
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards the next generation of biomedicines by site-selective conjugation
Qi-Ying
Hu
*a,
Francesco
Berti
b and
Roberto
Adamo
*b
aNovartis Institutes for Biomedical Research (NIBR), 100 Technology Square, Cambridge, MA 02139, USA. E-mail: qiying.hu@novartis.com; Tel: +1 617 871 7607
bGSK Vaccines (former Novartis Vaccines & Diagnostics), Via Fiorentina 1, 53100 Siena, Italy. E-mail: roberto.x.adamo@gsk.com; Tel: +39 0577 539393
First published on 21st January 2016
Abstract
Bioconjugates represent an emerging class of medicines, which offer therapeutic opportunities overtaking those of the individual components. Many novel bioconjugates have been explored in order to address various emerging medical needs. The last decade has witnessed the exponential growth of new site-selective bioconjugation techniques, however very few methods have made the way into human clinical trials. Here we discuss various applications of site-selective conjugation in biomedicines, including half-life extension, antibody–drug conjugates, conjugate vaccines, bispecific antibodies and cell therapy. The review is intended to highlight both the progress and challenges, and identify a potential roadmap to address the gap.
 Qi-Ying Hu | Qi-Ying Hu received his PhD in organic chemistry at National University of Singapore in 2001, and was a postdoctoral fellow with Professor E. J. Corey at the Harvard University from 2002 to 2004. He subsequently took a laboratory head and an investigator role at Novartis Institutes for Biomedical research (NIBR) at Cambridge, MA, USA. He has established drug discovery chemistry research in both small molecules and biologics across wide disease areas. He has (co)invented several clinical candidates, and novel bioconjugation methods, including the selective tyrosine conjugation, and disulfide conjugation. He is currently leading novel chemistry-edited biotherapeutics projects. |
 Francesco Berti | Francesco Berti got his PhD in Chemical Sciences in 2002 from the University of Siena (Italy), working firstly on magnetic resonance technologies. While completing his doctoral studies, in 2001 he moved to Novartis Vaccines (now GSK Vaccines) where he worked on the preparation and structural characterization of a meningococcal ACW135Y polysaccharide-protein conjugate vaccine, which was eventually licensed in US and Europe in 2010 (Menveo®). His current role is Head of Vaccine Chemistry and Formulation Department at GSK Vaccines. Over the last 12 years he has been working on the research and development of several carbohydrate-based vaccines, tackling a variety of infective diseases. |
 Roberto Adamo | Roberto Adamo obtained his PhD in Pharmaceutical Science from the University of Catania (Italy) in 2003, with a thesis on the synthesis of biologically relevant inositols. After a post-doctoral fellowship at the NIH in Bethesda, USA, under the supervision of Dr P. Kovac, he moved as EU Marie Curie fellow to the group of J. P. Kamerling at the Utrecht University, Netherlands. In 2007 he joined Novartis Vaccines (now GSK Vaccines), where he is currently the Head of the Carbohydrate Chemistry Laboratory, and the Leader of the Conjugation & Synthesis Platform. His research interests focus on the synthesis of glycans, glycoconjugates and glyconanoparticles to be used for carbohydrate-based therapeutics. |
1. Introduction
Bioconjugation strategies for the covalent crosslinking of a synthetic or semisynthetic molecule (e.g. drugs, carbohydrates, peptides and other bio- or synthetic polymers) to a biomolecule (e.g. proteins, nucleotides or polysaccharides) have attracted increasing attention in the biopharmaceutical field.1 Bioconjugates represent an emerging class of medicines, which offer therapeutic opportunities overtaking those of the individual components. The history of medical application of bioconjugates can be traced back to as early as 1920s, when Avery and Goebel reported that a non-immunogenic bacterial capsular polysaccharide can stimulate an immune response upon covalent conjugation to a protein carrier.2 It took a long journey before this remarkable observation could be finally translated into ProHIBIT®, the first licensed conjugate vaccine against Haemophilus influenza type b (Hib) for the US market, in 1987 (Fig. 1). In parallel, bioconjugation for half-life extension of therapeutic proteins has been extensively investigated, and led to the launch of a PEGylated Adenosine deaminase Adagen® as a remedy for severe combined immunodeficiency disease in the US in 1990. Later in 1994, PEGylated asparaginase Oncaspar® was licensed for the treatment of pediatric leukemia, expanding the scope of bioconjugation to anti-tumor therapy.3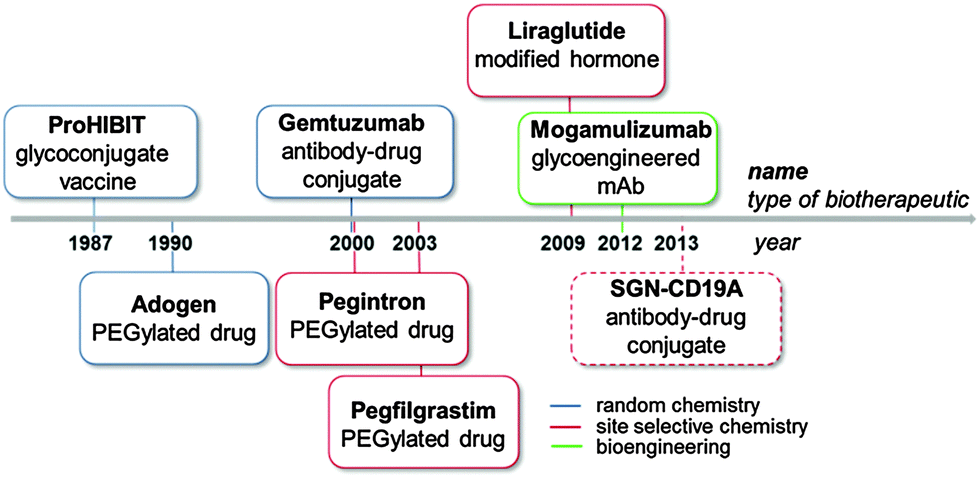 | ||
| Fig. 1 Important milestones achieved with bioconjugate medicines show how the scientific discoveries in protein modifications through chemical approaches and bioengineering over the last twenty years are impacting the pharma industry. Licensed biomedicines are indicated with a full frame line, products in clinical trials with a dotted frame line. | ||
In the same decade, the idea of targeted therapy conceived by Ehrlich a century ago was realized first by the registration of several antibody-based imaging agents,4 and then by the FDA approval of the antibody–drug conjugate (ADC) Gemtuzumab ozogamicin (Mylotarg®, Wyeth-Pfizer) in 2000.5
Unfortunately, the compound was withdrawn from the market in 2010 due to its marginal benefits. The concept resurged with improved technology and two other ADCs (Adcetris®, Seattle Genetics/Takeda, and Kadcyla®, Genentech-Roche/ImmunoGen) were approved by FDA in 2010.
Currently, there are more than 40 ADCs at various stages of clinical trials,6 and some of them will hopefully make their way into market.
In 2009, the first site selectively modified biotherapeutic, the hormone Liraglutide (Victoza®) developed by Novo Nordisk, was licensed, representing a breakthrough for bioconjugate medicines.7
The recent introduction of the first glycoengineered monoclonal antibody (mAb) (Mogamulizumab, Poteligeo®, Kyowa Hakko Kirin Co)8 in Japan highlighted the potential of precisely modified therapeutics also via an engineered cell line (Fig. 1).
In 2014 four bioconjugate medicines, namely the white blood cell booster Neulasta®, the glycoconjugate vaccine Prevnar13®, and the antidiabetics Levemir® and Victoza®, ranked at relevant positions in the list of the Top 50 pharmaceutical products based on annual global sales (Table 1).9 The success of bioconjugates is driving the development of a variety of biomolecules to address unmet medical needs in different disease areas.10
| Rank | Brand names | Indication | Sales 2013 ($m) |
|---|---|---|---|
| 16 | Neulasta® | Neutropenia | 4596 |
| 17 | Prevnar13® | Pneumococcal disease | 4464 |
| 39 | Levemir® | Type 1, 2 diabetes | 2454 |
| 42 | Victoza® | Type 2 diabetes | 2318 |
Over the recent years, the requirement of conjugation strategies ensuring improved pharmacokinetic and/or pharmacodynamic properties, increased efficacy and the safer profile of bioconjugate medicines has promoted the development of many in vitro and in vivo site selective methods.
These techniques offer great opportunities for the design of tailored biopharmaceuticals tackling therapeutic challenges, but also introduce new challenges that need to be overcome before fulfilling their promise. This review is intended to cover the recent applications of site-selective bioconjugation methods in various therapeutic areas. After a brief introduction of general tactics for the site-selective protein modification, we discuss the application of these techniques in different classes of protein-based pharmaceuticals, including long acting proteins, antibody–drug conjugates, conjugate vaccines, and cell therapies. Particular focus is given to examples with in vivo or clinical data to elucidate opportunities and challenges towards successful translation in humans.
2. Tactics in regioselective conjugation
Generally protein-based bioconjugate medicines involve the coupling of different classes of molecules which include primarily (a) polymers to extend the circulating half-life of protein therapeutics;11 (b) cytotoxic anti-cancer drugs12,13 that are coupled to mAbs for selective delivery to cancer cells; or (c) glycans that can be linked to proteins in order to (i) induce an anti-carbohydrate response,14 (ii) enhance the immunological activity of antigens,15 or (iii) modulate the pharmacokinetic and/or pharmacodynamic properties of antibodies.16 In addition, small molecules (immunopotentiators) targeting specific receptors have also been conjugated to modulate the immune activity of antibodies or protein antigens.17,18Classic procedures to modify proteins typically target the most abundant surface residues, including K, D/E or C residues.19 The ε-amino group of K can be directly coupled by different methods, mainly by reductive amination or amide bond formation.20 Alternatively, a variety of bifunctional linkers can be used to modify the protein for further incorporation of the target payload/glycan. Likewise, carboxylic acids of D/E residues can react with amines by condensing agents or modified with bifunctional linkers.14 Generally these methods are inherently associated with low regioselectivity which results in unpredictable variability of the product quality.
Differently from K and D/E, C can be targeted in a selective manner by forming, for example, mixed disulfides or by alkylation with suitable electrophiles, such as α-halocarbonyls (e.g., iodoacetamide)21 and Michael acceptors (e.g., maleimides or vinyl sulfones).22 On the other hand, C is typically presented in disulphide bridges, and its involvement could have deleterious results on the protein structure.
In general, the use of random conjugation methods can introduce changes on protein conformation, producing detrimental effects on cell–biomolecule interactions.23
One of the key requirements to achieve optimal efficacy of a protein therapy is the preservation of the original protein functionality upon conjugation. In addition, the payload potency and loading, linker, and immunogenicity of the payload or linker are integral factors to be considered in optimisation.18,19
Site-selective conjugation methods hold the central role in this regard.1,19,24 Performing a regioselective chemical reaction on a protein and maintaining its integrity is highly challenging, primarily due to the distinct requirements for manipulating a protein as compared to small organic molecules. Protein conjugation uses water as the sole solvent at nearly neutral pH. The reaction temperature is usually below 40 °C, and the reactant concentration is lower than mM. Therefore, the reaction typically requires high kinetics with compatibility to water and extensive functionalities on the protein. Conceptually, the selectivity can be accessed by targeting the most differentiating canonical amino acid(s) on proteins, or pre-installing an orthogonal functionality by protein engineering (Scheme 1).
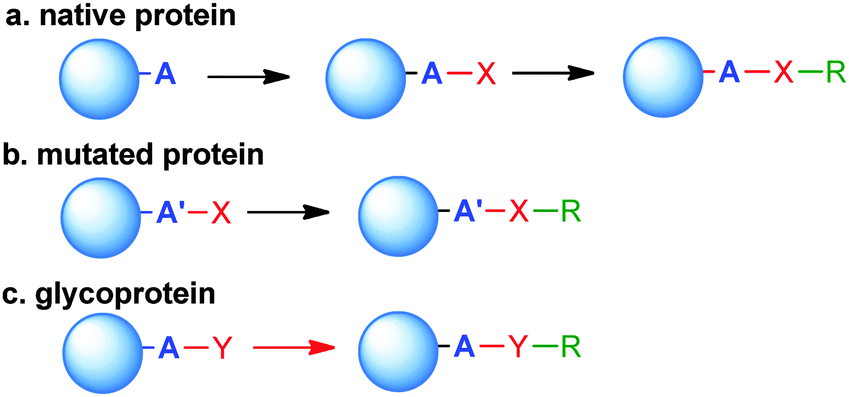 | ||
| Scheme 1 General approaches to site-selective bioconjugates. (a) An endogenous amino acid residue (A) with unique reactivity is modified with functionality (X) ready for further coupling with payload or glycan (R). (b) The functionality X is already present in a residue A′ genetically introduced in the protein. (c) Post translational modifications (typically glycan) occurring at certain positions can be targeted to introduce the substituent R. | ||
2.1 Selective bioconjugation reactions targeting canonical amino acids
There are 20 types of canonical amino acids in proteins. Among these, H, D, R, C, Q, E, K, N, S, Y, T, and W residues and N- or C-terminus are potentially reactive. Traditional conjugation methods are mainly addressed to the most reactive residues (e.g., K), due to the favourable reaction kinetics. The high nucleophilicity of the amine in the K side chain and the relatively good surface exposition of K residues in water soluble proteins often pose significant challenges to achieve good regioselectivity. Many excellent methods have been developed for site directed modification, and we will briefly introduce the major strategies (Scheme 2).24,25 It is known that the N-terminal amine has slightly higher pKa than the corresponding ε-amine group of K. Under slightly acidic conditions, N-terminal amine retains good reactivity, while lysines are less prone to reaction due to protonation. A series of strategies have been developed by taking advantage of this feature, including reductive amination, acylation, and pyridoxal phosphate (PLP) mediated transamination reaction.26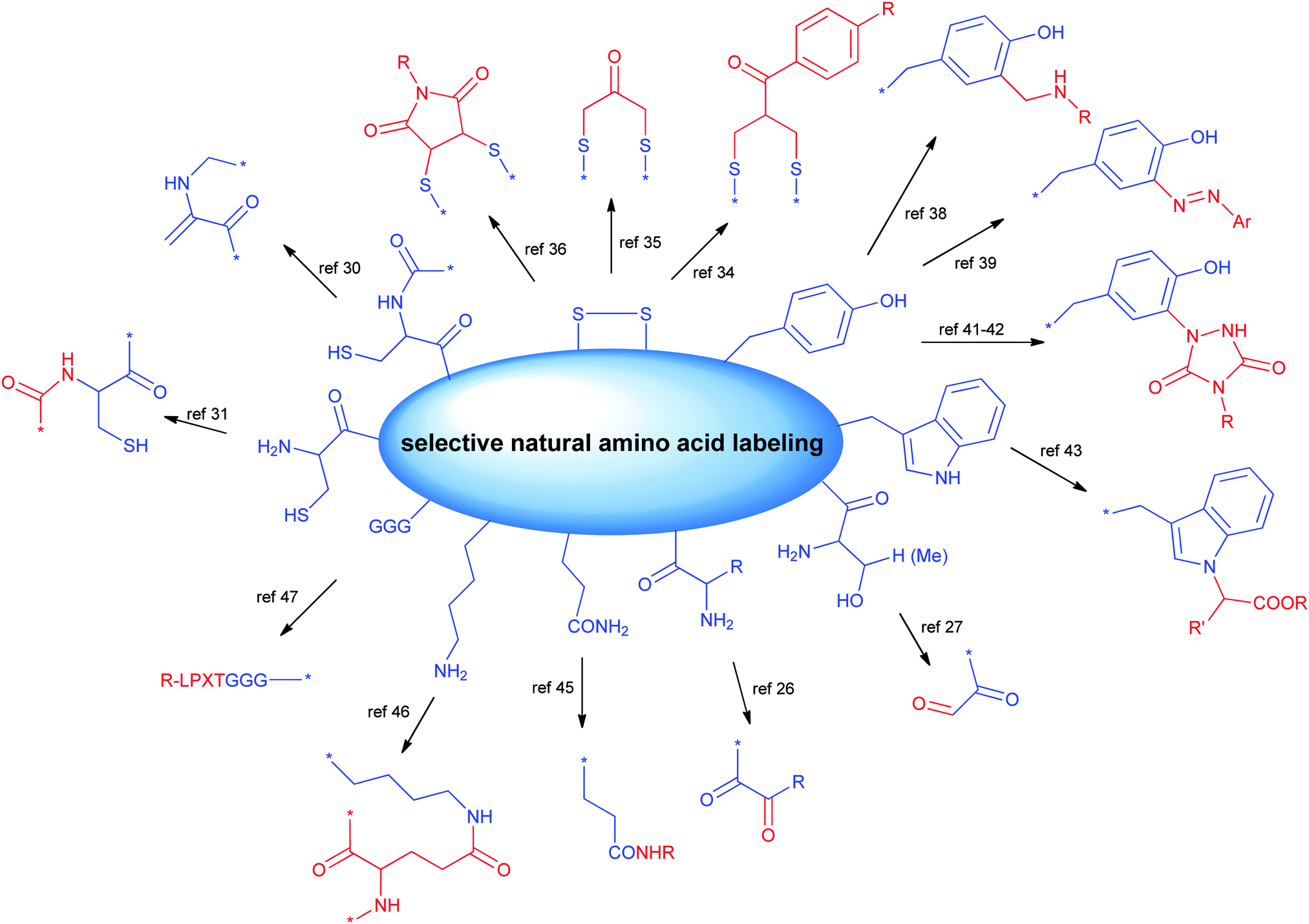 | ||
| Scheme 2 Major site-selective conjugation methods targeting canonical amino acids. | ||
N-terminal S or T present a unique adjacent amino alcohol moiety, which can undergo efficient oxidative cleavage to aldehyde by treating with sodium periodate.27 The newly formed aldehyde is a great orthogonal chemical handle for the subsequent manipulation.
C is traditionally a highly favourable conjugation site due to its outstanding reactivity and low population. Besides the reactions mentioned above, the thiol group of C28 can undergo different modifications, including the disulfide exchange reaction to form mixed disulfides29 or SeS-derivatives,30 and the oxidative elimination of C to generate a dehydroalanine, which is a useful acceptor for Michael-type reactions with thiol nucleophiles.31 N-terminal C is suitable for native chemical ligation with a thioester to form an amide linkage.32 This reaction has been frequently used in the semi-synthesis of proteins.33
Most proteins have no free cysteine on the surface, and most of them are present in disulphide bonds, which are potentially crucial to the protein tertiary structure. A few excellent strategies selectively cleave the disulfide bond first, and subsequently reconnect two cysteines together by the introduction of a short covalent bridge between them.30,31 The bridge also serves as the attachment point for payloads. Bisulfone linkers,34 dihaloacetone35 or dibromomaleimides36,37 represent the frontier in this direction.
Aromatic amino acids Y or W have lower population than K, as well as lower exposition on the surface. The reactivity of these aromatic amino acids has been explored for site-selective conjugation in recent years. A seminal example was the reaction of formaldehyde with the electron-rich aniline to give the iminium intermediate which then undergoes a Mannich condensation with the phenol of tyrosine residues to provide bioconjugates.38 Alternatively, diazonium salts can react with the phenol group to provide ortho-substituted tyrosine residues.39 Among the different proposed methods,40 a unique class of compounds which have been proven useful for the modification of protein therapeutics is given by triazolinediones which condense with tyrosine residues.41 This reaction has been shown to proceed with high selectivity towards tyrosine when the tris(hydroxymethyl)aminomethane (Tris) buffer is used, to trap the isocyanates derived from the in situ degradation of the triazolidinones that would direct the reaction to the lysine residues instead.42
Attempts to target W by transition metal catalysed reaction based on rhodium carbenoids yielded mixtures of N- and C-adducts.43 However, the highly acidic conditions might limit its applicability.
Modification of certain residues, such as K, Q and G, can be selectively achieved by chemoenzymatic methods.44 These strategies target residues within an enzyme recognition sequence introduced by protein engineering (Scheme 2).
