the University of Oxford. He received his MSci from the University of Glasgow, during which time he undertook a 12-month placement at Roche Basel. Under the supervision of Prof. Michael Willis, his DPhil research is focused on the study of organosulfur chemistry. His research interests lie in the development of novel synthetic methodology and its application to the practical total synthesis of natural products and other medicinally relevant targets. |
 S. R. Ellis | Sam Ellis is a DPhil student in the Synthesis for Biology and Medicine CDT at the University of Oxford. He received his MChem from the University of Oxford, where he carried out his part II research with Prof. Jeremy Robertson on studies towards the total synthesis of guanacastane diterpenoids. His research interests include enantioselective organocatalytic transformations and their applications to the synthesis of natural products. His current research under the supervision of Prof. Darren Dixon is focused on the total synthesis of Daphniphyllum alkaloids. |
 T. A. Fleming | Tom is conducting early drug discovery research in the field of DNA damage/repair. In his work in the Brown research group, he combines novel fluorogenic assays for high-throughput screening, biophysical studies and computational techniques to discover hit compounds and develop chemical probes for human DNA repair machinery. As part of the Synthesis for Biology & Medicine CDT, Tom's project is a close collaboration between several industrial partners, thereby accelerating research progress and creating open-access drug discovery tools and knowledge. |
 M. S. W. Richardson | Melodie Richardson is part of the first cohort of the Synthesis for Biology and Medicine CDT in the University of Oxford, and is now in Professor Timothy Donohoe's group. Her DPhil project is working towards the total synthesis of pectenotoxin-4 and is co-supervised by Dr Chris Tame, GSK, Stevenage. She achieved a first class MChem degree at Pembroke College, Oxford in the summer of 2014. Her Part II research project, supervised by Prof. Jeremy Robertson, was on the synthesis of drug molecules for the treatment of ERF-related craniosynostosis in collaboration with Professor Andrew Wilkie at the WIMM. |
 D. J. Dixon | Darren J. Dixon obtained his BA, MA and DPhil (supervised by Professor Stephen Davies) from the University of Oxford. After a postdoctoral appointment with Professor Steve Ley FRS he was appointed to the Staff of the Department of Chemistry, University of Cambridge in 2000. In 2004 he took a Senior Lectureship at The University of Manchester and was promoted to Reader in 2007. In 2008, he moved to his current position of Professor of Chemistry, University of Oxford. His honors include an EPSRC Leadership Fellowship, the RSC Catalysis in Organic Chemistry Award, the AstraZeneca Research Award and Novartis Lectureship. |
1. Introduction
Enantioselective desymmetrisation is a powerful method for generating complexity from relatively simple achiral or meso starting materials. With the appropriate choice of reagents and catalysts, multiple stereocentres can be set in one reaction with high enantioselectivity, making such strategies especially attractive in complex natural product synthesis. It is therefore of no surprise that research into developing new enantioselective desymmetrisation reactions has been gaining in popularity in recent years. In particular, the advent of organocatalysis has opened up many exciting new opportunities, providing fundamentally different reactivity modes to more traditional metal-catalysed desymmetrisations.1The first major review of enantioselective desymmetrisation was published by Willis in 1999.2 Since then, a number of substrate- and catalyst-focused reviews have been published.3–11 This review seeks to provide an overview of recent developments (since 2010) of organocatalytic enantioselective desymmetrisations. Earlier contributing work will be covered where relevant to the discussion. This review will not cover reactions that are catalysed by transition metals or enzymes; for a comprehensive coverage of enzyme-catalysed enantioselective desymmetrisation reactions, see the review by Gotor and co-authors.12
The International Union of Pure and Applied Chemistry (IUPAC) defines desymmetrisation as “the loss of one or more symmetry elements […] in the conversion of a prochiral molecular entity into a chiral one.”13 However, this broad definition does not distinguish between the creation of enantiomerically enriched or racemic products,14 and includes enantioselective addition reactions to enantiotopic faces of prochiral substrates (such as alkenes and carbonyl compounds), which are not covered here (vide infra).
For the purposes of this review, we define an enantioselective desymmetrisation reaction as one which converts an achiral or meso compound to an enantiomerically enriched chiral product by the destruction of an improper symmetry element (most commonly σ, but also i, or Sn), provided that the point of reaction does not lie on that symmetry element (Scheme 1a). This definition therefore precludes the differentiation of enantiotopic faces, such as the enantioselective addition to alkenes (V to VI) or carbonyl compounds (VII to VIII), as the point of reaction lies on the symmetry element that is destroyed as a consequence of the reaction (Scheme 1b).
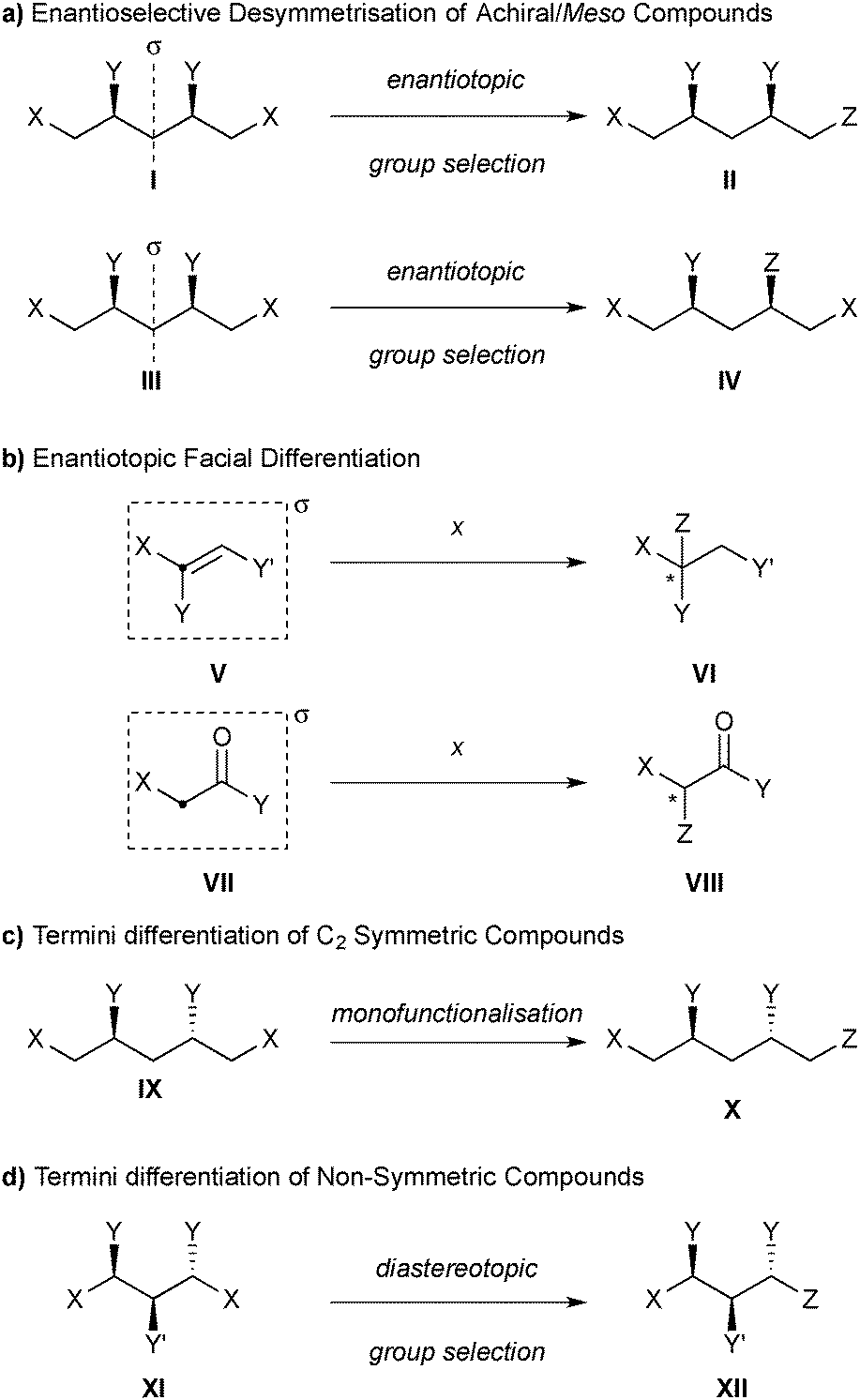 | ||
| Scheme 1 (a) Illustration of the definition of enantioselective desymmetrisation for the purposes of this review, and how it differs from (b) enantiotopic facial differentiation, and from termini differentiation of (c) C2-symmetric and (d) non-symmetric compounds, which are not covered by this review. | ||
We will not cover the termini differentiation of C2-symmetric compounds (IX to X, Scheme 1c), as this can be achieved by mono-functionalisation, nor the termini differentiation of non-symmetric compounds (XI to XII, Scheme 1d), as this is achieved with diastereotopic group selection.2,15
This review will be organised by substrate, according to the functional group undergoing desymmetrisation. Substrates for which there are fewer examples are collected together in the miscellaneous section towards the end of the document.
By showcasing the power of enantioselective desymmetrisation, we hope to catalyse the development of novel desymmetrisation processes, and enable their use in efficient and selective synthesis of natural products and other important synthetic targets.
2. Anhydrides
Cyclic meso-anhydrides are highly popular substrates for enantioselective desymmetrisation by nucleophilic ring opening. A variety of nucleophiles, particularly alcohols, have been used to open cyclic anhydrides and the resulting carboxylic acid functionality can undergo a variety of transformations, underscoring the synthetic utility of this strategy. Typically, high enantioselectivities can be obtained using succinic anhydrides, which are often the substrate of choice. Glutaric anhydrides tend to be more challenging and often give rise to inferior enantioselectivities using the same catalysts. Nevertheless, catalysts have been identified that achieve excellent selectivity for the desymmetrisation of both succinic and glutaric anhydrides. Previous reviews on this area include the seminal review by Willis,2 a short highlight by Spivey et al.,16 a more in depth review by Deng et al.,17 and stereoselective anhydride openings with a comprehensive history on the area by Bolm and co-workers.18 More recently, Díaz de Villegas et al. published a review on enantioselective organocatalysed anhydride desymmetrisation,19 and Connon et al. reviewed organocatalytic asymmetric additions to meso-anhydrides.20 These reviews should be referred to for comprehensive coverage up to 2010. This section will provide an update of the field's advances since these reviews.Cinchona alkaloid derivatives have been used extensively in enantioselective desymmetrisation, particularly with anhydride substrates. An account by Song et al. reviewed a wide range of cinchona-based sulfonamide bifunctional catalysts for the desymmetrisation of cyclic meso-anhydrides using alcohols.21 The account addressed the concept, scope and practical applications of cinchona-based sulfonamide organocatalysts, including solid-supported variations, with catalyst loadings ranging from 30 mol% to as low as 0.5 mol%. Marcelli and Heimstra also reviewed the use of cinchona alkaloids in asymmetric organocatalysis, with a relevant section on the desymmetrisation of anhydrides.22
The mechanism through which cinchona alkaloids catalyse the desymmetrisation of anhydrides is still under debate. Deng et al. attempted to elucidate the mechanism of methanolysis on cis-2,3-dimethyl succinic anhydride 1 (Scheme 2b).23 They analysed their experimental results, showing a first-order dependence in the catalyst and anhydride. Methanol also showed first-order dependence at low concentrations; however, zero-order dependence was found when excess amounts of methanol were used. This kinetic data favours general base catalysis over a nucleophilic catalysis mechanism (Scheme 2a).
 | ||
| Scheme 2 (a) Mechanistic cycles for nucleophilic and general base catalysis. (b) Methanolysis of cis-2,3-dimethyl succinic anhydride 1. (c) The app-closed conformation of the catalysts, considered the reactive conformation for desymmetrisation by Deng. Catalyst C-2 is “locked” in the app-closed conformation. (d) Proposed transition state for methanolysis. | ||
Exploring the use of conformationally rigid cinchona alkaloids, the authors identified catalyst C-2 – “locked” in the app-closed conformation depicted in Scheme 2c – which desymmetrised anhydride 1 in similarly high enantioselectivities as the conformationally-free catalyst C-1 (93% ee vs. 96% ee, Scheme 2c). This suggested the active conformation of the cinchona alkaloid is in the app-closed form. They then proposed a transition state model to explain the observed stereoselectivity (Scheme 2d). The methyl group of the methanol points away from the C6′–OMe methyl group of the catalyst, whilst the anhydride methyl groups are pointing away from the alcohol–catalyst complex to minimise steric interactions. This allows attack at the pro-S carbonyl via the stereoelectronically preferred Bürgi–Dunitz angle.
Aviyente et al. investigated the mechanistic aspects of the methanolysis of meso-anhydrides catalysed by chiral amines via density functional theory (DFT) analysis.24 They analysed both the nucleophilic and general base-catalysed pathways (Scheme 2a) of anhydride 3 with chiral amine C-3 as the catalyst (Scheme 3a). Conformational analysis of the catalyst C-3 showed that the structure with the substituents equatorial was the lowest energy conformer (Scheme 3b), and this was subsequently used for the mechanistic analysis of methanolysis.
 | ||
| Scheme 3 (a) Methanolysis of anhydride 3 with amino alcohol C-3 in Aviyente's DFT studies. (b) Lowest energy conformation of the chiral amino alcohol catalyst. (c) Nucleophilic catalysis and (d) general base catalysis transition states of methanolysis of anhydride 3 with amino alcohol C-3. | ||
In the nucleophilic pathway, the transition state structure (Scheme 3c) was calculated to be a late transition state with a high energy barrier of 62.7 kcal mol−1 in the gas phase (59.5 kcal mol−1 in toluene), suggesting this pathway was unlikely to be operative.
The ring opening reaction in the alternative general base catalysed pathway could proceed via a concerted or a stepwise mechanism. The concerted pathway has the nucleophilic attack of methanol occurring simultaneously with the anhydride ring opening, whereas the stepwise pathway has the formation of a tetrahedral intermediate before ring opening. Calculations of both pathways showed the lowest activation barrier was 35.9 kcal mol−1 in the gas phase (36.1 kcal mol−1 in toluene) for the concerted pathway (Scheme 3d). This amounts to a 26.8 kcal mol−1 in the gas phase (23.4 kcal mol−1 in toluene) free energy difference between the transition states of the nucleophilic and general base pathways, suggesting the general base pathway is more likely to occur.
Aviyente et al. proceeded to show the effect of excess methanol and the polarity of the solvent on reaction energies to better explore the concerted base-catalysed reaction. Peripheral methanol helps stabilise charge as the ring opens, slightly lowering the activation energy compared to a singular methanol molecule used in the previous calculations. Increasing solvent polarity from toluene to acetone (dielectric constant, ε 2.38 and 20.56 respectively) slightly increases the activation energy for the concerted base-catalysed reaction. However, attempts to analyse the stereochemical outcome of the reaction predicted the formation of the wrong enantiomeric product obtained from experimental observations.
Mandoli and Uccello-Baretta et al. also explored the mechanism of methanolysis of anhydride 5 (Scheme 4a) with mono- and bis-quinidine organocatalysts through NMR studies.25 Their experimental evidence supported the hypothesis of the cinchona alkaloids acting as general-base catalysts rather than nucleophilic catalysts, as no defined molecular aggregates involving the alkaloid catalyst and the anhydride substrate, such as the catalyst–anhydride adduct present in the nucleophilic catalyst pathway (Scheme 2a), was observed from DOE and ROE correlations. However, large shifts in the 1H NMR resonances around the quinuclidine site of the catalyst, the proposed site of interaction with MeOH in the general base mechanism (Scheme 2a), in the presence of MeOH-d4 were observed, suggesting a strong interaction between the catalyst and MeOH. No significant shift in resonances were observed in the catalyst in the presence of anhydride. They noted that the conformation of the catalyst appeared to change on increasing the reactant alcohol concentration, converting the prevalent resting state Closed(2) (equivalent to the app-closed conformation on Scheme 2c) into Open(3) (Scheme 4b).26 This resulted in the increase of the ROE dipolar interaction between H9 and H8, showing a reduction of the dihedral angle from Closed(2) to Open(3). Moreover, they proposed that the functionalised C9 OH could have a more active role in the catalytic process rather than just enforcing the preferential reactive conformation. However, the role remains unclear.
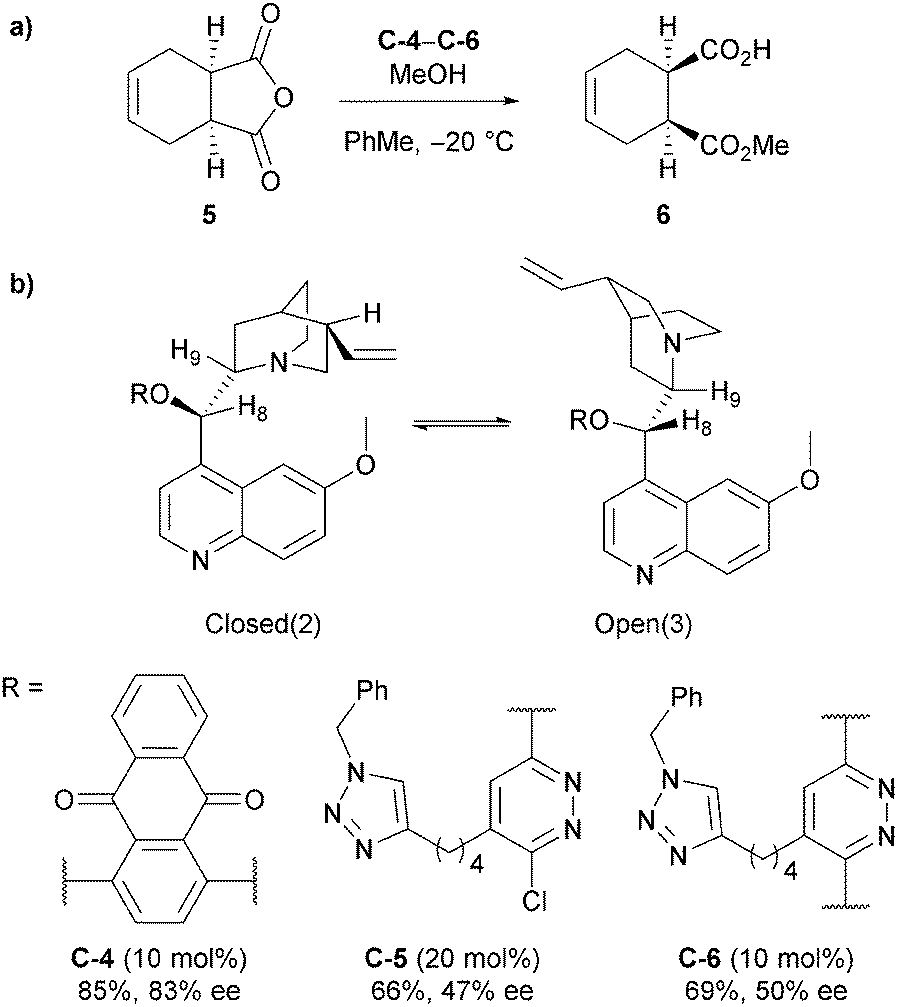 | ||
| Scheme 4 (a) Methanolysis of cis-1,2,3,6-tetrahydrophthalic anhydride 5. (b) Closed(2) resting conformation and Open(3) conformation of the catalysts stabilised by polar protic solvents proposed by Mandoli and Uccello-Baretta and co-workers. | ||
Mandoli and Uccello-Baretta et al. then followed up their NMR study by examining monomeric and dimeric anthraquinone and phenanthryl catalysts, focusing mainly on enantiodiscrimination of the racemic half esters which falls outside of the scope of this review.27 However, it is worth noting that they also found the Open(3) conformer (Scheme 4b) was prevalent in solution via ROESY analysis of catalyst C-4.
A computational study by Wong et al. proposed an alternative transition state model for the methanolysis of succinic anhydrides. They proposed the developing oxyanion from the nucleophilic addition into the anhydride is stabilised by an oxyanion hole created by the DHQD-derived cinchona alkaloid catalyst.28 The oxyanion hole consists of the C6–OMe, C5–H and C9–H forming C–H⋯O interactions, which stabilise the developing negative charge in the gauche-open conformation (Fig. 1). This differs to the previous study by Deng et al.,23 as they did not take into account the potential for such an oxyanion hole stabilisation.
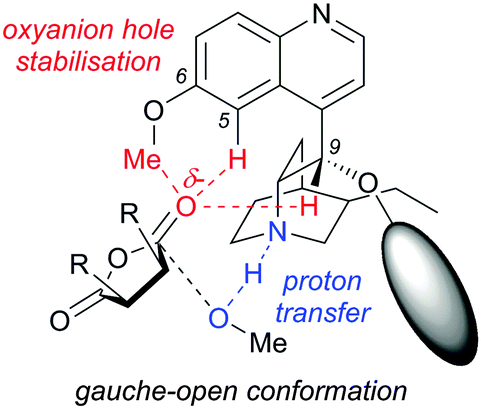 | ||
| Fig. 1 Transition state proposed by Wong for the methanolysis of succinic anhydrides, with an oxyanion hole stabilising the developing negative charge on the anhydride oxygen in the gauche-open conformation. | ||
Aviyente et al. more recently carried out DFT calculations for the methanolysis of anhydrides using cinchona alkaloids quinine C-7 and quinidine C-8 as the catalysts.29 Building upon the previous studies, they confirmed the most stable conformer was Open(3) (Scheme 4b) for the general-base catalysed pathway for methanolysis of anhydride 3 with both quinine and quinidine (Scheme 5a) and this was used in further calculations. Analysis of transition states focusing on the C9–OH and C6–OMe groups found the oxyanion of the tetrahedral intermediate, generated from attack of methanol, is stabilised by hydrogen bonding with these features of the catalyst, similar to those proposed by Wong and co-workers.28 To mimic the reaction conditions, Aviyente and co-workers modelled the use of excess methanol (3 equivalents) in the stabilisation of the transition structure. One molecule of methanol forms a bridging hydrogen bond to the catalyst hydroxyl and the carbonyl oxygen of the attack site, whilst the second methanol participates in hydrogen bonding to the carbonyl oxygen of the resulting carboxylic acid (Scheme 5b). The assistance of the excess methanol was incorporated into their distortion/interaction model (also known as activation-strain model) calculations, showing the prevalence of an early transition state leading to a substantial increase in calculated enantiomeric ratios.
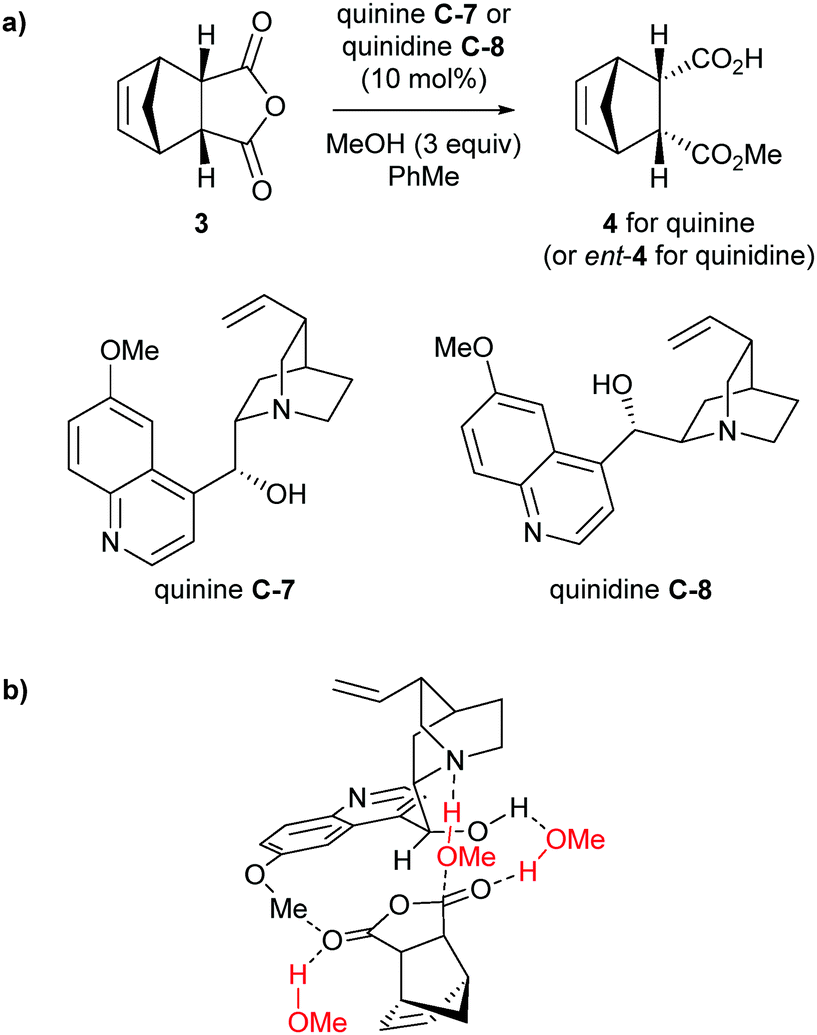 | ||
| Scheme 5 (a) Methanolysis of anhydride 3 used in DFT calculations by Aviyente. (b) Transition structure for quinine C-7 with excess methanol. Methanol molecules are coloured in red. | ||
Hameršak et al. recently reported that substoichiometric catalyst loading (10 mol%) inverted the enantioselectivity of the catalyst for the alcoholysis of 3-substituted glutaric anhydrides, compared to superstoichiometric loading (160 mol%).30 In an attempt to verify what causes the switch in enantioselectivities, organic acids were added. They noted that higher pKa aliphatic acids increased the enantioselectivity, whereas the structures of the aromatic acids were more important than the pKa for enantiocontrol. This was explained by π–π interactions between the aromatic ring of the acid additive and the quinoline moiety, which could additionally stabilise the catalyst conformation. As an extension of this discovery, the scope of this reaction using different organic acids was explored.31 Phenylacetic acid substructures achieved the best enantioselectivities, and it was noted that the enantioselectivity of the ring-opening reaction was no longer dependent on catalyst loading. From a synthetic standpoint, this method is advantageous as only one catalyst is required to access both enantiomers of the product by the simple addition, or omission, of an organic acid (Scheme 6).
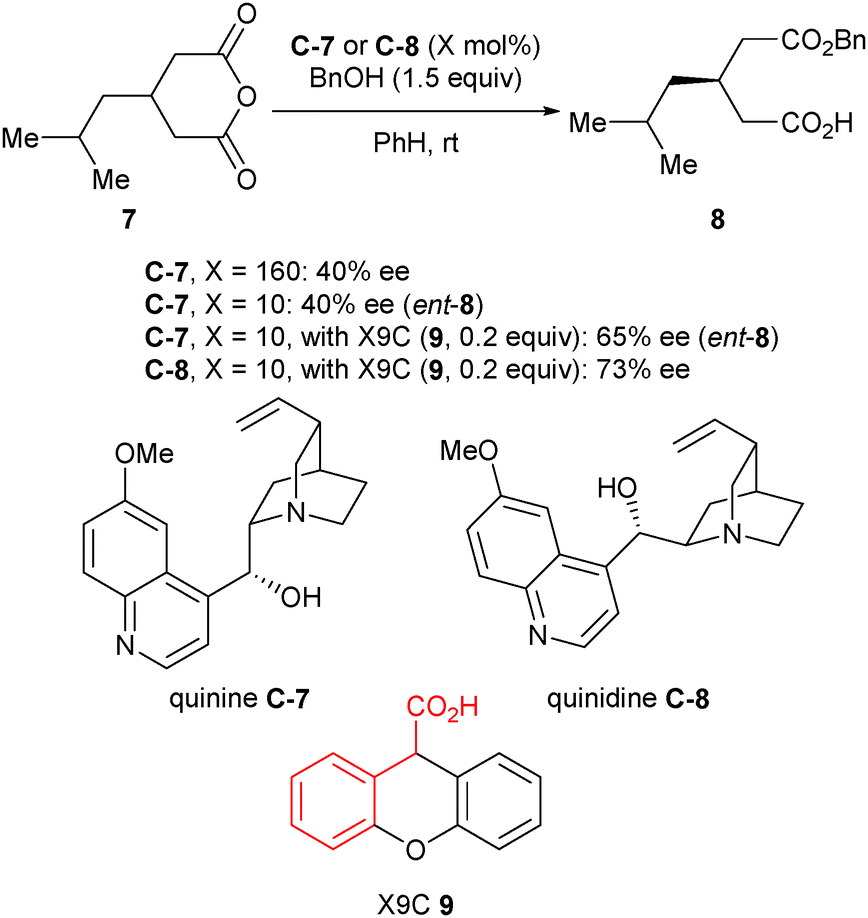 | ||
| Scheme 6 Enantioselective alcoholysis of anhydrides by Hameršak using quinine C-7 or quinidine C-8 and xanthene-9-carboxylic acid (X9C, 9). Red indicates phenyl acetic acid moiety. | ||
In an attempt to explain the switch in enantioselectivity, a computational study compared the lowest energy conformers of the reaction with and without an acid additive.31 For computational tractability, the substituent at the 3-position of the anhydride was fixed as a methyl group, and the acid used was acetic acid (Fig. 2). In the absence of the acid additive, the lowest energy transition state conformer was predicted to be stabilised by two hydrogen-bonds between the alcohol and quinuclidine amine, and the pro-S carbonyl with the catalyst OH. As the pro-S carbonyl was involved in the H-bonding with the catalyst –OH, the more-flexible pro-R carbonyl was attacked by methanol. Upon addition of acid, it participates in hydrogen-bonding with the quinuclidine and alcohol, with the pro-R carbonyl hydrogen-bonding to the catalyst OH. This allows the pro-S carbonyl to be attacked by methanol, reversing the enantioselectivity, in agreement with the experimental data.
 | ||
| Fig. 2 Computational study carried out by Hameršak showing lowest energy conformers with and without the acid additive to demonstrate the switch in enantioselectivity; the red carbonyl is attacked by the methanol. | ||
Song et al. explored the use of cinchona-derived sulfonamide catalysts in desymmetrising glutaric anhydrides.32 They noted that thiourea and squaramide cinchona-based catalysts were prone to self-aggregation due to their bifunctional H-bond donating and accepting nature, which results in a strong dependence of reactivity and enantioselectivity on both concentration and temperature. However, their sulfonamide variant did not form H-bonded self-aggregates even in the solid state. Sulfonamide catalyst C-9 with methanol was shown to desymmetrise various glutaric anhydrides with a substituent on the C3 position of the anhydride; tolerating protected alcohols, alkyl groups and aryl groups (Scheme 7a). They also used allyl and benzyl alcohol to desymmetrise anhydride 16 in high yields and high to excellent enantioselectivities (Scheme 7b).
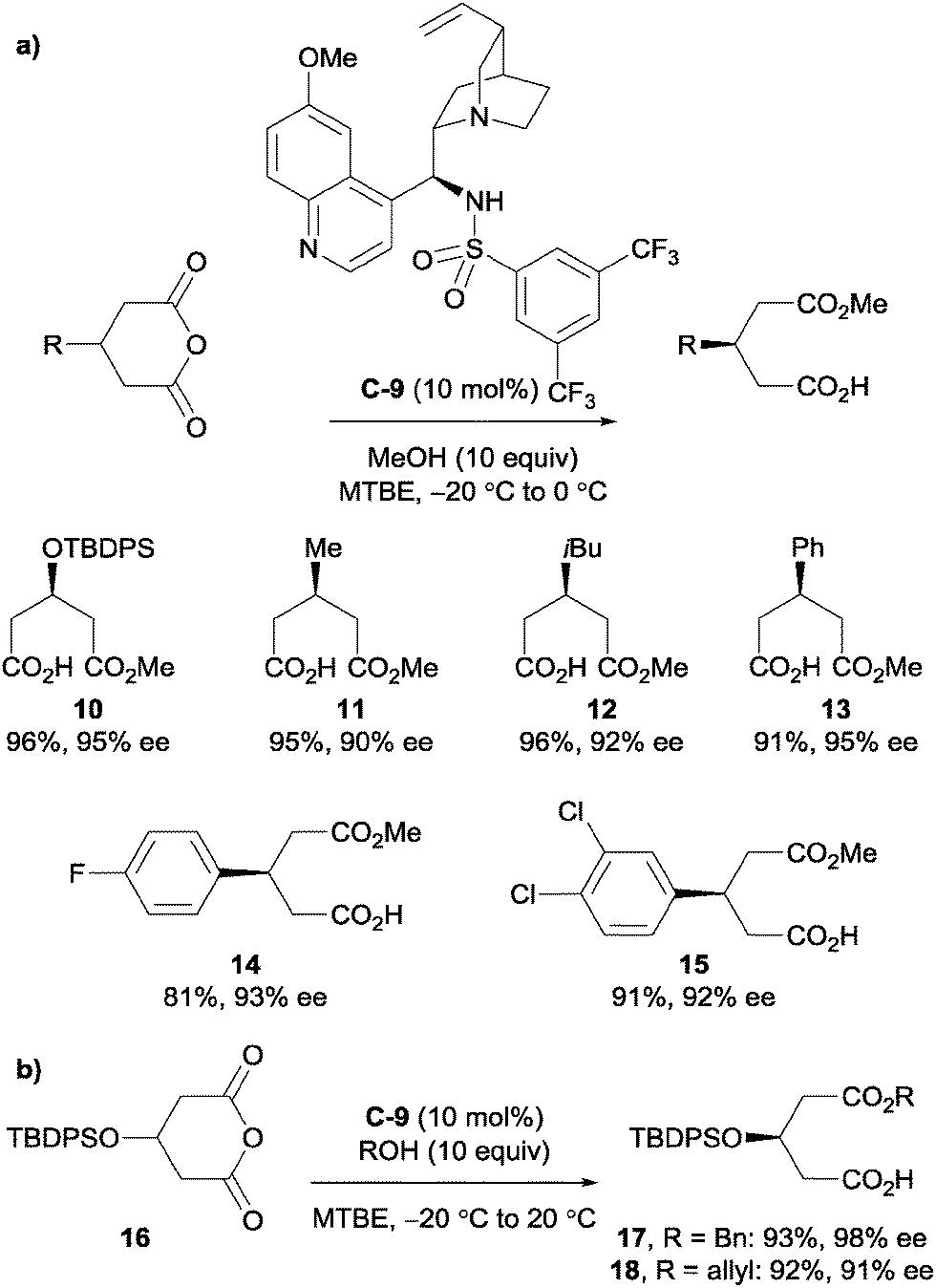 | ||
| Scheme 7 Cinchona-derived sulfonamide catalyst C-9 used by Song in the enantioselective desymmetrisation of (a) various glutaric anhydrides via methanolysis; and (b) anhydride 16 using allylic and benzylic alcohols. | ||
The authors further investigated the self-aggregation of bifunctional catalysts via DOSY (diffusion-ordered spectroscopy) NMR.33 Various bifunctional catalysts were analysed in the methanolysis of anhydride 19 (Scheme 8a), all showing a decrease in enantioselectivity with increasing reaction concentration or decreasing temperature. Analysis of diffusion coefficients using 19F-DOSY experiments, albeit for a Michael reaction, indicated that catalyst concentrations of <0.002 M for C-12 and C-13 are required to minimise dimerisation of the catalysts. They then developed a novel self-association free dimeric cinchona alkaloid catalyst C-14 to methanolytically desymmetrise anhydride 21, and showed no appreciable effect of concentration on the enantioselectivity (Scheme 8b).
 | ||
| Scheme 8 (a) Methanolytic desymmetrisation of anhydride 19 used to demonstrate dependency of enantioselectivity on temperature and concentration using the catalysts shown. (b) Self-association free catalyst C-14 for the methanolytic desymmetrisation of glutaric anhydride 21, showing no enantioselective dependency on reaction concentration. | ||
Furthermore, Song and co-workers went on to utilise this self-association free catalyst to methanolytically desymmetrise various meso-glutaric anhydrides.34 The chiral bis-squaramide catalyst C-14 is sterically bulky enough to suppress self-aggregation. It was noted that the enantioselectivity imparted by this catalyst in the methanolysis of 21 was not significantly dependent on the concentration of the reaction, unlike its monomeric counterparts. These observations were supported by detailed DOSY studies, showing very little change in the diffusion coefficient (D) of catalyst C-14 at different concentrations (D = 5.12 × 10−10 m2 s−1 at 10 mM to D = 5.08 × 10−10 m2 s−1 at 20 mM). Protected alcohols and aryl groups at the 3-position of the glutaric anhydrides were well tolerated; desymmetrised products were obtained in high yields (91–98%) and with high enantioselectivities (89–93% ee, Scheme 9).
 | ||
| Scheme 9 Self-association free catalyst C-14 developed by Song for the methanolytic desymmetrisation of 3-substituted glutaric anhydrides. | ||
Chen et al. used cinchona-derived alkaloids in their desymmetrisation of succinic anhydrides using primary nitroallylic alcohols.35 The use of thiourea catalyst C-15 accessed one enantiomer of the half ester, while using the amino catalyst C-16 accessed the enantiomeric product (Scheme 10). Various aryl groups on the terminal end of the allylic alcohol were explored, as well as a range of bicyclic succinic anhydrides. The half ester products were obtained in good yields (56–90%) and with good to excellent enantioselectivities (68–99% ee).
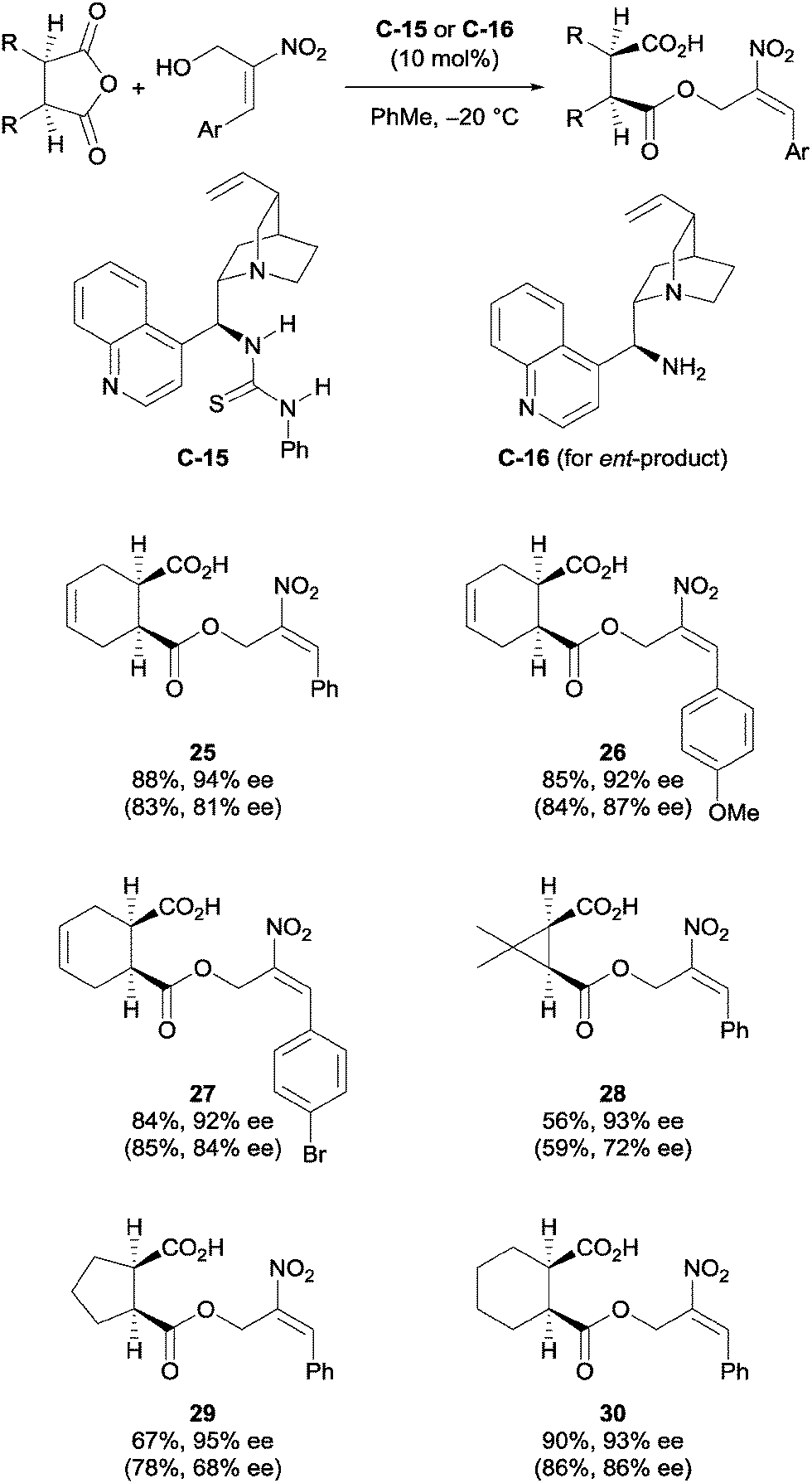 | ||
| Scheme 10 Desymmetrisation of succinic anhydrides using primary nitroallylic alcohols by Chen. Values in parentheses are for the enantiomeric product obtained using catalyst C-16. | ||
The authors also combined kinetic resolution with enantioselective desymmetrisation of glutaric anhydrides using nitroallylic alcohols.36 Each enantiomer of the racemic secondary nitroallylic alcohol, (±)-ethyl 2-hydroxy-3-nitro-4-phenylbut-3-(E)-enoate, could be kinetically resolved by reacting with 3-substituted glutaric anhydrides using cinchonidine-derived thiourea catalyst C-15 (Scheme 11). The (R)-configured substrates of the secondary alcohol react faster with the anhydride under the optimised catalyst conditions, leaving the less-reactive (S)-configured substrate enantioenriched. As a kinetic resolution, the maximum yield of either the (S)-secondary alcohol or the desymmetrised anhydride can only be 50%, based upon the quantity of racemic alcohol used. Lower temperature (−20 °C) increased selectivity but this resulted in long reaction times (3–6 days). Nevertheless, both the (R)-secondary alcohol and the desymmetrised anhydride could be obtained in high yields in good to excellent diastereoselectivity and enantioselectivity. The catalyst is thought to operate via a general acid/base catalysis mechanism, by simultaneously deprotonating the alcohol and activating the anhydride by hydrogen bonding in the transition state.
 | ||
| Scheme 11 Kinetic resolution of a racemic secondary nitroallylic alcohol combined with enantioselective desymmetrisation of glutaric anhydrides developed by Chen. The ees given are for the major diastereoisomer. Values in parentheses are for the resolved (S)-alcohol. | ||
Connon et al. developed a kinetic resolution of secondary thiols in combination with the enantioselective desymmetrisation of 3-substituted glutaric anhydrides.37 Their main focus was on the kinetic resolution of thiol rac-37, however they demonstrated the capability of desymmetrising 3-substituted glutaric anhydrides using a cinchona-derived sulfonamide catalyst C-17 (Scheme 12). The observed diastereoselectivity was good, and both the desymmetrised anhydride and the resolved thiol were obtained in high ees (98% and 90% ee respectively).
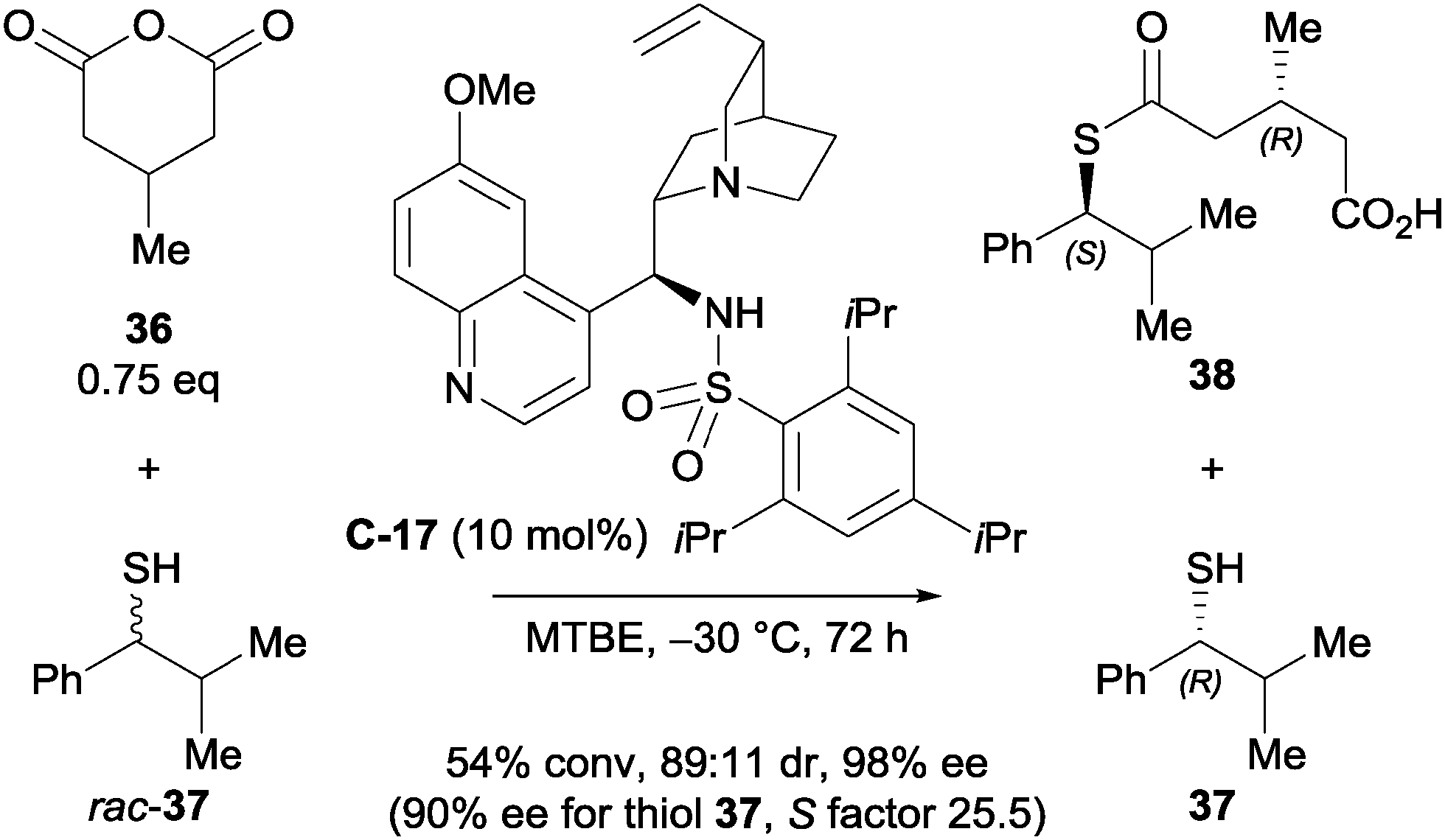 | ||
| Scheme 12 Kinetic resolution of a secondary thiol with concomitant desymmetrisation of a 3-substituted glutaric anhydride by Connon. | ||
Bolm et al. developed novel, non-alkaloid based catalysts for the desymmetrisation of anhydrides.38 Inspired by the core structure of cinchona alkaloids, they developed β-amino alcohol catalysts (for example catalyst C-18) that could enantioselectively catalyse the methanolysis of anhydride 3 (Scheme 13a). Various other small molecules with a similar motif, such as a 1,2-diamino group (imines C-19 and C-23, sulfonamides C-20 and C-24, thioureas C-21 and C-25, and squaramide C-22) were explored as alternative catalysts. The methanolytic desymmetrisation of anhydride 3 using these catalysts generally proceeded with good to high enantioselectivities and high yields with 10 mol% loading. These novel catalysts were also applied to the thiolysis of anhydride 5, however lower yields and enantioselectivities were observed (Scheme 13b).
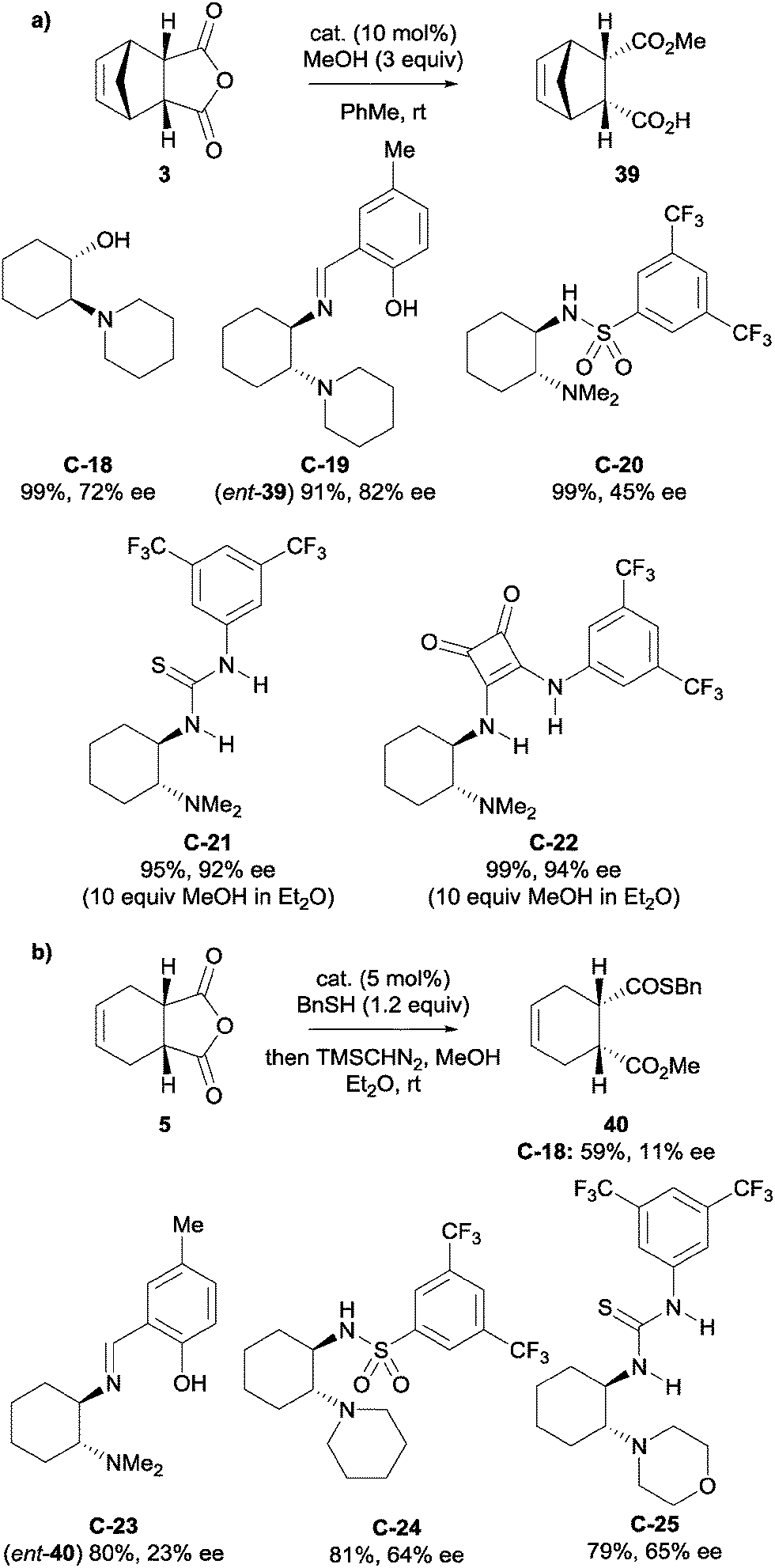 | ||
| Scheme 13 (a) Asymmetric methanolysis of anhydride 3 and (b) asymmetric thiolysis of anhydride 5, using various low-molecular weight catalysts developed by Bolm. | ||
Pedrosa et al. also developed thiourea catalysts to desymmetrise anhydrides.39 Their thioureas were derived from α-amino acids, and they showed that using the other enantiomer of the thiourea catalyst switched the enantioselectivity – not unexpectedly – of the half ester product for anhydride 3 (Scheme 14a). A range of bicyclic and tricyclic succinic anhydrides were desymmetrised using thiourea catalyst C-26 in typically high yields and good to excellent enantioselectivities. One example of desymmetrisation of glutaric anhydride 36 afforded a slightly lower enantioselectivity at 82–84% ee compared to the typical >90% ee observed for the majority of the succinic anhydrides (Scheme 14b). Allylic and benzylic alcohols were used as alternative alcohols to methanol for the desymmetrisation of anhydride 3, in 98% ee and 74% ee respectively (Scheme 14c). Based on calculations performed previously by other groups,40–42 they proposed the transition state model for catalyst C-26 whereby the thiourea forms hydrogen bonds to the pro-R carbonyl of the anhydride, and the methanol proton is hydrogen bonded to the pendant tertiary amino group (Scheme 14d).
 | ||
| Scheme 14 (a) Methanolysis of various meso-anhydrides using an α-amino acid derived thiourea catalyst; (b) an example of a glutaric anhydride using the same method; (c) use of different alcohols in the desymmetrisation; (d) transition state structure proposed by Pedrosa for the desymmetrisation of anhydride 3 with methanol. | ||
List et al. developed a new structural motif for bifunctional organocatalysis based upon BINOL-derived phosphoric acids.43 The catalyst contains a Brønsted acid site to activate the electrophile, such as the anhydride carbonyl, and a Brønsted basic site to activate pronuclophiles, such as the alcohol for the desymmetrisation (Scheme 15a). Optimised bifunctional molecule C-27 catalysed the asymmetric methanolysis of various succinic anhydrides in high yields (81–97%) and with good to excellent enantioelectivities (82–98% ee). Pleasingly, the enantiomer of the catalyst was shown to produce the enantiomeric half ester product for anhydride 46 in a similar yield and ee (Scheme 15b). They also explored the use of alternative alcohols to open cyclobutane-fused succinic anhydride (50–52, Scheme 15b), in high to excellent ees. Their methodology was then utilised in the formal synthesis of the monoterpene natural product (+)-grandisol 54, providing access to intermediate 53 in 3 steps from half ester 46 (Scheme 15c).
 | ||
| Scheme 15 (a) Design of a BINOL-derived phosphoric acid bifunctional catalyst by List including Brønsted acidic and basic sites to activate electrophiles and nucleophiles respectively; (b) examples of alcoholytic desymmetrisation of succinic anhydrides using BINOL-derived catalyst C-27, with the values in parentheses for product ent-46 using the (R)-enantiomer of the catalyst; (c) utility of the methodology in the formal synthesis of (+)-grandisol, 54. | ||
A recent example of the use of solid-supported cinchona alkaloids in the methanolysis of anhydrides was shown by Mandoli et al.44 Polymer-bound anthraquinone derivative C-28, synthesised by the Huisgen cycloaddition of a polymer-supported azide with a terminal alkyne attached to an anthraquinone derivative, was utilised in the desymmetrisation of tetrahydrophthalic anhydride 5 in reasonable yields (48–59%) with good ee (70–72% ee, Scheme 16). The observed enantioselectivity represented a slight improvement on previous solid-supported catalyst examples,45,46 however it did not surpass the 92% ee achieved on the same substrate using a quinine anthraquinone ether derivative on silica gel.47
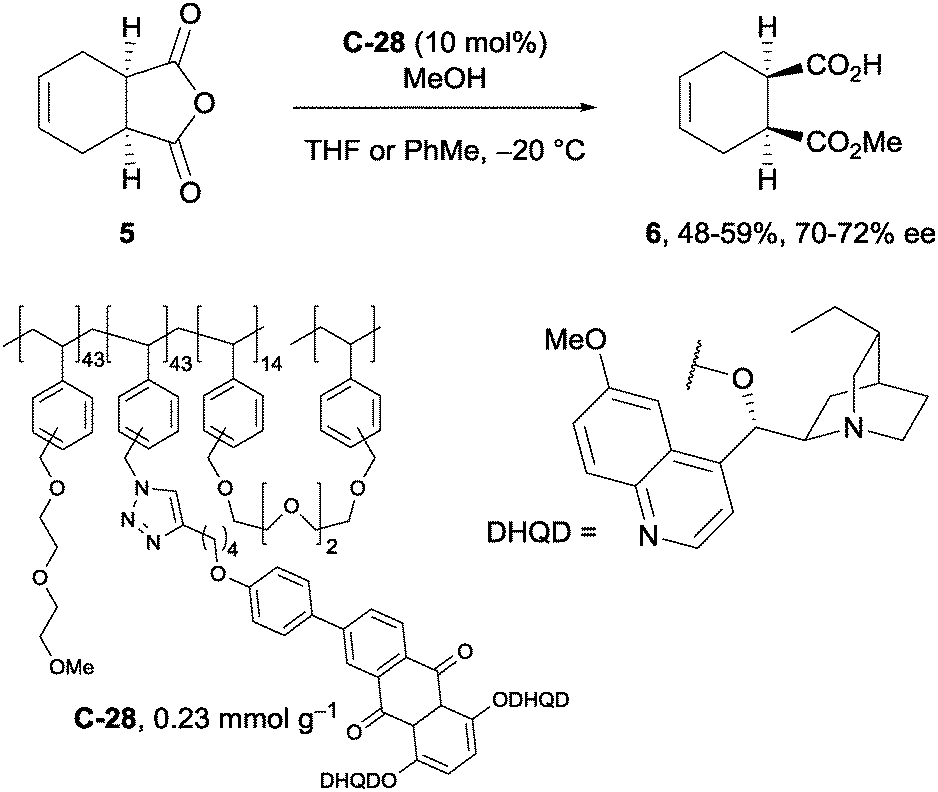 | ||
| Scheme 16 Utility of polymer-bound dimeric dihydroquinidine anthraquinone catalyst C-28 in the methanolysis of tetrahydrophthalic anhydride 5 by Mandoli. | ||
Yashima et al. synthesised various novel helical phenylacetylene polymers bearing cinchona alkaloid derivatives to use as catalysts for reactions catalysed by cinchona alkaloids, including the methanolysis of anhydride 19 (Scheme 17).48 However, comparing the enantioselectivities of the polymer vs. the monomer, they found the polymer bound alkaloids gave slightly lower ees than their monomeric counterparts.
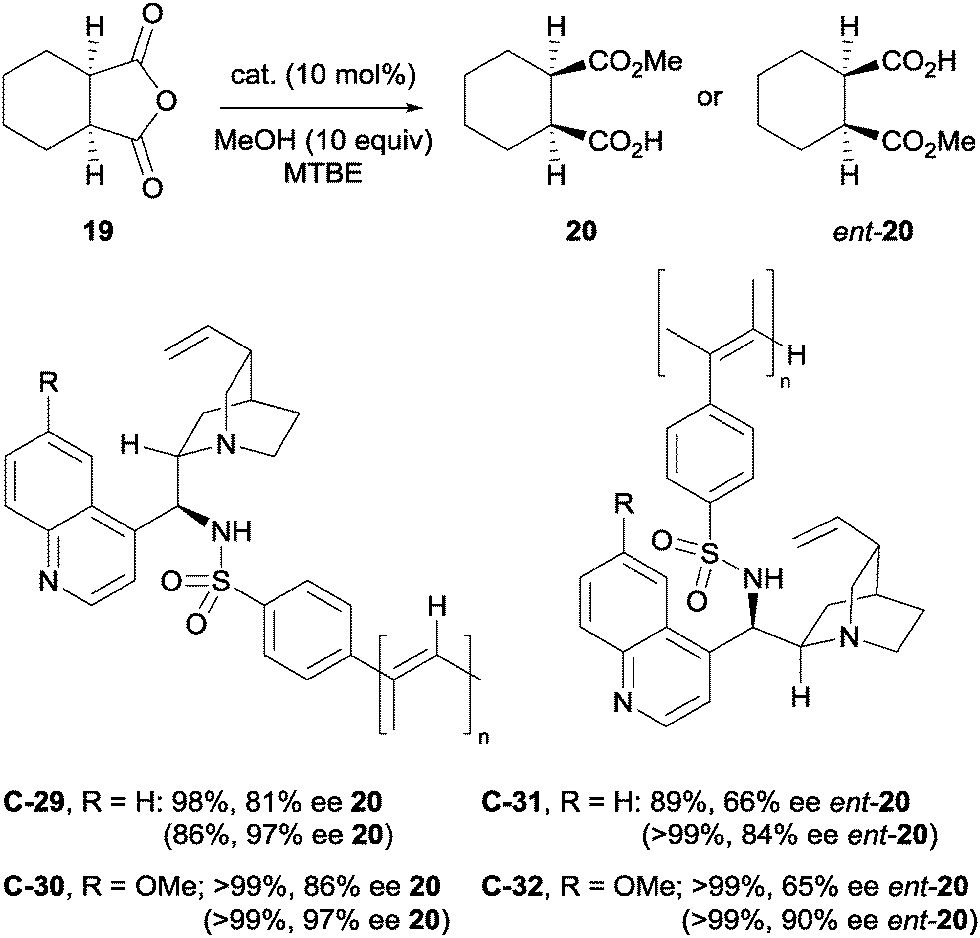 | ||
| Scheme 17 Novel helical phenylacetylene polymers bearing cinchona alkaloids, developed by Yashima for the methanolysis of anhydride 19. The values in parentheses indicate the yields and ees obtained using the monomeric counterpart. | ||
Connon et al. attempted to develop a novel method of immobilising chiral organocatalysts on magnetic nanoparticles.49 After initial experiments on the opening of a few succinic anhydrides with a non-immobilised organocatalyst to find one suitable for immobilisation, they attached a magnetic nanoparticle away from the catalytic site (the acidic sulfonamide proton and the quinuclidine nitrogen). They showed it could desymmetrise succinic anhydride 5 with methanol in good ee (100% conversion, 80% ee, Scheme 18), and that catalyst C-33 was stable for up to 20 cycles with little decrease in catalyst activity (97% conversion, 77% ee). However, in comparison with the non-immobilised catalyst, the enantioselectivity was inferior (96.5% ee). As a control experiment, they found that the magnetic nanoparticle could catalyse the methanolysis on its own, thus accounting for the loss of enantioselectivity on immobilisation.
