
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of N⋯O non-covalent interactions involving σ-holes: “electrostatics” or “dispersion”†
Rahul
Shukla
and
Deepak
Chopra
*
Crystallography and Crystal Chemistry Laboratory, Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-pass Road, Bhauri, Bhopal-462066, Madhya Pradesh, India. E-mail: dchopra@iiserb.ac.in; Fax: + 91 755 6692392
First published on 29th September 2016
Abstract
In this article, the existence of N⋯O noncovalent interactions was explored in per-halo substituted ammonia–water complexes. Optimized geometry at the MP2/aug-cc-pVTZ level shows that the N⋯O distance in all complexes is less than the sum of the vdW radii of N and O. The strength of these contacts was directly dependent on the extent of chlorine substitution on N or O atoms. Also, the level of theory and the basis set employed for the binding energy calculations have a direct effect on the strength of the N⋯O contacts. Energy decomposition analysis reveals that dispersion was the major contributor towards the stability of these contacts followed by electrostatic energy. The topological analysis further confirmed the existence of N⋯O contacts due to the presence of a bond critical point between the N and the O atom in all the complexes. These contacts have characteristics of a σ-hole interaction with the NBO analysis revealing that the primary charge transfer in all the complexes is occurring from O(lp) to σ*(N–X) orbitals, confirming these interactions to be predominantly in the category of pnicogen bonds.
Introduction
Investigation of noncovalent interactions is an important aspect of supramolecular chemistry1–5 as well as biology.6–10 In this regard, the hydrogen bond interaction11,12 is the most studied non-covalent interaction which has been extensively studied both experimentally and theoretically.13–17 However, the current focus has now shifted towards the investigation of noncovalent interactions involving “σ-holes”, which are a region of positive electrostatic potential on an atom that can act as an electrophile and is capable of interacting noncovalently with an electron rich nucleophile resulting in “σ-hole bonding”.18,19 σ-Hole bonding has been explored extensively in the case of halogen bonds,20 where it has been established that these σ-hole interactions are highly directional and stabilized.21–25 The concept of σ-hole bonding is now expanded well beyond the realm of halogen bonding and has been used to define other noncovalent interactions such as pnicogen bonds,26–28 chalcogen bonds,29–31 tetrel bonds,32–36 and aerogen bonds.37–39 Recent reviews on σ-hole interactions have demonstrated the growing importance of these interactions and have also discussed different methods which can be employed to study such interactions.21,40Pnicogen bonds which involve Group-V elements have been studied extensively since P⋯P interactions were first reported in orthocarborane derivatives through NMR studies.41,42 Recent reports have also pointed towards the importance of pnictogen bonds in crystal engineering.43–45 Studies have shown that pnicogen bonds are directional and stabilized interactions.26–28,46 While there are multiple reports of pnicogen bonds, involving P, As, and Sb as the electrophile,47–52 pnicogen bonds involving N as an electrophile are comparatively limited. However, it has been established both experimentally and computationally that under a suitable electronic environment N can have a σ-hole character and can participate in the formation of pnicogen bonds.44,53,54 Also, while there are reports of P/As/Sb⋯O pnicogen bonds,55,56 there is no study on N⋯O pnicogen bonds involving σ-holes. However, N⋯O bonds involving π-bonds have been reported in the literature.57 Similarly, chalcogen bonds which involve Group-VI elements as electrophilic centers have been studied extensively in the case of chalcogen–chalcogen58–60 interactions. Besides chalcogen–chalcogen interactions, chalcogen bonding with other non-chalcogen nucleophiles has also been studied in crystal structures.61–63 Most of the studies on chalcogen bonds have been centered on S and Se atoms but the possibility of O participation in chalcogen bonds has not been explored previously. Also, in the various ab initio analyses of chalcogen bonds, there are studies where N can act as a nucleophile in the formation of S⋯N and Se⋯N chalcogen bonds64–66 and hence it is of interest to study N⋯O contact in this regard. Also, the fact that both pnicogen and chalcogen are capable of behaving both as an electron-acceptor and as an electron-donor, during the formation of non-covalent interactions makes the study of N⋯O contact important.67–69