Transglutaminases (TGases), a family of widely expressed enzymes, have been used to selectively label Q45 or K.46 Sortases from Staphylococcus aureus or Streptococcus pyogenes have also been applied for specific conjugation of the N-terminal (G)n (n ≥ 3) or C-terminal LPXTG sequence.47
Other enzymes potentially suited for protein modifications include: (i) Escherichia coli biotin ligase (BirA), capable of recognizing and biotinylating an engineered 15-residue ‘acceptor peptide’ (AP) sequence, which can be fused to the N-terminus or C-terminus of any target protein;48 (ii) E. coli lipoic acid ligase (LplA) attaching lipoic acid analogs to the LplA acceptor peptide;49 (iii) Protein Farnesyl Transferase (PFTase) and Protein Geranylgeranyl Transferase (PGGTase) catalyzing protein prenylation.50 In addition, enzymes catalyzing glycosylation of S/T residues with GlcNAc can be the starting point for further chemoenzymatic glycan modification.51 A series of glycosyltransferases, including sialyltransferases or galactosyltransferases, have also been employed to generate site-specifically modified biotherapeutics.52
A plethora of methods have been described for site specific chemical or chemoenzymatic incorporation of probes, radioactive agents or fluorophores that we are not discussing here, but that could offer a starting point for bioconjugation of small/macro-molecules.24 Many of these techniques can potentially be used in an orthogonal manner to place copies of the conjugated molecules or even different types of molecules.53
2.2 Regioselectivity via preinstalled functionality by molecular biology
Methods targeting native amino acids are generally substrate-dependent, and the selectivity obtained on one substrate is often not transferable to another substrate. The choice of conjugation sites is also limited. On the other hand, molecular biology methods introduce functionalities suitable for site-specific conjugation, potentially at any position of choice. Currently, several variants in this class of modifications have been developed (Scheme 3).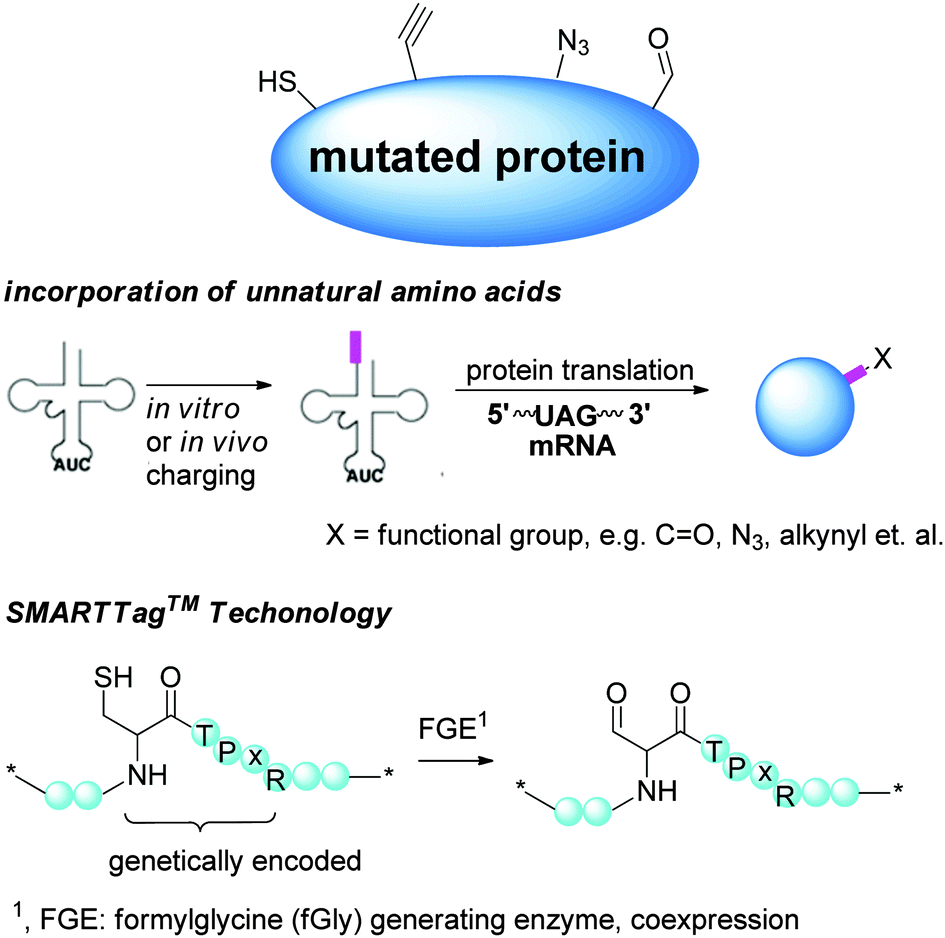 | ||
| Scheme 3 Regioselectivity via preinstalled functionality by molecular biology. | ||
In recent years, this method has been demonstrated to be successful in the formation of homogeneous covalent protein–protein, protein–small molecules and glycan–protein linkages, heterodimeric protein conjugates,61 antibody–drug conjugates62 and glycoconjugates.63,64
Very recently, quadruplet-decoding transfer RNAs have been developed to enable encoding multiple pairs of distinct uAAs into a single protein.63 This technique represents a major breakthrough in this field.
uAA incorporation by open cell free synthesis (OCFS) has been tackled. In 2009, Goerke and Swartz successfully employed an Methanocaldococcus jannaschii aminoacyl tRNA synthetase (aaRS)-tRNA for cell free incorporation of the uAAs para-azido-L-phenylalanine (pAzF) into dihydrofolate reductase.65 In spite of the initial low product yield, the concept holds potential to overcome some limitations associated with the cell based expression system. Taking advantage of an M. jannaschii tyrosyl tRNA derived synthetase/uAA pairs in an E. coli-based cell-free expression system reported by Otting et al.,66 a method enabling a more robust cell-free based expression of uAA-containing proteins has been optimized.67
Selenocysteine can be engineered into proteins, and provides an alternative uAA as the site-specific conjugation handle for generating homogeneous biotherapeutics.68 Selenocysteine is recognized as the 21st amino acid and its specific incorporation is directed by the UGA codon.69 Unique tRNAs that have complementary UGA anticodons are aminoacylated with serine. Conversion of the seryl-tRNA into selenocysteyl-tRNA, followed by specific binding to a special elongation factor, leads to ribosomal-mediated synthesis of selenoproteins.
2.3 Modification of inserted functionalities
One of the essential requisites in the design of bioconjugate medicine is the choice of a selective chemistry enabling the orthogonal connection of the coupling partner (small molecule/glycan) with the functionalized protein (Scheme 4).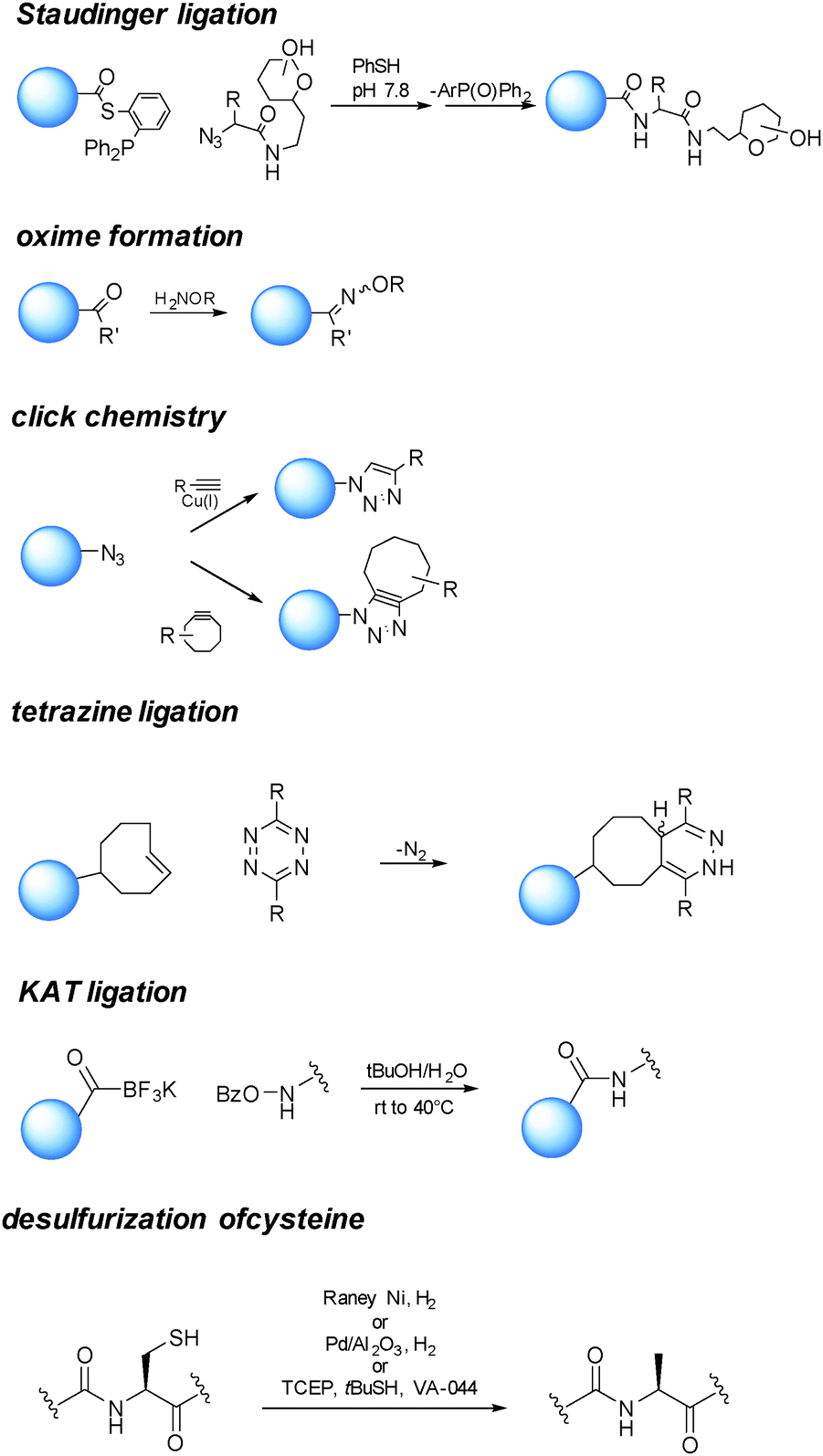 | ||
| Scheme 4 Examples of reactions enabling orthogonal connection of the coupling partner (small molecule/glycan) with the functionalized protein. | ||
Among the different proposed ligation strategies,73,74 chemical ligation between an N-terminal C and a partner containing an α-thioester group can generate an amide bond at the ligation junction,75via an initial trans-thioesterification followed by spontaneous intramolecular S to N acyl shift. This reaction has given access to glycosylphosphatidylinositol (GPI)-anchored glycoproteins.76 This approach generates a free cysteine, which is usually undesirable to a therapeutic protein. Several methods have been developed to selectively remove the free thiol, such as hydrogenation in the presence of RANEY® Ni or Pd on Al2O3.77 Very recently, Wan and Danishefsky reported a powerful radical desulfurization, which is highly efficient and general in the total synthesis of (glyco)proteins.78
The reaction of a phosphine and an azide to form an iminophosphorane, described in 1919 by Staudinger and Meyer,79 has also been harnessed as a bioconjugation tool.80 Since in the original reaction a phosphine oxide remained as part of the product, “traceless” variants of the reaction have been developed,81,82 finding application in the preparation of glycoproteins and modified proteins.83
The sulfhydryl from cysteine can be exploited to produce adducts with payloads by displacement of halogens,84 thiol–ene addition with olefins,85 Michael type reactions with maleimides,86 or exchange with disulfide/selenenylsulfide to provide dithioesters.87 Similarly to the sulfhydryl of C, the selenol group of selenocysteine can be modified by the formation of mixed Se–S ethers.26,85
The Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition reaction of azides and alkynes has gained particular attention since their first report in 2001.88,89 This reaction ensures orthogonality with the amino acid residues.
Cu(I) salts are known to be cytotoxic, and this has prevented their use for imaging studies in living organisms. Although it has been estimated that the amount used to catalyze the cycloaddition reaction for the preparation of bioconjugate medicines is far below the proposed permitted daily exposure,90 a valid alternative is represented by the strain promoted version of the reaction.91,92
A number of substituted cycloalkynes has been currently used for fast cycloaddition with azides.93,94 The catalyst-free inverse-electron-demand Diels–Alder cycloaddition between 1,2,4,5-tetrazine and trans-cyclooctene (TCO) is another highly selective and efficient ligation reaction.95,96 The need of Cu(I) catalysis has been observed when a glycan partner is condensed.64
Reaction of a carbonyl with a hydrazide or an aminooxy group has also been proven useful in bioconjugation strategies. Particularly oximes, which can be efficiently formed under acidic conditions or at closely neutral pH by aniline catalysis,32 are thermodynamically stable comparably to the corresponding hydrazones.
Bode and coworkers have developed a highly efficient amide-forming ligation of potassium acyltrifluoroborates and hydroxylamines in water (KAT Ligation). The method has showed potential in the synthesis of proteins.97
The utility of the described chemistries has also been proven in a large number of examples in combination with uAAs. For instance positioning of a tyrosine with a ketone handle has been used for hydrazine/oxime formation,98 insertion of azidohomoalanine has been followed by coupling of a ligand through click chemistry99 or Staudinger ligation,100 dehydroalanine has been incorporated as a useful intermediate for Michael type additions,101 and the 4-iodo-L-phenylalanine-containing protein has been chemoselectively modified by means of a Mizoroki–Heck reaction to create C–C bonds.102
2.4 Post-translational protein modifications
N-linked glycosylation of proteins is the most abundant post translational protein modification and, therefore, has been targeted for its potential to deliver site specifically modified glycoproteins, and more recently to link payloads to functionalized sugar residues (Scheme 5).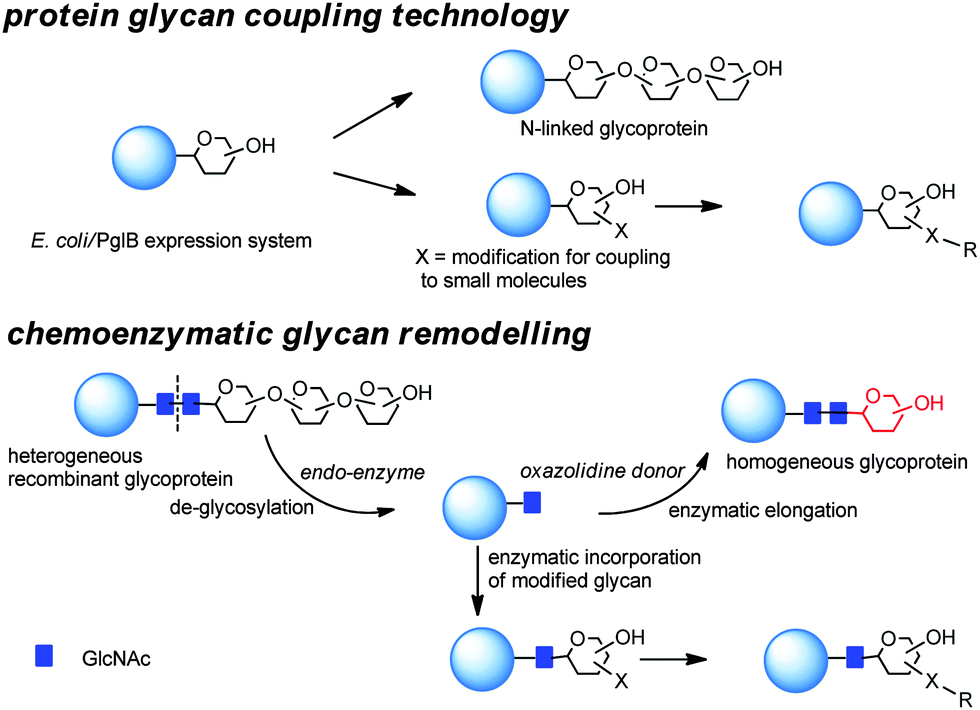 | ||
| Scheme 5 Bioengineering approaches for the preparation of homogeneous glycoproteins. | ||
Generally in eukaryotes, the oligosaccharide is preassembled on the lipid carrier dolichyl pyrophosphate at the membrane of endoplasmic reticulum and then selectively transferred to asparagine residues within the sequence NXST of nascent polypeptide chains.103 Bacterial and eukaryotic N-linked glycosylation pathways are, however, homologous processes. In particular, Campylobacter jejuni possesses a general N-linked glycosylation system where the oligosaccharide is assembled on the lipid carrier undecaprenyl-pyrophosphate (Und-PP) at the cytoplasmic side of the inner membrane, and translocated to the periplasm by the ABC transporter homologue PglK.104 Finally, the oligosaccharyltransferase (OTase) PglB transfers the oligosaccharide from the lipid carrier to the acceptor proteins.
The assembly of the O-antigen constituting the outer component of the LPS of Gram-negative bacteria involves, according to the so called “Wzy-dependent mechanism”, the synthesis of repeating subunits on the lipid carrier Und-PP at the cytoplasmic side of the inner membrane. Once completed, O-antigen subunits are flipped across the cytoplasmic membrane, polymerized by the Wzy polymerase in the periplasmic space, and transferred to the lipid A core by the WaaL ligase.105 Alternatively, the formation of a polymeric O antigen by reactions can occur at the cytosolic face of the cytoplasmic membrane in the “ABC transporter-dependent” pathway.105
The nascent polysaccharide chain is transported across the inner membrane by an ATP-binding cassette transporter, and subsequently ligated to the lipid A core.106 In Escherichia coli, the WecA UDP-GlcNAc:undecaprenylphosphate GlcNAc-1-phosphate transferase can initiate either assembly pathways.107 The C. jejuni N-glycosylation machinery can be functionally transplanted to E. coli.108 PglB expressed in a WaaL mutant strain of E. coli can efficiently accept diverse Und-PP-linked glycans as substrates.109 By using this glycosylation machinery, a variety of polysaccharides can be potentially transferred to recombinant proteins, enabling the one pot biosynthesis of glycoproteins.109 This approach appears to be suited for the incorporation of a limited but precise number of glycans, with variable length. Similarly to engineering of cysteines or uAAs, the attachment site is given by the NXST tag, and therefore the connectivity point can be theoretically varied to find the optimal portion of the protein for modification.
Glycoengineering has also been used to generate human carbohydrate structures on the surface of recombinant Gram-negative bacteria, such as E. coli and Salmonella enterica. In particular, polymers of the ubiquitous glycan Galβ1-4GlcNAc, a typical motif in N-glycosylated mammalian proteins, were expressed and used as acceptors for fucosylation leading to polymers of Lewis X antigens.110 Glycoengineered lipooligosaccharides (LOSs) allowed studying of pro-inflammatory responses in murine dendritic cells.
Alternative N-glycosylation systems with different peculiarities have been recently discovered. The NGT tag found in Actinobacillus pleuropneumoniae, which is involved in the biosynthesis of autotransporter adhesins mediating adhesion to the host cells, a crucial property for colonization and pathogenesis, has been found to yield homogeneous glycoforms modified with glucose (Glc), either at S or T residues, representing a valuable starting material for further transglycosylation reactions.111 Attempts to engineer yeasts to produce defined glycosylated proteins have been conducted with low success in a mutant strain of Saccharomyces cerevisiae.112 O-linked113 and S-linked114,115 modifications have also been proposed as viable alternatives to N-glycosylation.
Chemical manipulations of certain sugar residues can be used alone or in association with enzymes to achieve site-selective bioconjugation. For instance, cis-diol on sialic acid (NeuNAc) can undergo selective oxidative cleavage to aldehyde under mild conditions, offering a functional group for protein modification.116
The use of the enzymes involved in post translational modifications and the corresponding substrate mimetics have been combined for chemoenzymatic remodelling of glycoproteins (Scheme 5).117 Initial heterogeneous glycoform mixtures are treated with an endoglycosidase (“Endo”) to trim off the variable portions of the oligosaccharides attached to the first GlcNAc residue of the N-glycosylated sites. Subsequent enzyme-mediated transfer of a synthetic glycan, in the form of activated glycan oxazoline, to the GlcNAc moiety by an endoglycosynthase mutant provides a homogeneous glycopeptide or glycoprotein.118 Different endoglycosidases (EndoH or EndoS)119,120 with complementary potential have been demonstrated to be able to degrade heterogeneous glycans to a single N-linked GlcNAc residue. This has been extended by transglycosylation using either Endo-M, (from Mucor hiemalis),121 Endo-A (from Arthrobacter protophormiae),122 Endo-CE from Caenorhabditis elegans,123 and Endo-BH from alkaliphilic Bacillus halodurans C-125.124 Enzymatic installation of defined glycans at a predetermined glycosylation site of peptides during the solid-phase peptide synthesis (SPPS) has allowed the preparation of libraries of homogeneous glycoconjugates.125
Selective post-translational modifications, such as glycosylation, occur in the majority of mammalian proteins. Mammalian cell lines can be re-engineered to express glycoforms which can be used for further glycan remodelling.
For example, in the so called GlycoDelete approach126 an endoT from the fungus Hypocrea jecorina was first targeted to the Golgi apparatus of 293SGnTI(−) cells, human embryonic kidney (HEK) 293S cells which were engineered by deletion of GnTI encoded by the gene MGAT1 to produce glycoproteins bearing Man5GlcNAc2 N-glycans (293SGnTI(−) cells). The attained GlcNAc N-glycan ‘stumps’ were then selected by specific lectins and finally modified by galactosyltransferases and sialyltransferases. By this approach, instead of dozens of different glycoforms normally produced by mammalian cells, glycoproteins incorporating primarily a Gal-GlcNAc disaccharide or its α-2,3-sialylated trisaccharide derivative and some of the monosaccharide intermediate were obtained.
Release of the variable oligosaccharides linked at the conserved N297 of antibodies has also been used to next remodel the glycan or chemically modify it for incorporation of small molecules.127 In some examples, sugar analogues can be efficiently incorporated into a protein, when incubated in the cell media leading to modified glycosylation patterns.128
3. Site-selective bioconjugate medicines
3.1 Half-life extension of protein therapeutics
Protein therapeutics are typically administrated by an invasive injection route, and the patient compliance is often an issue. In addition, the cost for the production of protein therapeutics is typically high, and frequent injections inevitably increase the total cost of the treatment. Many efforts have been devoted to the development of strategies for the extension of the circulating half-life.129 The clearance of protein therapeutics occurs primarily via renal filtration, but is also related to their potential proteolytic degradation, and the potential antidrug immune response. The clearance is dependent on the hydrodynamic size of the protein. Typically, molecules with a molecular weight equal or above 60 kDa are unable to pass through the renal glomerular capillaries into the Bowman's capsule, remaining in circulation.130 Various conjugation strategies have been developed to increase the hydrodynamic size of protein therapeutics, including (i) polymer (e.g. polyethylene glycol) conjugation; (ii) fatty acid conjugation; (iii) IgG or Fc conjugation; and (iv) albumin conjugation.7,131A central question associated with any of the above strategies is how to maximally maintain the protein activity after conjugation. Here site-selective bioconjugation appears to be the logic choice.
