
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed-linker UiO-66: structure–property relationships revealed by a combination of high-resolution powder X-ray diffraction and density functional theory calculations†
Marco
Taddei
 a,
Davide
Tiana
b,
Nicola
Casati
c,
Jeroen A.
van Bokhoven
ad,
Berend
Smit
be and
Marco
Ranocchiari
*a
a,
Davide
Tiana
b,
Nicola
Casati
c,
Jeroen A.
van Bokhoven
ad,
Berend
Smit
be and
Marco
Ranocchiari
*a
aLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institute, 5232 Villigen-PSI, Switzerland. E-mail: marco.ranocchiari@psi.ch
bLaboratory of Molecular Simulation, EPFL, Rue de l'Industrie 17, 1951 Sion, Switzerland
cSwiss Light Source, Paul Scherrer Institute, 5232 Villigen-PSI, Switzerland
dInstitute for Chemical and Bioengineering, ETH Zürich, Vladimir Prelog Weg 1, 8093 Zürich, Switzerland
eDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA
First published on 6th December 2016
Abstract
The use of mixed-linker metal–organic frameworks (MIXMOFs) is one of the most effective strategies to modulate the physical–chemical properties of MOFs without affecting the overall crystal structure. In many instances, MIXMOFs have been recognized as solid solutions, with random distribution of ligands, in agreement with the empirical rule known as Vegard's law. In this work, we have undertaken a study combining high-resolution powder X-ray diffraction (HR-PXRD) and density functional theory (DFT) calculations with the aim of understanding the reasons why UiO-66-based amino- and bromo-functionalized MIXMOFs (MIXUiO-66) undergo cell expansion obeying Vegard's law and how this behaviour is related to their physical–chemical properties. DFT calculations predict that the unit cell in amino-functionalized UiO-66 experiences only minor expansion as a result of steric effects, whereas major modification to the electronic features of the framework leads to weaker metal–linker interaction and consequently to the loss of stability at higher degrees of functionalization. For bromo-functionalized UiO-66, steric repulsion due to the size of bromine yields a large cell expansion, but the electronic features remain very similar to pristine UiO-66, preserving the stability of the framework upon functionalization. MIXUiO-66 obtained by either direct synthesis or by post-synthetic exchange shows Vegard-like behaviour, suggesting that both preparation methods yield solid solutions, but the thermal stability and the textural properties of the post-synthetic exchanged materials do not display a clear dependence on the chemical composition, as observed for the MOFs obtained by direct synthesis.
Introduction
Metal–organic frameworks (MOFs) are some of the most compelling classes of materials currently investigated because they combine crystallinity and large porosity with impressive chemical and structural versatility.1–4 Mixed-linker metal–organic frameworks (MIXMOFs or MTVMOFs) are a subclass of MOFs incorporating two or more linkers having similar sizes and different functional groups.5,6 The resulting crystal structure is isoreticular to that observed when only one type of ligand is used, but the physical–chemical properties of the material are modulated by the relative amounts of functional groups attached to the framework.7,8 In some cases, new properties can emerge from the concomitant presence of two or more functionalities exposed inside the pores, which can be especially useful in selective gas sorption and separation9,10 or catalysis.11–15MIXMOFs based on several topologies have been obtained, notably MOF-5,7,12,16–19 MIL-53,20–22 MIL-101,17,23,24 DMOF-1,25,26 UiO-6627–30 and ZIF-8.31–33 Most MIXMOFs are regarded as homogeneous solid solutions, with the linkers randomly distributed throughout the framework, rather than being segregated in phase-pure domains.34–36 Apart from a few exceptions,25 this behaviour is generally observed at any relative ratio of ligands. Information on the random distribution of linkers in MIXMOFs is an important aspect, especially for enabling their use as catalysts for cascade reactions or processes where the vicinity of the active sites is key to ensure catalytic activity,11 or to precisely tailor the pore size to improve the sieving properties.32,33
One of the pieces of evidence normally used to support the solid solution hypothesis in MIXMOFs is the observation of a gradual shift of reflections in the powder X-ray diffraction (PXRD) patterns associated with varying ligand ratios.16,20 The shift is due to the adaptation of the structure to the presence of linkers with different properties, leading to either shrinkage or expansion of the unit cell. This is a typical behaviour of solid solutions, i.e. metal alloys or mixed-metal oxides, which is described by the empirical rule known as Vegard's law.37–39 In the case of heterogeneous distributions arising from scarce compatibility between monomers, such as core–shell architectures or segregation into single phase domains, the PXRD pattern would clearly display the simultaneous presence of two well-defined sets of reflections associated with each single crystalline phase.6 More challenging is the situation where short-range compositional inhomogeneity in a long-range ordered matrix exists, which can be hardly distinguished from a completely homogeneous solid solution by conventional X-ray diffraction techniques and requires other approaches.36,40–42
The agreement to Vegard's law for inorganic materials has been related to factors such as the ionic radius, volume of valence electrons, Brillouin zone overlaps and electrochemical factor.39 Moreover, the regular variation of the unit cell is often accompanied by a similar behaviour of the physical properties of the materials, e.g. melting temperature43 and band gap.44 In the case of MIXMOFs, little is known so far about the factors responsible for the shrinkage or expansion of the unit cell upon changes in the composition. This lack of insight can be ascribed to the fact that MOFs, being organic–inorganic coordination compounds, are much more complex systems to study than metal alloys or ionic compounds. However, the existence of characteristic structural trends in MIXMOFs might as well correlate with the modulation of physical–chemical properties and it is therefore of high interest to investigate whether these trends can be associated with specific structure–property relationships.
In the present work, we performed a thorough fundamental study by coupling high-resolution PXRD (HR-PXRD) with density functional theory (DFT) calculations on UiO-66 MIXMOFs (MIXUiO-66) based on either terephthalic acid (BDC) and 2-aminoterephthalic acid (ABDC) or BDC and 2-bromoterephthalic acid (BBDC) (Fig. 1).
 | ||
| Fig. 1 Molecular structures of the ligands employed in this work. | ||
Two series of amino- and bromo-functionalized samples were obtained by direct synthesis (DS_MIXUiO-66), whereas two other series containing ABDC and BDC were prepared by postsynthetic ligand exchange (PSE_MIXUiO-66) starting from UiO-66 and UiO-66-NH2, respectively. HR-PXRD was exploited to assess whether MIXUiO-66 obeyed Vegard's law and can be regarded as solid solutions and if the preparation method had any relevant influence on the distribution of ligands in the crystal structure. DFT calculations were essential for unraveling the causes of the observed structural trends and for relating them to the thermal and chemical stability of the MIXUiO-66. We chose to investigate MIXUiO-66 containing either ABDC or BBDC because these two linkers have complementary features: the amino group in ABDC is a small-sized electron-donor, and the bromo substituent in BBDC is instead a sterically demanding electron-withdrawer. In addition, amino- and bromo-functionalized UiO-66 display characteristic reactivity and are ideal platforms for post-synthetic modification.45–47
Experimental section
DS_MIXUiO-66
DS_MIXUiO-66-NH2 was prepared according to the procedure reported by Chavan et al.:27 ZrCl4 (333 mg, 1.43 mmol) was dissolved in N,N-dimethylformamide (DMF, 37 mL) and H2O (0.033 mL, 1.83 mmol). Then, BDC and/or ABDC were added in a stoichiometric amount with respect to ZrCl4 (see Table S1 for details on the amounts, ESI†) and the mixture was sonicated until it became clear. Five reaction solutions were prepared in this way, containing 0%, 25%, 50%, 75% and 100% ABDC, respectively. Each solution was introduced into a glass vial, sealed and kept for 72 h in an oven at 100 °C. After the reaction was completed, the content of every vial was centrifuged and the solid was washed with DMF (10 mL), soaked in DMF (10 mL) for 16 h, washed with acetone (2 × 10 mL), soaked in acetone (10 mL) for 16 h and finally dried in a vacuum oven at 70 °C. The mixed-linker samples were named according to the amount of ABDC used in the synthesis as follows: DS_MIXUiO-66-NH2-25, DS_MIXUiO-66-NH2-50 and DS_MIXUiO-66-NH2-75.DS_MIXUiO-66-Br was prepared according to the same procedure, just by replacing ABDC with the same molar amount of BBDC in the reaction mixture (Table S2, ESI†). The mixed-linker samples were named according to the amount of BBDC used in the synthesis as follows: DS_MIXUiO-66-Br-25, DS_MIXUiO-66-Br-50 and DS_MIXUiO-66-Br-75.