In this article, we explore the possibility of the existence of the unusual N⋯O non-covalent bond in a per-halo substituted ammonia–water complex, i.e. X1X2X3N⋯OY1Y2 [X1/X2/X3/Y1/Y2 = F, Cl] complex, through ab initio calculations. The reason we replaced all the hydrogen atoms with halogens was to eliminate the possibility of formation of strong O/N–H⋯N/O hydrogen bonds. The aim of this study was aimed at the following questions.
(1) Can N⋯O non-covalent bonds exist in an appropriate electronic environment?
(2) If N⋯O contacts in these complexes do exist, whether it will still be a σ-hole interaction, given the presence of strong electron withdrawing halogen atoms attached to both N and O simultaneously?
(3) Can such N⋯O contacts be categorized as a pnicogen bond or a chalcogen bond?
Computational details
The optimization of all the complexes was performed at the Møller–Plesset theory (MP2) level with the aug-cc-pVTZ level basis set using Gaussian09.70 All complexes were observed to be true minima with no imaginary frequency. All further calculations in this study were performed by utilizing the coordinates of the optimized geometry. Counterpoise-corrected binding energies were computed at the MP2/aug-cc-pVDZ level for all the complexes by taking into account the basis set superimposition error (BSSE).71 In addition to this, BSSE corrected binding energies were also evaluated at MP2/aug-cc-pVTZ and MP2/aug-cc-pVQZ levels to study the effect of basis set on binding energies. In addition to this, BSSE corrected binding energies were further evaluated at the CCSD(T) level using the aug-cc-pVDZ basis set. Binding energies were further extrapolated to the Complete Basis Set (CBS) limit,72 which is based on the idea that correlation energy is roughly proportional to X−3 for basis sets of the aug-cc-pVXZ type.73 This method has been employed in recent studies also.74–77| ΔEMP2/CBS = (64ΔEMP2/aug-cc-pVQZ − 27ΔEMP2/aug-cc-pVTZ)/37 |
| ΔECCSD(T)/CBS = EMP2/CBS + (ΔECCSD(T)/aug-cc-pVDZ − ΔEMP2/aug-cc-pVDZ) |
Results and discussion
The Molecular Electrostatic Potential (MEP) map for all the monomers is illustrated in Fig. 1. The blue and red regions correspond to positive electrostatic and negative electrostatic regions, respectively. In the case of NF3, N has a negative electrostatic region corresponding to lone pairs of electrons. This region of minimum on N becomes more negative varying from −0.92 to −12.30 kcal mol−1 as F attached to N is successively replaced with Cl. Along the F–N bond, the presence of a σ-hole on N is clearly evident in NF3. Upon gradual replacement of F attached to N with chlorine, the magnitude of the σ-hole on N decreases from 22.30 kcal mol−1 to 6.63 kcal mol−1; the trend being in the order NF3 > NF2Cl > NFCl2 > NCl3. This decrease in the magnitude of the σ-hole accompanied by an increase in magnitude of the negative electrostatic region can be attributed to the decrease in the electron withdrawing capability as F is replaced by Cl.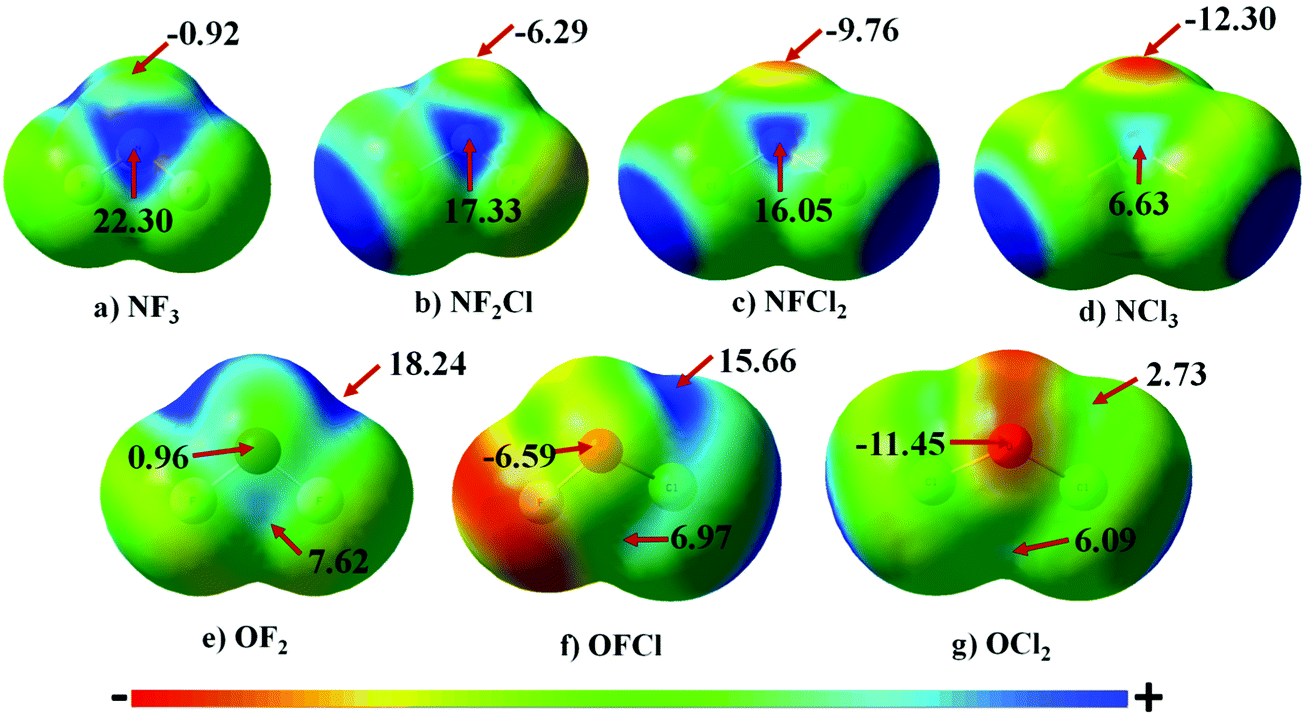 | ||
| Fig. 1 Electrostatic potential maps for the electrostatic positive regions of monomers on the 0.001 a.u. isodensity surface with potentials ranging from −12 kcal mol−1 (red) to 12 kcal mol−1 (blue). All energy values are reported in kcal mol−1. | ||
In the case of OF2, there are two regions of σ-hole on O atom. The first region is along the F–O bond having a maxima of 18.24 kcal mol−1 while the other is perpendicular to the F–O bond having a maxima of 7.62 kcal mol−1. These two regions of σ-hole are interconnected to each other in the case of OF2, which results in a region of positive electrostatic region of low magnitude on O [0.96 kcal mol−1] surrounded by a positive electrostatic region of large magnitudes. The successive replacement of F by Cl results in the strength of the σ-holes in both the regions on the O atom decreasing gradually. Also, the region of negative electrostatic potential is clearly evident in the case of OFCl and OCl2 monomers. In addition to this, in the case of OCl2, the σ-hole perpendicular to the Cl–O bond is more prominent [6.09 kcal mol−1] than that of the σ-hole along the Cl–O bond [2.73 kcal mol−1].
As evident from ESP analysis of the monomers, both N and O have both negative electrostatic as well as positive electrostatic (σ-hole) potential regions and hence it was interesting to explore the possibility of N⋯O noncovalent interactions. A total of 12 dimer combinations were possible using the monomers as shown in Scheme 1.