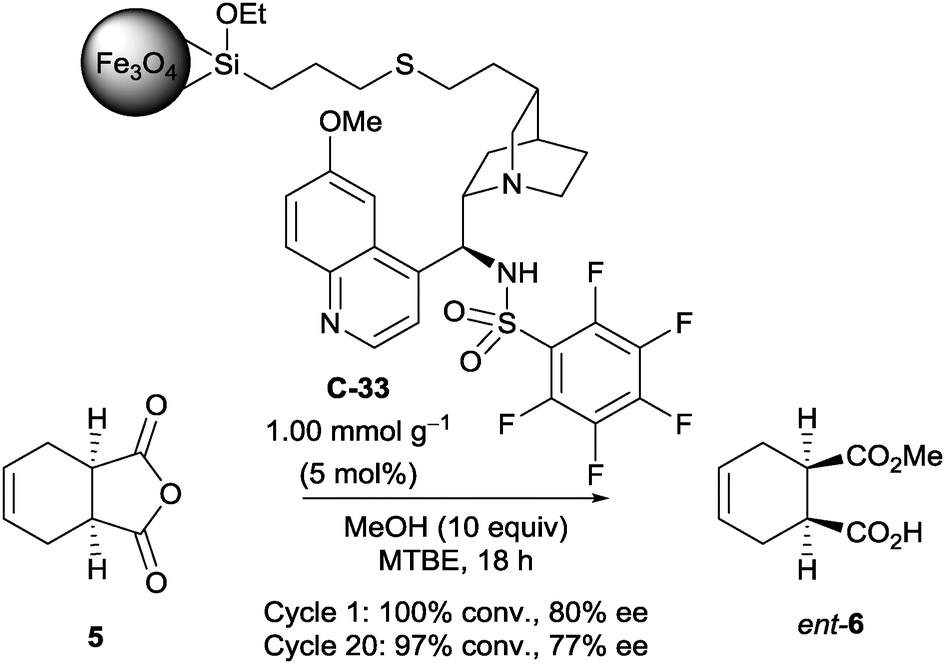 | ||
| Scheme 18 Connon's sulfonamide catalyst C-33, immobilised on a magnetic nanoparticle, used in the methanolysis of succinic anhydride 5. | ||
List et al. immobilised a cinchona alkaloid derivative onto a nylon textile by photochemical irradiation to examine the capabilities of textile-supported catalysis.50 A range of meso-anhydrides were successfully desymmetrised using methanol and immobilised catalyst C-34 (Scheme 19). The immobilised catalyst was shown to be recyclable multiple times without decreasing enantioselectivity, and the reaction was scaled up to gram-scale for product 10 using an iterative continuous flow reactor.
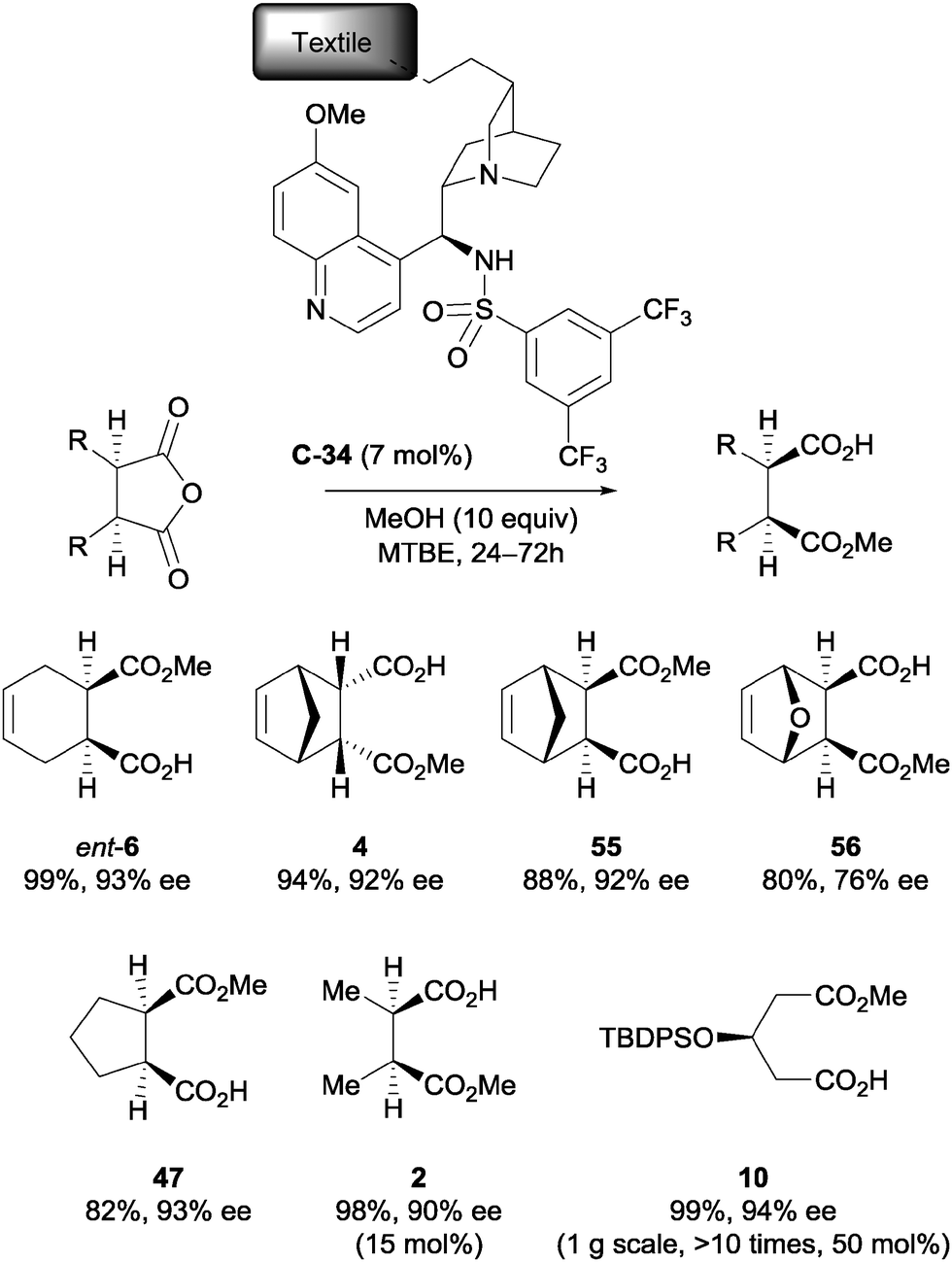 | ||
| Scheme 19 List's nylon textile immobilised sulfonamide catalyst C-34, used in the methanolysis of various anhydrides. | ||
2.1 Anhydride desymmetrisation in synthesis
Enantioselective desymmetrisation of anhydrides via alcoholysis, or less commonly thiolysis, is a well-established approach for the enantioselective synthesis of small chiral molecules, particularly drug compounds.Huang et al. developed the first asymmetric synthesis of a 4-aryl-substituted 5-carboxy-3,4-dihydropyrin-2-one derivative, a core structure of compounds with promising biological activity for the treatment of rheumatoid arthritis and chronic pain.51 After optimisation of the reaction conditions developed by Connon et al.,52 they achieved the desymmetrisation of anhydride 57 in 80% ee with ten equivalents of methanol and 2 mol% of catalyst C-12 (Scheme 20a). Treatment of the reaction product with toluene and hexane enriched the enantiomeric excess to 96% ee. Formylation adjacent to the ester and cyclisation with ammonium acetate in acetic acid afforded product 58 in 48% yield over the three steps with >95% ee. Ester hydrolysis afforded the desired core 59 in 86% yield. This was taken to pilot plant scale by Roche.53
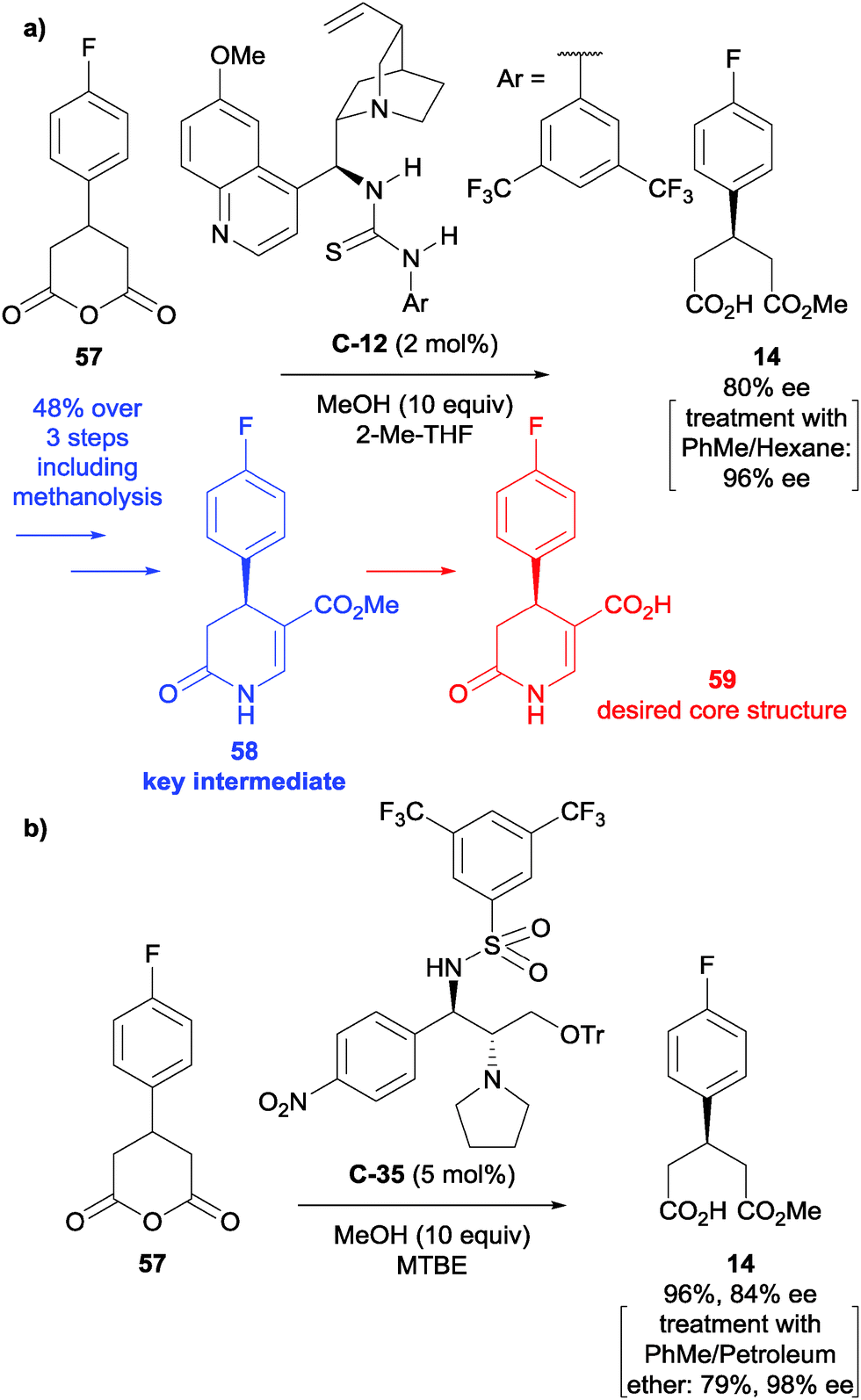 | ||
| Scheme 20 (a) Use of cinchona-derived thiourea catalyst C-12 in the synthesis of the desired dihydropyridinone core by Huang; (b) novel sulfonamide catalyst C-35 used in the synthesis of a key intermediate for P2X7 antagonists by Yang. | ||
Yang et al. developed novel sulfonamide catalyst C-35 for the methanolysis of various succinic and glutaric anhydrides.54 Following optimisation, this was subsequently used in the synthesis of the same key intermediate 58 towards P2X7 receptor antagonists, desymmetrising the same glutaric anhydride as Huang and co-workers (Scheme 20b). The desymmetrisation step was achieved in a 96% yield with 84% ee initially. This could be further improved upon treatment with toluene and petroleum ether to achieve a 79% yield and 98% ee of the desired half ester.
As part of their long-standing interest in the synthesis of (+)-biotin, Chen et al. used cinchona alkaloid-based sulfonamide catalyst C-9 in one of their syntheses of (+)-biotin 65.55 Their initial starting point was based upon previous work by Song et al.,40 and with subsequent optimisation of the reaction conditions and the use of a bulkier alcohol (trans-cinnamyl alcohol vs. methanol), they could achieve the desymmetrisation of meso-anhydride 60 in 98% yield and 92% ee albeit with stoichiometric quantity of catalyst C-9 (Scheme 21a). This was taken forward to access (+)-biotin 65 in a further 6 steps via the Roche lactone 64.
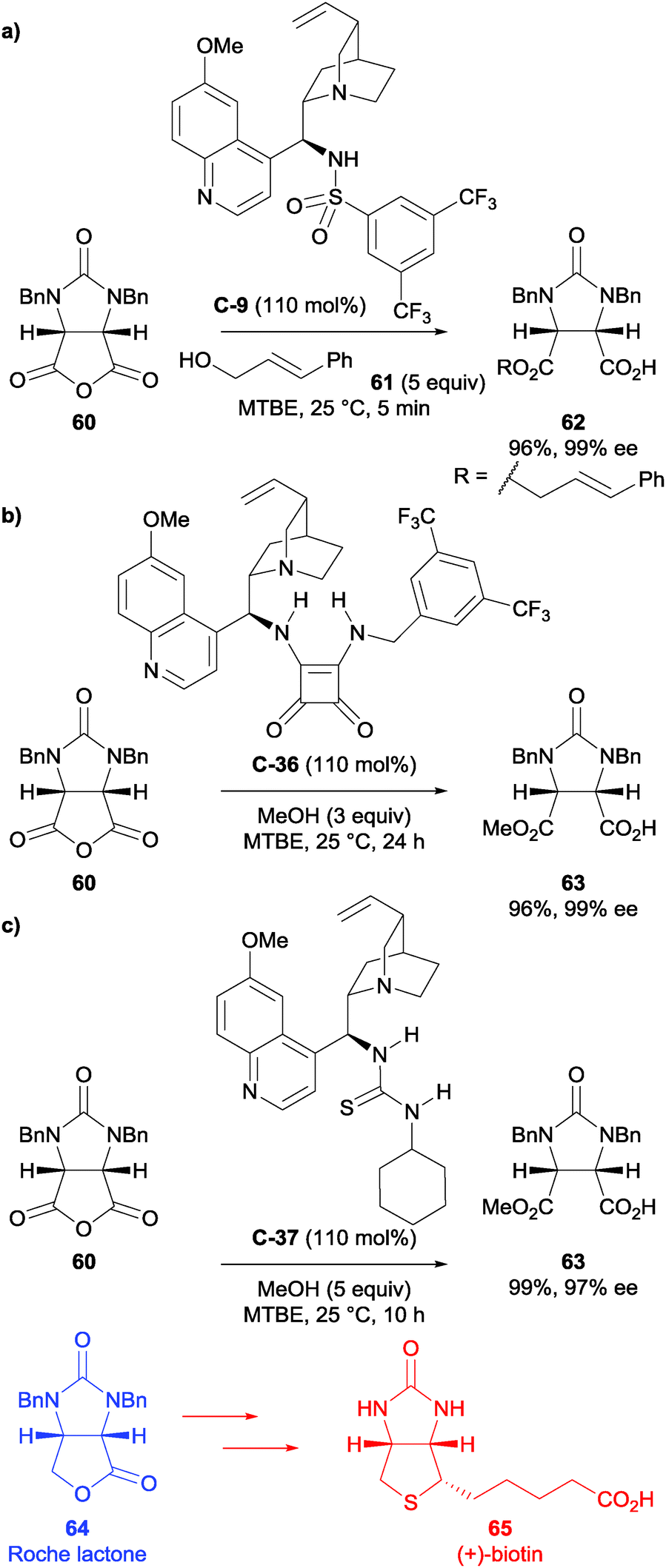 | ||
| Scheme 21 Cinchona alkaloid derived catalysts (a) sulfonamide C-9, (b) squaramide C-36, and (c) thiourea C-37, used by Chen in the desymmetrisation of succinic anhydride 60 in synthesis of (+)-biotin 65via the Roche lactone intermediate 64. | ||
The authors also used squaramide catalyst C-36 in the methanolysis of meso-succinic anhydride 60 (Scheme 21b).56 This could be achieved in an excellent 96% yield and 99% ee, however stoichiometric use of the squaramide catalyst was required. The desymmetrised product was taken on further to obtain (+)-biotin 65 in a 48% overall yield.
Finally, the authors applied thiourea catalyst C-37 (Scheme 21c),57 which was not as enantioselective as squaramide C-36 (97% ee) but furnished the desired product in 99% yield. The catalyst C-37 was used in excess (110 mol%) as lower catalyst loadings were not as enantioselective, however they successfully demonstrated that they could retrieve and reuse the thiourea catalyst. This intermediate was taken forward and (+)-biotin 65 was synthesised in an overall yield of 44%.
Ivšić et al. utilised alcoholytic desymmetrisation methodology in their synthesis of GABOB 67 (Scheme 22).58 The catalytic use of the sulfonamide C-38 with benzyl alcohol on 3-(tert-butyldimethylsiloxy)glutaric anhydride 21 afforded the benzyl ester 66 in 79% yield and 90% ee. Poorer yields and enantioselectivities were obtained when the anhydride was opened with cinnamyl alcohol, or when the alcohol protecting group on the anhydride was changed to either benzyl or benzyloxymethyl acetal. Following the desymmetrisation reaction, a Curtius rearrangement and deprotection led to GABOB 67 in good yield. It should be noted that the GABOB intermediates had several orthogonal protecting groups that could be selectively removed in the presence of the others for further manipulations.
 | ||
| Scheme 22 Alcoholytic desymmetrisation by Ivšić in the synthesis of (S)-GABOB 67, using the sulfonamide derivative of quinine C-38. | ||
Xu and Qing et al. utilised previous methodology reported by Bolm et al.59 to synthesise the Hepatitis C virus protease inhibitor Telaprevir 70.60 Quinidine C-8 in stoichiometric quantity (110 mol%) was used to access the desired half ester 47 (Scheme 23). From the half ester, the desired proline derivative can be accessed in 8 steps. The proline derivative 69 was accessed in a total yield of 27% from the half ester 47, and was taken forward in the synthesis to afford Telaprevir 70 in an overall yield of 16% (11 LLS).
 | ||
| Scheme 23 Quinidine C-8 catalysed methanolysis of anhydride 68 in the synthesis of Telaprevir 70. | ||
Hamersak and Ivšić used anhydride desymmetrisation in the synthesis of both enantiomers of Rolipram 73.61 Based on their previous work (described above),31 the addition of xanthene-9-carboxylic acid (X9C, 9) was used to reverse the enantioselectivity of the organocatalysts quinine C-7 or quinidine C-8. The alcoholytic desymmetrisation of anhydride 71 to afford half ester 72 proceeded in high yields with both pseudo-enantiomeric catalysts with or without X9C 9, although enantioselectivities were moderate. (62–73% ee, Scheme 24). However, the enantiomeric excess could be significantly improved via resolution with (S)-phenylethylamine to 95% ee as the corresponding salt. This allowed access to both enantiomers of the desymmetrised anhydride with either catalyst. These materials were taken forward into a one-pot extended Curtius reaction to afford both (R)- and (S)-Rolipram 73.
 | ||
| Scheme 24 Desymmetrisation strategy towards the synthesis of both enantiomers of Rolipram 73. Addition of xanthene-9-carboxylic acid (X9C, 9) reverses the enantioselectivity of the organocatalyst used. Resolution with (S)-phenylethylamine can increase enantioselectivity up to 95% ee. | ||
Guingant et al. used the desymmetrisation of succinic anhydrides 68 and 75 in their total syntheses of (+)-Brefeldin C 74, (+)-nor-Me Brefeldin A 77 and (+)-4-epi-nor-Me Brefeldin A 78.62 The asymmetric methanolysis of meso-anhydride 68 had previously been reported by Bolm et al. (93%, 95% ee),59 so upon replication of the reported procedure, they accessed the half ester ent-47 in 99% and 94% ee (Scheme 25a). The desymmetrised product was utilised to access (+)-Brefeldin C 74 in a total of 15 steps in 4.6% yield from anhydride 68.
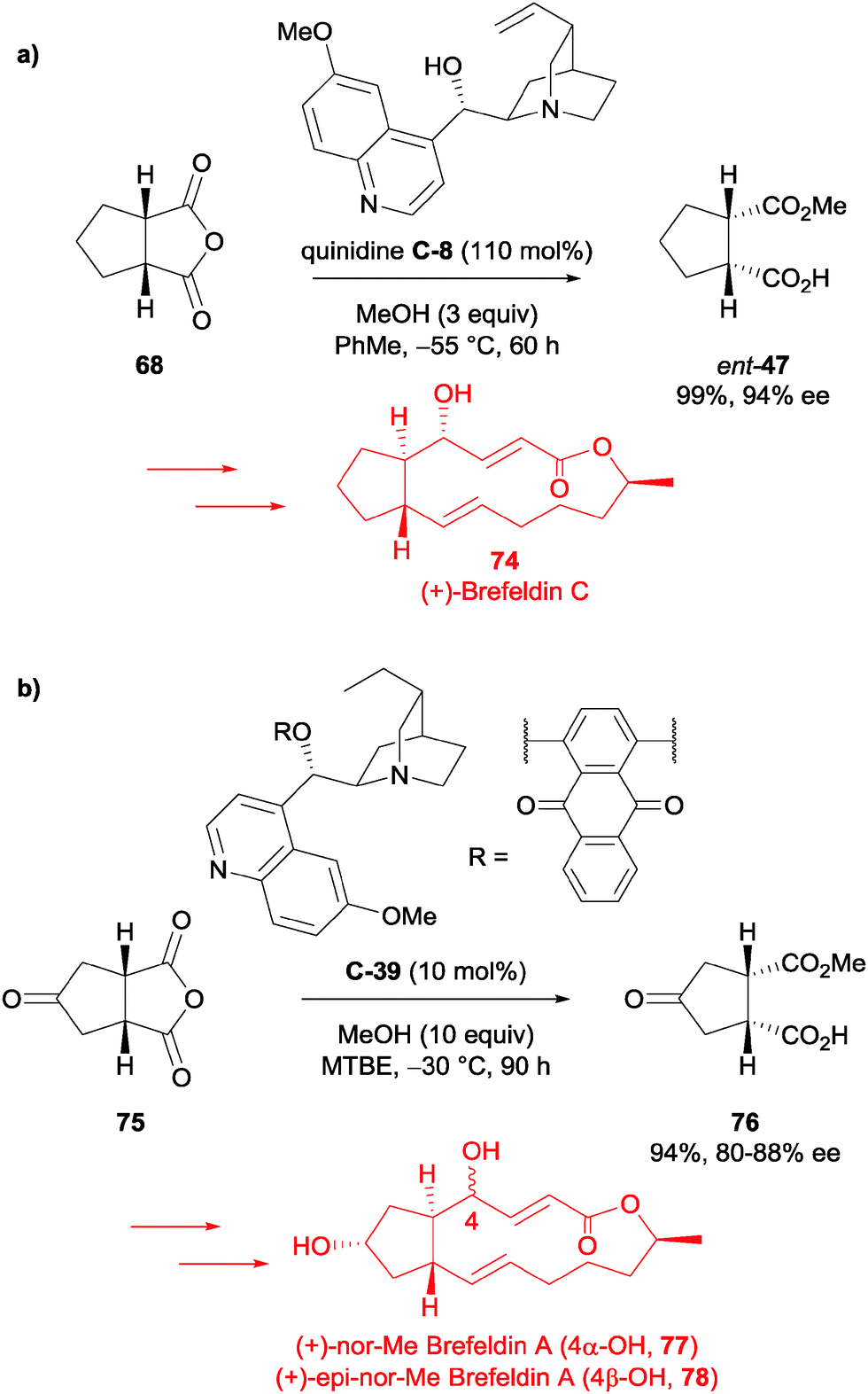 | ||
| Scheme 25 Asymmetric methanolysis by Guingant of (a) anhydride 68 in the synthesis of (+)-Brefeldin C 74; (b) anhydride 75 in the synthesis of (+)-nor-Me Brefeldin A 77 and (+)-4-epi-nor-Me Brefeldin A 78. | ||
However, for (+)-nor-Me Brefeldin A 77 and (+)-4-epi-nor-Me Brefeldin A 78, the desymmetrisation of the analogous anhydride 75 was not as successful. Under the same desymmetrisation conditions, the half ester product 76 was obtained in a moderate 60% ee. Nevertheless, modifying a procedure from a patent using the desired substrate,63 they could reproducibly access the desired half ester 76 in 94% yield and 80–88% ee using catalyst C-39 (Scheme 25b). Half ester 76 was taken forward to access both (+)-nor-Me Brefeldin A 77 and (+)-4-epi-nor-Me Brefeldin A 78 in 18 steps from anhydride 75, in around 1.5% yield.
Chen et al. also used asymmetric methanolysis in their synthesis of Lipitor (81, Pfizer).64 The bifunctional sulfonamide catalysts used in this study were derived from (1S,2S)-2-amino-1-(p-nitrophenyl)propane-1,3-diol, a by-product of the industrial production of chloramphenicol. Optimisation of the methanolysis reaction conditions allowed an enantioselectivity of 90% ee to be achieved at 10 mol% loading of chosen catalyst C-35 (Scheme 26). The corresponding desymmetrised acid 80 was utilised in an efficient 14-step synthesis of Lipitor 81. This route is a potentially useful alternative to the currently employed industrial route.
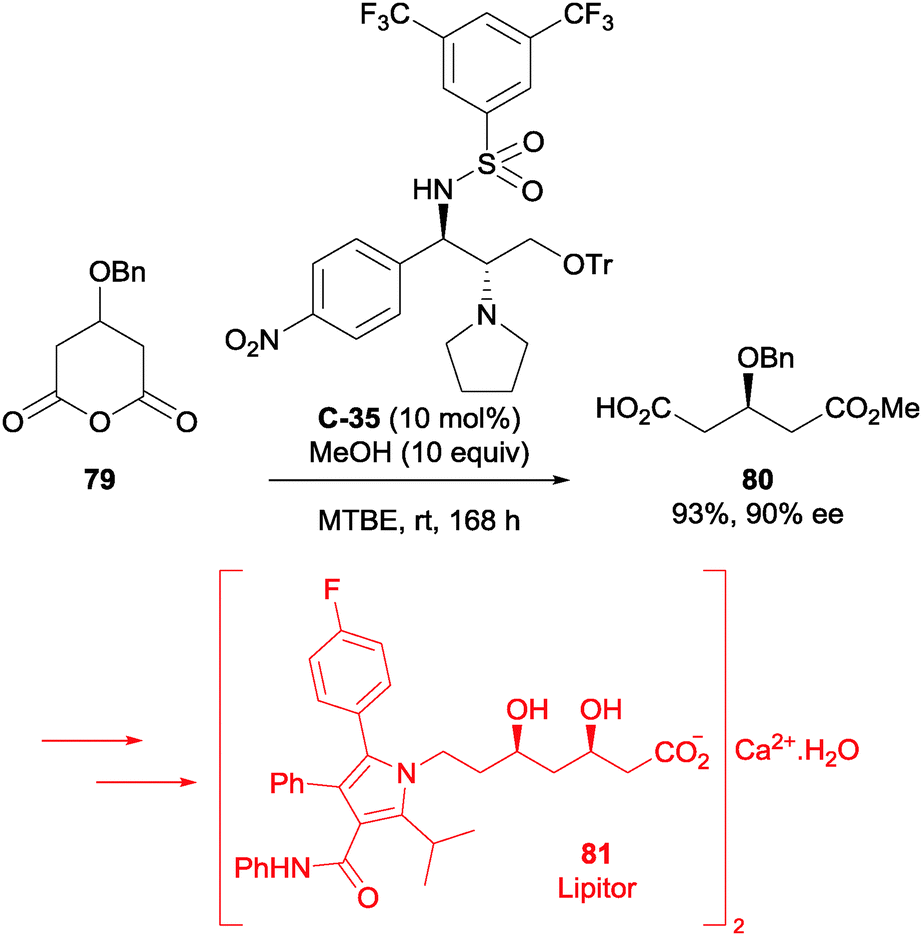 | ||
| Scheme 26 Methanolysis of glutaric anhydride 79 by Chen using bifunctional sulfonamide catalyst C-35 in the synthesis of Lipitor 81. | ||
The authors also utilised a related urea-containing catalyst in their synthesis of vitamin D3 analogues.65 Their approach to the A-ring precursor of Calcifediol 84 featured enantioselective alcoholysis of anhydride 5. After screening several catalysts and optimising the reaction conditions, they desymmetrised anhydride 5 using methanol and urea-containing catalyst C-40 in 93% yield and 90% ee (Scheme 27). Upon screening the conditions, they noted a significant improvement in enantiomeric excess upon dilution (69% ee at 0.1 mol dm−3 to 88% ee at 0.025 mol dm−3 at 5 mol% catalyst loading). This could be further be improved by a single recrystallisation from methyl tert-butyl ether, giving 82 in 87% yield and 96% ee. The desired half ester 82 was taken forward in the synthesis to access the A-ring precursor 83 in a total of 13 steps.
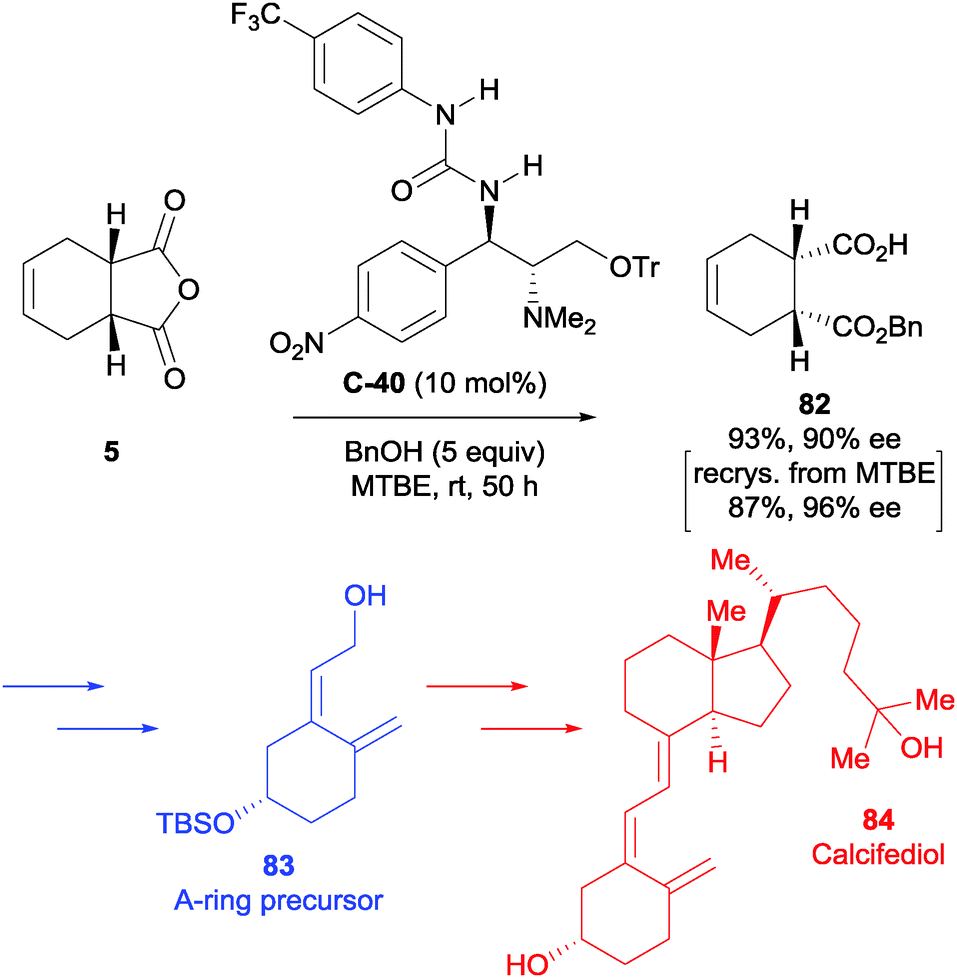 | ||
| Scheme 27 Synthesis of the A-ring precursor 83 to Calcifediol 84 using urea-based catalyst C-40 in the desymmetrisation of anhydride 5 by Chen. | ||
Enantioselective thiolysis was utilised in the synthesis of pregablin, an anticonvulsant drug. A review on organocatalytic carbon–sulfur bond-forming reactions by Enders et al. has a relevant section on the desymmetrisation of anhydrides using thiols.66 After initial development of the reaction conditions using benzyl mercaptan as the nucleophile, organocatalyst C-41, and subsequent esterification of the resulting acid with TMSCHN2, Chen et al. showed the method's generality with various anhydride substrates.67 It was then applied to glutaric anhydride 7, without the subsequent esterification, in 89% yield with 94% ee (Scheme 28a). This was used to synthesise to pregablin 86 in a further 4 steps in an overall 38% yield.
 | ||
| Scheme 28 (a) Thiolysis of glutaric anhydride 7 in the synthesis of pregablin 86 by Chen; (b) alcoholysis of glutaric anhydride 7 in the synthesis of pregablin analogues by Hameršak. | ||
Hameršak et al. synthesised various pregablin derivatives with carbohydrate, serotonin, tryptamine and adamantyl amine moieties.68 Glutaric anhydride 7 was desymmetrised using cinnamyl alcohol as developed in their previous report on pregablin synthesis (Scheme 28b).69 The half ester 87 was accessed in 72% ee under the quinine C-7 reaction conditions, however they could increase the enantiopurity to 97% ee upon recrystallisation with (S)-phenylethylamine. This method could access various pregablin analogues in a further 3 steps.
Landais et al. developed the first route to optically active polyesters via enantioselective desymmetrisation of meso-glutaric anhydrides (Scheme 29).70 In contrast to monofunctional analogues, polymerisation of anhydrides requires strict control of stoichiometry between the anhydride and the diol used to achieve the highest polymer weight. Initial optimisations determined that benzylic and linear alcohols were good candidates to react with bis-anhydrides. Monitoring by 1H NMR spectroscopic analysis, they noted the reaction did not reach completion after 7 days, which was believed to be due to the presence of CO2H groups sequestering the bifunctional catalyst C-9. Nevertheless, the CO2H groups did not react with the alcohol groups of the diols under the reaction conditions, therefore no cross-linking was observed. A number of polyesters were prepared with the degree of polymerisation (linear nmax) up to 14, with some degree of cyclic by-product also formed. To determine the average enantioselectivity for each step, the polymer had to be degraded to the corresponding bis-lactone for HPLC analysis, with the average ees ranging between 52–92% ee.
 | ||
| Scheme 29 Step-growth polymerisation of bis-cyclic anhydrides in the synthesis of chiral polyesters by Landais; reagents and conditions: (i) ROH (1 equiv.), C-9 (10 mol%), THF, 25 °C; (ii) EtOCOCl, NMM, THF then NaBH4, 0 °C, 15 h; (iii) 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 10 mol%), THF, 25 °C, 15 h. | ||
3. Epoxides, oxetanes, aziridines and azetidines
3.1 Epoxides
Epoxides are readily synthesised from alkenes, usually in a single stereospecific transformation, making them a readily available substrate for enantioselective desymmetrisation. The ring strain and polarisation of the epoxide functional group predisposes it towards elimination reactions with bases and addition reactions with nucleophiles, potentially leading to a range of synthetically useful products. Hodgson has reviewed epoxides and their chemistry previously in 1996.71 Although the field has significantly advanced since then, the reader is referred to this article for more detailed discussion of epoxide desymmetrisation strategies. Since then, in 2013, the organocatalytic enantioselective desymmetrisation of epoxide substrates (alongside aziridines) has been reviewed by Wang,72 and a recent review of organocatalytic enantioselective reactions of epoxides by Meninno & Lattanzi also discusses some epoxide desymmetrisation approaches.73Most commonly, epoxides are exploited in desymmetrisation strategies via nucleophilic ring-opening transformations. Sun et al. have demonstrated this approach, conducting enantioselective ring-opening reactions on meso-epoxides with heterocyclic thiol nucleophiles, achieving moderate to high yields with enantiomeric excesses reaching 86% (Scheme 30).74
 | ||
| Scheme 30 Desymmetrisation of meso-epoxides using chiral Brønsted acid catalysis by Sun. | ||
The reported substrate scope was broad, and enantioselectivities up to 84% ee were achieved with five or six-membered ring epoxide substrates. Cyclooctene oxide and diaryl-substituted epoxides were also shown to react with moderate enantioenrichment but required elevated temperatures.
The authors propose the origin of enantioselectivity arises from steric interactions between the 2,4,6-triisopropylphenyl groups of the catalyst and the R groups of the epoxide. The catalyst activates the epoxide while stabilising the thione form of the nucleophile through hydrogen bonding, resulting in highly enantioselective ring opening (Fig. 3). Chiral phosphoric acid catalysts feature heavily throughout this review but are themselves reviewed in depth elsewhere by Rueping et al.75
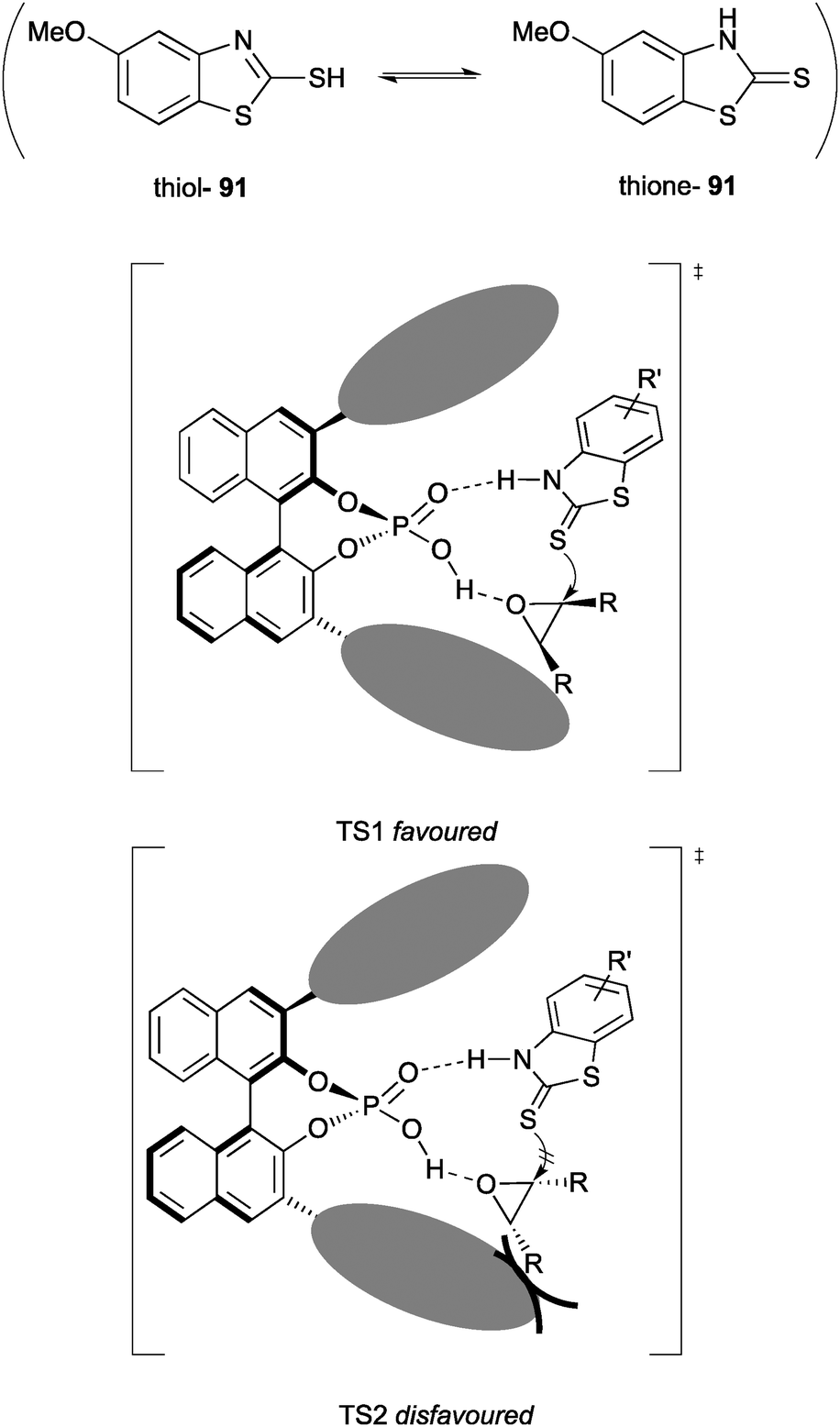 | ||
| Fig. 3 Transition state models proposed by Sun to explain the origin of asymmetric induction by chiral phosphoric acid catalysis in the desymmetrisation of meso-epoxides. | ||
Sun and co-workers also reported a two-step process to protect the resultant free hydroxyl group with a silyl protecting group and reveal the thiol for use in further reactions. The reaction was shown to proceed without erosion of enantioselectivity (Scheme 31) and significantly increased the utility of this desymmetrisation technique.
 | ||
| Scheme 31 Cleavage of the heterocyclic nucleophile from the enantioenriched epoxide products without loss of optical purity. | ||
Organocatalytic enantioselective desymmetrisation of meso-epoxides with thiocarboxylic acids has been demonstrated by List and co-workers.76 Using confined chiral phosphoric acid catalyst C-43, ring-opening of the epoxide followed by transesterification at elevated temperatures afforded O-protected hydroxythiols in good yields (63–98%) and high enantioselectivities (72–96% ee) (Scheme 32). The intermediate thioester products could also be isolated, generally with slightly improved yields and enantioselectivities. Both thiobenzoic acid and thioacetic acid could be used, and good results were obtained with both (hetero)cyclic and acyclic substrates. Poorer selectivity was obtained when R1 = Ph. The authors propose that the chiral phosphoric acid catalyst and thiocarboxylic acid undergo heterodimeric association by hydrogen bonding.
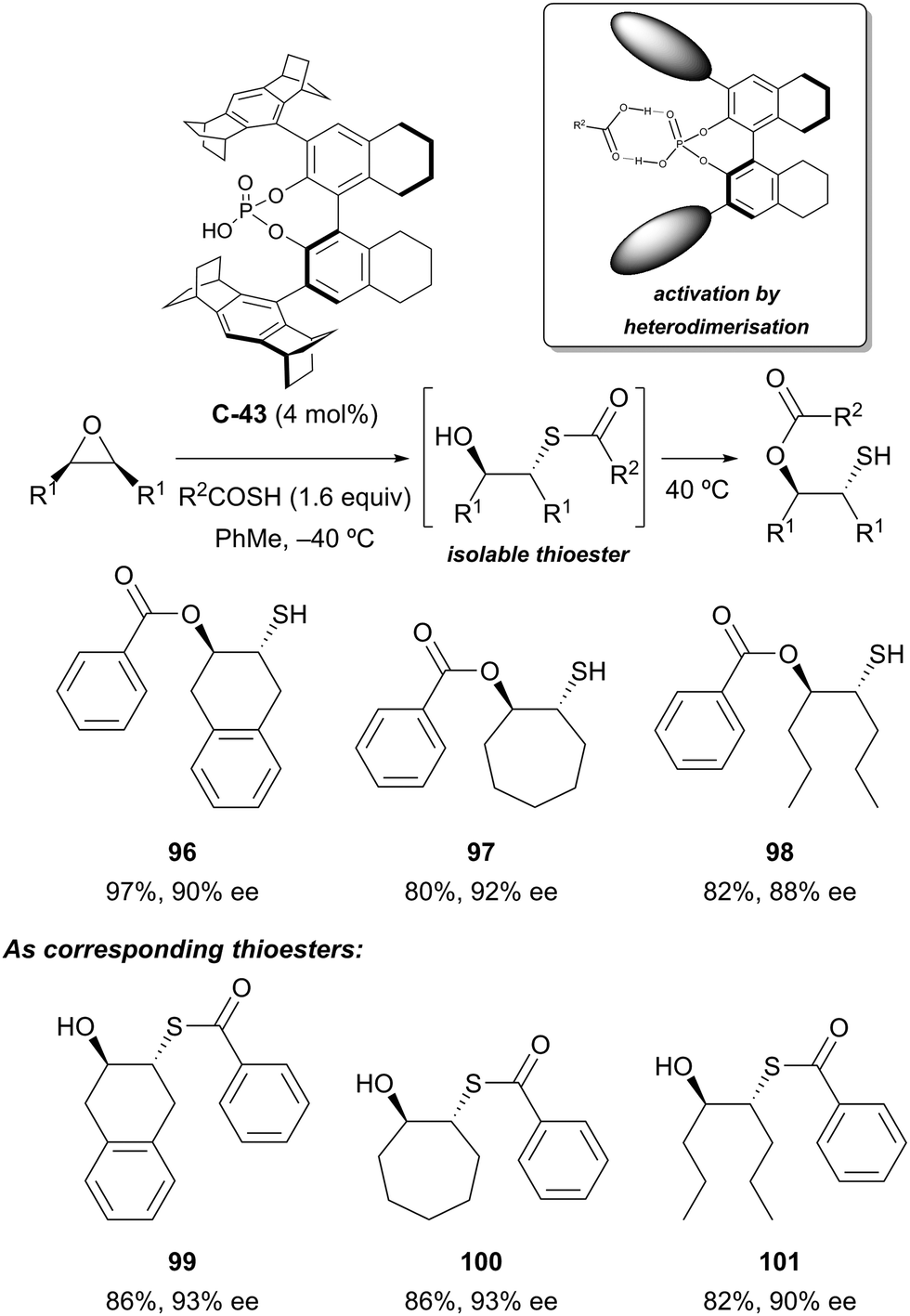 | ||
| Scheme 32 Asymmetric organocatalysed cascade reaction developed by List to desymmetrise meso-epoxides. | ||
The List group exploited the same chiral phosphoric acid catalyst C-43 in the desymmetrisation of meso-epoxide substrates by nucleophilic ring opening with benzoic acid (Scheme 33).77 The monoprotected anti-1,2-diols were obtained in good yields and enantioselectivities. A key advantage of using benzoic acid as a nucleophile is the compatibility with the prior peracid-mediated epoxidation of an alkene and subsequent hydrolytic cleavage, enabling a one-pot highly enantioselective organocatalytic anti-dihydroxylation process.
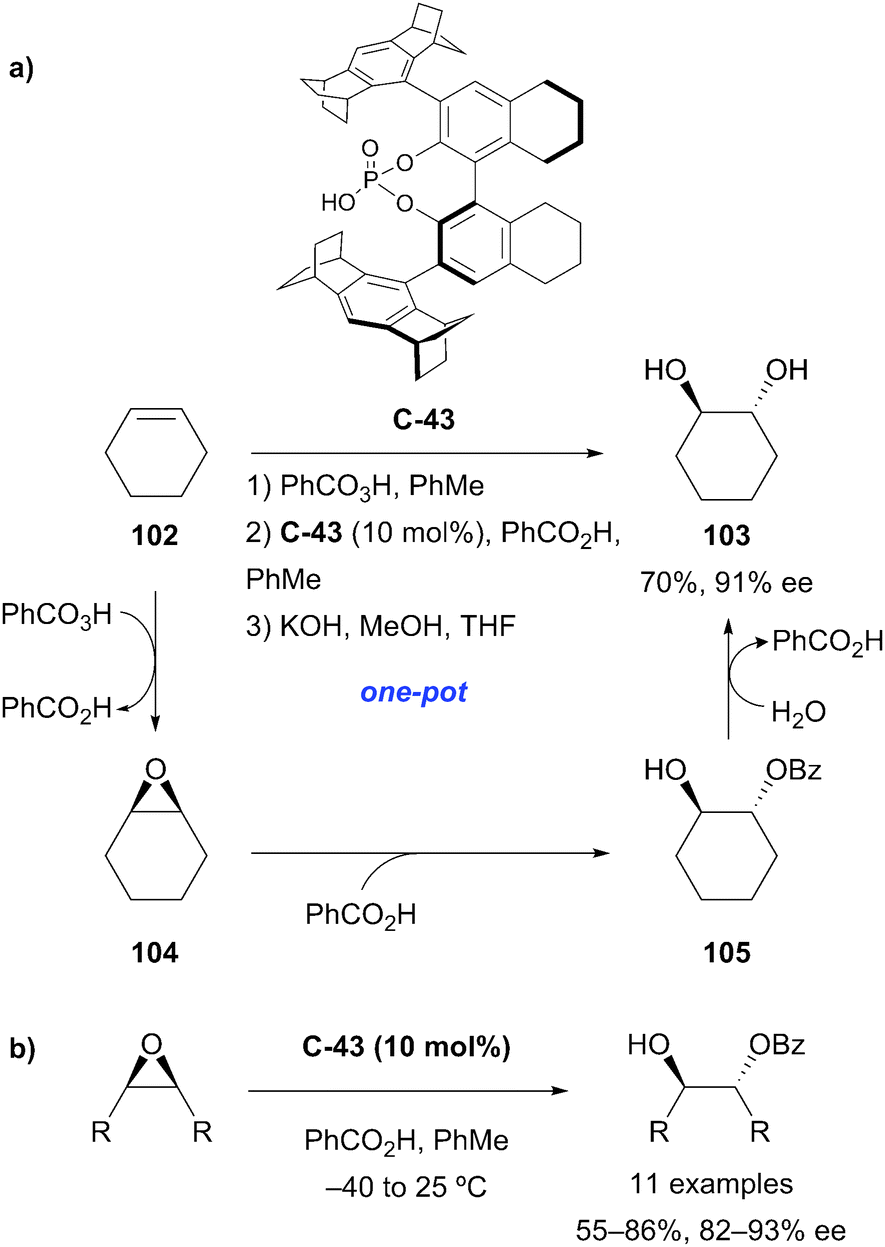 | ||
| Scheme 33 (a) One-pot epoxidation-enantioselective desymmetrisation-deprotection developed by List to achieve anti-dihydroxylation of olefins. (b) Desymmetrisation of meso-epoxides using benzoic acid to afford mono-protected diols. | ||
Many variations on the binaphthyl motif with atropisomeric chirality have been explored in asymmetric organocatalysis. Kotani et al. reported enantioselective desymmetrisation of meso-epoxides with a BINAP phosphine oxide catalyst C-44.78 The reaction afforded 1,2-chlorohydrins in good yields but with varying levels of enantioselectivity. With a range of acyclic symmetric cis-alkene substrates, Kotani achieved a one-pot epoxidation and subsequent desymmetrisation, obtaining chiral chlorohydrins in good yields and with reasonable levels of enantioselectivity (Scheme 34). The desymmetrisation of cyclic and heterocyclic substrates was found to produce lower levels of enantioselectivity.
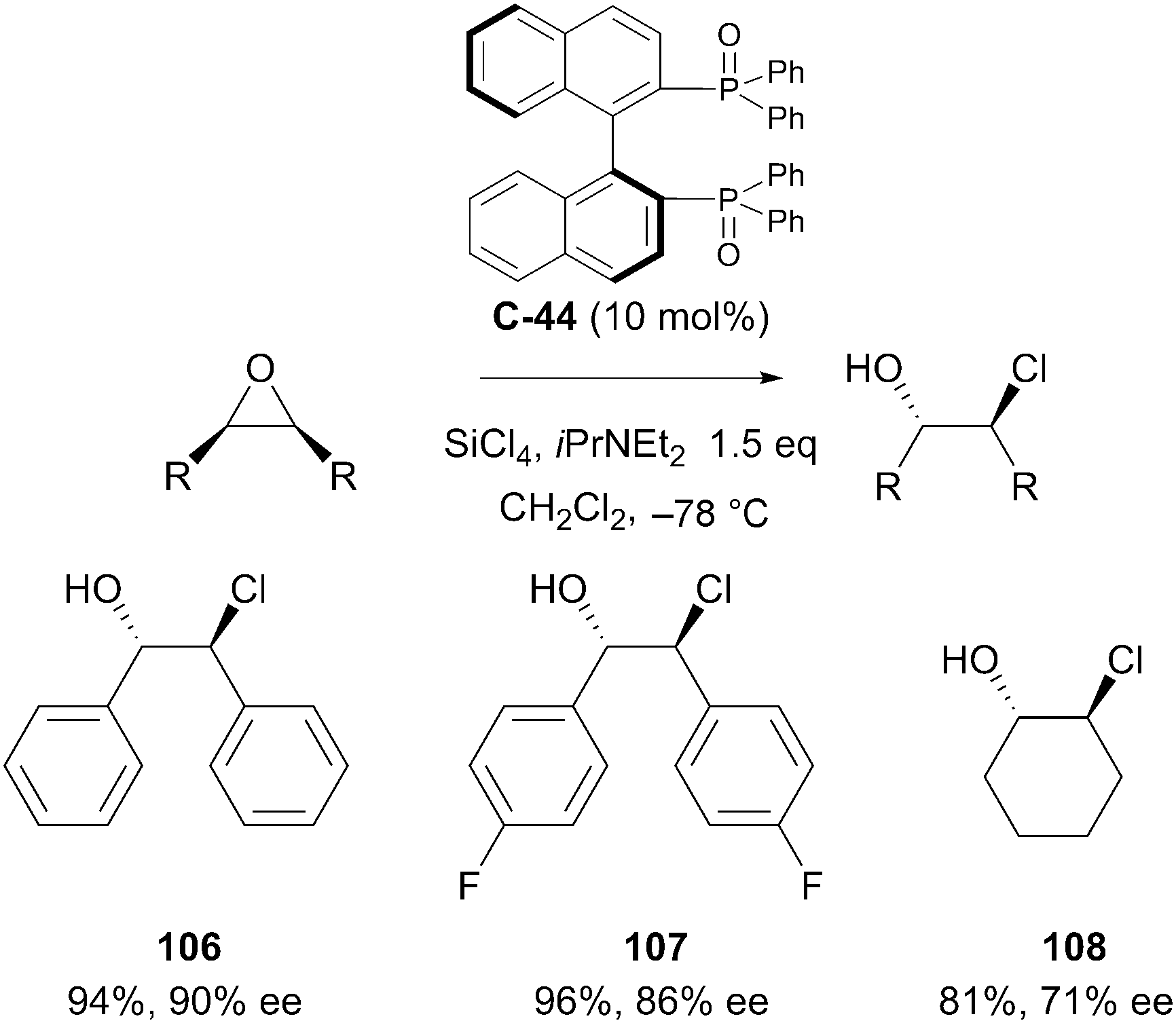 | ||
| Scheme 34 Desymmetrisation of biaryl meso-epoxides under (S)-BINAP oxide catalysis reported by Kotani. | ||
Ramanathan et al. have also reported the desymmetrisation of epoxides to afford 1,2-chlorohydrins. In the presence of the novel, conformationally rigid bipyridine N,N′-dioxide organocatalyst C-45, treatment of epoxides with tetrachlorosilane and Hünig's base at −30 °C afforded chlorohydrins in good yields and poor to high enantioselectivities. Notably, the catalyst loading was very low at 0.5 mol% (Scheme 35).79
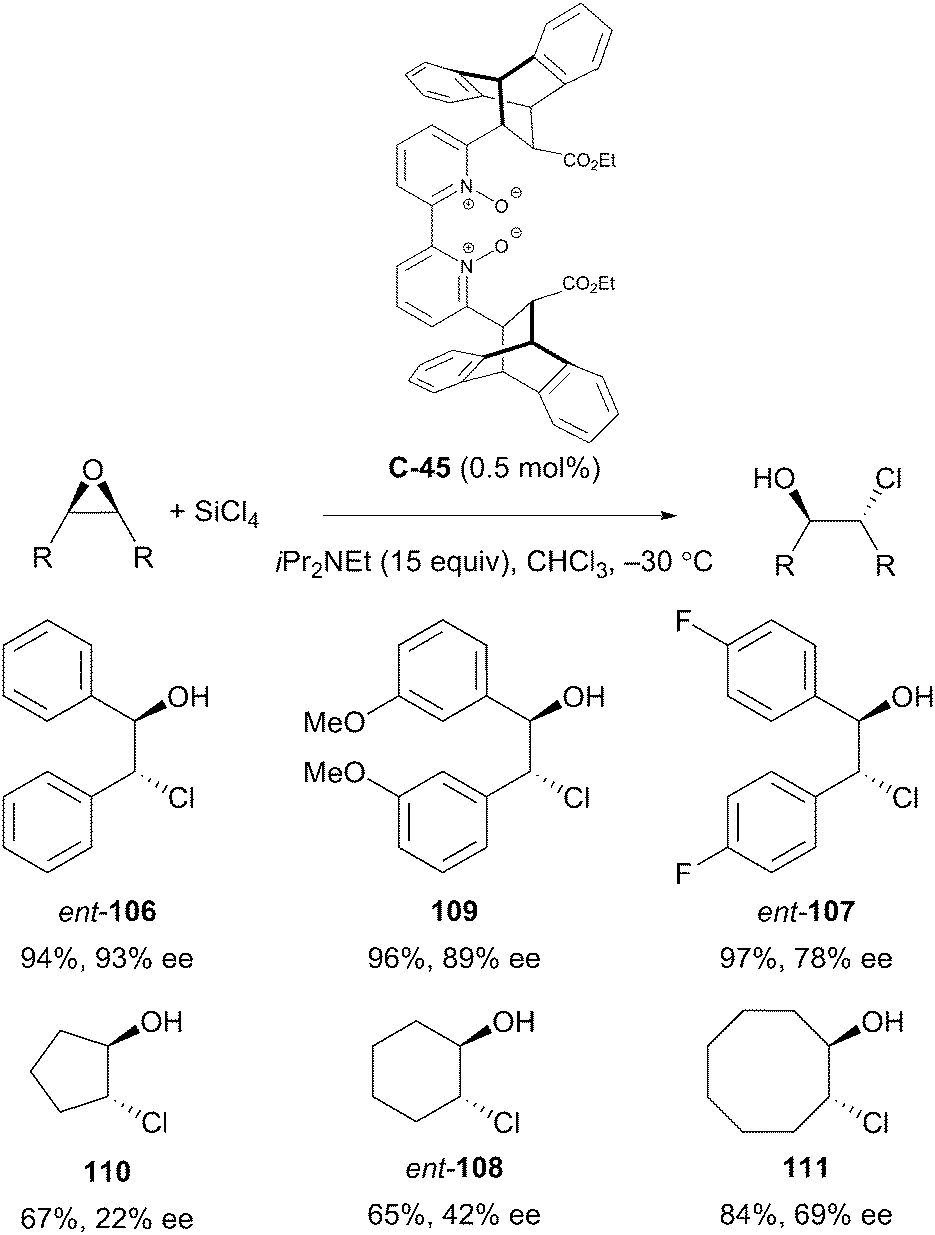 | ||
| Scheme 35 Desymmetrisation of a variety of symmetrically substituted cis-stilbene oxide derivatives by nucleophilic epoxide ring opening reported by Ramanathan. | ||
Bala et al. employed a peptidyl thiourea organocatalyst C-46 to effect the desymmetrisation of cis-stilbene oxide derivatives by epoxide ring opening with N-phenylpiperazine-based nucleophiles.80 The process afforded several chiral β-amino alcohols in good yields (>80%) and with modest enantioselectivities (55–79% ee). Yields and enantioselectivities varied according to substitution patterns of the cis-stilbene substrates. Steric hindrance by ortho-substitution of the epoxide phenyl rings, or interaction between the peptidyl catalyst and nitro substituents at any position (ortho, meta or para) resulted in loss of reactivity and enantioselectivity (Scheme 36).