PSE_MIXUiO-66
PSE was performed by slightly modifying the literature procedure.29ABDC (18 mg, 0.1 mmol; 36 mg, 0.2 mmol; 72 mg, 0.4 mmol, respectively) was introduced into three 20 mL glass vials, 2.5 mL of 1 M KOH was added, and then the solution was neutralized with 1 M HCl. 70 mg of UiO-66 (0.2 mmol BDC) was added to each vial. The vials were sealed and kept at 80 °C for 5 days. The content of each vial was then centrifuged and the solid was washed with water (5 mL), washed with DMF (5 mL), soaked in DMF (5 mL) for 16 h, washed with acetone (2 × 5 mL), soaked in acetone (5 mL) and finally dried in a vacuum oven at 70 °C. The samples prepared using this procedure were named PSE_MIXUiO-66-NH2(42%), PSE_MIXUiO-66-NH2(66%) and PSE_MIXUiO-66-NH2(86%), where the numbers in brackets are the amounts of ABDC after exchange calculated by quantitative NMR analysis.
BDC (17 mg, 0.1 mmol; 34 mg, 0.2 mmol; 68 mg, 0.4 mmol, respectively) was introduced into three 20 mL glass vials, 2.5 mL of 1 M KOH was added, and then the solution was neutralized with 1 M HCl. 78 mg of UiO-66-NH2 (0.2 mmol ABDC) was added to each vial. The vials were sealed and kept at 80 °C for 5 days. Workup was performed as described above. The samples prepared using this procedure were named PSE_MIXUiO-66-NH2(65%), PSE_MIXUiO-66-NH2(57%) and PSE_MIXUiO-66-NH2(47%), where the numbers in brackets are the amounts of ABDC after exchange calculated by quantitative NMR analysis.
Digestion of samples for NMR analysis
Quantitative NMR analysis was performed on a Bruker Avance III spectrometer (500 MHz). 20 mg of solid were digested for 24 h in 1 mL of 0.1 M NaOH in D2O. The NMR tubes were then loaded with the solution, which was filtered to avoid the presence of solid particles in dispersion.Thermogravimetric analysis
Thermogravimetric analysis was performed on a Mettler-Toledo instrument. About 10 mg of samples was loaded in an alumina crucible and heated up to 700 °C at a rate of 5 K min−1 with an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 N2/O2 mixture. UiO-66 and UiO-66-NH2 were also analyzed under a pure N2 stream.
:20 N2/O2 mixture. UiO-66 and UiO-66-NH2 were also analyzed under a pure N2 stream.
Gas sorption analysis
Nitrogen sorption isotherms at 77 K were recorded on a Micromeritics 3Flex analyzer. The samples (about 30 mg) were activated overnight under vacuum at 150 °C prior to analysis. The BET numbers were calculated in the 0.003–0.035 P/P0 range, where all the criteria defined by Gomez-Gualdron et al.48 were fulfilled.HR-PXRD data collection and treatment
HR-PXRD data were collected at the Material Science (MS) beamline at Swiss Light Source (SLS),49 using 16 keV radiation for the amino-functionalized samples and 12.4 keV for the bromo-functionalized samples. Glass capillaries with a diameter of 0.5 mm were used for all the standards and samples. Evacuation was performed on capillaries loaded with powders and glass wool as a stopper. The samples were inserted in a Schlenk tube and kept under vacuum at 120 °C for 72 h. Then, argon was flowed inside the Schlenk tube and the capillaries were sealed with a flame and wax without exposing them to moist air. In situ evacuation was performed on a capillary connected to a vacuum pump via a sample stage equipped with a Swagelok connection.Whole powder pattern fitting on all standards and samples was performed by the Pawley method using the program TOPAS 5.0. Si-640d was employed as a standard to determine the exact wavelength, zero error, and horizontal and vertical displacement parameters. Na2Ca3Al2F14 (NAC) was employed as a standard to determine the instrumental profile function. For MIXUiO-66 samples, the lattice parameter, crystal size and strain were refined.
Computational studies