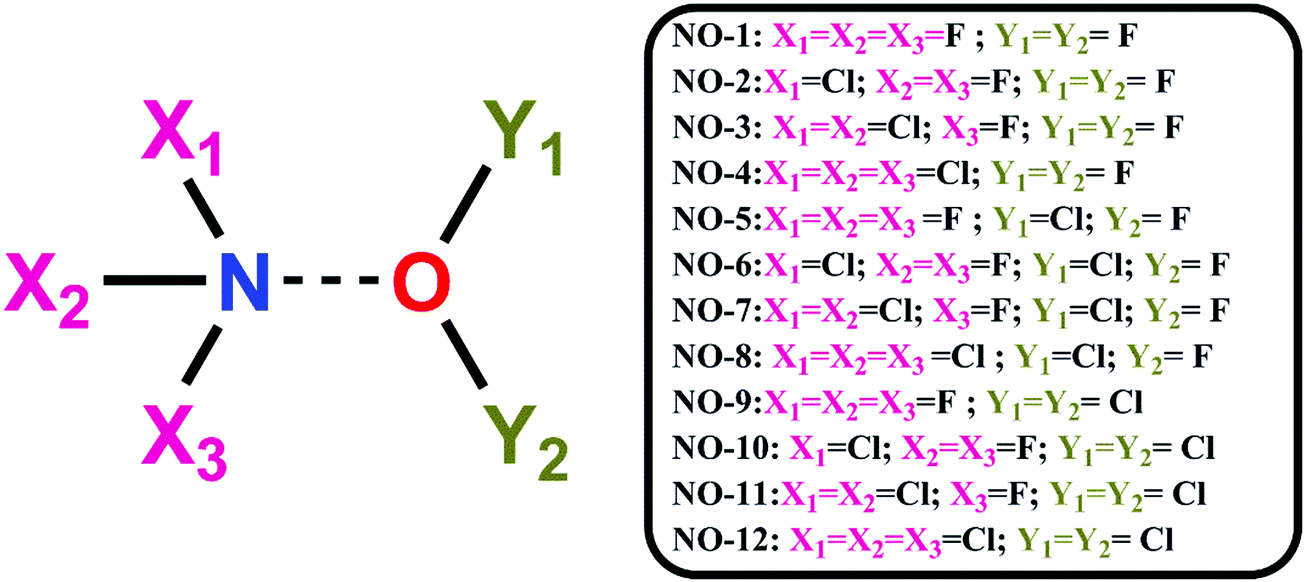 | ||
| Scheme 1 Nomenclatures of twelve complexes investigated in this study. | ||
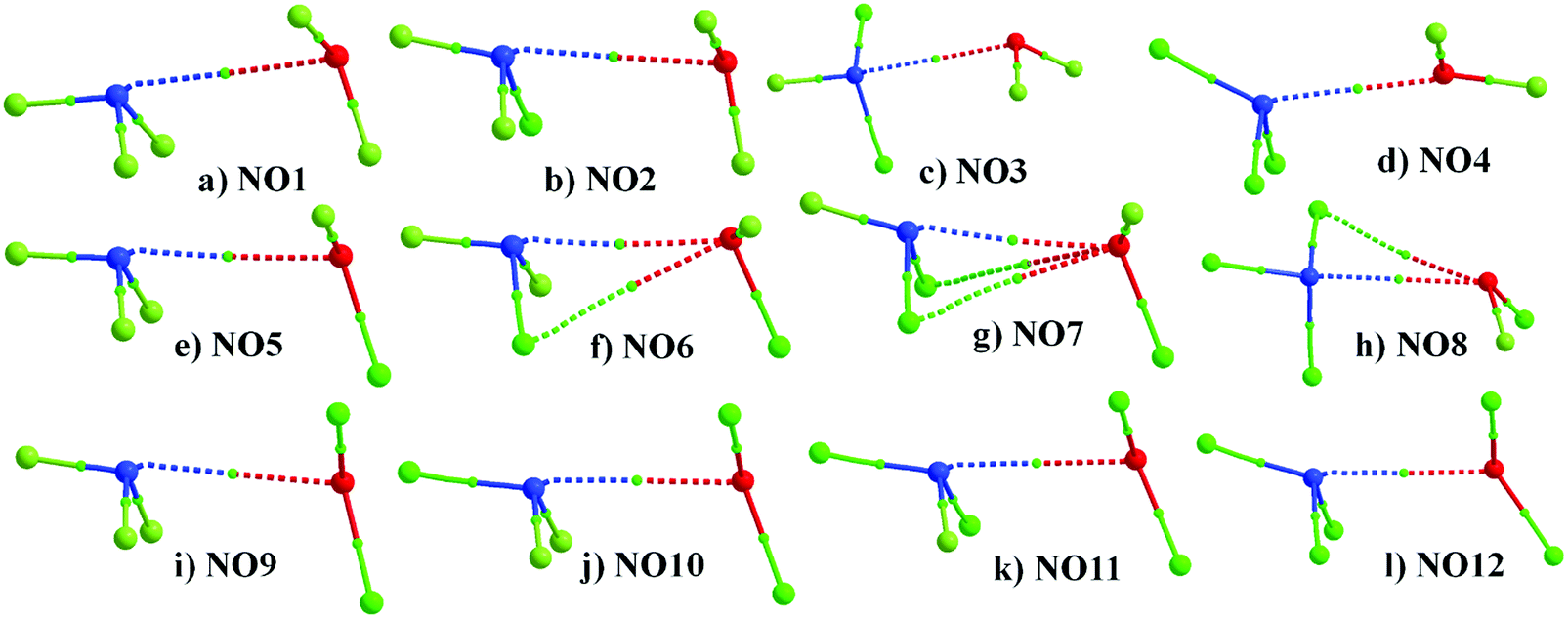
The 12 possible dimers were optimized and a true minimum was obtained in all the cases where it was evident that N and O are approaching towards each other for the formation of N⋯O noncovalent bonds [Fig. 2]. The N⋯O distance across all complexes ranged from 2.753 Å in NO4 to 3.042 Å in NO1, which is less than the sum of the vdW radii of nitrogen and oxygen [3.07 Å].90 A comparison between NO1 to NO4; NO5 to NO8; NO9 to NO12 reveals that there is decrease in N⋯O distance as F attached to N is replaced by Cl. The X–N⋯O angle was highly directional in these complexes, which corresponds to the N⋯O contact having characteristics of pnicogen bonds.68 The presence of pnicogen bond character, in some cases, even though both the interacting monomers have substantial positive σ-hole character is a case of counterintuitive interactions. Counterintuitive interactions are interactions between two positive or two negative electrostatic regions in the ground states and have been studied extensively in a recent study.91 The decrease in the N⋯O distance with Cl substitution was accompanied by a decrease in ∠X–N⋯O as F attached to N is replaced by Cl (Table 1). This decrease in ∠X–N⋯O with Cl substitution is accompanied by an increase in ∠Y–O⋯N. However, the magnitude of ∠X–N⋯O was much higher compared to ∠Y–O⋯N in all the cases except for NO4. In NO4, ∠Y–O⋯N (160°) was observed to be more directional than ∠X–N⋯O (142°) [Table 1]. A comparison with our previous study68 reveals that this points towards the presence of significant chalcogen bond characteristics in addition to features of pnicogen bonds in the case of NO4. This also explains why the N⋯O distance was observed to be the shortest in the case of NO4 as it has characteristics of two kinds of σ-hole interactions. Similarly, a comparison of NO1 with NO5, NO9; NO2 with NO6, NO10; NO3 with NO7, NO11; and NO4 with NO8, NO12 reveals that the replacement of F attached to O with Cl also results in a slight decrease in the N⋯O distance except in complexes involving NCl3 [Fig. 2].
| Complex | N⋯O dist. (Å) | ∠X–N⋯O (°) | ∠Y–O⋯N (°) | Bond path length [BPL] (Å) | ρ (e Å−3) | ∇2ρ (e Å−5) | |Vb|/Gb |
|---|---|---|---|---|---|---|---|
| NO1 | 3.042 | 174 | 102 | 3.071 | 0.039 | 0.676 | 0.710 |
| NO2 | 3.022 | 168 | 103 | 3.047 | 0.043 | 0.721 | 0.724 |
| NO3 | 2.957 | 161 | 121 | 2.981 | 0.051 | 0.845 | 0.725 |
| NO4 | 2.753 | 142 | 160 | 2.771 | 0.074 | 1.310 | 0.726 |
| NO5 | 2.999 | 177 | 108 | 3.028 | 0.043 | 0.756 | 0.718 |
| NO6 | 2.992 | 171 | 111 | 3.018 | 0.046 | 0.786 | 0.727 |
| NO7 | 2.964 | 167 | 115 | 2.987 | 0.052 | 0.851 | 0.734 |
| NO8 | 2.942 | 162 | 117 | 2.957 | 0.056 | 0.916 | 0.740 |
| NO9 | 2.958 | 174 | 108 | 2.986 | 0.050 | 0.832 | 0.734 |
| NO10 | 2.939 | 170 | 109 | 2.962 | 0.055 | 0.886 | 0.743 |
| NO11 | 2.921 | 165 | 110 | 2.939 | 0.059 | 0.950 | 0.751 |
| NO12 | 2.864 | 159 | 122 | 2.879 | 0.068 | 1.098 | 0.757 |
 | ||
| Fig. 2 Geometries of the optimized complexes. Distances are in Å and angles are in degrees (°). | ||
The counterpoise corrected binding energies of all the optimized structures were evaluated at the MP2/aug-cc-pvDZ level and the results show that the binding energies ranged from −0.54 kcal mol−1 for NO1 to −2.12 kcal mol−1 for NO12 (Table 2). It was also observed that the stability of different complexes was directly correlated with the nature of substitution on the N or O atom. As shown in Fig. 3, the increase in chlorine substitution on N leads to an enhancement in the stability of the complexes. Similarly, an enhancement in stability is also observed when F attached to O is replaced with the Cl atom keeping the substitution on the N atom unchanged. The trends observed in this study are in accordance with previous studies wherein it was observed that the presence of a heavier halogen atom in the complex results in a higher magnitude of the binding energy for σ-hole complexes.74 Also, a comparison of the trend in binding energies with geometrical parameters of optimized complexes shows that the smaller N⋯O distance is correlated with high binding energies. However, this increase in binding energy with increased Cl substitution was also accompanied by a decrease in ∠X–N⋯O and an increase in ∠Y–O⋯N (Tables 1 and 2). The weak nature of these interactions also suggests that a cautious approach should be adopted to discuss the N⋯O contacts.
