
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical characterization of the pyranose 2-oxidase variant N593C shows a complete loss of the oxidase function with full preservation of substrate (dehydrogenase) activity†
Dagmar
Brugger
ab,
Leander
Sützl
ab,
Kawah
Zahma
a,
Dietmar
Haltrich
ab,
Clemens K.
Peterbauer
ab and
Leonard
Stoica
*a
aFood Biotechnology Laboratory, University of Natural Resources and Life Sciences Vienna, Muthgasse 11, 1190 Vienna, Austria. E-mail: leonard.stoica@rub.de
bBioToP – The Doctoral Programme on Biomolecular Technology of Proteins, Muthgasse 18, 1190 Vienna, Austria
First published on 28th October 2016
Abstract
This study presents the first electrochemical characterization of the pyranose oxidase (POx) variant N593C (herein called POx-C), which is considered a promising candidate for future glucose-sensing applications. The resulting cyclic voltammograms obtained in the presence of various concentrations of glucose and mediator (1,4-benzoquinone, BQ), as well as the control experiments by addition of catalase, support the conclusion of a complete suppression of the oxidase function and oxygen reactivity at POx-C. Additionally, these electrochemical experiments demonstrate, contrary to previous biochemical studies, that POx-C has a fully retained enzymatic activity towards glucose. POx-C was immobilized on a special screen-printed electrode (SPE) based on carbon ink and grafted with gold-nanoparticles (GNP). Suppression of the oxygen reactivity at N593C-POx variant is a prerequisite for utilizing POx in electrochemical applications for glucose sensing. To our knowledge, this is the first report presented in the literature showing an absolute conversion of an oxidase into a fully active equivalent dehydrogenase via a single residue exchange.
Introduction
The flavoprotein pyranose 2-oxidase (POx; pyranose: oxygen 2-oxidoreductase; synonym: glucose 2-oxidase; EC 1.1.3.10) from Trametes multicolor (TmPOx; synonym, Trametes ochracea)1 was previously tested for glucose detection by immobilization within an Os-hydrogel film as electron-relay.1,2 A major drawback of using POx for glucose sensing, however, is its high affinity towards oxygen, as determined by Decamps.3 POx has a Km value towards oxygen of 0.46 mM, whereas the affinity of GOx from Aspergillus niger (AnGOx) for oxygen is 6-fold lower as expressed by the higher Michaelis constant Km of 2.9 mM. Oxygen reduction at POx thus interferes with the overall sensitivity towards glucose for a POx-based biosensor operating at low potentials, excluding the approach of detecting the enzymatically produced H2O2 by oxidation at potentials above +600 mV vs. Ag|AgCl.Aside from this limitation, POx presents several advantages over other oxidoreductases more commonly used for glucose sensing such as GOx and GDH.4 Similar to GOx, POx is also a member of the glucose–methanol–choline (GMC) oxidoreductase family,5,6 with obvious structural similarities. However, TmPOx shows an affinity towards D-glucose that is 72 times higher than that of AnGOx (Km values of 3.1 mM and 225 mM, respectively),3 which complies perfectly with the concentration range of clinical interest. Also, the enzymatic efficiency (vmax/Km) of POx towards D-glucose (9 U mM−1 × mg) is an order of magnitude higher than that of AnGOx (0.75 U mM−1 × mg), thus at least similar current densities from a POx biosensor are expected. In contrast to GOx, which is anomerically restricted to catalyze only the oxidation of β-D-glucose,7 POx converts both α- and β-D-glucose.3 The FAD cofactor is covalently bound to the polypeptide chain of POx,6,8 whereas the FAD cofactor of GOx is only tightly entrapped within the apoenzyme, and GDH relies on a freely diffusing PQQ cofactor.9
As shown in Scheme 1, homotetrameric TmPOx10 catalyzes the regioselective oxidation of aldopyranoses (D-glucose being its favoured substrate) at position C-2 forming the corresponding 2-ketoaldoses or osones.11 This catalytic oxidation of a sugar substrate by POx does not involve proton exchange and thus does not lead to a pH-shift, as is the case with GOx, which results in the formation of gluconic acid through hydrolysis of the primary oxidation product D-glucono-1,5-lactone following C1-oxidation of β-D-glucose.12 During this first half-reaction, the FAD-cofactor of POx is reduced to FADH2 (eqn (1)). The following half-reaction involves the reoxidation of the FADH2 cofactor by reducing molecular oxygen to H2O2 (eqn (2)).13 Possible alternative electron acceptors for POx are quinones (eqn (3)), certain radicals or chelated metal ions.10 Considering the immobilization of POx at polarized interfaces as depicted in Scheme 1, the resulting products of the enzymatic reaction are quasi-selectively oxidized at the electrode surface (see eqn (4) and (5)).