Quantum Espresso v5.350 was used for all calculations. The geometry optimizations were performed at the gamma point using the DFT (density functional theory) PBEsol51 norm-conserving pseudopotential52 at a cut-off energy of 100 Ry (see Fig. S1 in the ESI† for the convergence). The convergence threshold was set to 10−6 and 10−5 for the energy and force, respectively, whilst the pressure threshold was set to 10−4. The electronic structure was converged up to 10−9. To reduce the computational cost, the calculations were performed using the UiO-66 primitive unit cell (114 atoms).Results and discussion
DS_MIXUiO-66
DS_MIXUiO-66 samples were prepared starting from reaction mixtures containing 0, 25, 50, 75 and 100% of functionalized ligands. Quantitative 1H-NMR analysis was performed to determine the relative amounts of ligands in the MIXUiO-66, showing that the incorporation of both ABDC and BBDC into the solid was slightly preferential with respect to BDC (Tables S3 and S4, ESI†). Fig. 2 shows the HR-PXRD patterns for both the series of samples.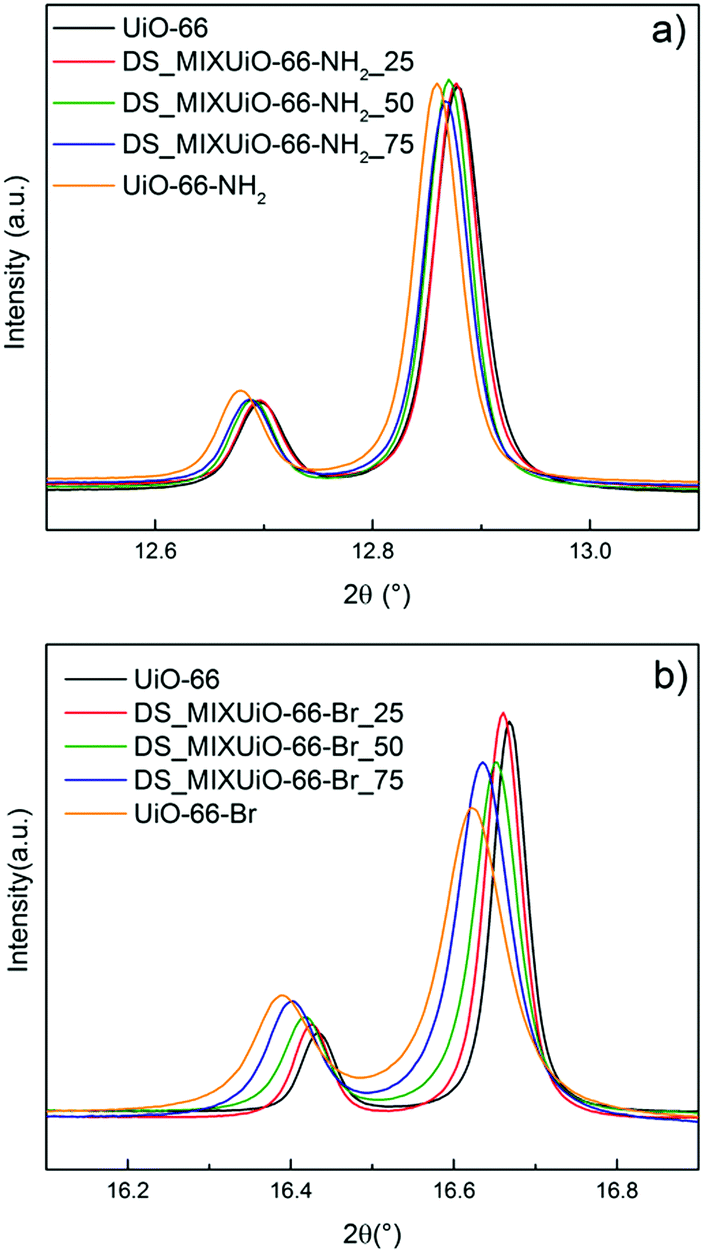 | ||
| Fig. 2 (531) and (006) reflections in the HR-PXRD patterns of DS_MIXUiO-66-NH2 (a, collected using 16 keV radiation) and DS_MIXUiO-66-Br (b, collected using 12.4 keV radiation) samples obtained from reaction mixtures containing 0% (black), 25% (red), 50% (green), 75% (blue) and 100% (orange) ABDC or BBDC. Intensities were scaled to allow easier visualization of the peak shift. | ||
UiO-66 crystallizes in the cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, therefore a single lattice parameter a defines its unit cell and all the reflections in the PXRD pattern shift upon cell expansion or contraction. The data clearly showed that both the series of samples underwent cell expansion upon increasing the amount of functionalized ligands in the framework, with the reflections being shifted to a lower 2θ angle (Fig. 2). Table 1 and Fig. 3 report the results of the Pawley refinement of the HR-PXRD patterns.
m space group, therefore a single lattice parameter a defines its unit cell and all the reflections in the PXRD pattern shift upon cell expansion or contraction. The data clearly showed that both the series of samples underwent cell expansion upon increasing the amount of functionalized ligands in the framework, with the reflections being shifted to a lower 2θ angle (Fig. 2). Table 1 and Fig. 3 report the results of the Pawley refinement of the HR-PXRD patterns.
| Sample name | a (Å) |
|---|---|
| DS_MIXUiO-66-NH 2 | |
| UiO-66 | 20.7419(2) |
| DS_MIXUiO-66-NH2-25 | 20.745(1) |
| DS_MIXUiO-66-NH2-50 | 20.753(1) |
| DS_MIXUiO-66-NH2-75 | 20.761(2) |
| UiO-66-NH2 | 20.774(2) |
| DS_MIXUiO-66-Br | |
| UiO-66 | 20.7419(2) |
| DS_MIXUiO-66-Br-25 | 20.746(4) |
| DS_MIXUiO-66-Br-50 | 20.758(3) |
| DS_MIXUiO-66-Br-75 | 20.779(3) |
| UiO-66-Br | 20.795(2) |
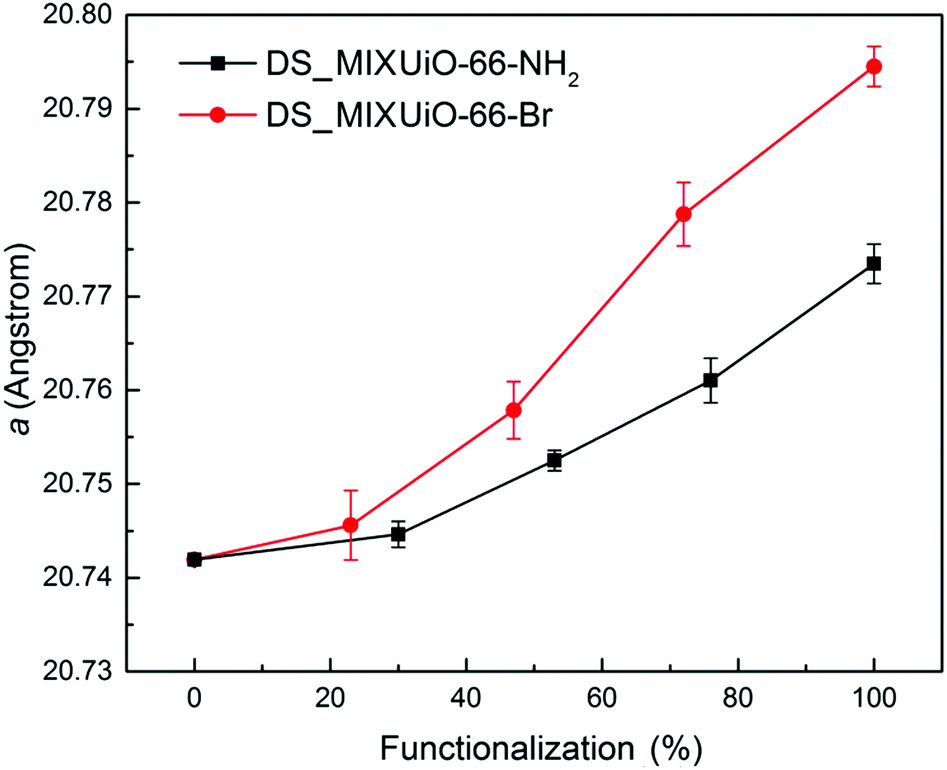 | ||
| Fig. 3 Refined lattice parameter of DS-MIXUiO-66-NH2 (black) and DS-MIXUiO-66-Br (red). | ||
The unit cell parameter a for the amino-functionalized MIXUiO-66 increased from 20.7419(2) Å to 20.774(2) Å when the amount of ABDC in the framework increased from 0% to 100%. A similar regular trend was observed for bromo-functionalized MIXUiO-66, although in this case the cell expansion from UiO-66 to UiO-66-Br was larger, with a reaching a value of 20.795(2) Å for the fully functionalized material. Based on these results, the samples appear to obey Vegard's law, suggesting that any potential local inhomogeneity does not play a significant role. Recently, Chavan and co-workers synthesized for the first time a series of MIXUiO-66 using both BDC and ABDC as ligands.27 They assessed the solid solution nature of the materials by means of coupled thermogravimetric-differential scanning calorimetric (TG-DSC) analysis, which displayed only one heat signal related to framework decomposition shifting to lower temperature with an increasing amount of ABDC in the framework. Our findings strengthen the solid solution hypothesis, expanding it to bromo-functionalized MIXUiO-66.