| MP2/aug-cc-pVDZ | MP2/aug-cc-pVTZ | MP2/aug-cc-pVQZ | CCSD(T)/aug-cc-pVDZ | CBS/MP2 | CBS/CCSD(T) | |
|---|---|---|---|---|---|---|
| NO1 | −0.54 | −0.7 | −0.75 | −0.54 | −0.79 | −0.79 |
| NO2 | −0.7 | −0.94 | −1.02 | −0.6 | −1.08 | −0.98 |
| NO3 | −0.97 | −1.26 | −1.37 | −0.7 | −1.45 | −1.18 |
| NO4 | −1.37 | −1.65 | −1.77 | −0.89 | −1.86 | −1.38 |
| NO5 | −0.87 | −1.1 | −1.18 | −0.83 | −1.24 | −1.2 |
| NO6 | −1.15 | −1.52 | −1.65 | −0.9 | −1.74 | −1.49 |
| NO7 | −1.39 | −1.88 | −2.06 | −0.89 | −2.19 | −1.69 |
| NO8 | −1.47 | −1.98 | −2.16 | −0.95 | −2.29 | −1.77 |
| NO9 | −1.12 | −1.43 | −1.54 | −0.97 | −1.62 | −1.47 |
| NO10 | −1.2 | −1.55 | −1.68 | −0.95 | −1.77 | −1.52 |
| NO11 | −1.63 | −2.13 | −2.31 | −1.1 | −2.44 | −1.91 |
| NO12 | −2.12 | −2.77 | −3.01 | −1.19 | −3.18 | −2.25 |
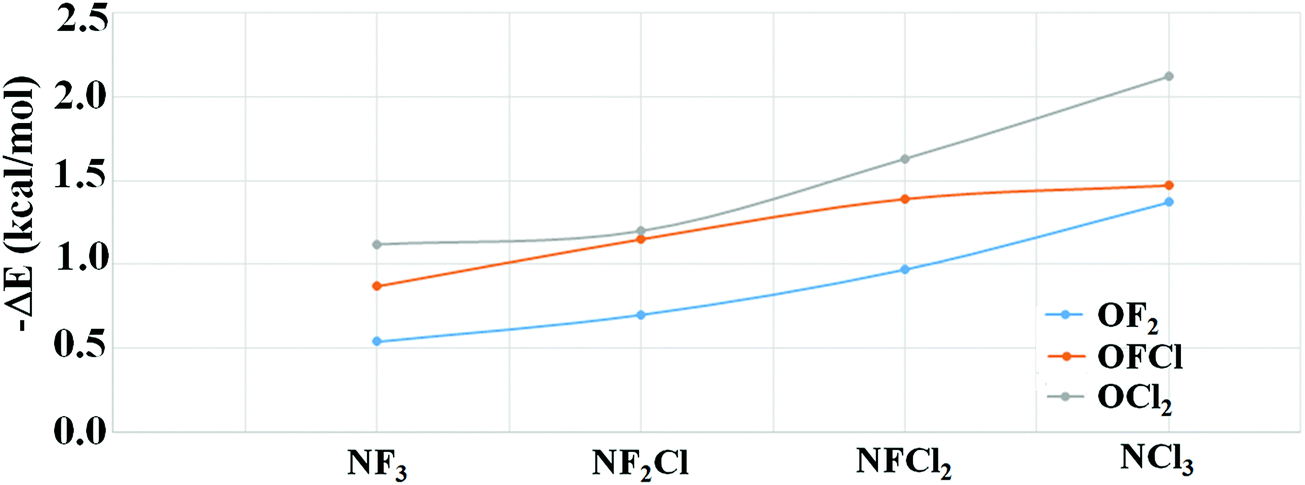 | ||
| Fig. 3 Change in magnitude of binding energy calculated at MP2/aug-cc-pVDZ with the increasing Cl substitution. | ||
To get deeper insights into the nature of N⋯O noncovalent interactions, we performed energy decomposition analysis using the LMO-EDA module present in GAMESS to evaluate the contribution of different energy components towards the strength of these noncovalent interactions at the MP2/aug-cc-pVDZ level. The total energies for each complex are decomposed into the electrostatic energy (Eelec), exchange-repulsion energy (Eex-rep), polarization energy (Epol), and the dispersion energy (Edisp). Eelec, Epol, and Edisp contribute towards the stability of the interaction while Eex-rep contributes towards the destabilization. The result shows that the magnitude of each stabilizing energy component increases simultaneously with the increase in Cl substitution on the N or O atom (Table S1, ESI†). In all the complexes formed, dispersion was the major contributor followed by contribution from electrostatic and polarization, respectively. The percentage contribution of dispersion was at least 60% towards stability in all the complexes except for NO4 where the contribution was calculated to be 57.5% [Fig. 4]. The percentage contribution was evaluated by adding the magnitudes of Eelec, Epol and Edisp for a given complex and then dividing the individual magnitude with the total sum. The contribution of electrostatic energies ranged from 21.7% in NO1 to 33.3% in NO4. The contribution of polarization was calculated to be 4% to 6.1% except for NO4 where the contribution of polarization was 9.1%. The highest percentage contribution of electrostatics and polarization combined with the lowest percentage contribution of dispersion towards the stability of NO4 can be attributed to the dual character of pnicogen and chalcogen bonds in the complex. In contrast to previously studied chalcogen and pnicogen bonds,66,68,74 the contribution of the electrostatic energy was observed to be less than that of dispersion energy. This shows that the characteristics of N⋯O contacts are quite different from the previously studied σ-hole interactions.