The conjugation of a large polymer to a therapeutic protein can increase the hydrodynamic size of the resulting conjugate, and eliminate the potential renal clearance via filtration. Polyethylene glycol (PEG) is the most extensively applied polymer in many bioconjugate therapeutics. The large flexible PEG can potentially interfere with the protein binding to its target. Therefore, the regioselectivity of conjugation is critical to maximally maintain the protein activity. Some representative examples will be discussed below.
Under denaturing conditions and in the absence of reducing agents, thiol PEG has been shown to target selectively C17 rather than the other four cysteine residues involved in two disulfide bridges.133 After conjugation, the protein was refolded by eliminating the denaturant through dialysis or gel chromatography.
At a near-neutral pH, maleimide–PEG has also been proven to be almost exclusively attached to the thiol group of C17.134 In another example, C residues were introduced by mutagenesis for conjugation to maleimide–PEGs.135 Good selectivity was achieved, due to the fact that C17 is more buried in comparison to the bioengineered ones. However, major drawbacks associated with cysteine modifications are the impact on the 4 helix structure of G-CSF, which is stabilized by disulfide bonds and it is known to be essential for the therapeutic activity, the need for renaturation, and the tendency to form aggregates following C modification. In addition, cytokines are typically very sensitive to changes in the structure with respect to immunogenicity. Therefore, other approaches have been pursued.
K is another common conjugation site of choice for PEGylation. However, all 4 K residues of G-CSF are located around the key receptor binding regions. Thus, targeting these sites result in significant reduction (often by 10–100 fold) of the bioactivity136 and an increased amount (and cost) of drug is required to achieve the same benefit. Consequently, alternative strategies are preferred.
N-terminal M has an α-amino group with pKa around 7.6–8.0.137 In contrast, the pKa value of the ε-amino group of K is 10–10.2. The different pKa values can be utilized for the site-specific PEGylation. Scientists at Amgen studied this route by acylation with carboxymethyl-N-hydroxysuccinimidyl (NHS) ester functionalized mPEG, or by reductive alkylation by mono-functional mPEG propionaldehyde (Scheme 6). Both PEGylated G-CSF showed excellent regiospecificity, and well maintained physical and biological properties compared to the parent G-CSF.
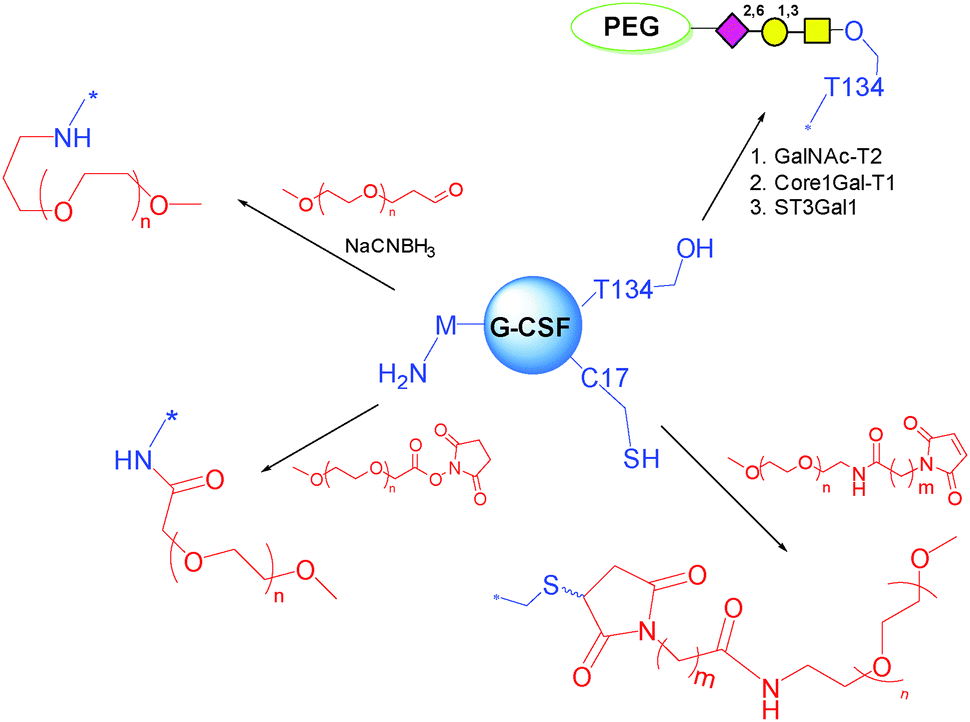 | ||
| Scheme 6 Methods for PEGylation of G-CSF. | ||
Importantly, the alkylated conjugate showed 4 times slower aggregation rate than the corresponding acylated conjugate, due to the unaltered PI value.138 It was selected for the further development into Pegfilgrastim, which was approved by FDA in 2002 (Neulasta®).
Recently, it has been shown that the mPEGs of high molecular weight demonstrated better N-terminal site-specific selectivity, separation purity and improved production yield.139
T134 is the naturally occurring glycosylation site on G-CSF. This site is remote from the active one, and has been targeted for PEGylation. Based on this concept, scientists at Neose Technologies Inc. have developed an excellent approach called GlycoPEGylation (Scheme 6). The method involves a sequence of enzymatic GalNAc O-glycosylation at specific S and T residues of recombinant aglycosylated proteins, followed by enzymatic transfer of Gal and NeuNAc bearing a 20 kDa PEG to the initially introduced GalNAc.51
Teva Biopharmaceuticals has developed this product as Lipegfilgrastim or Lonquex®, which has been licensed by EMA in 2013, and marketed in Germany. PEGylated filgrastim has a human half-life of 15 to 80 hours, much longer than the parent filgrastim (3–4 hours). Therefore, it can be dosed once-per-chemotherapy cycle administration instead of the daily injection of filgrastim. In addition, patients dosed with the PEGylated form also observed lower incidence of febrile neutropenia than patients receiving filgrastim. Overall, the PEGylated filgrastim demonstrated superior efficacy, safety profile, and also offered convenience of administration.140,141 It has received great uptake by physicians. In 2014, the global sales of Pegfilgrastim topped $5.9 billion.
Pegasys® was prepared by reacting IFN-α 2a with 40 KDa 2-branched mono-methoxypoly(ethylene glycol)-succinimidyl carbonate (mPEG) in sodium borate buffer (pH 9) (Scheme 7A).144 The position of PEGylation was determined by isomer separation, peptide mapping, sequencing and mass spectrometry.
 | ||
| Scheme 7 PEGylation of different residues of IFN. | ||
Interestingly, 96% PEGylation was on K31, K121, K131, and K134 among total 11 lysines. In an early study, the PEGylation with 5 kDa PEG resulted in isomers at all 11 lysines. Steric hindrance appears to be the key driver to the regioselectivity. It is also worth to note that the N-terminal cysteine does not appear to be a conjugation site, perhaps due to less nucleophilicity and higher steric hindrance. This conjugate has ∼7% of native IFN activity as tested in vitro. On the other hand, the in vivo efficacy was markedly improved due to the sustained exposure. The compound was approved by FDA in 2002, and currently marketed by Genentech-Roche.
Scientists at Enzon investigated PEGylation of interferon-α 2b with succidimidyl carbonate PEG at various pHs (Scheme 7B). It was found that PEG preferentially attached to H34 when the reaction was performed at pH 6.5.145 The regioselectivity on imidazole was also determined by NMR, and the N1 position was identified as the attachment point. A 12 kDa PEG was selected for further development. The H34 PEGylated IFN-α 2b was found to be stable at pH 6.8 for 1 month at room temperature. However, the final product was formulated in lyophilized powder, which needs to be reconstituted prior to injection. This conjugate was approved by FDA in 2000, and was marketed as PEG-Intron®.
PEG-Intron® and PEGasys® have been compared extensively and the published data showed numerous interesting features in terms of efficacy, pharmacokinetic, safety, and cost.146 In particular, PEGasys exhibits a more prolonged pharmacokinetic, a lower administration dose and a better cure rate for hepatitis C compared to PEG-Intron®.
Balan et al. at University of London developed an appealing strategy for the site-selective PEGylation using native disulfide bonds as the attachment points without the aid of protein re-engineering (Scheme 7C).147 The conjugation inserted a 3-atom bridge between two cysteines, and therefore the tertiary structure was maintained. In the PEGylation of IFN, both disulfides were labelled, giving a mixture of 2 regioisomers and the double PEGylated IFN. The mono-PEGylated IFN was isolated, and showed >50% activity retention in in vitro assay. The compound has been planned to enter into clinical trials.
In contrast to PEGylation of IFN-α, PEGylation of IFN-β 1a at the N-terminal methionine with 20 kDa mPEG-O-2-methylpropionaldehyde gave nearly the single N-terminal regioisomer (Scheme 7D).148 The resulting conjugate (Plegridy®) fully retained in vitro antiviral activity. This clearly supports the importance of modifying sites distal to receptor binding site(s).149 This biotherapeutic has been approved by FDA in 2014 as a multiple sclerosis treatment.
Xu et al. at Amgen explored the utility of the cysteine mutation approach for FGF21 PEGylation.151
The approach utilizes protein engineering to incorporate free cysteines at strategically attachment points. The selection of multiple positions for maleimide-mediated PEGylation enabled to systematically develop the structure–activity relationship. In this study it was found that the conjugation site is relevant to the conjugation efficiency and activity. Furthermore, the study unveiled a seemingly correlation between the PEGylation site and vacuole formation potential. Potential vacuolization in kidneys is considered as a safety concern, and would potentially lead to kidney failure. FGF21 aims to be a chronic treatment for diabetic patients. Hence accumulation of PEG associated vacuoles needs careful monitoring in the target population. The study suggested that certain PEGylation sites have non-detectable vacuolization with respect to the moderate vacuolization observed for other sites. Overall, the cysteine mutation method has flexibility and efficiency to enable an evaluation of various sites for PEGylation. Noteworthily, FGF21 has an endogenous disulfide, therefore the newly introduced cysteine should avoid disulfide scrambling (Scheme 8). This is perhaps true to many proteins with endogenous disulfides.
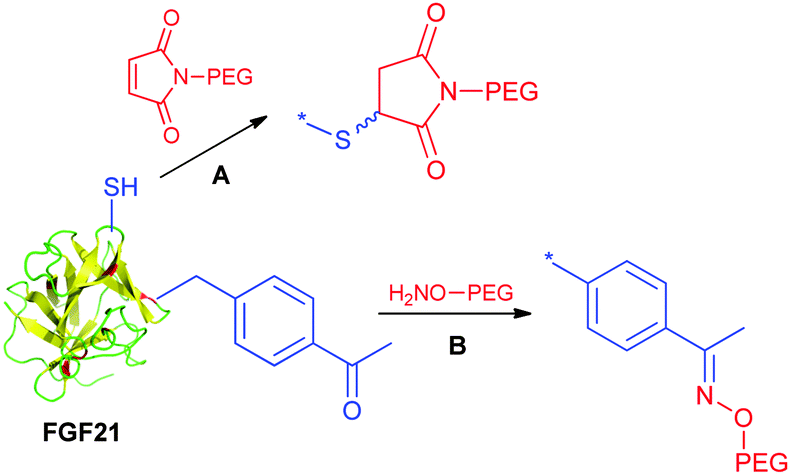 | ||
| Scheme 8 Cysteine mutation approach for FGF21 PEGylation. | ||
Song et al. recently developed recombinant human FGF21 variants by strategically introducing cysteine residues via site-directed mutagenesis. Their approach was based on a solid-phase nickel affinity PEGylation strategy, where the engineered surface exposed cysteine residues of immobilized proteins were used as a platform for efficient and site-selective conjugation with PEG–maleimide (Scheme 9). This method offered an improved PEGylation yield and a streamlined purification process.152 Incorporation of uAAs has also been exploited by Ambrx for selective PEGylation of FGF21 (Scheme 8B).153 Using the homology modeling and structure-based design, specific sites were identified in human FGF21 for site-specific PEGylation ensuring the preservation of receptor binding regions. The in vitro activity of the PEGylated FGF21 analogs was dependent on the site of PEG placement and corresponded to the one anticipated by the binding model. Site-specific PEGylated analogs demonstrated in vivo dramatic increase of the circulating half-life and enhanced efficacy.
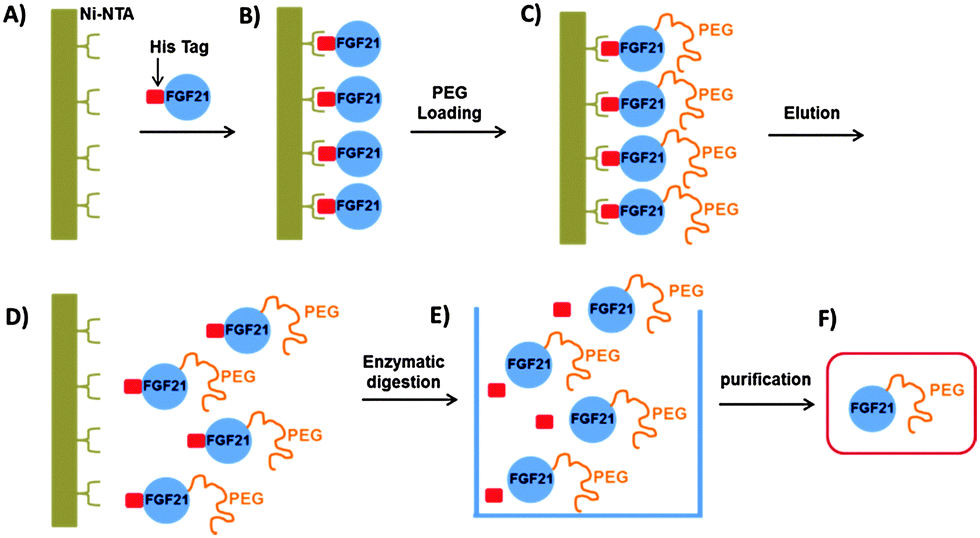 | ||
| Scheme 9 Solid-phase nickel affinity PEGylation strategy. Adapted from ref. 152. | ||
3.2 Modulating the half-life of hormones
Site-selective modifications have also been applied to tune the pharmacokinetic properties of hormones, such as insulin and glucagon, involved in the control of the glucose levels in the bloodstream. Insulin, composed of two polypeptide chains (A and B) joined by disulfide bridges, is a key diabetic treatment. One limitation in the use of insulin is its short half-life in the circulatory system. Glycosylation could be one of the possible strategies to achieve long-acting insulin formulations. However the expression system in mammalian cells of glycosylated forms is highly uncontrolled in terms of the structure of the glyco chain, glycosylation site, and number of glycans. To ensure controlled site-specific glycosylation a chemoenzymatic method was developed,156 involving the introduction of mono-, di-, and trisialyloligosaccharides to mutant insulins through enzymatic reactions. Sugar chains were first attached by transglutaminase (TGase) at an accessible N-terminal glutamine residue of the B-chain, and then sialylated by α-2,6-sialyltransferase (R2,6-SiaT). Sia2,6-di-LacNAc-Ins(BF1Q) and Sia2,6-tri-LacNAc-Ins(B-F1Q), displaying two and three sialyl-N-acetyllactosamines, respectively, were administered to hyperglycemic mice. Both branched glycoinsulins showed prolonged glucose-lowering effects compared to native or lactose-carrying insulins, showing that NeuNAc is important in obtaining a prolonged effect. Sia2,6-tri-LacNAc-Ins(B-F1Q) (Chart 1), in particular, induced a significant delay in the recovery of Glc levels and was elected as the most efficacious form to prevent insulin shock. This effect was explained with the multivalent effect of the sialooligosaccharides on the stability of insulin in the blood stream and low affinity for the insulin receptor.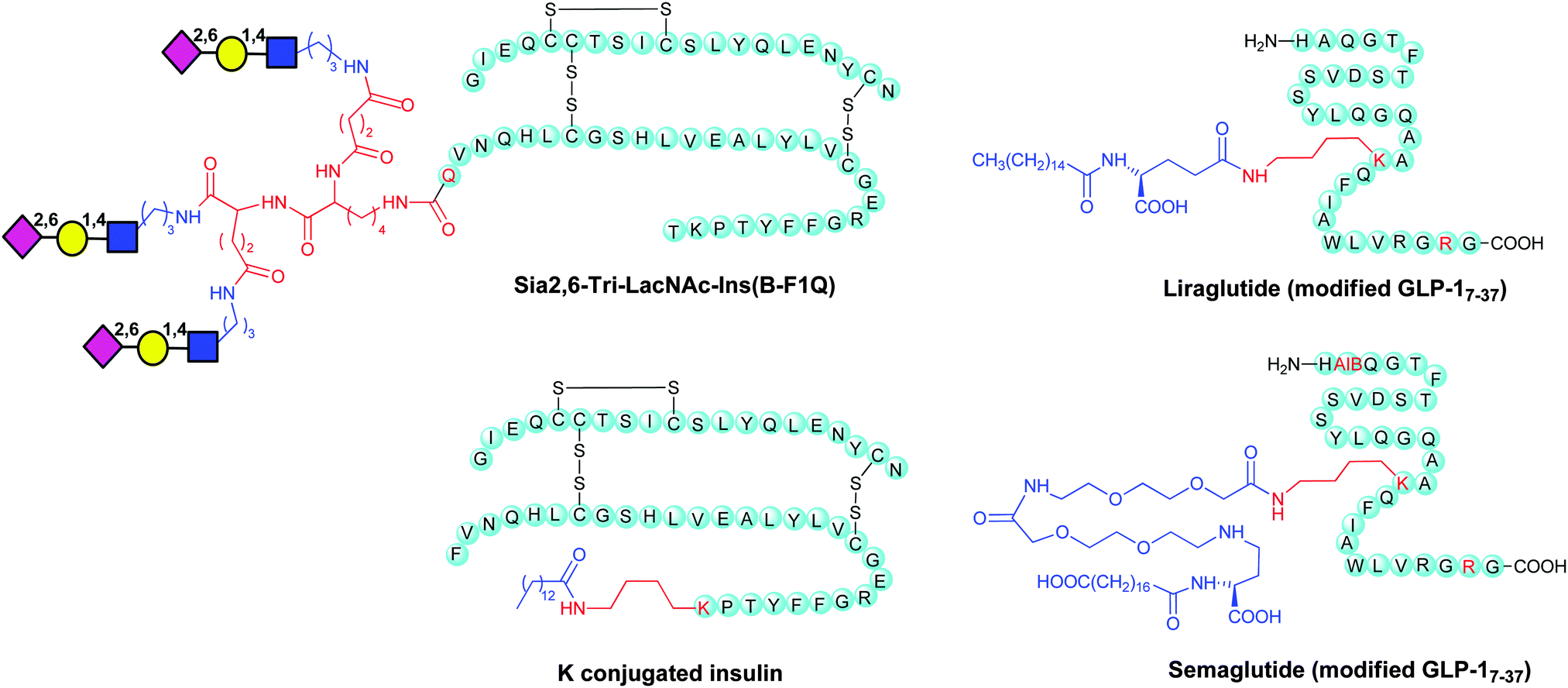 | ||
| Chart 1 Examples of modified insulin and GLP-1. | ||
Glucagon-like peptide 1 (7–36) amide (GLP-1) has been attracting considerable attention as a therapeutic agent for the treatment of type 2 diabetes.157 By a glycoengineering strategy glycosylated analogues of GPL-1 with N-acetylglucosamine (GlcNAc), N-acetyllactosamine (LacNAc), and α-2,6-sialyl N-acetyllactosamine (sialyl LacNAc) were chemoenzymatically prepared. Addition of sialyl LacNAc to GLP-1 greatly improved in vitro stability against the proteolytic activity of dipeptidyl peptidase-IV (DPP-IV) and neutral endopeptidase (NEP) as compared to the native type, thus extending the blood glucose-lowering activity in vivo. The di- and triglycosylated analogues with sialyl LacNAc showed further prolonged blood glucose lowering activity.
An elegant example of a combination of bioengineering and chemical modification is given by the synthesis of liraglutide. This is a biologically active medicine that mimics a natural product, the native human Glucagon Like Peptide-1 (GLP-1). GLP-1 is a 30 amino acid peptide hormone, naturally produced by intestinal L-cells, which regulates insulin.
Native human GLP-1(7–37) has a plasma half-life of approximately 2 minutes. Liraglutide (Victoza®) replaced K34 of the native GLP-1(7–37) to R, and attached a 16-carbon fatty-acid chain with a glutamic acid spacer to K26 (Chart 1).
This biotherapeutic has been developed by Novo Nordisk and is currently manufactured using recombinant DNA technology and cultured in Saccharomyces cerevisiae yeast.158
The fatty acid binds albumin in circulation, and avoids fast renal clearance, and protease mediated degradation. This results in an extended plasma half-life of 13 hours, which makes the modified hormone possible for once-daily administration.159 Liraglutide was first licensed by EMA in 2009, and then by FDA in 2010.