In order to separate the effect of the solvation on the unit cell expansion from the real effect of the functionalization, the amino-functionalized samples were evacuated for 72 h at 120 °C in a vacuum. Under these conditions, most of the molecules of solvent trapped in the pores were removed, while avoiding dehydroxylation of the clusters, which occurs at higher temperature and is known to cause cell contraction.15 The cell underwent expansion upon solvent removal, but the trend observed for the as-synthesized samples was maintained (Fig. 4 and Table S5, ESI†). In this case a increased from 20.7520(4) Å for UiO-66 to 20.798(2) Å for UiO-66-NH2, evidencing how the extent of the cell expansion was slightly larger than that in the as-synthesized samples (0.046 Å vs. 0.032 Å, respectively). An in situ HR-PXRD experiment was performed while evacuating a capillary containing non-functionalized UiO-66 to further assess the effect of solvent removal (Fig. S2, ESI†). The sample was initially kept at room temperature and vacuum was applied to the capillary: after 30 minutes the cell parameter increased from 20.750(3) Å to 20.756(3) Å, indicating that part of the solvent had already been removed. Upon continuing evacuation for further 30 minutes, a reached a steady value of 20.758(3) Å. When the temperature was increased to 70 °C, the cell parameter further expanded to 20.764(2) Å, reaching a steady value of 20.767(3) Å at 100 °C, suggesting that all the solvent had been removed. To get rid of the effect of thermal expansion, the sample was then cooled down to room temperature under vacuum and a shrank to 20.763(2) Å. The expansion upon complete removal of the solvent was 0.013 Å, in good agreement with the value observed when the sample was evacuated ex situ (0.010 Å). The slightly lower expansion observed for the sample evacuated ex situ was attributed to the non-complete removal of solvent. This result confirms the reliability of the data collected on the evacuated samples.
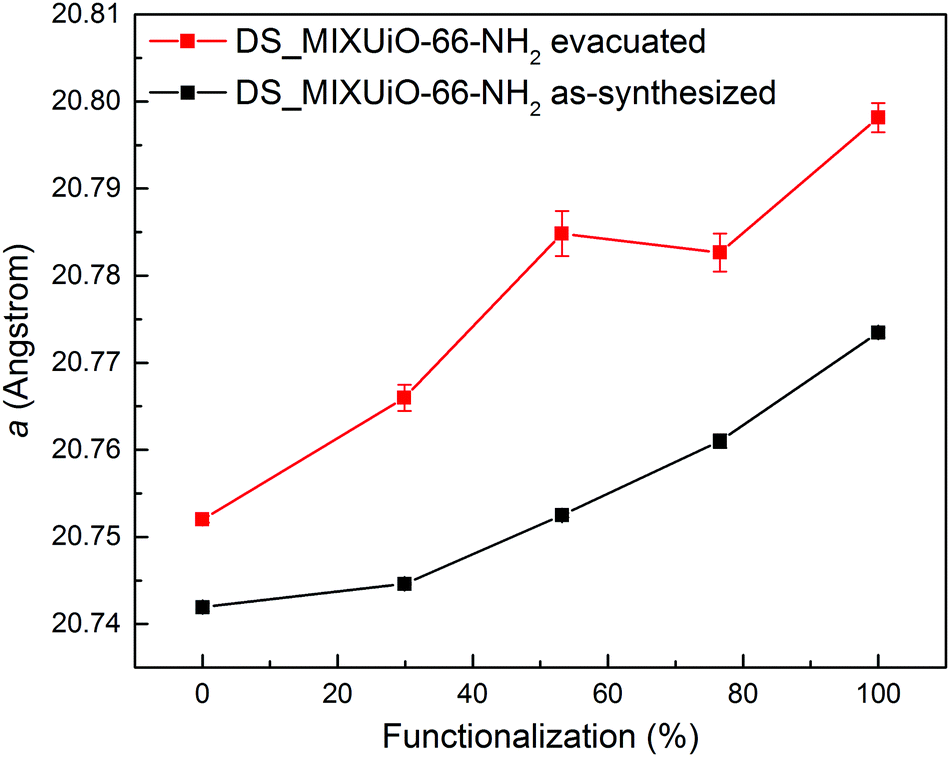 | ||
| Fig. 4 Comparison of the refined lattice parameters for DS_MIXUiO-66-NH2 samples as-synthesized (black) and after evacuation at 120 °C for 72 h under vacuum (red). | ||
Computational studies were performed to investigate the reason of the unit cell expansion. Starting from the experimental structure of pristine UiO-66, NH2- and Br-groups were consecutively added to each single BDC linker (six BDC linkers are present in the UiO-66 primitive cell) and fully optimized (i.e. relaxing the atomic positions and lattice parameters, see the ESI† for the optimized geometries). Fig. 5 shows the calculated highest occupied crystalline orbitals (HOCOs) for UiO-66, UiO-66-NH2 and UiO-66-Br. UiO-66 is composed of Zr6O4(OH)4 clusters and BDC organic ligands; its HOCO is a combination of non-bonding O p-orbitals which resembles that found in ZrO2 (Fig. S3, ESI†). However, in UiO-66 also “organic” oxygen atoms of the carboxylate are present which contribute to the orbital (Fig. 5, top) resulting in a “hybrid organic–inorganic” orbital that stabilizes the framework. Functionalizing the ligand with amino groups significantly changed the electronic features of the compound, as recently described also by Hendrickx et al.53 and Flage-Larsen et al.54 As the BDC ligands were replaced with ABDC ligands, new orbitals were populated; the new HOCOs were localized on the phenyl ring and on the amino group of the functionalized ligands (Fig. 5, middle) and the nature of the HOCO changed becoming mainly “organic” in nature. As the new orbital got more and more populated increasing the percentage of the functionalized ligand, the band gap shrank and the interaction between the organic ligands and the clusters became weaker, explaining the loss of stability of UiO-66-NH2 compared to that of the pristine UiO-66 found by thermogravimetric analysis.54,55 The mean Zr–O bond lengths in UiO-66-NH2 and UiO-66 were virtually the same (2.269 Å vs. 2.270 Å), excluding a straightforward correlation between the strength of the linker–metal interaction and the Zr–O bond length. Steric effects between NH2 and the O lone pairs seemed not to be relevant at any ABDC loading, resulting in a very small expected cell expansion (Table 2). As found by experiments, a larger cell expansion for the bromo-functionalized MIXUiO-66 was predicted by calculations (Table 2). Upon functionalizing the ligand with Br instead of NH2, new HOCOs were populated but a different situation occurred. In this case the new populated HOCOs were closer to the configuration of pristine UiO-66 (i.e. of “hybrid” nature) than they were in the amino-functionalized samples. The presence of Br, which has a large atomic volume due to its lone pairs, yielded steric repulsion between the Br and O p-orbitals (Fig. 5, bottom), inducing an increase of the cell volume without affecting the stability, as revealed by thermogravimetric analysis, discussed herein. Fig. 6 compares the relative change in the volume (V/V0) of the predicted and experimental unit cells with the increase of the functionalized ligand.