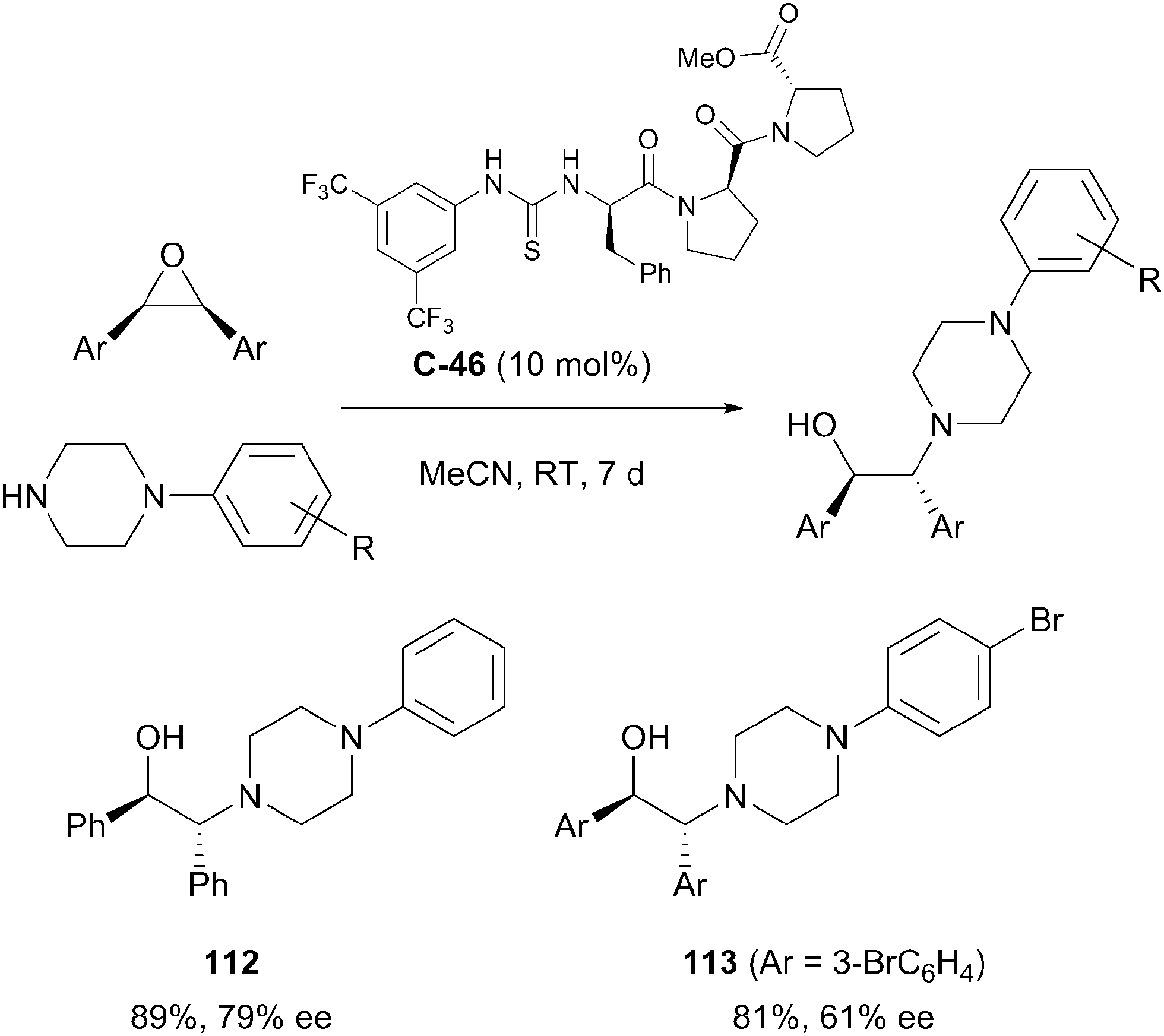 | ||
| Scheme 36 Biaryl epoxide desymmetrisation by Bala employing thiourea organocatalysis to afford enantioenriched 1,2-aminoalcohols. | ||
3.2 Oxetanes
Oxetanes substituted in the 3-position also undergo desymmetrisation via nucleophilic ring opening.81 Although there are relatively few examples, in the presence of a chiral Brønsted acid, this can lead to a range of useful products in high yields and with high selectivity. To this end, Sun and co-workers have reported the desymmetrisation of prochiral 3,3-disubstituted oxetanes to produce a range of oxygen-containing heterocycles in generally excellent yields and enantioselectivities.82 In the presence of chiral phosphoric acid catalyst C-47, the strained oxetane ring underwent intramolecular nucleophilic ring opening by a pendant alcohol, allowing access to a range of chiral 1,4-dioxanes, as well as tetrahydropyrans and an interesting spirocyclic 1,4-oxathiane (Scheme 37).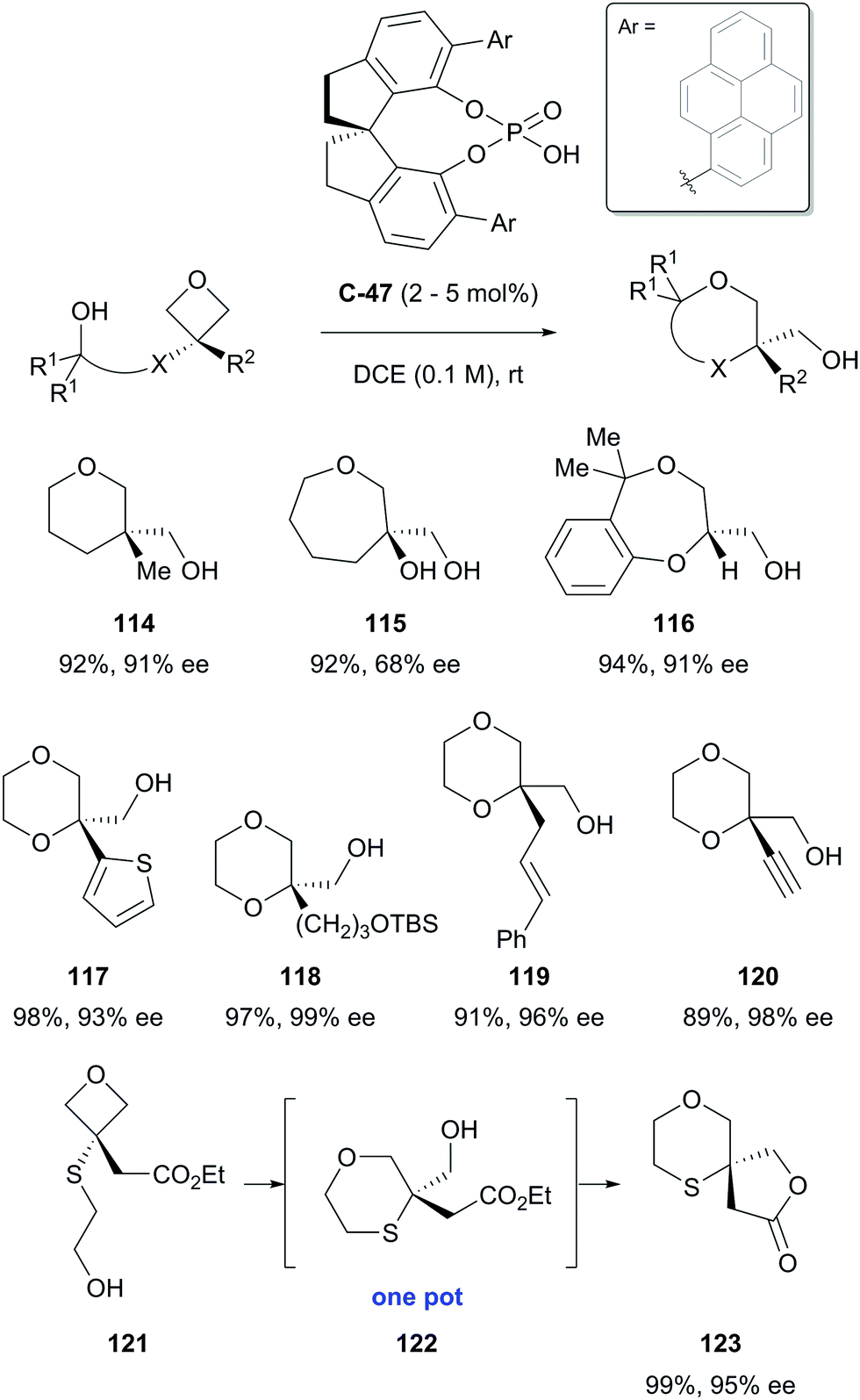 | ||
| Scheme 37 Desymmetrisation of oxetanes by Sun, affording a variety of oxa-heterocycles in excellent yields and enantioselectivities. | ||
The Sun group also published the successful desymmetrisation of prochiral 3-substituted oxetanes by ring-opening with a thiol nucleophile.83 Under chiral phosphoric acid catalysis, the standard reaction conditions afforded a wide range of highly-functionalised primary alcohols in excellent yields and with high levels of enantioselectivity, consistently greater than 90% ee. The desymmetrisation of 3-substituted oxetane substrates led to a wide range of primary alcohol products bearing tertiary chiral centres, whilst 3,3-disubstituted substrates led to generation of products bearing challenging quaternary centres. High levels of enantioenrichment were generally observed, with a low catalyst loading of 2.5 mol% (Scheme 38).
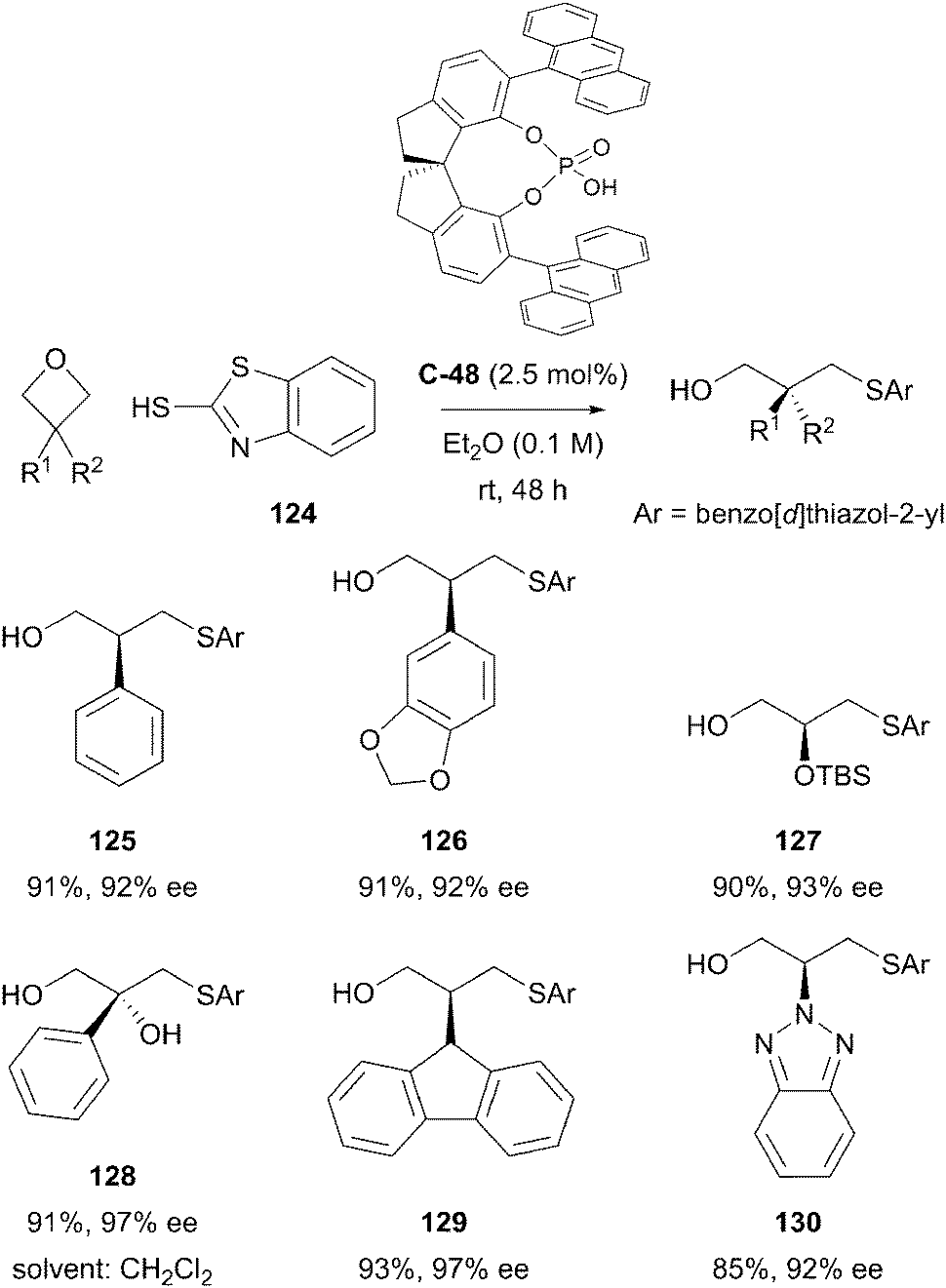 | ||
| Scheme 38 Desymmetrisation of prochiral 3-substituted oxetanes with a nucleophilic thiol under chiral phosphoric acid catalysis by Sun. | ||
Sun and co-workers have also reported the use of a prochiral 3-aryl oxetane as a directing group in a three-component hetero-Diels–Alder reaction to form complex nitrogenous polycylic products, generating four chiral centres in the process (Scheme 39).84 In the presence of a chiral phosphoric acid catalyst, an indole was employed as an electron-rich dienophile in an aza-Diels–Alder reaction with the imine formed in the reaction between an aniline and aromatic aldehyde. An initial experiment with benzaldehyde, 3,5-dimethoxyaniline and indole provided a complex mixture of products; however, when the benzaldehyde was substituted in the ortho-position with an ether to act as a hydrogen bond acceptor, the desired product was obtained in good yield and excellent diastereoselectivity, albeit in only 2–5% ee. Changing the ether to a more rigid oxetane resulted in a remarkable increase in enantioselectivity, giving the hexacycle in an impressive 98% ee and desymmetrising the 4-membered ring in the process. A wide range of substituents, including methyl, halogen, alcohol and ether groups were well-tolerated on the indole, primarily in the 5- and 6-positions. Electron-donating groups were exclusively used to decorate the aniline, while the aromatic aldehyde component could be adorned with both electron-donating and withdrawing groups. Indeed, the phenyl ring could be replaced with a thiophene while retaining great enantioselectivity, albeit at the cost of some diastereoselectivity. Furthermore, it was possible to form a chiral quaternary centre in moderate ee using a 3,3-disubstituted oxetane. Also noteworthy are the use of low catalyst loadings (2.5 mol%) and the ease of purification (simple filtration of the sparingly soluble product).
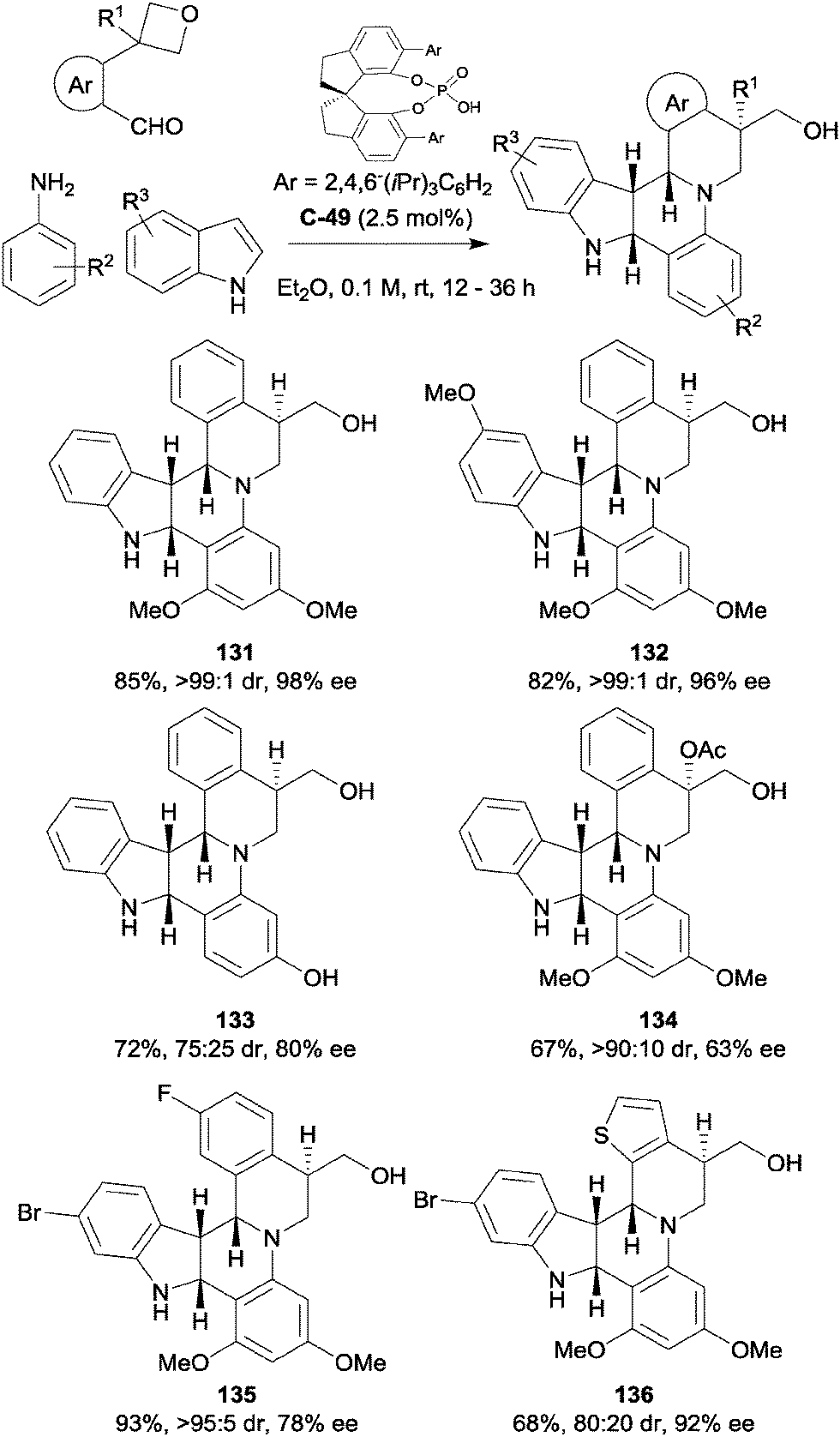 | ||
| Scheme 39 Synthesis of alkaloid-like hexacyclic products via an inverse electron demand Diels–Alder reaction directed by a prochiral 3-substituted oxetane reported by Sun. | ||
3.3 Aziridines and azetidines
The aziridine group is structurally similar to the epoxide, and nucleophilic ring opening is therefore also a common desymmetrisation strategy for these substrates. The aziridine nitrogen atom is generally protected in desymmetrisation reactions and the nature of nitrogen protecting group adds an extra variable to reaction design. Symmetric aziridines represent simple precursors for the production of challenging chiral scaffolds by enantioselective desymmetrisation and there has been significant progress since the earliest examples from over two decades ago.72List and co-workers have developed a novel approach towards the nucleophilic ring opening of meso-aziridines.85 The heterodimerisation of carboxylic acids with chiral phosphoric acids was exploited to develop what the authors describe as a new activation principle for organocatalysis. Heterodimerisation serves to increase both the acidity of the phosphoric acid (S-TRIP) and the nucleophilicity of the carboxylic acid, allowing the desymmetrisation of aziridines in excellent yields (70 to 99%) and enantioselectivities (85% to >99% ee) (Scheme 40). Both benzoic acid and acetic acid could be employed, although 7–10 equivalents were necessary to encourage association with S-TRIP, thereby preventing catalyst degradation by direct reaction with the aziridine starting material. The reaction proceeded well using a range of cyclic substrates, with slightly lower enantioselectivities for simple alkyl substituted aziridines (142).
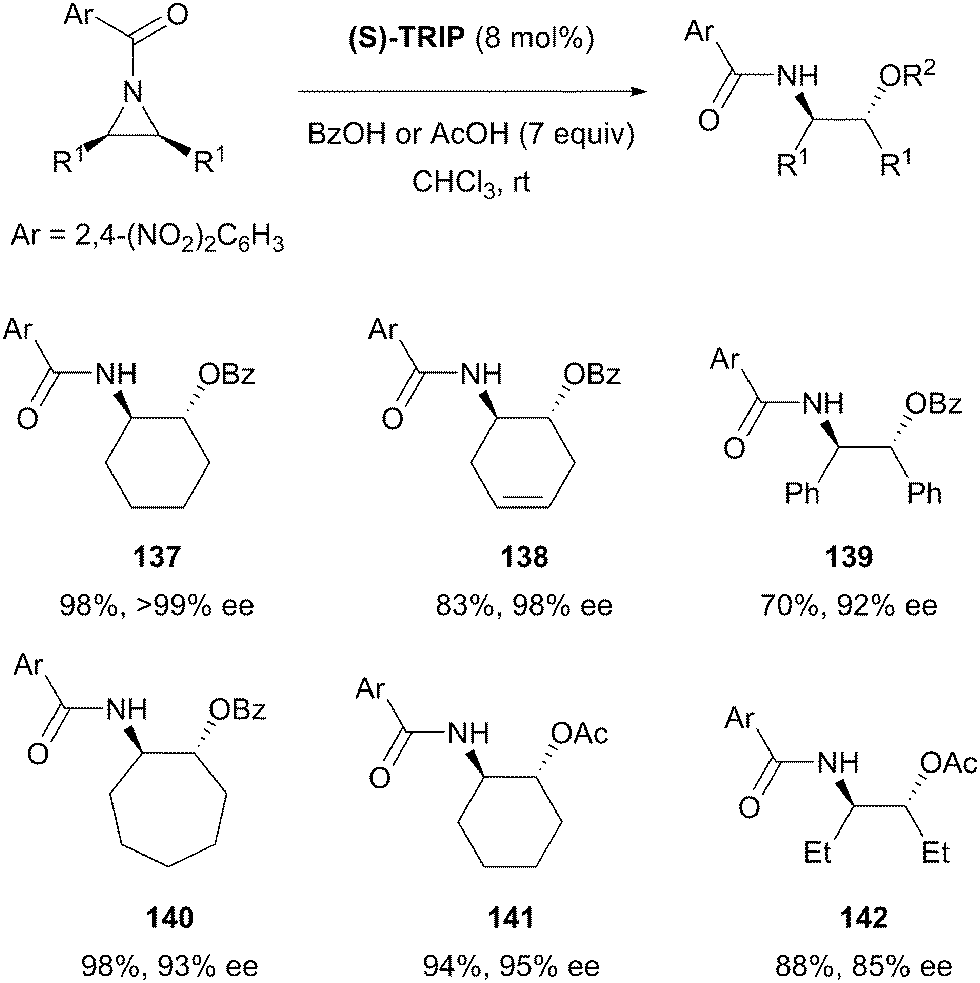 | ||
| Scheme 40 Use of a chiral phosphoric acid and achiral co-catalyst as a nucleophile activator in enantioselective desymmetrisation of meso-aziridines by List. | ||
Ooi and co-workers employed a related strategy to facilitate the enantioselective desymmetrisation of meso-aziridines with halide ions;86 the interaction of chiral triazolium chloride C-50 in combination with trimethylsilylchloride or -bromide resulted in the formation of a tight onium silicate ion pair, which acted as an effective source of nucleophilic halide. Ring opened trans-β-haloamine derivatives were obtained in high yields (83–97%) and with excellent stereocontrol (86–97% ee) (Scheme 41). The use of electron-withdrawing substituents on aryl groups at key sites on the catalyst proved crucial to achieving the highest enantioselectivities; the authors believe this increases the anion-binding ability of the catalyst, thereby forming a tighter ion pair with the silicate intermediate.
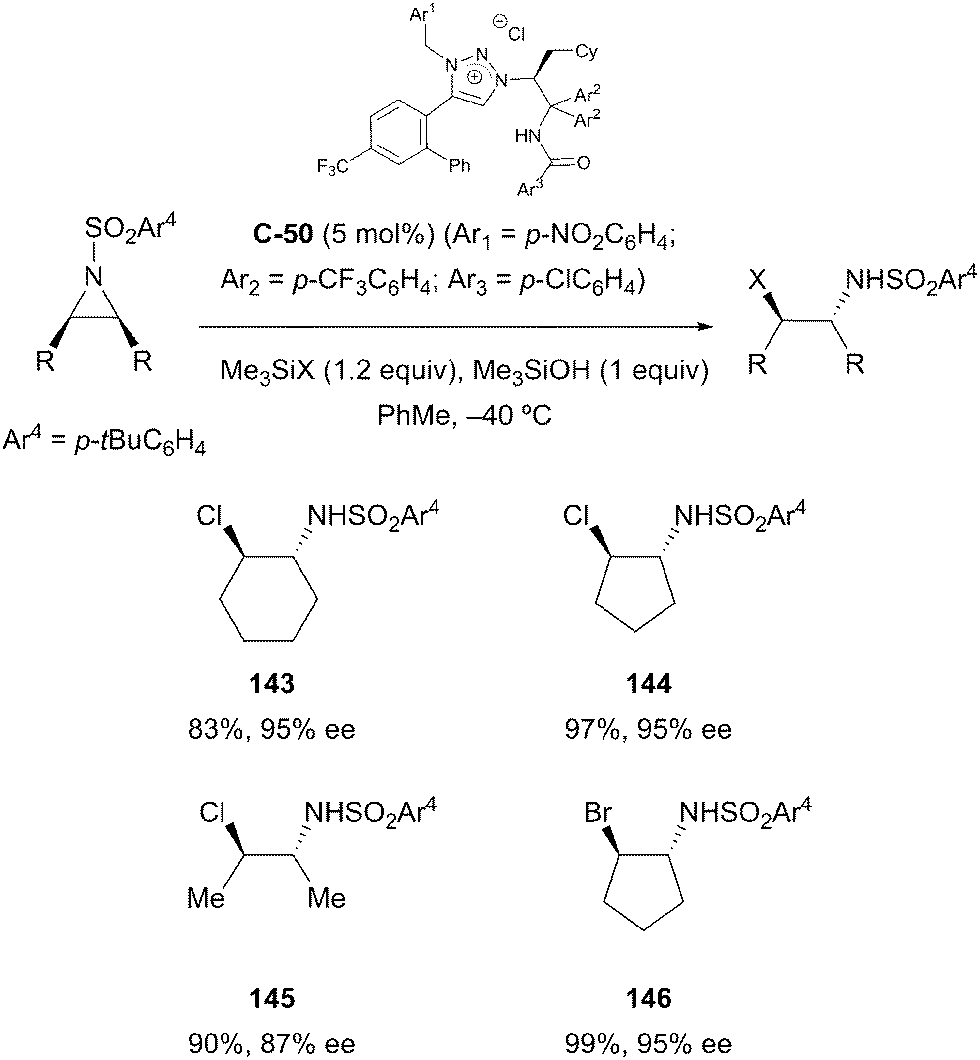 | ||
| Scheme 41 Triazolium catalysed desymmetrisation of aziridines by Ooi. | ||
Chiral Brønsted acid catalysis has also been implemented by Della Sala and co-workers to effect the desymmetrisation of meso-aziridines with a benzeneselenol nucleophile.87 The desymmetrisation afforded enriched β-aminoselenide compounds that are scarcely accessible from the chiral pool, and which can serve as useful synthetic intermediates. Products were obtained from a broad range of substrates in good yields and with high levels of enantioselectivity, up to 98% ee (Scheme 42a).
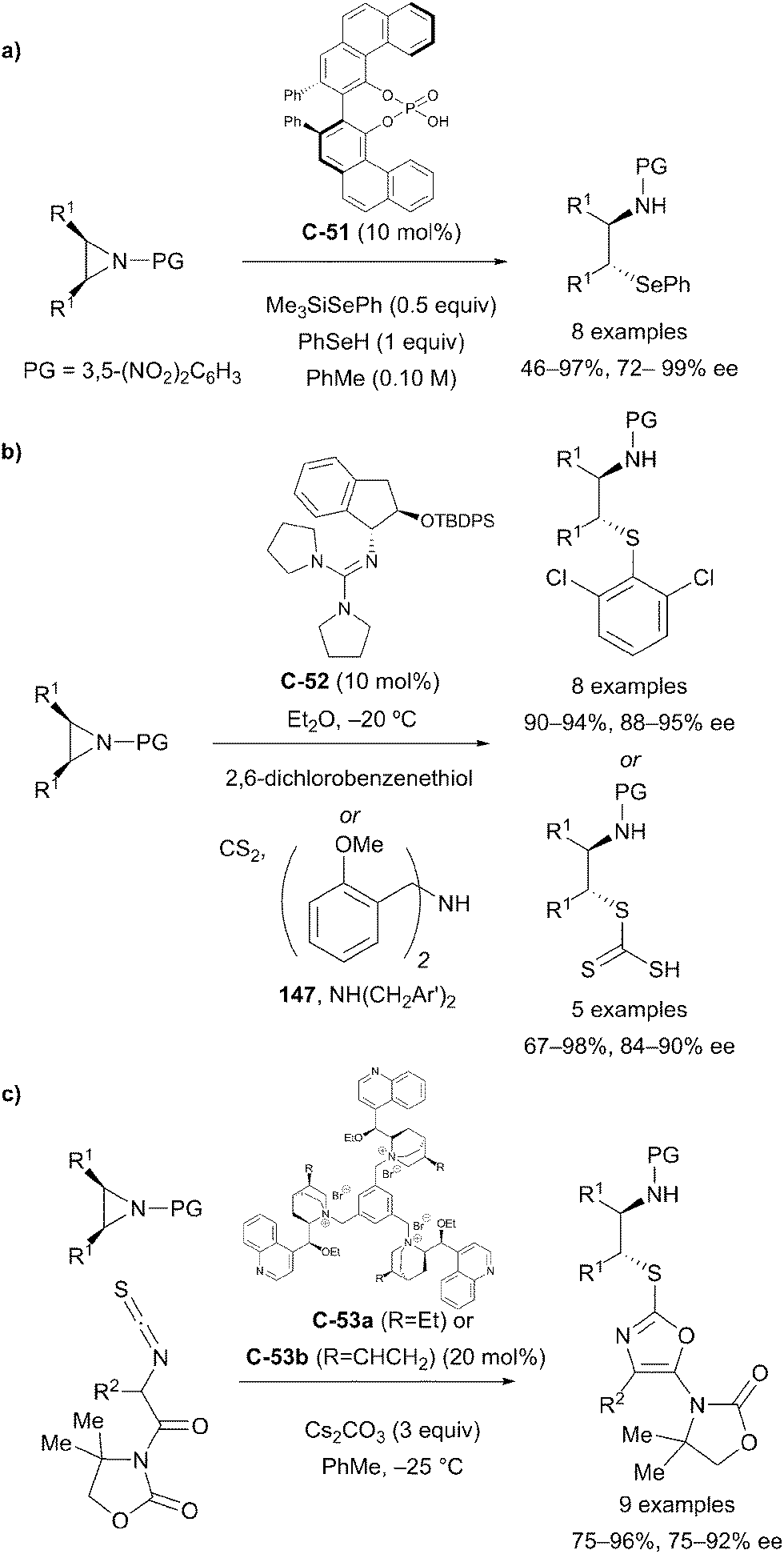 | ||
| Scheme 42 Catalytic enantioselective ring-opening reactions of meso-aziridines, by: (a) Della Sala using chiral phosphoric acid catalyst C-51; (b) Tan using guanidine catalyst C-52; (c) Wang, using trimeric cinchonidine salts C-53a and C-53b. | ||
Tan et al. have developed an aminoindanol-derived guanidine catalyst for the asymmetric nucleophilic ring opening of meso-aziridines.88 The 3,5-dinitrobenzoyl-protected symmetric aziridine substrates were subjected to nucleophilic attack with a 2,6-dichlorobenzenethiol nucleophile. The reaction afforded enantioenriched 1,2-aminosulfide products in excellent yields and with high levels of enantioselectivity (Scheme 42b).
The authors proposed two possible mechanisms for the reaction and conducted DFT calculations to explain the origin of enantioselectivity. A lowest energy transition state structure for the (R,R) product for a mechanism of nucleophilic attack by the thiolate selectively at one aziridine carbon atom of the catalyst-associated substrate was found, showing hydrogen bond activation of the aziridine and simultaneous positioning of substrate and catalyst for minimal steric interactions and enantioselective nucleophilic attack (Fig. 4).
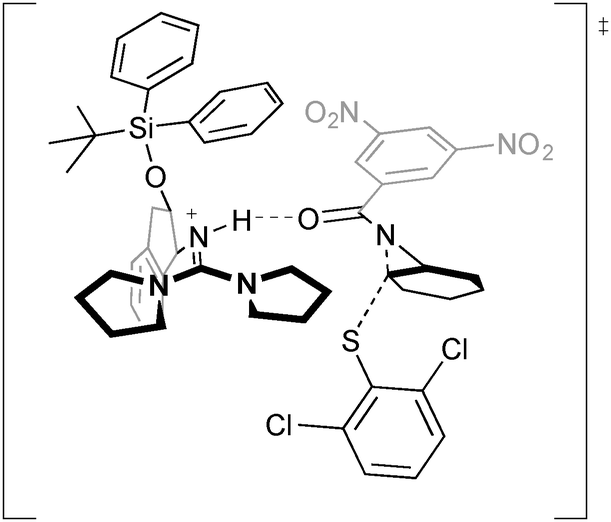 | ||
| Fig. 4 Hydrogen bonding activation for the desymmetrisation of aziridines, described by Tan using DFT calculations. | ||
The Wang group achieved the desymmetrisation of a range of cyclic meso-aziridine substrates by nucleophilic ring opening using trimeric cinchonidine catalysts C-53a and C-53b.89 Reaction of meso-aziridines with α-isothiocyanato imides afforded 2-thiooxazoles in high yields (75 to 96%) and with high enantioselectivity (75 to 92% ee, Scheme 42c).
Shang reported the enantioselective desymmetrisation of meso-aziridines via nucleophilic ring opening with aromatic thiols.90 A selection of chiral bifunctional quaternary phosphonium salt catalysts were screened. Under the optimised conditions, the reaction afforded a range of enantioenriched β-amino sulfides. Products were obtained in high yields but with only moderate enantioselectivity and substrate scope was limited in both the nucleophilic and aziridine partner (Scheme 43).
 | ||
| Scheme 43 Enantioselective desymmetrisation via nucleophilic ring-opening of meso-aziridines was reported by Shang. | ||
Despite the relative stability of the azetidine ring towards nucleophilic ring opening, Sun et al. have reported the successful enantioselective desymmetrisation of a range of azetidines.91 Using chiral phosphoric acid catalyst C-55 (5 mol%), an impressive range of enantioenriched, protected amines could be prepared via ring opening with 2-mercaptobenzothiazole 91b in excellent yields (73–98%) and enantioselectivities (76 to >99% ee, Scheme 44). A number of 3,3-disubstituted azetidines could also be desymmetrised to provide products with quaternary stereocentres, although enantioselectivity was generally lower for these substrates. The benzothiazole moiety could be removed by displacement of the thiol with sodium methoxide, increasing the synthetic utility of the method. The authors state that this is the first catalytic enantioselective intermolecular desymmetrisation of azetidine substrates.
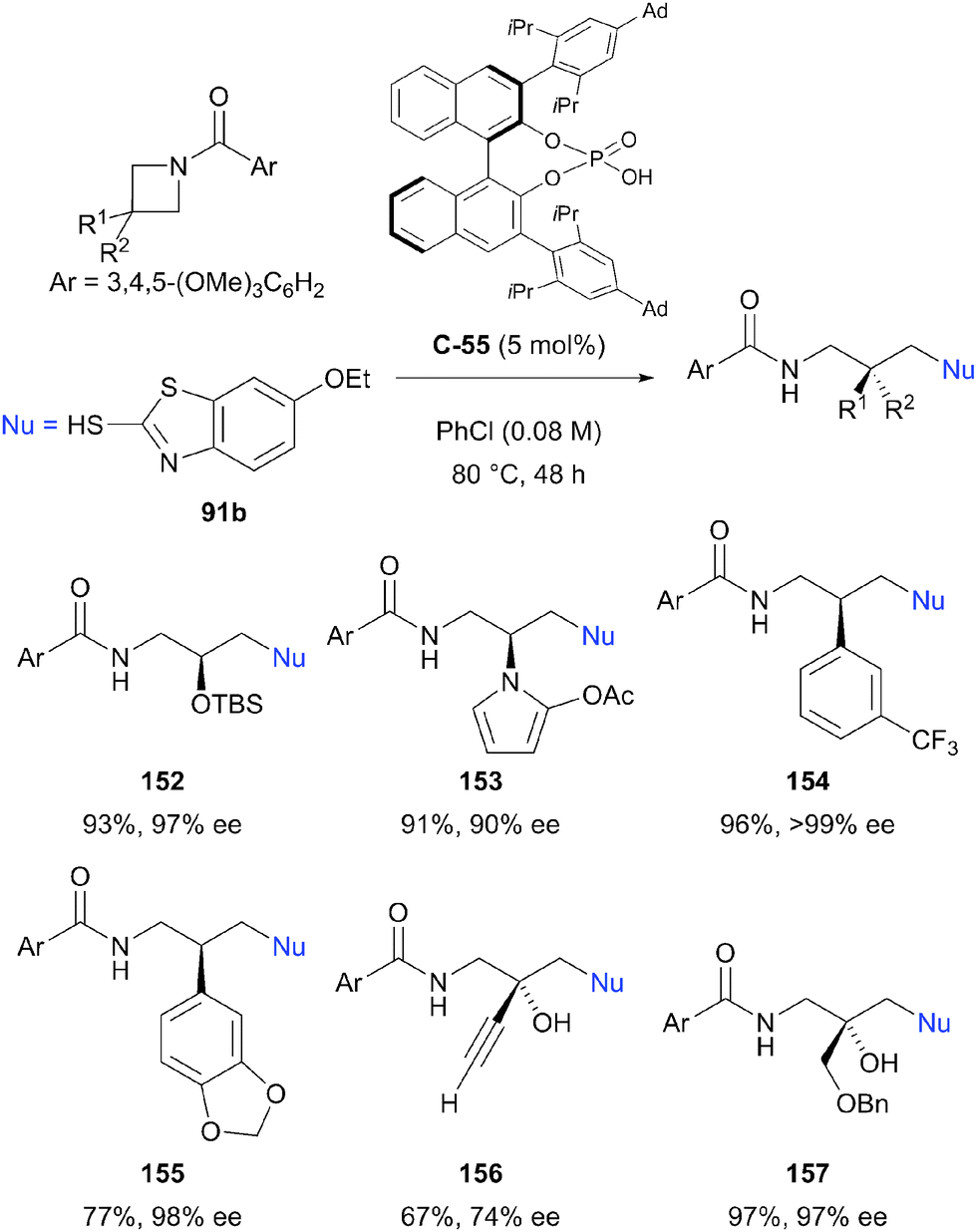 | ||
| Scheme 44 The first example of enantioselective intermolecular desymmetrisation of azetidines by nucleophilic ring opening reported by Sun. | ||
4. Diols and polyols
Symmetric diols are a highly studied substrate for organocatalytic enantioselective desymmetrisation reactions, and have received considerable attention over the past decade. This powerful transformation quickly and efficiently affords building blocks with multiple contiguous stereocentres from simple and often inexpensive starting materials. Symmetric diols undergo a range of desymmetrisation reactions, most commonly acylation, oxidation, and silylation, though other derivatisation methods such as phosphorylation and sulfonylation have been demonstrated.92 While the organocatalysed desymmetrisation of diols has been the subject of recent reviews by Diaz-de-Villegas5 and Xu,6 this review will highlight more recent advances.Some of the earliest work in the field by Vedejs identified chiral phosphines, such as C2-symmetric bis(phospholane) C-56, first developed by Burk et al.,93 as catalysts for the enantioselective acylation of several meso-1,2-diols (Fig. 5a).94 Monoacetates and monobenzoates were obtained in low to good conversion (10–84%) and low but promising enantioselectivity (19–81% ee) over eight examples using C-56. Interestingly, the authors noted the occurrence of racemisation of the monoacetate (159) of cis-cyclopentane-1,2-diol via an intramolecular acetyl transfer. This led to diminishing enantioselectivity as conversion increased (28% ee at 67% conversion, compared to 52% ee at 10% conversion), an effect not observed with other substrates.
 | ||
Fig. 5 Early work on the enantioselective acylation of meso-1,2-diols by (a) Vedejs, (b) Oriyama, and (c) of meso-1,3-diols by Oriyama; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) values represent conversion. values represent conversion. | ||
Later, Oriyama and co-workers reported good yields (73–89%) and greatly improved enantioselectivities (60–97% ee) for the enantioselective benzoyl protection of several meso-1,2-diols using chiral diamine catalyst C-57a at loadings as low as 0.5 mol% (Fig. 5b).95 The authors suggest that the two nitrogen atoms activate benzoyl chloride in a bidentate manner, creating a chiral environment responsible for the stereochemical outcome of the reaction. Furthermore, the authors applied a similar diamine catalyst C-57b to the enantioselective monobenzoylation of meso-1,3-diols (Fig. 5c). This was achieved with similarly high levels of enantioselectivity (85–98% ee), however yields were poorer (22–33%), providing an early indication of the problems facing substrates in which the enantiotopic hydroxyl groups are further separated from one another.96
4.1 Acylation of diols
Some of the earliest attempts to desymmetrise diols were achieved via enantioselective acylation. Since the early work of Vedejs and Oriyama (Fig. 5), the enantioselective acylation of diols has received considerable attention, and the area has developed substantially as a result. An array of catalyst classes has been shown to facilitate the reaction (including diamines, dimethylaminopyridines, isothioureas, N-heterocyclic carbenes (NHC), cinchona alkaloid-derived phosphinites, and phosphoric acids) with varying degrees of success.In 2003, Fujimoto and co-workers disclosed the first application of phosphinite derivatives of the cinchona alkaloids to catalyse asymmetric reactions.97 The authors developed phosphinite catalyst C-58, consisting of a Lewis basic phosphorus centre to activate the acylating agent, and a Brønsted basic quinuclidine fragment capable of hydrogen bonding and trapping protons (Fig. 6). Using the corresponding phosphinic acid catalyst, only racemic product was obtained; the authors suggest that this demonstrates the importance of the trivalent phosphinite group.
 | ||
| Fig. 6 Bifunctional cinchona alkaloid-derived phosphinite catalyst developed by Fujimoto for the enantioselective acylation of meso-1,2-diols. | ||
Cinchona alkaloid-derived phosphinite catalyst C-58 was highly effective for the enantioselective benzoylation of several meso-1,2-diols (Scheme 45).97 Excellent yields (80–99%) and good to excellent enantioselectivities (76–94% ee) were obtained for a small range of substrates, representing a significant improvement on the work of Vedejs and Oriyama. Additionally, cyclopentane-1,2-diol was desymmetrised to monobenzoate 166 in good yield and ee, with no apparent acyl transfer causing racemisation. The addition of Hünig's base was highly important, resulting in a considerable increase in yield (83% compared to 34% without; using cinchonidine catalyst analogue). Although successful, the restriction of acylating agent to benzoyl chloride (other reagents such as tBuCOCl, (PhCO)2O, and o-MeC6H4COCl resulted in poor or no reactivity) and relatively high catalyst loading of 30 mol% are considerable drawbacks to the method.
 | ||
| Scheme 45 Enantioselective benzoylation of meso-1,2-diols using cinchonine-derived phosphinite catalyst by Fujimoto. | ||
The transformation was later reported by Kundig and co-workers using quinine-derived diamine catalyst C-59 at considerably lower loadings of 2 mol% (Scheme 46).98 Some products, such as monobenzoylated cyclohexane-1,2-diol 161, are obtained with slightly improved yield and enantioselectivity compared to the work of Oriyama and Fujimoto, however in general the approach did not offer a substantial improvement, especially in terms of enantioselectivity. The authors attribute the lower yields and enantioselectivities for 1,2-diphenylethane-1,2-diol (ent-160) and 1,2,3,4-tetrahydronapthylene-2,3-diol (168) to their poorer solubility in THF, the reaction solvent.
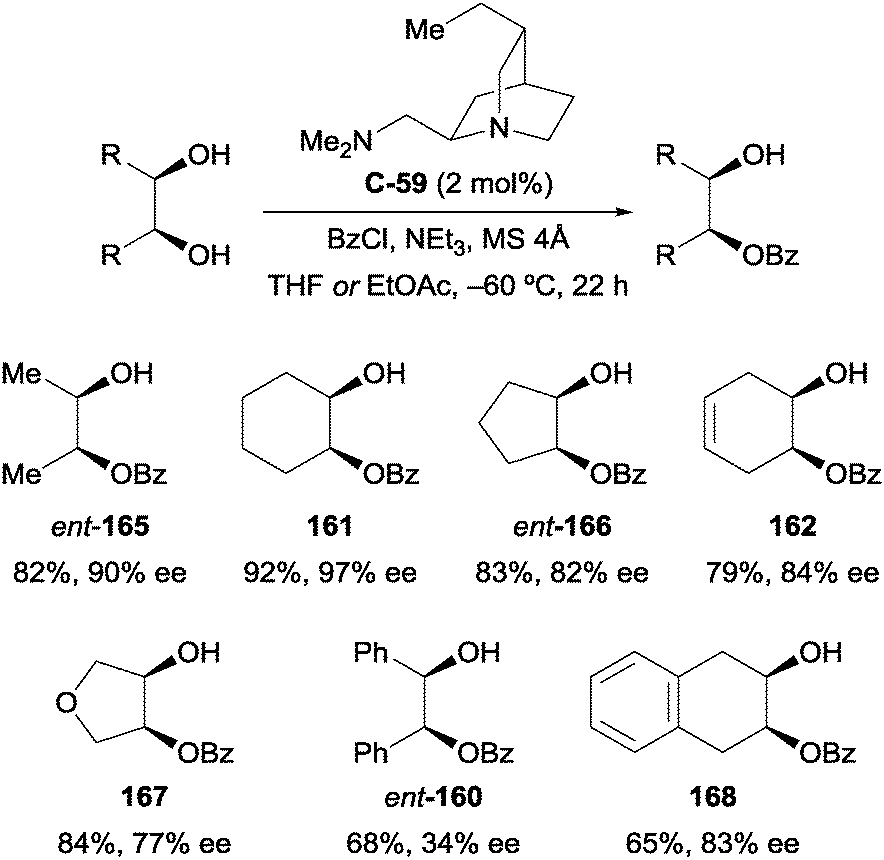 | ||
| Scheme 46 Enantioselective benzoylation of meso-1,2-diols using quinine-derived diamine catalyst by Kundig. | ||
The authors determined that formation of the corresponding diester occurs by a competing kinetic resolution, whereby acylation of the residual hydroxyl group on the minor enantiomer is fast, resulting in enrichment of the major enantiomer. This was demonstrated by subjecting a racemic mixture of monobenzoylated cyclohexane-1,2-diol (rac-161) to the reaction conditions, after which the major enantiomer (161) was enriched (76% ee), and there was significant formation of the corresponding diester (169, 44% yield; Scheme 47).
 | ||
| Scheme 47 Kundig's kinetic resolution of monobenzoylated products leading to formation of diester, and enrichment of the starting material. | ||
More recently, Paradies and co-workers used the cinchona alkaloid derived phosphinite catalyst C-60 (the quinidine analogue of C-58) under similar conditions as those described by Fujimoto to carry out enantioselective desymmetrisation of meso-4,6-protected myo-inositol derivatives.99 Typically, synthetic strategies to obtain inositol phosphates rely on extensive use of protecting groups. Using similar conditions to that of Fujimoto (Scheme 45), quinidine-derived phosphinite catalyst C-60 was used to enantioselectively benzoylate several 4,6-protected myo-inositol derivatives in excellent yield and enantioselectivity (Scheme 48). The reaction was tolerant of several different protecting groups, allowing the synthesis of multiple orthogonally protected enantiopure myo-inositol derivatives (171–174). The reaction was less selective when carried out on 2,4,6-protected inositol, affording the 2,3,4,5-protected derivative 174 with poor enantioselectivity (15% ee). This result suggests that interaction of the C2 hydroxyl group with the quinuclidine scaffold is essential for high enantioselectivity.
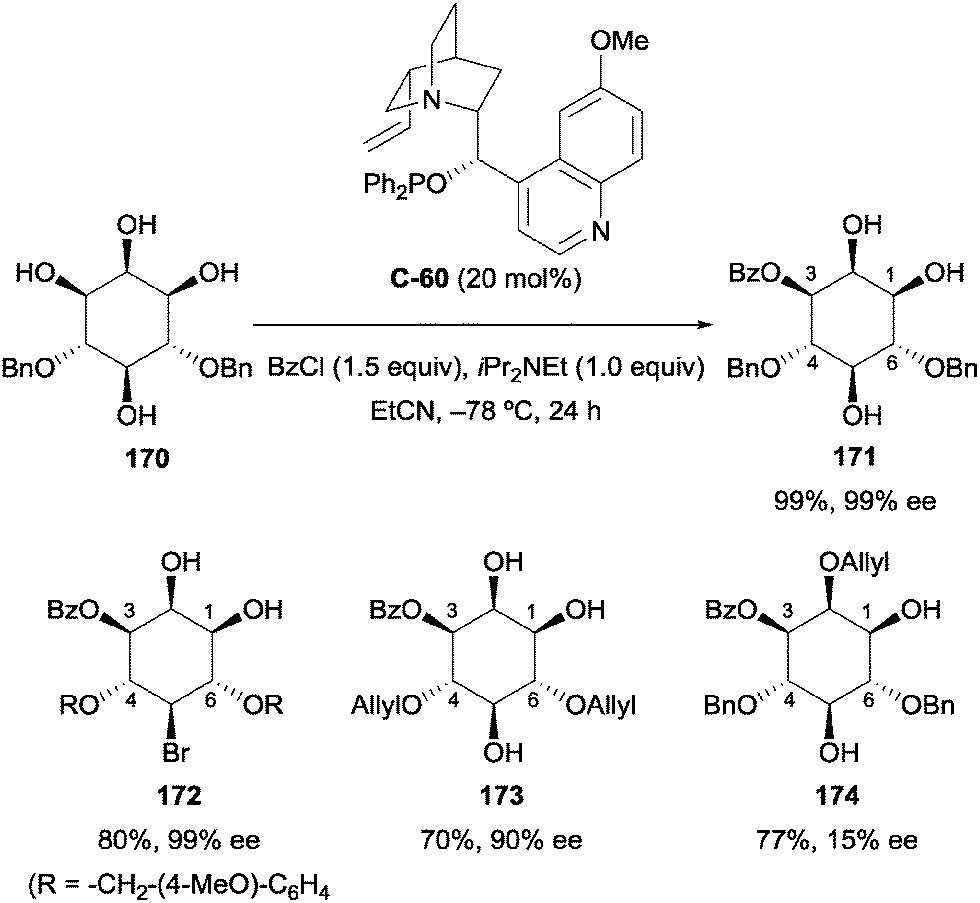 | ||
| Scheme 48 Desymmetrisation of 4,6-protected myo-inositol using chiral phosphinite catalyst by Paradies. | ||
Another class of organocatalysts which has seen significant development in recent years is the oligopeptides.3,4 In 2005, Miller and co-workers used peptide catalyst C-61 to carry out the enantioselective acylation of a range of achiral glycerol derivatives with good to high levels of enantioselectivity (up to 97%; Scheme 49).100 The highest enantioselectivities required a larger excess of Ac2O, which resulted in considerable formation of the corresponding achiral diester, limiting the usefulness of the method. Desymmetrisation of meso secondary 1,3-diols to afford monoacetates 176 and 177 resulted in poorer enantioselectivity (43% and 17% ee respectively), which the authors speculate could be due to additional syn-pentane interactions disfavouring adoption of a reactive conformation.
 | ||
| Scheme 49 Enantioselective desymmetrisation of achiral 1,3-diols using chiral diamine catalyst by Miller; aconversion to diester in parentheses; b Ac2O (2 equiv.) used. | ||
Schreiner and co-workers recently developed a more broadly applicable oligopeptide catalyst for the enantioselective monoacylation of 1,2-diols.101 Catalyst C-62 (Scheme 50) features a π-methyl-histidine (N-methyl imidazole) residue, the catalytic moiety responsible for the desymmetrisation, and an inert adamantane amino acid unit, which improves both rigidity and solubility. Oligopeptide catalyst C-62 was effective at desymmetrising a small range of cyclic and acyclic substrates by mono-acylation in moderate to excellent yield (45–97%) and moderate to high enantioselectivity (38–90% ee). The authors noted the occurrence of intramolecular acyl transfer for some substrates, which manifested itself in an erosion of ee caused by racemisation during workup. For example, the ee of the cyclohexane-1,2-diol monoacetate determined before workup was 88%, which decreased substantially to only 60% afterwards.
 | ||
| Scheme 50 Enantioselective desymmetrisation/oxidation of 1,2-diols using oligopeptide catalyst developed by Schreiner; ayields and ees after desymmetrisation step in parentheses. | ||
In order to overcome this limitation, the authors sought to oxidise the remaining alcohol in situ to prevent the problematic acyl transfer. The addition of TEMPO led to rapid oxidation, affording α-acetoxy ketones 178–181 in good yield and good enantioselectivity, with ees comparable to those observed after the desymmetrisation step (Scheme 50).
The catalyst was less successful when applied to 1,3-, 1,4-, and 1,5-diols, with the monoacylated products being obtained with poor enantioselectivity (4–39% ee) (Fig. 7).102
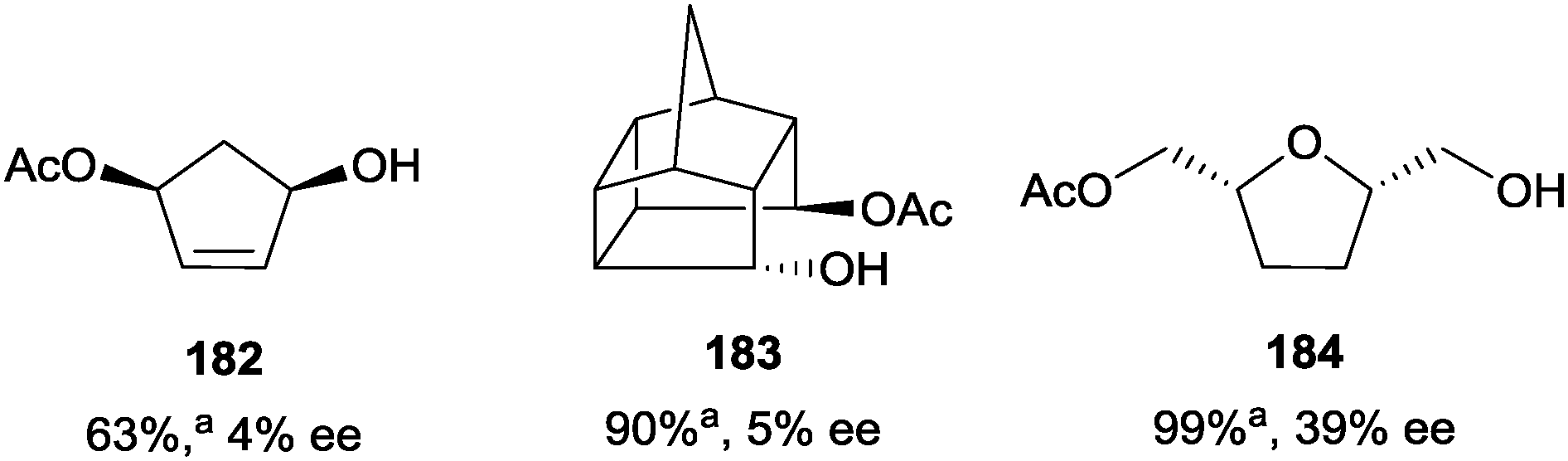 | ||
| Fig. 7 Enantioselective acylation of 1,3-, 1,4-, and 1,5-diols using Schreiner's oligopeptide catalyst C-62; aValues represent conversions measured by GC. | ||
As a further development of their one-pot desymmetrisation–oxidation strategy, Schreiner and co-workers explored a novel multicatalyst route. This led to the design of several oligopeptide catalysts containing independent catalyst moieties for both desymmetrisation and oxidation. By appending a TEMPO moiety at the terminus of the peptide catalyst (C-63, Scheme 51), the authors were able to achieve good yields and moderate enantioselectivities in a tandem desymmetrisation/oxidation reaction.103 Additionally, the rate of oxidation was increased using oligopeptide catalyst C-63 compared to the use of free TEMPO. For example, oxidation of cyclohexane-1,2-diol monoacetate to α-acetoxy ketone 178 took 1 h to reach >99% yield with catalyst C-63, while taking 3 h with free TEMPO.
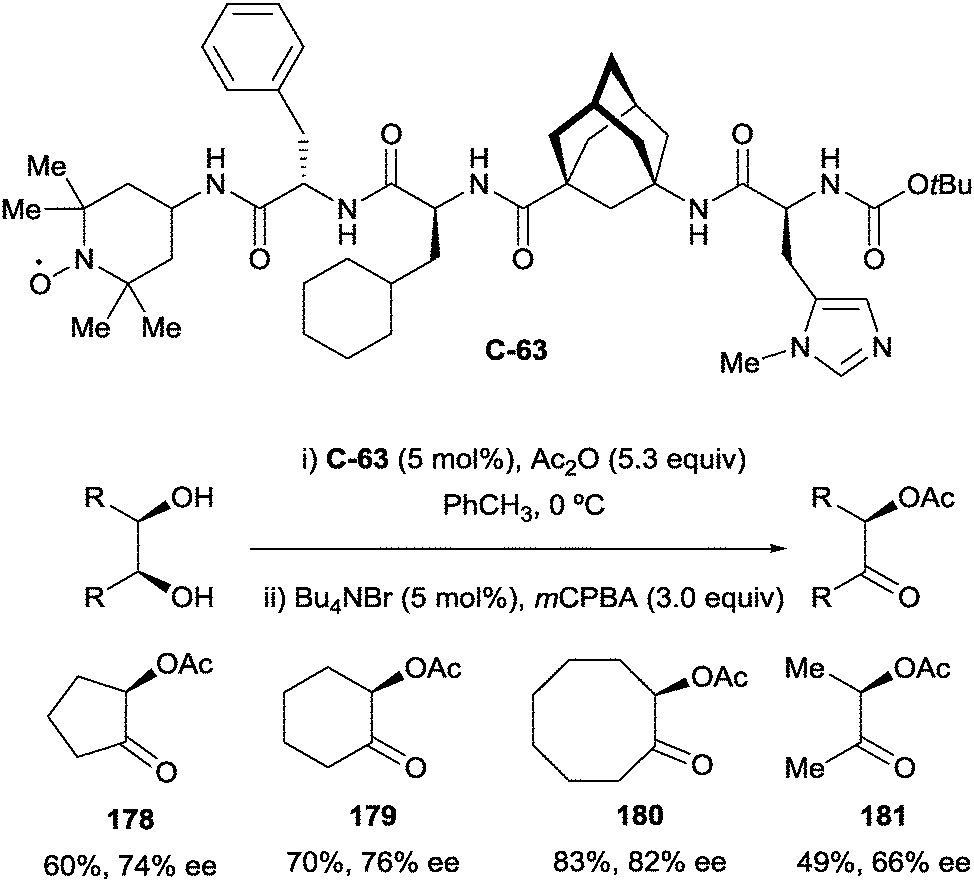 | ||
| Scheme 51 Tandem enantioselective desymmetrisation/oxidation using oligopeptide catalyst bearing TEMPO moiety developed by Schreiner. | ||
In general, while the yields and enantioselectivites using a TEMPO catalytic moiety do not surpass those using free TEMPO, this work presents a powerful and exciting idea of creating extended networks of catalytic moieties on oligopeptide backbones, to carry out diverse transformations with a single catalyst in one pot.
The examples given so far demonstrate that methods to enantioselectively acylate meso-1,2-diols are often less effective when applied to meso-1,3-diols. This is often attributed to the greater distance between the two hydroxyl groups, and the increased degrees of freedom which that often entails, making enantiotopic differentiation more challenging.
Ishihara and co-workers have also developed a amino acid-derived catalyst (C-64) for the enantioselective acylation of achiral and meso glycerol derivatives (Scheme 52).104 The catalyst afforded monoacylation products in moderate yields (up to 74%), typically with high levels of enantioselectivity (86–99% ee). Some substrates proved less effective and selective, such as methyl- and benzyl protected amines 188 and 189 (55% and 11% ee respectively), and all-syn derivative 190 (4% ee).