Replacement of Q8 in Liraglutide with α-aminoisobutyric acid (AIB) and slight modification of the fatty acid side led to the development of semaglutide (Chart 1), which has improved the circulating half-life suitable for once weekly dosing. It is interesting that it is also pursued as an oral GLP-1 agonist in Phase III clinical trials for type 2 diabetes.7
An approach similar to that employed in liraglutide was pursued by Novo Nordisk for the development a half-life extended insulin (Detemir or Levemir®) by deletion of the B-30 threonine and coupling with a 14-C fatty acid at the C-terminal lysine on the B-chain (Chart 1).160
Cysteine modification has also been exploited to obtain site-specific conjugates of dicoumarol, an oral anticoagulant that interferes with the metabolism of vitamin K (4-hydroxycoumarin) and is known to bind tightly to human serum albumin (HSA). By this method long-acting and highly biologically active GLP-1 derivatives were obtained.161 PEGylation represents another feasible strategy for site-selective modification of hormones. PEGylation of glycosyl modifications of the recombinant form of human TSH, a gonadotropin that stimulates the thyroid gland to secrete thyroid hormones, has also been designed with the scope of prolonging the short half-life of rhTSH in the circulation avoiding a multidose regimen.162 Periodate oxidation of NeuNAc or Gal residues was employed for targeting PEG to the three N-linked glycosylation sites on the protein. Conjugates of different PEG sizes and the number of incorporated copies were screened to eventually identify a 40 kDa mono-PEGylated NeuNAc-mediated conjugate, which exhibited a 3.5-fold more prolonged action than rhTSH in rats, as a 5-fold lower affinity was more than compensated by a 23-fold extension of the circulation half-life.
Incorporation of uAA p-acetylphenylalanine (pAcF) at distinct locations of the human growth hormone (hGH) allowed site-specific PEGylation to produce homogeneous hGH variants. A mono-PEGylated mutant hGH modified at residue 35 demonstrated favorable pharmacodynamic properties in GH-deficient rats.163
3.3 IgG conjugation
IgGs have a long half-life (21 days) due to the FcRn recycling effect and the size. Therefore, many efforts have been directed to genetically fuse Fc to various peptides/proteins to extend the half-life. However, the genetic fusion strategy might not be applicable when therapeutic peptides deriving from synthesis or containing uAAs need to be used. Several chemical fusion strategies have been developed to overcome this limitation. Scientists at Biogen Inc. utilized native chemical ligation (NCL) to chemically fuse a synthetic Atrial Natriuretic Peptide (ANP) to the N-terminus of Fc (Scheme 10).164 ANP is released into circulation by the cardiac muscle when the heart undergoes increased atrial stretching. ANP regulates water and salt excretion, and consequently blood volume and pressure. Fusion of Fc and ANP was designed to enhance the half-life of ANP via FcRn recycling.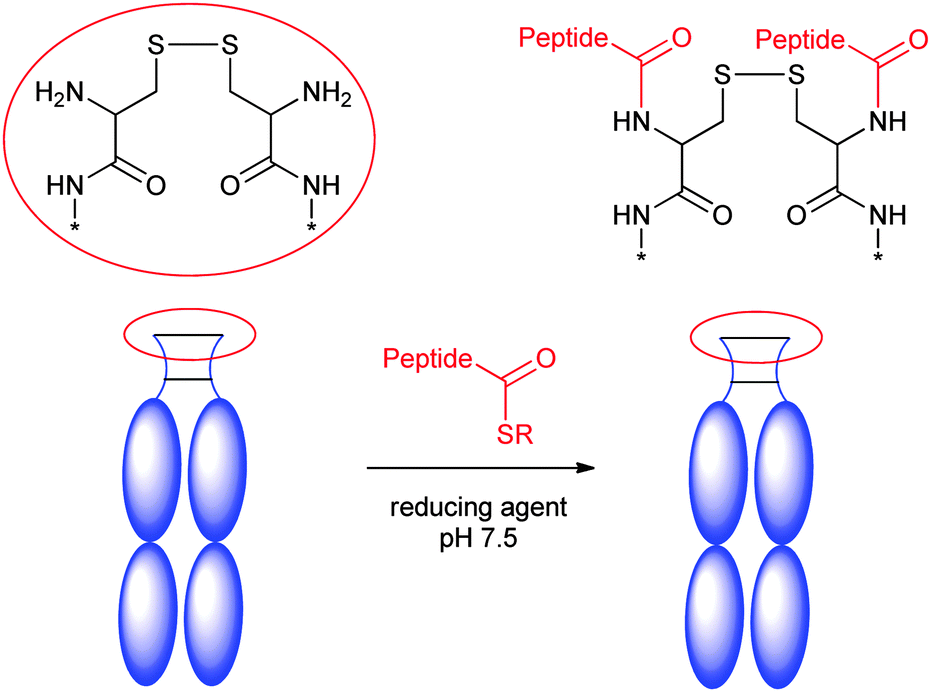 | ||
| Scheme 10 Fusion of Fc with synthetic ANP. Adapted from ref. 164. | ||
Synthetic ANP peptides were synthesized with thioesters at either the N- or C termini, and subsequently linked to the N-terminus of recombinant Fc using NCL. The desired half-life extension was observed in rat.
An interesting approach to prolong the half-life of therapeutics is the conjugation to red blood cells.
Shi et al. used sortase ligation to selectively label the cell surface with various peptides and proteins bearing a LPXTGG tag (Scheme 11). In this example red blood cells function as a carrier and extend the circulation time up to 28 days.165
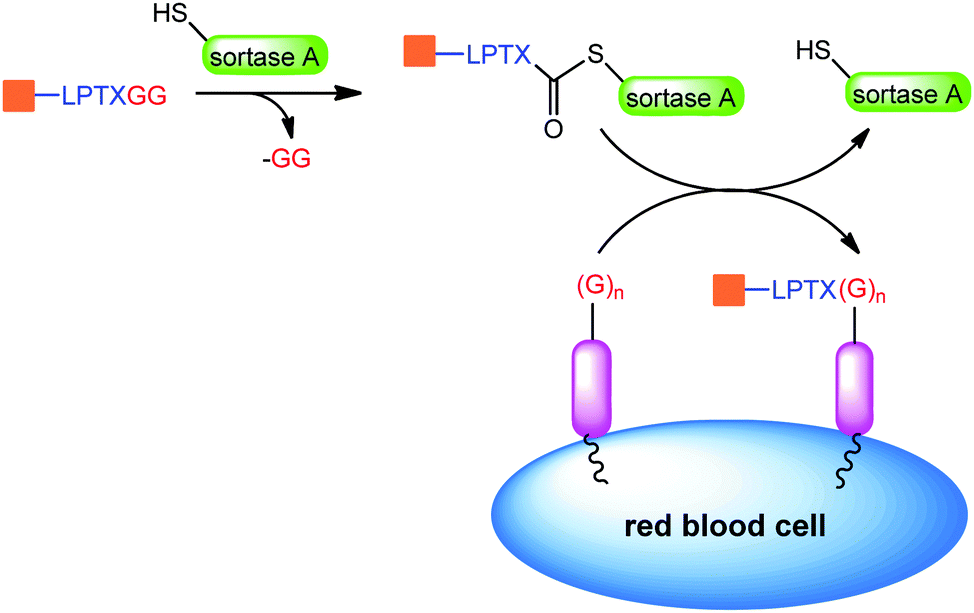 | ||
| Scheme 11 Sortase ligation on red blood cells. Adapted from ref. 165. | ||
4. Antibody–drug conjugates and empowered antibodies
Antibody–drug conjugates (ADCs) have emerged as a new cancer treatment. Currently, there are two approved drugs on the market and about 40 ADCs at various stages of clinical trials.17,18 The promise to revolutionize the cancer treatment also propelled the development of new conjugation techniques.127 Furthermore, some of these methods together with other technologies have been explored to enhance the function of traditional antibodies, generally referred to as empowered antibodies.4.1 Lysine conjugation
Early ADCs were prepared by conjugation at lysines. There are about 90 lysines on IgGs, and selectivity is challenging to be obtained. However, scientists at Immunogen Inc. have proven that the regioselectivity can be controlled consistently. For example, a robust lysine conjugation strategy was developed to attach the maytansinoid drug, DM1 to the humanized monoclonal IgG1 antibody huN901 at lysines.166Interestingly, mapping of the modified residues suggested that the higher selectivity was achieved among lysines with similar surface exposure.166 The strategy was used for the synthesis of Kadcyla®, which was approved by FDA in 2013.167
4.2 Thiol–maleimide addition at interchain cysteines
ADCs typically utilize immunoglobin G (IgG) as the targeting agent. Interestingly, most of the IgG1s have 4 conserved interchain disulfides, which are much more exposed on the surface than other intrachain disulfides. In addition, they are distal from the antigen complementary binding regions (CDRs). Scientists at Seattle Genetics utilized this feature in the preparation of ADCs. The strategy is based on partial reduction of all 4 interchain disulfides, followed by maleimide–thiol addition to introduce payloads (Scheme 12A).168 Cathepsin B–cleavable peptide linkers were used to attach a potent and very stable antimitotic agent monomethyl auristatin E (MMAE) to mAbs, thus ensuring peptidase mediated release of the payload. The strategy usually yielded a mixture of site-controlled conjugates with various drug to antibody ratios (DARs) from 0 to 8. After conjugation, the covalent linkages between light or heavy chains were disrupted, nevertheless the conjugate showed adequate structural integrity.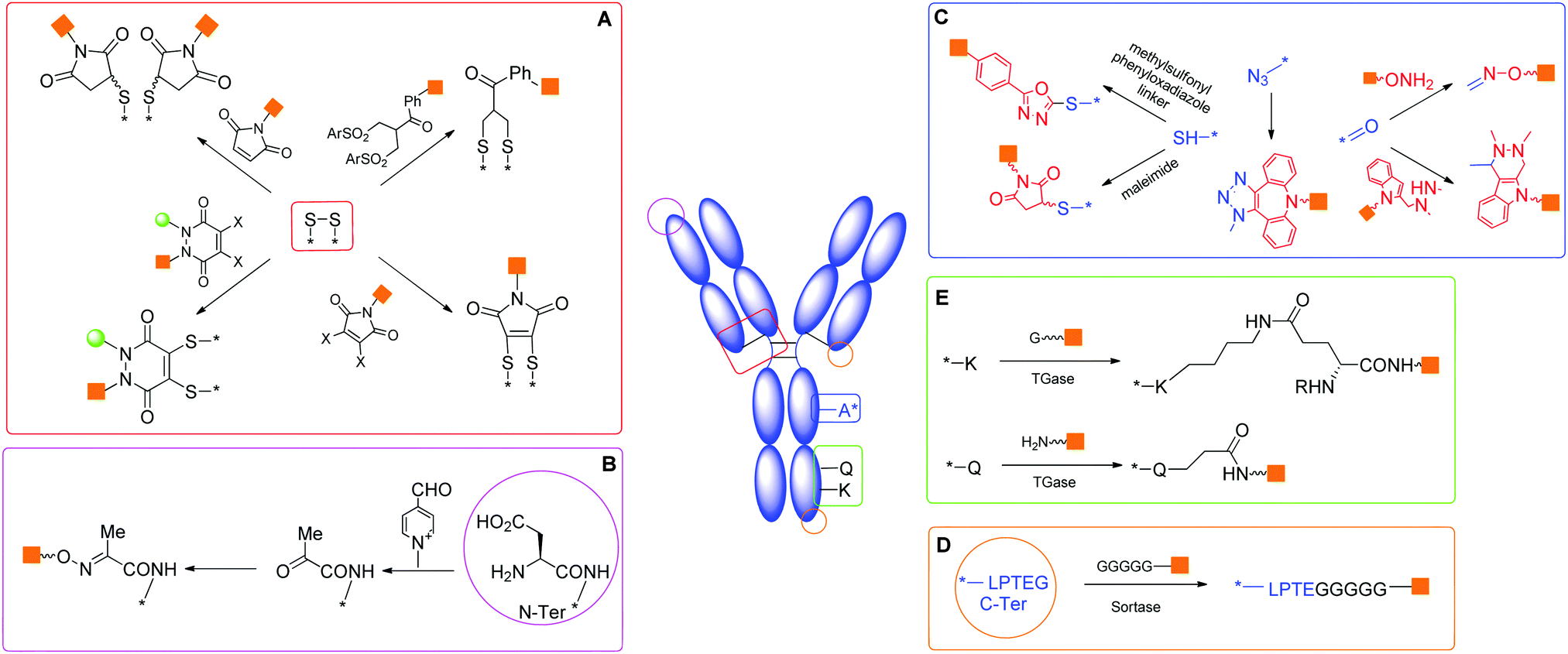 | ||
| Scheme 12 Strategies for modification of mAb or Fab. | ||
Various protocols involving full reduction-partial oxidation, or partial reduction to release interchain cysteines have been evaluated. The resulting conjugates have also been fractionated to enable in vivo evaluation of the impact of drug-to-antibody ratios (DARs). It was found that higher DAR (6 or 8) species have rapid clearance and led to undesired toxicity. DAR 2 or 4 appear to be the optimal ratios.169 The native interchain disulfide bonds as the choice of antibody conjugation sites has been validated in the development of Adcetris® by Seattle Genetics-Takeda.170 The method avoided the need for protein re-engineering to control the regioselectivity.
4.3 Modification of interchain disulfides
It is largely believed that the interchain disulfides might be important to maintain the antibody tertiary structure. Many efforts have been devoted to conjugate at disulfides without breaking the covalent linkages, and several methods have been reported.In 2013, a group at University College London described an elegant approach to prepare the homogeneous antibody fragment (Fab) by conjugation through disulfide bridging (Scheme 12A).171 By using di-bromo or di-thiol substituted maleimides,86,172,173 labelling occurred specifically at the C-terminal disulfide.
The same group recently disclosed the application of a similar protocol to the preparation of full IgG conjugates (Scheme 12A).174,175 Sequential reduction and disulfide bridging gave a mixture of IgG conjugates with a DAR ranging from 0 to 4 together with the half-antibody. In contrast, an in situ protocol avoided the generation of the half-antibody, providing conjugates with a DAR from 0 to 4 in high yield. This group has also developed a similar conjugation reagent, called dibromo-1,2-dihydro-pyridazine-3,6-diones (DBPDs).176
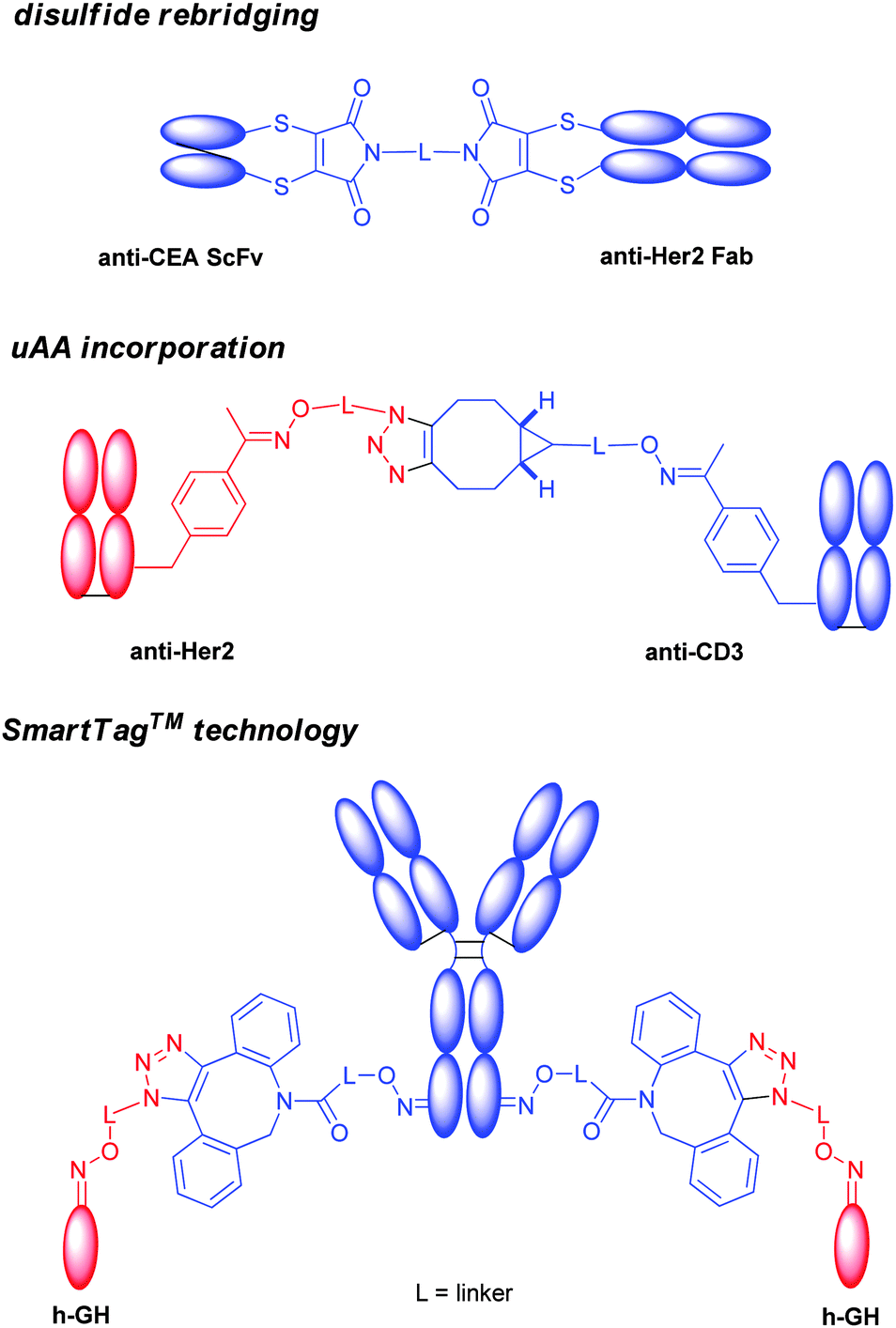
Two nitrogen atoms can carry different orthogonal clickable handles (plag-and-play approach) for sequential introduction of dual payloads (Scheme 12A).177 The reagent was demonstrated to be efficient in the conjugation of the Fab antibody fragment with excellent homogeneity. An anti-Her2 full IgG conjugate was also exploited, and showed good in vitro potency. The rigidity of the maleimide bridge enables the successful detection of antigens with a spin labeled antibody fragment by continuous-wave electron paramagnetic resonance (cw-EPR), therefore immuno-biosensors for EPR-based detection of antibody-antigen interactions (called spinostics) were designed.171 This type of conjugation is currently pursued by Igenica and Thiologics. A disulfide rebridging reagent employing sulfonyl leaving groups was used by Polytherics to develop a homogeneous MMAE-trastuzumab conjugate (Scheme 12A).34 The method gave a homogeneous and stable conjugate with a DAR of 4 as the major product, and together with a small portion of DAR 3 and DAR 5 conjugates.
Anti-Her2 ADC was prepared, showing a clear dose–response based on drug loading with the DAR 4 conjugate having the highest potency in vitro and a much higher efficacy in vivo compared with the lower DAR conjugates. Furthermore, the DAR 4 conjugate demonstrated superior efficacy compared to trastuzumab-DM1 (T-DM1, Kadcyla®), as evaluated in a low HER2 expressing the JIMT-1 xenograft model. Good homogeneity and stability have been demonstrated by various emerging disulfide bridging technologies. More in vivo and clinical evaluation are necessary to better understand the potential of these new technologies.
4.4 N-terminal conjugation
Francis and co-workers utilized the transamination method to chemically introduce a ketone at the N-terminus of a Fab (Scheme 12B).26 This reaction occurs upon exposure to pyridoxal 5′-phosphate (PLP) under mild conditions in buffered aqueous solution. The resulting pyruvamide derivatives can be further elaborated with functionalized alkoxyamines to give oximes. A key advantage of this strategy is its selectivity for the N-terminal amino group with no participation of lysine residues, thus affording antibody conjugates modified in a limited number of locations. Recently, Francis et al. further optimized the condition by using N-methylpyridinium-4-carboxaldehyde instead of PLP for the transamination reaction.178 The condition has been applied to full IgG, and yielded antibody conjugates with 2 or 4 payloads. It is worth noting that the N-terminus is the antigen binding region CDR, and the conjugation at this site might not be compatible to some antibodies.4.5 ADCs with mutated conjugation sites
Many interesting features have been unveiled during this exploration. By comparing the properties of a THIOMAB–drug conjugate directed against ovarian cancer antigen MUC16 with an ADC prepared by conventional cysteine conjugation to MMAE using the same antibody–payload combination, scientists at Genentech found that even though the homogeneous conjugate carried half the amount of cytotoxic payload, it was as potent and efficacious as the conventional ADC in both in vitro and in vivo models.181
Interestingly, the THIOMAB appeared better tolerated by both rats and cynomolgus monkeys than was the conventional ADC, and an improved therapeutic index was achieved using the site-specific conjugation method. This study highlighted that site-selective methods not only provide unique biophysical and therapeutic properties, but also that the actual site of conjugation on the antibody backbone could have a major influence on the in vivo behaviour of an ADC molecule. It was later reported that the highly solvent accessible site of a MMAE-thiotrastuzumab variant rapidly lost conjugated thiol-reactive linker-payloads in plasma due to the exchange with the free thiols of albumin. This exchange led to a lower efficacy in vivo. In contrast, a partial accessible site with a positive charged environment accelerated the succinimide ring opening, and prevented the thiol exchange reaction. This finding provided critical insight into the rational selection of the conjugation site.182 Nevertheless, these features apparently added by site-selective conjugation will need further validation in patients.