| FAD + aldopyranose → FADH2 + 2-keto-aldopyranose | (1) |
| FADH2 + O2 → FAD + H2O2 | (2) |
| FADH2 + benzoquinone → FAD + hydroquinone | (3) |
| hydroquinone → benzoquinone + 2H+ + 2e− | (4) |
| 2H2O2 + 2H+ + 2e− → H2O | (5) |
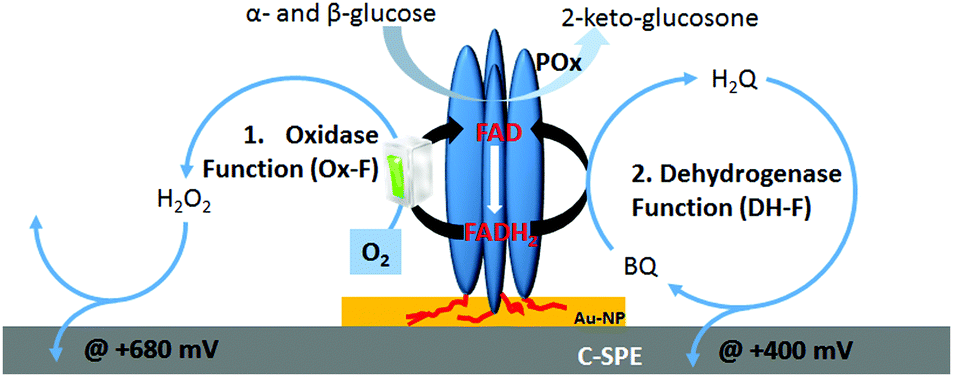 | ||
| Scheme 1 Schematic representation of immobilised POx homotetramer onto SPE electrode and the main electrochemical processes at the electrode, (1) oxidase function (Ox-F) and (2) dehydrogenase function (DH-F), undergoing at various polarization potentials in presence of catalytic oxidation of glucose at POx. | ||
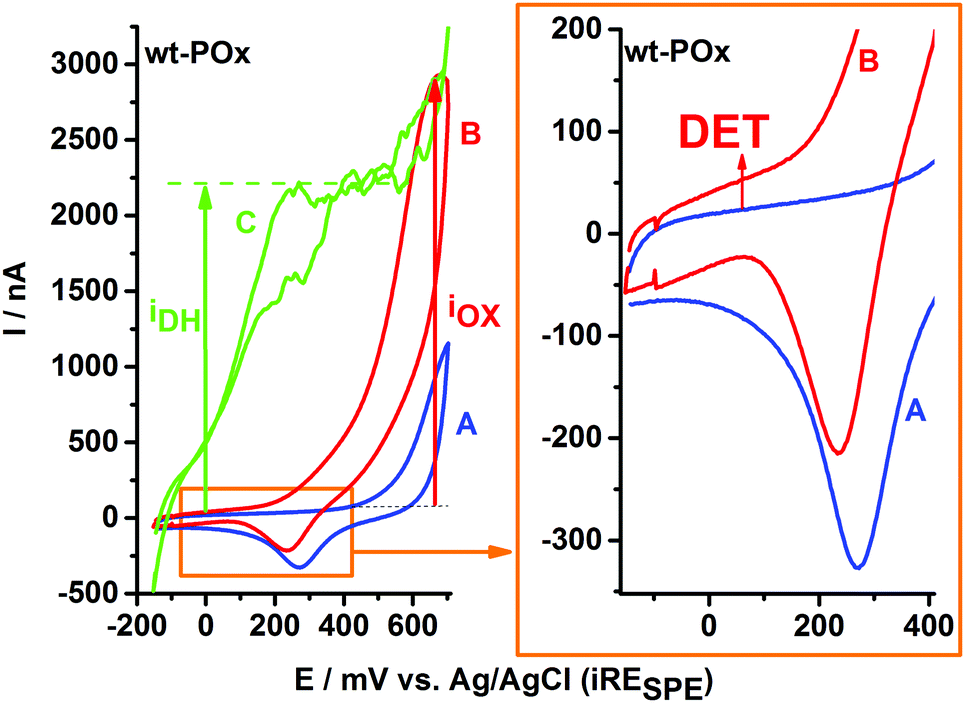 | ||
| Fig. 1 (left plot) Cyclic voltammograms recorded for wt-POx in various conditions: (A) in buffer; (B) in presence of 300 mM glucose; and (C) in presence of 300 mM glucose and 700 μM 1,4-benzoquinone (BQ). (right plot) Preliminary evidences of direct electron transfer (DET): magnification of CVs plot in the low potential range shows an upper shift in the oxidative direction in the presence of glucose (CV-B) in comparison to CV-A recorded in buffer. | ||
The gold–carbon electrode surface of a screen-printed electrode plays a dual role for the chemisorption of POx on gold-nanoparticles via existing His-tags and for recycling of 1,4-benzoquinone (enzyme mediator) between the reduced FAD cofactor of POx and the polarized carbon electrode.