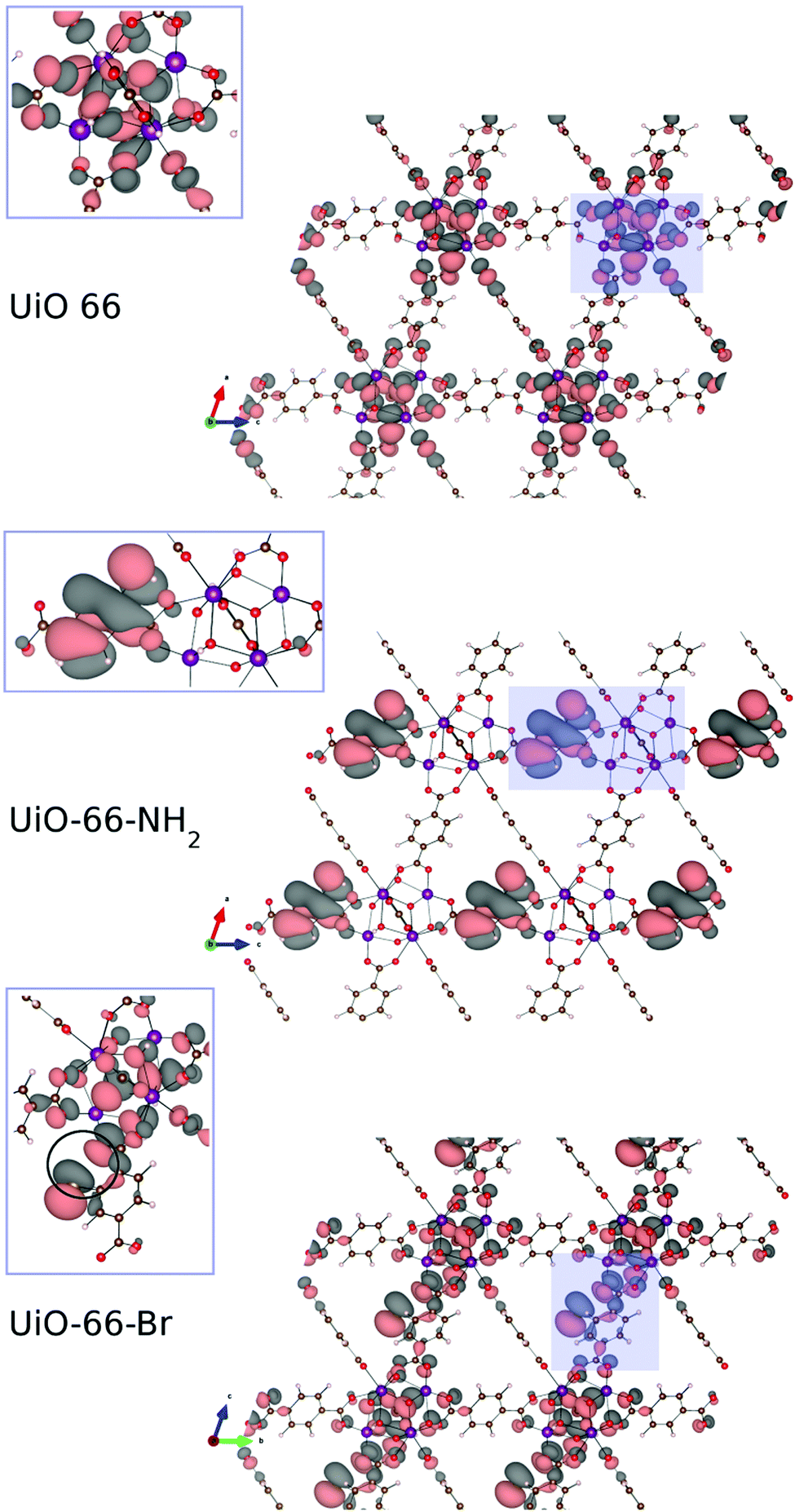 | ||
| Fig. 5 Calculated highest occupied crystalline orbitals (HOCOs) for UiO-66 (top), UiO-66-NH2 (middle) and UiO-66-Br (bottom). The zoomed insets show how the orbital is spread between the inorganic and organic part for UiO-66 and UiO-66-Br, while for UiO-66-NH2 it is predominantly of organic nature. For UiO-66-Br a steric repulsion (pointed out inside the circle in the inset) between the Br and O lone pair arises. Images of the crystalline orbitals were obtained using the VESTA software.56 Adopted colour scheme: Br light brown, C dark brown, H light pink, N blue, O red, and Zr purple. Isovalue: 5 × 10−4. | ||
| Functionalization (%) | Unit cell volume MIXUiO-66-NH2 (Å3) | Unit cell volume MIXUiO-66-Br (Å3) |
|---|---|---|
| 0 | 2308.83 | 2308.83 |
| 17 | 2309.68 | 2310.65 |
| 33 | 2309.87 | 2312.36 |
| 50 | 2309.91 | 2314.17 |
| 67 | 2310.19 | 2315.53 |
| 83 | 2310.26 | 2317.47 |
| 100 | 2310.46 | 2318.45 |
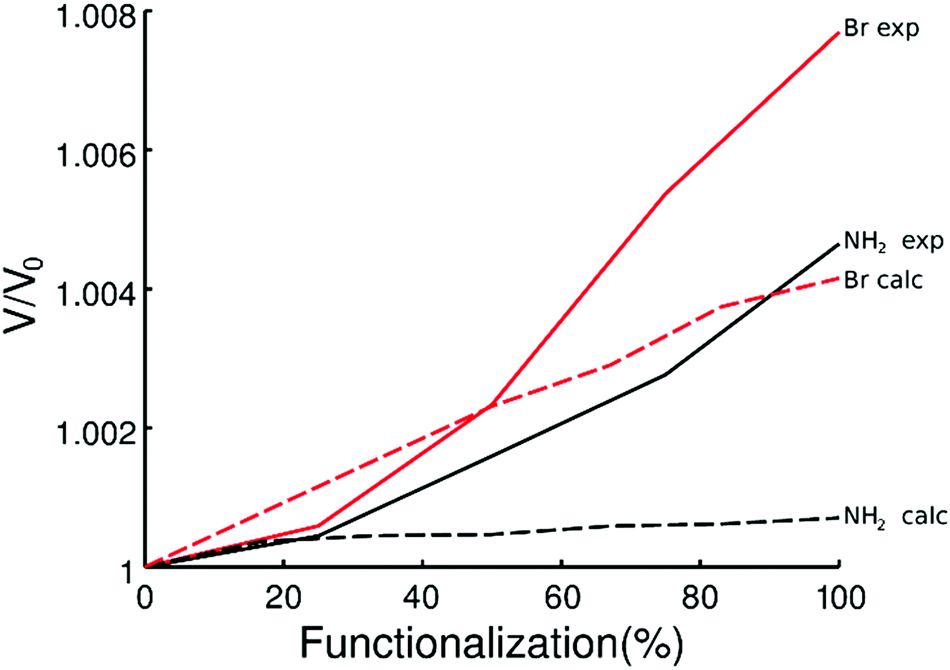 | ||
| Fig. 6 Predicted (dashed line) and experimental (continuous line) volume change as a function of the amount of functionalized ligands for MIXUiO-66-NH2 (black) and MIXUiO-66-Br (red). | ||
The calculations seemed to underestimate the cell expansion, especially for the MIXUiO-66-NH2 series, and the discrepancy between experimental and calculated trends became larger with increasing functionalization degree. This can be explained remembering that calculations are performed at 0 K and do not include any vibrations. Hence we can attribute the discrepancies to the thermal motion which adds some steric repulsion between the functionalized ligand and the framework. This steric contribution seems to be increasingly larger as the functionalization degrees increase. Nevertheless, a good agreement in the expansion trend (i.e. linear expansion with increasing functionalization, larger cell expansion for the Br-functionalized UiO-66) is found, corroborating the idea that the functionalized ligands are in fact randomly incorporated into the framework and that the solid solution hypothesis is reliable. This also suggested that defects, in the form of either missing-linkers (as revealed by TGA)57 and/or missing-clusters (as revealed by weak superlattice reflections in the PXRD patterns),58 were not the major contributors to the observed unit cell expansion.