Seattle Genetics Inc. reported the use of engineered cysteine mutant antibody for the preparation of site-controlled anti-CD70 ADC. A highly hydrophobic pyrrolobenzodiazepine (PBD) dimer was employed as the cytotoxic payload. It was found that the conjugation at interchain cysteines produced an unacceptable level of aggregation. In contrast, the site-controlled conjugate showed minimal aggregation, accompanied by good efficacy and tolerability in the animal model.183 The method enabled the preparation of an anti-CD33 ADC bearing the pyrrolobenzodiazepine (PBD) dimer payload. The ADC (SGN-CD33A) was the first reported site-controlled ADC entering into clinical trials.184
ADCs derived from THIOMABs typically utilize thiol–maleimide addition. As mentioned above, the conjugate can undergo thiol exchange reactions with free thiols in circulation, leading to premature toxin release.185 In order to overcome this limitation, many strategies have been developed besides the careful selection of the cysteine mutation site. More stable linkages have been generated using methylsulfonyl phenyloxadiazole compounds (Scheme 12C).186 The substitution effect has also been evaluated, and it was found that the introduction of the electro-withdrawing group adjacent to the maleimide nitrogen atom can promote ring opening to prevent the drug detachment.
After formation of an intermediate hydrazonium ion, intramolecular alkylation with the nucleophilic indole of the linker generates a stable C–C bond with final site-specific attachment of the payload.
By means of the recombinant DNA-based eukaryotic protein expression system using Chinese hamster ovary (CHO) cells developed Schultz and coworkers,62 pAF residues were genetically encoded into mAbs against 5T4 (A1) or Her2, and monomethyl auristatin D (MMAD) was subsequently incorporated by oximation (Scheme 12C).190 The resulting constructs with DAR of 2 demonstrated superior in vitro efficacy and selectivity, and in vivo pharmacokinetics and efficacy in rodent models when compared with conventional random cysteine conjugated ADCs with a DAR of 4.
In another study, pAcF was site specifically incorporated at A114 of the heavy chain of an antibody against Her2, and the corresponding ADC was prepared.191 The resulting site specific anti-Her2 ADC exhibited in vitro cytotoxicity and in vivo tumor regression comparable to a control made by random interchain cysteine conjugation. However superior in vitro serum stability and preclinical toxicology profiles in rats were observed.
PAcF has been also used for site-specific incorporation into IgG directed to CXCR4, a protein highly expressed in the majority of metastatic cancers, and conjugated to an auristatin through a hydrolytically stable oxime linkage.192 The resulting homogeneous ADC showed pronounced in vitro cytotoxicity to CXCR4+ cancer cells and eliminated pulmonary lesions from in a lung-seeding tumor mouse model derived from human osteosarcoma cells, without significant off-target toxicity.
Incorporation of pAMF into mAbs has been achieved by a cell-free protein expression system based on a novel variant of the Methanococcus jannaschii tyrosyl tRNA synthetase (TyrRS).67 DBCO–PEG–monomethyl auristatin (DBCO–PEG–MMAF) was coupled to an anti-Her2 IgG bearing pAMFs using strain-promoted azide–alkyne cycloaddition (SPAAC, Scheme 12C). The obtained ADC was proven highly potent in in vitro cellular assays.
The recent production of the clinical amount of an anti-Her2 ADC ARX788 from Ambrx indicated the process scalability for mAbs incorporating uAAs.193
4.6 Enzyme mediated conjugation for ADCs
Antibody modification is achievable also by enzymatic methods. mAbs are typically glycosylated at N297. It was found that N297Q mutation in the hinge region of mAb chCE7 gives origin to the aglycosylated form, increases the flexibility of the C/E loop (Q295–T299), and enhances accessibility of transglutaminase (TGase) mediated conjugation at Q295.46 This strategy was recently sharpened to introduce enzymatically bio-orthogonal thiol or azide linkers onto the mAb trastuzumab for the following attachment of suitable MMAE-derivatives by thiol–maleimide and strain-promoted azide–alkyne cycloaddition, respectively (Scheme 12D).194Homogeneous modified antibody–drug conjugates with DAR 2 were obtained. Alternatively, Rinat-Pfizer incorporated a glutamine tag (LLQG) into a variety of surface accessible regions of an anti-EGFR (epidermal growth factor receptor) IgG1 antibody to find positions that allow efficient conjugation by a transglutaminase from Streptoverticillium mobaraense and maintain the favorable physical properties of antibodies (Scheme 12D).195 By this screening, sites were found conveying optimal conjugation efficiency, while retaining favorable antibody biophysical properties. Further characterization by high-resolution mass spectrometry of an amino-polyethylene glycol-6-propionyl monomethyl auristatin D (AmPEG6-MMAD) conjugate with glutamine tags in the C-terminus of the heavy chain (C16 HC), C-terminus of light chain (C16 LC), and both in the light and heavy chains (C16 LC HC) identified an unintended conjugation site (Q295), which carried approximately 1.3% of the conjugated drug.196 Accordingly, a Q295N mutant was made to eliminate this off-target conjugation, and yielded highly homogeneous conjugates that were more than 99.8% site-specific. The resulting ADC with a DAR of 3.8 is currently moving to clinical trials. Preparation of ADC through the bacterial enzyme sortase A (SrtA) mediated conjugation has also been described (Scheme 12E).197
In one example, the anti-Her2 Fab of the clinically-validated antibody trastuzumab was fused with the plant toxin gelonin.198 LPETG was fused at the C-terminus of the Fab heavy chain, and the toxin was equipped with a Gly2 sequence at its N-terminus, distal to the toxin active site in the C-terminal region. An antibody-toxin fusion was subsequently prepared by SrtA mediated conjugation. Sortase catalysed conjugation is currently explored by NBE therapeutics for the introduction of payloads in a regioselective manner.
4.7 Targeting glycan as the conjugation site
Glycosylation of the Fc region of human IgGs occurs at a conserved N-glycosylation site within the CH2 domain, where glycans are linked to N297. The carbohydrate chain attached at this site is usually comprised of a complex glycan composed of GlcNAc and mannose (Man), and followed by variable addition of Gal, NeuNAc, fucose (Fuc), as well as bisecting GlcNAc residues (Scheme 13). This site can be targeted for conjugation by different approaches. | ||
| Scheme 13 Typical N-glycosylation pattern in human IgG and approaches for its modification. | ||
The process was successfully used to modify three antibodies with different small molecules, including trastuzumab and two cytotoxic agents, with an average loading of ∼1.6 cytotoxic agents per antibody molecule.
Modification of an anti-CD22 monoclonal antibody by the commercially available sialyltransferase ST6Gal1 and CMP NeuNAc with an azide at the C-9 position (N3NeuNAc) enabled the selective insertion of cytotoxic drug doxorubicin bearing dibenzylcyclooctynol (DIBO) via SPAAC (Chart 2).200 The anti-CD22 antibody linked to doxorubicin exhibited dose-dependent cytotoxicity. The conjugated drug was slightly less active than the unmodified form, indicating the efficient cleavage of the hydrazine linkage.
 | ||
| Chart 2 Bispecific constructs. | ||
By these methods, ADCs with a DAR of up to 4 can be achieved. One limitation of these approaches is the incapability to introduce modifications in the portion of antibodies (5–17%) which is usually only mannosylated. To overcome these limits, recently a strategy has been proposed (Scheme 13A) as (i) trimming of all glycan isoforms (complex, hybrid, high-mannose) by an endoglycosidase, which renders available the core GlcNAc; (ii) enzymatic transfer of a Gal residue harboring an azide in the acetamide group for further conjugation; and (iii) the use of copper-free click chemistry with bicyclononyne (BCN), a cyclooctyne with minimal lipophilicity to reduce aggregation.201
The first modification of the glycan pattern was achieved in the anti-neuroblastoma chimeric IgG1 chCE7 by tetracycline-regulated expression in Chinese hamster ovary cells of β-(1,4)-N-acetylglucosaminyltransferase III (GnTIII), a glycosyltransferase catalyzing formation of bisected oligosaccharides.202 An optimal range of GnTIII expression was found for the production of mAb with enhanced antibody-dependent cellular cytotoxicity (ADCC) in vitro. Removal of core Fuc by knocking down α-1,6-fucosyltransferase has also been shown to selectively and significantly augment binding affinity to FcγRIII, and resulted in an 100-fold increase in ADCC activity.203 A humanized and glycoengineered anti-CD20 mAb, GA101 (Obinutuzumab) was developed by glycoengineering the carbohydrates of the Fc region using recombinant glycoengineering antibody production technology (GlycoMAb; Glycart-Roche) to enrich mAb with bisected afucosylated Fc region-carbohydrates.204 It has been recently approved by EMA and FDA for the treatment of chronic lymphocytic leukemia. Humanization of the rat ICR62 antibody by glycoengineering the Fc region to contain bisected afucosylated carbohydrates has also led to the development of GA201,205 a novel anti-EGF receptor (EGFR) monoclonal antibody with enhanced ADCC properties. An approach based on the inhibition of Fuc incorporation into the carbohydrate chains of mAbs by means of sugar mimetics (SEA technology) has been pursued by Seattle Genetics. Enhanced ADCC activity in preclinical models was obtained for some candidates, and SEA-CD40 is in Phase I clinical trials for the treatment of solid tumors.206
Currently a variety of alternative production systems for glyco-optimized proteins, including yeast, duck, rat, algae, moss and tobacco cell lines is available.8 For example, GlycoFi (now a part of Merck BioVentures) has designed and engineered several yeast cell lines (mainly Pichia pastoris) to perform the major steps of the human N-glycosylation pathway. Therefore, the technology provided a general platform to deliver proteins, monoclonal antibodies and derivatives (Fab fragments, Fc fusion proteins, immuno-conjugates) with defined glycans as potential pharmaceuticals.
Biobetter versions of trastuzumab, cetuximab, rituximab and infliximab derived from these technologies are in development. The recent approval in Japan of mogamulizumab (Poteligeo®) for the treatment of relapsed or refractory CCR4-positive adult T cell leukemia–lymphoma represents the first glyco-engineered antibody to reach a major market, and is a milestone in the development of empowered therapeutic antibodies by glycoengineering. Detailed discussion of biotechnological approaches for glycoengineering antibodies is outside the scope of the present review, and for this topic we redirect the readers to recent reviews in the field.8,207 Combination of bioengineering and chemical modification of mAbs is expected to enrich the variety of protein therapeutics.
Remodelling of the oligosaccharides at the N297 residue of antibodies has been pursued as a possible strategy for incorporating drugs at defined positions by chemical or chemoenzymatic modifications.127 A mutant galactose transferase has been developed by Qasba's group to introduce 2-keto modified galactose, which is in turn connected to the payload (Scheme 13B).208 Incorporation of the unnatural sugar 6-thiofucose in the N-glycan has allowed Michael-type addition with cytotoxic molecules (Scheme 13B).209
4.8 Bispecific antibodies
Bispecific antibody is composed of two CDRs from distinct antibodies, and can bind simultaneously two different antigens. Alongside many excellent molecular biology methods, site-selective conjugation offered a rapid and flexible way to assemble this type of format.A group at university college London reported the use of bis-dibromomaleimide for the synthesis of a homogeneous bispecific antibody, by crosslinking an anti-CEA single chain antibody (ScFv) to an anti-Her2 Fab (Chart 2).210
Schultz et al. reported the use of unnatural amino acid modified Fabs for the preparation of a bispecific antibody. PAcF has been site-specifically incorporated into each Fab. Oxime ligation was used to introduce an azido or alkynyl group to each Fab, which was subsequently cross-linked together by copper free click chemistry (Chart 3).211 Recently, comparison of bispecific antibodies composed of anti-Her2 IgG or Fab site-specifically conjugated to anti-CD3 Fab via genetically encoded pAcF showed that different valencies did not significantly affect antitumor efficacy, whereas the presence of an Fc domain enhanced cytotoxic activity, although it triggered antigen-independent T-cell activation.212
 | ||
| Chart 3 Defined glycoconjugate vaccines. | ||
The SmartTag™ technology also found great applicability to crosslink proteins. Aldehyde was introduced site-specifically to a full length human IgG, which was in turn functionalized by a strained alkynyl group (Chart 3). The other partner, the growth hormone (h-GH), was labelled similarly with an azido group. The subsequent copper free click chemistry successfully produced a heterobifunctional protein.213
Schultz et al. also developed a strategy to form tetrameric anti-Her2 Fab.61 Herceptin Fab mutant incorporating pAcF was expressed and conjugated to the toxin saporin (Sap 6), endowed with genomic DNA fragmentation activity, through a bifunctional aminooxy–maleimide linker that was selectively coupled to both the keto group of pAcF in anti-Her2 and the thiol group of cysteine in Sap 6.
The use of a bispecific antibody to simultaneously target CD3 on T cells and tumor-associated antigens to recruit cytotoxic T cells to cancer tissue has been revisited by the same group. A small molecule DUPA binding to the prostate-specific membrane antigen was selectively conjugated to a mutant anti-CD3 Fab at the incorporated pAcF.214 The conjugate proved potent in vitro and in vivo activity (prophylactic and treatment xenograft mouse models) combined with a good serum half-life.
5. Conjugate vaccines
5.1 Conjugation of glycan antigens
Glycoconjugate vaccines represent an important class of pharmaceuticals, which guarantee the prevention and even eradication of bacterial infections, such as pneumonia or meningitis in children.215,216Unconjugated bacterial polysaccharides are T-cell-independent antigens, and are unable to induce a persistent memory response. In contrast polysaccharides covalently linked to proteins bind to polysaccharide-specific receptors on the surface of APCs and, after intracellular processing, can engage T cells following the re-exposition of digested peptides in complex with Major Histocompatibility Complex class II (MHCII).217 Glycoconjugation is, therefore, a fundamental step in order to ensure memory response and boost effect to the vaccine. Recent isolation of carbohydrate specific T-cell clones indicated that the sugar portion of glycopeptides, originated by intracellular digestion of glycoconjugates, may be directly involved in T cell activation.218 This implies that the conjugation site might originate a variety of different glycopeptides, of which the relative efficiency in determining the T-cell response is unknown. It is still not clear whether the T cell activating peptides and glycopeptides coexist within APCs and compete for T cell activation. In both scenarios the connectivity to the protein is a parameter which merits further exploration.219 Current carbohydrate-based vaccines are prepared from heterogeneous mixtures of sugars linked by unspecific methods to the carrier protein giving complex mixtures of products. The immunogenicity of glycoconjugates is influenced by a series of interconnected features, some of which are related to the sugar (length, non-end terminal residues, exposition of charged functional groups, and the number of sugar copies linked to the protein), and others to the conjugation chemistry (type of linker, length, etc.).14 The complexity of randomly prepared glycoconjugates has not made possible to apply a systematic approach to decipher how these parameters influence the activity of this class of biopharmaceuticals and to fully understand their mechanism of action.19
Recently, different methods for the chemical or enzymatic assembly of defined oligosaccharides have rendered feasible the synthesis of complex carbohydrates. A first important proof-of-concept for the sustainability of a vaccine based on synthetic saccharide antigens is witnessed by the release on the market of the Cuban vaccine against Haemophilus influenzae type b in humans.220 While synthetic methods are aiding unveiling the key carbohydrate requirements needed for optimal activity,221 the effects of the conjugation site and linkers have been scarcely explored.
A case study commissioned by WHO estimated a cost of $200–500 million for bringing a new vaccine from the concept stage to the market.222 Interestingly, although a major expense would be the clinical development of the product, relevant factors affecting the cost of glycoconjugate vaccines have been identified in (i) the source of the carbohydrates; (ii) development of a commercially feasible conjugation chemistry process; (iii) manufacturing of the product, which typically include scale-up of production, filling and/or freeze-drying of the biotherapeutic, packaging, storage, and distribution of the finished product.223
The use of site-selective approaches would confer to vaccine conjugates higher batch-to-batch consistency and robust structure-biological activity correlation when compared to classic methods. The better defined chemico-physical characteristics of the vaccines would result in improved quality controls during the process development, and the reduced number of routine controls for product release, giving indisputable advantages in terms of quality standards and manufacturing costs.
5.2 Chemical approaches
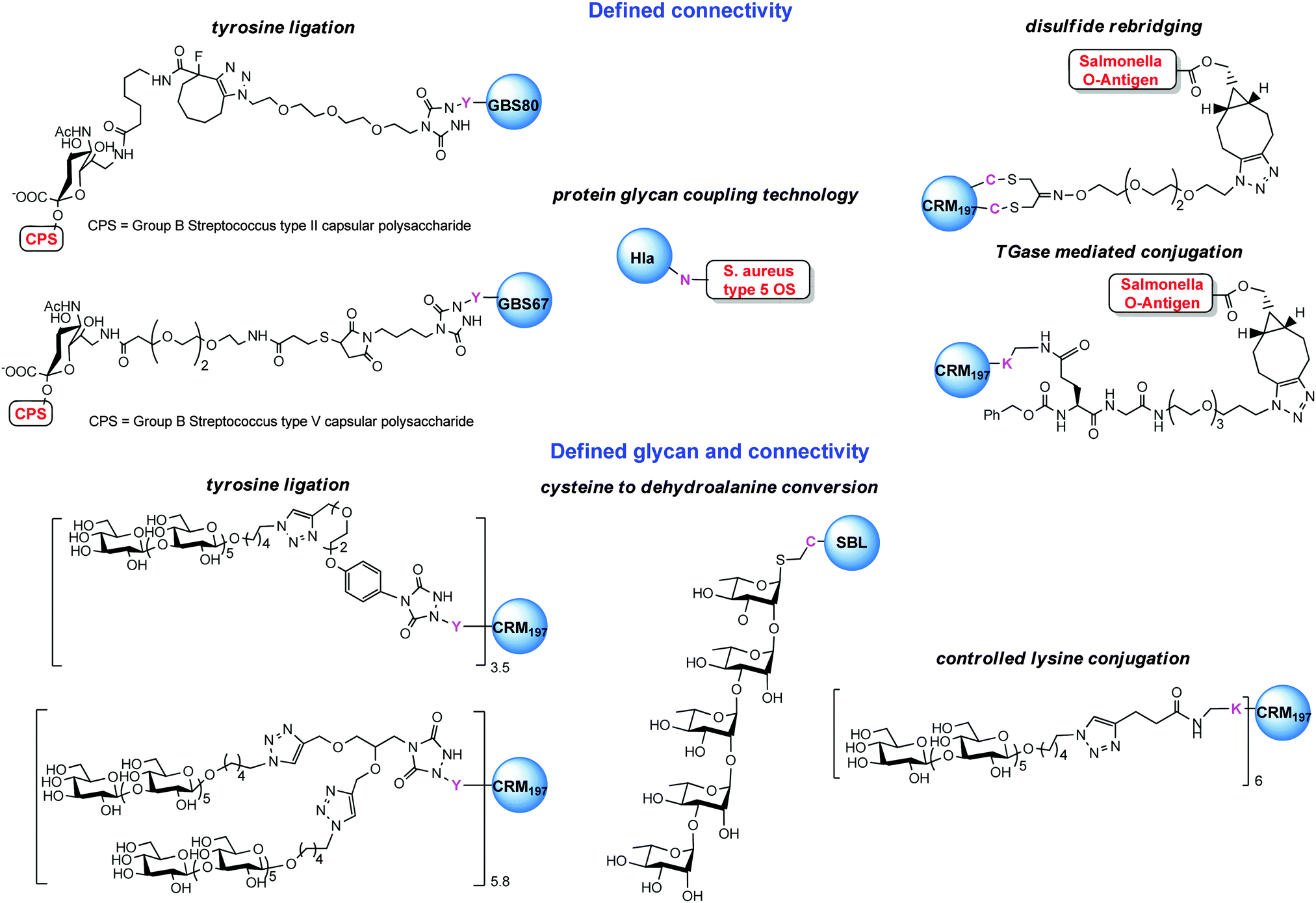
A first strategy for production of homogeneous glycoconjugates was based on the coordination of both carbohydrate synthesis and conjugation methodology.224 This approach features glycosyl disulfides as versatile donors in complex carbohydrate synthesis, providing strategic access to glycosyl thiols for site-selective attachment to the cysteine residues of the protein carrier. By this approach the thiol polyrhamnoside O-antigen of Klebsiella pneumonia was bound through a thioether linkage to the dehydroalanine generated on the subtilisin protein (SBL) mutant S156C (Chart 3). Multimeric copies of sugars were also linked by thiol–ene coupling to the Qb virus-like bacteriophage particle. In these early examples the capability to induce in vivo an anti-saccharide immune response was not examined.In general homogeneous proteins are attractive candidates to have well characterized products and to correlate the immunogenicity with a single attachment site. On the other hand this could lead to an increased dose of administered protein. In some cases it is known that an overdose of carrier protein can suppress the efficacy of following administrations.225 A potential solution to this issue was anticipated by GSK Vaccines (former Novartis Vaccines & Diagnostics) and Novartis Institutes for Biomedical Research (NIBR) in the tyrosine ligation.42
The reaction of triazolinediones with the four more exposed tyrosine residues of the genetically detoxified diphtheria toxin mutant CRM197 was exploited. CRM197 was chosen as protein since it is present in registered vaccines and cannot be easily engineered, and therefore chemical manipulations appeared to be very attractive. The choice of the reaction medium was crucial to direct the reaction away from the lysine residues, and the use of Tris buffer enabled the insertion of an alkyne linker onto Y27, Y46, Y358 and Y380. Following CuACC of a β-(1,3)-glucan hexasaccharide bearing an azide spacer allowed installing defined sugars at predetermined sites. In a following study the construction of a glycoconjugate with double copies of the β-(1,3)-glucan antigen on the same tyrosine residues was accomplished.90
Interestingly, a conjugate with a controlled number of hexasaccharides onto the more surface available lysine residues of CRM197 was attained by careful optimization of a two-step click chemistry based conjugation approach (Chart 3). CRM197 possesses 39 lysine residues, of which 19 are surface exposed. By ESI MS analysis of digested labelled CRM197 it was observed that the reaction of alkyne/azide N-hydroxysuccinimide linker with protein proceed with a pronounced regioselectivity on some of the lysine residues (namely K103, K221 and K242, K236, K498 and K526) as long as not more than six linker moieties were attached to the protein.226 This finding was rationalized by means of computational calculations based on the crystal structure of CRM197. A good correlation of the empirically modified lysines was found with the calculated solvent accessibility of the residues and the amino acidic contour of the modified sites.227 The two constructs having tyrosines modified by one or two β-(1,3)-glucans, respectively, and the glycoconjugate derivatized at the more surface accessible lysines were compared in mice with the same sugar randomly attached to CRM197 and to a CRM197 conjugate of laminarin. The latter is a natural glucan that was previously shown to be highly protective against systemic and mucosal C. albicans infections when conjugated to CRM197. Surprisingly, the tyrosine conjugate exhibited very high immunogenicity which was comparable to the longer and more complex laminarin conjugate but which was not further increased by linking two sugar antigens at the same residues. The defined laminarin conjugate induced the antibodies with the strongest inhibition activity against host cell adherence in the set. This indicated that the efficacy of the glycoconjugates was depending on a balance of sugar loading and conjugation sites.90
When the tyrosine directed approach was next tested with larger charged polysaccharides, such as the capsule of Group B Streptococcus (Streptococcus agalactiae), a pathogen related to neonatal infections, the copper catalyzed click chemistry showed not to be the optimal approach for linking the polymer.228 Further improvements of the conjugation efficiency were achieved by the use of strain promoted click chemistry. Vaccine candidates carrying the capsular polysaccharides from type II and V GBS could be bound with high yields to pilus proteins GBS80 or GBS67 from the same pathogen, thus opening the path to conjugates where the protein is used with the dual role of the carrier and the antigen.