In a previous biochemical study,14 site saturation mutagenesis and microtiter plate screening was used to identify POx variants with reduced oxidase activity but with preserved dehydrogenase activity, among them being the variant N593C. As seen in the crystal structure of TmPOx, residue N593 is involved in the electron transfer between the sugar substrate and the C(4a) position of the flavin6 and is located directly in the active site of TmPOx. In this work, we demonstrate the electrochemical consequences of the replacement of asparagine (N) at position 593 with cysteine (C), resulting in a complete shut-down of the oxidase function of POx.
Materials and methods
Chemicals
All chemicals were of the highest commercially available purity and purchased from Merck (Darmstadt, Germany) or Sigma Aldrich (St. Louis, MO), unless otherwise stated. Gold Nanoparticle (110GNP) modified Screen-Printed Carbon Electrodes (SPCEs) (DRP-110GNP) were purchased from DropSense (Llanera, Spain). All solutions were prepared using a 50 mM sodium phosphate buffer (pH 6.5).Generation of wild-type TmPOx and its mutant N593C
The generation of mutant N593C (herein denoted POx-C) and its counter-partners N593S and N593K, as well as the purification of the enzyme variants were carried out repetitive and separate by two co-authors (DB and LS) according to previous publications.15,16 After concentration and washing steps, both POx enzymes (wt-POx and POx-C) were stored at 8 °C. The activity assay, applied to wild-type POx (wt-POx) and the mutant N593C (POx-C) was previously described.17 Protein concentration was determined by the Bradford assay18 using the BioRad Protein Assay Kit with bovine serum albumin as standard. The final enzyme solutions wt-POx and POx-C had a protein concentration of 23 mg mL−1 and 6.1 mg mL−1, respectively.Electrode preparation and electrochemical set-up
The screen-printed electrode (SPE) platform consists of three electrodes: a 4 mm diameter disk-shaped carbon electrode as working electrode (WE), a printed silver-paste band as reference electrode (RE) and a 1 mm board carbon band as counter electrode (CE). The immobilization of wt-POx or POx-C onto the screen-printed electrode was performed by addition of a constant amount of the biocomponent (50 μg; approx. 2 or 8 μL enzyme solution diluted in 50 mM phosphate buffer pH 6.5). Chemisorption of the POx enzymes on the Au-NP was secured by the His6-tag of POx. After modification with the enzyme solution, the SPE were dried and cured overnight at 8 °C.All electrochemical measurements were carried out in 50 μL buffer droplets of appropriate composition, placed in a way to completely cover the three electrodes of the SPE and to preserve an identical, hemispherical geometry of the electrochemical cell. Since the entire volume of the electrochemical cell is only 50 μL, a rapid oxygen transfer from the gas phase and its diffusion towards the electrode are assured, together with the accumulation of any enzymatically generated H2O2 in the proximity of the electrode, thus securing a very sensitive electrochemical detection. In order to remove any weakly adsorbed enzyme from SPE and to achieve similar starting conditions for each modified SPE, four potential cycles were initially applied for each SPE by running twice two cycles of CV, including a buffer exchange step in between them. Stability of enzyme chemisorption permitted the run of repetitive experiments with various droplet compositions, using the same SPE sensor. All experiments were repeated at least three times on different SPEs, in order to qualitatively validate the observed bioelectrochemical behaviour of the immobilized enzymes.