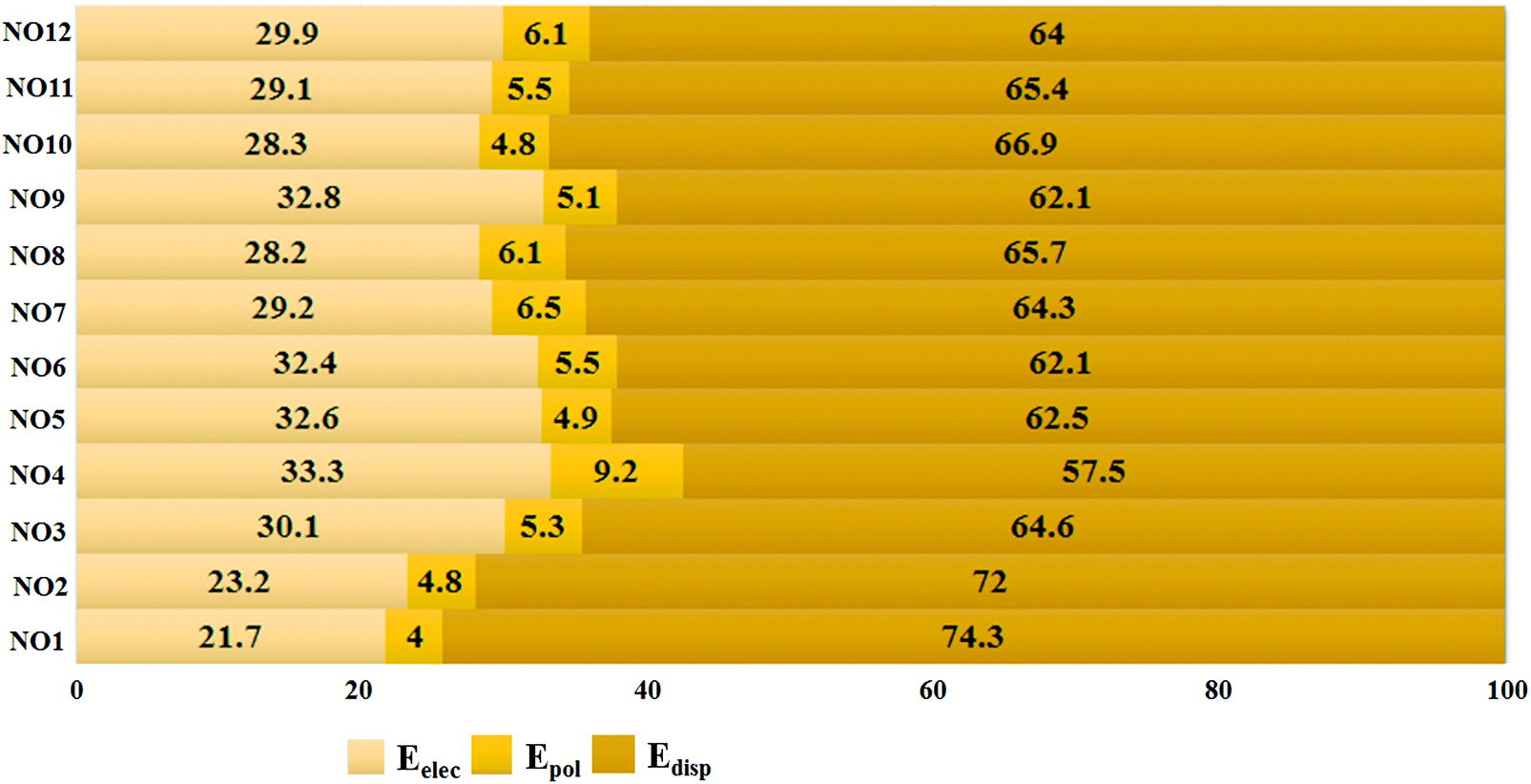 | ||
| Fig. 4 Percentage contribution of Eelec, Epol, and Edisp towards the total binding energies for each complex. | ||
Since, the binding energy of the complex can depend on the basis set used, we also evaluated the BSSE corrected binding energies at higher basis sets (Table 2). The result shows that there is an enhancement in the binding energy as we go from double-ξ to triple-ξ and subsequently to quadruple-ξ at the MP2 level. The enhancement in the magnitude of binding energies was more prominent while going from double-ξ to triple-ξ as compared to going from triple-ξ to quadruple-ξ (Fig. 5). Extrapolation of these values to the CBS limit at the MP2 level results in a slight increase in the magnitude of the binding energies. Binding energies evaluated at CCSD(T)/aug-cc-pVDZ results in a decrease in binding energies as compared to the values computed at the MP2 level using the same basis set. Extrapolation to the CBS limit at the CCSD(T) level results in an enhancement in the magnitude of the binding energies in all the complexes. The magnitude of the binding energies at the CCSD(T)/CBS limit was closer to those observed at the MP2/aug-cc-pVTZ level.
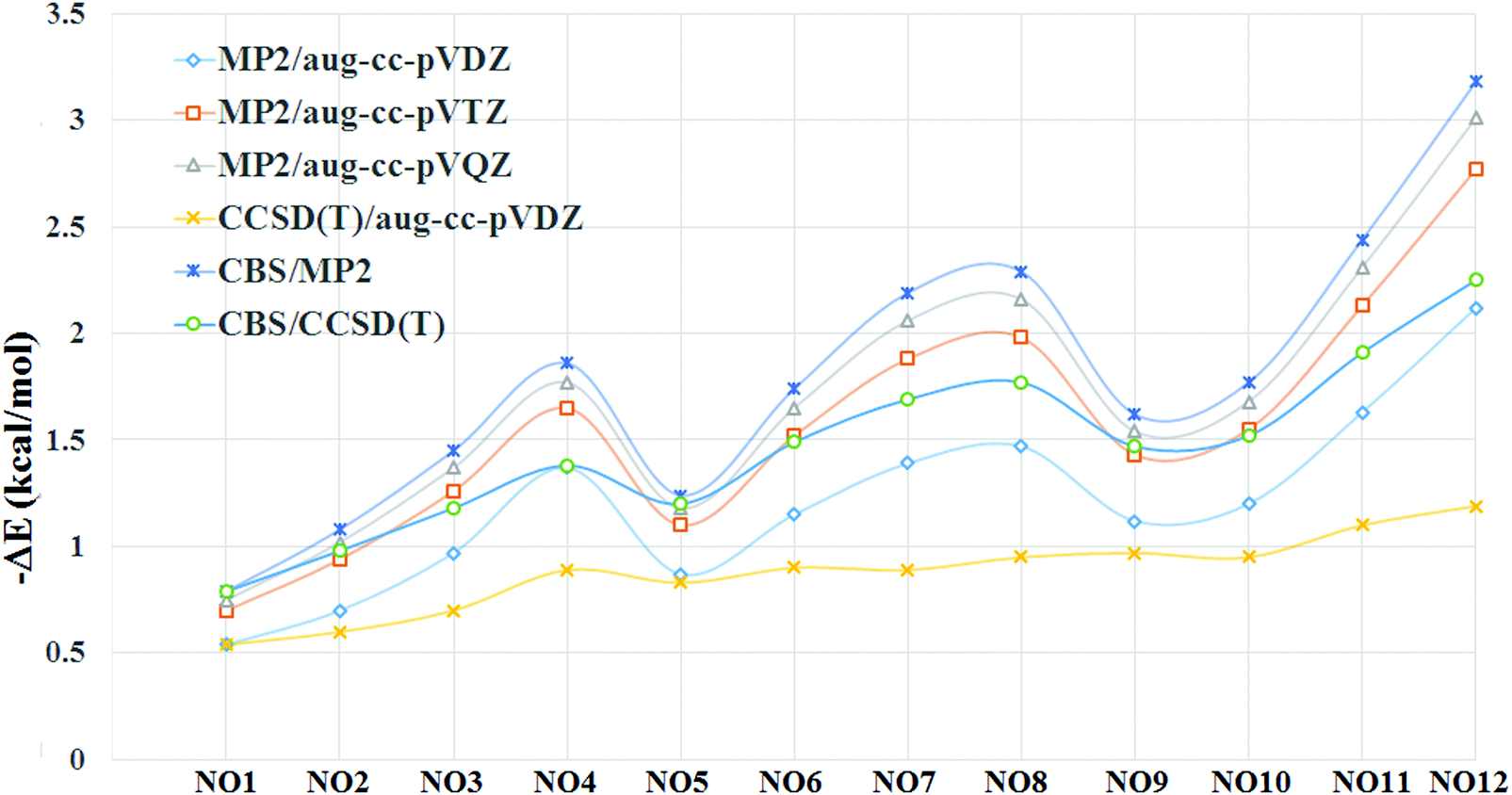 | ||
| Fig. 5 Variation of binding energies at different levels of theory and/or basis sets. | ||
In numerous studies, the Quantum Theory of Atoms in Molecules, developed by late Prof. R. F. W. Bader, has been widely used for determining the topological properties of different kinds of σ-hole interactions. For a better understanding of the N⋯O noncovalent bonds, AIM analysis was performed on all the complexes at the MP2/aug-cc-pVTZ level. The molecular graph of all the twelve complexes is shown in Fig. 6. The presence of a Bond Critical Point [BCP] between N and O is clearly evident in all the complexes, further confirming the existence of these interactions. Table 1 presents the value of Bond Path Length [BPL], electron density (ρ), Laplacian (∇2ρ) of electron density, local potential energy (Vb), and local kinetic energy (Gb) at N⋯O BCPs. The magnitude of the N⋯O bond path length (BPL) ranged from 2.771 to 3.071 Å, which was similar to the bond length of the N⋯O contacts. Here it is important to understand that although N⋯O bond length and bond path length have similar magnitudes, the origin of these values are different.92 The magnitude of ρ at N⋯O BCP ranged from 0.039 to 0.074 e Å−3 and the ∇2ρ were all positive with values ranging from 0.676 to 1.310 e Å−5 across all the complexes. The small magnitude of ρ and positive ∇2ρ show that the N⋯O contacts are closed-shell interactions. The magnitude of both ρ and Laplacian was in the range proposed by Koch and Popelier.93 Espinosa et al.94 suggested that the |Vb|/Gb ratio is a very good indicator for determining the behavior of noncovalent interactions. If |Vb|/Gb < 1, the bond is an electrostatic interaction; if 1 < |Vb|/Gb < 2, the bond will have a partial covalent nature; and when |Vb|/Gb > 2, it points towards the formation of a covalent bond. In our systems, in all the cases at N⋯O BCP, |Vb|/Gb < 1, which indicates the presence of electrostatic characteristics in N⋯O contacts. In addition to the presence of BCPs between N and O in all complexes, additional BCPs were observed between O and Cl in the case of NO6, NO7 and NO8 (Fig. 6). The magnitude of ρ and ∇2ρ at the O⋯Cl BCP was observed to be less than the corresponding N⋯O contact present in the same complexes (Table S2, ESI†).