A vaccine obtained by conjugation of PSII to GBS80 (Chart 3) was demonstrated to induce anti-carbohydrate antibodies comparable to the same polysaccharide conjugated to CRM197.229
Anti-glycan and anti-protein antibodies were effective in inducing bacterial killing in vitro of strain expressing either the PSII capsule or GBS80, and conferred protection to the offspring of the vaccinated mice, indicating that this type of vaccine can be used for maternal immunization in order to prevent infections of newborns. Likewise, a glycoconjugate made combining the GBS67 pilus protein and PSV capsular polysaccharide was proven effective in mice.230 In this study a combination of tyrosine ligation and thiol–maleimide addition enabled the preparation of an efficacious vaccine, avoiding the production of anti-linker antibodies.
Selective conjugation to protein as a carrier and an antigen appears to be very appealing for the development of future vaccines, since the repetitive use of classic proteins in vaccination schedules, which nowadays require different doses of the same vaccine and concomitant administration of multiple vaccines could result in diminished response against the antigen.225 Recently, a Cbz-Gln-Gly (ZQG) linker bearing azide was seen compatible to microbial transglutaminase (mTGase) catalyzed lysine modification.227,230 Control of the pH enabled us to achieve selectivity at K37/39 of CRM197 (Scheme 14). Extended reaction time and more acidic pH led to additional modification of K33. The protein was next coupled to the Salmonella O-antigen to create vaccines with defined connectivity.
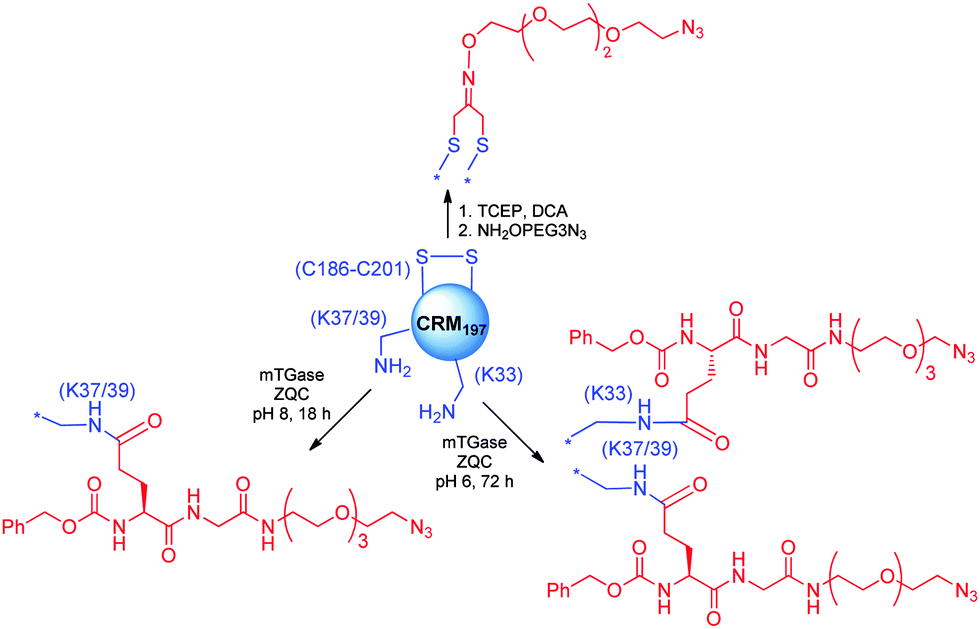 | ||
| Scheme 14 Selective modification of CRM197 by pH-controlled mTGase catalyzed lysine modification and disulfide rebridging with DCA (1,3-dichloroacetone). | ||
CRM197 presents two disulfide bridges: the C461–C471 bond is buried inside the protein, while C186–C201 is well exposed. TCEP reduction of the latter disulfide allowed selective modification of the protein with 1,3-dichloroacetone (Scheme 14), which was used for modification with an aminooxy linker bearing an azide for strain promoted click chemistry with the Salmonella O-antigen.227
The novel constructs (Chart 3) were tested in comparison with a large set of conjugates prepared with multiple copies of the sugar at defined sites. Very importantly, the conjugate at the disulfide showed superior immunological activity than the one at K37/39, clearly demonstrating that the attachment site was impacting the vaccine efficacy. This study highlights the paramount role of the novel selective conjugation methods in understanding the biological functions of modified proteins.
The observation that a lower degree of glycan incorporation might be compensated by the use of longer oligosaccharides, which express multiple copies of the minimal epitope (the glycan portion responsible for raising functional antibodies),231 let us foresee that a balance of defined attachment sites and optimized saccharide length could give rise to effective homogenous vaccines.
5.3 Bioengineered glycoprotein vaccines
The protein glycan coupling technology developed by GlycoVaxyn has recently found application in the delivery of a series of glycoconjugate vaccine candidates. Genes encoding S. aureus capsular polysaccharide (CP) biosynthesis PglB, and a protein carrier (detoxified Pseudomonas aeruginosa exoprotein A or S. aureus α toxin Hla) were coexpressed in E. coli. Recombinant proteins N-glycosylated with S. aureus serotype 5 or 8 CPs were purified from E. coli.232 Rabbits and mice immunized with the glycoprotein vaccines produced antibodies that were active in vitro in functional assays. Active and passive immunization strategies targeting the CP protected mice against bacteremia, and vaccines targeting Hla protected against lethal pneumonia. The CP–Hla bioconjugate vaccine (Chart 3) protected against both bacteremia and lethal pneumonia, providing broad-spectrum efficacy against staphylococcal invasive disease.The same technology has been applied to design bioconjugate vaccines for the prevention of bacillary dysentery in children caused by Shigella dysenteriae, Shighella flexneri and Shighella sonnei. A Phase I trial of a monovalent vaccine against S. dysenteriae O1 has been completed in Switzerland, while Phase I development of the vaccine against S. flexneri is underway in the US.233
Not all the antigenic bacterial cell surface polysaccharides are accessible to the bacterial oligosaccharyltransferase PglB. This is the case for the Vi antigen of Salmonella enterica serovar Typhi, consequently the glycoengineering of vaccines against typhoid fever is not feasible. To circumvent this limitation, the O-antigen of the E. coli O121 wbqG mutant was used to express a Vi-like polysaccharide which showed cross reactivity with antibodies raised against the Vi polysaccharide.234
Interestingly, while the mutant O-antigen structure could be efficiently transferred to acceptor proteins using the bacterial N-glycosylation system, the immunogenicity of the resulting conjugates against S. enterica was very poor. This indicated that a different epitope was expressed in E. coli, suggesting that the oligosaccharide chain length was too short in order to induce anti Vi antibodies. In general efficiency of the oligosaccharide transfer by PglB and the number of sugar moieties incorporated into the protein and polymerized within the O-antigen chain may currently represent limiting factors for this technology. The same approach has been recently shown applicable for the preparation of diagnostic tools for pathogen detection. For instance, the structural identity of Yersinia enterocolitica O9 and Brucella abortus O-antigens was exploited to generate magnetic beads coated with recombinant glycoprotein which were used as diagnostics of brucellosis, one of the most common zoonotic diseases with over half a million new cases annually.235 Noteworthily, injection of the glycoprotein into mice generated an IgG response that recognized the O-antigen of B. abortus, although this response was not protective against a challenge with a virulent strain. Similarly a recombinant glycoprotein antigen, an N-formylperosamine O-polysaccharide–protein conjugate (OAg–AcrA) was used for the development of an indirect immunoassay leading to the diagnosis of bovine brucellosis.236 Expression of glycoproteins from E. coli O157, O145 and O121 has enabled also the development of an indirect ELISA (glyco-iELISA) which clearly discriminates between healthy children and patients infected with Shiga toxin-producing E. coli (STEC), a life-threatening condition characterized by hemolytic anemia, thrombocytopenia and acute renal failure.237
This technology could, therefore, also provide diagnostics for clinical testing of carbohydrate-based vaccines.
5.4 Chemoenzymatic assembly of glycoconjugate vaccines
Strategies for site selective conjugation of defined glycans have been proven to be crucial tools also towards the development of an anti HIV vaccine. HIV-1 utilizes a high density of glycans to limit host antibody recognition of proteins. However, the high density limits glycan processing and the resulting oligomannose structures can be recognized by broadly neutralizing antibodies isolated from HIV-1 infected patients.HIV-1 is characterized by an atypical and highly dense glycoprotein envelope which consists of a trimer of a gp120 and gp41 heterodimer. Each gp120 subunit has an average of 25 N-linked glycosylation sites that render it one of the most heavily glycosylated proteins known. The glycans expressed in gp120 are predominantly Man8-9GlcNAc2 structures.238,239
These oligomannose-type glycans form a cluster on the envelope surface, often referred to as ‘the mannose patch’ or ‘intrinsic mannose patch’ (IMP), that is present across all viral clades. Recently the crystal structure of a stabilized Env trimer mimic has been resolved, confirming the close proximity of the N-linked glycans on HIV-1.240 The abundance of oligomannose-type glycans is further increased in the context of the intact trimer and on the virion surface leading to a ‘trimer associated mannose-patch’ (TAMP).241 Although the Env glycosylation takes place using the host cell machinery, protein–glycan and glycan–glycan interaction occurring at the interface of monomers within the trimer create a non-self glycan motif on gp120 which may be a target for vaccine development. Importantly, broadly neutralizing antibodies recognize glycan-reactive quaternary epitopes located primarily in the first, second and third variable regions (V1V2 and V3) of gp120.
In this context, efforts were addressed to the synthesis of defined N-glycosylated V1V2 peptides by a chemoenzymatic method based on the installation of a GlcNAc moiety at the predetermined glycosylation site during solid-phase peptide assembly.
Synthetic glycans, in the form of activated glycan oxazolines, were transferred to the GlcNAc moiety by an endoglycosynthase mutant which controls the formation of the native β-(1,4) glycosidic linkage between the two core GlcNAc moieties.125 By this highly convergent approach the synthesis of 25 V1V2 glycopeptides containing high mannose or complex-type N-glycans was accomplished. Antibody binding studies by SPR elected the insertion of a Man5GlcNAc2 glycan at the N160 position to be essential for PG9 and PG16 recognition (Chart 4). These studies also revealed a critical role of a terminal sialylated complex-type N-glycan at the secondary glycosylation site (N156 or N173) for recognition by PG9 and PG16. A chemoenzymatic glycosylation remodeling method was also applied for the synthesis of selectively fluorinated glycoproteins.242 The chemically assembled fluoroglycan oxazoline was used as a donor substrate for endoglycosidase (ENGase)-catalyzed transglycosylation to a GlcNAc-protein. Interestingly, it was observed that at the C-6 of the 6-branched mannose moiety in the Man3GlcNAc core resulted in significantly enhanced reactivity of the substrate in enzymatic transglycosylation. Fluorinated glycoforms of ribonuclease B (RNase B) synthesized by this approach aided the elucidation of specific carbohydrate–protein interactions with lectin concanavalin A (Con A). 6-OH on the α-1,6-branched Man moiety was demonstrated to be important for Con A recognition.
 | ||
| Chart 4 Design of HIV glycopeptides made by controlled glycosylation. | ||
These studies highlight the potential of well-defined glycoconjugates in deciphering relevant biological functions, and possibly in the selection of vaccine candidates.243
5.5 Adjuvant conjugation to protein antigens
The magnitude and quality of the immune response directed to vaccine antigens can be modulated by a variety of adjuvants. Adjuvants can differ in their mechanism of action, safety, potency, and capacity to elicit different types of immune responses.244 Among the adjuvants, Toll like receptor 2 (TLR2) agonists represent a promising class of molecules, which have showed efficacy and low toxicity in clinical studies.245–247 Moyle et al. described the attachment of synthetic lipopeptides, obtained by linking Pam2- and Pam3Cys to lipid core peptides, to the cysteine residue of a recombinant protein antigen through Michael type addition.248In a follow-up work, the site-specific attachment of three synthetic TLR2 ligands (lipid core peptide (LCP), Pam2Cys, and Pam3Cys) was realized by Native Chemical Ligation (NCL) of α-thioester groups onto engineered protein antigens and the N-terminal cysteine of modified lipid adjuvant peptides (Scheme 15).249 Using this approach, a small library of broadly protective multi-antigenic vaccines against Group A Streptococcus (GAS, Streptococcus pyogenes) was generated, to select the best vaccine candidate. The lipid components favored self-assembly of nanoparticles in PBS.
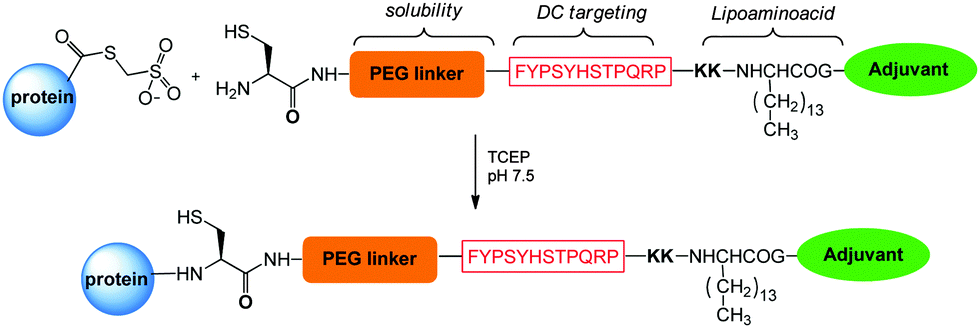 | ||
| Scheme 15 Site selective attachment of adjuvant to recombinant protein antigens via NCL. | ||
These formulations elicited in mice specific anti-antigen antibodies, covering the top 20 circulating strains in developed countries. This study was an important proof-of-concept for subunit protein vaccine antigens modified with adjuvants at precise positions.
6. Cell therapy
Cell therapy has been increasingly used for tissue replacement and regeneration. Overcoming the potential immune response, and the migration of the injected cells to the intended organ (homing) are key to the success of this therapy. Enzymatic edition of cell surface glycans can be used to reprogram cell surface carbohydrate antigens, and modulate the immune response. One paradigmatic example of this approach is the reshaping of blood group carbohydrate antigens to avoid rejection of blood cells during transfusion (Scheme 16).250 | ||
| Scheme 16 Site-selective modifications in cell therapy. | ||
Inadequate homing is considered as the cause of many failures in bone marrow transplantation. It was found that enzymatic enrichment of cell surface sialofucosylated motifs on mesenchymal stromal cells (MSCs) can significantly enhance the homing process by increased recognition of marrow vessels expressing vascular E-selectin, a lectin with high specificity for sialofucosylated determinants. This approach is currently evaluated for the treatment of several rare genetic disorders (Scheme 16).251
This concept has been extended to anti-tumour therapy, where simultaneous generation of fucose deficient endogenous antitumor antibodies and non-fucosylated surface glycans of neutrophils has been proven to augment the activities of cancer vaccines.252
Besides glycan modification, site-selective conjugation can find application in this context. A successful example is the use of the sortase mediated conjugation of peptides or proteins tagged with an LPTEG motif to the exposed N-terminal glycines of components of the cell surface.253 Chimeric antigen receptors (CARs) are composed of an extracellularly displayed targeting moiety specific for a tumor associated antigen, linked to a cytoplasmic signaling domain that mimics the receptor engagement and drives signal transduction. The binding of the target protein on a tumor cell via CAR receptors induces T cell activation, followed by tumor killing via T cell mediated cytoxicity.254 This strategy has encountered the success in clinical evaluation, however a major drawback associated with genetic cell manipulations for therapeutic purposes is the risk of lymphocyte transformation, and even de novo tumor formation.
Ploegh and coworkers proved that the transpeptidase sortase A from S. aureus is suitable for the conjugation of single domain antibodies to activate CD8 T cells (Scheme 16). This study opens up new perspectives in the use of site selective conjugation methods to modify the cell surfaces for therapeutic applications.
7. Conclusions and future outlook
At present mAbs, vaccines and recombinant proteins constitute the top three product categories among the biologic medicines under clinical development.255Bioconjugates bearing features from biomolecules and synthetic medicines hold great potential for the prevention and treatment of various illnesses, such as cancer, metabolic or autoimmune disorders, microbial infections and cancer, and to tackle intractable diseases (Table 2). However, since its first debut in the late 1980s, the development of bioconjugate medicines has been considerably slower than the corresponding monoclonal antibodies or protein therapeutics.
| Name | Therapeutic target | Site-selective conjugation approach | Development phase | Ref. |
|---|---|---|---|---|
| Neulasta®/Pegfilgrastim | G-CSF | pH-controlled modification of N-terminal methionine | Commercial | 138 |
| Lonquex®/Lipegfilgrastim | G-CSF | Glycoengineering | Commercial | 51 |
| Pegasys® | IFN-β | Controlled lysine conjugation | Commercial | 144 |
| PEG-Intron® | IFN-α 2b | Histidine conjugation | Commercial | 145 |
| Plegridy® | IFN-β 1a | N-terminal modification | Commercial | 148 |
| Cimzia®/Certolizumab pegol | anti TNF-α Fab | Bioeengineering and cysteine modification | Commercial | 154 |
| Victoza®/Liraglutide | GLP-1 | Lysine modification | Commercial | 7 |
| Semaglutide | GLP-1 | Lysine modification | Phase III | 7 |
| Levemir®/Insulin Detemir | Insulin | Bioeengineering and C-terminal modification | Commercial | 160 |
| SGN-CD33A | anti CD33A ADC | Cysteine modification of THIOMAB | Phase I | 184 |
| ARX788 | anti Her2 ADC | mAb incorporating uAA | Entering Phase I | 193 |
| anti-EGFR-AmPEG6-MMAD | anti EGFR ADC | mTGase catalysed conjugation of glutamine | Phase I | 195 |
| Poteligeo®/Mogamolizumab | anti CCR4 mAb | Commercial | 8 | |
| Obinutuzumab/GA101 | anti CD20 mAb | Glycoengineering | Commercial | 204 |
| GA201 | anti EGFR mAb | Entering Phase I | 205 | |
| Shighella bioconjugates | Shighella infection | Recombinant glycoprotein | Phase I | 233 |
The manufacturing of bioconjugates is more sophisticated, and includes the protein expression by the biological system, the chemical synthesis of linker and payloads, and the chemical conjugation. Each step requires rigorous quality control to ensure batch consistency and regulatory compliance. The regulatory process in turn involves multiple parties within the agency. The control of site-specificity in conjugation holds great promise to accelerate the development of bioconjugate medicines, because of the potential optimal biological outcome, the ease of manufacturing and regulatory process.
Site-selective conjugation was initially explored in the PEGylation of therapeutic proteins to maximally maintain the potency of the parent protein. The impact of attachment site or linkage on the conjugate physical property was also demonstrated. The development of the antibody–drug conjugate fuelled the growth of many new site-selective bioconjugation methods, and unveiled many new features. For example, the site of choice is relevant to the stability of linkers, and hence impacts the pharmacokinetics and tolerability of the drug. The strategic selection of the attachment point can also enable the incorporation of hydrophobic payloads that would be challenging by other methods. Furthermore, the site controlled conjugation allows the introduction of multiple types of payloads in a defined manner, and potentially offer new therapeutic opportunities.
More recently, site-controlled glycoconjugate vaccines demonstrated outstanding immunological activity with few, short, but defined oligosaccharides. Certain attachment points appear to be more efficacious than others. This exciting progress will potentially accelerate the development of timeline for glycoconjugate to fight various deadly infectious diseases. Certainly, clinical evidence and regulatory success are still needed. We expect that the transition from statistic conjugates to site-selective conjugates will follow a similar path of PEGylation.