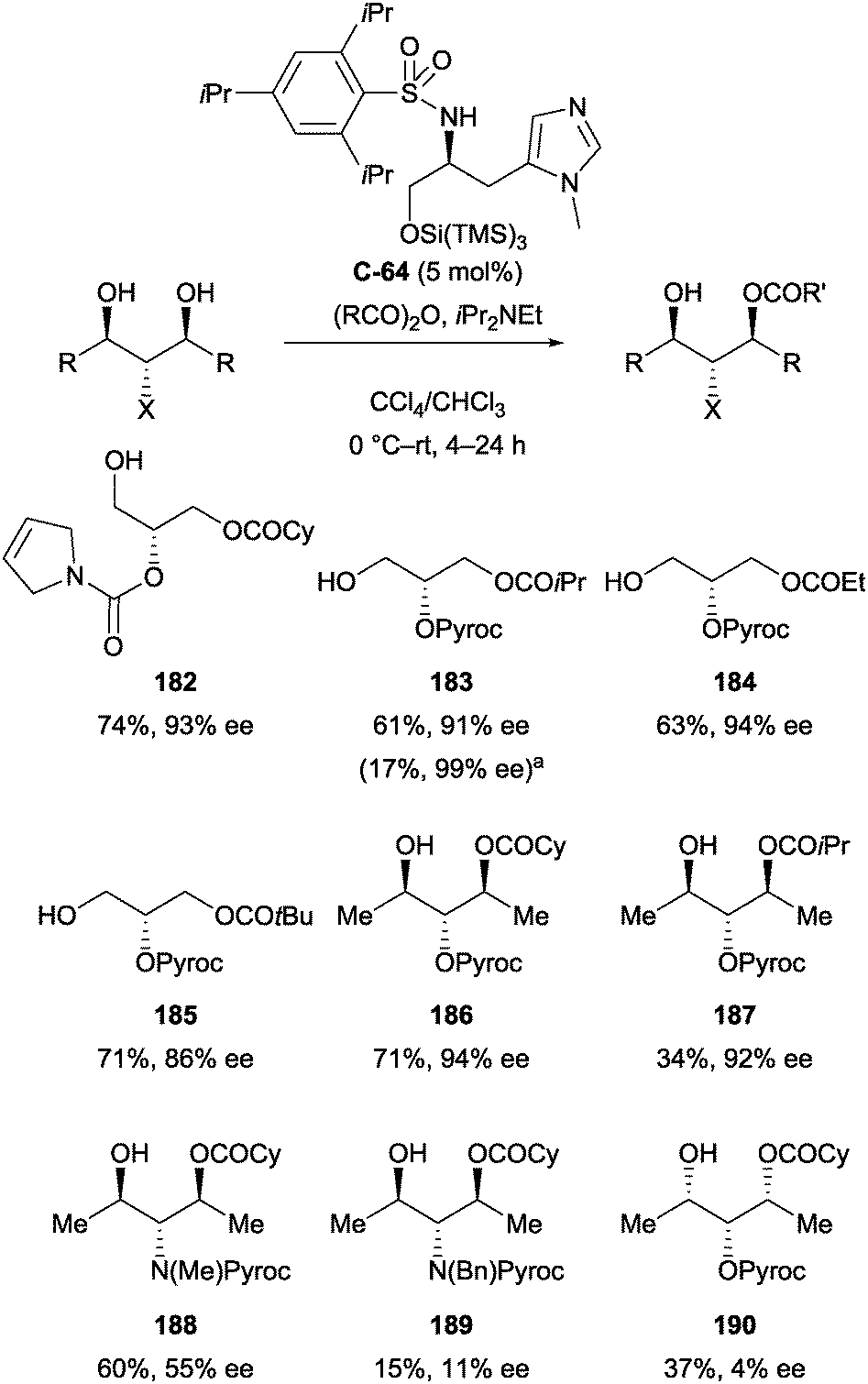 | ||
| Scheme 52 Desymmetrisation of achiral and meso glycerol derivatives using catalyst C-64 by Ishihara; a−20 °C, 3 h. | ||
S. Yamada and co-workers demonstrated enantioselective acylation using dimethylaminopyridine (DMAP) derivative C-65, achieving moderate to good yields (31–87%).105 Enantioselectivity was varied (18–97% ee) owing to the diverse nature of substrates investigated. High enantioselectivity was observed for monoacylation of 1,2- (191), acyclic 1,4- (193), and 1,6-diols (195), while 1,3- and cyclic 1,4-diols proceeded with much poorer enantioselectivity (Scheme 53).
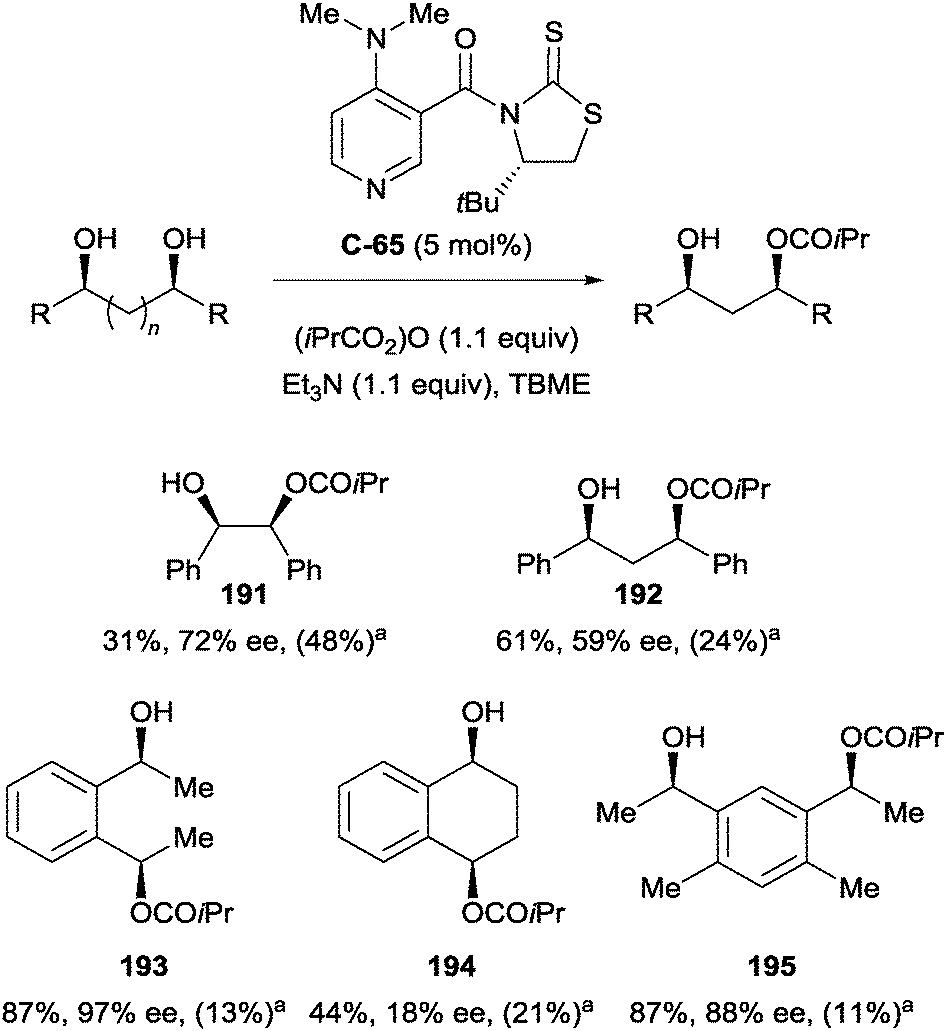 | ||
| Scheme 53 Enantioselective acylation by S. Yamada on range of diols using DMAP derived catalyst; ayield of diester in parentheses. | ||
The high yield of the corresponding diester of 191 suggests the occurrence of acyl transfer and subsequent second acylation resulting in kinetic resolution, in line with the findings of Kundig and co-workers (vide supra, Scheme 47). The authors suggest that the cause for low enantioselectivity in the case of cyclic 194vs. acyclic 193 could be due to conformational differences of the hydroxyl groups. The application of C-65 to a broader range of diols separates this work from other examples, which are typically limited to one class of diols.
The authors carried out a thorough analysis to rationalize the stereochemical outcome of the reaction. X-ray studies supported the idea of π–cation interactions between the pyridinium ring and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond, as the S–Py distance is reduced upon formation of the pyridinium salt (Fig. 8a). Additionally, NMR studies suggested a predominance of acylated conformer I over II, whereas in the case of the pyridine analogue no such preference was observed. DFT energy level calculations supported this result, and provide an explanation for facial selectivity (Fig. 8b). Finally, the authors suggest that enantiotopic differentiation is caused by a difference in steric interactions of one hydroxyl group over the other.
S bond, as the S–Py distance is reduced upon formation of the pyridinium salt (Fig. 8a). Additionally, NMR studies suggested a predominance of acylated conformer I over II, whereas in the case of the pyridine analogue no such preference was observed. DFT energy level calculations supported this result, and provide an explanation for facial selectivity (Fig. 8b). Finally, the authors suggest that enantiotopic differentiation is caused by a difference in steric interactions of one hydroxyl group over the other.
 | ||
| Fig. 8 Rationalisation for stereochemical outcome in the kinetic resolution of racemic alcohols and the enantioselective desymmetrisation of meso-diols by S. Yamada. | ||
Bressy and co-workers have recently demonstrated a much improved method for the enantioselective acylation of meso-1,3-diols using an isothiourea catalyst.106 Isothiourea catalysts have been used previously in enantioselective desymmetrisations, for example Birman and co-workers used catalyst BTM (C-66) to carry out the enantioselective acylation of 1,7-diol lobelanidine 196 (en route to a synthesis of (−)-lobeline 198) in excellent yield (92%) and enantioselectivity (>99% ee, Scheme 54).107
 | ||
| Scheme 54 Enantioselective acylation of 1,7-diol using isothiourea catalyst BTM en route to the synthesis of (−)-lobeline, by Birman; ayield of diester in parentheses. | ||
In catalyst optimisation studies, the authors found that Smith's HyperBTM catalyst (C-67) was more effective than BTM (C-66) under the same conditions (96% ee compared to 82% ee). The monoacylated product could be obtained in greater yield and enantioselectivity while reducing reaction time, temperature, and catalyst loading (Scheme 55).
 | ||
| Scheme 55 Catalyst optimisation by Bressy, demonstrating the increased efficiency of HyperBTM over BTM; areaction carried out at −20 °C with 2 mol% C-67 in 2.5 h. | ||
The authors demonstrated the method on a range of olefinic 1,3-diols with good yields (46–91%) and enantioselectivity (84–98%; Scheme 56). The non-olefinic and non-aromatic substrates (R = PhCH2CH2– 204, and CH3CHCH– 205) resulted in poorer yields (38% and 46% respectively) while the former also suffered from considerably poorer enantioselectivity (21% ee). These results suggest the importance of both olefinic and aromatic groups on the efficiency and stereochemical outcome of the reaction. The high enantioselectivities observed are driven by an in situ kinetic resolution, whereby the residual hydroxyl group on the minor enantiomer rapidly undergoes second acylation. The authors termed this process chiroablative kinetic resolution, in line with the observation has been made previously by Kundig and others (vide supra, Scheme 47).
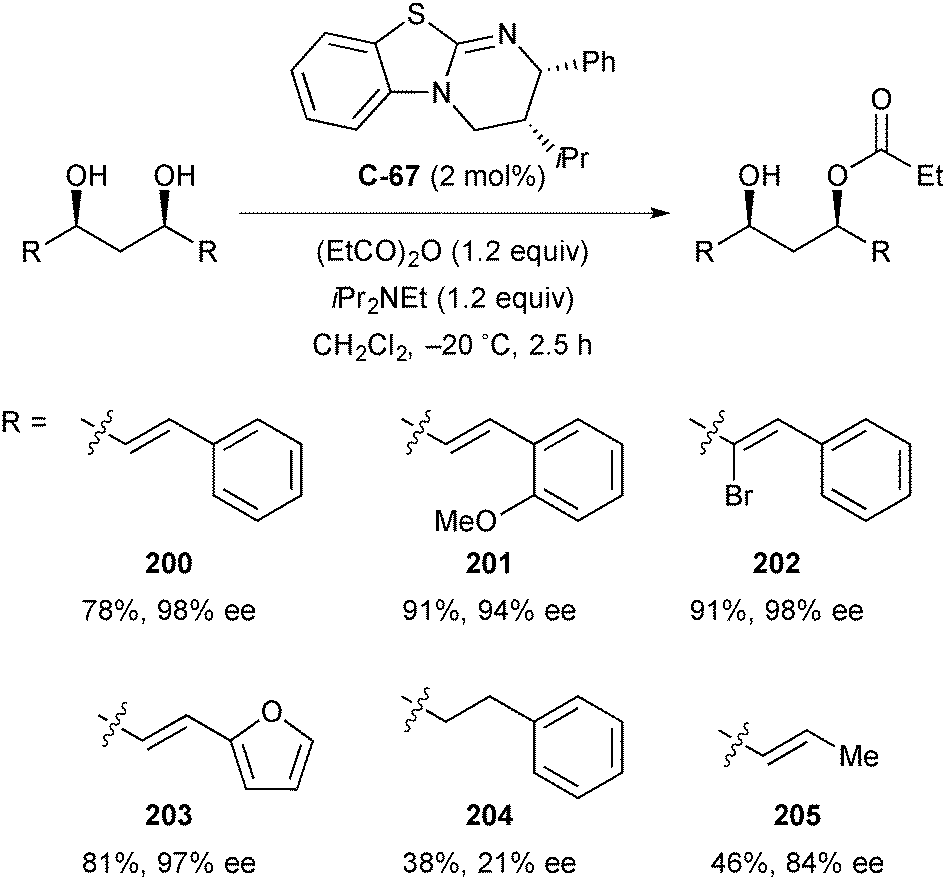 | ||
| Scheme 56 Enantioselective acylation of meso-1,3-diols by desymmetrisation/chiroablative kinetic resolution by Bressy. | ||
The authors validated the kinetic resolution hypothesis by monitoring the change in ee over the course of the reaction. The results showed good correlation between increasing formation of the diester with enantioenrichment of the monoester. Furthermore, the desymmetrisation was applied to a total synthesis of (−)-diospongin A (207, Scheme 57) using the enantiomeric form of BTM, catalyst C-68, demonstrating the versatility of the method in synthesizing either enantiomer with high selectivity.
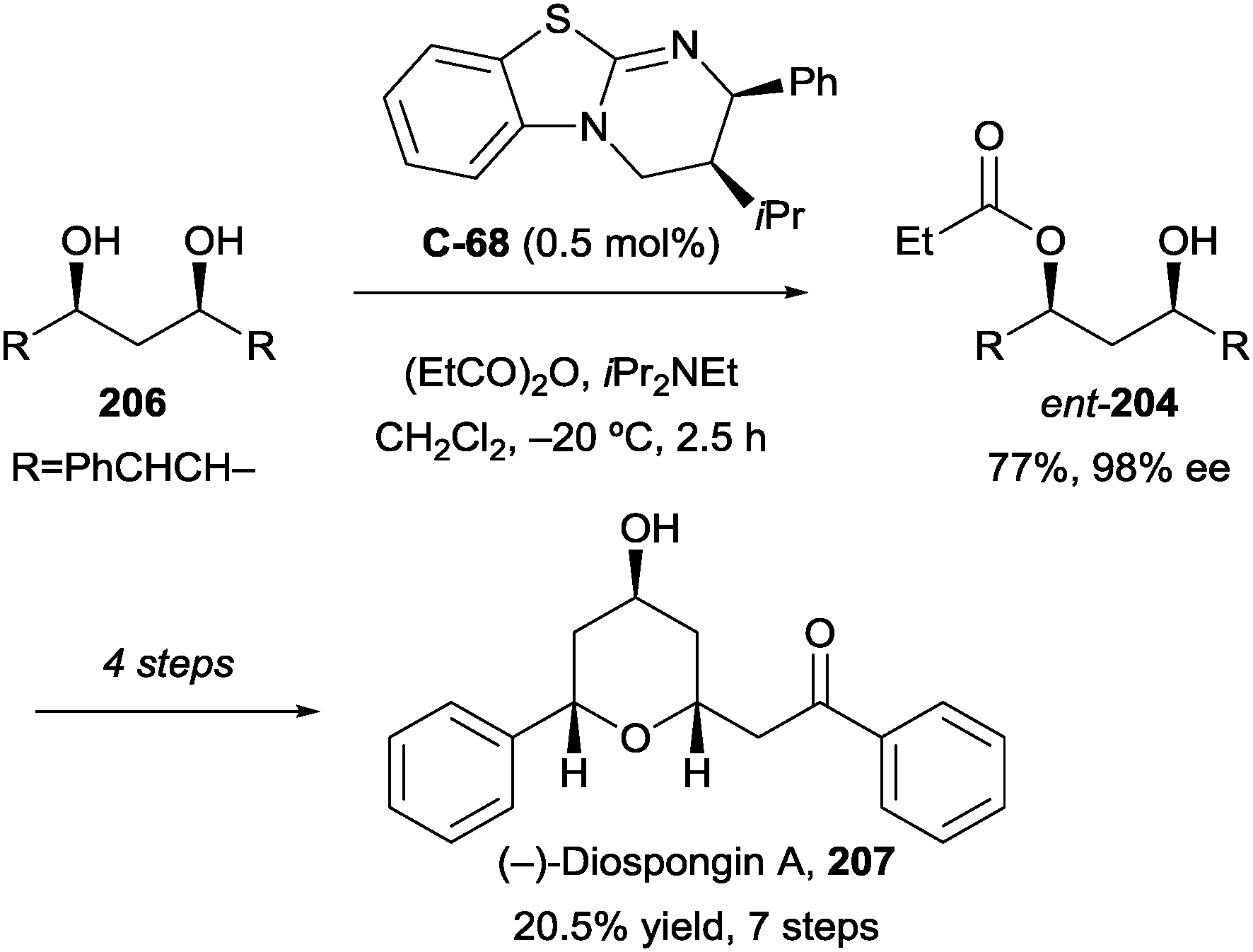 | ||
| Scheme 57 Application of desymmetrisation/kinetic resolution to the synthesis of (−)-diospongin A by Bressy. | ||
K. Yamada and co-workers developed a strategy for the kinetic resolution of racemic diols using NHC catalysts with carboxylate salts.108 The authors proposed (based on DFT calculations) that, during the kinetic resolution of racemic 1,2-diols the carboxylate salt deprotonates one hydroxyl group, while the other stabilises this transition state by hydrogen-bonding (Fig. 9).
 | ||
| Fig. 9 Transition state for NHC–carboxylate salt kinetic resolution of rac-diols, proposed by K. Yamada, illustrating the participation of both hydroxyl groups. | ||
This hypothesis was validated by demonstrating that the presence of an additional hydrogen-bond donor was necessary for the reaction to proceed (Scheme 58a), with no reaction observed for the monoalkylated substrate 209 (R = Me). The authors then applied the method to the enantioselective desymmetrisation of 2-amino-meso-1,3-diol 214 with excellent yield (98%) and enantioselectivity (>99% ee) (Scheme 58b), demonstrating the excellent level of control that NHC catalyst C-69 affords.
 | ||
| Scheme 58 (a) Kinetic resolution of racemic 1,2-diols and (b) enantioselective acylation of 2-amino-meso-1,3-diol using chiral NHC catalyst with carboxylate salt co-catalyst by K. Yamada; ano reaction. | ||
Zheng and co-workers have developed a procedure for the enantioselective acylation of meso-1,3-diols via the enantioselective oxidation of benzylidene acetals to the monoester using DMDO and chiral phosphoric acid proton transfer catalysts (Scheme 59).109
 | ||
| Scheme 59 Optimisation of acetal group for Zheng's enantioselective acylation of meso-1,2-diols. | ||
During screening of chiral phosphoric acids, the authors identified (S)-TRIP (C-70) as a highly efficient catalyst, resulting in excellent enantioselectivity when using 2-phenylpropane-1,3-diol as the model substrate (Scheme 59). The optimal acetal was found to be p-methoxyphenyl (PMP), resulting in better yields and enantioselectivities compared to phenyl. With the aid of DFT calculations, the authors proposed that the origin of enantioselectivity arises from attractive aryl–aryl interactions between the substrate PMP group and the 2,4,6-triisopropylphenyl group of the catalyst.
The procedure was very effective on a range of substrates, achieving excellent yields and enantioselectivity in most cases (Scheme 60). For 2-aryl substituted-1,3-diols, both electron-withdrawing and electron-donating substituents at the ortho-, meta-, and para-positions of the 2-aryl group were well tolerated. Monoalkyl substrates 2-benzyl-propane-1,3-diol (223) and 2-tert-butyl-1,3-propanediol (224) underwent the transformation in good yields but with reduced enantioselectivity (80 and 74% ee respectively) compared to 2-aryl substituted examples.
 | ||
| Scheme 60 Enantioselective acylation of 1,3-diols via benzylidene acetals using (S)-TRIP (C-70) by Zheng. | ||
The authors also demonstrated that the method could be applied to 2,2-disubstituted-1,3-diols to furnish chiral quaternary centres in similarly high yields and enantioselectivities (Scheme 60). Additionally, the procedure could be carried out on gram-scale, providing an effective and reliable two-step route to enantioenriched monoprotected 1,3-diols.
Chen and co-workers have recently demonstrated the first example of enantioselective desymmetrisation by acetalization of 1,3-diols.110 Acetal-tethered 1,3-diols underwent trans-acetalization in the presence of chiral phosphoric acid catalyst C-71 to furnish a range of aryl substituted THF derivatives (228–233). The process generates two stereocentres in excellent yield, diastereo- and enantioselectivity in most cases. A modified phosphoric acid catalyst (C-72) was necessary to achieve syntheses of analogous THP derivatives (234, 235), but excellent yields and enantioselectivities were also achieved on a limited range of substrates (Scheme 61).
 | ||
| Scheme 61 Enantioselective acetalization of 1,3-diols using chiral phosphoric acid catalysts by Chen; an = 1, catalyst C-71; bn = 2, catalyst C-72. | ||
4.2 Oxidation of 1,2-diols
Examples of organocatalytic methods for the enantioselective oxidation of meso-1,2-diols are limited. The transformation was first reported in 2006 by C. G. Zhao and co-workers using Shi's oxazolidinone catalysts.111 This afforded mono-oxidation products of aryl substituted meso-1,2-diols in good yield and enantioselectivity, arising from preferential oxidation of the (S)-centre (Scheme 62).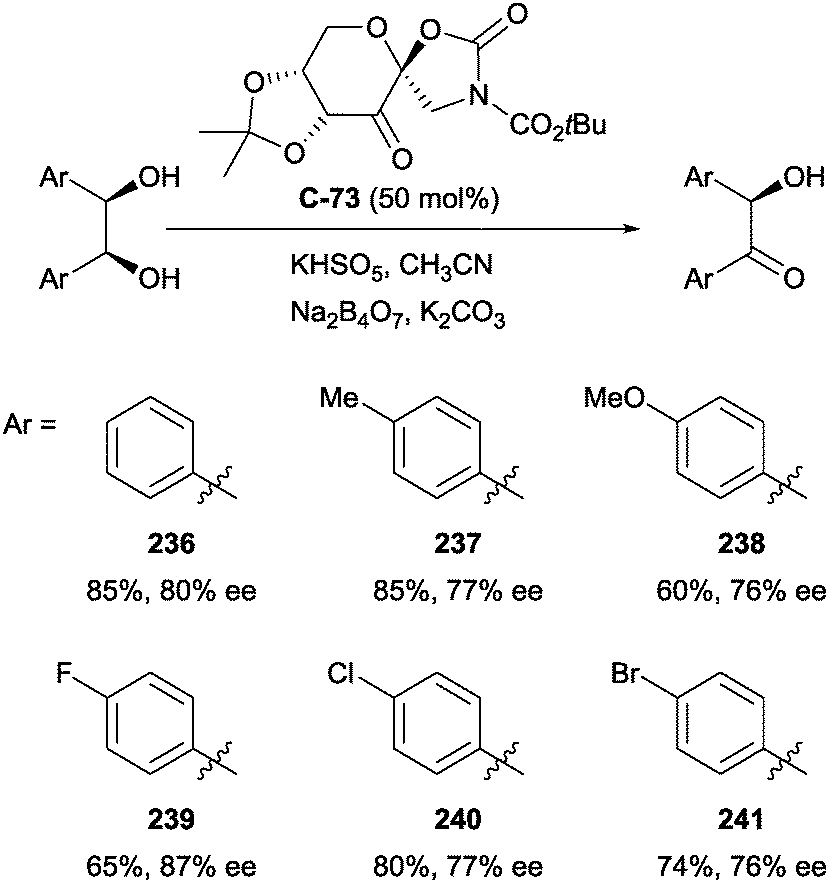 | ||
| Scheme 62 Enantioselective oxidation of aryl substituted meso-1,2-diols by C. G. Zhao. | ||
More recently, the use of quinine-derived urea catalyst C-74 to carry out enantioselective oxidation of meso-diols has been reported by Y. Zhao and co-workers, affording mono-oxidation products in high yields with good selectivity (Scheme 63).112 The authors initially investigated the use of quinine-derived amides as ligands for the copper-catalysed oxidation of meso-diols, using N-bromosuccinimide (NBS) as the oxidant. However, the reaction proceeded with much higher enantioselectivity in the absence of copper.
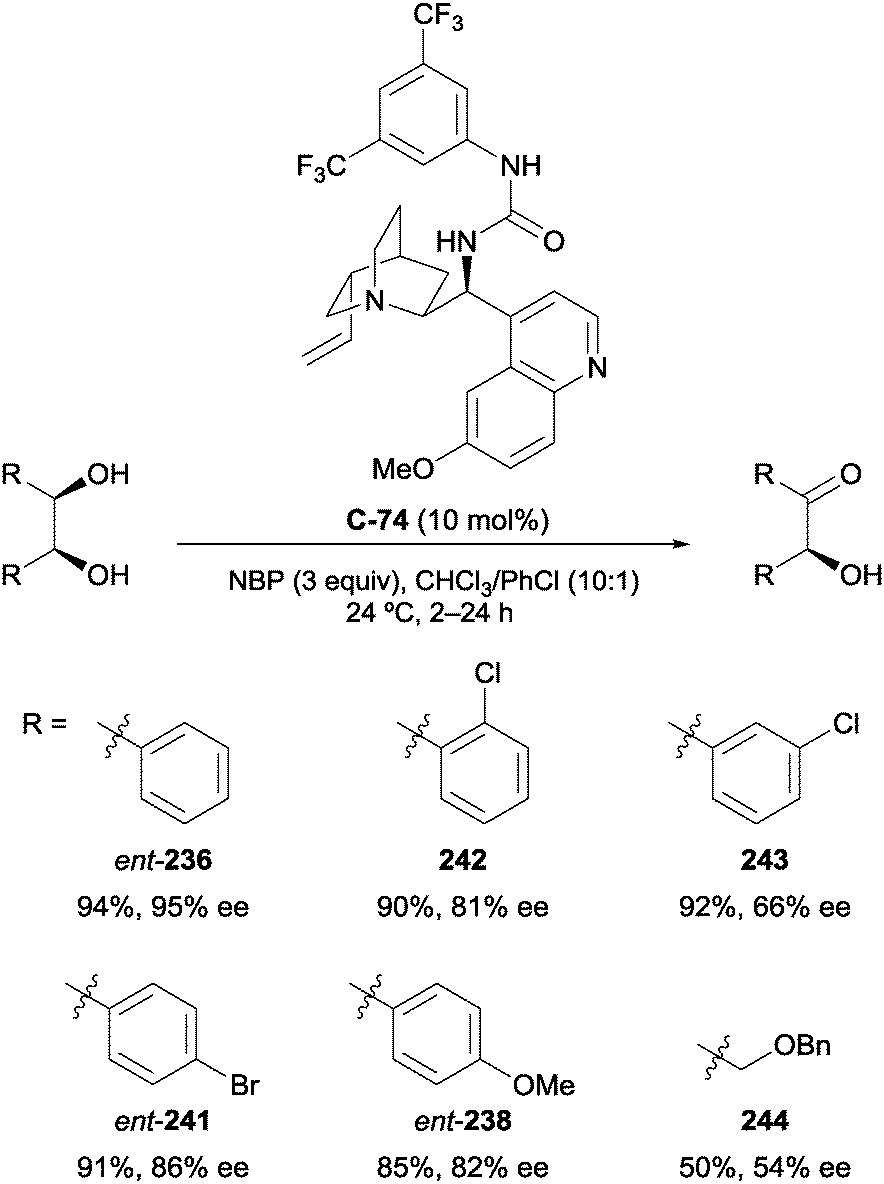 | ||
| Scheme 63 Enantioselective oxidation of meso-1,2-diols by Y. Zhao using bifunctional quinine-derived urea catalyst C-74. | ||
Modification of the catalyst to include a urea moiety and electron withdrawing aromatic ring, in conjunction with the use of N-bromophthalimide (NBP) as the optimal oxidant, resulted in excellent yields (50–94%) and moderate to excellent enantioselectivity (54–95% ee). Dialkyl substituted meso-diols were poorer substrates compared to benzylic systems, proceeding with reduced yield (50%) and lower enantioselectivity (54%) (244, Scheme 63). The authors speculated that the high enantioselectivities arose from hydrogen bonding between the diol, phthalimide, and urea moieties, creating a highly organized transition structure.
4.3 Silylation of diols
Pioneering work by Hoveyda and co-workers recently demonstrated the enantioselective monosilylation of meso-1,2- and 1,3-diols using N-methylimidazole bifunctional catalyst C-75, though high loadings and long reaction times were required to achieve good conversion and selectivity (Scheme 64a).113 The system was later applied to 1,2,3-triols as part of the synthesis of Cleroindicins C, D (251), and F (Scheme 64b).114 Tan and co-workers have also developed an N-methylimidazole catalyst capable of delivering different silyl groups to a range of substrates in good yield and enantioselectivity (Scheme 64c).115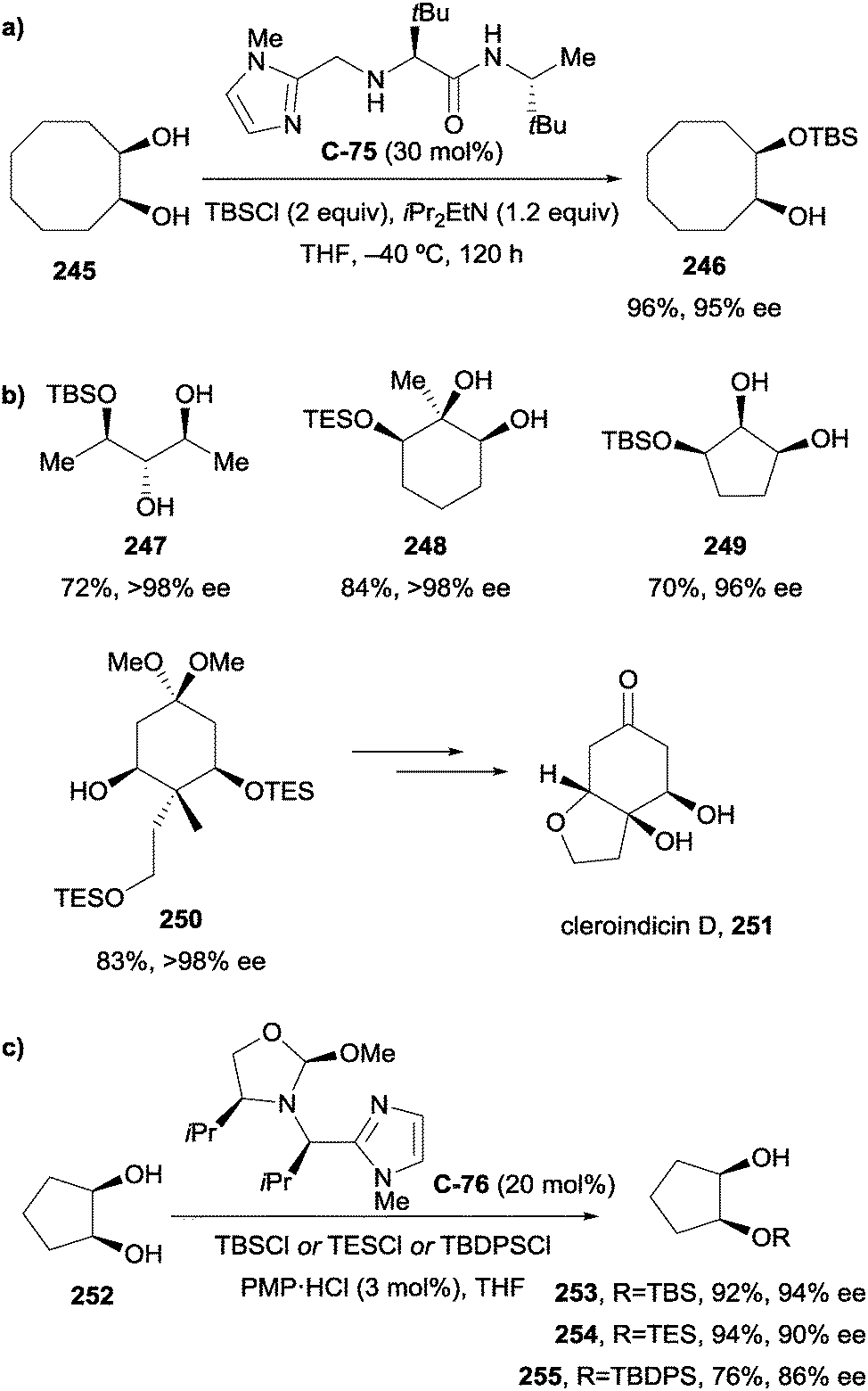 | ||
| Scheme 64 Enantioselective silylation of (a) 1,2-diols and (b) 1,3-diols using N-methylimidazole bifunctional catalysts developed by Hoveyda, and (c) 1,2-diols developed by Tan. | ||
The enantiodetermining transition state originally proposed by Hoveyda and co-workers, whereby the catalyst operates in a bifunctional manner, has since been examined computationally by the authors. These studies, using the silylation of methanol mediated by imidazole as a model system, indicate that in the most optimal case, two imidazole units take part in the reaction, and are arranged in an anti-orientation (Fig. 10). If this holds true for catalyst C-75, then it is unlikely to operate efficiently as a bifunctional catalyst due to geometric constraints preventing the anti-orientation. Previous studies by the authors suggested that the catalyst was acting as a Brønsted base, activating the diol, and so a compatible Lewis basic co-catalyst was sought to activate the electrophile.
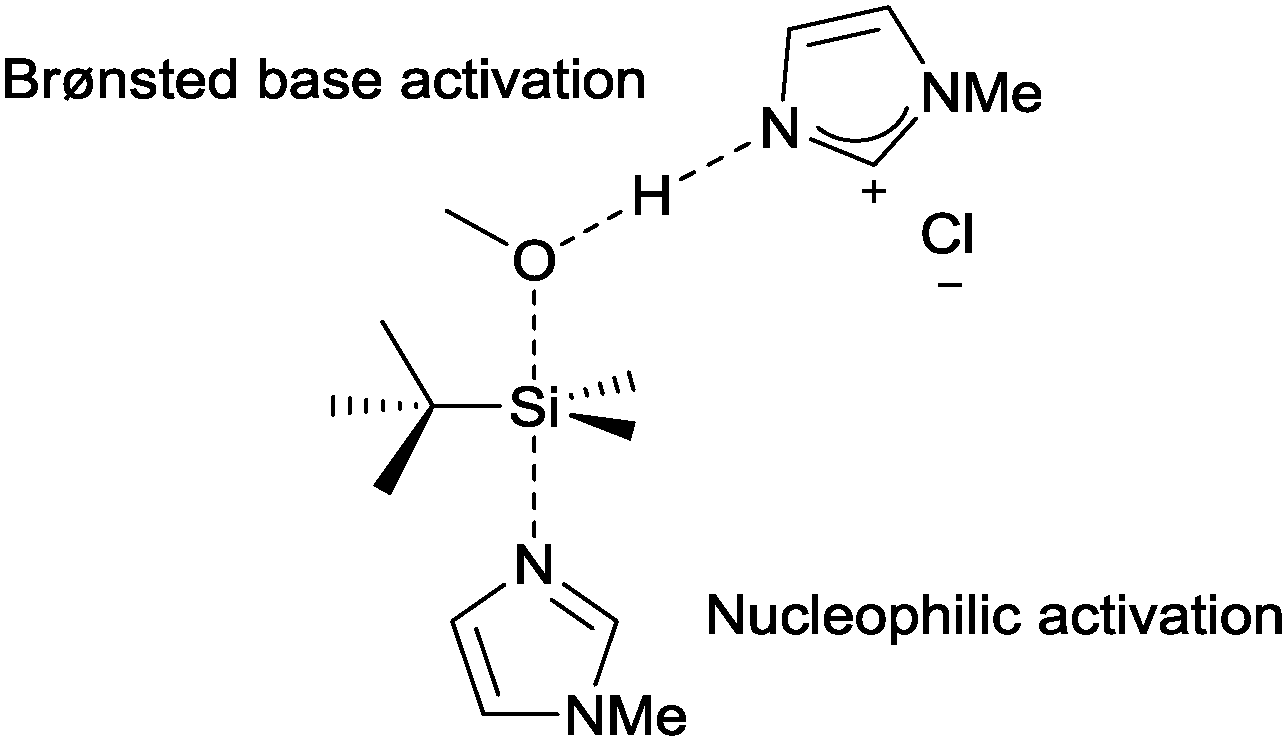 | ||
| Fig. 10 Computational studies by Hoveyda demonstrating optimal anti-orientation of Brønsted and Lewis base activators. | ||
5-Ethylthiotetrazole (256) was found to be the optimal achiral co-catalyst, and the resulting system selectively silylated a range of 1,2- and 1,3-diols in excellent yields (78–97%) and enantioselectivity (88–95% ee) with lower loadings of C-75 (20 mol%, Scheme 65) compared to previous work (30 mol%). Acyclic substrates (260) suffered from reduced yields (74%) though this improved significantly (93%) with a longer reaction time of 12 h. Additionally, the process was amenable to larger scale (1 g) synthesis at low catalyst loadings (5 mol% of C-75 and 256) giving 246 in excellent yield (96%) and enantioselectivity (93%).116
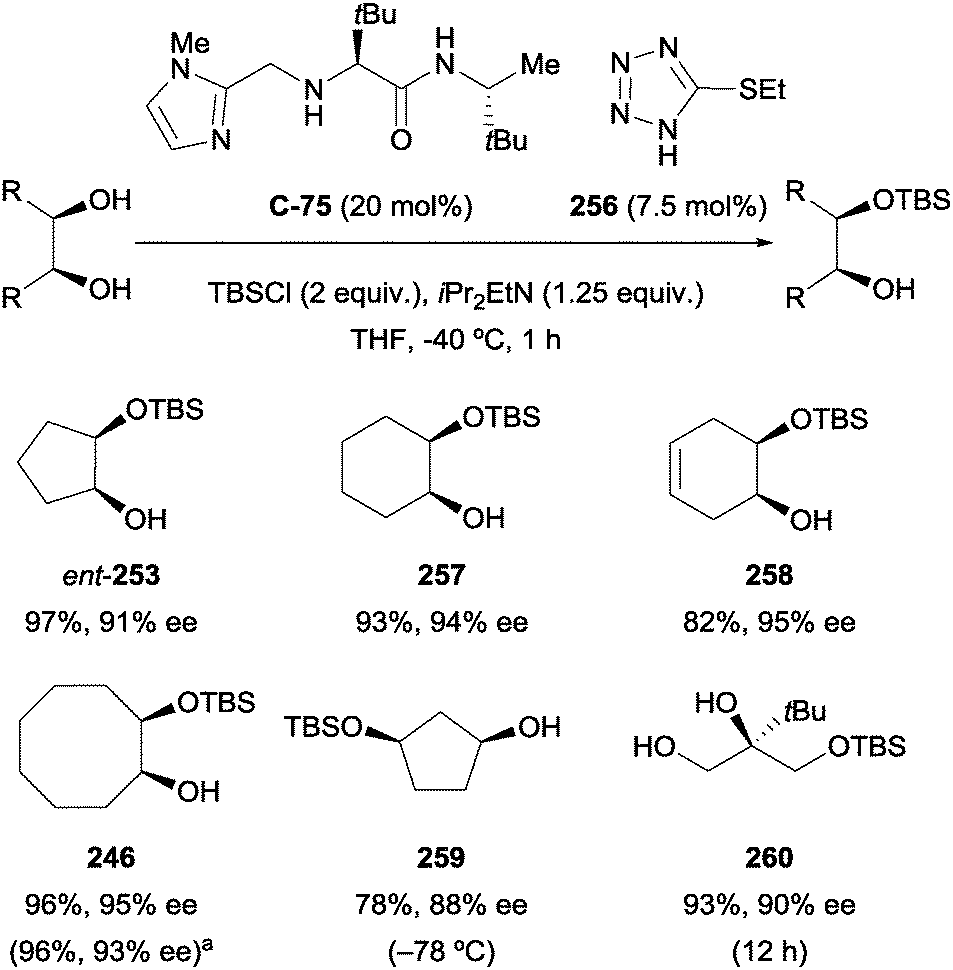 | ||
| Scheme 65 Hoveyda's use of achiral 5-ethylthiotetrazole co-catalyst as a nucleophilic activator in the enantioselective silylation of 1,2-diols; a1 g scale reaction using C-75 (5 mol%) and 256 (5 mol%). | ||
More recently, List and co-workers have developed a Brønsted acid catalyzed silylation of meso-1,2-diols using basic silyl reagents.117 The use of acid-activated basic silyl reagents, such as allylsilanes118 and hexamethyldisilazane (HMDS),119 has been previously demonstrated, however this represents the first time the transformation has been carried out enantioselectively.
Initial studies using TRIP (C-70) as the catalyst identified HMDS as an optimal silyl source. Further investigations found that the STRIP catalyst (C-71) was superior, and optimized conditions using meso-hydrobenzoin as the model susbtrate provided the mono-protected species 261 in good yield (84%) and enantioselectivity (90% ee, Scheme 66). A range of aryl-substituents was tolerated with similarly high yields and enantioselectivities. A limitation of this method, however, is the long reaction times needed to achieve good yields on some substrates, for example para-fluorophenyl product 265 required 10 d. The authors observed that para-substitution of the aryl groups led to improved yield and enantioselectivity (262, 96%, 90% ee) compared to ortho- and meta-substitution. The mono-protection of cyclohexane-1,2-diol resulted in an excellent yield, however enantioselectivity was much lower (266, 51% ee) compared to aryl substrates, suggesting the importance of the aromatic rings in determining selectivity.
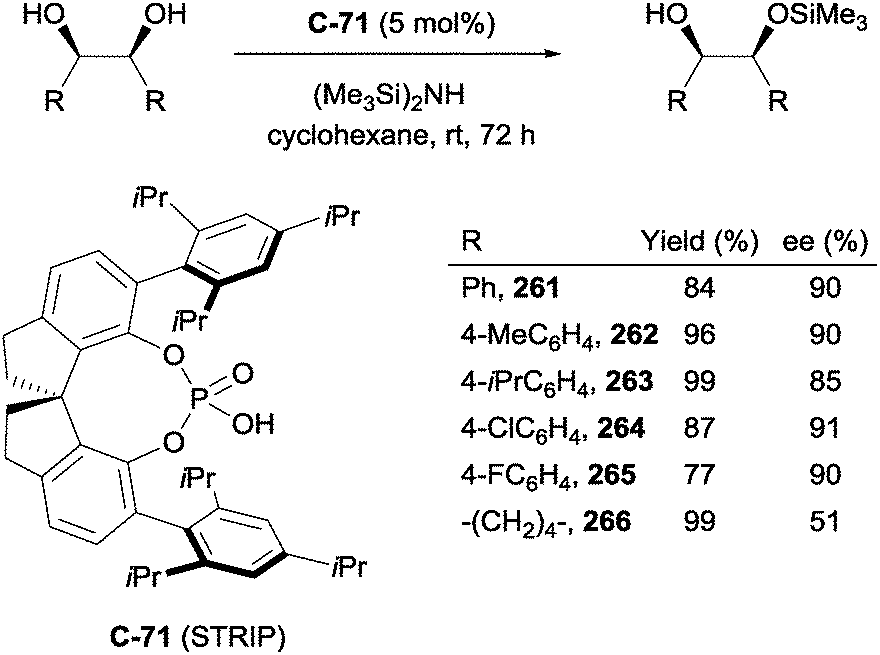 | ||
| Scheme 66 Enantioselective silylation of meso-diols with electrophilic silylating agents under chiral phosphoric acid catalysis by List. | ||
Song and co-workers also demonstrated the desymmetrising silylation of some meso-hydrobenzoin derivatives using their general method for the silylative kinetic resolution of benzyl alcohols (HMDS with BINOL-based polyether catalysts) at very low catalyst loadings (1 mol%) with high enantioselectivity.120 Together, these strategies provide extremely powerful, mild, and broadly applicable methods for the enantioselective silyl protection of symmetric diols.
4.4 Enantioselective desymmetrisation of olefinic diols
Yeung and co-workers have developed several effective strategies for the enantioselective desymmetrisation of olefinic 1,3-diols. The authors developed C2-symmetric sulfide catalyst C-76, which was found to be highly effective at catalyzing the enantioselective bromoetherification of olefinic 1,3-diols (Scheme 67).121 In general, diastereoselectivity was improved for trisubstituted olefins compared to disubstituted olefins (R2 = H). Excellent yields and drs were obtained for a range of trisubstituted olefins with good enantioselectivity and in most cases, a range of substituted aryl groups were tolerated well. Other substrates proved more challenging, with ortho-substitution of aryl rings on trisubstituted olefins resulting in near absence of enantioselectivity (271), while a good level of selectivity was retained in the case of a disubstituted olefin (272).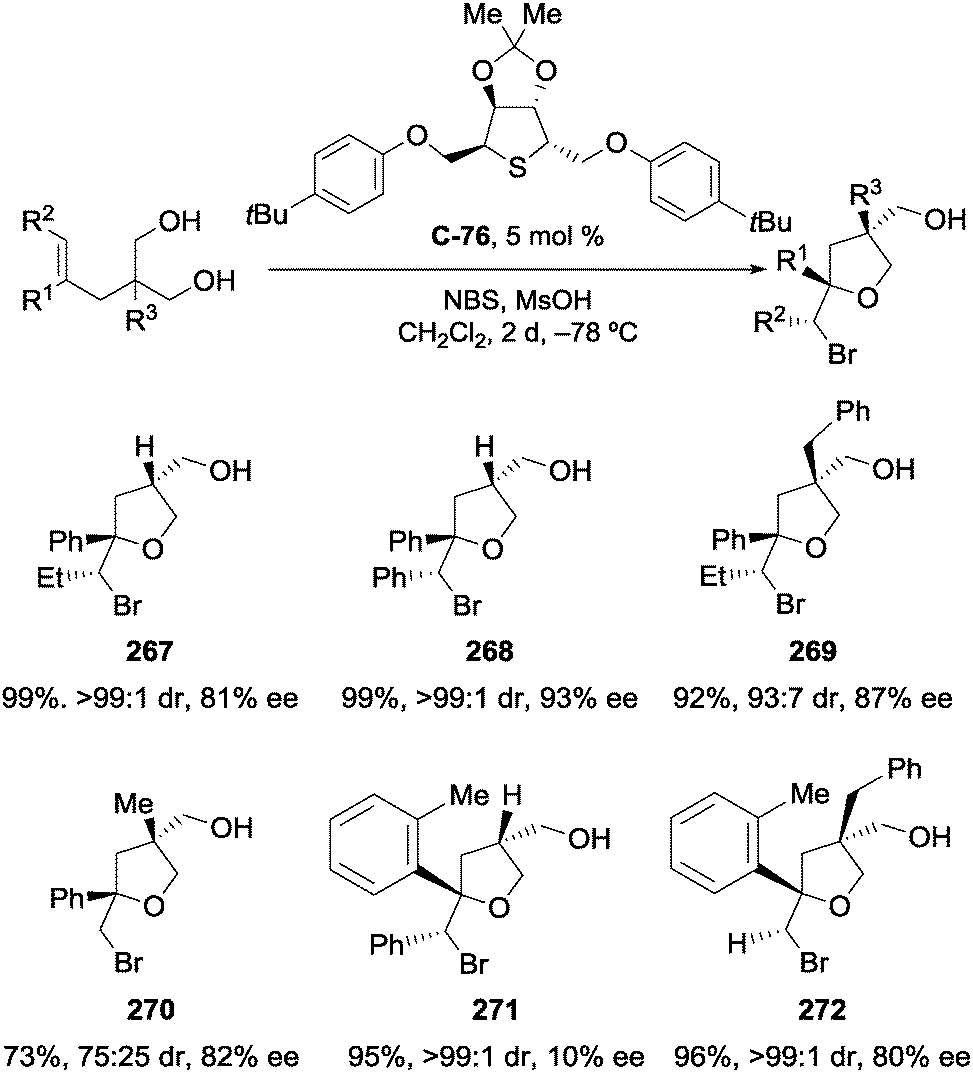 | ||
| Scheme 67 Yeung's enantioselective bromoetherification of olefinic 1,3-diols using C2-symmetric sulfide catalyst C-76. | ||
Mechanistically, the authors believe that the sulfide activates NBS to form an activated chiral bromonium complex to deliver the bromide to the olefin enantioselectively (Fig. 11). The presence of MsOH was necessary for the reaction to occur with high diastereo- and enantioselectivity, but its exact role in the mechanism is unclear.
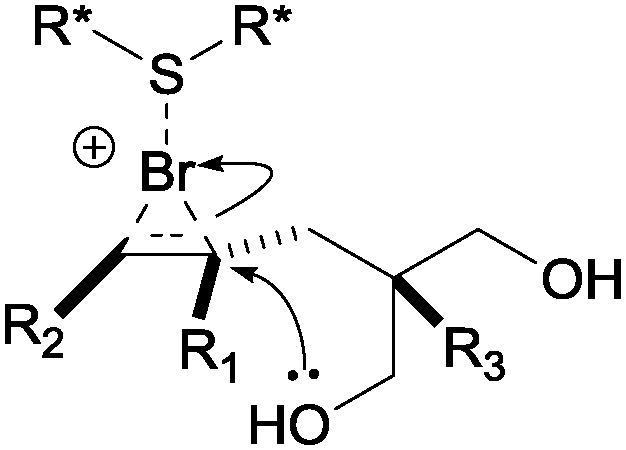 | ||
| Fig. 11 Formation of chiral bromonium ion with C2-symmetric sulfide catalyst C-76 for enantioselective bromoetherification of olefinic 1,3-diols as described by Yeung. | ||
The bromoetherification of symmetric diolefinic 1,3-diols was also demonstrated with the use of quinine-derived catalyst C-77, in high yields with good diastereo- and enantioselectivity (Scheme 68).122 The procedure worked well on mixed olefins, and addition of a further equivalent of bromide source (NBS) led to the formation of spirocycles 274, 276, and 278 in excellent yield and diastereoselectivity in most cases.
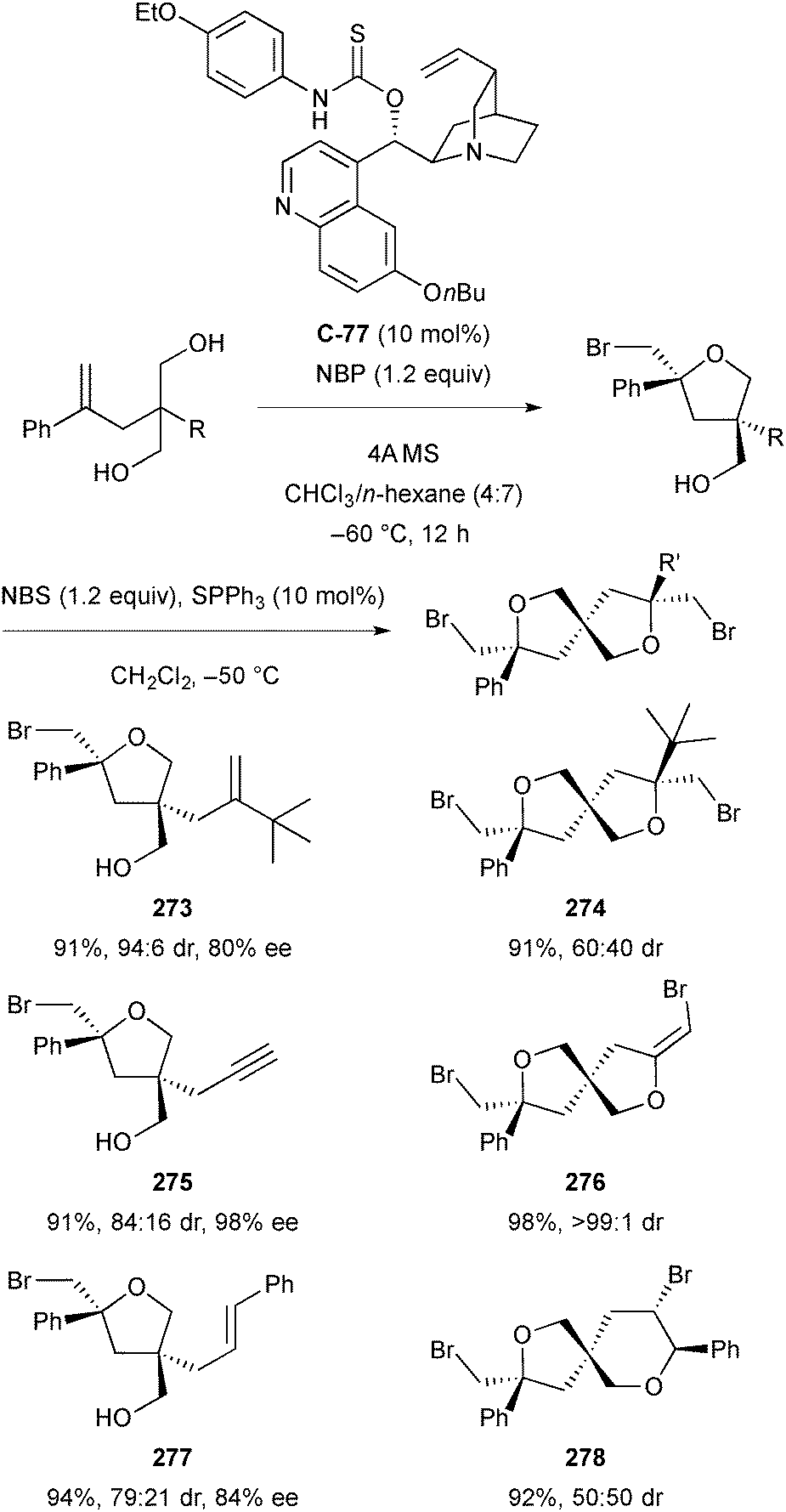 | ||
| Scheme 68 Enantioselective bromoetherification of diolefinic 1,3-diols using thioamide catalyst C-77 leading to the formation of spirocycles by Yeung. | ||
The bifunctional structure of aminothiocarbamate catalyst C-77 controls both the diastereoselectivity and enantioselectivity of the reaction. The authors propose that hydrogen bonding occurs between the quinuclidine nitrogen and the 1,3-diol, forming a six-membered pseudo chair-like transition state. The bulkier substituent occupies the pseudoequatorial position, and the amino-thiocarbamate restricts the geometry of the enolate, resulting in a high degree of selectivity.
The desymmetrisation of 1,8-diols containing a central alkene via haloetherification has been demonstrated in two closely related publications by Hennecke.123,124 The reaction could be catalyzed by various BINOL phosphates; a triphenylsilyl substituted variant C-78 introduced as a sodium salt proved to be optimal. Its proposed mode of action is ion pairing with a haliranium intermediate and simultaneous activation of one of the enantiotopic hydroxyl groups through hydrogen bonding (Scheme 69). (Z)-Alkenediols provided bromo- and iodoetherification products with generally moderate to good yields and enantioselectivities. Substrates with tertiary diols as well as homologous diols leading to pyrans upon haloetherification were also explored, however enantioselectivity was low in these cases.
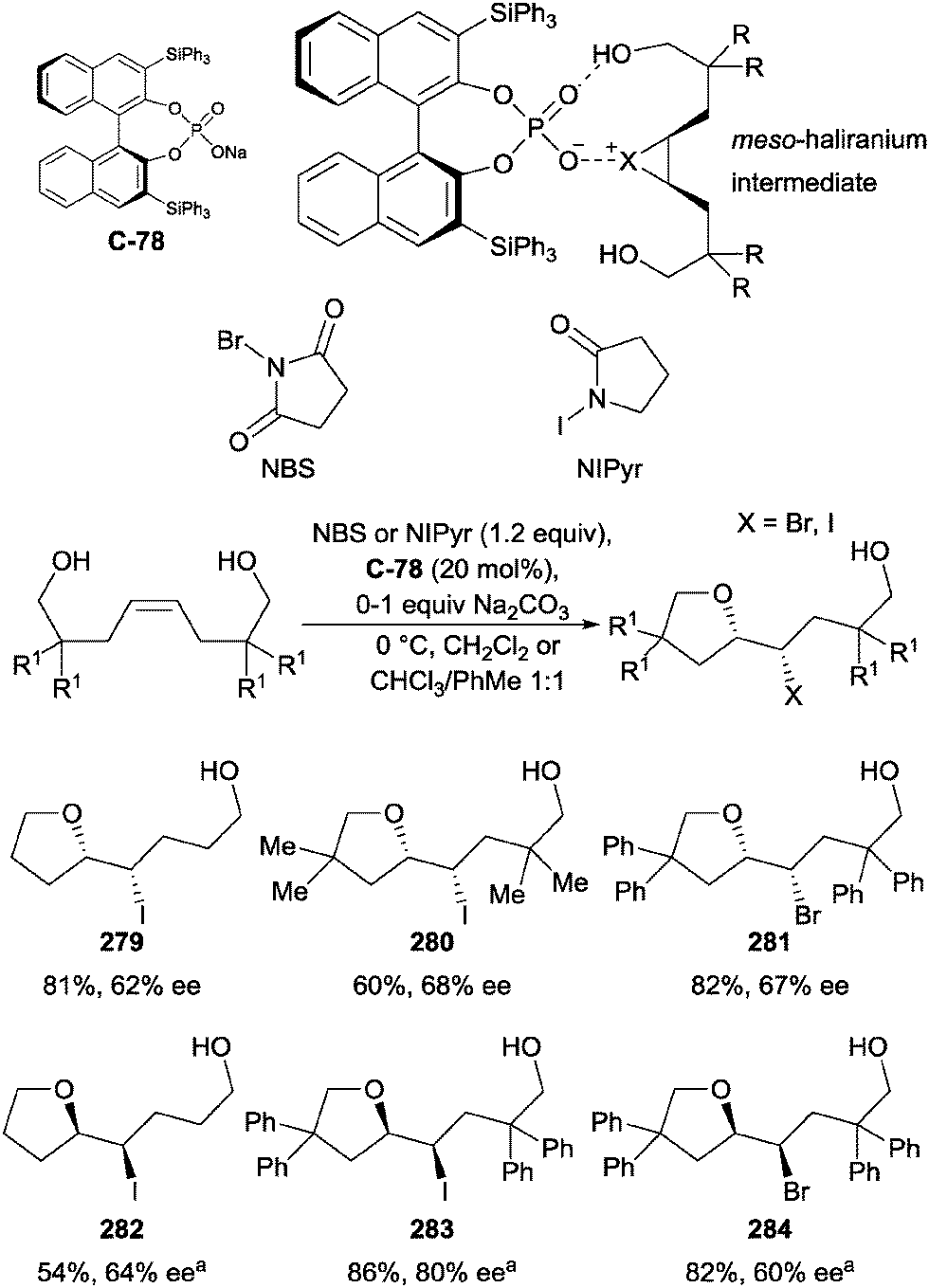 | ||
| Scheme 69 Desymmetrisation of alkenediols through halolactonisation by Hennecke, and the proposed activation of the meso-haliranum diol formed from (Z)-alkenediol; a(Ra) enantiomer of the catalyst was used. | ||
Some enantioselectivity (16–38% ee) was also observed with (E)-alkenediols even though they afford chiral haliranium intermediates (whereas (Z)-alkenediols afford meso-haliraniums). The authors proposed that one stereoisomer of the reversibly formed enantiomeric haliranium species was preferentially cyclised by the catalyst.
5. Alkenes, dienes and diynes
Compounds containing two symmetrically arranged alkene, alkyne or allene groups or a single, symmetrically substituted group of this type are viable targets for enantioselective desymmetrisation. Most of the organocatalytic desymmetrisation methods that have been applied to these substrates are electrophilic additions such as haloetherifications. Many of these are catalysed by acidic binaphthol derivatives that can form ion pairs with reactive haliranium intermediates. Some pre-2014 examples of these reactions have been presented in a recent review by U. Hennecke and M. Wilking.125Desymmetrisation of bis-alkenoic acids through asymmetric protolactonisation was recently reported by K. Ishihara.126 This method was a secondary finding in a study focused on the kinetic resolution of racemic α-substituted carboxylic acids through protolactonisation. The reaction was catalysed by a chiral phosphonium species obtained by in situ protonation of substituted binaphthol-based chiral phosphate ester C-79 (Scheme 70a). A catalytic amount of chlorosulfuric acid was used in combination with the chiral catalyst to form a reactive phosphonium salt. This species preferentially delivers a proton to a single enantiotopic face of the alkene, giving rise to stereoselectivity. The proposed structure of the transition state for protolactonisation of alkenoic acids is shown in Scheme 70a. In the reactive conformation, the Re face of the alkene is aligned with the proton source. In the case of the (R)-α-substituted acid, a steric clash occurs between its major group at the α-position and a triphenylsilyl group on the catalyst, thus increasing the energy barrier for the reaction. The pathway to the (S)-enantiomer does not experience the same steric hindrance, leading to kinetic resolution.
 | ||
| Scheme 70 (a) Catalyst structure and the proposed alignment of (R)- and (S)-alkenoic acid with the chiral phosphonium species. (b) Kinetic resolution and desymmetrisation of γ,δ-unsaturated carboxylic acids by Ishihara. (c) Desymmetrising protolactonisation of bis-alkenoic acids; aabsolute configuration not determined. | ||
Application of the same catalytic system to desymmetrisation of bis-alkenoic acids was studied on two examples (Scheme 70c), providing a γ- and δ-lactone. Good yields and moderate enantioselectivities were achieved; γ-lactone 288 was obtained in 78% yield with 76% ee, whereas the δ-lactone 286 formed in 82% yield and with 79% ee.