 | ||
| Fig. 6 Molecular graph showing the existence of N⋯O BCP in all complexes. | ||
Natural Bond Orbital (NBO) analysis was also performed to evaluate the donor–acceptor orbital interactions and associated second-order perturbation energies E(2) for the N⋯O interactions (Table S3, ESI†). In all the complexes, O(lp) to σ*(N–X) was the major orbital interaction between the monomers of the complexes. However, the magnitude of this orbital interaction was quite low with values ranging from 0.28 to 0.66 kcal mol−1. But this shows that all complexes were predominantly pnicogen bonds where the σ-hole present on N was interacting noncovalently with the lone pair of electrons present on the O atom. In the case of NO4, the contribution of N(lp) to σ*(O–Y) was also observed to be significant with a magnitude of 0.38 kcal mol−1, showing the presence of chalcogen bond characteristics in addition to the pnicogen bond character (Table S3, ESI†). This is in accordance with the high directionality of ∠Y–O⋯N in addition to ∠X–N⋯O observed in the case of NO4. This further confirms the notion that directionality of noncovalent interactions plays a direct role in orbital interactions. The charges obtained for monomer fragments participating in complex formation from NBO analysis were equal and opposite in all cases (Table S3, ESI†).
NCI analyses around the N⋯O bond critical point for the least stable (NO1) and the most stable (NO12) contacts were performed. It is based on the concept of the presence of the electron density between interacting atoms, which can be obtained from experimental and theoretical calculations. The reduced density gradient [RDG = |∇ρ|/2(3π2)1/3ρ4/3] can be used to describe the nature of interaction between two atoms. NCI at each RDG point can be characterized by using the NCI descriptor based on sign(λ2)ρ. The plot of ρ*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sign(λ2) against RDG can be used to distinguish between stabilizing [ρ*sign(λ2) < 0] and destabilizing [ρ*sign(λ2) > 0] interactions.86–88Fig. 7(a) and (b) shows the presence of interacting isosurface regions between N and O in the case of NO1 and NO2. The plot of ρ*sign(λ2) against RDG clearly demonstrates that the N⋯O contacts are stabilized interactions due to ρ*sign(λ2) < 0 (Fig. 7(c) and (d)).
sign(λ2) against RDG can be used to distinguish between stabilizing [ρ*sign(λ2) < 0] and destabilizing [ρ*sign(λ2) > 0] interactions.86–88Fig. 7(a) and (b) shows the presence of interacting isosurface regions between N and O in the case of NO1 and NO2. The plot of ρ*sign(λ2) against RDG clearly demonstrates that the N⋯O contacts are stabilized interactions due to ρ*sign(λ2) < 0 (Fig. 7(c) and (d)).