TGA of the materials is shown in Fig. 7. As already reported by Chavan et al.,27 the stability of the framework of DS_MIXUiO-66-NH2 samples decreased as the loading of ABDC increased (Fig. 7a). A similar behaviour was also observed for other topologies.16,20 In the case of MIXUiO-66, the loss of stability can be imputed to the decreasing strength of the metal–linker interaction when moving from pure UiO-66 to pure UiO-66-NH2, as evidenced by the calculations discussed above. Performing the analysis under a 100% nitrogen stream (Fig. S4, ESI†), the same trend was observed, ruling out the possibility that the stability loss was due to easier combustion of the organic part when more ABDC was embedded in the framework. On the other hand, Fig. 7b shows that the thermal stability of DS-MIXUiO-66-Br samples was minimally affected by the presence of BBDC, suggesting that steric effects did not have a significant impact on the strength of the ligand–metal interaction. Again, this is in good agreement with the prediction of our calculations. The amount of defects calculated from TGA was about 9% for all the MIXUiO-66-NH2 samples (except for MIXUiO-66-NH2-25, for which we found 0%), whereas for MIXUiO-66-Br it ranged from 5% for MIXUiO-66-Br-50 to 13% for MIXUiO-66-Br-75 and UiO-66-Br up to 20% for MIXUiO-66-Br-25. The very similar decomposition temperatures of MIXUiO-66-Br samples, despite defectivity ranging between 5 and 20%, do not display a significant influence of defects on the stability. Our findings unravel the reasons behind the observed thermal stability of amino- and bromo-functionalized UiO-66 materials, which were previously the only object of speculations by Kandiah et al.59 The observations by the same authors on the lower stability to hydrolysis of UiO-66-NH2 if compared to UiO-66 and UiO-66-Br can also be rationalized based on the weaker interaction between ABDC and the clusters. It is indeed known that in most MOFs hydrolysis occurs at the metal–linker bonds, which are the chemically weakest points.60 Nitrogen sorption isotherms (Fig. S5, S6 and Tables S6, S7, ESI†) display ideal type 1 behaviour and a regular decrease of accessible volume with increasing loading of either ABDC or BBDC.
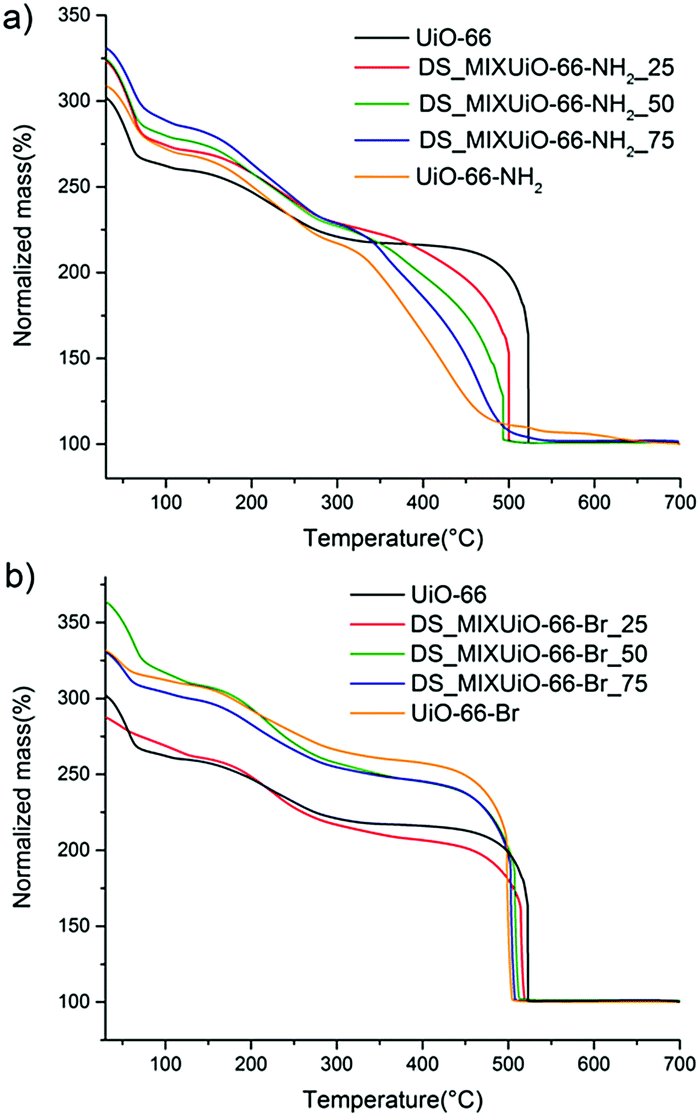 | ||
| Fig. 7 Thermogravimetric curves of DS_MIXUiO-66-NH2 (a) and DS_MIXUiO-66-Br (b). | ||
PSE_MIXUiO-66
PSE is an alternative method to prepare MIXUiO-66 by soaking phase-pure materials in a solution of the desired ligand in a polar solvent.61 Some materials that cannot be obtained using other synthetic methods have been prepared via PSE, promoting it as a very powerful tool in MOF synthesis.62–65 Kim and co-workers29 used aerosol time-of-flight mass spectrometry (ATOFMS) to prove that single UiO-66 microcrystals that underwent PSE contained both the ligands. This evidence, together with the observation that crystallinity was not affected, ruled out the possibility that the MOF underwent dissolution and recrystallization into two distinct crystalline phases. However, the process of PSE is still not fully understood and it is not clear whether the degree of substitution is uniform throughout the microcrystals or if there is a gradient due to increased diffusion limitations in the inner part of the crystals that might generate domains with considerably different compositions.We prepared a series of MIXUiO-66-NH2 (hereafter PSE_MIXUiO-66-NH2_A) by soaking UiO-66 in aqueous solutions containing 0.5, 1.0 and 2.0 equivalents of ABDC, and another series (hereafter PSE_MIXUiO-66-NH2_B) by soaking UiO-66-NH2 in aqueous solutions containing 0.5, 1.0 and 2.0 equivalents of BDC. Quantitative 1H-NMR analysis revealed that the degree of exchange of ABDC into UiO-66 (42%, 66% and 86%, when 0.5, 1.0 and 2.0 equivalents of ABDC were used, respectively) was higher than that of BDC into UiO-66-NH2 (35%, 43%, 53%, when 0.5, 1.0 and 2.0 equivalents of BDC were used, respectively). This confirmed once again the higher coordinating capability of ABDC with respect to BDC (Table S8 and Fig. S7, ESI†). Complete exchange was not achieved even when a two-fold excess of free ligand was used, suggesting that an equilibrium state was reached as the concentration of the ligand expelled from the MOF in the solution increased.