Another example of electrophile-induced desymmetrising cyclisation of alkenes is the halolactonisation of unsaturated carboxylic acids. Methods of this type were reported by Hennecke and co-workers. Bromolactonisation of bis-alkynoic acids was initially described,127 followed by cyclisations of bis-alkenoic and bis-allenoic acids.128 The lactonisation proceeds via a bromonium cation which undergoes intramolecular nucleophilic attack by carboxylate. Basic organocatalysts used in this work were proposed to act by binding the substrate carboxylic acid groups via hydrogen bonding, leading to their activation in a chiral environment.
The most successful catalyst was the dimeric cinchona alkaloid-derived (DHQD)2PHAL C-80 (Scheme 71) which has been previously used in asymmetric halolactonisations.129 Its central phthalazine motif was found to be important for effective stereocontrol. With bis-alkynoic acids it afforded high yields (76–98%) and generally good enantioselectivity (70–96% ee) across a range of substrates using NBS as the “Br+” source (Scheme 71a). Terminal alkynes were found to react with slightly lower enantioselectivity than alkynes bearing aliphatic or aromatic substituents R2 substituents. The identity of R1 had little effect on the efficiency or the stereochemical outcome.
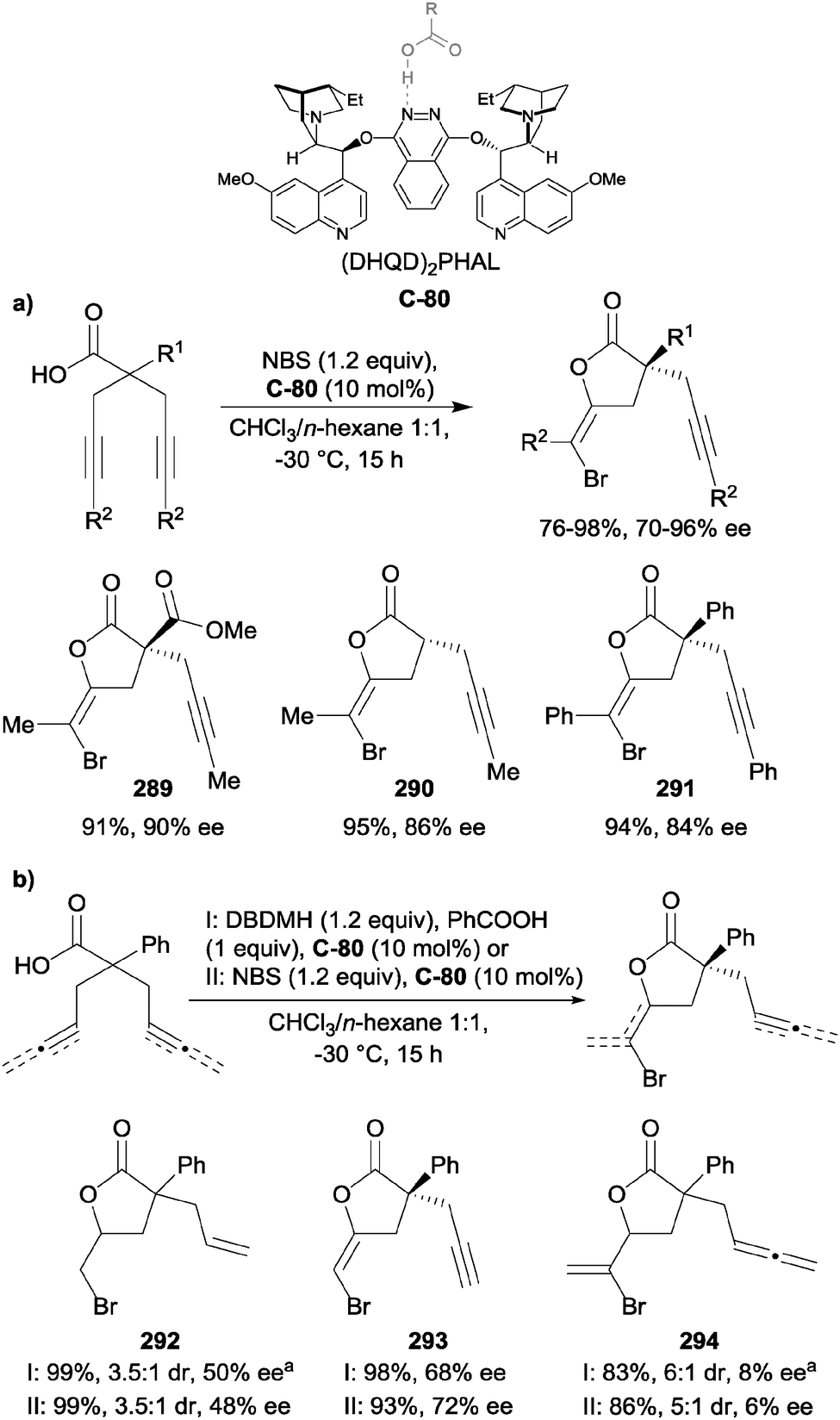 | ||
| Scheme 71 Desymmetrising bromolactonisations of bis-alkenoic, bis-alkynoic and bis-allenoic carboxylic acids by Hennecke; aabsolute configuration not determined. | ||
Organocatalytic methods developed for bis-alkynoic acids were also applied to bis-alkenoic and bis-allenoic analogs, in which an additional stereocentre is established upon halolactonisation. With the (DHQD)2PHAL catalyst and either NBS or 1,3-dibromo-5,5-dimethylhydantoin (DBDMH), these substrates could be cyclised in high yields (Scheme 71b). Stereocontrol was however only moderately effective. The desymmetrisation of the bis-allenoic acid proceeded in high yields and good drs, but ees were very low.
Asymmetric bromoetherification catalysed by (DHQD)2PHAL C-80 was also applied by T. Kan et al. to symmetric cyclohexa-1,4-dienes bearing a carboxylic acid group.130 The cyclizations provided β- and γ-lactones in moderate to high yields and with variable but generally good enantioselectivity (Scheme 72a). A strong advantage of this process is that it establishes three stereogenic centres diastereospecifically. Together with its good performance for both β- and γ-lactones, this distinguishes the presented work as a powerful methodology. The presence of an unprotected hydroxyl group (R1 = CH2OH) resulted in racemic product, whilst silyl protected alcohols reacted with good yields and enantioselectivity. In terms of R2 substitution, a methyl group was well tolerated, while an OMOM substituent was found to decrease the yield.
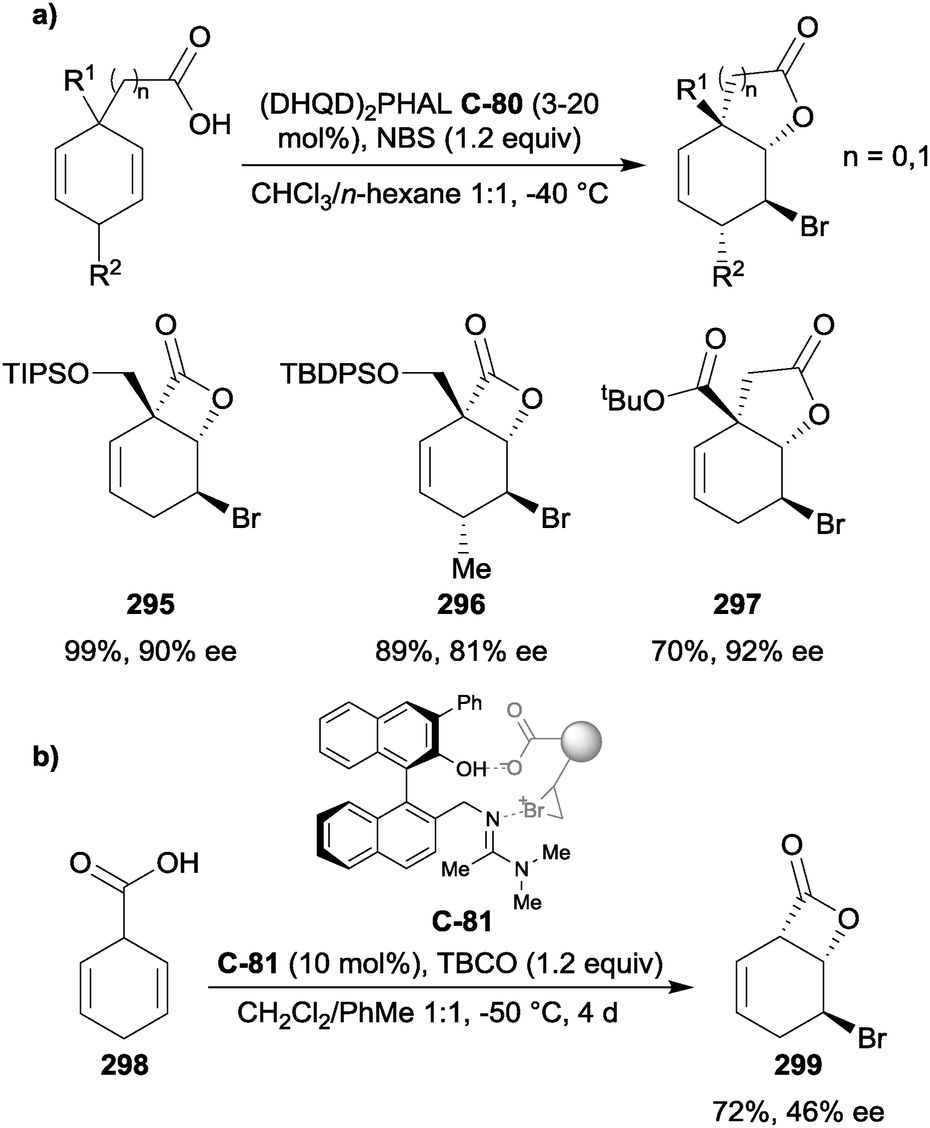 | ||
| Scheme 72 (a) Bromoesterification of cyclohexa-1,4-dienes catalysed by (DHQD)2PHAL by Kan; (b) an example of the same reaction catalysed by a bifunctional binaphthol derivative by Martin; TBCO = 2,4,4,6-tetrabromocyclohexadienone. | ||
An analogous β-lactone formation was reported as a single example in a study by S. Martin and co-workers focusing on non-desymmetrising stereoselective bromolactonisations.131 In this case the reaction was catalysed by a bifunctional binaphthyl derivative C-81, substituted with an acidic phenol OH and a basic amidine group (Scheme 72b). The authors propose that the amidine stabilises the bromonium intermediates whereas the phenolic OH associates with the carboxylic acid through hydrogen bonding, therefore placing the substrate in a chiral environment. With 72% yield and 46% ee this method has a lower performance than the (DHQD)2PHAL-catalysed process in the previous example (Scheme 72a), although the related non-desymmetrising reactions in this work displayed a very good degree of stereoselectivity.
A process of asymmetric oxysulfenylation and oxyselenylation of alkenes was applied to desymmetrisations of cyclic alkenes by Y. Shi and co-workers.132 This reaction is catalysed by Brønsted acids; stereocontrol is possible when chiral acids such as CSA (camphorsulfonic acid) or binaphthol-based N-triflyl phosphoramides are used. Initially a cyclic sulfonium or selenonium species forms in a reaction between the alkene and an “S+/Se+” donor such as N-thiosuccinimide 300 (Scheme 73a). The resulting cation is thought to form an ion pair with a chiral anion formed from the acidic catalyst.
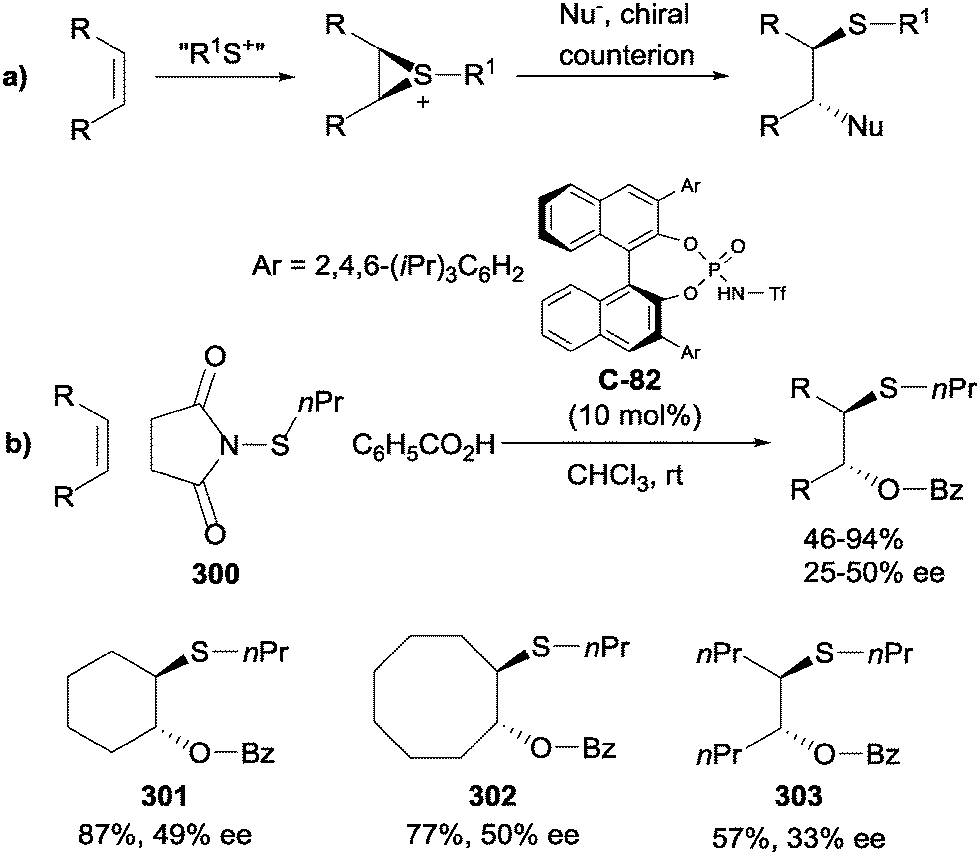 | ||
| Scheme 73 (a) Electrophilic sulfenylation of symmetrically substituted alkenes (b) asymmetric oxysulfenylation of alkenes using phosphoramide catalyst C-82 by Shi. | ||
The conditions were optimised for the desymmetrisation of cyclohexene and then tested on cyclic alkenes from cyclopentene to cyclooctene as well as some acyclic symmetric alkenes (Scheme 73b). Variable yields (46–94%) and low to moderate enantioselectivity (25–50% ee) were observed. Although the diastereospecific formation of two stereogenic centres is a considerable strength of this method, its efficiency and enantiocontrol are currently only moderate.
6. Ketones and diketones
Catalytic enantioselective α-addition to ketones has experienced a surge in popularity since the renaissance of organocatalysis in 2000.133–135 However, despite the intense research efforts devoted to the field, it was only several years later that this methodology was combined with enantioselective desymmetrisation. Early examples include Barbas's α-hydroxylation of prochiral 3,4,4,5-tetrasubstituted cyclohexanones,136 Cheng's asymmetric Michael addition of 4-substituted cyclohexanones to β-nitrostyrenes,137 and Gong's desymmetrising aldol reaction.138 Organocatalysis remains an exciting and active field, with a plethora of novel catalysts being developed, many of which have found application in desymmetrisation reactions.Tian and co-workers have demonstrated enantioselective desymmetrisation on 4-substituted cyclohexanones via α-alkylation, employing a chiral imidazolidinone catalyst to distinguish between the two prochiral faces of the cyclohexanones (Scheme 74).139 Loss of 4-toluenesulfonamide from thioxanthene 304 produced a carbocation which was subsequently captured by the cyclohexanone-derived enamine, giving trans-2,4-disubstituted-cyclohexanones in excellent yields and drs and good ees. The desymmetrisation was, however, shown for just three prochiral cyclohexanones and one electrophile. A catalytic (10 mol%) amount of trifluoroacetic acid was used to boost reactivity, although reactions still needed three days to reach completion.
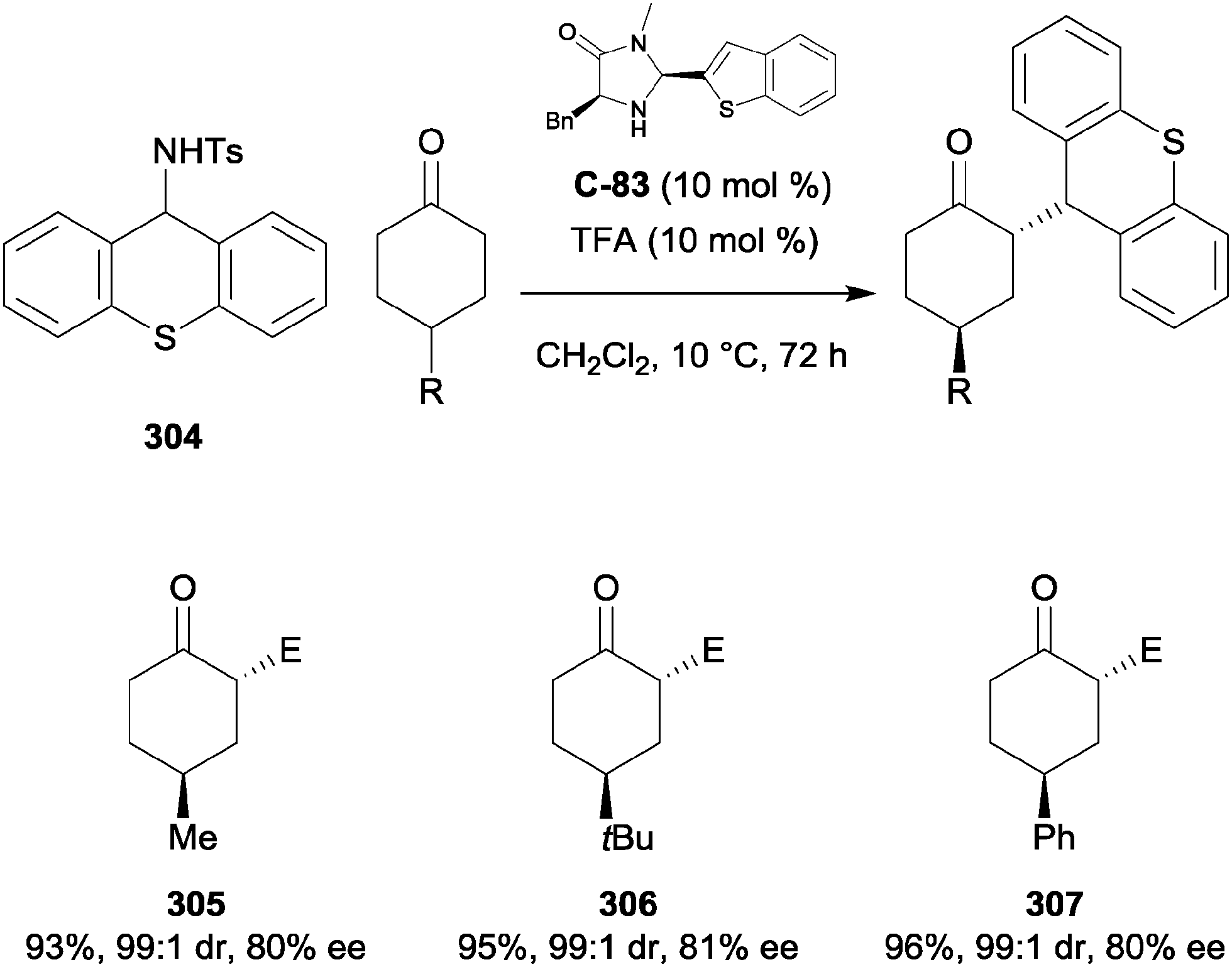 | ||
| Scheme 74 Catalytic enantioselective α-alkylation of 4-substituted cyclohexanones using chiral amine catalyst C-83 and benzylic sulfonamide 304 by Tian. | ||
Zhang and co-workers reported a similar reaction, again using 4-substituted cyclohexanones and enamine organocatalysis, this time in the form of a functionalised chiral ionic liquid, which they proposed stabilises charged intermediates formed during the reaction (Scheme 75).140 Catalytic phthalic acid (37.5 mol%) encouraged the loss of water from the dibenzylic alcohol 308, forming the stabilised 4,4′-bis(dimethylaminophenyl)methane carbocation, which reacted with the transient enamine nucleophilic intermediate to give trans-2,4-disubstituted cyclohexanones in high yields, generally good drs and moderate to good ees. Wider substrate scope was demonstrated for the cyclohexanones, with alkyl, aryl, and oxygenated substituents tolerated in the 4-position, albeit with lower enantio- and diastereoselectivity when R = OH and R = OAc respectively. High catalyst loadings of 25 mol% were employed. Kokotos and co-workers also demonstrated the same reaction on 4-methylcyclohexanone using pyrrolidine-derived catalyst C-85 with equivalent diastereoselectivity and lower catalyst loading, but with poorer enantioselectivity and in markedly lower yield.141
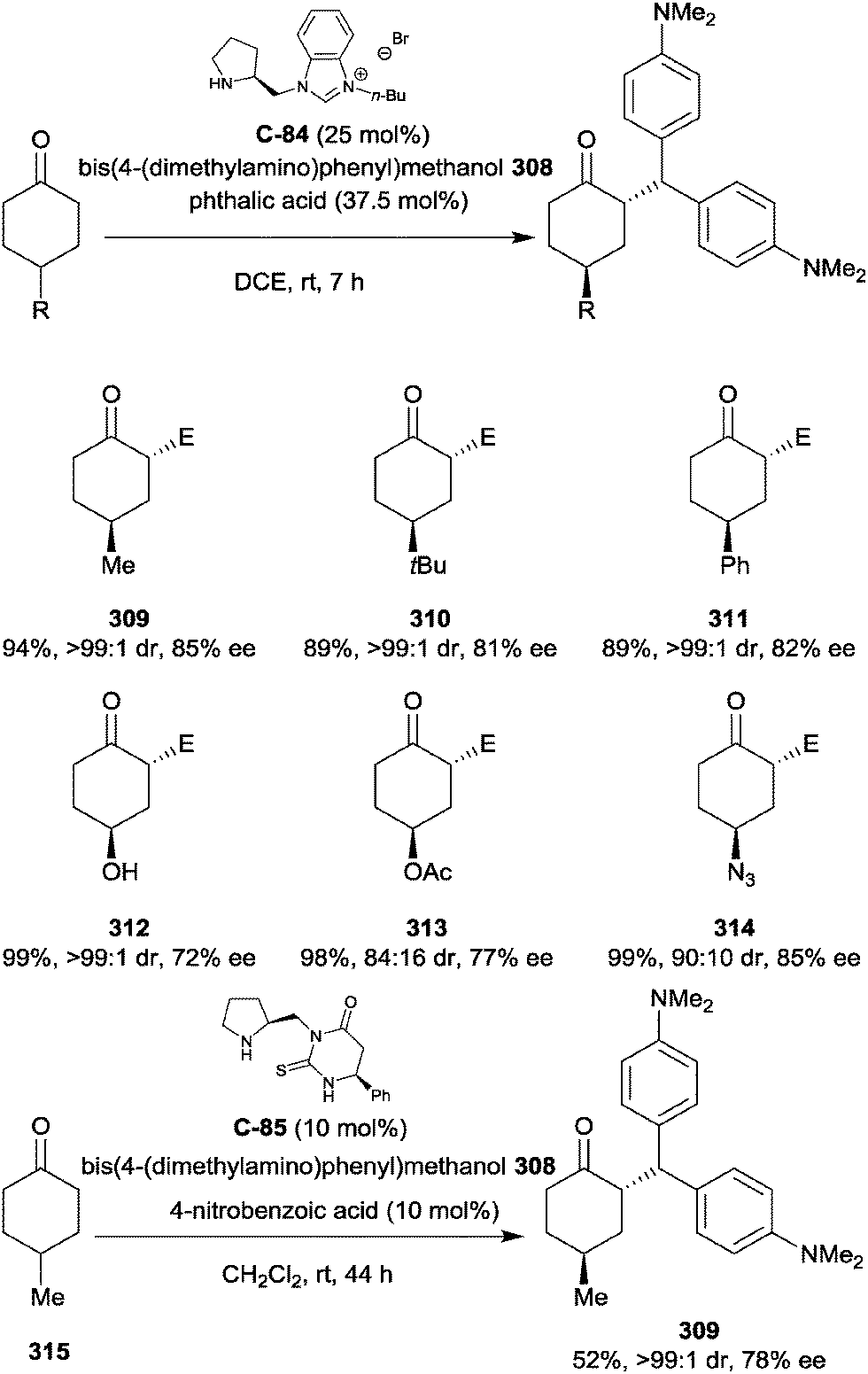 | ||
| Scheme 75 Enantioselective SN1 α-alkylation of 4-substituted cyclohexanones using benzylic alcohol 308 by Zhang and Kokotos. | ||
Melchiorre and co-workers have developed a photo-organocatalytic approach to the enantioselective α-alkylation of cyclic ketones (Scheme 76).142 Using cinchona alkaloid-based primary amine catalyst C-86, the cyclohexanone-derived enamine formed a donor–acceptor complex with the electron-poor aromatic ring; subsequent irradiation with visible light led to the formation of benzyl and iminium radicals, which could combine in a highly enantioselective manner. Although only one desymmetrisation example was shown, yields and selectivities were excellent. Use of phenacyl bromides as alkylating agents with cyclohexanones was also possible; this approach therefore shows promise for the development of novel desymmetrisation reactions.
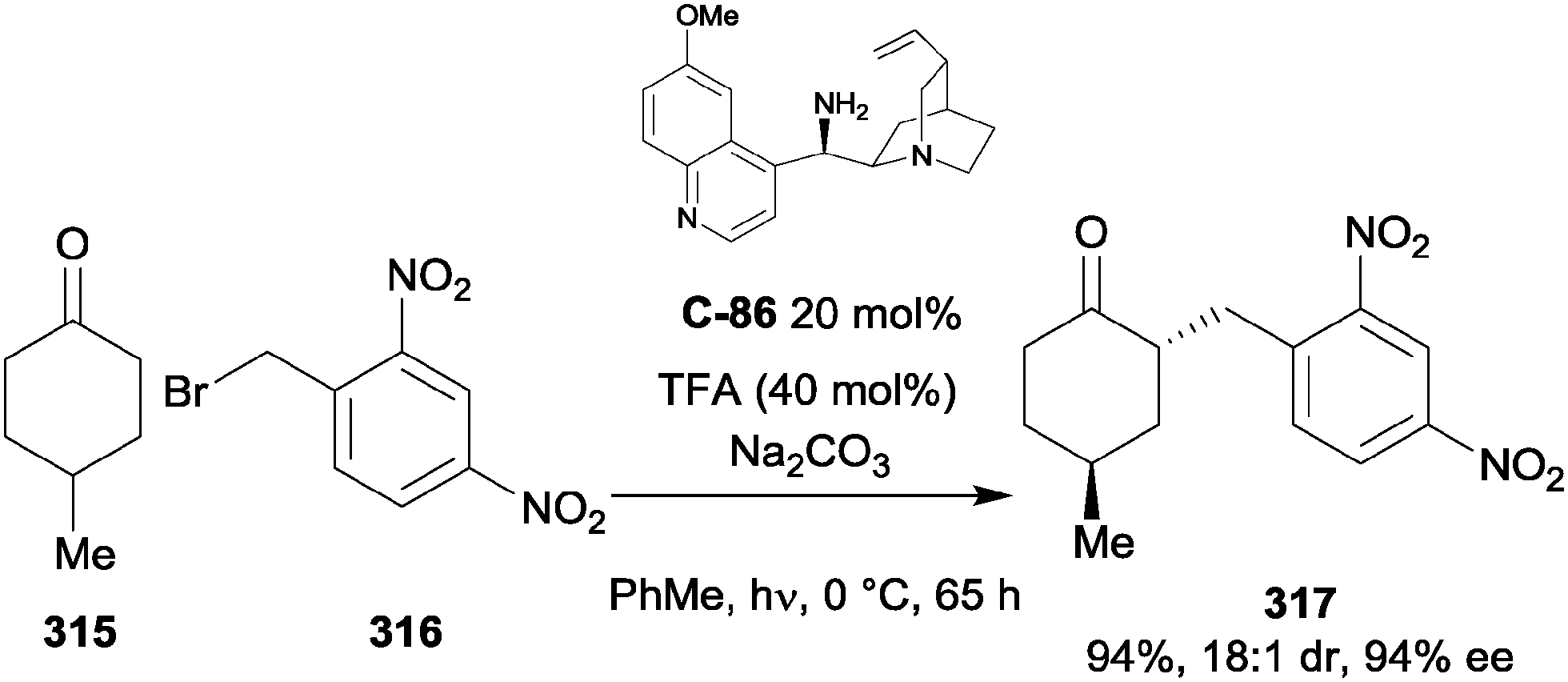 | ||
| Scheme 76 Desymmetrisation of 4-methylcyclohexanone 315via photo-alkylation with electron deficient benzyl bromide 316 by Melchiorre. | ||
6.1 Aldol reactions
The aldol reaction is a mild and powerful method of forming C–C bonds; many catalytic enantioselective variants of the reaction have been developed using chiral metal complexes and small organic molecules, with L-proline used in the first organocatalytic aldol reactions. Enantioselective desymmetrisation has the potential to further increase the utility of the aldol reaction, with the ability to form three stereocentres in high enantiomeric excess from simple starting materials. Electron deficient aromatic aldehydes, such as nitrobenzaldehydes, are popular electrophiles for such reactions due to their lack of α-hydrogens, precluding self-condensation, and high reactivity. Moreover, the nitro group is adept at accepting hydrogen bonds, which can aid selectivity with suitable organocatalysts. A large number of organocatalysts have been developed since 2010 for enantioselective aldol reactions, with many exemplifying desymmetrisation via the reaction of 4-methylcyclohexanone 315 with 4-nitrobenzaldehyde 318 (Scheme 77).23,143–150 Excellent yields and selectivities have been achieved in particular by Fu and co-workers.143 | ||
| Scheme 77 Organocatalysts employed in the enantioselective aldol desymmetrisation of 4-methylcyclohexanone 315 with 4-nitrobenzaldehyde 318. | ||
Enantioselective desymmetrising aldol reactions have also been carried out on less activated benzaldehydes. Ramachary and co-workers have reported the desymmetrisation of 4-methylcyclohexanone via reaction with 2-alkynyl benzaldehydes using prolinamide catalyst C-96 and benzoic acid as an additive (Scheme 78).151 Excellent yields and selectivities were obtained for the two examples shown, although they were achieved by conducting the reaction in neat ketone at low temperature (−35 °C). The opposite enantiomers could be prepared in similar yields and selectivities using the enantiomeric form of the catalyst.
 | ||
| Scheme 78 Enantioselective desymmetrisation of 4-methylcyclohexanone 315 with 2-alkynyl benzaldehydes under the action of prolinamide catalyst C-96 by Ramachary. | ||
Lipshutz and co-workers have developed a water-soluble proline-containing surfactant C-97 which can act as a catalyst in the catalytic asymmetric aldol reaction of cyclohexanones (Scheme 79).152 The catalyst remains in the aqueous layer after extraction of the product with ethyl acetate; addition of starting materials to the water then restarts the reaction, termed in-flask catalyst recycling by the authors. The authors state that reaction likely occurs not in water but in a hydrophobic pocket created by the catalyst, and hence water-insoluble ketones may be used. Although diastereoselectivity was moderate in all cases, the scope of the reaction did extend beyond nitrobenzaldehydes, with 3-bromobenzaldehyde giving excellent enantioselectivities.
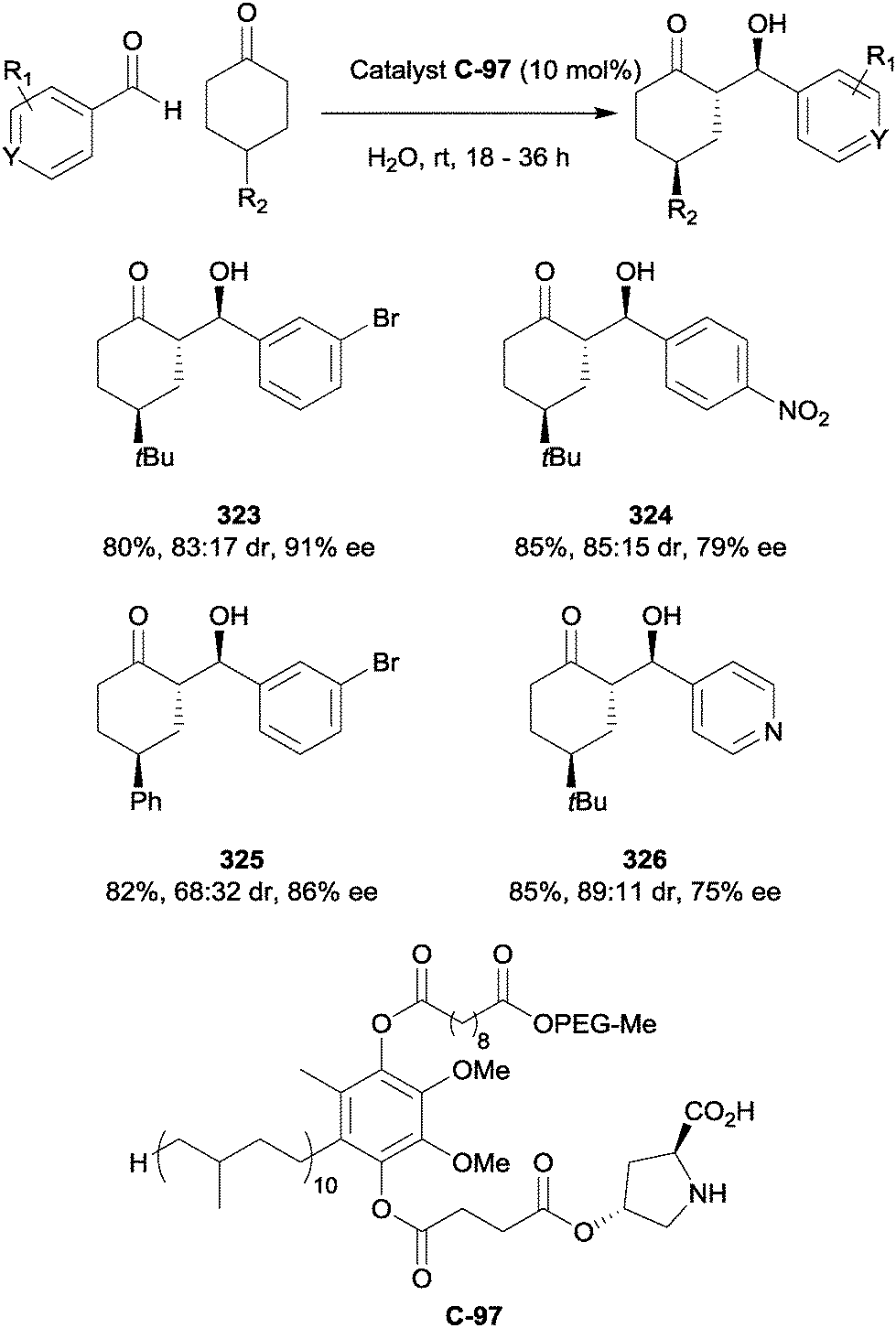 | ||
| Scheme 79 Aldol desymmetrisation of prochiral cyclohexanones under aqueous conditions using proline-derived surfactant C-97 as a catalyst (PEG = poly(ethylene glycol)) by Lipshutz. | ||
Nájera and co-workers have reported the use of binam–prolinamide organocatalyst C-98 in the desymmetrisation of 4-substituted cyclohexanones via a solvent-free aldol reaction with 2,2-dimethoxyacetaldehyde (Scheme 80).153 The reaction proceeded in high yields, diastereo- and enantioselectivities for the three desymmetrisation substrates tested, giving predominantly the anti,anti-aldol products. However, long reaction times were necessary for complete conversion. The authors have reported the use of the same catalyst for the analogous reactions with glyoxylic acid and ethyl glyoxylate, although yields and selectivities were generally more modest for these substrates.154–156
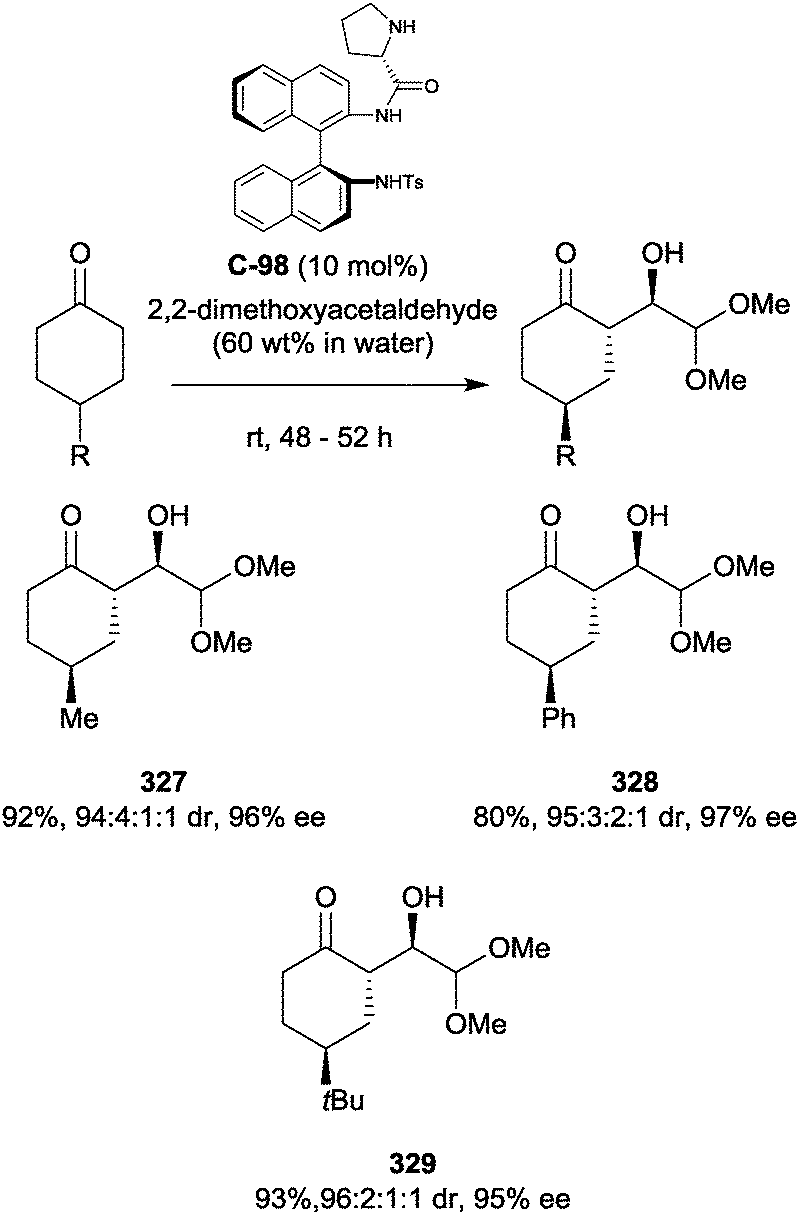 | ||
| Scheme 80 Enantioselective desymmetrisation of 4-substituted cyclohexanones via aldol reaction with 2,2-dimethoxyacetaldehyde in the presence of (Sa)-binam-sulfo-L-prolinamide by Nájera. | ||
Martín-Rapún and co-workers have reported the desymmetrisation of 4-methylcyclohexanone via an anti-selective Mannich addition to an α-iminoester using an immobilised pyrrolidine-derived organocatalyst (Scheme 81).157 The reaction proceeded with good yields and excellent selectivities and has applications to continuous flow processes.
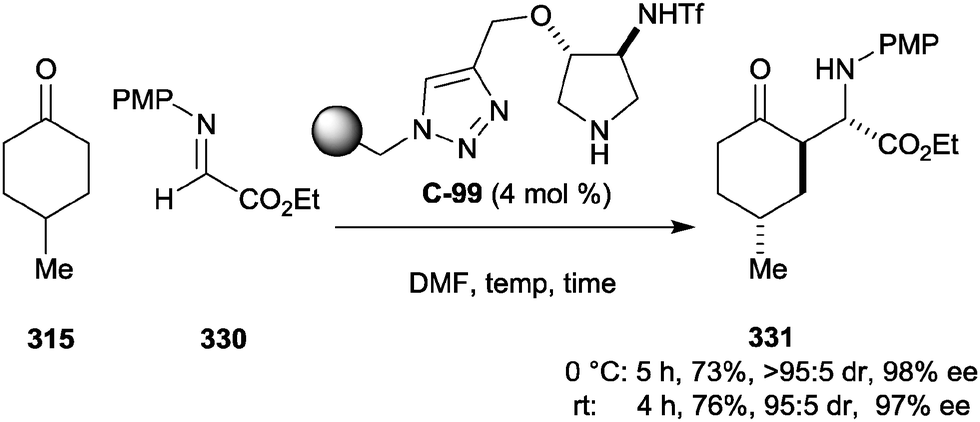 | ||
| Scheme 81 Martín-Rapún's anti-Mannich desymmetrisation of 4-methylcyclohexanone 315 using supported pyrrolidine catalyst C-99. | ||
Seidel and co-workers have reported the first example of a catalytic enantioselective Friedländer reaction, using ortho-aminobenzaldehydes and 4-substituted cyclohexanones (Scheme 82).158 During initial screening, L-proline was quickly found to be a suitable secondary amine catalyst, providing the desired quinoline in good yield and moderate ee. trans-4-Hydroxyproline gave a slight improvement in ee, and protection of the oxygen with a tert-butyldimethylsilyl group maintained this selectivity while improving solubility in nonpolar solvents. Solvent screening revealed aromatic solvents provided good selectivity, while those with basic centres such as pyridine and N-methylimidazole significantly increased reaction rate. A 3:7 v/v solution of toluene and pyridine at −25 °C proved optimal, giving the desired products in excellent ees and moderate to high yields after stirring for 3 days. Less electron-poor, and hence less electrophilic, ortho-aminobenzaldehydes were run at a higher temperature (−5 °C) to increase reactivity. No o-aminobenzaldehydes with electron-donating groups were shown. Products from this reaction could be used to prepare enantioenriched analogues of the drug tacrine, formerly used to treat Alzheimer's disease.
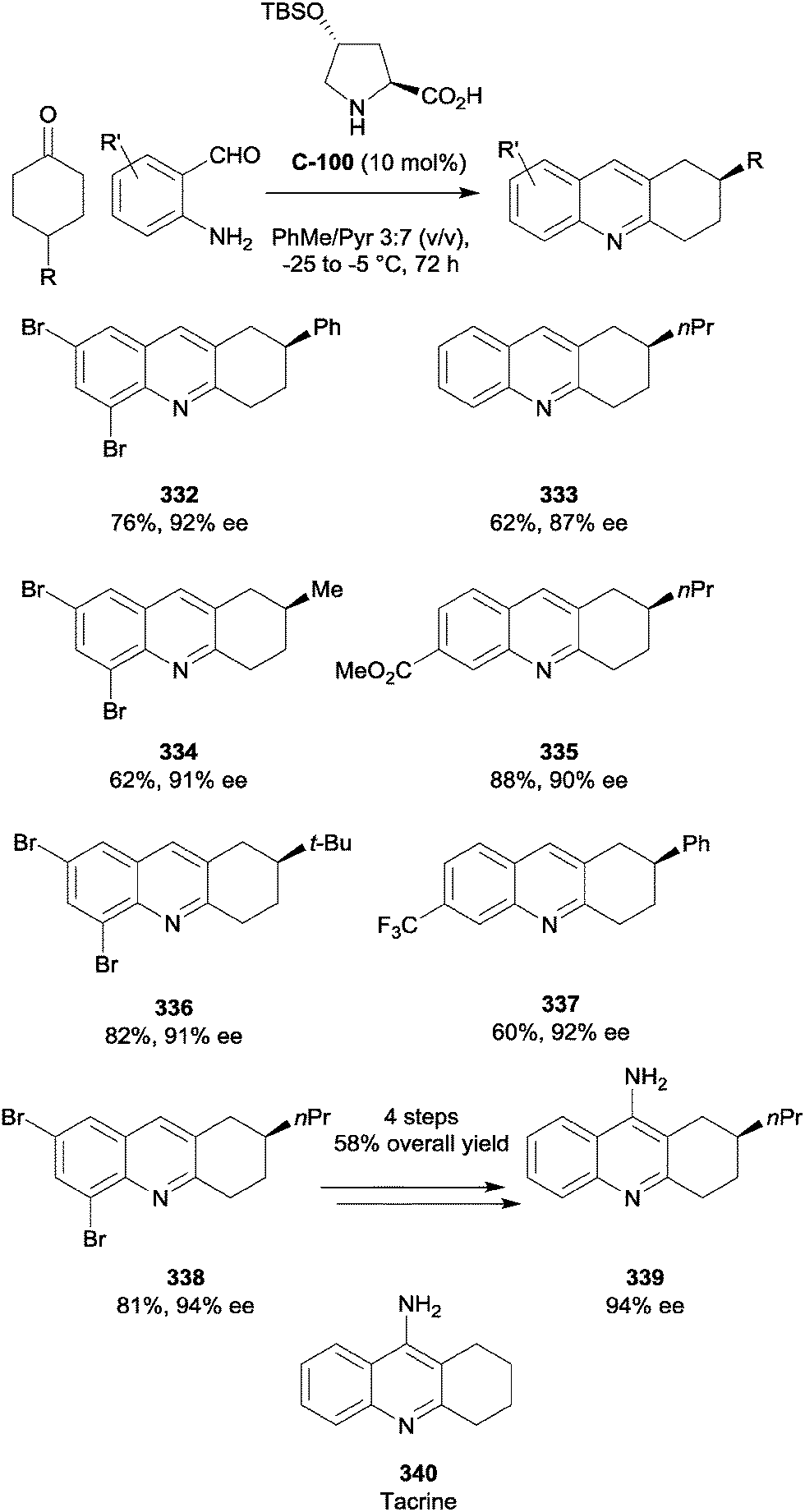 | ||
| Scheme 82 Proline-derived catalyst C-100 promoted catalytic enantioselective Friedländer reaction of prochiral cyclohexanones with o-aminobenzaldehydes reported by Seidel, enabling the synthesis of chiral derivatives of the drug Tacrine. | ||
Gong and co-workers succeeded in extending the scope of the reaction to electron rich substrates using a chiral phosphoric acid catalyst C-101 in combination with achiral aromatic amine 343 (Scheme 83).159 Under these conditions, even 2-amino-4,5-dimethoxybenzaldehyde underwent reaction with 4-phenylcyclohexanone in an impressive 90% ee, although the yield deteriorated to 50%, compared to 86% (87% ee) for substitution with a single methoxy group in the 5-position. In 2013, Nájera and co-workers increased the scope of the ketone partner using prolinamide-derived catalyst C-98, demonstrating the desymmetrisation of cyclohexanones substituted with an ester and a bulky silyl ether in the 4-position, proceeding with high yields and ees under the solvent free conditions.160
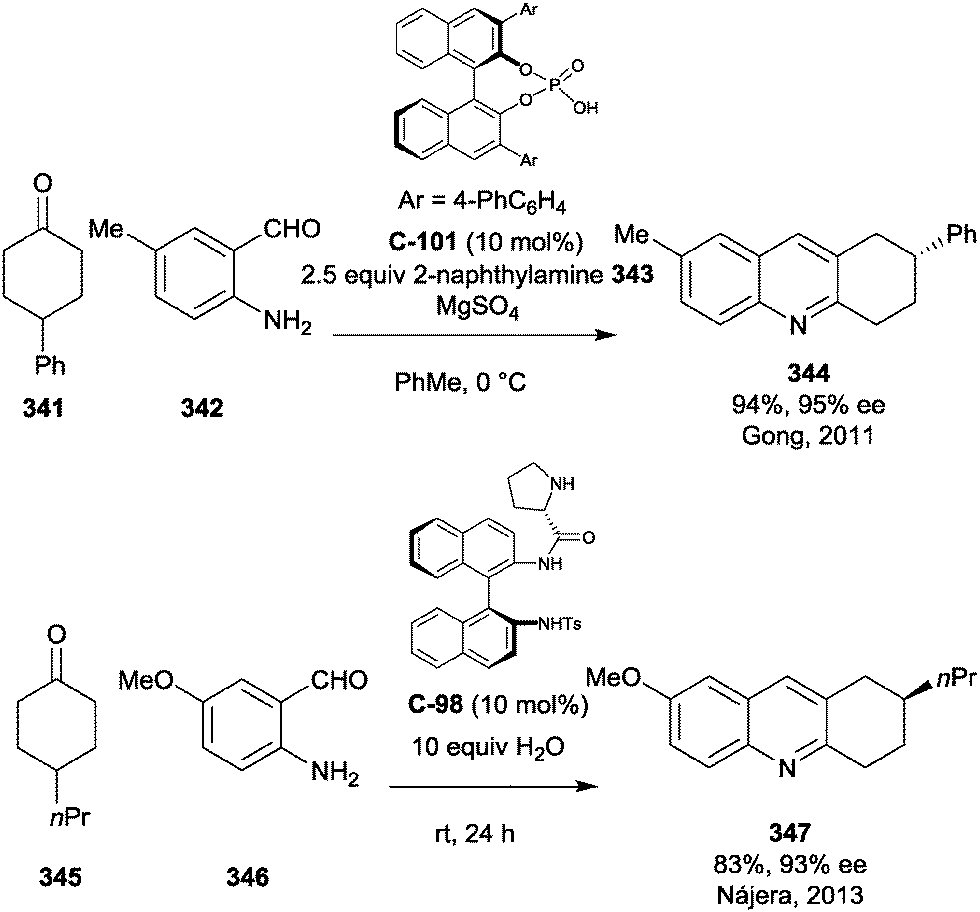 | ||
| Scheme 83 Desymmetrisation of 4-substituted cyclohexanones via enantioselective Friedländer reaction with o-aminobenzaldehydes under the action of proline- and binol-derived organocatalysts C-101 (Gong) and C-98 (Nájera). | ||
The Friedländer synthesis is not the only classical heterocycle forming reaction to have benefited from recent developments in organocatalysis and desymmetrisation; in 2011, List and co-workers reported the first example of a catalytic asymmetric Fischer indole synthesis (Scheme 84).161 Using novel spirocyclic phosphoric acid catalyst C-48, benzyl protected phenylhydrazones derived from 4-substituted cyclohexanones were converted into their corresponding 3-substituted tetrahydrocarbazoles in high yields and generally high enantioselectivity. Interestingly, the use of BINOL-derived phosphoric acid catalysts such as C-102 gave very low ees on similar substrates during optimisation, demonstrating the utility of STRIP-type catalyst C-48. The use of a benzyl group with a sterically demanding substituent in the para-position led to higher selectivity, therefore a 4-iodobenzyl (PIB) group was employed. Substitution on the aniline ring, however, usually led to lower enantioselectivity. A number of diversely substituted aryl groups were tolerated in the 4-position of the cyclohexyl ring, while R1 = Me and OBz gave high ees of 90% and 97% respectively; bulky alkyl groups eroded the selectivity significantly, however, with the 4-tert-butyl substrate giving 60% ee. The ion exchange resin Amberlite® CG50 removed ammonia from the reaction; this allowed the use of a catalytic amount of the chiral phosphoric acid. Under normal conditions, superstoichiometric quantities of acid are often required due to salt formation with the ammonia by-product. The authors state that the catalyst is responsible for increasing the rate of hydrazone–enehydrazine tautomerisation, with the benzyl protecting group playing a pivotal role in increasing the basicity of the aniline nitrogen. Hydrogen bond interactions between the enehydrazine and the phosphoric acid determine the enantioselectivity of the reaction.
 | ||
| Scheme 84 List's desymmetrisation of hydrazone derivatives of prochiral cyclohexanones via organocatalytic Fischer indole synthesis. PIB = para-iodobenzyl. | ||
Chen and co-workers have reported a three-component reaction of 1,3-indanedione, benzaldehydes and 4-substituted cyclohexanones (Scheme 85).162 Catalysed by a bifunctional quinine-derived thiourea organocatalyst, the quadruple reaction generates significant molecular complexity, forming polycyclic products in variable yields, moderate to excellent drs and high ees. Electron-donating and -withdrawing groups were tolerated on the aromatic aldehyde, and substitution pattern could be varied with no apparent detrimental effect on conversion or selectivity. Less variation was displayed for the cyclohexanone, however, with only relatively bulky groups being employed in the 4-position. Diastereoselectivity dropped significantly to 2:1 when R1 = iPr, compared to >19:1 for the tert-butyl analogue. The authors propose that the enantioselectivity arises from the preferential approach of a 2-arylidene-1,3-dione intermediate to the less hindered face of the reactive enolate intermediate; the reactive partners are held in place through hydrogen bonding to the bifunctional organocatalyst.
 | ||
| Scheme 85 Formation of sterically hindered polycyclic compounds via the organocatalysed multicomponent reaction of indanedione, prochiral cyclohexanones and aromatic aldehydes, described by Chen. | ||
Bencivenni and co-workers have reported the benzoyloxylation of 4-phenylcyclohexanone using benzoyl peroxide (Scheme 86).163 Using 10 mol% of quinine-derived organocatalyst C-103 and salicylic acid as an additive, the reaction proceeded in good yield with complete enantioselectivity. However, no diastereodiscrimination was observed, likely due to the small steric demand of the benzoyl group. The authors state that the hydroxyl group on salicylic acid is likely key to reactivity, due to hydrogen bonding to the carbonyl oxygens on benzoyl peroxide.
 | ||
| Scheme 86 Desymmetrisation of 4-phenylcyclohexanone via α-benzoyloxylation with benzoyl peroxide and quinidine-derived organocatalyst C-103 by Bencivenni. | ||
6.2 Michael additions
The Michael reaction is one of the most widely used methods for forming C–C bonds, and as such there has been considerable interest in developing enantioselective organocatalytic variants. There has been much progress in the enantioselective α-alkylation of ketones with Michael acceptors via enamine catalysis. β-Nitrostyrenes were the first electrophile used in this reaction, due to the high reactivity of the electron-poor olefin, the consistent regioselectivity, and particularly the ability of the nitro group to participate in hydrogen bonding to organocatalysts, aiding stereocontrol. The use of this activated Michael acceptor in catalytic enantioselective reactions with ketones was pioneered by List and Enders in the early 2000s.133,134 An early example of its involvement in enantioselective desymmetrisation was shown by Córdova in 2006, albeit only with 4-methylcyclohexanone and in very low diastereoselectivity.164 The development of more selective proline-derived organocatalysts has seen higher ees achieved in reactions of a variety of ketones, including desymmetrisations of 4-substituted cyclohexanones. Nitrostyrenes remain the most popular Michael acceptors, but catalytic enantioselective desymmetrising additions to other alkenes have recently appeared.Peddinti has reported two systems for the enantioselective desymmetrising Michael addition of ketones to β-nitrostyrene 363 (Scheme 87).165,166 High catalyst loadings were employed in both cases and enantioselectivities were moderate. Higher enantioselectivities were obtained by the groups of Zhou167 and Kokotos168 with their thiophosphoramidate and fluoropyrrolidine catalysts C-104 and C-107, respectively. Diastereoselectivities did not, however, match those of the work previously carried out by Xiao in 2009 (up to >99:1).169
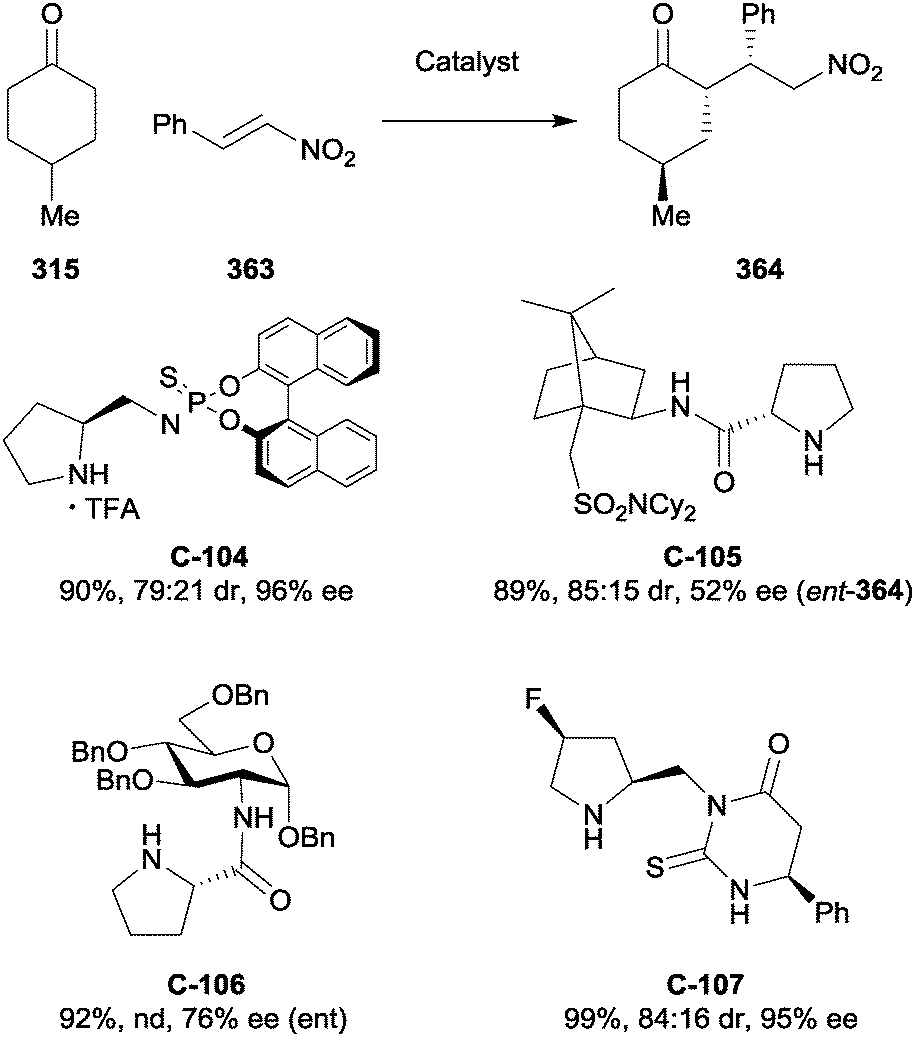 | ||
| Scheme 87 Organocatalysts employed by Kokotos, Peddinti, and Zhou for the enantioselective desymmetrisation of 4-methylcyclohexanone via Michael addition to β-nitrostyrene. | ||
In a related process, nitrodienes have been employed as Michael acceptors by Xu and co-workers using a pyrrolidinyl-thioimidazole catalyst.170 Catalyst loading was low at just 5 mol%, but addition of 5 mol% chiral thioureidic acid in place of the more common benzoic acid was necessary to obtain the highest ees. 4-tert-Butylcyclohexanone was effectively desymmetrised in excellent yields and ees and with good diastereoselectivity (Scheme 88). The authors postulate that enantioselectivity arose from the formation of a complex between the imidazolium and thioureidic acid, which in turn participated in hydrogen bonding to the nitro group. The same group has extended this methodology to the reaction of bromonitrodienes in similarly high ees and yields using a related prolinethiol ether catalyst and para-trifluoromethylbenzoic acid additive. Diastereoselectivities were more modest, although this could be due to the use of less sterically demanding methyl and ethyl groups in the 4-position of the cyclohexanone (Scheme 88).
 | ||
| Scheme 88 Desymmetrisation of prochiral cyclohexanones via Michael addition to nitrodienes and bromonitrodienes reported by Xu. | ||
Enantioselective desymmetrisation processes have also been performed using other electron deficient Michael acceptors. Barros and co-workers demonstrated the ability of pyrrolidine catalyst C-111 to promote Michael additions of 4-substituted cyclohexanones to vinyl bis(phosphonate) 375 (Scheme 89).171 The process gave good yields, but the enantio- and diastereoselectivity of the transformation proved disappointing in most cases.