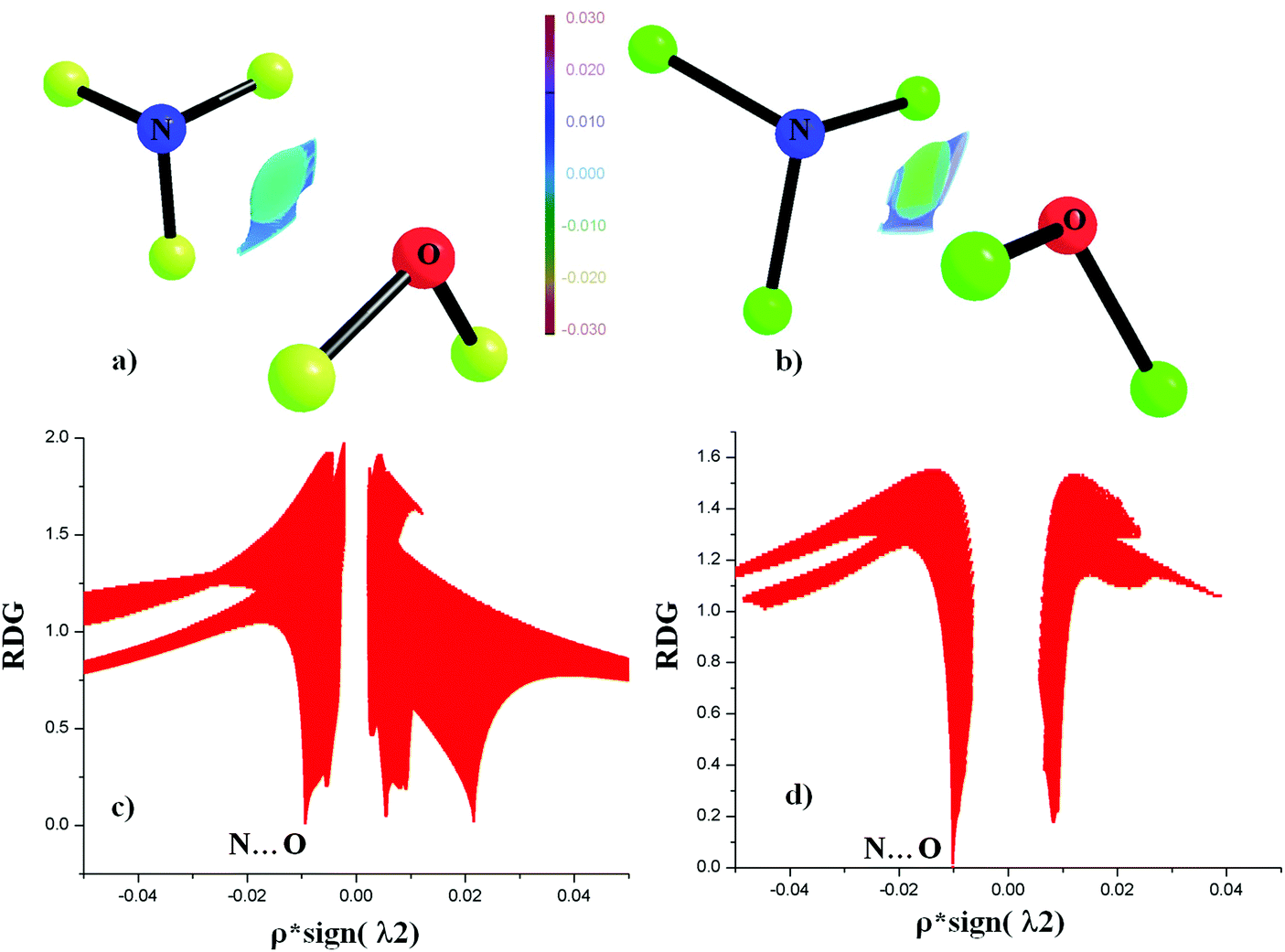 | ||
| Fig. 7 RDG isosurface for N⋯O contact for (a) NO1 and (b) NO2. The color is plotted in the range of −0.03 ρ*sign(λ2) 0.03 a.u. Plot of reduced density gradient (RDG) versus electron density multiplied by the sign of the second Hessian eigen value [ρ*sign(λ2)] for (c) NO1 (d) NO2. | ||
Summary
In this study, we have explored the existence of N⋯O noncovalent interactions in per-halo substituted ammonia–water complexes, i.e. X1X2X3N⋯OY1Y2 [X1/X2/X3/Y1/Y2 = F, Cl] through ab initio calculations and explored their nature and characteristics through different methods. All the complexes investigated in this study confirmed the existence of stabilized and highly directional N⋯O contacts. The existence of these contacts was further confirmed by the existence of a bond critical point between N and O in all complexes. The |Vb|/Gb ratio shows the presence of electrostatic behavior in these interactions, which is complemented by the result of energy decomposition analysis showing the substantial contribution of electrostatic energy towards the stability of these complexes. However, the contribution of dispersion energy towards stabilization was more prominent in all the cases, which are also supported by NCI analysis. Also, the analysis of ESP confirms the existence of σ-hole on N and O atoms and NBO analysis point towards the fact that N⋯O contacts indeed fall into the category of σ-hole interaction due to a major charge transfer from O(lp) to σ*(N–X) resulting in the formation of pnicogen bonds.Acknowledgements
We thank the reviewers for their suggestions towards the improvement of the manuscript. R. S. thanks DST-INSPIRE for the PhD Scholarship. R. S. also thanks Prof. T. N. Guru Row for providing computational facility in his laboratory. D. C. thanks IISER Bhopal for infrastructural facility and DST-SERB for research funding.References
- M. Raynai, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2011, 43, 1660–1733 RSC.
- A. Tkatchenko, D. Alfé and K. S. Kim, J. Chem. Theory Comput., 2012, 8, 4317–4322 CrossRef CAS PubMed.
- G. R. Desiraju, Chem. Commun., 1997, 1475–1482 RSC.
- H. S. Schneider, T. Schiestel and P. Zimmermann, J. Am. Chem. Soc., 1992, 114, 7698–7703 CrossRef CAS.
- H. R. Khavasi, A. Ghanbarpour and A. A. Tehrani, RSC Adv., 2016, 6, 2422–2430 RSC.
- N. J. Zondlo, Nat. Chem. Biol., 2010, 6, 567–568 CrossRef CAS PubMed.
- V. Ball and C. Maechling, Int. J. Mol. Sci., 2009, 10, 3283–3315 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CrossRef CAS.
- H. Zhu, I. Sommer, T. Lengauer and F. S. Domingues, PLoS One, 2008, 3, e1926 Search PubMed.
- M. Gurusaran, M. Shankar, R. Nagarajan, J. R. Helliwell and K. Sekar, IUCrJ, 2014, 1, 74–81 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- A.-C. Uldry, J. M. Griffin, J. R. Yates, M. Pérez-Torralba, M. D. S. Maria, A. L. Webber, M. L. L. Beaumont, A. Samoson, R. M. Claramunt, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 945–954 CrossRef CAS PubMed.
- D. Dey, T. P. Mohan, B. Vishalakshi and D. Chopra, Cryst. Growth Des., 2014, 14, 5881–5896 CAS.
- P. Panini and D. Chopra, Cryst. Growth Des., 2014, 14, 3155–3168 CAS.
- J. Contrera-Garcia and W. Yang, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed.
- B. Nepal and S. Scheiner, J. Phys. Chem. A, 2014, 118, 9575–9587 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and P. Lane, Int. J. Quantum Chem., 2007, 107, 3046–3052 CrossRef CAS.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed and references therein.
- A. R. Voth, P. Khuu, K. Oishi and P. S. Ho, Nat. Chem., 2009, 1, 74–79 CrossRef CAS PubMed.
- D. J. R. Duarte, G. L. Sosa, N. M. Peruchena and I. Alkorta, Phys. Chem. Chem. Phys., 2016, 18, 7300–7309 RSC.
- N. Cheng, F. Bi, Y. Liu, C. Zhang and C. Liu, New J. Chem., 2014, 38, 1256–1263 RSC.