HR-PXRD patterns of all the samples clearly displayed the presence of a single phase (Fig. S8 and S9, ESI†). As for DS_MIXUiO-66-NH2, there was no evidence of the presence of significant local inhomogeneity. Table 3 and Fig. 8 show the results of the Pawley refinements on the HR-PXRD patterns.
| Sample name | a (Å) |
|---|---|
| PSE_MIXUiO-66-NH2_A(42%) | 20.763(7) |
| PSE_MIXUiO-66-NH2_A(66%) | 20.771(3) |
| PSE_MIXUiO-66-NH2_A(86%) | 20.780(6) |
| PSE_MIXUiO-66-NH2_B(65%) | 20.7643(7) |
| PSE_MIXUiO-66-NH2_B(57%) | 20.7627(2) |
| PSE_MIXUiO-66-NH2_B(47%) | 20.7628(7) |
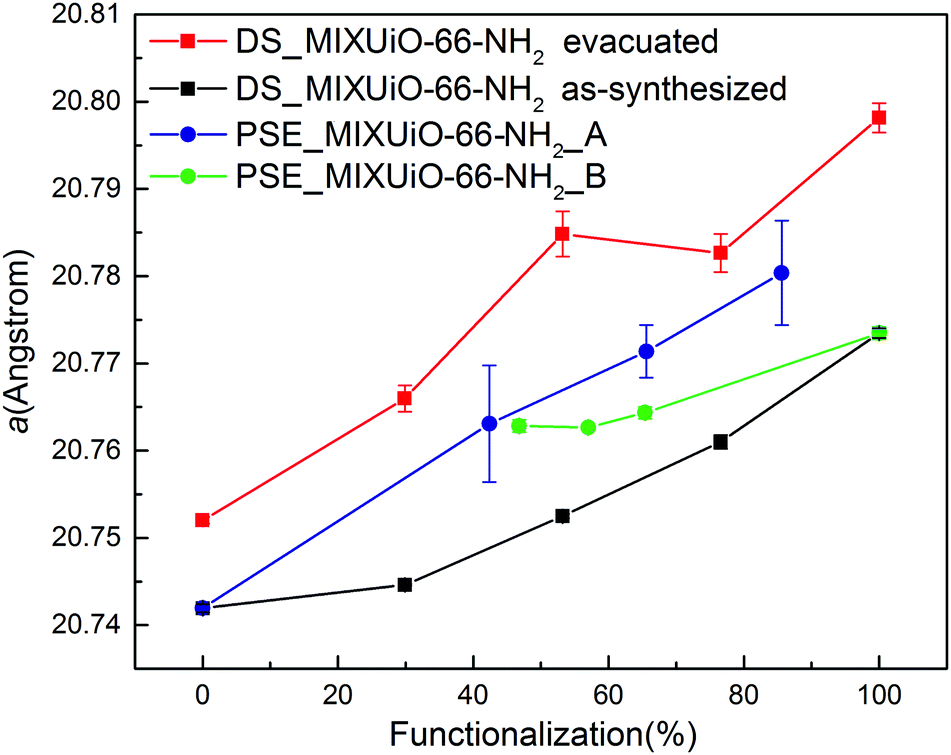 | ||
| Fig. 8 Refined lattice parameter of PSE_MIXUiO-66 series (blue) and PSE_MIXUiO-66-NH2 series (green) compared with as-synthesized DS_MIXUiO-66-NH2 (black) and evacuated DS_MIXUiO-66-NH2 (red). | ||
The lattice parameter of PSE_MIXUiO-66-NH2_A grew larger as more ABDC was introduced into the framework. In the case of PSE_MIXUiO-66-NH2_B, the lattice parameter decreased as the framework incorporated more BDC, although sample PSE_MIXUiO-66-NH2_B(47%) slightly deviated from the ideal Vegard behaviour. Comparison of the absolute values of the lattice parameter for PSE_MIXUiO-66-NH2_A, PSE_MIXUiO-66-NH2_B and DS_MIXUiO-66-NH2 reveals that the samples prepared by PSE have larger a than expected from analysis of the as-synthesized materials obtained by DS, but still smaller than the evacuated ones. This difference can be mainly attributed to the effect of the solvent: TGA of PSE_MIXUiO-66-NH2_A and PSE_MIXUiO-66-NH2_B (Fig. S10 and S11, ESI†) displays on average a smaller weight loss than DS_MIXUiO-66-NH2 at temperature lower than 250 °C, where the removal of solvent molecules from the pores occurs. This is most likely a result of the different preparation procedures. DS was carried out in DMF and the workup involved the same solvent and acetone. As a result, two distinct weight loss steps below 250 °C can be seen in the TG curves of these samples (Fig. 7a). PSE was performed in water and the workup procedure involved soaking in both DMF and acetone. A single weight loss step below 250 °C suggested that water was preferentially adsorbed inside the pores (Fig. S10 and S11, ESI†).
The thermal stability of the framework of all materials obtained by PSE was between those of pure UiO-66 and pure UiO-66-NH2 (Fig. S10 and S11, ESI†). However, there was no clear dependence of the thermal stability on the amount of ABDC. Similarly, the gas sorption isotherms did not display any evident trend for the surface area and porosity (Fig. S12 and S13, ESI†). These results suggest that MIXUiO-66 prepared by PSE, although being phase-pure and seemingly obeying Vegard's law, could undergo partial amorphization or occlude free linkers inside the pores, which were not completely removed during the workup procedure, thus affecting their thermal stability and textural properties.
Conclusions
We performed a thorough fundamental investigation on amino- and bromo-functionalized MIXMOFs based on the UiO-66 topology prepared by either direct synthesis or post-synthetic ligand exchange. The samples obtained by direct synthesis displayed the ideal behaviour of solid solutions. HR-PXRD analysis revealed that they obeyed Vegard's law, undergoing cell expansion with increasing relative amount of ABDC and BBDC. No clear evidence of important short-range inhomogeneity was detected in the diffraction patterns. This proved that the ligands were randomly embedded in the framework. DFT calculations were exploited to support the solid–solution hypothesis and to identify the factors responsible for cell expansion, with the aim of disclosing specific structure–property relationships. The experimental trends of cell expansion were correctly predicted by theory and the thermal and chemical stability of the framework could be explained on the basis of the electronic features of the MOFs. The unit cell of MIXUiO-66-NH2 slightly expanded as a consequence of a small steric contribution, while the metal–linker interaction was significantly weakened, causing loss of stability. MIXUiO-66-Br underwent a larger unit cell expansion due to the presence of bulky bromine atoms, however the electronic features remained very similar to those of pristine UiO-66, without affecting the stability of the framework. MIXMOFs prepared by post-synthetic exchange did not display the same behaviour, suggesting that different synthesis methods lead to materials with different properties. The findings reported in this work highlight the intimate correlation existing between the fine structural details, electronic features and physical–chemical properties of MIXMOFs, paving the way for a rational design of these materials with precise features.Acknowledgements
The authors thank Doris Sutter for NMR analysis. This work made use of the software package GNU Octave, and the authors are grateful for the support of the Octave development community.66 The authors acknowledge Paul Scherrer Institute for provision of the synchrotron radiation beamtime, proposal 20121129. The authors thank the Energy System Integration (ESI) platform at Paul Scherrer Institute for funding. This work was supported by a grant from the Swiss National Supercomputing Center (CSCS) under Project no. s611. DT and BS were partially supported by the National Center of Competence in Research (NCCR) “Materials' Revolution: Computational Design and Discovery of Novel Materials (MARVEL)” of the Swiss National Science Foundation (SNSF) and by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 666983).Notes and references
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. D. Burrows, CrystEngComm, 2011, 13, 3623 RSC.
- D. N. Bunck and W. R. Dichtel, Chem. – Eur. J., 2013, 19, 818–827 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- H. Furukawa, U. Muller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- J. G. McDaniel, K. Yu and J. R. Schmidt, J. Phys. Chem. C, 2013, 117, 17131–17142 CAS.