Cyclic voltammetry (CV) experiments were performed at a Metrohm AUTOLAB B.V. potentiostat (PGSTAT204), and using NOVA 1.10 as controlling software for setting the experimental conditions and for recording/displaying the current output. For all CVs, the applied potential at WE was swept for two complete cycles with a scan rate of 10 mV s−1, starting from −150 mV and up to +700 mV versus an internal pseudo-reference electrode. All measurements were carried out at room temperature. Due to the short time of individual experiments, no special considerations were given to droplet evaporation.
The experiments in absence of oxygen were performed in a glove box system (Whitley DG250 Anaerobic Workstation) under nitrogen pressure and after repetitive degassing of the solutions before inclusion to the inert environment.
The raw data were further plotted using the Origin software (OriginLab Corporation).
Results and discussion
Fig. 1 exemplifies the typical sequence of experiments applied to characterize the bioelectrochemistry of immobilized POx showing a SPE modified with wt-POx. CV-A represents the background current recorded in buffer. Due to the presence of Au-NP, CV-A shows a typical signal for gold oxidation, which in the case of Au-NP starts at potentials above +500 mV, as well as its corresponding Au2O reduction peak at approx. +280 mV, characteristic for adsorbed species. Despite the overlap of the Au oxidation peak with the potential range for H2O2 oxidation, the trace electrochemistry of Au-NP (CV-A) can be easily neglected and discriminated from the CV-B, recorded in the presence of 300 mM glucose. Since H2O2 is a reaction product of glucose oxidation at wt-POx, it will accumulate in the vicinity of the electrode, due to the reduced volume (50 μL) of the electrochemical cell, leading to the corresponding oxidation wave visible in CV-B. Thus the function of Au-NP is not limited to an anchoring role for POx onto the electrode, but it also facilitates a better sensitivity towards H2O2 detection compared to carbon electrodes. This IOX current for H2O2 oxidation shows an onset potential around +250 mV and a peak of approx. 2.3 μA at +650 mV.In Fig. 1 (right plot), a zoom-in over the low potential range reveals an upper shift of the current waves, visible for both scanning directions in the presence of glucose, which is typical for a direct electron transfer (DET) process from wt-POx to the electrode. This has a value of approx. 30 nA. This DET current is visible for each POx-modified electrode, irrespective of whether wt-POx or POx-C are used, and thus seems to represent a true DET and not simply an experimental artifact. However, the magnitude of the current (30 nA) suggests that this DET phenomenon is only very low in comparison to the total catalytic current of approx. 2.3 μA (IOX) generated at the same electrode in the presence of glucose. In the presence of 700 μM 1,4-benzoquinone (BQ) as electron mediator, and 300 mM glucose as electron donor, the biocatalytic current (IDH in CV-C) generated by POx and recorded at the electrode by oxidation of hydroquinone at the carbon electrode starts sharply at approx. −100 mV and reaches a steady state of approx. 2.3 μA already at +400 mV. Since the value of IDH and IOX are rather equal in the case of wt-POx, we can conclude that both electron acceptors, oxygen and the mediator BQ, are equally preferred by wt-POx and therefore the oxidase function (Ox-F) at wt-POx has a similar proportion as the dehydrogenase function (DH-F). However, the two processes are competing with each other, and the proportion of each is determined only by the concentrations of substrates. Higher concentrations of mediator (BQ) do not lead to higher IDH currents, which suggests that the IOX current is not limited by the availability of oxygen, but only by the kinetics of the glucose oxidation step (kcat) and the number of available catalytic centers (IDH = IOX ∼ [Enz] × kcat).
The above sequence of experiments was repeated with wt-POx under anaerobic conditions (under nitrogen atmosphere) using glove-box system. In absence of oxygen, the addition of glucose does not produce any deviation of the current from background CV recorded in buffer only (see ESI,† SI-1). This demonstrates one more time that the oxidative current obtained in presence of glucose and oxygen is purely due to oxidation of H2O2 which is enzymatically formed by wt-POx and accumulated within the electrolyte drop. However, addition of mediator to the enzymatic system under anaerobic condition, but in presence of glucose, leads to a similar biocatalytic current, proving conserved activity of wt-POx.
The same experimental sequence was repeated in presence of oxygen with an N593C-POx (POx-C) modified SPE, and the resulting CVs are presented in Fig. 2 (right plot), whereas the characteristics of a wt-POx modified electrode (as described above) are presented for comparison in the left plot.