- M. Kolář, J. Hostaš and P. Hobza, Phys. Chem. Chem. Phys., 2014, 16, 9987–9996 RSC.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem. – Eur. J., 2011, 17, 6034–6038 CrossRef CAS PubMed.
- L. Guan and Y. Mo, J. Phys. Chem. A, 2014, 118, 8911–8921 CrossRef CAS PubMed.
- S. Scheiner, Acc. Chem. Res., 2013, 46, 280–288 CrossRef CAS PubMed.
- W. Wang, B. Ji and Yu. Zhang, J. Phys. Chem. A, 2009, 113, 8132–8135 CrossRef CAS PubMed.
- S. Tsuzuki and N. Sato, J. Phys. Chem. B, 2013, 117, 6849–6855 CrossRef CAS PubMed.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 125, 12543–12547 CrossRef.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824–1834 RSC.
- D. Mani and E. Arunan, Phys. Chem. Chem. Phys., 2013, 15, 14377–14383 RSC.
- S. P. Thomas, M. S. Pavan and T. N. Guru Row, Chem. Commun., 2014, 50, 49–51 RSC.
- E. C. Escudero-Adán, A. Bauzá, A. Frontera and P. Ballester, ChemPhysChem, 2015, 16, 2530–2533 CrossRef PubMed.
- A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed.
- A. Bauzá and A. Frontera, Phys. Chem. Chem. Phys., 2015, 17, 24748–24753 RSC.
- A. Bauzá and A. Frontera, ChemPhysChem, 2015, 16, 3625–3630 CrossRef PubMed.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed and references therein.
- W. E. Hill and L. M. Silva-Trivino, Inorg. Chem., 1978, 17, 2495–2498 CrossRef CAS.
- W. E. Hill and L. M. Silva-Trivino, Inorg. Chem., 1979, 18, 361–364 CrossRef CAS.
- P. Politzer, J. S. Murray, G. V. Janjić and S. D. Zarić, Crystals, 2014, 4, 12–31 CrossRef.
- S. Sarkar, M. S. Pavan and T. N. Guru Row, Phys. Chem. Chem. Phys., 2015, 17, 2330–2334 RSC.
- A. Bauzá, D. Quiñonero, P. M. Deyá and A. Frontera, CrystEngComm, 2013, 15, 3137–3144 RSC.
- S. J. Grabowski, Chem. – Eur. J., 2013, 19, 14600–14611 CrossRef CAS PubMed.
- Y. Chen, L. Yao and X. Lin, Comput. Theor. Chem., 2014, 1036, 44–50 CrossRef CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2016, 17, 1608–1614 CrossRef PubMed.
- D. Setiawan, E. Kraka and D. Cremer, J. Phys. Chem. A, 2015, 119, 1642–1656 CrossRef CAS PubMed.
- C. Trujillo, G. Sánchez-Sanz, I. Alkorta and J. Elguero, New J. Chem., 2015, 39, 6791–6802 RSC.
- G. Sánchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, Comput. Theor. Chem., 2015, 1053, 305–314 CrossRef.
- J. E. D. Bene, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2014, 118, 2360–2366 CrossRef PubMed.
- S. Scheiner, Chem. Phys. Lett., 2011, 514, 32–35 CrossRef CAS.
- G. Tripathi, K. Badi-uz-zama and G. Ramanathan, Chem. Phys. Lett., 2016, 653, 117–121 CrossRef CAS.
- M.-X. Liu, H.-Y. Zho, Q.-Z. Li and J.-B. Cheng, J. Mol. Model., 2016, 22, 10 CrossRef PubMed.
- I. Alkorta, J. E. Del Bene and J. Elguero, Crystals, 2016, 6, 19 CrossRef.
- G. Sánchez-Sanz, C. Trujillo, M. Solimannejad, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2013, 15, 14310–14318 RSC.
- C. Bleiholder, D. B. Werz, H. Köpper and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef CAS PubMed.
- D. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638–10639 CrossRef CAS PubMed.
- C. Bleiholder, R. Gleiter, D. B. Werz and H. Köpper, Inorg. Chem., 2007, 46, 2249–2260 CrossRef CAS PubMed.
- S. P. Thomas, D. Jayatilaka and T. N. Guru Row, Phys. Chem. Chem. Phys., 2015, 17, 25411–25420 RSC.
- E. S. Yandanova, D. M. Ivanov, M. L. Kuznetsov, A. G. Starikov, G. L. Starova and V. Y. Kukushkin, Cryst. Growth Des., 2016, 16, 2979–2987 CAS.
- I. Khan, P. Panini, S. U.-D. Khan, U. A. Rana, H. Andleeb, D. Chopra, S. Hameed and J. Simpson, Cryst. Growth Des., 2016, 16, 1371–1386 CAS.
- U. Adhikari and S. Scheiner, ChemPhysChem, 2011, 514, 36–39 CAS.
- U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2014, 118, 3183–3192 CrossRef CAS PubMed.
- R. Shukla and D. Chopra, J. Phys. Chem. B, 2015, 119, 14857–14870 CrossRef CAS PubMed.
- M. D. Esrafili and N. Mohammadirad, J. Mol. Model., 2015, 21, 176 CrossRef PubMed.
- R. Shukla and D. Chopra, Phys. Chem. Chem. Phys., 2016, 18, 13820–13829 RSC.
- H. Zhou, Q. Li, W. Li and J. Cheng, New J. Chem., 2015, 39, 2067–2074 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS.
- B. K. Mishra, S. Karthikeyan and V. Ramanathan, J. Chem. Theory Comput., 2012, 8, 1935–1942 CrossRef CAS PubMed.
- V. P. N. Nziko and S. Scheiner, Phys. Chem. Chem. Phys., 2016, 18, 3581–3590 RSC.
- D. Quiñonero, Molecules, 2015, 20, 11632–11659 CrossRef PubMed.
- D. Quiñonero, A. Bauzá, G. Sánchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, New J. Chem., 2016 10.1039/C6NJ01334A.
- G. Sánchez-Sanz, C. Trujillo and I. Alkorta, Comput. Theor. Chem., 2016, 1090, 171–179 CrossRef.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, Advances in electronic structure theory: GAMESS a decade later, in Theory and Applications of Computational Chemistry: the First Forty Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, pp. 1167–1189 Search PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- A. E. Reed, F. Weinhold, L. A. Curtiss and D. J. Pochatko, J. Chem. Phys., 1986, 84, 5687–5705 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed.
- G. Saleh, L. L. Presti, C. Gatti and D. Ceresoli, J. Appl. Crystallogr., 2013, 46, 1513–1517 CAS.
- G. Saleh, C. Gatti, L. L. Presti and J. Contreras-Garc, Chem. – Eur. J., 2012, 18, 15523–15536 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- C. B. Hubschle and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 238–240 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. S. Murray, Z. P.-I. Sheilds, P. G. Seybold and P. Politzer, J. Comput. Sci., 2015, 10, 209–216 CrossRef.
- R. F. W. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05899j |
| This journal is © the Owner Societies 2016 |