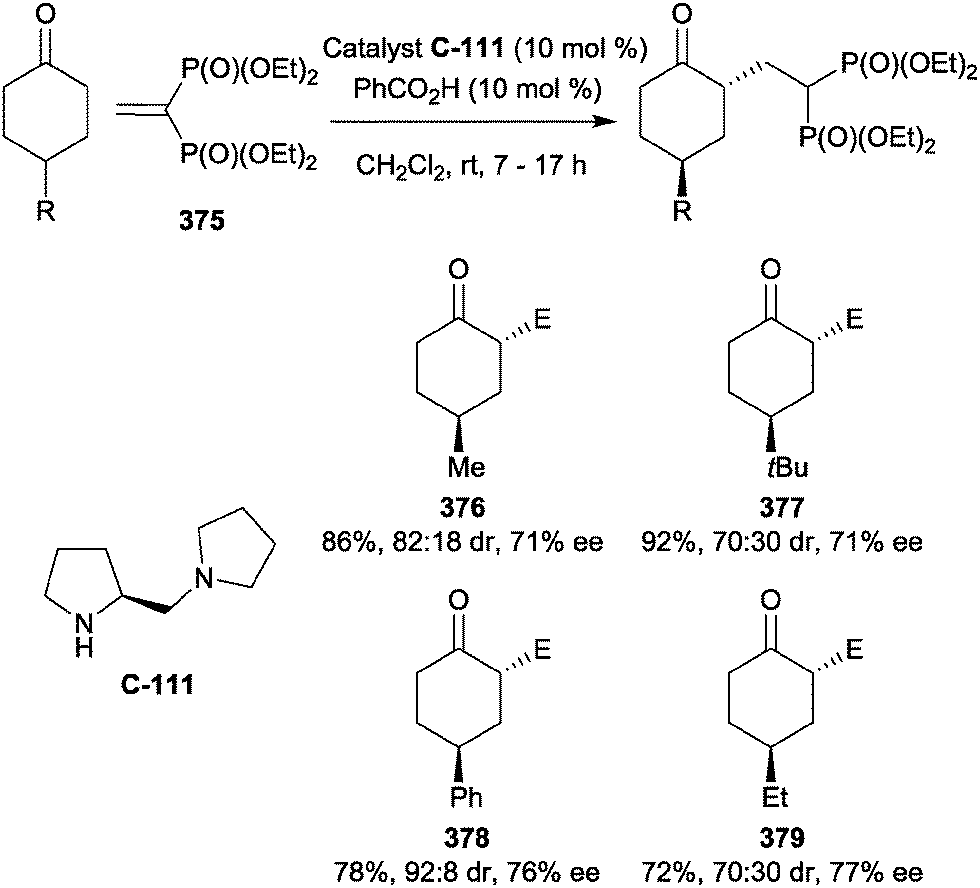 | ||
| Scheme 89 Desymmetrising Michael addition of 4-substituted cyclohexanone derivatives to vinyl(bisphosphonate) 375 by Barros. | ||
Chen and co-workers reported the use of a novel camphor-derived pyrrolidine catalyst C-112 for the enantioselective Michael addition of 1,1-bis(phenylsulfonyl)ethylene 380 to 4-substituted cyclohexanones (Scheme 90).172 Incorporating a thiourea moiety and synthesised in six steps from functionalised L-proline and camphor derivatives, in the presence of catalytic benzoic acid the bifunctional organocatalyst facilitated the formation of trans-2,4-disubstituted cyclohexanones in moderate to excellent yields, poor drs and good ees. The opposite enantiomer could be obtained using cinchonidine-derived catalyst C-113, with similarly low diastereoselectivity but generally improved yields and ees. Catalyst loadings were, however, uniformly high at 20 mol%.
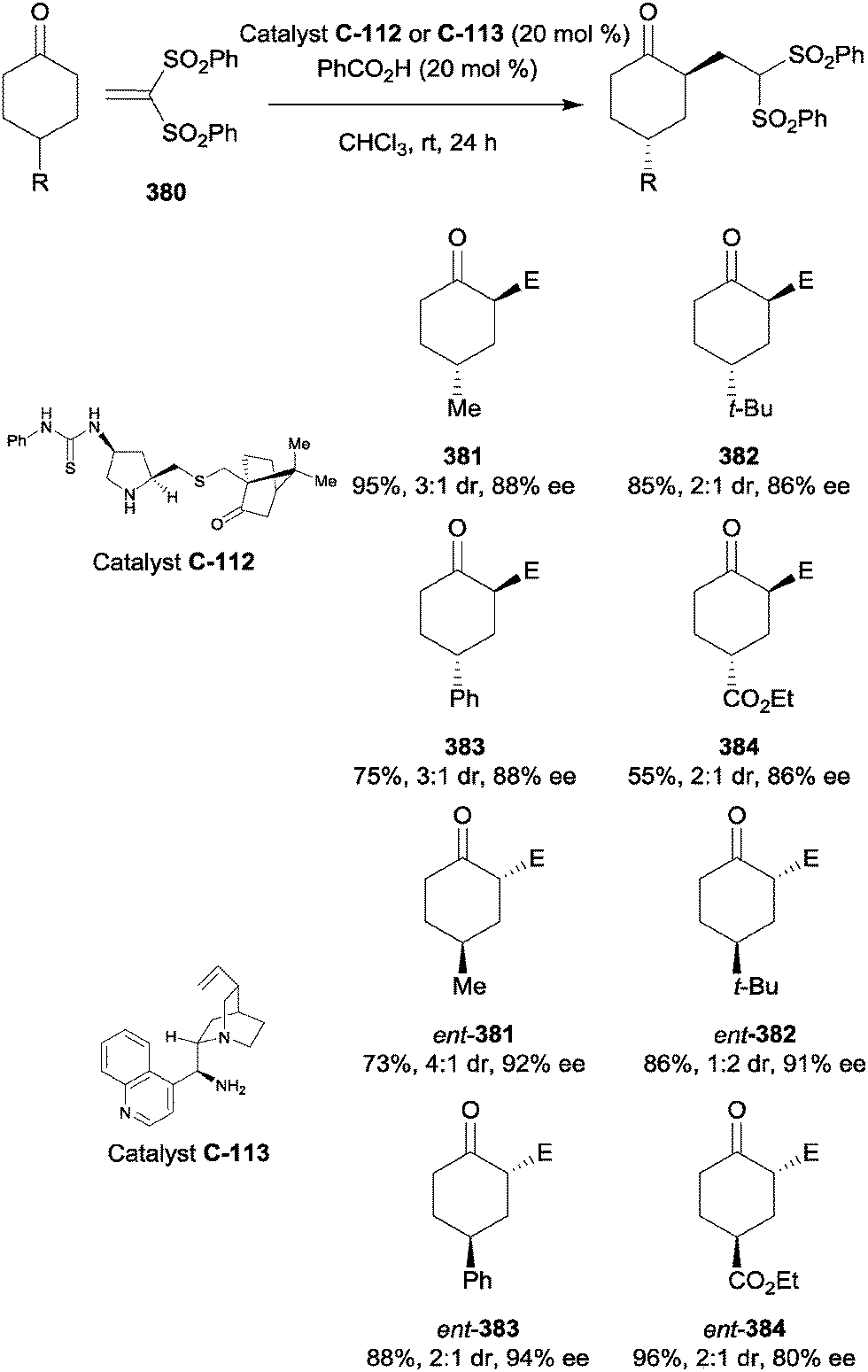 | ||
| Scheme 90 Enantioselective α-alkylation of 4-substituted cyclohexanones via Michael addition to vinyl sulfone 380 by Chen. | ||
Dixon and co-workers have reported the first desymmetrisation of a 4-substituted cyclohexanone via a catalytic asymmetric intramolecular Michael addition, simultaneously developing a novel preparation of the morphan (2-azabicyclo[3.3.1]nonane) ring system,173 present in a large number of biologically active alkaloid natural products (Scheme 91).174 Initial investigations revealed the ability of primary amines to facilitate the reaction, and a catalyst screening identified the cyclohexanediamine-derived organocatalyst C-114 as providing optimal yields and selectivities. Catalytic amounts of benzoic acid were found to increase the rate of reaction. Yields and selectivities were universally high across a broad range of substrates. Low catalyst loadings were employed, although long reaction times were necessary for full conversion. Interestingly, when the reaction was conducted using a (Z)-configured α,β-unsaturated ester, the same product was isolated in racemic form. Computational studies elucidated the origin of the enantiocontrol, and resulted in the discovery of the novel thiourea catalyst C-115, which proved equally as efficient as the more complex catalyst C-114, suggesting the tert-leucine moiety does not play a significant role in stereoinduction.
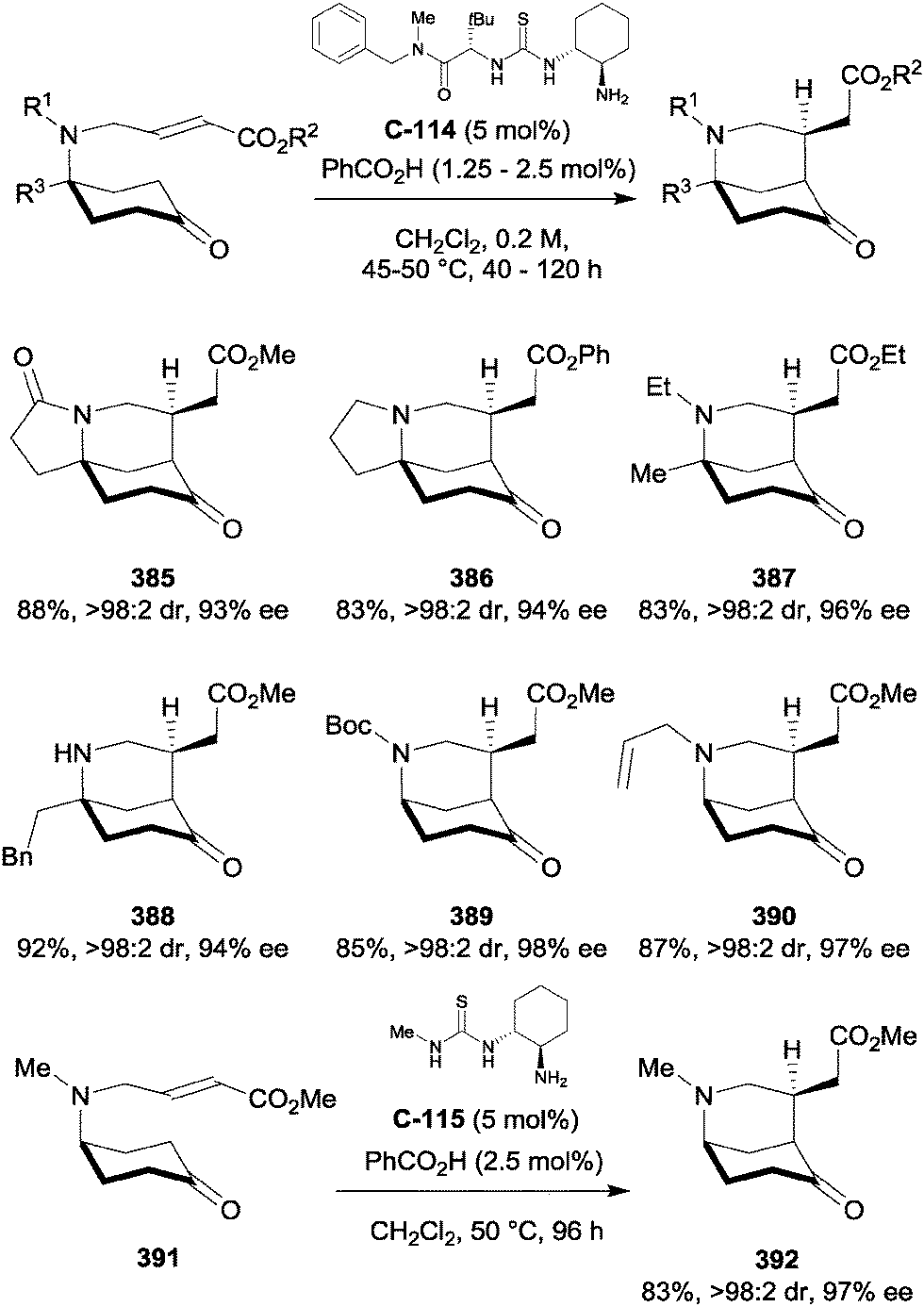 | ||
| Scheme 91 Dixon's asymmetric construction of the morphan core via desymmetrising intramolecular Michael addition. | ||
6.3 Asymmetric deprotonation
An example of desymmetrisation via an asymmetric deprotonation process was reported by Levacher and co-workers.175 In combination with N,O-bis(trimethylsilyl)acetamide 393, acting as both a base and silylating agent, cinchona alkaloid-derived chiral quaternary ammonium salt catalyst C-116 was employed to obtain silyl enol ethers in low ees from 4-substituted cyclohexanones (Scheme 92). Although the enantioselectivity was poor in all cases, the authors argue that this is compensated for by the use of low catalyst loading (5 mol%) and only one reagent for both deprotonation and silylation. | ||
| Scheme 92 Asymmetric silyl enol ether formation from 4-substituted cyclohexanones in the presence of quinine-derived organocatalyst C-116 by Levacher. | ||
6.4 Cyclobutanone substrates
In contrast to 4-substituted cyclohexanones, there has been relatively little research into the enantioselective organocatalytic desymmetrisation of prochiral cyclobutanones. Early examples by the groups of Murahashi176 and Ding177 were limited to enantioselective Baeyer–Villiger oxidation. More recently, an interesting ring expansion of 3-substituted cyclobutanones has been reported by Frongia and co-workers (Scheme 93).178 In an attempt to form α-aminoxylated cyclobutanones,136 a 4-arylcyclobutanone was reacted with nitrosobenzene using L-proline as an organocatalyst. The reaction instead furnished 4-substituted-5-hydroxy-γ-lactams, via expansion of a strained 4-membered ring. The best results were obtained with the proline-derived tetrazole catalyst C-117. Yields and enantioselectivities were low, while diastereoselectivity varied widely.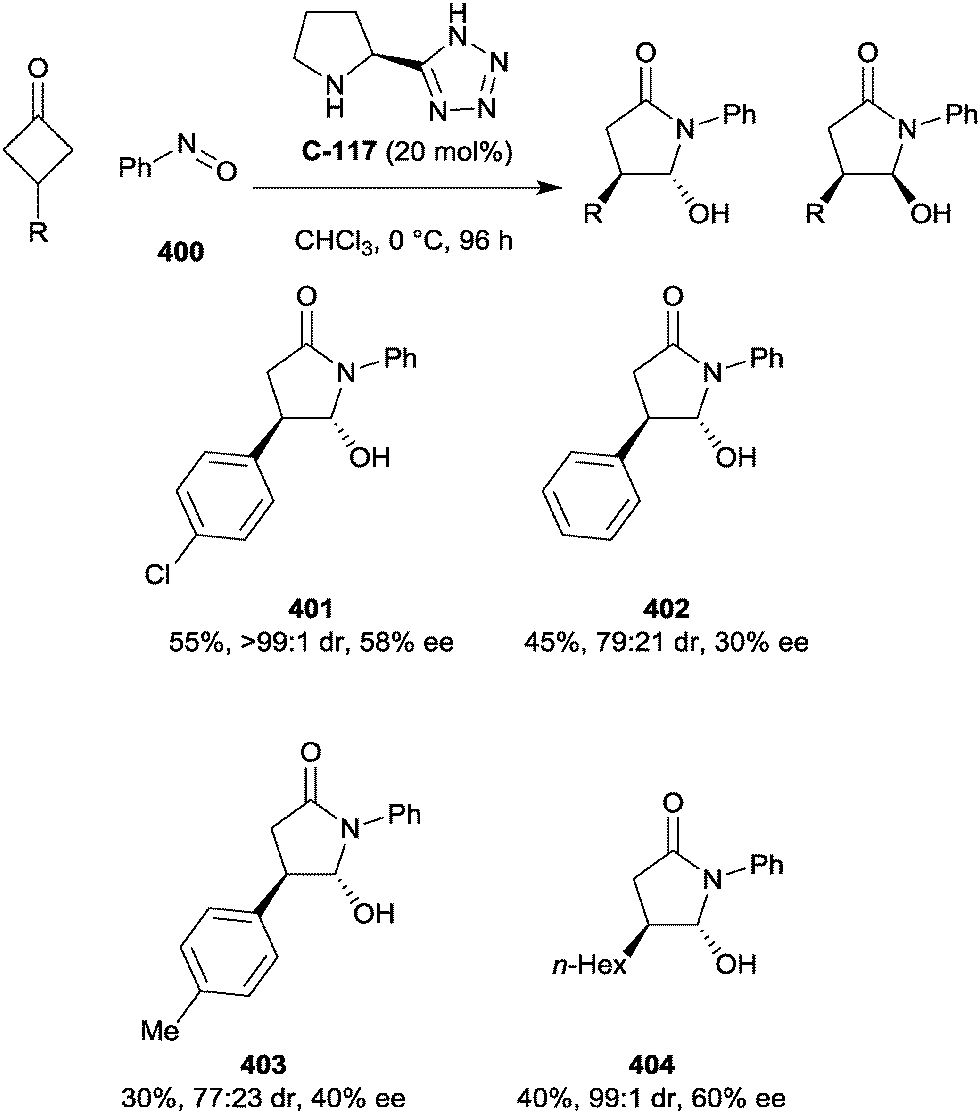 | ||
| Scheme 93 Desymmetrisation of prochiral cyclobutanones via ring expansion reaction with nitrosobenzene by Frongia. | ||
Frongia and co-workers have also developed the desymmetrisation of prochiral cyclobutanones via an organocatalysed aldol reaction (Scheme 94).179 Initial investigations revealed proline as a suitable catalyst, with the sulfonamide derivative C-118 giving optimal diastereo- and enantioselectivities. Substrate scope was, however, limited to electron-poor aryl aldehydes, with nitro substitution giving the best selectivities. More electron-rich and ortho-substituted benzaldehydes failed to give any product. Both aryl and alkyl substituents were tolerated in the 3-position of the cyclobutanone. Long reaction times and relatively high catalyst loadings were required, with a large excess of the cyclobutanone (20 equivalents) being used in all cases.
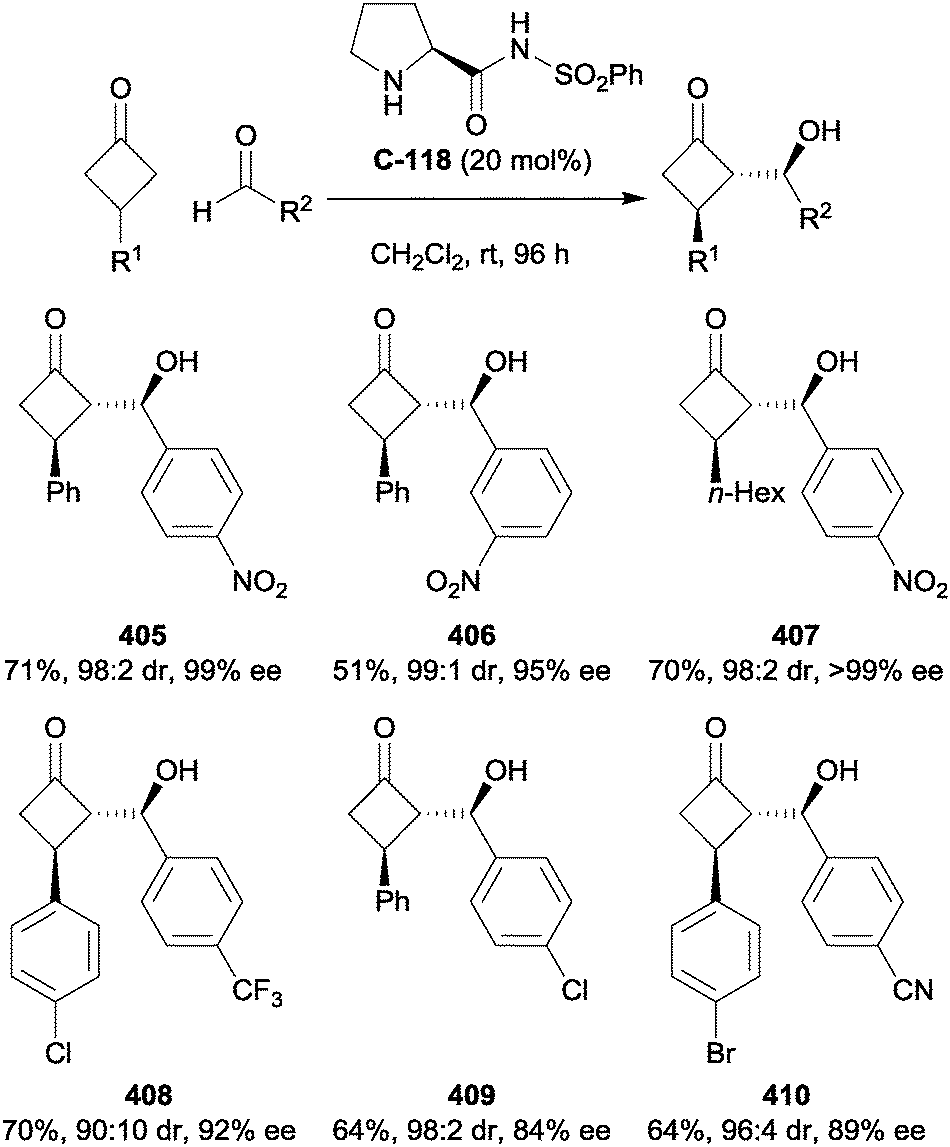 | ||
| Scheme 94 Aldol desymmetrisation of 3-substituted cyclobutanones by Frongia. | ||
The same group has extended the enantioselective desymmetrisation of 3-substituted cyclobutanones to include organocatalysed Michael addition to nitroalkenes, stated by the authors to be the first example of such a reaction (Scheme 95).180 The products, containing three contiguous stereocentres, were obtained in moderate to good yields, variable drs and poor ees. Both alkyl and aryl substitution were possible on the nitro-olefin and the cyclohexanone. Long reaction times were necessary for full conversion in all cases. The authors speculate that the nitro group is involved in hydrogen bonding to the enamine proton and the thiourea moiety on the catalyst and that this orientation of the reactive partners is responsible for stereoinduction.
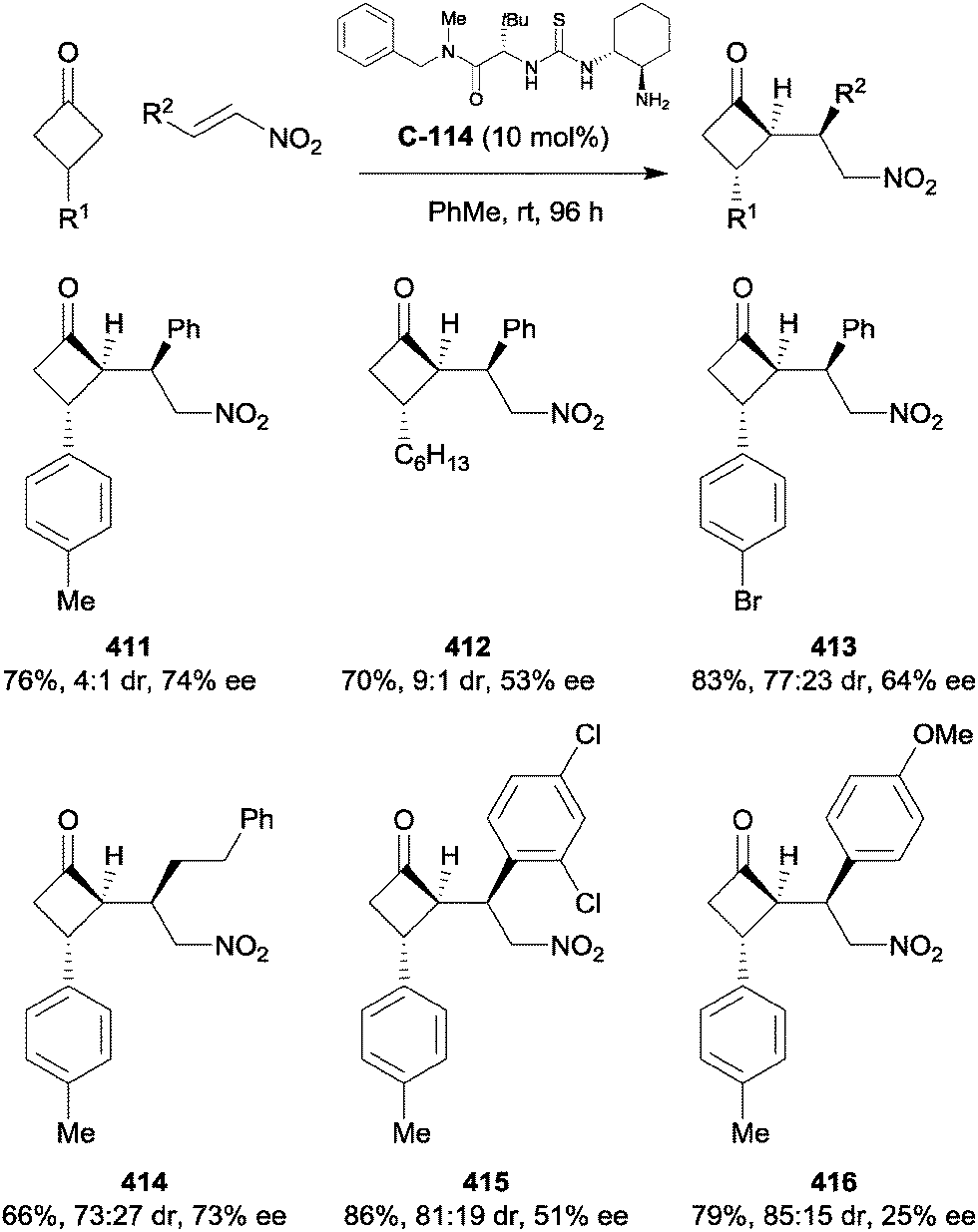 | ||
| Scheme 95 Organocatalytic enantioselective desymmetrisation of prochiral cyclobutanones via Michael addition to nitroalkenes by Frongia. | ||
6.5 Diketone substrates
Prochiral 1,3-diketones are also useful substrates for enantioselective desymmetrisation. Indeed, one of the earliest examples of organocatalysis, the Hajos–Parrish–Eder–Sauer–Wiechert reaction, involves the desymmetrisation of a cyclic 2,2-disubstituted-1,3-diketone using L-proline.181,182 Scheidt and co-workers published an example of the desymmetrisation of 2,2-disubstituted-1,3-diketones using NHC catalysis in 2007,183 and later applied it to the total synthesis of the sesquiterpene natural products Bakkenolide I, J and S.184Romo and co-workers have reported an organocatalytic enantioselective process to form tricyclic β-lactones via the desymmetrisation of cyclic 1,3-diketones, the first such “nucleophile-catalysed aldol lactonisation” (NCAL) reaction on cyclic diones (Scheme 96).185 In an improvement over previous stoichiometric NCAL reactions, inexpensive toluenesulfonyl (tosyl) chloride was used as an activator in place of the modified Mukaiyama reagent 2-bromopropylpyridinium chloride. Furthermore, (S)-HBTM (homobenzotetramisole) C-119 was employed as a nucleophilic catalyst, enabling the reaction by combining with the carboxylic acid functionality to form an ammonium enolate, which could then undergo aldol lactonisation with stereoselective attack at a ketone. Addition of LiCl resulted in increased yields, but generally at the cost of some enantioselectivity. Enantioselectivity was proposed to arise from a nO → σC–S* non-bonding interaction. The authors state that lithium cation could coordinate both the ketone and ammonium enolate oxygens and the sulfur on the catalyst, activating the ketone and forming a chairlike bicyclic transition state. Test reactions performed with and without catalyst and lithium chloride revealed a complex process in which the additive was seemingly involved in promoting both racemic and asymmetric pathways, potentially explaining its impact on yield and selectivity. Possible achiral mechanisms for the NCAL reaction include formation of a ketene and subsequent [2+2] cycloaddition. The tricyclic products formed could be derivatised to various diverse carbon frameworks, which the authors suggest may find use in total synthesis.
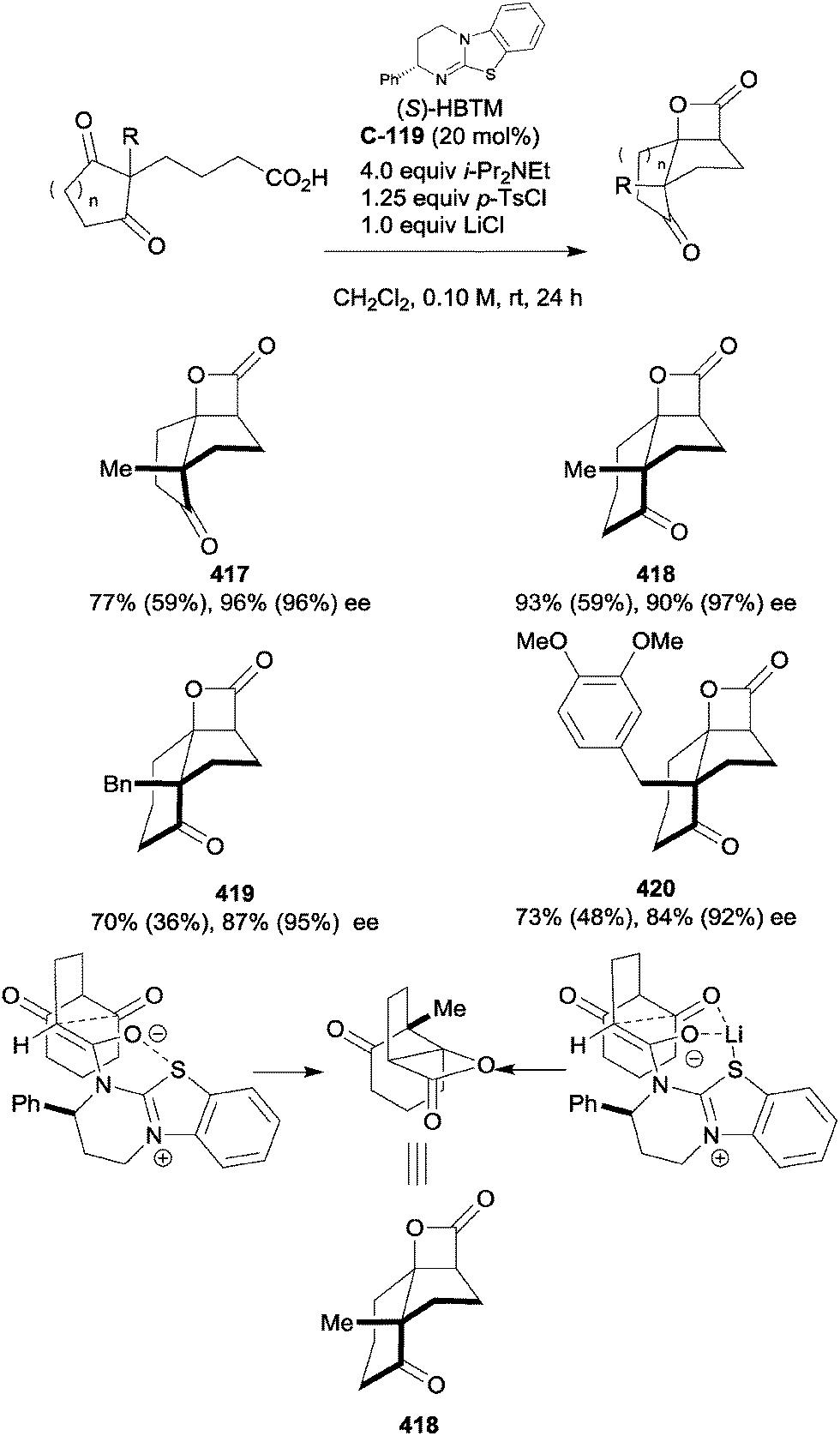 | ||
| Scheme 96 Enantioselective intramolecular nucleophile-catalysed aldol lactonisation reaction of cyclic 1,3-diketones with a pendant carboxylic acid catalysed by (S)-HBTM C-119, reported by Romo; Parentheses indicate yields and ees obtained without the use of LiCl additive. | ||
Lam and co-workers have disclosed an efficient method for the construction of enantioenriched bicyclo[3.2.1]octanes and bicyclo[3.3.1]nonanes via the desymmetrisation of cyclic 2,2-disubstituted 1,3-diketones (Scheme 97).186 Employing chiral phosphoric acid catalyst C-42 to encourage enolisation and subsequently, the authors propose, bind both the enol and pendant enone functionality, the molecule could be coaxed into undergoing an asymmetric intramolecular Michael addition, giving the desired products in generally good yields and high ees. Diastereomeric products where the ketone was in the axial rather than equatorial position were formed in some reactions, but were easily separable by chromatography. A variety of aryl and heteroaryl enones were tolerated by the reaction, with little variation in yield and selectivity regardless of the electron-withdrawing or -releasing substituents, or Lewis basic atoms in the ring. The presence of a para-nitro group did, however, erode enantioselectivity appreciably (72% ee). Yields and enantioselectivity were retained when R2 = tBu, although when a secondary alkyl substituent on the enone was employed (R2 = CH2CH2OBn), yields and diastereoselectivity dropped significantly. Aryl and allyl groups were well-tolerated at the 2-position of the cyclic diketone, albeit with more moderate yields for the former. One example containing an α,β-unsaturated amide (n = 1, R1 = Me, R2 = NMe2) was also shown; the Michael product could be formed in moderate yield and ee using higher temperatures, increased catalyst loading and longer reaction time. Surprisingly, no reaction was observed for the analogous α,β-unsaturated benzyl ester.
 | ||
| Scheme 97 Lam's formation of bicyclo[3.n.1]alkanes via enantioselective intramolecular Michael addition of cyclic 2,2-disubstituted dione enones; values in parentheses indicate yields and ees of minor diastereomer. | ||
Akiyama and co-workers have developed the first desymmetrisation of a 2,2-disubstituted cyclopentane-1,3-dione via an intramolecular reductive amination, forming cis-fused pyrrolidine and piperidine derivatives in good to high enantioselectivities (Scheme 98).187 The presence of the meta-hydroxyphenyl group on the nitrogen was found to be pivotal; analogues with oxygen substituents in the 2- and 4-positions gave low conversions and selectivities, as did the meta-methoxyaniline. It is thought that hydrogen bonding between the phosphoric acid catalyst, the hydroxyl group and the ketone governs the enantioselectivity of the reaction. The reaction showed a strong dependence on the steric bulk of the indanedione for the formation of fused piperidines; the methyl derivative proceeded in 87% yield at 0 °C, while the ethyl and benzyl derivatives required heating to 50 °C to achieve lower yields and selectivities. For R = Ph, only traces of product were detected even after heating to 80 °C. Cyclisation to form the fused pyrrolidine products proved more favourable, however, with high yields and selectivities even when R = Bn.
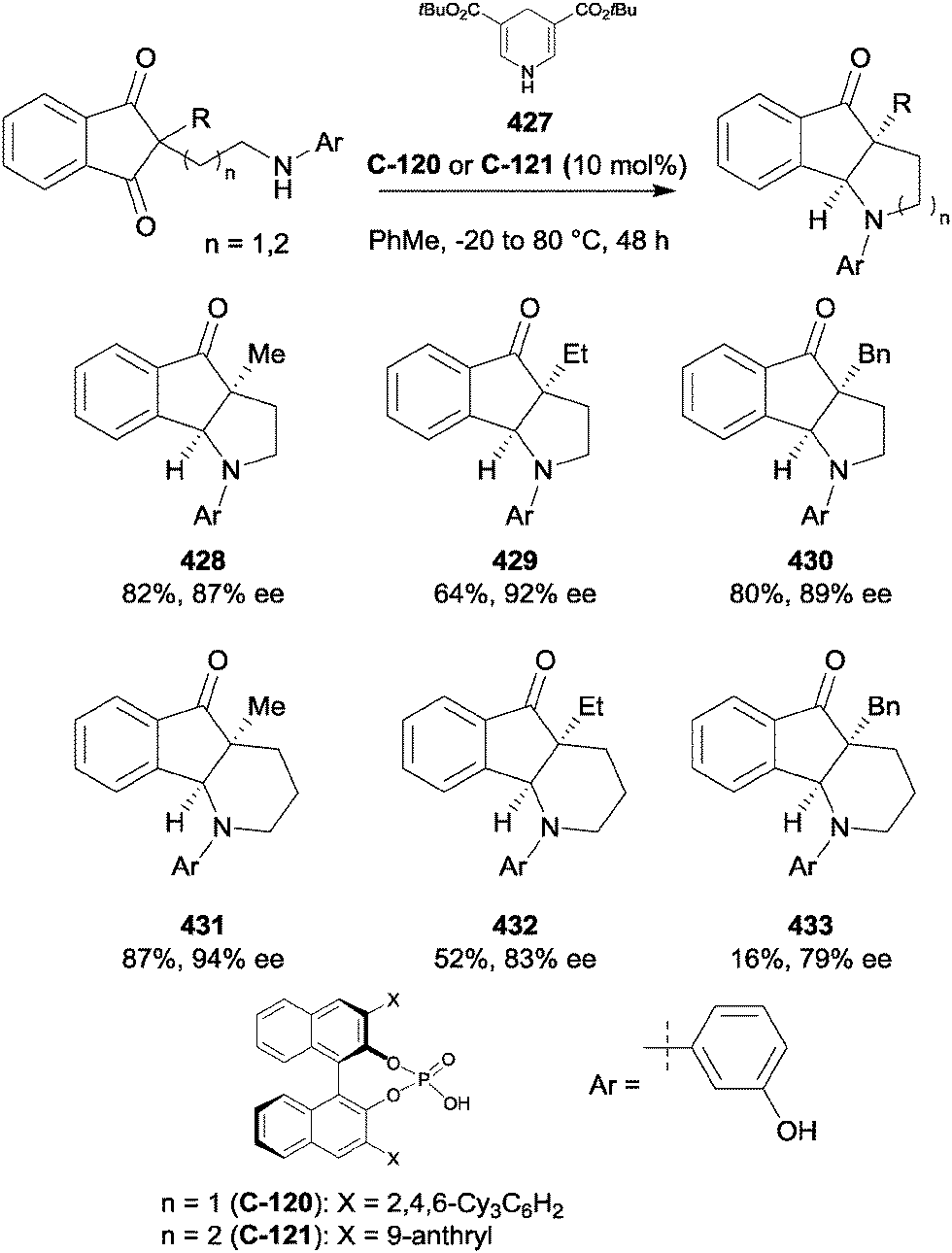 | ||
| Scheme 98 Akiyama's chiral phosphoric acid catalysed desymmetrisation of prochiral indanediones via intramolecular reductive amination. | ||
Feng and co-workers have developed a method for the enantioselective desymmetrisation of 5-substituted cyclohexane-1,3-diones with (Z)-bromonitrostyrenes, forming bicyclic dihydrofuran products in good yields, moderate drs and good to high ees (Scheme 99).188 Stereocontrol of the tandem Michael-alkylation reaction is attained using the novel chiral N,N′-dioxide organocatalyst C-PrPr2C-122, thought by the authors to bind both the enol tautomer of the dione via hydrogen bonding to the amine oxide, and similarly the amide N–H of the catalyst to the nitro group of the Michael acceptor. The scope of the reaction was somewhat limited; styrenes with substituents on all positions of the aromatic ring gave similar results, although no strongly activating or deactivating groups were shown. 3-Thienyl and 2-naphthyl groups were also tolerated on the nitroalkene, while only phenyl, 4-fluorophenyl and 2-furyl substituents were tested on the cyclohexanedione, with furan giving the highest ees, consistently around 90%.
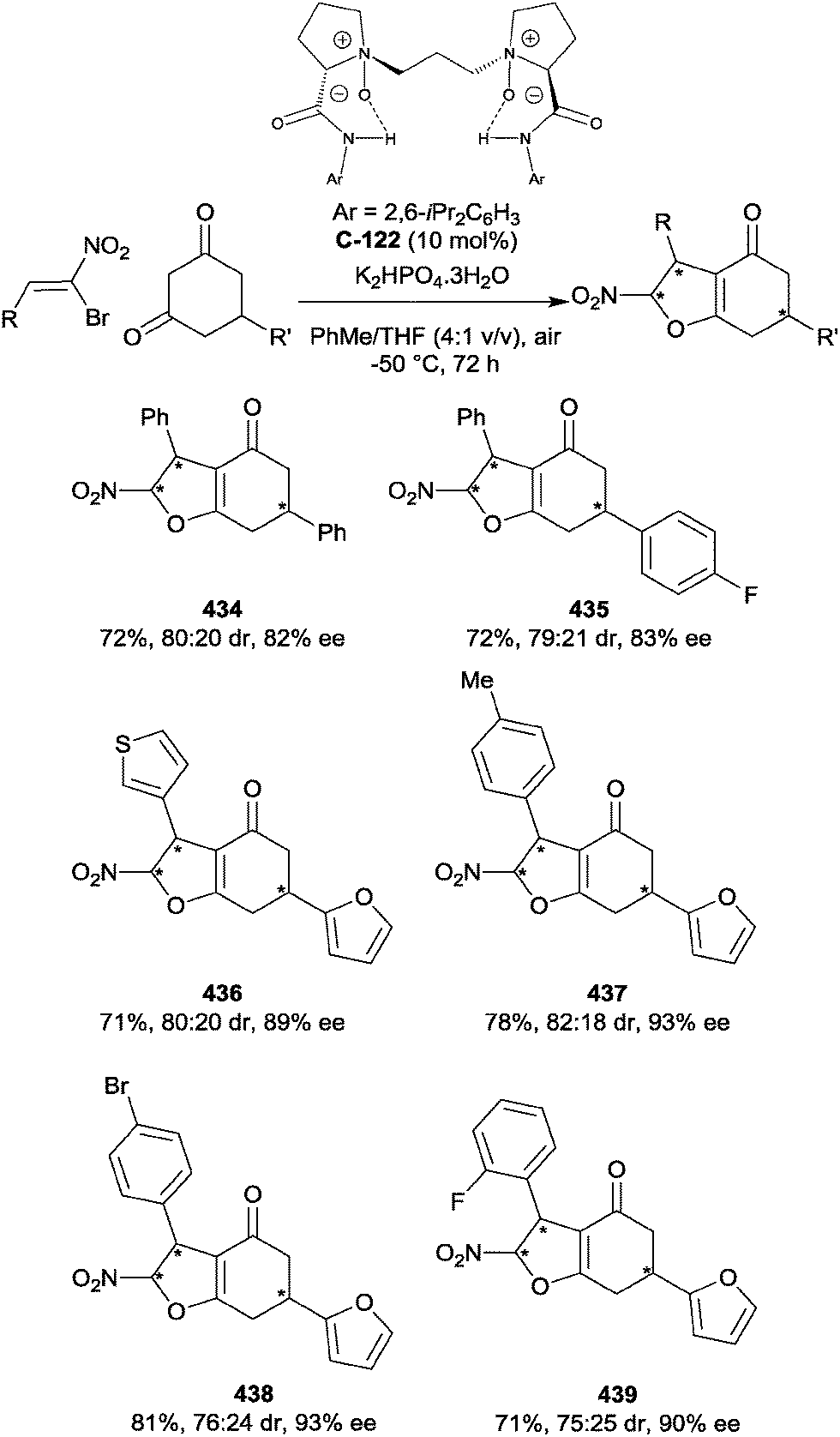 | ||
| Scheme 99 Tandem Michael-alkylation reaction of nitrobromostyrenes with 5-(hetero)aryl cyclohexane-1,3-diones, catalysed by Feng's chiral N,N′-dioxide catalyst C-122. | ||
Rueping and co-workers have reported a method to synthesise enantioenriched tetrahydroxanthenes via the desymmetrisation of 5-substituted cyclohexane-1,3-diones (Scheme 100).189ortho-Quinone methides were formed in situ by the BINOL phosphoric acid catalysed dehydration of hydroxybenzyl alcohols, with subsequent Michael addition of the 1,3-diketone and elimination of a second equivalent of water giving the tetrahydroxanthene products. Diastereo- and enantioselectivities for the 5-aryl cyclohexanediones were universally excellent, although straight chain alkyl substitution led to a slight erosion in ees and significantly decreased diastereoselectivity. The authors note that the formation of such a 1,5-relationship between stereocentres is unprecedented using chiral Brønsted acid catalysis. The phosphoric acid is proposed to have multiple roles in the reaction: forming the quinone methide, enolisation of the cyclohexadienone, and promoting the Michael addition and determining the enantioselectivity via binding of both intermediate enone and quinone methide.
 | ||
| Scheme 100 Desymmetrisation of prochiral cyclohexane-1,3-diones to form tetrahydroxanthenes reported by Rueping. | ||
7. Enones and dienones
Organocatalytic enantioselective desymmetrisation of symmetric cyclohexadienones has been furthered by the efforts of several research groups. The unsaturated carbon–carbon bonds of the dienone are often exploited in Michael addition reactions. With the use of chiral organocatalysts, additions can often be achieved with high enantioselectivity. The desymmetrisation of achiral 2,5-cyclohexadienones under metal and metal-free catalysis was reviewed in 2014 by Kalstabakken and Harned.190 The asymmetric construction of chiral cyclohexenone derivatives by organocatalysis was reviewed by Li et al. in 2014.191Fang and Wang recently reviewed the advances in asymmetric organocatalysis mediated by bifunctional catalysts bearing multiple hydrogen bond donors.192 Wang et al. employed amine–thiourea catalyst C-123 to achieve the desymmetrisation of a cyclohexadienone bearing a spirocyclic oxindole at the C4-position. This achieved the construction of an all-carbon quaternary centre adjacent to a sulfur-substituted tertiary centre.193 Wang demonstrated that the system tolerated a range of aromatic thiols, maintaining enantiomeric excesses of greater than 80% across the tested substrates (Scheme 101).
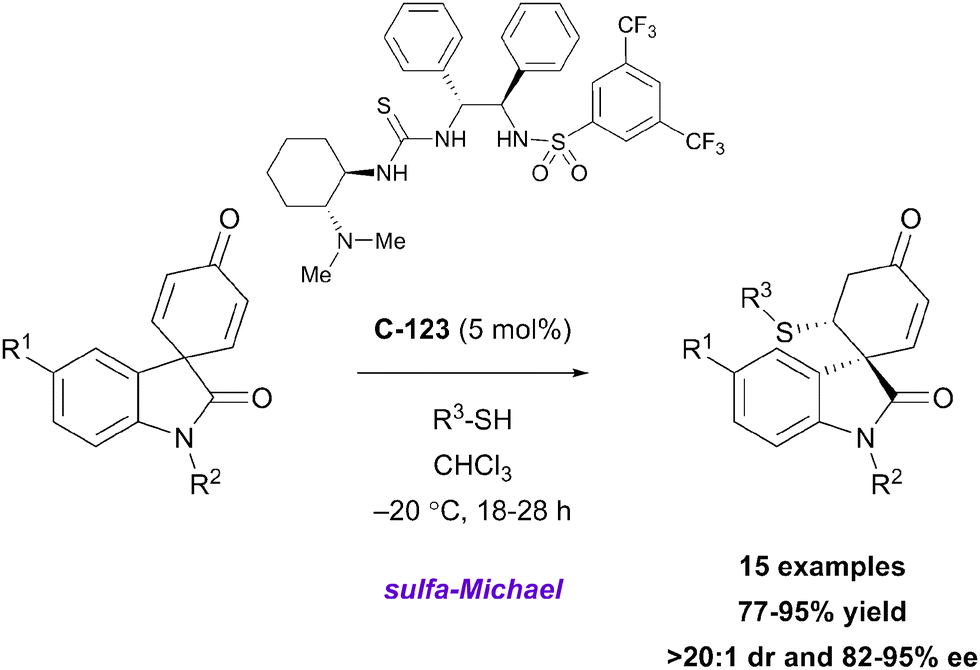 | ||
| Scheme 101 Desymmetrisation of spirocyclic cyclohexadienones by bifunctional thiourea-catalysed sulfa-Michael additions reported by Wang. | ||
Wang further functionalized the molecule, installing a second chiral centre by the addition of a second thiol to the remaining enone from the opposite face (Scheme 102).
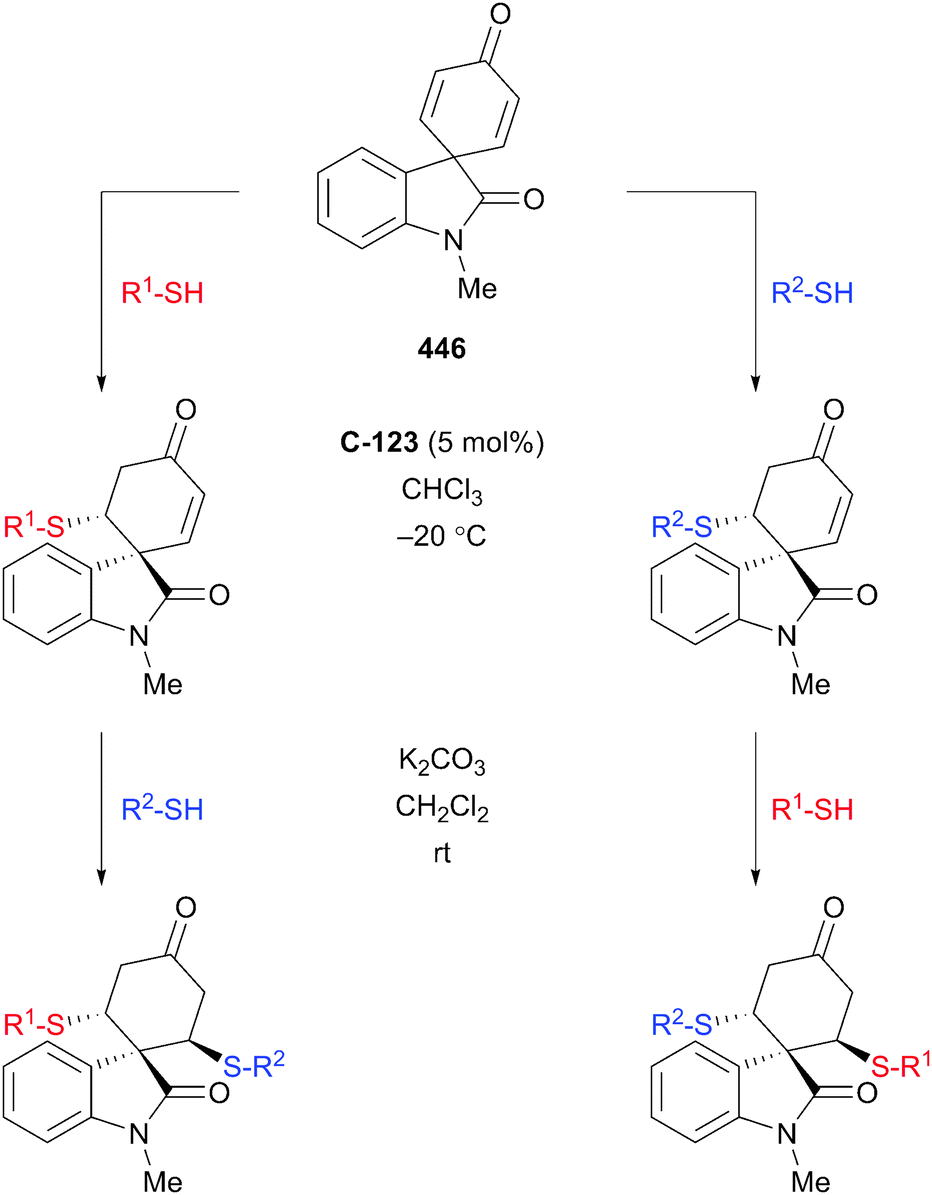 | ||
| Scheme 102 Regioselective functionalisation of spiro-oxindole via sequential sulfa-Michael additions by Wang. | ||
The application of the intramolecular Michael reaction to the desymmetrisation of 4,4-disubstituted cyclohexadienones has yielded a range of enantioenriched polycyclic products, which could serve as useful intermediates in natural product syntheses. You and co-workers have developed this reaction for Michael additions with carbon, nitrogen and oxygen nucleophiles (Scheme 103a). The authors employed cinchonine-derived thiourea catalyst C-124 to effect the desymmetrisation of cyclohexadienones containing a pendant bisphenylsulfonyl methylene group (Scheme 103b).194 The strategy afforded a range of fused cyclohexenone derivatives in high yields and with high degrees of enantioselectivity.
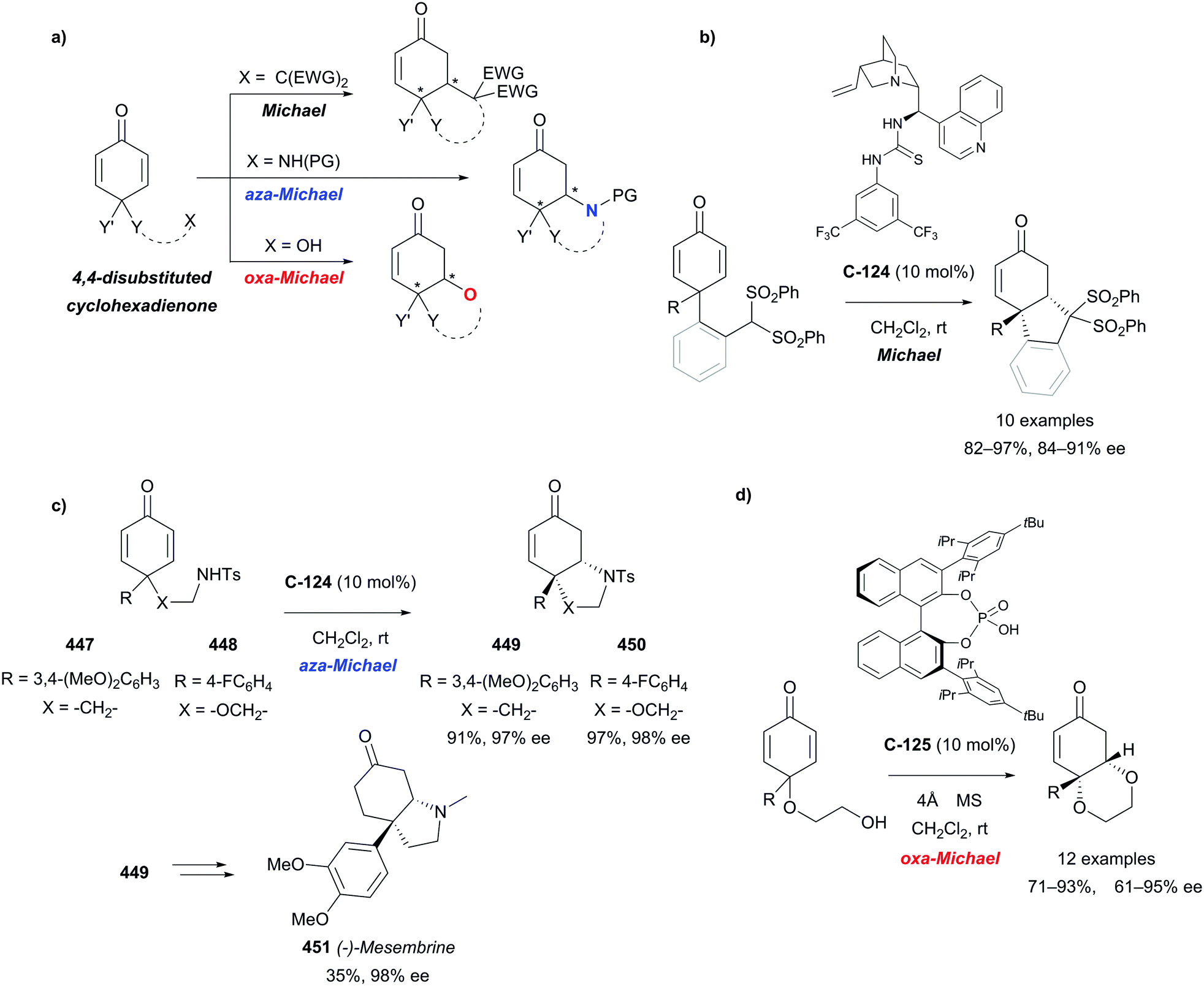 | ||
| Scheme 103 (a) Asymmetric, organocatalysed Michael, aza-Michael and oxa-Michael additions reported by You in the desymmetrisation of prochiral 4,4-disubstituted cyclohexadienones. (b) Generation of enantioenriched carbocycles by intramolecular Michael reactions. (c) Synthesis of chiral morpholine and pyrrolidine heterocycles, including (−)-Mesembrine, by intramolecular aza-Michael reactions. (d) Intramolecular oxa-Michael reactions affording useful precursors for natural product syntheses including Cleroindicins C and F. | ||
You et al. also applied thiourea C-124 to an aza-Michael desymmetrisation, affording bicyclic pyrrolidine and morpholine derivatives in high yields (75-97%, one example 26%) and with excellent enantioselectivity (80–98% ee). This strategy was successfully applied to the total synthesis of (−)-Mesembrine, which was obtained in 35% yield and 98% ee in 5 steps from pyrrolidine 449 (Scheme 103c).195 The You group also demonstrated the use of chiral phosphoric acid catalyst C-125 for intramolecular oxa-Michael reactions to generate fused bicyclic 1,4-dioxanes in similarly high yields and enantioselectivity (Scheme 103d).196
Harned et al. used quaternary cinchona alkaloid-based ammonium salt C-126 for the enantioselective desymmetrisation of 4,4-disubstituted cyclohexadienones by phase-transfer catalysis.197 Desymmetrisation of the meso-cyclohexadienones proceeded via an intramolecular Michael reaction between the α,β-unsaturated enone and the activated methylene group. Whilst only moderate yields and levels of enantioselectivity were obtained, in the case of brominated substrates, the method afforded unusual tricyclic cyclopropanes (Scheme 104).
 | ||
| Scheme 104 Harned reported moderate enantioselectivities with a cinchona alkaloid phase-transfer catalyst, with the generation of unusual cyclopropane tricycles when X = Br. | ||
Ye et al. achieved excellent yields and high levels of enantioselectivity in an intramolecular oxa-Michael desymmetrisation of cyclohexadienones with the use of a simple diamine catalyst C-127 (Scheme 105a).198 The reported substrate scope demonstrated tolerance of alkyl, aryl and alkoxy substitution in the 4-position of the cyclohexadienone, resulting in the production of a range of enantioenriched 1,4-dioxane derivatives. Complex bicyclo[2.2.2]octan-2-one structures bearing multiple stereocentres could be generated via a vinylogous Michael addition to the resulting α,β-unsaturated ketone (Scheme 105b).
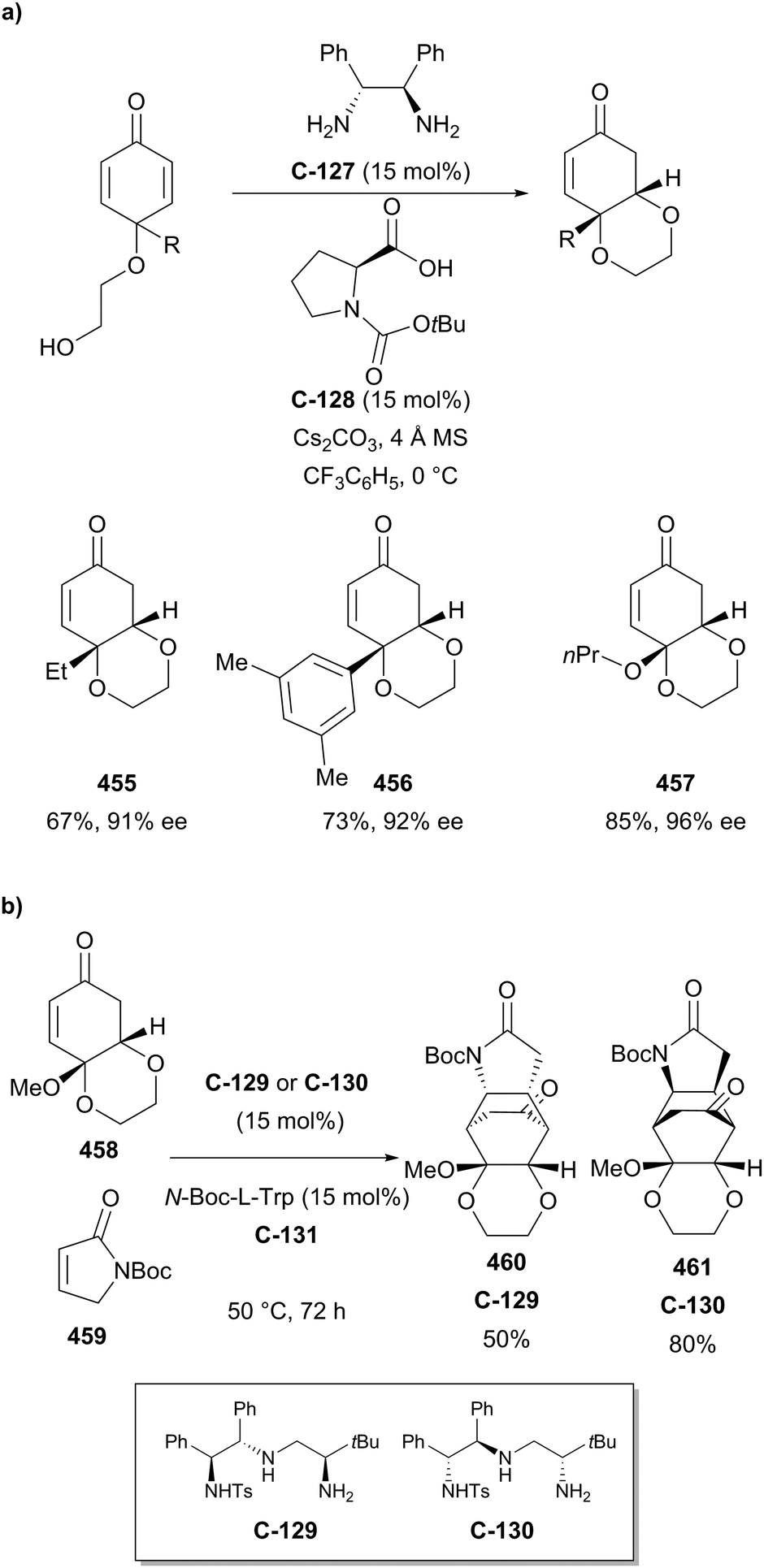 | ||
| Scheme 105 (a) Work by Ye on the desymmetrisation of cyclohexadienones with chiral amine catalysis produced enantioenriched 1,4-dioxane derivatives. (b) Further derivatisation affords complex bridged tetracyclic compounds. | ||
You et al. have demonstrated the use of N-heterocyclic carbene catalysis in the enantioselective desymmetrisation of 4-substituted cyclohexadienones. When substituted in the 4-position with an ortho-benzaldehyde, cyclohexadienones could be desymmetrised via intramolecular Michael addition of the corresponding Breslow intermediate. Use of triazolium salt C-132 afforded tricyclic carbocycles in excellent yields and with good levels of enantioselectivity (Scheme 106a).199 Additionally, α-substituted cyclohexadienones underwent desymmetrisation with the same catalyst to afford complex nitrogen-containing tricyclic structures.200 Products were obtained in very high yields (up to 96%) and with excellent enantioselectivity (Scheme 106b).
 | ||
| Scheme 106 Desymmetrisation of cyclohexadienones by You, using D-camphor-derived triazolium salt C-132 as an effective catalyst for the production of chiral tricycles via intramolecular Stetter reaction. | ||
Cascade reactions of 4-substituted cyclohexadienones have been used in desymmetrisation strategies to rapidly produce highly functionalised chiral molecules. An example is the reaction of quinols201 or quinamines202 with α,β-unsaturated aldehydes. The process begins with an initial oxa- or aza-Michael addition (Scheme 107a). The resulting intermediate undergoes an intramolecular Michael cyclisation which affords highly substituted bicyclic compounds. Desymmetrisation of the diene occurs in the second step, although a stereocentre is also established in the initial aza-Michael stage. Both steps are catalysed by proline-derived Jørgensen-Hayashi type catalyst C-133203 which forms chiral iminium/enamine intermediates with aldehydes. Four stereogenic centres are established in these processes; achieving this in one experimental step comprised of two reactions promoted by a single catalyst is an excellent result.