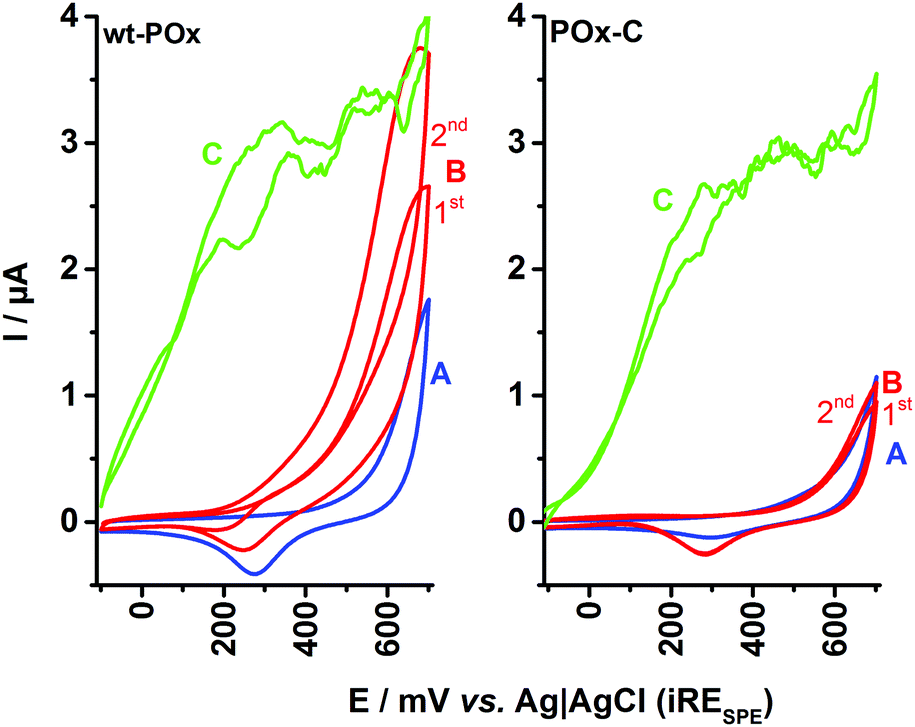 | ||
| Fig. 2 Comparative cyclic voltammograms recorded for SPEs modified with wt-POx (left plot) and POx-C variant N593C (right plot) for various conditions: (A) in 50 mM phosphate buffer pH 6.5 (CV-A – blue line); (B) in the presence of 300 mM D-glucose (CV-B – red line); and (C) in the presence of 300 mM D-glucose and 700 μM 1,4-benzoquinone (CV-C – green line). Experimental conditions: scan rate 10 mV s−1, applied potential −150 to +700 mV vs. Ag/AgCl. 1st and 2nd represent the first and second potential CV-scans. | ||
In the case of wt-POx, the addition of glucose leads to an oxidation current for the enzymatically produced H2O2, as observed also in Fig. 1. Consecutive CV cycles at the wt-POx modified electrode, in the presence of glucose, will produce an accumulation of H2O2 in the droplet, visible by an increase of the peak current (IOX) at +650 mV, between the first and second scans. However, in the case of POx-C modified SPE, the shape of the CV recorded in the presence of glucose and oxygen and in absence of the mediator remains identical to the voltammogram obtained in the presence of buffer, irrespective of the number of cycles applied. Due to the small volume of the electrochemical cell (50 μL), this result implies a total absence of H2O2, which could have been produced during glucose oxidation at POx-C. This indicates complete loss of the oxidase function at POx-C.
The full preservation of the activity of immobilized POx-C towards glucose is demonstrated by the addition of mediator (700 μM BQ) to the droplet (Fig. 2, left plot, CV-C). Thus, the mediator offers an alternative to oxygen reduction as well as an efficient pathway by shuttling the electrons from FADH2–POx-C to the carbon surface of SPE. The MET current for POx-C has a similar shape and a maximum IDH value of ∼3 μA at +400 mV, as recorded also for wt-POx. These observations support the hypothesis that POx-C has an intact catalytic activity and affinity for its sugar substrate comparable to wt-POx. In other words, the POx-C variant presents a pure dehydrogenase activity by completely suppressing the oxidase function visible at wt-POx.