Bioconjugate medicine is on the verge of entering a new era with very intense ongoing research activities towards better version of biologics or new classes, including glycoengineered antibodies, bispecific antibodies, and chemically engineered cell therapies. The development will witness significant acceleration with the maturation of technologies, manufactory process, characterization, and regulatory path. However, the realization of the promise requires further development of site-selective conjugation to facilitate the realization of the promise. We believe that the following questions hold priority:
(1) Can a homogeneous conjugate provide adequate biological advantages over the corresponding recombinant (fusion) protein or statistic conjugates?
(2) Can a homogeneous conjugate medicine be consistently prepared through a time and cost effective manufacturing process?
(3) Can we develop proper strategies to manage or minimize potential undesirable properties introduced by new technologies, e.g. immunogenicity, distribution, accumulation and the consequent biological activities of the released payload-linker?
(4) Can we define the optimal application scope for a given method or technology?
(5) Can we develop a proper regulatory strategy for these hybrid biologics to ensure the adequate compliance and efficient clinical and regulatory path?
We believe that the full potential of bioconjugate medicines to improve current therapies and tackle unmet medical needs will be maximally appreciated in due course.
Notes and references
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–883 CrossRef CAS PubMed.
- O. T. Avery and W. F. Goebel, J. Exp. Med., 1931, 54, 437–447 CrossRef CAS PubMed.
- C. H. Fu and K. M. Sakamoto, Expert Opin. Pharmacother., 2007, 8, 1977–1984 CrossRef CAS PubMed.
- A. M. Wu, in Drug Delivery in Oncology: From Basic Research to Cancer Therapy, ed. F. Kratz, P. Senter and H. Steinhagen, 1st edn, 2012, pp. 411–439 Search PubMed.
- I. Sassoon and V. Blanc, Methods Mol. Biol., 2013, 1045, 1–27 Search PubMed.
- R. A. Larson, E. L. Sievers, E. A. Stadtmauer, B. Löwenberg, E. H. Estey, H. Dombret, M. Theobald, D. Voliotis, J. M. Bennett, M. Richie, L. H. Leopold, M. S. Berger, M. L. Sherman, M. R. Loken, J. J. van Dongen, I. D. Bernstein and F. R. Appelbaum, Cancer, 2005, 104, 1442–1452 CrossRef CAS PubMed.
- M. Lorenz, A. Evers and M. Wagner, Bioorg. Med. Chem. Lett., 2013, 23, 4011–4018 CrossRef CAS PubMed.
- A. Beck and J. M. Reichert, MAbs, 2012, 4, 419–425 CrossRef PubMed.
- http://www.drugs.com/stats/top100/2013/sales .
- S. J. Webb, Nat. Biotechnol., 2011, 29, 297–298 CrossRef CAS PubMed.
- S. Jevševa, M. Kusterle and M. Kenig, Methods Mol. Biol., 2012, 901, 233–246 Search PubMed.
- A. Papachristos, N. Pippa, C. Demetzos and G. Sivolapenko, Drug Delivery, 2015, 27, 1–5 CrossRef PubMed.
- F. Mack, M. Ritchie and P. Sapra, Semin. Oncol., 2014, 41, 637–652 CrossRef CAS PubMed.
- P. Costantino, R. Rappuoli and F. Berti, Expert Opin. Drug Discovery, 2011, 6, 1045–1067 CrossRef CAS PubMed.
- A. Donadei, S. Gallorini, F. Berti, D. T. O'Hagan, R. Adamo and B. C. Baudner, Mol. Pharmaceutics, 2015, 12, 1662–1672 CrossRef CAS PubMed.
- K. Zheng, C. Bantog and R. Bayer, MAbs, 2011, 3, 568–576 CrossRef PubMed.
- G. Li, S. Wang, X. Xue, X. Qu and H. Liu, Drug Discoveries Ther., 2013, 7, 178–184 CAS.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- R. Adamo, A. Nilo, B. Castagner, O. Boutureira, F. Berti and G. J. L. Bernardes, Chem. Sci., 2013, 4, 2995–3008 RSC.
- S. J. Hou, R. Saksena and P. Kovac, Carbohydr. Res., 2008, 343, 196–210 CrossRef CAS PubMed.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS PubMed.
- J. Morales-Sanfrutos, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2010, 75, 4039–4047 CrossRef CAS PubMed.
- C.-S. Goh, D. Milburn and M. Gerstein, Curr. Opin. Struct. Biol., 2004, 14, 1–6 CrossRef PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- E. Baslé, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef PubMed.
- R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS PubMed.
- R. Fields and H. B. F. Dixon, Biochem. J., 1968, 108, 883–887 CrossRef CAS PubMed.
- J. M. Chalker, G. J. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- O. Boutureira, G. J. Bernardes, M. Fernandez-Gonzalez, D. C. Anthony and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1432–1436 CrossRef CAS PubMed.
- G. J. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS PubMed.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- R. M. Hofmann and T. W. Muir, Curr. Opin. Biotechnol., 2002, 13, 297–303 CrossRef CAS PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- Q.-Y. Hu and H. Imase, Inter. Pat. Appl., WO 2014/083505 A083501, 2014 Search PubMed.
- F. Bryden, A. Maruani, H. Savoie, V. Chudasama, M. E. B. Smith, S. Caddick and R. W. Boyle, Bioconjugate Chem., 2014, 25, 611–617 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- D. W. Romanini and M. B. Francis, Bioconjugate Chem., 2008, 19, 153–157 CrossRef CAS PubMed.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406–7413 CrossRef CAS PubMed.
- L. H. Jones, A. Narayanan and E. C. Hett, Mol. BioSyst., 2014, 10, 952–969 RSC.
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- Q.-Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827–3832 RSC.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- D. Rabuka, Curr. Opin. Chem. Biol., 2010, 14, 790–796 CrossRef CAS PubMed.
- C. W. Lin and A. Y. Ting, J. Am. Chem. Soc., 2006, 128, 4542–4543 CrossRef CAS PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grunberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- J. M. Antos, G. L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- M. Fernandez-Suarez, H. Baruah, L. Martinez-Hernandez, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef CAS PubMed.
- J. W. Wollack, J. M. Silverman, C. J. Petzold, J. D. Mougous and M. D. Distefano, ChemBioChem, 2009, 10, 2934–2943 CrossRef CAS PubMed.
- S. DeFrees, Z. G. Wang, R. Xing, A. E. Scott, J. Wang, D. Zopf, D. L. Gouty, E. R. Sjoberg, K. Panneerselvam, E. C. Brinkman-Van der Linden, R. J. Bayer, M. A. Tarp and H. Clausen, Glycobiology, 2006, 16, 833–843 CrossRef CAS PubMed.
- R. M. Schmaltz, S. R. Hanson and C.-H. Wong, Chem. Rev., 2011, 111, 4259–4307 CrossRef CAS PubMed.
- G. Grotenbreg and H. Ploegh, Nature, 2007, 26, 993–995 CrossRef PubMed.
- M. Steiner, I. Hartmann, E. Perrino, G. Casi, S. Brighton, I. Jelesarov, G. J. L. Bernardes and D. Neri, Chem. Sci., 2013, 4, 297–302 RSC.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS PubMed.
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182–188 CAS.
- W. H. Zhang, G. Otting and C. J. Jackson, Curr. Opin. Struct. Biol., 2013, 23, 581–587 CrossRef CAS PubMed.
- A. J. de Graaf, M. Kooijman, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2009, 20, 1281–1295 CrossRef CAS PubMed.
- J. A. Johnson, Y. Y. Lu, J. A. Van Deventer and D. A. Tirrell, Curr. Opin. Chem. Biol., 2010, 14, 774–780 CrossRef CAS PubMed.
- K. L. Kiick, J. C. M. van Hest and D. A. Tirrell, Angew. Chem., Int. Ed., 2000, 16, 2148–2152 CrossRef.
- B. M. Hutchins, S. A. Kazane, K. Staflin, J. S. Forsyth, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Chem. Biol., 2011, 18, 299–303 CrossRef CAS PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinhac, B. Felding-Habermannd, V. V. Smiderc and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- K. Wang, A. Sachdeva, D. J. Cox, N. M. Wilf, K. Lang, S. Wallace, R. A. Mehl and J. W. Chin, Nat. Chem., 2014, 6, 393–403 CrossRef CAS PubMed.
- T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssingerm, Bioconjugate Chem., 2015, 26, 802–806 CrossRef CAS PubMed.
- A. R. Goerke and J. R. Swartz, Biotechnol. Bioeng., 2009, 102, 400–416 CrossRef CAS PubMed.
- K. Ozawa, K. V. Loscha, K. V. Kuppan, C. T. Loh, N. E. Dixon and G. Otting, Biochem. Biophys. Res. Commun., 2012, 418, 652–656 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS PubMed.
- X. Li, J. Yang and C. Rader, Methods, 2014, 65, 133–138 CrossRef CAS PubMed.
- D. L. Hatfield and V. N. Gladyshev, Mol. Cell. Biol., 2002, 22, 3565–3576 CrossRef CAS PubMed.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS PubMed.
- P. Agarwal, J. van der Weijden, E. M. Sletten, D. Rabuka and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 46–51 CrossRef PubMed.
- P. Agarwal, R. Kudirka, A. E. Albers, R. M. Barfield, G. W. de Hart, P. M. Drake, L. C. Jones and D. Rabuka, Bioconjugate Chem., 2013, 24, 846–851 CrossRef CAS PubMed.
- P. Thapa, R.-Y. Zhang, V. Menon and J.-P. Bingham, Molecules, 2014, 19, 14461–14483 CrossRef PubMed.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef PubMed.
- T. K. Tiefenbrunn and P. E. Dawson, Biopolymers, 2010, 94, 95–106 CrossRef CAS PubMed.
- C. F. Becker, X. Liu, D. Olschewski, R. Castelli, R. Seidel and P. H. Seeberger, J. Am. Chem. Soc., 2008, 47, 8215–8219 CAS.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141 CrossRef CAS PubMed.
- R. Kleineweischede and C. P. Hackenberger, Angew. Chem., Int. Ed., 2008, 47, 5984–5988 CrossRef CAS PubMed.
- R. S. Swanwick, A. M. Daines, L. H. Tey, S. L. Flitsch and R. K. Allemann, ChemBioChem, 2005, 6, 1338–1340 CrossRef CAS PubMed.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- L. Castañeda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187–8189 RSC.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tørnoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- R. Adamo, Q.-Y. Hu, A. Torosantucci, S. Crotti, G. Brogioni, M. Allan, P. Chiani, C. Bromuro, D. Quinn, M. Tontini and B. Berti, Chem. Sci., 2014, 5, 4302–4311 RSC.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- J. C. van Hest and F. L. van Delft, ChemBioChem, 2011, 12, 1309–1312 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182–2184 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS PubMed.
- A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683–5686 CrossRef CAS PubMed.
- V. W. Cornish, K. M. Hahn and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 8150–8151 CrossRef CAS.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef CAS PubMed.
- G. J. L. Bernardes, L. Linderoth, K. J. Doores, O. Boutureira and B. G. Davis, ChemBioChem, 2011, 12, 1383–1386 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- K. Kodama, S. Fukuzawa, H. Nakayama, T. Kigawa, K. Sakamoto, T. Yabuki, N. Matsuda, M. Shirouzu, K. Takio, K. Tachibana and S. Tokoyama, ChemBioChem, 2006, 7, 134–139 CrossRef CAS PubMed.
- V. S. Terra, D. C. Mills, L. E. Yates, S. Abouelhadid, J. Cuccui and B. W. Wren, J. Med. Microbiol., 2012, 61, 919–926 CrossRef CAS PubMed.
- C. Alaimo, I. Catrein, L. Morf, C. L. Marolda, N. Callewaert, M. A. Valvano, M. F. Feldman and M. Aebi, EMBO J., 2006, 25, 967–976 CrossRef CAS PubMed.
- C. R. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS PubMed.
- D. Bronner, B. R. Clarke and C. Whitfield, Mol. Microbiol., 1994, 14, 505–519 CrossRef CAS PubMed.
- P. D. Rick, G. L. Hubbard and K. Barr, J. Bacteriol., 1994, 176, 2877–2884 CAS.
- M. Wacker, D. Linton, P. G. Hitchen, M. Nita-Lazar, S. M. Haslam, S. J. North, M. Panico, H. R. Morris, A. Dell, B. W. Wren and M. Aebi, Science, 2002, 298, 1790–1793 CrossRef CAS PubMed.
- M. Wacker, M. F. Feldman, N. Callewaert, M. Kowarik, B. R. Clarke, N. L. Pohl, M. Hernandez, E. D. Vines, M. A. Valvano, C. Whitfield and M. Aebi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7088–7093 CrossRef CAS PubMed.
- M. Mally, C. Fontana, S. Leibundgut-Landmann, L. Laacisse, Y. Y. Fan, G. Widmalm and M. Aebi, Mol. Microbiol., 2013, 87, 112–131 CrossRef CAS PubMed.
- A. Naegeli, C. Neupert, Y. Y. Fan, C. W. Lin, K. Poljak, A. M. Papini, F. Schwarz and M. Aebi, J. Biol. Chem., 2014, 289, 2170–2179 CrossRef CAS PubMed.
- N. F. Parsaie, M. Aebi, G. Bernhard and A. D. Frey, Appl. Environ. Microbiol., 2013, 79, 997–1007 CrossRef PubMed.
- A. Faridmoayer, M. A. Fentabil, D. C. Mills, J. S. Klassen and M. F. Feldman, J. Bacteriol., 2007, 189, 8088–8098 CrossRef CAS PubMed.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78–80 CrossRef CAS PubMed.
- H. Wang, T. J. Oman, R. Zhang, C. V. G. De Gonzalo, Q. Zhang and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 84–87 CrossRef CAS PubMed.
- Q. Zhou, J. E. Stefano, C. Manning, J. Kyazike, B. Chen, D. A. Gianolio, A. Park, M. Busch, J. Bird, X. Zheng, H. Simonds-Mannes, J. Kim, R. C. Gregory, R. J. Miller, W. H. Brondyk, P. K. Dhal and C. Q. Pan, Bioconjugate Chem., 2014, 25, 510–520 CrossRef CAS PubMed.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- L. X. Wang, Carbohydr. Res., 2008, 343, 1509–1522 CrossRef CAS PubMed.
- A. L. Tarentino, T. H. J. Plummer and F. Maley, J. Biol. Chem., 1974, 249, 818–824 CAS.
- J. J. Goodfellow, K. Baruah, K. Yamamoto, C. Bonomelli, B. Krishna, D. J. Harvey, M. Crispin, C. N. Scanlan and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 8030–8033 CrossRef CAS PubMed.
- L.-X. Wang, M. Tang, T. Suzuki, K. Kitajima, Y. Inoue, S. Inoue, J.-Q. Fan and Y. C. Lee, J. Am. Chem. Soc., 1997, 119, 11137–11146 CrossRef CAS.
- W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L. X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef CAS PubMed.
- T. Kato, K. Fujita, M. Takeuchi, K. Kobayashi, S. Natsuka, K. Ikura, H. Kumagai and K. Yamamoto, Glycobiology, 2002, 12, 581–587 CrossRef CAS PubMed.
- K. Fujita, H. Takami, K. Yamamoto and K. Takegawa, Biosci., Biotechnol., Biochem., 2004, 68, 1059–1066 CrossRef CAS PubMed.
- M. N. Amin, J. S. McLellan, W. Huang, J. Orwenyo, D. R. Burton, W. C. Koff, P. D. Kwong and L.-X. Wang, Nat. Chem. Biol., 2013, 9, 521–526 CrossRef CAS PubMed.
- L. Meuris, F. Santens, G. Elson, N. Festjens, M. Boone, A. Dos Santos, S. Devos, F. Rousseau, E. Plets, E. Houthuys, P. Malinge, G. Magistrelli, L. Cons, L. Chatel, B. Devreese and N. Callewaert, Nat. Biotechnol., 2014, 32, 485–489 CrossRef CAS PubMed.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- L. Xia, J. M. McDaniel, T. Yago, A. Doeden and R. P. McEver, Blood, 2004, 104, 3091–3096 CrossRef CAS PubMed.
- R. E. Kontermann, Curr. Opt. Biotechnol., 2011, 22, 868–876 CrossRef CAS PubMed.
- M. J. Burne, T. M. Osicka and W. E. Comper, Kidney Int., 1999, 55, 261–270 CrossRef CAS PubMed.
- S. Jevsevar, M. i. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef CAS PubMed.
- K. Welte, J. Gabrilove, M. H. Bronchud, E. Platzer and G. Morstyn, Blood, 1996, 88, 1907–1929 CAS.
- F. M. Veronese, A. Mero, F. Caboi, M. Sergi, C. Marongiu and G. Pasut, Bioconjugate Chem., 2007, 18, 1824–1830 CrossRef CAS PubMed.
- S. Salmaso, S. Bersani, A. Scomparin, F. Mastrotto, R. Scherpfer, G. Tonon and P. Caliceti, Bioconjugate Chem., 2009, 20, 1179–1185 CrossRef CAS PubMed.
- M. S. Rosendahl, D. H. Doherty, D. J. Smith, A. M. Bendele and G. N. Cox, BioProcess Int., 2005, 3, 52–57 CAS.
- S. Bowen, N. Tare, T. Inoue, M. Yamasaki, M. Okabe, I. Horii and J. F. Eliason, Exp. Hematol., 1999, 27, 425–432 CrossRef CAS PubMed.
- T. Kuga, K. Komatsua, M. Yamasakia, S. Sekinea, H. Miyajia, T. Nishia, M. Satoa, Y. Yokooa, M. Asanob, M. Okabeb, M. Morimotob and S. Itoh, Biochem. Biophys. Res. Commun., 1989, 59, 103–111 CrossRef.
- J. E. Frampton and G. M. Keating, BioDrugs, 2005, 19, 405–407 CrossRef CAS PubMed.
- Y. Zhai, Y. Zhao, J. Lei, S. Zhiguo and M. Guanghui, J. Biotechnol., 2009, 142, 259–266 CrossRef CAS PubMed.
- Y. Kosaka, Y. Rai, N. Masuda, T. Takano, T. Saeki, S. Nakamura, R. Shimazaki, Y. Ito, Y. Tokuda and K. Tamura, Support Care Cancer, 2015, 23, 1137–1143 CrossRef PubMed.
- G. Kourlaba, M. A. Dimopoulos, D. Pectasides, D. V. Skarlos, H. Gogas, G. Pentheroudakis, A. Koutras, G. Fountzilas and N. Maniadakis, Support Care Cancer, 2015, 23, 2045–2051 CrossRef PubMed.
- G. Uze, G. Lutfalla and K. E. Mogensen, J. Interferon Cytokine Res., 1995, 15, 3–26 CrossRef CAS PubMed.
- J. J. Marler, J. B. Rubin, N. S. Trede, S. Connors, H. Grier, J. Upton, J. B. Mulliken and J. Folkman, Pediatrics, 2002, 109, E37 CrossRef PubMed.
- P. Bailon, A. Palleroni, C. A. Schaffer, C. L. Spence, W.-J. Fung, J. E. Porter, G. K. Ehrlich, W. Pan, Z.-X. Xu, M. W. Modi, A. Farid, W. Berthold and M. Graves, Bioconjugate Chem., 2001, 12, 195–202 CrossRef CAS PubMed.
- Y.-S. Wang, S. Youngster, J. Bausch, R. Zhang, C. McNemar and D. F. Wyss, Biochemistry, 2000, 39, 10634–10640 CrossRef CAS PubMed.
- G. Esmat, M. El Kassas, M. Hassany, M. Gamil and M. El Raziky, Liver Int., 2014, 34, 24–28 CrossRef CAS PubMed.
- S. Balan, J.-W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–75 CrossRef CAS PubMed.
- D. P. Baker, E. Y. Lin, K. Lin, M. Pellegrini, R. C. Petter, L.-L. Chen, R. M. Arduini, M. Brickelmaier, D. Wen, D. M. Hess, L. Chen, D. Grant, A. Whitty, A. Gill, D. J. Lindner and R. B. Pepinsky, Bioconjugate Chem., 2006, 17, 179–188 CrossRef CAS PubMed.
- A. Basu, K. Yang, M. Wang, S. Liu, R. Chintala, T. Palm, H. Zhao, P. Peng, D. Wu, Z. Zhang, J. Hua, M.-C. Hsieh, J. Zhou, G. Petti, A. J. Xiguang Li, M. Mendez, J. Liu, C. Longley, Z. Zhang, M. Mehlig, V. Borowski, M. Viswanathan and D. Filpula, Bioconjugate Chem., 2006, 17, 618–630 CrossRef CAS PubMed.
- N. Itoh, Cell Tissue Res., 2010, 342, 1–11 CrossRef CAS PubMed.
- J. Xu, J. Bussiere, J. Yie, A. Sickmier, P. An, E. Belouski, S. Stanislaus and K. W. Walker, Bioconjugate Chem., 2013, 24, 915–925 CrossRef CAS PubMed.
- L. Song, Y. Zhu, H. Wang, A. A. Belov, J. Niu, L. Shi, Y. Xie, C. Ye, X. Li and Z. Huang, Biomaterials, 2014, 35, 5206–5215 CrossRef CAS PubMed.