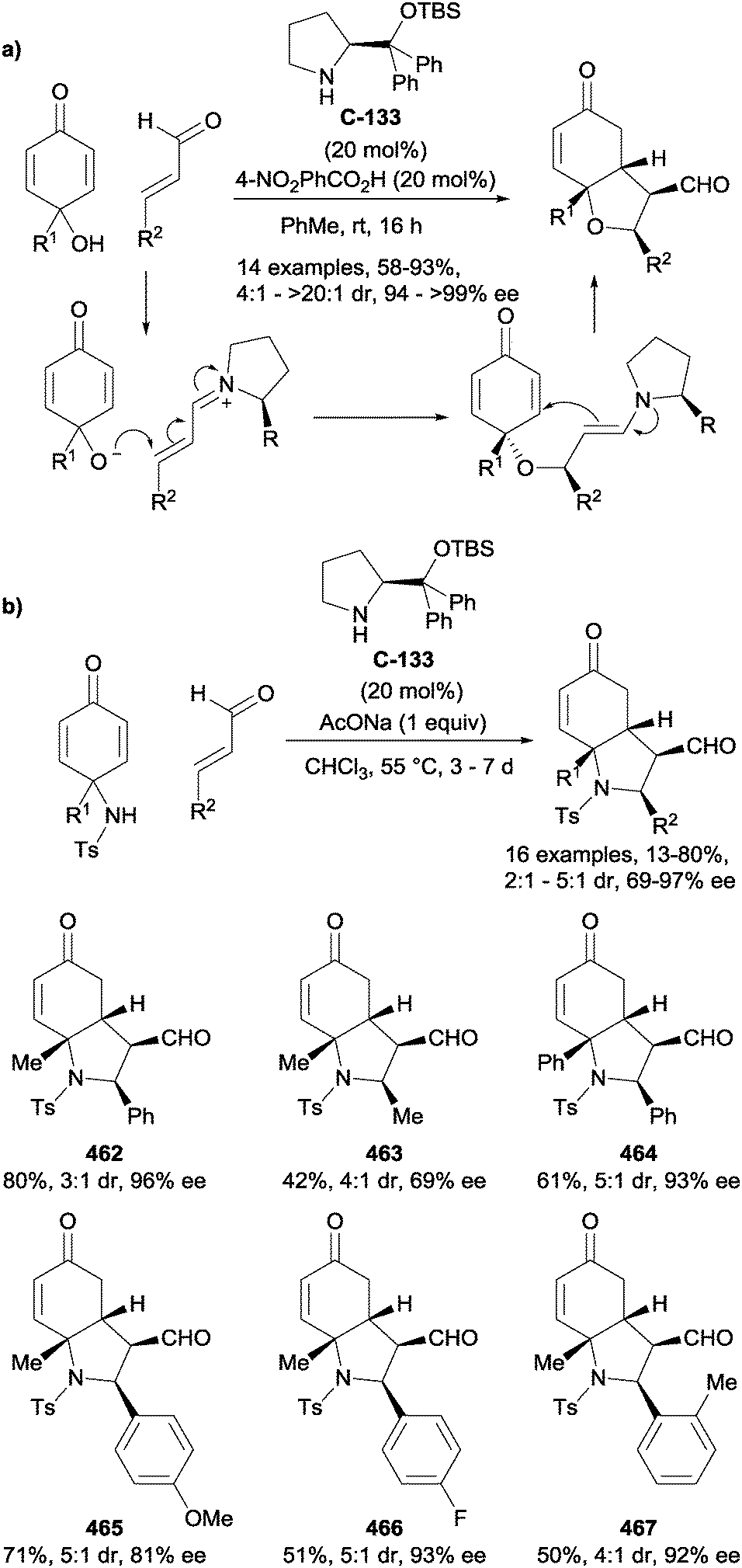 | ||
| Scheme 107 (a) Johnson's desymmetrisation of quinols through oxa-Michael–Michael addition cascade. (b) Analogous reaction of quinamines by Greck. | ||
The earlier report on quinols by J. Johnson et al. (Scheme 107a) showed generally good yields and stereocontrol, and in particular excellent enantioselectivity.201 Electron-neutral aromatic R2 substituents provided the best results, while electron-rich, strongly electron-deficient or heteroaromatic groups caused a decrease in diastereoselectivity to 5:1–9:1 dr. The same effect was observed with when R1 or R2 were bulky substituents.
In the later study on quinamines by C. Greck and co-workers (Scheme 107b) the target hydroindoles were obtained in moderate to good yields.202 Stereocontrol afforded low to moderate drs of 2:1–5:1, although enantiomeric excess of the major diastereoisomer was greater than 90% in most cases. Relatively large catalyst loadings (20 mol%) and long reaction times (7 d) were needed to promote the process. The substrate scope revealed that at R2 aromatic groups with electron-donating or neutral para-substituents gave the best results. ortho- and meta-substituted variants as well as electron-deficient aromatics led to lower yields (27–65%). A methyl group was favourable at R1 position; larger groups afforded decreased yields and diastereoselectivity (13%, 2:1 dr, 90% ee for R1 = Bu).
Another example of a desymmetrising hetero-Michael addition is Rovis and co-workers' reaction of peroxyquinols with aldehydes, which leads to 1,2,4-trioxane products bearing an endo-peroxide pharmacophore that is shared with the antimalarial natural product artemesinin.204 The first stage is the reversible formation of a racemic peroxyhemiacetal (Scheme 108). This intermediate then undergoes an enantioselectivity-determining oxa-Michael addition to the quinol. This step proceeded under Brønsted acid catalysis by a biindane phosphoric acid C-71; a thiourea cocatalyst C-134 was also employed. The authors propose that the second step of the reaction involves a dynamic kinetic resolution of the racemic peroxyhemiacetal intermediate. This was confirmed by isolating the peroxyhemiacetal and heating it in the presence of the chiral phosphoric acid catalyst C-71, which afforded the trioxane product as a single diastereomer with high enantioselectivity.
 | ||
| Scheme 108 Stereoselective desymmetrisation of peroxyquinols through hemiacetalisation – oxa-Michael addition sequence by Rovis. | ||
A large selection of aldehydes (R3) and a small scope of peroxyquinols varied at R1 and R2 were tested. The results were generally very good, with moderate to high yields and >90% ee achieved throughout. In addition, single diastereoisomers of products were formed, which is an excellent result considering that three stereogenic centres are determined in a single step. Aromatic aldehydes were found to react with comparatively lower yield (45–66%) than that achieved for aliphatic aldehydes (71–95%).
A related process is the aminolysis/aza-Michael addition sequence used for asymmetric desymmetrisation of the cyclohexadienone motif, developed by Fan and co-workers (Scheme 109).205 This method was developed en route to the Apocynaceae alkaloid synthesis to generate a hydrocarbazole structure and to establish a quaternary stereocentre through desymmetrisation. In the presence of thiourea organocatalyst C-135, attack of an amide by a primary amine liberated a stabilised carbamate anion, which subsequently underwent an intramolecular asymmetric aza-Michael addition to the cyclohexadienone. The cis-fused hydrocarbazole was formed with high enantioselectivity and generally moderate yields. The Takemoto-type bifunctional thiourea–tertiary amine organocatalyst C-135 has been proposed to simultaneously polarize the dienone through hydrogen bonding and increase nucleophilicity of the carbamate group.
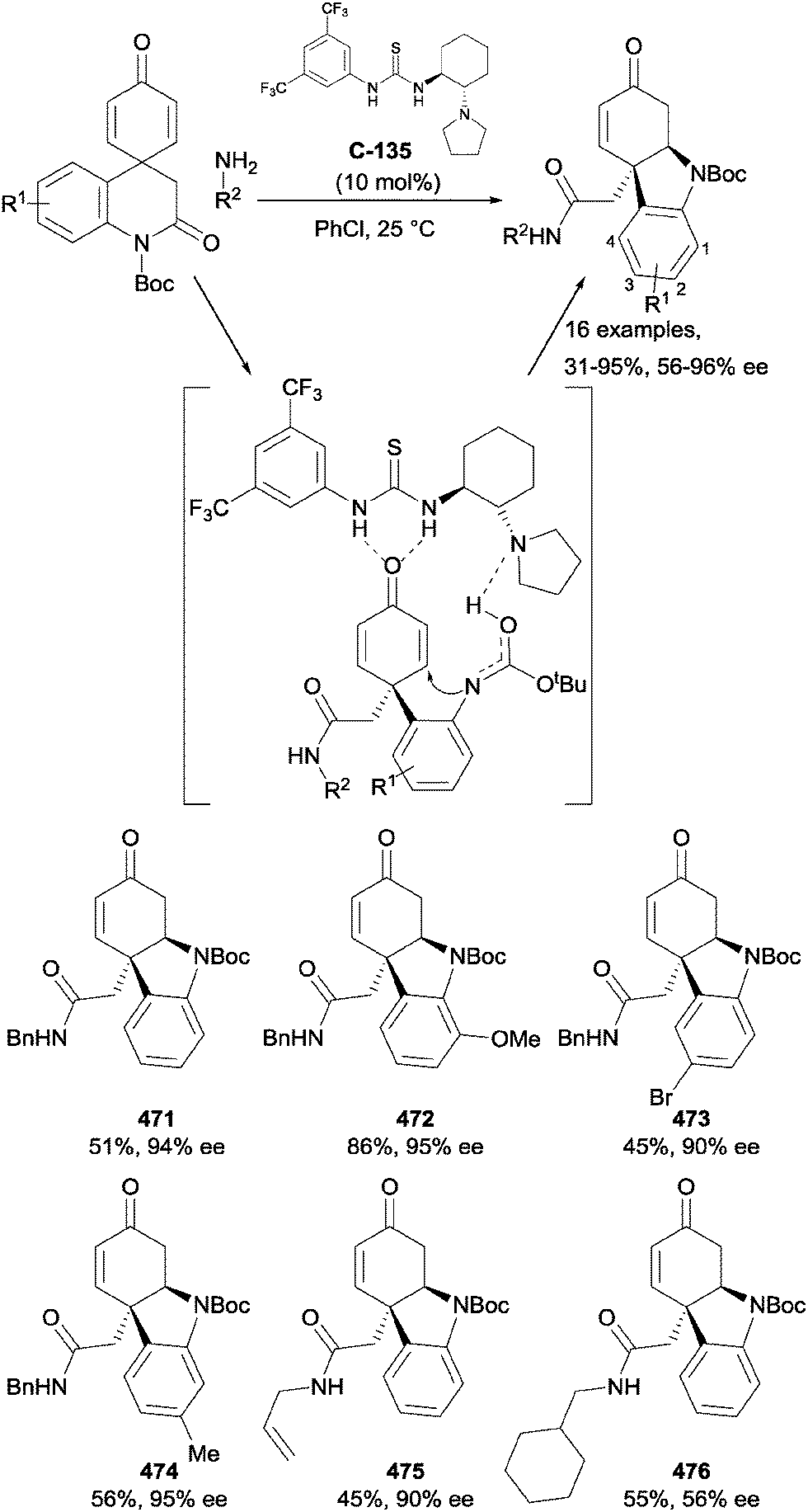 | ||
| Scheme 109 A step in the total synthesis of Apocynaceae alkaloids comprised of aminolysis and desymmetrising aza-Michael addition with an organocatalytic transition state proposed by Fan. | ||
Substitution of the aromatic ring with electron-withdrawing groups had a negative impact on yields, while substitution with the strongly electron-donating methoxy group gave an impressive 95% yield. Benzylic or allylic primary amine nucleophiles were necessary for selectivity; the use of cyclohexylmethylamine gave the desired product in just 56% ee, while homobenzylamine gave a slightly diminished ee of 82%, relative to 94% when benzylamine was reacted with the same para-dienoneimide. 1-Adamantoxycarbonyl-substituted amides could be used in place of the standard Boc-protected substrates; however, when the amide was substituted with a phenylsulfonyl group, the product was found to be racemic despite a dramatically reduced reaction time of 2 hours, a fact the authors attribute to the increased stability of the sulfonamide anion relative to the carbamate analogue. In other examples, long reaction times were generally necessary to achieve full conversion. The methodology was exemplified via the total synthesis of two hydrocarbazole-containing natural products, (+)-deethylibophyllidine and (+)-limaspermidine.
Gaunt et al. developed an electrophile-triggered catalytic dearomatisation of anisidine derivatives with a subsequent enantioselective desymmetrisation cascade that affords complex tricyclic structures (Scheme 110). The reported products contain one quaternary and two tertiary stereocentres and multiple functional elements for further downstream derivatisation. Excellent yields and levels of enantioselectivity were obtained across a range of substrates, however reducing the separation between amide and aldehyde by a single methylene unit resulted in significantly decreased enantioselectivity (481, 20% ee). The synthetic utility of the functionalised tricycles was highlighted by further transformations including Wittig olefination, Suzuki and Sonogashira cross-couplings, Buchwald–Hartwig coupling and reductive amination.206
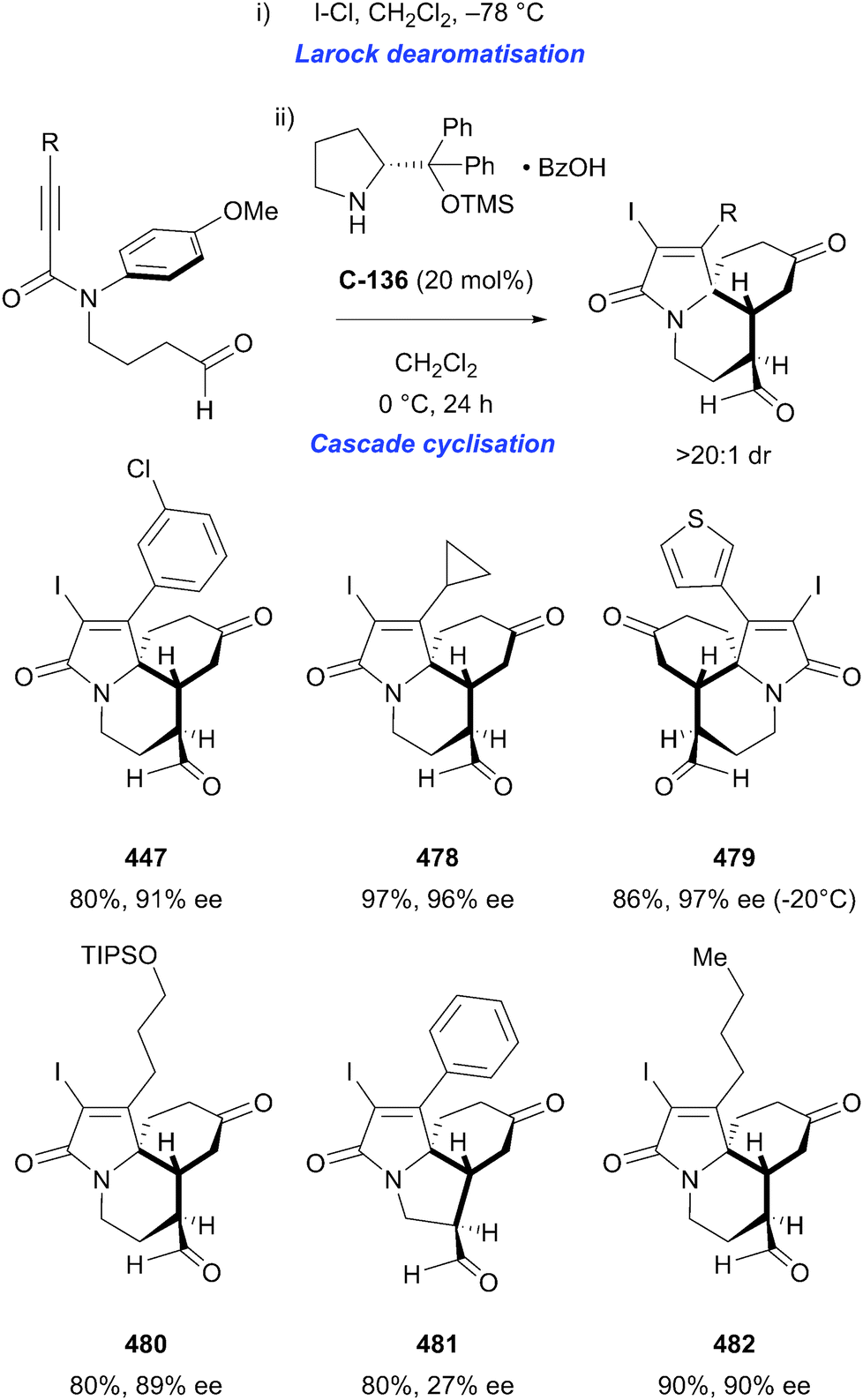 | ||
| Scheme 110 Gaunt's electrophile triggered Larock dearomatisation–desymmetrisation strategy. | ||
Gaunt and co-workers have also demonstrated enantioselective desymmetrisation in a related dearomatisation-cyclisation process.207 Various natural product-inspired scaffolds, including Erythrina alkaloid and morpholine core structures could be obtained via the sequential dearomative oxidative coupling of a phenol with a tethered electron rich arene, and subsequent organocatalytic Michael reaction of a pendant aldehyde to the resultant enone (Scheme 111). Proline-derived catalysts C-137 and C-138 were employed to effect the conjugate addition in moderate yields (35–60%) and with high enantioselectivity (84–>99%).
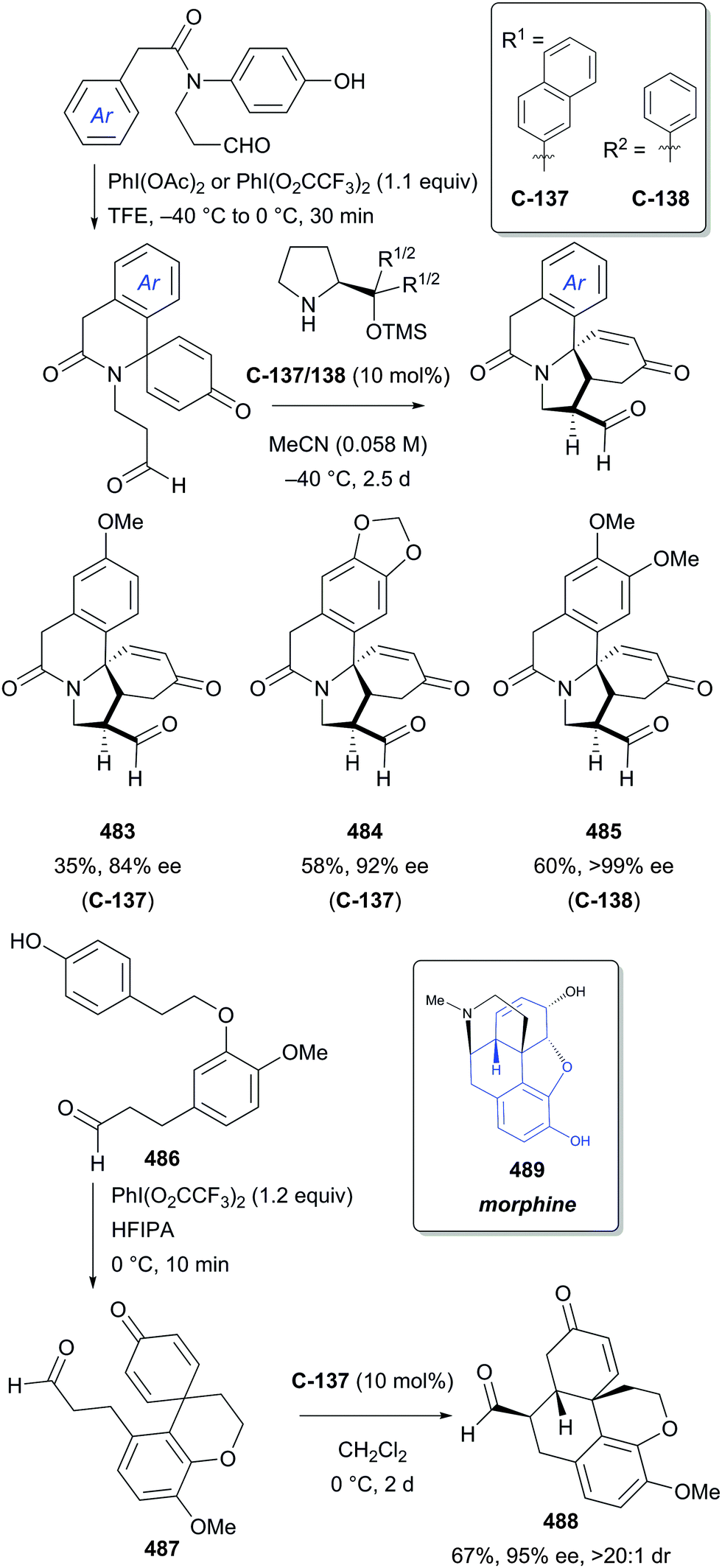 | ||
| Scheme 111 Gaunt's dearomatisation–cyclisation afforded various alkaloid skeletons including the (−)-morphine core. | ||
Kotsuki and co-workers have reported a method for the desymmetrisation of 4-substituted cyclohexadienones using diethyl malonate as a nucleophile, providing the cyclohexanone products in generally good yields, moderate drs and high ees.208 Reactions had to be carried out under high pressure (0.8 GPa) over two days using 30 mol% of thiourea organocatalyst C-139 to achieve full conversion. Several 4-methyl-4-aryl cyclohexadienones were reacted; enantio- and diastereoselectivity were retained upon substitution in the para-position of the aromatic ring regardless of the electronic properties of the group; ortho- and meta-substitution was not shown. Diastereoselectivity was, however, eroded from 5:1 to 2:1 when R2 = ethyl, showing the importance of the difference in size of the substituents to the face of nucleophilic addition (Scheme 112).
 | ||
| Scheme 112 Kotsuki's high pressure enantioselective Michael addition of diethyl malonate to 4,4-disubstituted cyclohexadienones in the presence of bifunctional thiourea catalyst C-139. | ||
The group propose transient iminium formation between the cyclic ketone and the chiral catalyst with the thiourea moiety of the catalyst coordinating the dicarbonyl oxygen atoms of the malonic ester and directing the Michael addition to one face (Scheme 113).
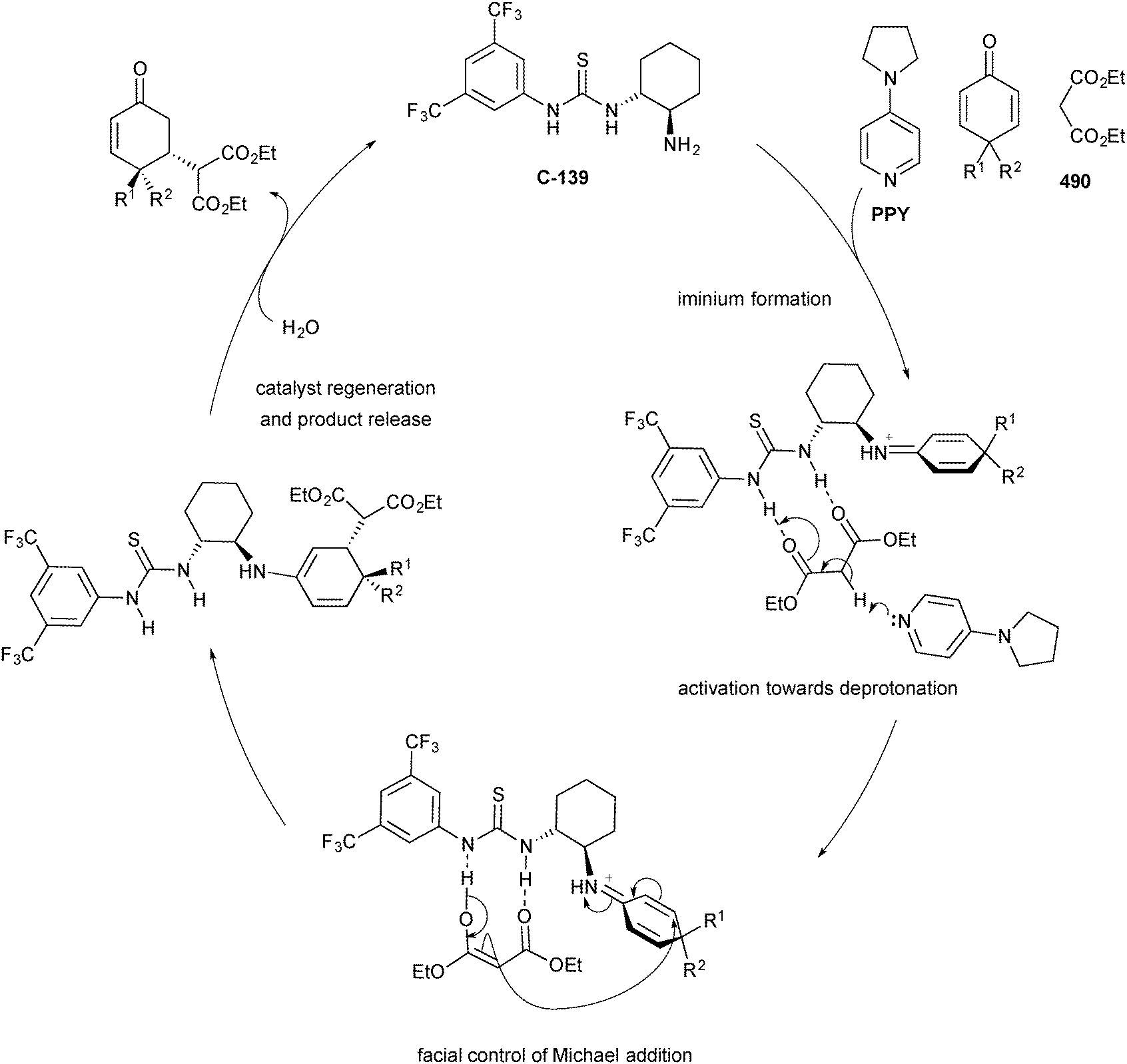 | ||
| Scheme 113 Catalytic cycle and origin of enantioselectivity proposed by Kotsuki for the enantioselective Michael addition of diethyl malonate to 4,4-disubstituted cyclohexadienones. | ||
Sasai, Enders and co-workers have developed a protocol for the enantioselective desymmetrisation of prochiral cyclohexadienones via intramolecular Rauhut-Currier reaction with a pendant enone.209,210 Using bifunctional phosphine catalyst C-140, which contains both Lewis basic and Brønsted acidic sites, a range of α-methylidene-γ-butyrolactones bearing quaternary stereocentres could be constructed with high levels of enantioselectivity, and in high yields (Scheme 114a). The formation of two contiguous quaternary centres, however, proved more challenging, as reflected in the lower yield and enantioselectivity reported for the desymmetrisation of the 3,3-disubstituted dienone to afford compound 499. The authors proposed a mechanism to explain the observed enantioselectivity, invoking hydrogen bonding between the acidic site of the catalyst and the phosphonium enolate intermediate. To avoid a steric clash between the catalyst's isopropyl substituent and the R2 group of the substrate, (S,S) intermediate B is formed preferentially (Scheme 114b).
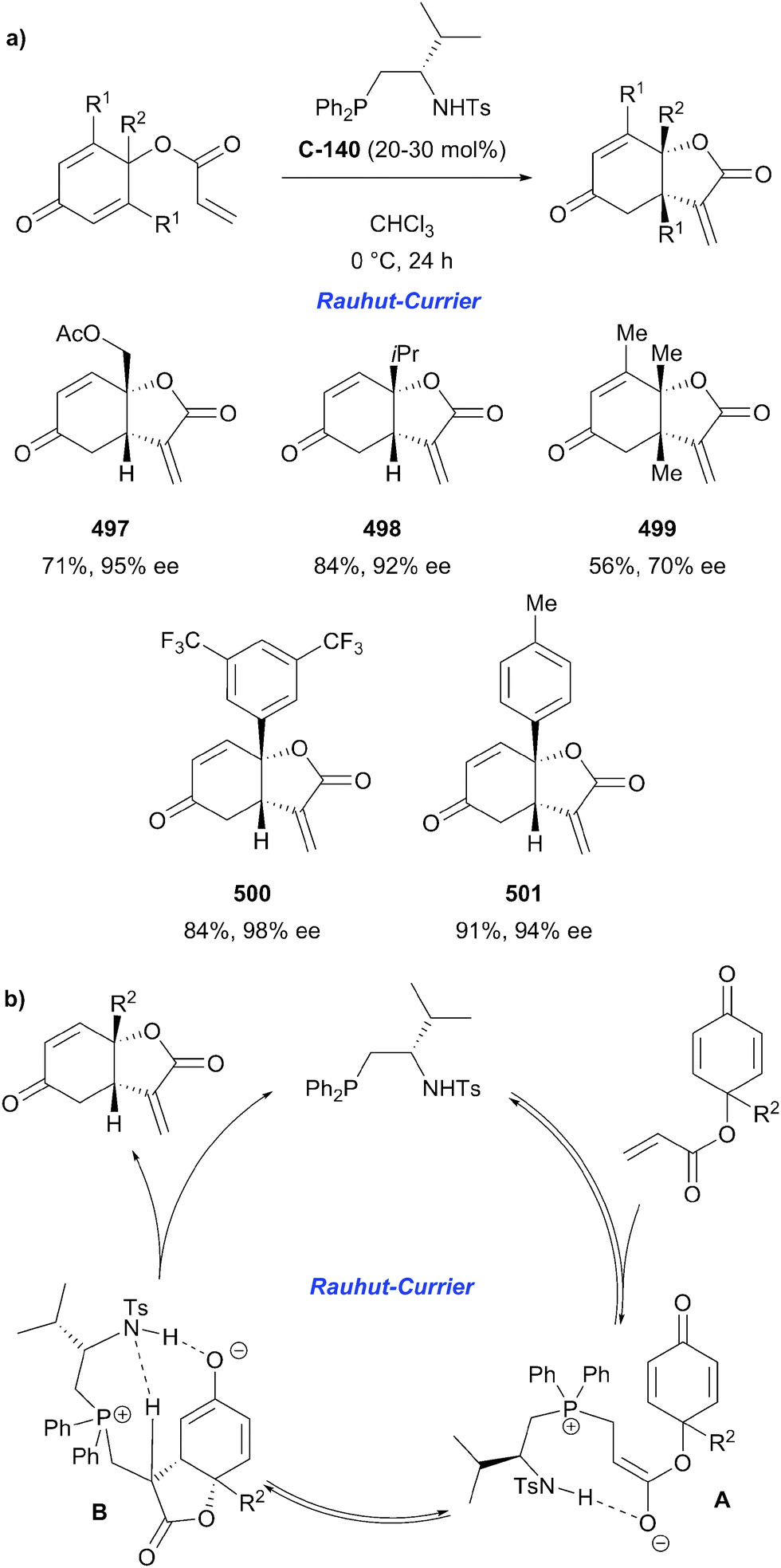 | ||
| Scheme 114 (a) Intramolecular enantioselective Rauhut–Currier cyclisations reported by Enders; (b) mechanism proposed by the authors. | ||
A novel class of chiral sulfinamide phosphine catalysts was reported by Zhang et al., and was applied to the enantioselective desymmetrisation of 4,4-disubstituted cyclohexadienones in intramolecular Rauhut–Currier reactions.211 Catalyst C-141a was found to effect the cyclisation, providing α-methylidene-γ-butyrolactones in high yields (81–95%) and with excellent enantioselectivity (96–99% ee) (Scheme 115). Importantly, even compound 499, bearing contiguous quaternary stereocentres, could be prepared in 98% ee. Diastereomeric catalyst C-141b could be employed to afford products from the opposite enantiomeric series in similar yields, but with reduced enantioselectivity (510 to 512).
 | ||
| Scheme 115 Zhang's intramolecular Rauhut–Currier reaction with sulfinamide catalyst C-141. | ||
Less common in the desymmetrisation of 4,4-disubstituted cyclohexadienones is the use of the Diels–Alder reaction. The intrinsic substrate control arising from 4,4-unsymmetrically disubstituted cyclohexadienones produces a degree of facial selectivity in the endo-Diels–Alder cycloaddition (Scheme 116).
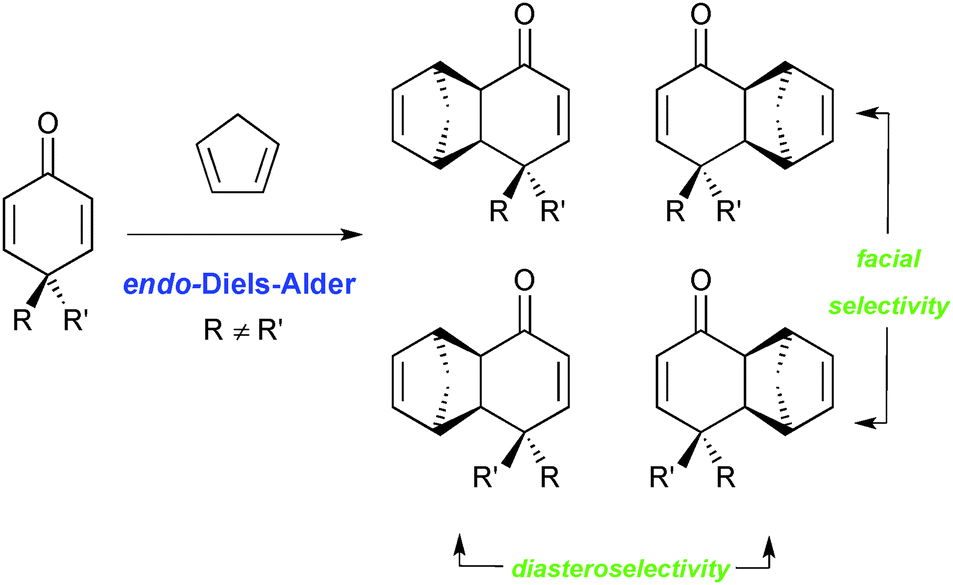 | ||
| Scheme 116 Facial and diastereoselectivity of the Diels–Alder cycloaddition applied to 4,4-disubstituted cyclohexadienones. | ||
Beyond enantioselective desymmetrisation, for broad coverage of asymmetric organocatalytic cyclisations and cycloadditions, the reader is directed to a comprehensive review on this subject by Rios and Moyano in 2011.212
Nishi et al. optimised the diastereoselectivity of the endo-Diels–Alder cycloaddition to a 4,4-disubstituted cyclohexadienone using chiral phosphoric acid organocatalysis.213 Under optimised reaction conditions, modest to good yields and diastereoselectivities were achieved for this challenging desymmetrisation, although the substrate scope is relatively narrow (Scheme 117).
 | ||
| Scheme 117 (a) Chiral phosphoric acid-catalysed intermolecular Diels–Alder cycloadditions described by Nishi; (b) DFT calculated lowest energy transition state demonstrating the bifunctional Brønsted acid–Lewis basic mode of catalysis and origins of diastereoselectivity. | ||
An asynchronous intramolecular [4+2] cycloaddition was reported by Alemán et al. in the desymmetrisation of cyclohexadienones, affording a range of tricyclic derivatives with high levels of enantioselectivity.214 A proline-derived organocatalyst was employed to effect the cyclisation. DFT calculations indicated that the reaction proceeds by an asynchronous concerted [4+2] cycloaddition, as opposed to a stepwise reaction. The desymmetrisation reaction was tolerant of a variety of acetal and hemi-aminal ether substrates. The broad range of tricyclic products represent useful 3D-templates and medicinal intermediates, and were obtained in good yields (Scheme 118). Fraile & Alemán have recently reviewed organocatalytic strategies for the production of chiral molecules by inter- and intramolecular [3+2] and [4+2] cycloadditions.215
 | ||
| Scheme 118 Chiral amine-catalysed [4+2] intramolecular cycloaddition-desymmetrisation of 4,4-disubstituted cyclohexadienones by Alemán. | ||
Mukherjee and co-workers have reported a method for the desymmetrisation of 2,2-disubstituted cyclopentene-1,3-diones via the vinylogous Michael addition of deconjugated butenolides, setting three stereocentres in the process, with one being an all-carbon quaternary centre (Scheme 119).216 Using bifunctional amine–thiourea catalyst C-144 (10 mol%), the nucleophilic attack of the deprotonated butenolide proceeded in universally excellent yields across the 31 examples shown. Enantio- and diastereoselectivities were mostly excellent, with simple, long-chain, bulky alkyl, allyl and benzyl groups all well-tolerated; direct aryl substitution on either component, however, led to moderate erosion of selectivity. The authors carried out “robustness screening”, running the reaction in the presence of various additives including potentially competing electrophiles such as aldehydes and Michael acceptors. Marginally lower enantioselectivities and longer reaction times were generally the only consequence of this, which the authors argue may be due simply to a decrease in effective catalyst concentration. Only basic amines derailed the reaction; primary and secondary amines being consumed, while tertiary amines simply inhibited the reaction without being used up. Addition of less basic aniline, in contrast, had only a slight negative impact on the yield.
 | ||
| Scheme 119 Enantioselective desymmetrisation of 2,2-disubstituted cyclopentenediones using deconjugated butenolides as nucleophiles in the presence of bifunctional amine–thiourea catalyst C-144, reported by Mukherjee. | ||
Mukherjee and co-workers have also reported the desymmetrisation of 2,2-disubstituted cyclopentene-1,3-diones via the Michael addition of nitroalkanes, aided by quinine-derived urea bifunctional organocatalyst C-145 (Scheme 120).217 Subsequent loss of nitrous acid resulted in an overall formal C(sp2)–H alkylation, stated by the authors to be the first example of nitroalkanes being used as an alkyl source. The authors also claim their reaction to be a “near-ideal” desymmetrization, with minimal alteration of the achiral starting compound in generating the additional stereocentre. A wide variety of 2-methyl-2-benzyl-cyclopentene-1,3-diones were probed, with almost universally high yields and ees obtained upon reaction with nitromethane. However, no strongly electron-withdrawing groups on the aromatic ring were tested, and substitution with chlorine at the 2-position lead to a markedly reduced ee (74%). 2-Methyl-2-allyl analogues could also be reacted successfully, although direct phenyl substitution eroded the ee to 67%. A number of nitroalkanes could also be employed, with ethyl, benzyl and homobenzyl giving good results. Further reaction with nitroalkanes was possible, providing unsymmetrical alkene products in good yields with no loss of enantioselectivity, although harsher conditions were required. The methodology was exemplified by the construction of the core of the natural product (+)-madindoline B in two steps from a symmetrical cyclopentene-1,3-dione. The authors argue that their reaction represents a significant advance over the previous state of the art to synthesise such molecules, which involve the use of sensitive metal alkyl reagents and a second oxidation step to restore the double bond functionality. Nitroalkanes, by contrast, are insensitive to moisture and widely available.
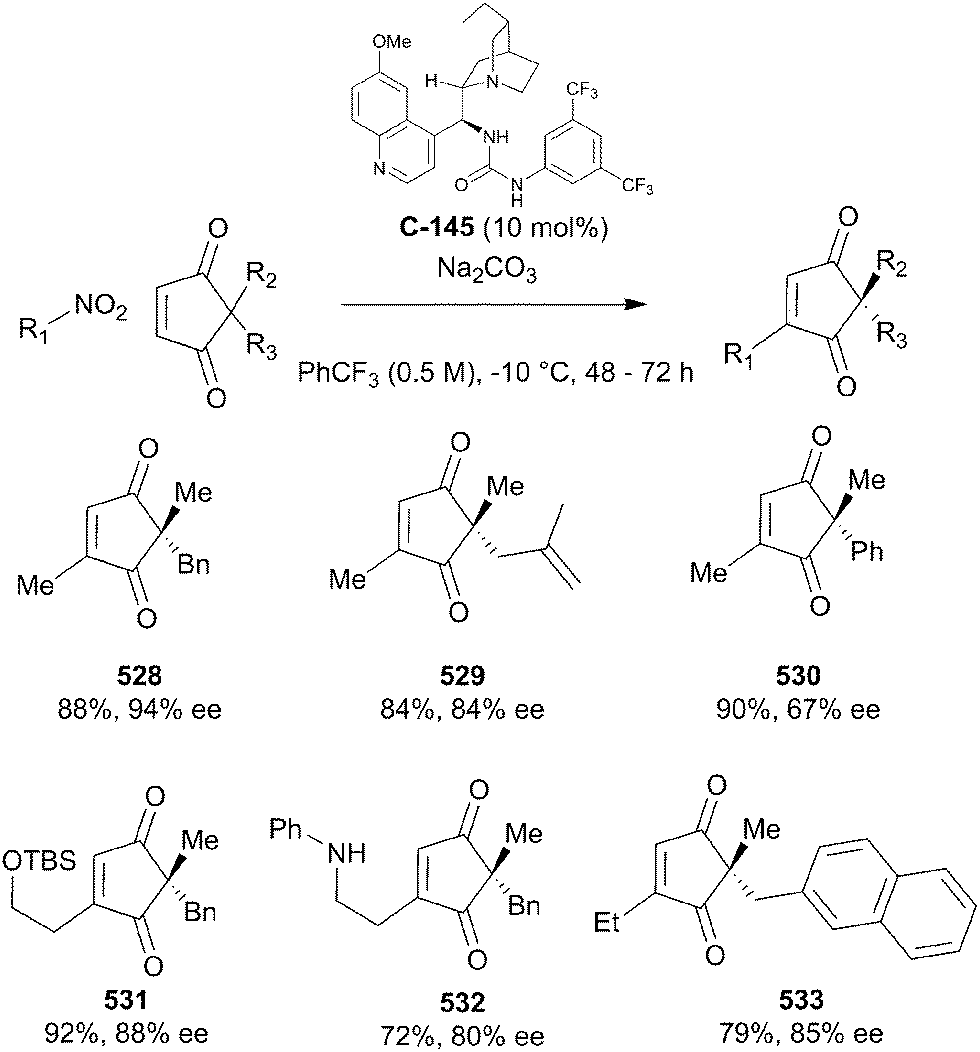 | ||
| Scheme 120 Enantioselective formal C(sp2)–H alkylation of cyclopentene-1,3-diones via Michael addition of nitroalkanes and subsequent base-induced loss of nitrous acid, reported by Mukherjee. | ||
A study by Kudo and co-workers demonstrated a desymmetrising Michael addition-like selective monohydrogenation of a ferrocenyl dienal through hydride transfer from a Hantzsch ester.218 The process is based on their previous kinetic resolution of prochiral ferrocenyl enals;219 it uses a solid-supported oligopeptide (14 amino acids) catalyst C-146 containing an N-terminal proline which is believed to form a reactive iminium adduct with the substrate (Scheme 121). The reaction induces planar chirality by desymmetrisation, producing the Rp enantiomer preferentially. The authors proposed that in the iminium adduct of the catalyst with pro-Rp enal chain the alkene is accessible for hydrogenation. The iminium intermediate formed from the pro-Sp enal, on the other hand, is shielded from the hydride donor by the ferrocene and the oligopeptide chain.
 | ||
| Scheme 121 Desymmetrising monohydrogenation of ferrocenyl bis(enals) by Kudo, giving rise to planar chirality. | ||
The monohydrogenated compound 535 was formed in good ee (73%) but only moderate yield (49%). A product of the subsequent intramolecular Michael cyclisation was also isolated when HCl was not added. Overall, this is an interesting example of planar chirality being induced relatively far from the aldehyde group that associates with the catalyst.
8. Miscellaneous substrates
8.1 Aldehydes
Gilmour and co-workers have developed an intriguing method for the enantioselective desymmetrisation of cyclopropane carbaldehydes via iminium catalysis (Scheme 122).220 By employing MacMillan's first-generation catalyst C-147, nucleophilic addition at the γ-position could be achieved, with subsequent capture of an electrophile at the α-position. They chose to exemplify this approach by employing nucleophilic and electrophilic sources of chlorine, to yield 1,3-dichloroaldehydes, constituting a formal addition of Cl2 across a cyclopropane. This process took place with generally high drs and moderate to good ees, showing little variability across fused bicyclic and simple alkyl substituted cyclopropanes. The cyclopropane carbaldehyde derived from cis-stilbene, however, displayed excellent diastereo- and enantioselectivity. Absolute configuration of the products was determined by derivatisation to the 3,5-dinitrobenzoate and subsequent X-ray crystal analysis. The authors state that the observed absolute configuration implies that nucleophilic addition of chloride takes place at C3, not C2.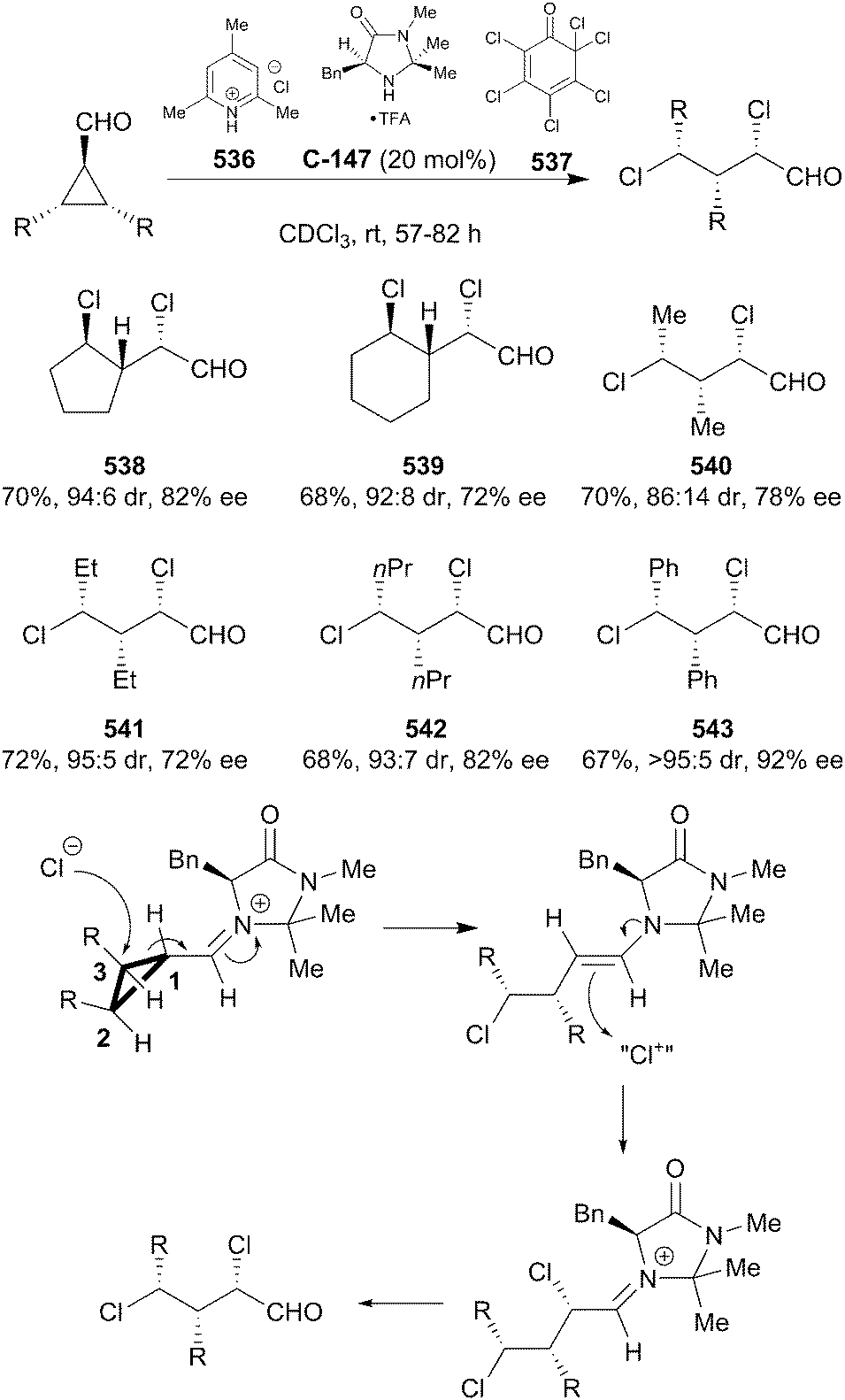 | ||
| Scheme 122 Synthesis of enantioenriched 1,3-dichloroaldehydes via desymmetrisation of cyclopropane carbaldehydes by Gilmour. Yields refer to product isolated after a three step sequence involving reduction of the aldehyde and subsequent acylation to form the 3,5-dinitrobenzoate. | ||
8.2 Dinitro substrates
Mukherjee et al. developed an enantioselective desymmetrisation of prochiral 1,3-dinitropropanes via organocatalytic allylic alkylation.221 Inspired by the work of Lu,222 Mukherjee screened various bifunctional catalysts containing a Lewis basic tertiary amine and a Brønsted acidic (thio)urea/squaramide moiety. They found catalyst C-148 was efficient at desymmetrising the dinitropropane under the reaction conditions, with >95% conversion in 10:1 dr and 97% ee (Scheme 123).
 | ||
| Scheme 123 Mukherjee's desymmetrisation of prochiral 1,3-dinitropropanes by organocatalytic allylic alkylation. Enantiomeric excess values are for the major diastereoisomer. Values in parentheses are for the enantiomeric product, obtained using the pseudo-enantiomeric catalyst C-149. | ||
The proposed mechanism for the reaction has the cinchona-derived catalyst react in an SN2′-type displacement of the OBoc group of the allylic carbonate. The active nucleophile, in this case the deprotonated 1,3-dinitropropane, undergoes a direct SN2 reaction, displacing the catalyst. They noted it was completely regioselective; no regioisomeric product, which would arise from an SN2′ (over SN2) reaction to displace the catalyst (Scheme 123), was detected.
Various aryl functionalities, including both electron-withdrawing and donating, as well as heteroaryl, acyclic and alkyl groups were well-tolerated, with good to excellent diastereoselectivity and excellent enantioselectivity. They also noted that the pseudo-enantiomeric cinchonine-derived catalyst C-149 could be used to obtain the opposite enantiomer ent-544 in excellent enantioselectivity, but with slightly reduced dr in 61% yield.
8.3 Diamines
Diamines and their derivatives are useful substrates for enantioselective desymmetrisation. This is typically achieved via an enzymatic resolution, which falls outside the scope of this review. However, recent advances have enabled the organocatalytic desymmetrisation of these compounds. Diamines are often mono-protected selectively, much like their diol counterparts, to leave access to one amine for further synthetic manipulations. This is difficult due to the nucleophilicity of amines; di-protection is often a problematic side-reaction.Seidel et al. published the first report in the area, utilising a dual small-molecule catalysis approach to selectively monobenzoylate cis-1,2-diamines.223 The simple nucleophilic catalyst, DMAP, was used in conjunction with a chiral anion receptor to form a chiral ion pair. This was successfully shown in previous work to kinetically resolve benzylic,224 propargylic,225 and allylic amines.226 Seidel et al. extended this to monobenzoylate various cis-1,2-diamines (Scheme 124), however their scope was limited to substrates possessing 1,2-diaryl groups. Nevertheless, good to high yields (60–82%) and enantioselectivities (71–95% ee) were achieved. It should be noted that they reported no intra- or inter-molecular acyl-transfer reactions that could potentially cause racemisation of the enantioenriched product.
 | ||
| Scheme 124 Seidel's monobenzoylation of cis-1,2-diamines using a chiral ion pair as the active acylating agent. | ||
Seidel later reviewed enantioselective acyl transfer reactions, with an emphasis on amines, which contained a section on the desymmetrisation of diamines.227
8.4 Imides
Imides have also found application in desymmetrisation reactions. However, in contrast to the ring opening strategies common to related anhydride substrates, imides are often enantioselectively reduced at one carbonyl. Furthermore, maleimides can be desymmetrised through Michael additions where the imide nitrogen is attached to a rotationally-restricted group. An overview of reductive catalytic enantioselective desymmetrisations of meso-compounds by Vidal-Ferran et al. contains a few examples of imide reductions, the majority of which utilise a metal which falls out of the scope of this review. However, the use of organocatalysts with borane to enantioselectively reduce one imide carbonyl has been demonstrated.228Bencivenni et al. developed a remote control of axial chirality of succinimides containing a bulky ortho-tert-butylphenyl substituent on nitrogen using a cinchona alkaloid-derived primary amine.229 Catalyst C-151 reacts with the enone, expelling water and forming the vinylogous enamine, which undergoes an aminocatalytic vinylogous Michael addition selectively on the opposite face to the tert-butyl group of the maleimide (Scheme 125a). However, the diastereoselectivity of the products was low due to epimerisation at the exocyclic chiral centre when R1 = H. Overall, the reaction results in the formation of two contiguous stereocentres and demonstrates remote control of axial chirality.
 | ||
| Scheme 125 Enantioselective desymmetrisation of maleimides by Bencivenni (a) via aminocatalytic vinylogous Michael addition, generating atropisomeric succinimides; (b) via an atropselective formal Diels–Alder. Values in parentheses are for pseudo-enantiomeric catalyst C-152 producing the enantiomeric product. | ||
Upon screening multiple primary amine catalysts and acidic co-catalysts, they found quinine-derived catalyst C-151 and N-Boc-L-phenylglycine 557 were optimal with the enone corresponding to product 558, achieving 75% yield with 70:30 dr in >99% ee for both diastereoisomers. The thermal stability was tested on succinimide 558, generating a new diastereoisomer upon prolonged heating at 130 °C from the rotation about the C–N bond, with a barrier to rotation of 31.9 kcal mol−1 corresponding to a half-life of 1000 years at 25 °C.
The ortho-tert-butyl group on the aryl ring of the maleimide is necessary to maintain a large barrier to rotation, which results in a chiral axis. Halogens, methoxy, phenyl and protected amine substituents were well-tolerated at the 4- and 5-positions of the aryl group of the maleimide, although any ortho-iodo, triethylsilyl and phenyl groups resulted in fast epimerisation of the chiral axis. Benzyl, alkyl and allyl groups at the R1 and R2 positions of the enone were tolerated, achieving moderate diastereoselectivities with excellent enantioselectivities.
A model was proposed to describe the P,R,S configuration obtained using the maleimide with the para-bromo substituent on the aryl ring, shown in the box in Scheme 125a. The Re-face of the γ-carbon of the enone approaches the bottom face of the CA carbon of the maleimide. The carbonyl group is activated by hydrogen bonding to the protonated quinuclidine to as depicted.
Bencivenni et al. then extended their work and developed an atropselective formal Diels–Alder reaction of N-arylmaleimides.230 Using the same cinchona alkaloid catalyst with a different acid catalyst, the extended enamine was formed, acting as the diene partner in the Diels–Alder reaction (Scheme 125b). It was also shown that using the pseudo-enantiomer C-152 of the catalyst C-151 could access the opposite enantiomer of the desired Diels–Alder adduct. A similar transition state was proposed, with one endo-TS (shown in the box in Scheme 125b) favoured over the other by the hydrogen bond stabilisation between the carbonyl of the maleimide and the protonated quinuclidine nitrogen.
Jones et al. developed an extension231 of their desymmetrisation strategy of maleimides by asymmetrically reducing one of the carbonyl groups using an oxazaborolidine catalyst.232 Direct desymmetrisation of the maleimide cannot be achieved due to its Cs symmetry from its secondary mirror plane, thus an anthracene moiety was attached via a Diels–Alder reaction. This removes the secondary mirror plane in the molecule, changing its symmetry to C2v, which can be desymmetrised.
Previous work identified BH3·THF as a suitable borane source and, after screening a number of oxazaborolidine catalysts, C-153 was found to give the highest enantioselectivities.231,233,234 The resulting hydroxylactam was converted to the more stable methoxyaminal by treatment with acid in methanol.
Optimal enantioselectivity was achieved with the N-PMP group (over the N-Bn and N-PMB groups, Scheme 126a) However, over-reduction of the N-PMP protected maleimide resulted in low yields of 50–60%. This was shown to be critical for high enantiomeric excess as the process was stereoablative: the enantiomeric purity was increased through selective over-reduction of the minor enantiomer of the hydroxylactam product of the first reduction. This was shown by reactions on hydroxylactam 572 with both enantiomers of catalyst C-153 and C-154. Initially, the hydroxylactam (S)-572 was in 34% ee, however upon treatment with the reaction conditions, the (1R,2S)-catalyst C-153 conditions increased the ee to 55% (Scheme 126b).
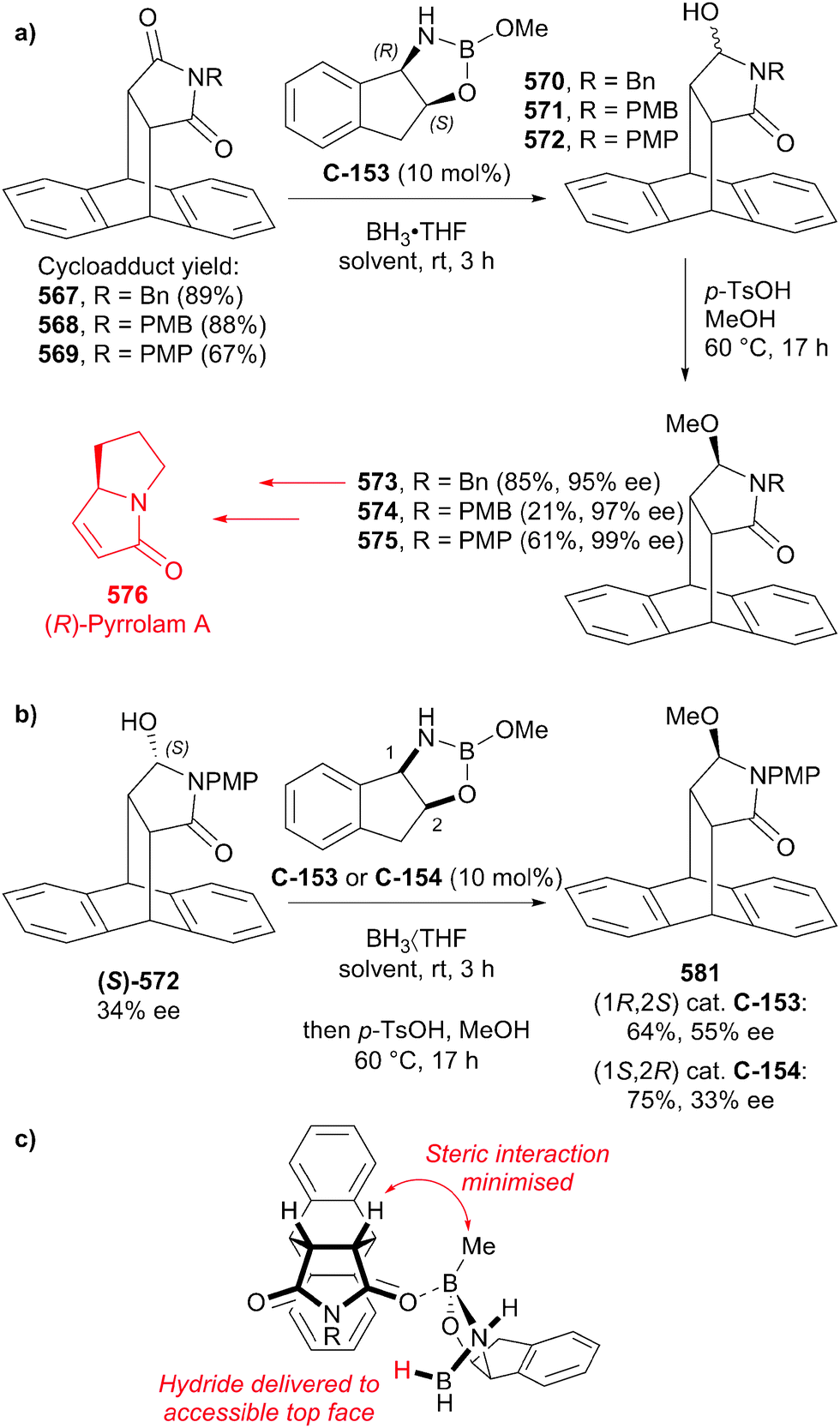 | ||
| Scheme 126 (a) Desymmetrisation by Jones using a temporary transformation to lower the symmetry of maleimides for asymmetric reduction; (b) evidence for the reaction being stereoablative; (c) model proposed by the authors for maleimide reduction. | ||
The authors proposed a model for the reduction, shown in Scheme 126c. The oxazaborolidine catalyst coordinates to the maleimide carbonyl, minimising steric interactions between the maleimide and the methyl group on the boron. The hydride is then delivered to the more accessible top face of the maleimide over the anthracene-blocked bottom face.