In order to confirm these conclusions, further control experiments were conducted in the presence of 20 U of catalase, with the results displayed in Fig. 3 (left plot for wt-POx and right plot for POx-C variant). Thus, the CV in buffer (A-curves) and in the presence of glucose (B-curves) are identical to the results shown in Fig. 2. When 20 U of catalase are added to the glucose-containing droplet, a clear collapse of the recorded current is visible for wt-POx to the level of the background current in buffer, whereas in the case of POx-C modified electrodes, no differences between CVs recorded in buffer, with glucose, or with glucose +20 U of catalase are visible. This observation indicates once again that (i) not even traces of H2O2 are produced by POx-C in the presence of glucose, and that (ii) for wt-POx, the peak current IOX at +650 mV is indeed a result of the electrochemical oxidation of H2O2.
 | ||
| Fig. 3 Control cyclic voltammograms recorded for SPEs modified with wt-POx (left plot) and POx-C (right plot) in presence of buffer (A), in the presence of 300 μM glucose (B), in presence of 300 mM glucose and 20 U catalase (C), and in presence of 300 mM glucose, 700 μM 1,4-benzoquinone and 20 U catalase (D). Experimental conditions: scan rate 10 mV s−1, applied potential −150 to +700 mV vs. Ag/AgCl. | ||
In the CV-D for POx-C, recorded after the addition of 700 μM BQ (mediator) and in the presence of 300 mM glucose and 20 U of catalase, a high peak current (1.7 μA) for IDH is visible for the oxidation of accumulated hydroquinone, unperturbed by any H2O2 in the droplet. This MET biocatalytic peak current from +200 mV further drops with increasing potential to a diffusion limited plateau (∼1.3 μA), equivalent to the one recorded for wt-POx. This set of experiments demonstrates unequivocally the dehydrogenase-like behaviour of the POx-C variant.
In order to confirm the special importance of the mutation N593C at POx, two other variants obtained in similar ways, were considered for similar investigations, such as N593S-POx and N593K-POx. The dependence of IDH in CV at +400 mV versus glucose concentration (see ESI,† SI-2) was measured in the range starting from 0.5 mM glucose up to 300 mM, and in presence of constant concentration of 1,4-BQ (700 μM), thus showing a typical Michaelis–Menten dependency. In comparison to N593C-POx (with 67% apparent substrate efficiency remaining, as seen in Table 1), these two replacements of the asparagine (N) residue with serine (S) (non-charged and hydrophilic aminoacid) or with lysine (K) (positively charged long-arm aminoacid) do not facilitate any significant enzymatic activity (apparent substrate efficiency of 5% and 0%, respectively) on the electrode for POx in presence of glucose and mediator. This difference is mainly due to strong influence on substrate specificity, Km.
| K m [mM] | I max [μA] | I max/Km [μA mM−1] | I max/Km [%] | |
|---|---|---|---|---|
| wt-POx | 2.3 ± 0.1 | 2.0 ± 0.02 | 0.876 | 100 |
| N593C-POx | 2.5 ± 0.5 | 1.5 ± 0.1 | 0.583 | 67 |
| N593S-POx | 19.2 ± 1.7 | 0.9 ± 0.02 | 0.04 | 5 |
| N593K-POx | 108 ± 20 | 0.18 ± 0.01 | 0.0016 | 0 |
Recording the catalytic currents at the same electrode at fixed concentration of glucose (300 mM) and at variable concentrations of BQ, a clear competitive inhibition effect between BQ and oxygen was observed for the wtPOx (see ESI,† SI-3), whereas for the variant N593C-POx no oxygen interferences are visible and thus the linear range for measurement of BQ is extended, with clear applicability in the field of enzymatic determination of neurotransmitters.
Horaguchi et al.19 constructed oxygen-interacting structural models of GOx based on superimposition of different crystal structures. This method is also applied here in order to rationally understand the effect of the amino acid exchange in N593C on blocking the oxygen reduction reaction at POx-C. For this purpose, the crystal structures of POx,6 pyranose dehydrogenase (PDH) showing a flavin C(4a)–oxygen adduct,20 and cholesterol oxidase (ChOx) with bound dioxygen21 were superimposed. Fig. 4(A) displays the wild-type amino acid asparagine at position 593 of TmPOx within a distance of only 1.1 Å from a possible position of O2, as modelled from the ChOx crystal structure, and within 3.9 Å to the covalently bound flavin C(4a)–oxygen adduct, as deployed from the PDH crystal structure. Images (B) and (C) in Fig. 4 present the possible positions of the cysteine replacing the asparagine at position 593.