- J. Mu, J. Pinkstaff, Z. Li, L. Skidmore, N. Li, H. Myler, Q. Dallas-Yang, A.-M. Putnam, J. Yao, S. Bussell, M. Wu, T. C. Norman, C. G. Rodriguez, B. Kimmel, J. M. Metzger, A. Manibusan, D. Lee, D. M. Zaller, B. B. Zhang, R. D. DiMarchi, J. P. Berger and D. W. Axelro, Diabetes, 2012, 61, 505–512 CrossRef CAS PubMed.
- N. Goel and S. Stephens, MAbs, 2010, 2, 137–147 CrossRef PubMed.
- A. Nesbitt, G. Fossati, M. Bergin, P. Stephens, S. Stephens, R. Foulkes, D. Brown, M. Robinson and T. Bourne, Inflammatory Bowel Dis., 2007, 13, 1323–1332 CrossRef PubMed.
- M. Sato, T. Furuike, R. Sadamoto, N. Fujitani, T. Nakahara, K. Niikura, K. Monde, H. Kondo and S. Nishimura, J. Am. Chem. Soc., 2004, 126, 14013–14022 CrossRef CAS PubMed.
- T. Ueda, K. Tomita, Y. Notsu, T. Ito, M. Fumoto, T. Takakura, H. Nagatome, A. Takimoto, S. Mihara, H. Togame, K. Kawamoto, T. Iwasaki, K. Asakura, T. Oshima, K. Hanasaki, S. Nishimura and H. Kondo, J. Am. Chem. Soc., 2009, 131, 6237–6245 CrossRef CAS PubMed.
- L. B. Knudsen, P. F. Nielsen, P. O. Huusfeldt, N. L. Johansen, K. Madsen, F. Z. Pedersen, H. Thøgersen, M. Wilken and H. Agersø, J. Med. Chem., 2000, 43, 1664–1669 CrossRef CAS PubMed.
- K. Raun, P. von Voss and L. B. Knudsen, Obesity, 2007, 15, 1710–1716 CrossRef CAS PubMed.
- P. Kurtzhals, S. Havelund, I. Jonassen, B. Kiehr, U. D. Larsen, U. Ribel and J. Markussen, Biochem. J., 1995, 15, 725–731 CrossRef.
- H. Jing, S. Lidan, C. Yingying, L. Zheng, H. Dandan, Z. Xiaoyun, Q. Hai and H. Wenlong, J. Med. Chem., 2013, 56, 9955–9968 CrossRef PubMed.
- A. Park, D. M. Honey, L. Hou, J. J. Bird, C. Zarazinski, M. Searles, C. Braithwaite, J. S. Kingsbury, J. Kyazike, K. Culm-Merdek, B. Greene, J. E. Stefano, H. Qiu, J. M. McPherson and C. Q. Pan, Endocrinology, 2013, 154, 1373–1383 CrossRef CAS PubMed.
- C. Ho, D. Tom, Y. J. Buechler, D. C. Litzinger, Z. Maio, A.-M. H. Putnam, V. S. Kraynov, B.-C. Sim, S. Bussell, T. Javahishvili, S. Kaphle, G. Viramontes, M. Ong, S. Chu, G. C. Becky, R. Lieud, N. Knudsen, P. Castiglioni, T. C. Norman, D. W. Axelroda, A. R. Hoffmane, P. G. Schultz, R. D. DiMarchig and B. E. Kimmelh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9060–9065 CrossRef PubMed.
- A. R. Mezo, K. A. McDonnell, S. C. Low, J. Song, T. J. Reidy, Q. Lu, J. V. Amari, T. Hoehn, R. T. Peters, J. Dumont and A. J. Bitonti, Bioconjugate Chem., 2012, 23, 518–526 CrossRef CAS PubMed.
- J. Shi, L. Kundrat, N. Pishesha, A. Bilate, C. Theile, T. Maruyama, S. K. Dougan, H. L. Ploegh and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10131–10136 CrossRef CAS PubMed.
- L. Wang, G. Amphlett, W. A. Blättler, J. M. Lambert and W. Zhang, Protein Sci., 2005, 14, 2436–2446 CrossRef CAS PubMed.
- U.S. Food and Drug Administration. 2013-02-22. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm340704.htm.
- S. O. Doronina, E. Brian, M. Toki, Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- M. M. Sun, K. S. Beam, C. G. Cerveny, K. J. Hamblett, R. S. Blackmore, M. Y. Torgov, F. G. Handley, N. C. Ihle, P. D. Senter and S. C. Alley, Bioconjugate Chem., 2005, 16, 1282–1290 CrossRef CAS PubMed.
- P. D. Senter and E. L. Sievers, Nat. Biotechnol., 2012, 30, 631–637 CrossRef CAS PubMed.
- F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. Kay, G. W. M. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2014, 3 DOI:10.3038/srep01525.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 CAS.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, S. V. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8183–8783 Search PubMed.
- A. Maruani, M. E. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 31 DOI:10.1038/ncomms7645.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- S. Panowski, S. Bhakta, H. Raab, P. Polakis and J. R. Junutula, MAbs, 2014, 6, 34–45 CrossRef PubMed.
- J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, L. G. D. Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S. F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- S. C. Jeffrey, P. J. Burke, R. P. Lyon, D. W. Meyer, D. Sussman, M. Anderson, J. H. Hunter, C. I. Leiske, J. B. Miyamoto, N. D. Nicholas, N. M. Okeley, R. J. Sanderson, I. J. Stone, W. Zeng, S. J. Gregson, L. Masterson, A. C. Tiberghien, P. W. Howard, D. E. Thurston, C.-L. Law and P. D. Senter, Bioconjugate Chem., 2013, 24, 1256–1263 CrossRef CAS PubMed.
- M. S. K. Sutherland, R. B. Walter, S. C. Jeffrey, P. J. Burke, C. Yu, H. Kostner, I. Stone, M. C. Ryan, D. Sussman, R. P. Lyon, W. Zeng, K. H. Harrington, K. Klussman, L. Westendorf, D. Meyer, I. D. Bernstein, P. D. Senter, D. R. Benjamin, J. G. Drachman and J. A. McEarchern, Blood, 2013, 122, 1455–1463 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- J. T. Patterson, S. Asano, X. Li, C. Rader, I. Barbas and F. Carlos, Bioconjugate Chem., 2014, 25, 1402–1407 CrossRef CAS PubMed.
- P. M. Drake, A. E. Albers, J. Baker, S. Banas, R. M. Barfield, A. S. Bhat, G. W. de Hart, A. W. Garofalo, P. Holder, L. C. Jones, R. Kudirka, J. McFarland, W. Zmolek and D. Rabuka, Bioconjugate Chem., 2014, 25, 1331–1341 CrossRef CAS PubMed.
- T. J. Hallam, E. Wold, A. Wahl and V. V. Smider, Mol. Pharmaceutics, 2015, 12, 1848–1862 CrossRef CAS PubMed.
- A. Flemming, Nat. Rev. Drug Discovery, 2014, 13, 178 CrossRef CAS PubMed.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Trana, Y. Suna, T. Phuong, R. Barnett, B. Hehli, F. Song, M. J. DeGuzmanb, S. Ensari, J. K. Pinkstaff, L. M. Sullivan, S. L. Biroc, H. Chod, P. G. Schultze, J. DiJoseph, M. Dougher, D. Ma, R. Dushin, M. Leal, L. Tchistiakova, E. Feyfanti, H.-P. Gerber and P. Sapra, Proc. Natl. Acad. Sci. U. S. A., 2013, 111, 1766–1771 CrossRef PubMed.
- D. Jackson, J. Atkinson, C. I. Guevara, C. Zhang, V. Kery, S.-J. Moon, C. Virata, P. Yang, C. Lowe, J. Pinkstaff, H. Cho, N. Knudsen, A. Manibusan, F. Tian, Y. Sun, Y. Lu, A. Sellers, X.-C. Jia, J. Ingrid, B. Anand, K. Morrison, D. S. Pereira and D. Stover, PLoS One, 2014, 9, e83865 Search PubMed.
- S. A. Kularatne, V. Deshmukh, J. Ma, V. Tardif, R. K. V. Lim, H. M. Pugh, Y. Sun, A. Manibusan, A. J. Sellers, R. S. Barnett, S. Srinagesh, J. S. Forsyth, W. Hassenpflug, F. Tian, T. Javahishvili, B. Felding-Habermann, B. R. Lawson, S. A. Kazane and P. G. Schultz, Angew. Chem., Int. Ed., 2014, 53, 11863–11867 CrossRef CAS PubMed.
- http://www.prnewswire.com/news-releases/fosun-pharma-hopu-investments-cel-healthcare-fund-and-wuxi-pharmatech-to-jointly-acquire-us-innovative-biotechnology-company-ambrx-300087450.html .
- P. Dennler, A. Chiotellis, E. Fischer, D. Brégeon, C. Belmant, L. Gauthier, F. Lhospice, F. o. Romagne and R. Schibli, Bioconjugate Chem., 2014, 25, 569–578 CrossRef CAS PubMed.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, G. F. Filzen, C. J. O'Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS PubMed.
- S. E. Farias, P. Strop, K. Delaria, M. Galindo Casas, M. Dorywalska, D. L. Shelton, J. Pons and A. Rajpal, Bioconjugate Chem., 2014, 25, 240–250 CrossRef CAS PubMed.
- C. S. Theile, M. D. Witte, A. E. M. Blom, L. Kundrat, H. L. Ploegh and C. P. Guimaraes, Nat. Protoc., 2013, 8, 1800–1807 CrossRef PubMed.
- P. Kornberger and A. Skerra, MAbs, 2014, 6, 354–366 CrossRef PubMed.
- K. Zuberbuhler, G. Casi, G. J. L. Bernardes and D. Neri, Chem. Commun., 2012, 48, 7100–7102 RSC.
- X. Li, T. Fang and G.-J. Boons, Angew. Chem., Int. Ed., 2014, 126, 1–5 CrossRef.
- R. van Geel, M. A. Wijdeven, R. Heesbeen, J. M. M. Verkade, A. A. Wasiel, S. S. van Berkel and F. L. van Delft, Bioconjugate Chem., 2015, 26, 2233–2242 CrossRef CAS PubMed.
- P. Umaña, J. Jean-Mairet, R. Moudry, H. Amstutz and J. E. Bailey, Nat. Biotechnol., 1999, 17, 176–180 CrossRef PubMed.
- F. Nimmerjahn and J. V. Ravetch, Science, 2005, 310, 1510–1512 CrossRef CAS PubMed.
- T. Robak, Curr. Opin. Invest. Drugs, 2009, 10, 588–596 CAS.
- C. A. Gerdes, V. G. Nicolini, S. Herter, E. van Puijenbroek, S. Lang, M. Roemmele, E. Moessner, O. Freytag, T. Friess, C. H. Ries, B. Bossenmaier, H. J. Mueller and P. Umaña, Clin. Cancer Res., 2013, 19, 1126–1138 CrossRef CAS PubMed.
- http://www.seattlegenetics.com/sea_technology .
- A. Beck, T. Wurch, C. Bailly and N. Corvaia, Nat. Rev. Immunol., 2010, 10, 345–352 CrossRef CAS PubMed.
- B. Ramakrishnan, E. Boeggeman, M. Pasek and P. K. Qasba, Methods Mol. Biol., 2011, 751, 281–296 CAS.
- N. M. Okeley, B. E. Toki, X. Zhang, S. C. Jeffrey, P. J. Burke, S. C. Alley and P. D. Senter, Bioconjugate Chem., 2013, 24, 1650–1655 CrossRef CAS PubMed.
- E. A. Hull, M. Livanos, E. Miranda, M. E. B. Smith, K. A. Chester and J. R. Baker, Bioconjugate Chem., 2014, 25, 1395–1401 CrossRef CAS PubMed.
- C. H. Kim, J. Y. Axup, A. Dubrovska, S. A. Kazane, B. A. Hutchins, E. D. Wold, V. V. Smider and P. G. Schultz, J. Am. Chem. Soc., 2012, 134, 9918–9921 CrossRef CAS PubMed.
- Y. Cao, Y. J. Axup, J. S. Y. Ma, R. E. Wang, S. Choi, V. Tardif, R. K. V. Lim, H. M. Pugh, B. R. Lawson, G. Welzel, S. A. Kazane, Y. Sun, F. Tian, S. Srinagesh, T. Javahishvili, P. G. Schultz and C. H. Kim, Angew. Chem., Int. Ed., 2015, 54, 7022–7027 CrossRef CAS PubMed.
- J. E. Hudak, R. M. Barfield, G. W. de Hart, P. Grob, E. Nogales, C. R. Bertozzi and D. Rabuka, Angew. Chem., Int. Ed., 2012, 51, 4161–4165 CrossRef CAS PubMed.
- C. H. Kim, J. Y. Axup, B. R. Lawson, H. Yun, V. Tardif, S. H. Choi, Q. Zhou, A. Dubrovska, S. L. Biroce, R. Marsdene, J. Pinstaffe, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17796–17801 CrossRef CAS PubMed.
- D. Pace, Expert Opin. Biol. Ther., 2013, 13, 11–33 CrossRef CAS PubMed.
- R. Rappuoli, Philos. Trans. R. Soc. London, Ser. B, 2011, 366, 2756–2758 CrossRef PubMed.
- F. Y. Avci and D. L. Kasper, Annu. Rev. Immunol., 2010, 28, 107–110 CrossRef CAS PubMed.
- F. Y. Avci, X. Li, M. Tsuji and D. L. Kasper, Nat. Med., 2011, 17, 1602–1610 CrossRef CAS PubMed.
- F. Berti and R. Adamo, ACS Chem. Biol., 2013, 8, 1653–1663 CrossRef CAS PubMed.
- V. Verez-Bencomo, V. Fernandez-Santana, E. Hardy, M. E. Toledo, M. C. Rodrıguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdes, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Torano, I. Sosa, I. Hernandez, R. Martınez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, Science, 2004, 305, 522–525 CrossRef CAS PubMed.
- C. Anish, B. Schumann, C. Lebev Pereira and P. H. Seeberger, Chem. Biol., 2014, 21, 38–50 CrossRef CAS PubMed.
- F. E. André, Dev. Biol., 2002, 110, 25–29 Search PubMed.
- L. Jódar, F. M. LaForce, C. Ceccarini, T. Aguado and D. M. Granoff, Lancet, 2003, 361, 1902–1904 CrossRef.
- E. J. Grayson, G. J. Bernardes, J. M. Chalker, O. Boutureira, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2011, 50, 4127–4132 CrossRef CAS PubMed.
- R. Dagan, J. Poolman and C. A. Siegrist, Vaccine, 2010, 28, 5513–5523 CrossRef CAS PubMed.
- S. Crotti, H. Zhai, J. Zhou, M. Allan, D. Proietti, W. Pansegrau, Q.-Y. Hu, F. Berti and R. Adamo, ChemBioChem, 2014, 14, 836–843 CrossRef PubMed.
- G. Stefanetti, Q.-Y. Hu, A. Usera, Z. Robinson, M. Allan, A. Singh, H. Imase, J. Cobb, H. Zhai, D. Quinn, M. Lei, A. Saul, R. Adamo, C. A. MacLennan and F. Micoli, Angew. Chem., Int. Ed., 2015, 54, 13198–13401 CrossRef CAS PubMed.
- A. Nilo, M. Allan, B. Brogioni, D. Proietti, V. Cattaneo, S. Crotti, S. Sokup, H. Zhai, I. Margarit, F. Berti, Q. Hu and R. Adamo, Bioconjugate Chem., 2014, 25, 2105–2111 CrossRef CAS PubMed.
- A. Nilo, L. Morelli, I. Passalacqua, B. Brogioni, M. Allan, F. Carboni, A. Pezzicoli, D. Maione, M. Fabbrini, M. R. Romano, Q.-Y. Hu, I. Margarit, F. Berti and R. Adamo, ACS Chem. Biol., 2015, 10, 1737–1746 CrossRef CAS PubMed.
- A. Nilo, I. Passalacqua, M. Fabbrini, M. Allan, A. Usera, F. Carboni, B. Brogioni, A. Pezzicoli, J. Cobb, M. R. Romano, I. Margarit, Q.-Y. Hu, F. Berti and R. Adamo, Bioconjugate Chem., 2015, 26, 1839–1849 CrossRef CAS PubMed.
- V. Poszgay, C. Chu, L. Pannell, J. Wolfe, J. B. Robbins and R. Schneerson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5194–5197 CrossRef.
- M. Wacker, L. Wang, M. Kowarik, M. Dowd, G. Lipowsky, A. Faridmoayer, K. Shields, S. Park, C. Alaimo, K. A. Kelley, M. Braun, J. Quebatte, V. Gambillara, P. Carranza, M. Steffen and J. C. Lee, J. Infect. Dis., 2014, 209, 1551–1561 CrossRef CAS PubMed.
- http://adisinsight.springer.com/drugs/800031702 .
- M. Wetter, M. Kowarik, M. Steffen, P. Carranza, G. Corradin and M. Wacker, Glycoconjugate J., 2013, 30, 511–522 CrossRef CAS PubMed.
- J. A. Iwashkiw, M. A. Fentabil, A. Faridmoayer, D. C. Mills, M. Peppler, C. Czibener, A. E. Ciocchini, D. J. Comerci, J. E. Ugalde and M. F. Feldman, Microb. Cell Fact., 2012, 11 DOI:10.1186/1475-2859-1111-1113.
- A. E. Ciocchini, D. A. Serantes, L. J. Melli, L. S. Guidolin, J. A. Iwashkiw, S. Elena, C. Franco, A. M. Nicola, M. F. Feldman, D. J. Comerci and J. E. Ugalde, Vet. Microbiol., 2014, 172, 455–465 CrossRef CAS PubMed.
- L. J. Melli, A. E. Ciocchini, A. J. Caillava, N. Vozza, I. Chinen, M. Rivas, M. F. Feldman, J. E. Ugalde and D. J. Comerci, J. Clin. Microbiol., 2015, 53, 528–538 CrossRef PubMed.
- E. P. Go, H. X. Liao, S. M. Alam, D. Hua, B. F. Haynes and H. Desaire, J. Proteome Res., 2013, 12, 1223–1234 CrossRef CAS PubMed.
- M. Crispin and K. J. Doores, Curr. Opin. Virol., 2015, 11, 63–69 CrossRef CAS PubMed.
- J. P. Julien, A. Cupo, D. Sok, R. L. Stanfield, D. Lyumkis, M. C. Deller, P. J. Klasse, D. R. Burton, R. W. Sanders, J. P. Moore, A. B. Ward and I. A. Wilson, Science, 2013, 342, 1477–1483 CrossRef CAS PubMed.
- K. J. Doores, C. Bonomelli, D. J. Harvey, S. Vasiljevic, R. A. Dwek, D. R. Burton, M. Crispin and C. N. Scanlan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13800–13805 CrossRef CAS PubMed.
- J. Orwenyo, W. Huang and L. X. Wang, Bioorg. Med. Chem., 2013, 21, 4768–4777 CrossRef CAS PubMed.
- H. Xin, S. Dziadek, D. R. Bundle and J. E. Cutler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13526–13531 CrossRef CAS PubMed.
- S. G. Reed, M. T. Orr and C. B. Fox, Nat. Med., 2013, 19, 1597–1608 CrossRef CAS PubMed.
- H. Lu, Front. Immunol., 2014 DOI:10.3389/fimmu.2014.00083.
- J. Schmidt, T. Welsch, D. Jager, P. F. Muhlradt, M. W. Buchler and A. Marten, Br. J. Cancer, 2007, 97, 598–604 CrossRef CAS PubMed.
- A. P. Basto and A. Leitão, J. Immunol. Res., 2014, 2014, 619410, DOI:10.1155/2014/619410.
- P. M. Moyle, J. Hartas, A. Henningham, M. R. Batzloff, M. F. Good and I. Toth, Nanomedicine, 2013, 9, 935–944 CrossRef CAS PubMed.
- P. M. Moyle, W. Dai, Y. Zhang, M. R. Batzloff, M. F. Good and I. Toth, Bioconjugate Chem., 2014, 25, 965–978 CrossRef CAS PubMed.
- Q. P. Liu, G. Sulzenbacher, H. Yuan, E. P. Bennett, G. Pietz, K. Saunders, J. Spence, E. Nudelman, S. B. Levery, T. White, J. M. Neveu, W. S. Lane, Y. Bourne, M. L. Olsson, B. Henrissat and H. Clausen, Nat. Biotechnol., 2007, 25, 454–464 CrossRef CAS PubMed.
- R. Sackstein, J. S. Merzaban, D. W. Cain, N. M. Dagia, J. A. Spencer, C. P. Lin and R. Wohlgemuth, Nat. Med., 2008, 14, 181–187 CrossRef CAS PubMed.
- N. M. Okeley, S. C. Alley, M. E. Anderson, T. E. Boursalian, P. J. Burke, K. M. Emmerton, S. C. Jeffrey, K. Klussman, C.-L. Law, D. Sussman, B. E. Toki, L. Westendorf, W. Zeng, X. Zhang, D. R. Benjamin and P. D. Senter, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5404–5409 CrossRef CAS PubMed.
- L. K. Swee, S. Lourido, G. W. Bell, J. R. Ingram and H. L. Ploegh, ACS Chem. Biol., 2014, 10, 460–465 CrossRef PubMed.
- M. Sadelain, R. Brentjens and I. Rivière, Curr. Opin. Immunol., 2009, 21, 215–223 CrossRef CAS PubMed.
- http://www.phrma.org/sites/default/files/pdf/biologicsoverview2013.pdf .
| This journal is © The Royal Society of Chemistry 2016 |