All three protecting groups gave the methoxyaminal in high enantiomeric excess, however the PMB group gave low yields due to poor conversion (53% of starting material). The N-PMP protected maleimide was utilised in the synthesis of (R)-Pyrrolam A 576. The anthracene moiety was removed in the final stage via flash vacuum pyrolysis in 69% yield (94% ee).
Jones et al. also desymmetrised glutarimides using a similar oxazaborolidine catalyst C-155.235 Selective reduction of one carbonyl of the 3-substituted glutarimides was carried out in moderate yields (51–61% yield) but with high enantioselectivity (82–92% ee, Scheme 127a).
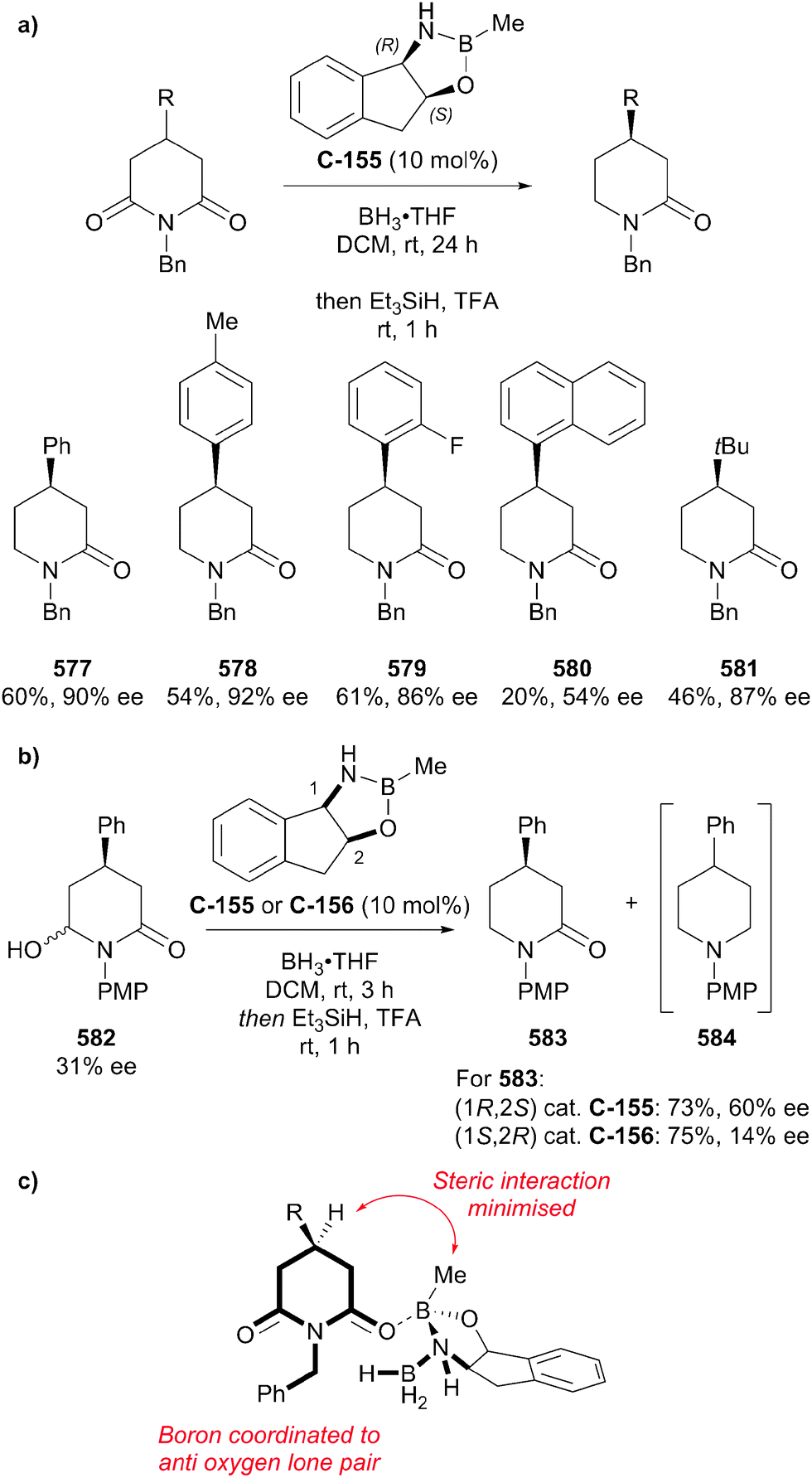 | ||
| Scheme 127 (a) Reductive desymmetrisation of N-benzyl protected glutaramides using oxazaborolidine catalyst C-155 by Jones; (b) evidence for the reaction being stereoablative; (c) model for glutaramide reduction. | ||
A similar stereoablative process was observed as in their previous work described above. When hydroxylactam 582 (31% ee) was re-subjected to the reaction conditions using C-155, an increase in enantiopurity to 60% ee was observed. However, the use of the (1S,2R) enantiomer C-156 resulted in erosion to 14% ee (Scheme 127b). This shows the catalyst selectively overreduces one enantiomer of the hydroxylactam.
A similar model to their previous work with the oxazaborolidine catalysts was proposed for the selectivity. The pre-transition state models minimise the steric repulsion between the B–Me group and the 3-substituent of the glutarimide, delivering the hydride selectively to one carbonyl over the other (Scheme 127c). This assumes the boron coordinates to the lone pair of the carbonyl anti to the amide C–N bond.
8.5 Biaryls
Conformationally restricted prochiral biaryls can be desymmetrised by selective functionalisation of one of their aromatic rings. Various methods of this type have been the subject of recent reviews,236,237 but relatively few organocatalytic examples are known. One such methodology is the electrophilic halogenation reported by T. Akiyama and co-workers.238,239 The authors used BINOL phosphate catalyst C-142 to direct a halogenating agent to the 4-position of highly electron-rich 2-arylresorcinol ring via a putative hydrogen bond network (Scheme 128). An asymmetric bromination method was initially reported,238 followed by analogous methods of chlorination and iodination.239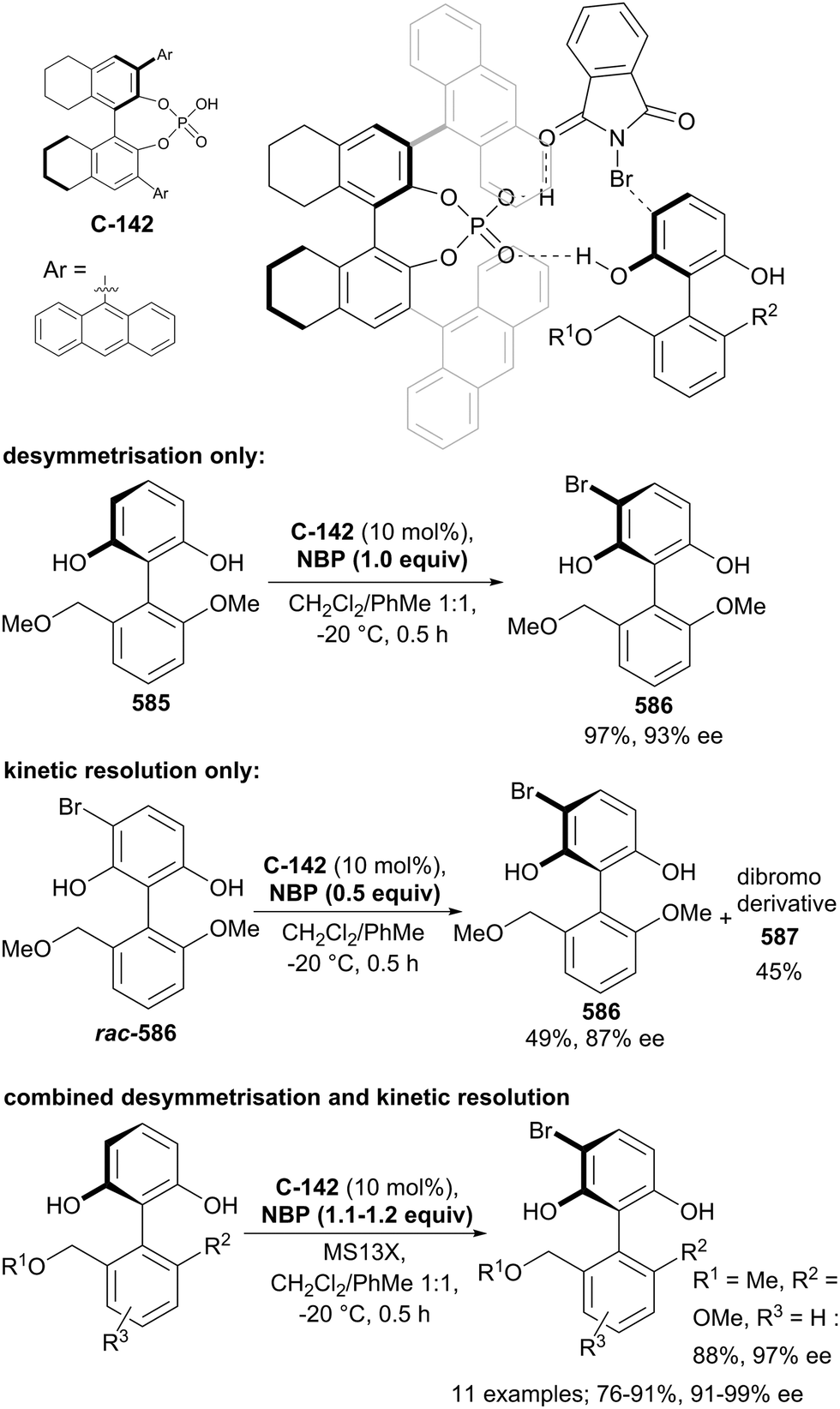 | ||
| Scheme 128 Combined desymmetrisation – kinetic resolution of prochiral biphenyls through atropselective halogenation by Akiyama. | ||
An interesting feature of this methodology is that it combines desymmetrisation with kinetic resolution. As shown in Scheme 129, the undesired atropisomer reacts with a second equivalent of NBP (N-bromophthalimide) faster than the desired one, thus enhancing enantiomeric excess of the monobrominated product. Enantioselectivity is therefore increased at the expense of yield when the brominating reagent NBP (N-bromophthalimide) is used in a small excess.
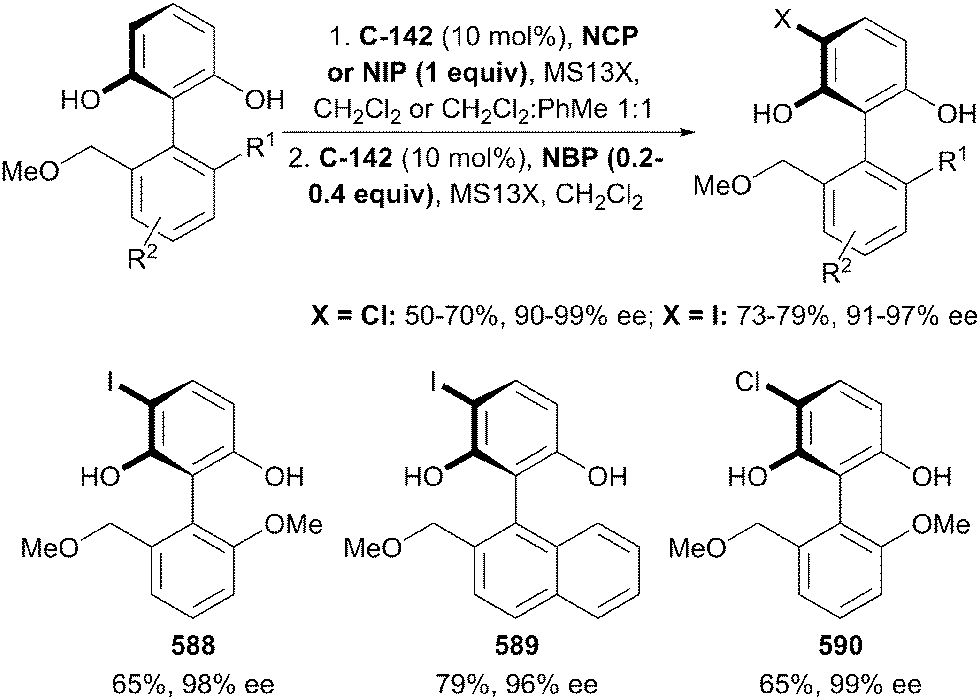 | ||
| Scheme 129 Asymmetric chlorination and iodination of biaryls with subsequent kinetic resolution through bromination by Akiyama. | ||
A range of substrates sharing the 1,3-dihydroxylated substitution on one aromatic ring and an alkoxymethyl group on the other were monobrominated with generally high yields and stereoselectivities. It was found that the hydroxyl groups of the resorcinol unit as well as the alkoxymethyl substituent on the lower ring were important for efficient and stereoselective reaction due to their participation in crucial hydrogen bonding interactions. Yield and enantioselectivity both dropped when these groups were masked or removed.
In the case of chlorination and iodination of the same biaryl substrates (Scheme 129), desymmetrisation using identical catalyst and conditions except for the substitution of NBP with N-chloro- or N-iodophthalimide, leads to high yields but only mediocre enantioselectivity (chlorination 34–69% ee, iodination 79–89% ee). It could however be improved by subjecting products to bromination conditions; as the undesired atropisomers were consumed, the ee was found to increase to ≥90%.
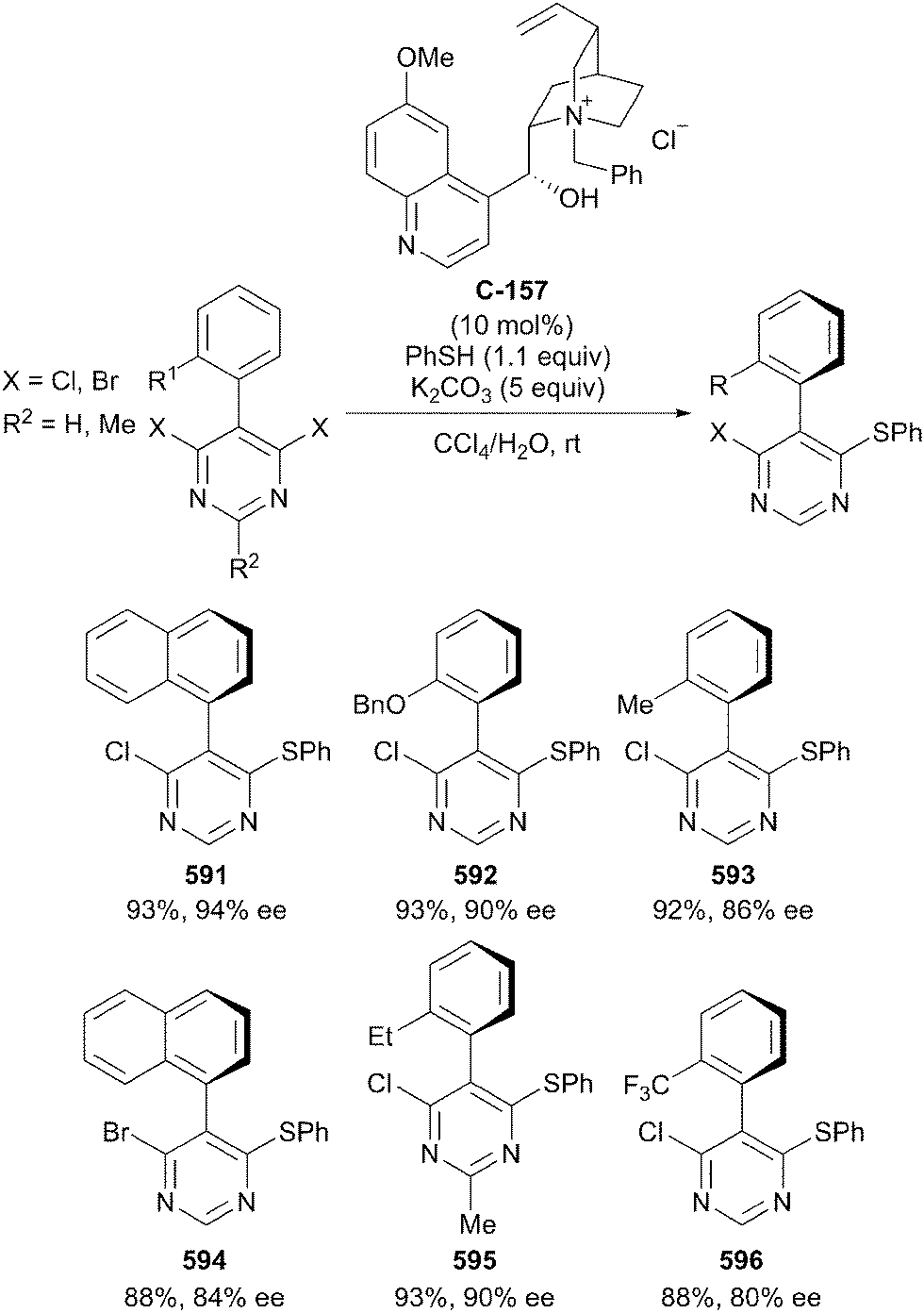 | ||
| Scheme 130 Smith's atropselective nucleophilic aromatic substitution reactions of dihalopyrimidine biaryls with thiophenol. | ||
Desymmetrisation of biaryls has also been accomplished through nucleophilic aromatic substitution by Smith and co-workers.240 In this example, a thiophenolate nucleophile undergoes an atropselective substitution reaction with a dihalopyrimidine. The reaction was done under phase transfer conditions with the use of quinine derivative C-157 (Scheme 130) which forms an ion pair with the thiophenolate nucleophile in nonpolar solvents.
 | ||
| Scheme 131 Desymmetrisation of diesters via chiral phosphoric acid-catalysed (a) lactonisation, by Petersen; (b) lactamisation, by Kawasaki. | ||
A range of dichloropyrimidine biaryls as well as a dibromopyrimidine compound reacted with generally high yields and enantioselectivity (85–97%, 74–94% ee in most examples). A slightly decreased enantioselectivity (80–84% ee) was observed for substrates bearing a naphthyl group. The steric bulk of the R1 substituent, which is important for conformational restriction of the biaryl, was found to influence the reaction; for the relatively bulky R1 = iPr (not shown) the yield was only 24% after 48 hours.
8.6 Diacids and diesters
An enantioselective desymmetrising lactonisation of derivatised malonates bearing a hydroxyl group in the γ- or δ-position has been recently reported by K. Petersen and co-workers.241 This approach is an alternative to halolactonisations and protolactonisations of alkenoic acids, also used as desymmetrising strategies. The catalyst is a binaphthol-based chiral phosphoric acid diester which activates the ester group in a chiral environment. This method originated from an earlier discovery of kinetic resolution of racemic hydroxyacids through asymmetric lactonisation.242 These preliminary studies demonstrated the superiority of BINOL-derived phosphoric acids over camphorsulphonic acid, TADDOL and thiourea catalysts. Substitution of the binaphthol scaffold with 2,4,6-triisopropylphenyl groups (C-70) was found to provide optimal enantioselectivity. The reaction conditions used for these kinetic resolutions were then applied to desymmetrisation.A small range of substituted di-tert-butyl malonates was converted into chiral γ-lactones (Scheme 131a). Yields above 90% were achieved in most cases, except for the benzyl-substituted malonate which afforded only 67%. High enantioselectivities were observed, with ees of 90–98%. A δ-lactone was also prepared using the same method with 84% yield and 86% ee. The best results (95% isolated yield, 98% ee) were achieved with the methyl-substituted malonate; this reaction was run on 1.9 g scale with 20 mg catalyst (0.4 mol%) to demonstrate the practicality of the method. Long reaction times (3–8 days) were generally needed for the reactions to reach completion.
A very similar method was used by T. Kawasaki and co-workers to achieve desymmetrising lactamisation of a prochiral diester en route to the natural product (−)-leuconoxine (Scheme 131b).243 In this case, a VAPOL-based phosphoric acid could be used to achieve good enantioselectivity at elevated temperature.
Dicarboxylic acids containing an alkene functionality in a central position can be desymmetrised through intramolecular halolactonisation. A bromolactonisation of this type was reported by Fujioka and co-workers244 using the previously developed C3-symmetric trisimidazoline catalyst C-159245–247 capable of chelating carboxylic acids through hydrogen bonding in a chiral environment (Scheme 132). A solvent system of 4:1 toluene/acetone was found to be optimal for using this interaction to achieve stereocontrol.
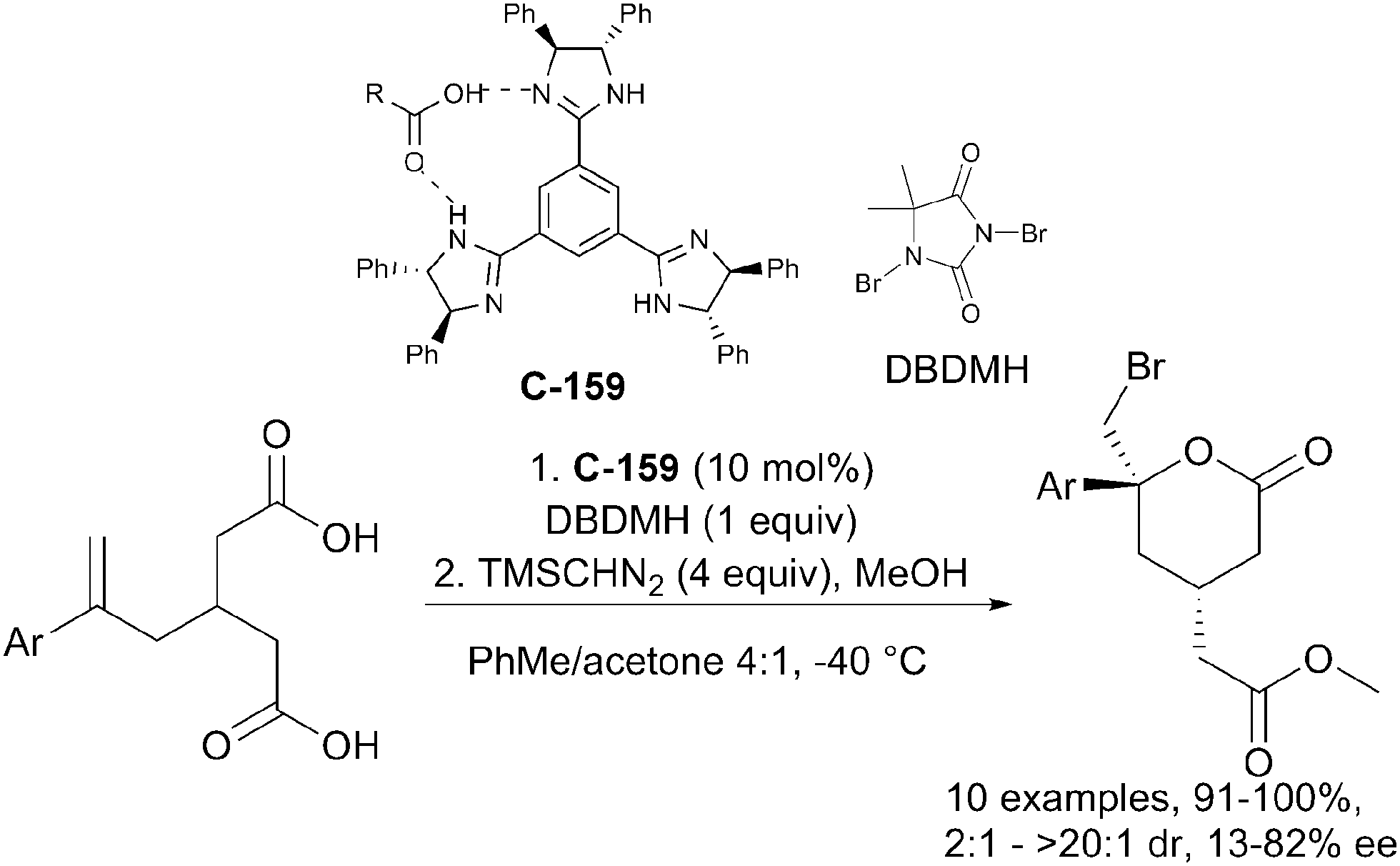 | ||
| Scheme 132 Fujioka's desymmetrising bromoesterification catalysed by a chiral trisimidazoline. | ||
Several alkenedicarboxylic acids were found to undergo bromolactonisation with high (>90%) yields. Stereocontrol was generally effective, with >20:1 dr and 66–82% ee achieved where Ar was an electron-neutral or slightly electron-deficient phenyl group. Both diastereoselectivity and enantioselectivity were lowered for electron rich Ar = 4-MeOC6H4 (2:1 dr, 13% ee) and for ortho-substituted Ar = 2-MeC6H4 (5:1 dr, 55% ee). Overall, this method displayed a good performance and is the first example of enantioselective desymmetrising bromolactonisation of alkenedicarboxylic acids.
9. Conclusions
Over the last two decades, enantioselective desymmetrisation has increasingly proven to be a powerful method for the generation of significant molecular and stereochemical complexity. In particular, organocatalytic enantioselective desymmetrisation is a burgeoning field, and one which has seen significant advances in the current decade. Although in some cases catalyst loadings are high, and reaction times can be long, impressive progress has been made with regards to enantioselectivity; in particular, very high ees have been achieved in the desymmetrisation of diols and anhydrides for a wide variety of substrates. Furthermore, in his 1999 review, Willis identified the catalytic formation of carbon–carbon bonds via enantioselective desymmetrisation as an area which had seen comparatively little progress compared to carbon–heteroatom bond formation.2 Many elegant organocatalytic reactions covered in this review, employing chiral bifunctional amine,169 (thio)urea,210 and phosphoric acid organocatalysts,181 exemplify the remarkable advances made since. C–C bonds can now be constructed in a highly selective manner, producing complex and synthetically valuable scaffolds. Further challenges and opportunities still await the imaginative organic chemist, however. In particular, the development of novel organocatalytic activation modes will be pivotal in ensuring current rate of progress is maintained. The methods detailed in this review will no doubt find use in the total synthesis of natural products due to their elegance and the unrivalled ability of enantioselective desymmetrisation to form multiple stereocentres with a high degree of selectivity in a single transformation.Acknowledgements
AB, TQD, SRE, TAF and MSWR are supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1). TQD also thanks the University of Oxford Pirie-Reid fund for a scholarship. DJD thanks the EPSRC for support.Notes and references
- D. W. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765–1784 RSC.
- H. Wennemers, Chem. Commun., 2011, 47, 12036–12041 RSC.
- C. E. Muller and P. R. Schreiner, Angew. Chem., Int. Ed. Engl., 2011, 50, 6012–6042 CrossRef PubMed.
- M. D. Diaz-de-Villegas, J. A. Galvez, R. Badorrey and M. P. Lopez-Ram-de-Viu, Chem. – Eur. J., 2012, 18, 13920–13935 CrossRef CAS PubMed.
- L. W. Xu, Y. Chen and Y. Lu, Angew. Chem., Int. Ed. Engl., 2015, 54, 9456–9466 CrossRef CAS PubMed.
- K. S. Petersen, Tetrahedron Lett., 2015, 56, 6523–6535 CrossRef CAS PubMed.
- M. S. Manna and S. Mukherjee, Org. Biomol. Chem., 2015, 13, 18–24 Search PubMed.
- S. Canesi, G. Maertens and M.-A. Ménard, Synthesis, 2014, 1573–1582 CrossRef.
- A. Quintavalla, L. Cerisoli and E. Montroni, Curr. Organocatal., 2014, 1, 107–171 CrossRef CAS.
- M. Wang, M. Feng, B. Tang and X. Jiang, Tetrahedron Lett., 2014, 55, 7147–7155 CrossRef CAS.
- E. Garcia-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110–PR180 CrossRef PubMed.
- G. P. Moss, Pure Appl. Chem., 1996, 68, 2193–2222 CrossRef CAS.
- K. Mikami and M. Lautens, New Frontiers in Asymmetric Catalysis, Wiley-Interscience, Hoboken, New Jersey, 2007 Search PubMed.
- S. R. Magnuson, Tetrahedron, 1995, 51, 2167–2213 CrossRef CAS.
- A. C. Spivey and B. I. Andrews, Angew. Chem., Int. Ed. Engl., 2001, 40, 3131–3134 CrossRef CAS.
- Y. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965–2983 CrossRef CAS PubMed.
- I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683–5712 CrossRef CAS PubMed.
- M. D. Díaz De Villegas, J. A. Gálvez, P. Etayo, R. Badorrey and P. López-Ram-De-Víu, Chem. Soc. Rev., 2011, 40, 5564–5587 RSC.
- Z. Rodriguez-Docampo and S. J. Connon, ChemCatChem, 2012, 4, 151–168 CrossRef CAS.
- H. Y. Bae and C. E. Song, Bull. Korean Chem. Soc., 2014, 35, 1590–1600 CrossRef CAS.
- T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS.
- H. Li, X. Liu, F. Wu, L. Tang and L. Deng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20625–20629 CrossRef CAS PubMed.
- B. Dedeoglu, S. Catak, K. N. Houk and V. Aviyente, ChemCatChem, 2010, 2, 1122–1129 CrossRef CAS PubMed.
- F. Balzano, R. P. Jumde, A. Mandoli, S. Masi, D. Pini and G. Uccello-Barretta, Chirality, 2011, 23, 784–795 CrossRef CAS PubMed.
- T. Bürgi and A. Baiker, J. Am. Chem. Soc., 1998, 120, 12920–12926 CrossRef.
- G. Uccello Barretta, A. Mandoli, F. Balzano, F. Aiello, B. De Nicola and A. Del Grande, Chirality, 2015, 27, 693–699 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed.
- B. Dedeoglu, S. Catak, A. Yildirim, C. Bolm and V. Aviyente, ChemCatChem, 2015, 7, 4173–4179 CrossRef CAS.
- T. Ivšić and Z. Hameršak, Tetrahedron: Asymmetry, 2009, 20, 1095–1098 CrossRef.
- T. Ivšić, J. Novak, N. Došlić and Z. Hameršak, Tetrahedron, 2012, 68, 8311–8317 CrossRef.
- S. E. Park, E. H. Nam, H. B. Jang, J. S. Oh, S. Some, Y. S. Lee and C. E. Song, Adv. Synth. Catal., 2010, 352, 2211–2217 CrossRef CAS.
- H. B. Jang, H. S. Rho, J. S. Oh, E. H. Nam, S. E. Park, H. Y. Bae and C. E. Song, Org. Biomol. Chem., 2010, 8, 3918–3922 Search PubMed.
- E. H. Nam, S. E. Park, S. Some, D. Y. Kim, H. Y. Bae and C. E. Song, Bull. Korean Chem. Soc., 2011, 32, 3127–3129 CrossRef CAS.
- Y. M. Chen, M. Amireddy and K. Chen, Tetrahedron, 2014, 70, 9064–9069 CrossRef CAS.
- S. Roy, K. F. Chen, R. Gurubrahamam and K. Chen, J. Org. Chem., 2014, 79, 8955–8959 CrossRef CAS PubMed.
- A. Peschiulli, B. Procuranti, C. J. O'Connor and S. J. Connon, Nat. Chem., 2010, 2, 380–384 CrossRef CAS PubMed.
- E. Schmitt, I. Schiffers and C. Bolm, Tetrahedron, 2010, 66, 6349–6357 CrossRef CAS.
- R. Manzano, J. M. Andrés, M. D. Muruzábal and R. Pedrosa, J. Org. Chem., 2010, 75, 5417–5420 CrossRef CAS PubMed.
- S. H. Oh, H. S. Rho, J. W. Lee, J. E. Lee, S. H. Youk, J. Chin and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 7872–7875 CrossRef CAS PubMed.
- S.-X. Wang and F.-E. Chen, Adv. Synth. Catal., 2009, 351, 547–552 CrossRef CAS.
- H. S. Rho, S. H. Oh, J. W. Lee, J. Y. Lee, J. Chin and C. E. Song, Chem. Commun., 2008, 1208–1210 RSC.
- V. N. Wakchaure and B. List, Angew. Chem., Int. Ed. Engl., 2010, 49, 4136–4139 CrossRef CAS PubMed.
- R. P. Jumde, A. Di Pietro, A. Manariti and A. Mandoli, Chem. – Eur. J., 2015, 10, 397–404 CAS.
- F. Bigi, S. Carloni, R. Maggi, A. Mazzacani, G. Sartori and G. Tanzi, J. Mol. Catal. A: Chem., 2002, 182–183, 533–539 CrossRef CAS.
- Y.-M. Song, J. Seok Choi, J. Woon Yang and H. Han, Tetrahedron Lett., 2004, 45, 3301–3304 CrossRef CAS.
- H. S. Kim, Y.-M. Song, J. S. Choi, J. W. Yang and H. Han, Tetrahedron, 2004, 60, 12051–12057 CrossRef CAS.
- H. Iida, Z. Tang and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2869–2879 CrossRef CAS.
- O. Gleeson, G.-L. Davies, A. Peschiulli, R. Tekoriute, Y. K. Gun'ko and S. J. Connon, Org. Biomol. Chem., 2011, 9, 7929–7940 Search PubMed.
- J.-W. Lee, T. Mayer-Gall, K. Opwis, C. E. Song, J. S. Gutmann and B. List, Science, 2013, 341, 1225–1229 CrossRef CAS PubMed.
- X. Huang, J. Zhu and S. Broadbent, Tetrahedron Lett., 2010, 51, 1554–1557 CrossRef CAS.
- A. Peschiulli, Y. Gun'ko and S. J. Connon, J. Org. Chem., 2008, 73, 2454–2457 CrossRef CAS PubMed.
- X. Huang, S. Broadbent, C. Dvorak and S.-H. Zhao, Org. Process Res. Dev., 2010, 14, 612–616 CrossRef CAS.
- H.-J. Yang, F.-J. Xiong, J. Li and F.-E. Chen, Chin. Chem. Lett., 2013, 24, 553–558 CrossRef CAS.
- F. Xiong, X. X. Chen and F.-E. Chen, Tetrahedron: Asymmetry, 2010, 21, 665–669 CrossRef CAS.
- X.-X. Chen, F. Xiong, H. Fu, Z.-Q. Liu and F.-E. Chen, Chem. Pharm. Bull., 2011, 59, 488–491 CrossRef CAS PubMed.
- F. Xiong, F. J. Xiong, W. X. Chen, H. Q. Jia and F. E. Chen, J. Heterocycl. Chem., 2013, 50, 1078–1082 CAS.
- T. Ivšić, I. Dokli, A. Rimac and Z. Hameršak, Eur. J. Org. Chem., 2014, 631–638 CrossRef.
- C. Bolm, I. Schiffers, C. L. Dinter and A. Gerlach, J. Org. Chem., 2000, 65, 6984–6991 CrossRef CAS PubMed.
- F. Zhang, X. Wen, Q.-L. Xu and H. Sun, Eur. J. Org. Chem., 2014, 8101–8109 CrossRef CAS.
- T. Ivšić and Z. Hameršak, Tetrahedron: Asymmetry, 2013, 24, 217–222 CrossRef.
- S. Archambaud, F. Legrand, K. Aphecetche-Julienne, S. Collet, A. Guingant and M. Evain, Eur. J. Org. Chem., 2010, 1364–1380 CrossRef CAS.
- L. Deng, Y. Chen and S. Tian, WO 0174741 (A2), 2001.
- X. Chen, F. Xiong, W. Chen, Q. He and F. Chen, J. Org. Chem., 2014, 79, 2723–2728 CrossRef CAS PubMed.
- H. Wang, L. Yan, Y. Wu, Y. Lu and F. Chen, Org. Lett., 2015, 17, 5452–5455 CrossRef CAS PubMed.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- H.-J. Yang, F.-J. Xiong, X.-F. Chen and F.-E. Chen, Eur. J. Org. Chem., 2013, 4495–4498 CrossRef CAS.
- Š. Horvat, Z. Hameršak, I. Stipetić and T. Jolas, Eur. J. Med. Chem., 2010, 45, 1447–1452 CrossRef PubMed.
- Z. Hameršak, I. Stipetić and A. Avdagić, Tetrahedron: Asymmetry, 2007, 18, 1481–1485 CrossRef.
- A. Martin, F. Robert, D. Taton, H. Cramail, J.-M. Vincent and Y. Landais, Chem. – Eur. J., 2014, 20, 11946–11953 CrossRef CAS PubMed.
- D. M. Hodgson, A. R. Gibbs and G. P. Lee, Tetrahedron, 1996, 52, 14361–14384 CrossRef CAS.
- P. A. Wang, Beilstein J. Org. Chem., 2013, 9, 1677–1695 CrossRef PubMed.
- S. Meninno and A. Lattanzi, Chem. – Eur. J., 2016, 22, 3632–3642 CrossRef CAS PubMed.
- Z. Wang, W. K. Law and J. Sun, Org. Lett., 2013, 15, 5964–5966 CrossRef CAS PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- M. R. Monaco, S. Prévost and B. List, J. Am. Chem. Soc., 2014, 136, 16982–16985 CrossRef CAS PubMed.
- M. R. Monaco, S. Prévost and B. List, Angew. Chem., Int. Ed. Engl., 2014, 53, 8142–8145 CrossRef CAS PubMed.
- S. Kotani, H. Furusho, M. Sugiura and M. Nakajima, Tetrahedron, 2013, 69, 3075–3081 CrossRef CAS.
- E. Gnanamani, N. Someshwar, J. Sanjeevi and C. R. Ramanathan, Adv. Synth. Catal., 2014, 356, 2219–2223 CrossRef CAS.
- S. S. Chimni, V. Kumar and N. Bala, Asian J. Org. Chem., 2014, 3, 700–705 CrossRef CAS.
- Z. Wang, Z. Chen and J. Sun, Org. Biomol. Chem., 2014, 12, 6028–6032 Search PubMed.
- W. Yang and J. Sun, Angew. Chem., Int. Ed. Engl., 2016, 55, 1868–1871 CrossRef CAS PubMed.
- Z. Wang, Z. Chen and J. Sun, Angew. Chem., Int. Ed. Engl., 2013, 52, 6685–6688 CrossRef CAS PubMed.
- Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed. Engl., 2013, 52, 2027–2031 CrossRef CAS PubMed.
- M. R. Monaco, B. Poladura, M. Diaz de Los Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed. Engl., 2014, 53, 7063–7067 CrossRef CAS PubMed.
- K. Ohmatsu, Y. Hamajima and T. Ooi, J. Am. Chem. Soc., 2012, 134, 8794–8797 CrossRef CAS PubMed.
- M. Senatore, A. Lattanzi, S. Santoro, C. Santi and G. D. Sala, Org. Biomol. Chem., 2011, 9, 6205–6207 Search PubMed.
- Y. Zhang, C. W. Kee, R. Lee, X. Fu, J. Y. Soh, E. M. Loh, K. W. Huang and C. H. Tan, Chem. Commun., 2011, 47, 3897–3899 RSC.
- Y.-M. Cao, F.-T. Zhang, F.-F. Shen and R. Wang, Chem. – Eur. J., 2013, 19, 9476–9480 CrossRef CAS PubMed.
- J. Zhang, D. Cao, H. Wang, G. Zhao and Y. Shang, Tetrahedron, 2015, 71, 1785–1791 CrossRef CAS.
- Z. Wang, F. K. Sheong, H. H. Y. Sung, I. D. Williams, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 5895–5898 CrossRef CAS PubMed.
- K. W. Fiori, A. L. Puchlopek and S. J. Miller, Nat. Chem., 2009, 1, 630–634 CrossRef CAS PubMed.
- M. J. Burk, J. E. Feaster and R. L. Harlow, Tetrahedron: Asymmetry, 1991, 2, 569–592 CrossRef CAS.
- E. Vedejs, O. Daugulis and S. T. Diver, J. Org. Chem., 1996, 61, 430–431 CrossRef CAS PubMed.
- T. Oriyama, K. Imai, T. Hosoya and T. Sano, Tetrahedron Lett., 1998, 39, 3021–3023 CrossRef.
- T. Oriyama, H. Taguchi, D. Terakado and T. Sano, Chem. Lett., 2002, 26–27 CrossRef CAS.
- S. Mizuta, M. Sadamori, T. Fujimoto and I. Yamamoto, Angew. Chem., Int. Ed. Engl., 2003, 42, 3383–3385 CrossRef CAS PubMed.
- E. P. Kundig, A. Enriquez Garcia, T. Lomberget, P. Perez Garcia and P. Romanens, Chem. Commun., 2008, 3519–3521 RSC.
- M. B. Lauber, C. G. Daniliuc and J. Paradies, Chem. Commun., 2013, 49, 7409–7411 RSC.
- C. A. Lewis, B. R. Sculimbrene, Y. Xu and S. J. Miller, Org. Lett., 2005, 7, 3021–3023 CrossRef CAS PubMed.
- C. E. Muller, D. Zell and P. R. Schreiner, Chem. – Eur. J., 2009, 15, 9647–9650 CrossRef PubMed.
- C. E. Muller, D. Zell, R. Hrdina, R. C. Wende, L. Wanka, S. M. Schuler and P. R. Schreiner, J. Org. Chem., 2013, 78, 8465–8484 CrossRef PubMed.
- C. E. Muller, R. Hrdina, R. C. Wende and P. R. Schreiner, Chem. – Eur. J., 2011, 17, 6309–6314 CrossRef PubMed.
- A. Sakakura, S. Umemura and K. Ishihara, Adv. Synth. Catal., 2011, 353, 1938–1942 CrossRef CAS.
- S. Yamada, T. Misono, Y. Iwai, A. Masumizu and Y. Akiyama, J. Org. Chem., 2006, 71, 6872–6880 CrossRef CAS PubMed.
- J. Merad, P. Borkar, T. Bouyon Yenda, C. Roux, J. M. Pons, J. L. Parrain, O. Chuzel and C. Bressy, Org. Lett., 2015, 17, 2118–2121 CrossRef CAS PubMed.
- V. B. Birman, H. Jiang and X. Li, Org. Lett., 2007, 9, 3237–3240 CrossRef CAS PubMed.
- S. Kuwano, S. Harada, B. Kang, R. Oriez, Y. Yamaoka, K. Takasu and K. Yamada, J. Am. Chem. Soc., 2013, 135, 11485–11488 CrossRef CAS PubMed.
- S. S. Meng, Y. Liang, K. S. Cao, L. Zou, X. B. Lin, H. Yang, K. N. Houk and W. H. Zheng, J. Am. Chem. Soc., 2014, 136, 12249–12252 CrossRef CAS PubMed.
- Z. Chen and J. Sun, Angew. Chem., Int. Ed. Engl., 2013, 52, 13593–13596 CrossRef CAS PubMed.
- K. Jakka and C. G. Zhao, Org. Lett., 2006, 8, 3013–3015 CrossRef CAS PubMed.
- Z. Q. Rong, H. J. Pan, H. L. Yan and Y. Zhao, Org. Lett., 2014, 16, 208–211 CrossRef CAS PubMed.
- Y. Zhao, J. Rodrigo, A. H. Hoveyda and M. L. Snapper, Nature, 2006, 443, 67–70 CrossRef CAS PubMed.
- Z. You, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed. Engl., 2009, 48, 547–550 CrossRef CAS PubMed.
- X. Sun, A. D. Worthy and K. L. Tan, Angew. Chem., Int. Ed. Engl., 2011, 50, 8167–8171 CrossRef CAS PubMed.
- N. Manville, H. Alite, F. Haeffner, A. H. Hoveyda and M. L. Snapper, Nat. Chem., 2013, 5, 768–774 CrossRef CAS PubMed.
- B. List, K. Hyodo, S. Gandhi and M. van Gemmeren, Synlett, 2015, 1093–1095 CrossRef.
- T. Morita, Y. Okamoto and H. Sakurai, Tetrahedron Lett., 1980, 21, 835–838 CrossRef CAS.
- C. A. Bruynes and T. K. Jurriens, J. Org. Chem., 1982, 47, 3966–3969 CrossRef CAS.
- S. Y. Park, J. W. Lee and C. E. Song, Nat. Commun., 2015, 6, 7512 CrossRef PubMed.
- Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627–5630 CrossRef CAS PubMed.
- D. W. Tay, G. Y. Leung and Y. Y. Yeung, Angew. Chem., Int. Ed. Engl., 2014, 53, 5161–5164 CAS.
- U. Hennecke, C. H. Mueller and R. Froehlich, Org. Lett., 2011, 13, 860–863 CrossRef CAS PubMed.
- C. H. Mueller, C. Roesner and U. Hennecke, Chem. – Asian J., 2014, 9, 2162–2169 CrossRef CAS PubMed.
- U. Hennecke and M. Wilking, Synlett, 2014, 1633–1637 CrossRef.
- M. Sakuma, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 2838–2841 CrossRef CAS PubMed.
- M. Wilking, C. Mueck-Lichtenfeld, C. G. Daniliuc and U. Hennecke, J. Am. Chem. Soc., 2013, 135, 8133–8136 CrossRef CAS PubMed.
- M. Wilking, C. G. Daniliuc and U. Hennecke, Synlett, 2014, 1701–1704 Search PubMed.
- D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS PubMed.
- K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016–6019 CrossRef CAS PubMed.
- D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128–11131 CrossRef CAS PubMed.
- H. Guan, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 2728–2735 CrossRef CAS.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425 CrossRef CAS PubMed.
- D. Enders and A. Seki, Synlett, 2002, 26–28 CrossRef CAS.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS PubMed.
- D. B. Ramachary and C. F. Barbas III, Org. Lett., 2005, 7, 1577–1580 CrossRef CAS PubMed.
- S. Luo, L. Zhang, X. Mi, Y. Qiao and J. P. Cheng, J. Org. Chem., 2007, 72, 9350–9352 CrossRef CAS PubMed.
- J. Jiang, L. He, S. W. Luo, L. F. Cun and L. Z. Gong, Chem. Commun., 2007, 736–738 RSC.
- Z. T. Weng, Y. Li and S. K. Tian, J. Org. Chem., 2011, 76, 8095–8099 CrossRef CAS PubMed.
- L. Zhang, L. Cui, X. Li, J. Li, S. Luo and J. P. Cheng, Chem. – Eur. J., 2010, 16, 2045–2049 CrossRef CAS PubMed.
- M. Trifonidou and C. G. Kokotos, Eur. J. Org. Chem., 2012, 1563–1568 CrossRef CAS.
- E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438 RSC.
- J. Xu, X. Fu, C. Wu and X. Hu, Tetrahedron: Asymmetry, 2011, 22, 840–850 CrossRef CAS.
- J.-W. Xu, X.-K. Fu, X.-Y. Hu and C.-L. Wu, Catal. Lett., 2011, 141, 1156–1163 CrossRef CAS.
- R. Pedrosa, J. M. Andrés, A. Gamarra, R. Manzano and C. Pérez-López, Tetrahedron, 2013, 69, 10811–10819 CrossRef CAS.
- T. P. Kumar, N. C. Vavle, V. Patro and K. Haribabu, Tetrahedron: Asymmetry, 2014, 25, 457–461 CrossRef CAS.
- S. Fotaras, C. G. Kokotos and G. Kokotos, Org. Biomol. Chem., 2012, 10, 5613–5619 Search PubMed.
- A. Martinez-Castaneda, B. Poladura, H. Rodriguez-Solla, C. Concellon and V. del Amo, Org. Lett., 2011, 13, 3032–3035 CrossRef CAS PubMed.
- S. Li, X. Fu and C. Wu, Res. Chem. Intermed., 2011, 38, 195–205 CrossRef.
- C. Wu, X. Fu and S. Li, Eur. J. Org. Chem., 2011, 1291–1299 CrossRef CAS.
- D. B. Ramachary, R. Mondal and R. Madhavachary, Org. Biomol. Chem., 2012, 10, 5094–5101 Search PubMed.
- B. H. Lipshutz and S. Ghorai, Org. Lett., 2012, 14, 422–425 CrossRef CAS PubMed.
- F. J. N. Moles, A. Bañón-Caballero, G. Guillena and C. Nájera, Tetrahedron: Asymmetry, 2014, 25, 1323–1330 CrossRef CAS.
- E. Gómez-Bengoa, C. Nájera, G. Guillena and F. Moles, Synthesis, 2014, 549–561 Search PubMed.
- F. J. N. Moles, G. Guillena and C. Nájera, RSC Adv., 2014, 4, 9963 RSC.
- C. Nájera, G. Guillena and F. Moles, Synlett, 2015, 656–660 Search PubMed.
- R. Martín-Rapún, S. Sayalero and M. A. Pericàs, Green Chem., 2013, 15, 3295–3301 RSC.
- L. Li and D. Seidel, Org. Lett., 2010, 12, 5064–5067 CrossRef CAS PubMed.
- L. Ren, T. Lei and L.-Z. Gong, Chem. Commun., 2011, 47, 11683–11685 RSC.
- A. Banon-Caballero, G. Guillena and C. Nájera, J. Org. Chem., 2013, 78, 5349–5356 CrossRef CAS PubMed.
- S. Muller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534–18537 CrossRef PubMed.
- Y. P. Chang, R. Gurubrahamam and K. Chen, Org. Lett., 2015, 17, 2908–2911 CrossRef CAS PubMed.
- M. S. Jadhav, P. Righi, E. Marcantoni and G. Bencivenni, J. Org. Chem., 2012, 77, 2667–2674 CrossRef CAS PubMed.
- Y. Xu, W. Zou, H. Sundén, I. Ibrahem and A. Córdova, Adv. Synth. Catal., 2006, 348, 418–424 CrossRef CAS.
- R. Rani and R. K. Peddinti, Tetrahedron: Asymmetry, 2010, 21, 2487–2492 CrossRef CAS.
- J. Agarwal and R. K. Peddinti, Tetrahedron Lett., 2011, 52, 117–121 CrossRef CAS.
- A. Lu, R. Wu, Y. Wang, Z. Zhou, G. Wu, J. Fang and C. Tang, Eur. J. Org. Chem., 2010, 2057–2061 CrossRef CAS.
- N. Kaplaneris, G. Koutoulogenis, M. Raftopoulou and C. G. Kokotos, J. Org. Chem., 2015, 80, 5464–5473 CrossRef PubMed.
- J.-R. Chen, Y.-Y. Lai, H.-H. Lu, X.-F. Wang and W.-J. Xiao, Tetrahedron, 2009, 65, 9238–9243 CrossRef CAS.
- Z. B. Li, S. P. Luo, Y. Guo, A. B. Xia and D. Q. Xu, Org. Biomol. Chem., 2010, 8, 2505–2508 Search PubMed.
- A. M. Faisca Phillips and M. T. Barros, Org. Biomol. Chem., 2012, 10, 404–412 Search PubMed.
- Y. M. Chen, P.-H. Lee, J. Lin and K. Chen, Eur. J. Org. Chem., 2013, 2699–2707 CrossRef CAS.
- F. Diaba and J. Bonjoch, Org. Biomol. Chem., 2009, 7, 2517–2519 Search PubMed.
- A. D. Gammack Yamagata, S. Datta, K. E. Jackson, L. Stegbauer, R. S. Paton and D. J. Dixon, Angew. Chem., Int. Ed. Engl., 2015, 54, 4899–4903 CrossRef CAS PubMed.
- A. Claraz, S. Oudeyer and V. Levacher, Tetrahedron: Asymmetry, 2013, 24, 764–768 CrossRef CAS.
- S. Murahashi, S. Ono and Y. Imada, Angew. Chem., Int. Ed. Engl., 2002, 41, 2366–2368 CrossRef CAS PubMed.
- S. Xu, Z. Wang, X. Zhang, X. Zhang and K. Ding, Angew. Chem., Int. Ed. Engl., 2008, 47, 2840–2843 CrossRef CAS PubMed.
- P. Piras, A. Frongia, F. Capitta, J. Ollivier and F. Secci, Synlett, 2010, 89–93 Search PubMed.
- D. J. Aitken, A. M. Bernard, F. Capitta, A. Frongia, R. Guillot, J. Ollivier, P. P. Piras, F. Secci and M. Spiga, Org. Biomol. Chem., 2012, 10, 5045–5048 Search PubMed.
- F. Secci, J. Ollivier, F. Capitta, A. Frongia, D. Aitken, P. Piras and R. Guillot, Synlett, 2014, 123–126 Search PubMed.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- U. Eder, G. Sauer and R. Weichert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- M. Wadamoto, E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 10098–10099 CrossRef CAS PubMed.
- E. M. Phillips, J. M. Roberts and K. A. Scheidt, Org. Lett., 2010, 12, 2830–2833 CrossRef CAS PubMed.
- C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed. Engl., 2010, 49, 9479–9483 CrossRef CAS PubMed.
- A. R. Burns, A. G. E. Madec, D. W. Low, I. D. Roy and H. W. Lam, Chem. Sci., 2015, 6, 3550–3555 RSC.
- K. Mori, A. Miyake and T. Akiyama, Chem. Commun., 2015, 51, 16107–16110 RSC.
- J. Feng, L. Lin, K. Yu, X. Liu and X. Feng, Adv. Synth. Catal., 2015, 357, 1305–1310 CrossRef CAS.
- C. C. Hsiao, H. H. Liao and M. Rueping, Angew. Chem., Int. Ed. Engl., 2014, 53, 13258–13263 CrossRef CAS PubMed.
- K. A. Kalstabakken and A. M. Harned, Tetrahedron, 2014, 70, 9571–9585 CrossRef CAS PubMed.
- X. Yang, J. Wang and P. Li, Org. Biomol. Chem., 2014, 12, 2499–2513 Search PubMed.
- X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185–1197 RSC.
- L. Yao, K. Liu, H.-Y. Tao, G.-F. Qiu, X. Zhou and C.-J. Wang, Chem. Commun., 2013, 49, 6078–6080 RSC.
- Q. Gu and S.-L. You, Org. Lett., 2011, 13, 5192–5195 CrossRef CAS PubMed.
- Q. Gu and S.-L. You, Chem. Sci., 2011, 2, 1519–1522 RSC.
- Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056–4057 CrossRef CAS PubMed.
- R. Tello-Aburto, K. A. Kalstabakken, K. A. Volp and A. M. Harned, Org. Biomol. Chem., 2011, 9, 7849–7859 Search PubMed.
- W. Wu, X. Li, H. Huang, X. Yuan, J. Lu, K. Zhu and J. Ye, Angew. Chem., Int. Ed. Engl., 2013, 52, 1743–1747 CrossRef CAS PubMed.
- M. Q. Jia and S. L. You, Synlett, 2013, 1201–1204 CAS.
- M. Q. Jia, C. Liu and S. L. You, J. Org. Chem., 2012, 77, 10996–11001 CrossRef CAS PubMed.
- M. T. Corbett and J. S. Johnson, Chem. Sci., 2013, 4, 2828–2832 RSC.
- L. Pantaine, V. Coeffard, X. Moreau and C. Greck, Org. Lett., 2015, 17, 3674–3677 CrossRef CAS PubMed.
- K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248–264 CrossRef CAS PubMed.
- D. M. Rubush, M. A. Morges, B. J. Rose, D. H. Thamm and T. Rovis, J. Am. Chem. Soc., 2012, 134, 13554–13557 CrossRef CAS PubMed.
- J.-Y. Du, C. Zeng, X.-J. Han, H. Qu, X.-H. Zhao, X.-T. An and C.-A. Fan, J. Am. Chem. Soc., 2015, 137, 4267–4273 CrossRef CAS PubMed.
- R. Leon, A. Jawalekar, T. Redert and M. J. Gaunt, Chem. Sci., 2011, 2, 1487–1490 RSC.
- A. E. Williamson, T. Ngouansavanh, R. D. M. Pace, A. E. Allen, J. D. Cuthbertson and M. J. Gaunt, Synlett, 2016, 116–120 CAS.
- N. Miyamae, N. Watanabe, M. Moritaka, K. Nakano, Y. Ichikawa and H. Kotsuki, Org. Biomol. Chem., 2014, 12, 5487–5855 Search PubMed.
- S. Takizawa, T. M.-N. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed. Engl., 2012, 51, 5423–5426 CrossRef CAS PubMed.
- S. Takizawa, T. M.-N. Nguyen, A. Grossmann, M. Suzuki, D. Enders and H. Sasai, Tetrahedron, 2013, 69, 1202–1209 CrossRef CAS.
- X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed. Engl., 2015, 54, 6874–6877 CrossRef CAS PubMed.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- R. Takagi and T. Nishi, Org. Biomol. Chem., 2015, 13, 11039–11045 Search PubMed.
- C. Martín-Santos, C. Jarava-Barrera, S. del Pozo, A. Parra, S. Díaz-Tendero, R. Mas-Ballesté, S. Cabrera and J. Alemán, Angew. Chem., Int. Ed. Engl., 2014, 53, 8184–8189 CrossRef PubMed.
- A. Fraile and J. Alemán, Synlett, 2015, 1940–1954 CAS.
- M. S. Manna and S. Mukherjee, Chem. Sci., 2014, 5, 1627 RSC.
- M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS PubMed.
- K. Akagawa, M. Akiyama and K. Kudo, Eur. J. Org. Chem., 2015, 3894–3898 CrossRef CAS.
- M. Akiyama, K. Akagawa, H. Seino and K. Kudo, Chem. Commun., 2014, 50, 7893–7896 RSC.
- C. Sparr and R. Gilmour, Angew. Chem., Int. Ed. Engl., 2011, 50, 8391–8395 CrossRef CAS PubMed.
- S. J. Singha Roy and S. Mukherjee, Chem. Commun., 2014, 50, 121–123 RSC.
- G.-Y. Chen, F. Zhong and Y. Lu, Org. Lett., 2011, 13, 6070–6073 CrossRef CAS PubMed.
- C. K. De and D. Seidel, J. Am. Chem. Soc., 2011, 133, 14538–14541 CrossRef CAS PubMed.
- C. K. De, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2009, 131, 17060–17061 CrossRef CAS PubMed.
- E. G. Klauber, C. K. De, T. K. Shah and D. Seidel, J. Am. Chem. Soc., 2010, 132, 13624–13626 CrossRef CAS PubMed.
- E. G. Klauber, N. Mittal, T. K. Shah and D. Seidel, Org. Lett., 2011, 13, 2464–2467 CrossRef CAS PubMed.
- D. Seidel, Synlett, 2014, 783–794 CrossRef CAS.
- H. Fernandez-Perez, P. Etayo, J. R. Lao, J. L. Nunez-Rico and A. Vidal-Ferran, Chem. Commun., 2013, 49, 10666–10675 RSC.
- N. Di Iorio, P. Righi, A. Mazzanti, M. Mancinelli, A. Ciogli and G. Bencivenni, J. Am. Chem. Soc., 2014, 136, 10250–10253 CrossRef CAS PubMed.
- F. Eudier, P. Righi, A. Mazzanti, A. Ciogli and G. Bencivenni, Org. Lett., 2015, 17, 1728–1731 CrossRef CAS PubMed.
- M. D. Barker, R. A. Dixon, S. Jones and B. J. Marsh, Tetrahedron, 2006, 62, 11663–11669 CrossRef CAS.
- B. J. Marsh, H. Adams, M. D. Barker, I. U. Kutama and S. Jones, Org. Lett., 2014, 16, 3780–3783 CrossRef CAS PubMed.
- R. A. Dixon and S. Jones, Tetrahedron: Asymmetry, 2002, 13, 1115–1119 CrossRef CAS.
- M. D. Barker, R. A. Dixon, S. Jones and B. J. Marsh, Chem. Commun., 2008, 2218–2220 RSC.
- I. U. Kutama and S. Jones, J. Org. Chem., 2015, 80, 11468–11479 CrossRef CAS PubMed.
- J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- G. Bencivenni, Synlett, 2015, 1915–1922 CrossRef CAS.
- K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964–3970 CrossRef CAS PubMed.
- K. Mori, M. Kobayashi, T. Itakura and T. Akiyama, Adv. Synth. Catal., 2015, 357, 35–40 CrossRef CAS.
- R. J. Armstrong and M. D. Smith, Angew. Chem., Int. Ed. Engl., 2014, 53, 12822–12826 CrossRef CAS PubMed.
- J. Wilent and K. S. Petersen, J. Org. Chem., 2014, 79, 2303–2307 CrossRef CAS PubMed.
- G. Qabaja, J. E. Wilent, A. R. Benavides, G. E. Bullard and K. S. Petersen, Org. Lett., 2013, 15, 1266–1269 CrossRef CAS PubMed.
- K. Higuchi, S. Suzuki, R. Ueda, N. Oshima, E. Kobayashi, M. Tayu and T. Kawasaki, Org. Lett., 2015, 17, 154–157 CrossRef CAS PubMed.
- K. Murai, J. Nakajima, A. Nakamura, N. Hyogo and H. Fujioka, Chem. – Asian J., 2014, 9, 3511–3517 CrossRef CAS PubMed.
- K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed. Engl., 2010, 49, 9174–9177 CrossRef CAS PubMed.
- K. Murai, T. Matsushita, A. Nakamura, N. Hyogo, J. Nakajima and H. Fujioka, Org. Lett., 2013, 15, 2526–2529 CrossRef CAS PubMed.
- K. Murai, N. Shimizu and H. Fujioka, Chem. Commun., 2014, 50, 12530–12533 RSC.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2016 |