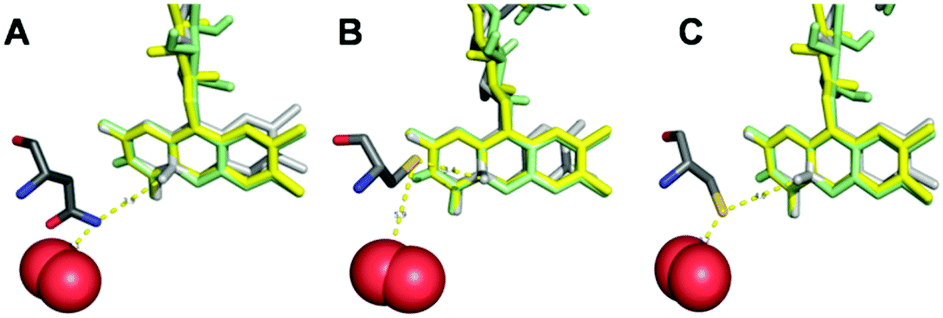 | ||
| Fig. 4 Interrelation of the residue at position 593 and the FAD-cofactor within the active site of Trametes multicolor POx and of its variant N593C. FAD from POx is shown in yellow, FAD from cholesterol oxidase (ChOx) in green and FAD from pyranose dehydrogenase (PDH) in light grey. The residue at position 593 is represented in dark grey and O2 in red. Image (A) shows the wild-type residue asparagine N593, images (B) and (C) display the two energetically favorable arrangements of the substituting cysteine residue (N593C) with respect to the FAD cofactor and to oxygen. The images were generated with PyMOL by employing coordinates from protein database (PDB) file 1tt0 for POx, file 4h7u for PDH and file 1mxt for ChOx (including O2). | ||
The PyMol software generated 75% probability for structure (B) with 3.4 Å distance of the sulfhydryl group in cysteine to the flavin C(4a)–oxygen adduct and 5.9 Å to O2; and 25% probability for structure (C) with 4.0 Å distance to the flavin C(4a)–oxygen adduct and 3.2 Å to O2. Irrespective of which conformation the cysteine residue at position 593 takes up, images B and C both suggest that this residue acts as an electronic screen between the donor FAD site and molecular oxygen as acceptor site of electrons. Furthermore, the cysteine residue does not have an acidic character (proton donor) in comparison to the replaced asparagine residue. The latter very likely carries a protonated amino group (proton donor) under physiological conditions, which can support the mechanism of oxygen reduction in the active site of POx, thus forming H2O2. Even though experimental evidence is lacking for these assumptions, these hypotheses can explain the lack of any oxidase functionality at POx-C, without altering the glucose reactivity as long as POx has a freely diffusing mediator as electron acceptor available in order to close the catalytic cycle, and thus explain the fully preserved dehydrogenase function at POx-C.
Thermal treatment of the modified electrodes for 1 h at 70 °C showed surprisingly high stability (∼50% for wtPOx and ∼30% for POx-C compared to untreated electrodes) of the enzymatic activity (see ESI,† SI-4). This thermal treatment induces however an increase of the direct electron transfer (DET) current recorded at low potentials for both enzymes in presence glucose, suggesting that only the tetrameric conformation of enzyme is most probably perturbed by the elevated temperature, however with no major loss of the enzymatic activity of the individual subdomains. This comes in contradiction to previous report22 which presented a complete loss of the enzyme activity in solution during less than 1 min exposure at 70 °C. The thermal resistance of the immobilized POx enzymes is an unexpected bonus for POx and a strong advantage for its application in the field of biomedical devices.
Conclusions
Electrochemical experiments demonstrate the complete switch-off of the oxidase function of pyranose 2-oxidase (POx) by only one single amino acid exchange at position 593, replacing asparagine (N) with cysteine (C). Contrary to the previous biochemical study on this variant,15 the electrochemical method used in this study is sufficiently sensitive to demonstrate in a volume of only 50 μL that the dehydrogenase function of POx remains unaltered by this amino acid exchange, and proves an unmodified reactivity of the variant towards glucose. To our knowledge, this is the first study presenting and demonstrating the transformation of an oxidase into a robust and catalytically efficient dehydrogenase. This study will represent the start of further investigations for understanding the reduction mechanism of molecular oxygen within the active site of oxidases, as well as to explore the applications of this newly engineered dehydrogenase in the field of biosensing, biofuel cell and biotechnology.Acknowledgements
This work was supported by the University of Natural Resources and Life Sciences Vienna (BOKU Doc Programme) and by the Austrian Science Fund (FWF) through individual grant P22094 (to CKP). DB and LS are members of the Doctoral Programme BioToP – Biomolecular Technology of Proteins (FWF grant W1224).Notes and references
- O. Spadiut, D. Brugger, V. Coman, D. Haltrich and L. Gorton, Electroanalysis, 2010, 22, 813–820 CrossRef CAS.