- T. Fukushima, S. Horike, Y. Inubushi, K. Nakagawa, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 4820–4824 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 CAS.
- X. Xu, J. A. van Bokhoven and M. Ranocchiari, ChemCatChem, 2014, 6, 1887–1891 CrossRef CAS.
- X. Xu, S. M. Rummelt, F. L. Morel, M. Ranocchiari and J. A. van Bokhoven, Chem. – Eur. J., 2014, 20, 15467–15472 CrossRef CAS PubMed.
- W. Kleist, F. Jutz, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 3552–3561 CrossRef CAS.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'Arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- W. Kleist, M. Maciejewski and A. Baiker, Thermochim. Acta, 2010, 499, 71–78 CrossRef CAS.
- F. L. Morel, M. Ranocchiari and J. A. van Bokhoven, Ind. Eng. Chem. Res., 2014, 53, 9120–9127 CrossRef CAS.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482–8486 CrossRef CAS PubMed.
- A. D. Burrows, L. C. Fisher, C. Richardson and S. P. Rigby, Chem. Commun., 2011, 47, 3380–3382 RSC.
- S. Marx, W. Kleist, J. Huang, M. Maciejewski and A. Baiker, Dalton Trans., 2010, 39, 3795–3798 RSC.
- Y. Jiang, J. Huang, S. Marx, W. Kleist, M. Hunger and A. Baiker, J. Phys. Chem. Lett., 2010, 1, 2886–2890 CrossRef CAS.
- T. Lescouet, E. Kockrick, G. Bergeret, M. Pera-Titus and D. Farrusseng, Dalton Trans., 2011, 40, 11359–11361 RSC.
- J. Bonnefoy, A. Legrand, E. A. Quadrelli, J. Canivet and D. Farrusseng, J. Am. Chem. Soc., 2015, 137, 9409–9416 CrossRef CAS PubMed.
- A. Beloqui Redondo, F. L. Morel, M. Ranocchiari and J. A. van Bokhoven, ACS Catal., 2015, 5, 7099–7103 CrossRef CAS.
- L. K. Cadman, J. K. Bristow, N. E. Stubbs, D. Tiana, M. F. Mahon, A. Walsh and A. D. Burrows, Dalton Trans., 2016, 45, 4316–4326 RSC.
- H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem. – Eur. J., 2005, 11, 3521–3529 CrossRef CAS PubMed.
- S. M. Chavan, G. C. Shearer, S. Svelle, U. Olsbye, F. Bonino, J. Ethiraj, K. P. Lillerud and S. Bordiga, Inorg. Chem., 2014, 53, 9509–9515 CrossRef CAS PubMed.
- Y. Sun, L. Sun, D. Feng and H. C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 6471–6475 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- C. H. Hendon, J. Bonnefoy, E. A. Quadrelli, J. Canivet, M. B. Chambers, G. Rousse, A. Walsh, M. Fontecave and C. Mellot-Draznieks, Chem. – Eur. J., 2016, 22, 3713–3718 CrossRef CAS PubMed.
- J. A. Thompson, C. R. Blad, N. A. Brunelli, M. E. Lydon, R. P. Lively, C. W. Jones and S. Nair, Chem. Mater., 2012, 24, 1930–1936 CrossRef CAS.
- J. A. Thompson, N. A. Brunelli, R. P. Lively, J. R. Johnson, C. W. Jones and S. Nair, J. Phys. Chem. C, 2013, 117, 8198–8207 CAS.
- K. Eum, K. C. Jayachandrababu, F. Rashidi, K. Zhang, J. Leisen, S. Graham, R. P. Lively, R. R. Chance, D. S. Sholl, C. W. Jones and S. Nair, J. Am. Chem. Soc., 2015, 137, 4191–4197 CrossRef CAS PubMed.
- T. Lescouet, E. Kockrick, G. Bergeret, M. Pera-Titus, S. Aguado and D. Farrusseng, J. Mater. Chem., 2012, 22, 10287 RSC.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- A. M. Katzenmeyer, J. Canivet, G. Holland, D. Farrusseng and A. Centrone, Angew. Chem., Int. Ed., 2014, 53, 2852–2856 CrossRef CAS PubMed.
- A. R. Denton and N. W. Ashcroft, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 3161–3164 CrossRef CAS.
- K. T. Jacob, S. Raj and L. Rannesh, Int. J. Mater. Res., 2007, 98, 776–779 CrossRef CAS.
- H. J. Axon and W. Hume-Rothery, Proc. R. Soc. A, 1948, 193, 1–24 CrossRef CAS.
- C. Michaelsen, Philos. Mag. A, 2006, 72, 813–828 CrossRef.
- C. Liu, T. Y. Luo, E. S. Feura, C. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2015, 137, 10508–10511 CrossRef CAS PubMed.
- K. C. Jayachandrababu, R. J. Verploegh, J. Leisen, R. C. Nieuwendaal, D. S. Sholl and S. Nair, J. Am. Chem. Soc., 2016, 138, 7325–7336 CrossRef CAS PubMed.
- J. R. Chelikowsky and K. E. Anderson, J. Phys. Chem. Solids, 1987, 48, 197–205 CrossRef CAS.
- R. E. Nahory, M. A. Pollack, W. D. Johnston and R. L. Barns, Appl. Phys. Lett., 1978, 33, 659 CrossRef CAS.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848 RSC.
- M. Servalli, M. Ranocchiari and J. A. Van Bokhoven, Chem. Commun., 2012, 48, 1904–1906 RSC.
- M. Kim, S. J. Garibay and S. M. Cohen, Inorg. Chem., 2011, 50, 729–731 CrossRef CAS PubMed.
- D. A. Gomez-Gualdron, P. Z. Moghadam, J. T. Hupp, O. K. Farha and R. Q. Snurr, J. Am. Chem. Soc., 2016, 138, 215–224 CrossRef CAS PubMed.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Boge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. Flechsig, F. Gozzo, B. Henrich, S. Jaggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Ludeke, R. Luscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitins, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- A. Dal Corso, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef CAS.
- K. Hendrickx, D. E. Vanpoucke, K. Leus, K. Lejaeghere, A. Van Yperen-De Deyne, V. Van Speybroeck, P. Van Der Voort and K. Hemelsoet, Inorg. Chem., 2015, 54, 10701–10710 CrossRef CAS PubMed.
- E. Flage-Larsen, A. Røyset, J. H. Cavka and K. Thorshaug, J. Phys. Chem. C, 2013, 117, 20610–20616 CAS.
- J. Portier, H. Hilal, I. Saadeddin, S. Hwang, M. Subramanian and G. Campet, Prog. Solid State Chem., 2004, 32, 207–217 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F. X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohen, J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed.
- H. Fei and S. M. Cohen, J. Am. Chem. Soc., 2015, 137, 2191–2194 CrossRef CAS PubMed.
- G. Nickerl, I. Senkovska and S. Kaskel, Chem. Commun., 2015, 51, 2280–2282 RSC.
- J. W. Eaton, D. Bateman and S. Hauberg, GNU Octave version 3.0.1 manual: a high-level interactive language for numerical computations, CreateSpace Independent Publishing Platform, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with amounts of reagents used in the syntheses; results of quantitative NMR analysis; HR-PXRD patterns; TG curves; nitrogen sorption isotherms; details of DFT calculations. See DOI: 10.1039/c6cp07801j |
| This journal is © the Owner Societies 2017 |