- F. Tasca, S. Timur, R. Ludwig, D. Haltrich, J. Volc, R. Antiochia and L. Gorton, Electroanalysis, 2007, 19, 294–302 CrossRef CAS.
- K. Decamps, I. J. Joye, D. Haltrich, J. Nicolas, C. M. Courtin and J. A. Delcour, Food Chem., 2012, 131, 1485–1492 CrossRef CAS.
- S. Ferri, K. Kojima and K. Sode, J. Diabetes Sci. Technol., 2011, 5, 1068–1076 CrossRef PubMed.
- (a) M. Albrecht and T. Lengauer, Bioinformatics, 2003, 19, 1216–1220 CrossRef CAS PubMed; (b) T. Wongnate and P. Chaiyen, FEBS J., 2013, 280, 3009–3027 CrossRef CAS PubMed.
- B. Martin Hallberg, C. Leitner, D. Haltrich and C. Divne, J. Mol. Biol., 2004, 341, 781–796 CrossRef PubMed.
- (a) B. E. P. Swoboda and V. Massey, J. Biol. Chem., 1965, 240, 2209–2215 CAS; (b) K. Kusai, I. Sekuzu, B. Hagihara, K. Okunuki, S. Yamauchi and M. Nakai, Biochim. Biophys. Acta, 1960, 40, 555–557 CrossRef CAS PubMed.
- P. Halada, C. Leitner, P. Sedmera, D. Haltrich and J. Volc, Anal. Biochem., 2003, 314, 235–242 CrossRef CAS PubMed.
- P. Dokter, J. Frank and J. A. Duine, Biochem. J., 1986, 239, 163–167 CrossRef CAS PubMed.
- C. Leitner, J. Volc and D. Haltrich, Appl. Environ. Microbiol., 2001, 67, 3636–3644 CrossRef CAS PubMed.
- S. Freimund, A. Huwig, F. Giffhorn and S. Köpper, Chem. – Eur. J., 1998, 4, 2442–2455 CrossRef CAS.
- R. Wilson and A. Turner, Biosens. Bioelectron., 1992, 7, 165–185 CrossRef CAS.
- M. Prongjit, J. Sucharitakul, B. A. Palfey and P. Chaiyen, Biochemistry, 2013, 52, 1437–1445 CrossRef CAS PubMed.
- D. Brugger, I. Krondorfer, C. Shelswell, B. Huber-Dittes, D. Haltrich, C. K. Peterbauer and C. M. Soares, PLoS One, 2014, 9, e109242 Search PubMed.
- D. Brugger, D. Haltrich and C. K. Peterbauer, New Biotechnol., 2014, 31, S21 CrossRef.
- D. Brugger, I. Krondorfer, K. Zahma, T. Stoisser, J. M. Bolivar, B. Nidetzky, C. K. Peterbauer and D. Haltrich, Biotechnol. J., 2014, 9, 474–482 CrossRef CAS PubMed.
- (a) O. Spadiut, C. Leitner, C. Salaheddin, B. Varga, B. G. Vertessy, T.-C. Tan, C. Divne and D. Haltrich, FEBS J., 2009, 276, 776–792 CrossRef CAS PubMed; (b) O. Spadiut, C. Leitner, T.-C. Tan, R. Ludwig, C. Divne and D. Haltrich, Biocatal. Biotransform., 2009, 26, 120–127 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- Y. Horaguchi, S. Saito, K. Kojima, W. Tsugawa, S. Ferri and K. Sode, Int. J. Mol. Sci., 2012, 13, 14149–14157 CrossRef CAS PubMed.
- T. C. Tan, O. Spadiut, T. Wongnate, J. Sucharitakul, I. Krondorfer, C. Sygmund, D. Haltrich, P. Chaiyen, C. K. Peterbauer, C. Divne and L. E. S. Netto, PLoS One, 2013, 8, e53567 CAS.
- P. I. Lario, N. Sampson and A. Vrielink, J. Mol. Biol., 2003, 326, 1635–1650 CrossRef CAS PubMed.
- O. Spadiut, T. C. Tan, I. Pisanelli, D. Haltrich and C. Divne, FEBS J., 2010, 277, 2892–2909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06009a |
| This journal is © the Owner Societies 2016 |