
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Initial steps for the thermal decomposition of alkaline-earth metal amidoboranes: a cluster approximation†
A. V.
Pomogaeva
 * and
A. Y.
Timoshkin
* and
A. Y.
Timoshkin
Inorganic Chemistry Group, Institute of Chemistry, St. Petersburg State University, Universitetskaya nab. 7/9, St. Petersburg, 199034, Russia. E-mail: avpomogaeva@cc.spbu.ru
First published on 20th October 2016
Abstract
A DFT study of thermal decomposition mechanisms of [M(NH2BH3)2]4 clusters with M = Mg, Ca, and Sr is presented. Multi-step reaction pathways leading to elimination of the first H2 molecule are explored at the M06/TZVP level of theory. For all studied M, the clusters adopt similar structures and exhibit similar transformations along the reaction pathways. Their activation energies decrease in the order Mg < Ca ≤ Sr. Four metal atoms in the cluster form a rigid planar construction that is found to be nearly unchanged during all transformations. Cleavage of the B–H bond in the environment of alkaline-earth metal atoms leads to the “capture” of the released H atom by neighboring metal atoms with the formation of a M3H moiety. While the activation energies for the cleavage of Hδ− can be as low as 14.3, 22.6 and 23.3 kcal mol−1 for M = Mg, Ca and Sr, respectively, barriers for the subsequent cleavage of Hδ+via destruction of the M3H moiety are about twice larger.
Introduction
Development of materials with properties appropriate for transportable hydrogen storage is an advanced field in modern chemistry.1,2 The excellent gravimetric and volumetric hydrogen density of ammonia borane and its derivatives attracts persistent interest in this class of compounds.3,4 It was found that alkali-metal amidoboranes LiNH2BH3 and NaNH2BH3 release more hydrogen at lower temperatures than pure ammonia borane and also suppress borazine release.5 However, release of a significant amount of ammonia was observed under thermal decomposition of alkali-metal amidoboranes.6–10Alkaline-earth amidoboranes Mg(NH2BH3)2 (MgAB),11 Ca(NH2BH3)2 (CaAB),12 and Sr(NH2BH3)2 (SrAB)13 are known laboratory species which also demonstrate improved dehydrogenation properties compared to ammonia borane. First, CaAB was obtained as a THF adduct,14 then pure CaAB was synthesized by ball milling of CaH2 and BH3NH3.12 These forms have different dehydrogenation properties. THF containing CaAB releases H2 mainly in the temperature range from 120 to 245 °C,14 while the solvent free CaAB eliminates hydrogen at ∼80 °C with peaks at 100 and 140 °C.12 H2 release from SrAB starts at about 60 °C and the decomposition becomes violent as the temperature increases to 93 °C. MgAB exhibits three overlapping dehydrogenation steps with the peak temperatures at 104, 162 and 223 °C.11 Activation energies for the three dehydrogenation steps are 20.1, 27.7 and 28.4 kcal mol−1. The third dehydrogenation step of MgAB is found to be mildly endothermic and the thermolysis of MgAB yields no volatile by-products.11 Dehydrogenation of CaAB also is not accompanied by the release of borazine.12 The release of B2H6 and NH3 was noticed during the thermal decomposition of SrAB.13
Experimental success in the synthesis of alkaline-earth amidoboranes has stimulated related theoretical studies. A number of works based on solid state theory are devoted to the prediction of crystal structure,15 dehydrogenation mechanisms,16 possible intermediate products of the dehydrogenation reaction,17 hydrogen diffusion pathways,18 and optical19 and elastic20 properties of metal amidoboranes (MAB). Advantages of MAB over pure ammonia borane (lower dehydrogenation barriers, a less exothermic overall dehydrogenation reaction and borazine suppression) are attributed to the ionic character of bonds in MAB and the catalytic role of the metal. However, the detailed dehydrogenation mechanism is still unclear. Kim et al.21 presented a comprehensive ab initio study of the mechanisms and kinetics of H2 release in monomers and dimers of MAB. They concluded that oligomerization (O-path) and non-oligomerization (D-path) pathways are competitive. The oligomerization pathway is found to be more favorable for alkali-metal amidoboranes. In contrast, the release of H2via direct transfer of H−, abstracted by the metal cation from the BH3 group, to H+ of the NH2 group of the same NH2BH3 unit is more favorable in the case of CaAB and MgAB. The activation energy of the release of the first H2 molecule from CaAB and MgAB is equal to 36 kcal mol−1 at the MP2/6-311++G** level of theory. Yuan et al.22 reported that the release of the first H2 molecule from the CaAB dimer is kinetically more favorable if it does not involve the Ca atom in the dehydrogenation transition state. No pathways of formation of an oligomeric [BH3NH2BH2NH2]− unit were found at the DFT(PBE/PAW) level of theory.22
These theoretical studies reveal the importance of intermolecular interactions for the particular development of chemical reactions in MAB. The crystal structure of these compounds features a network based on weak M–N and M–H(BH3) interactions. Cluster approximation is an intermediate approach between molecular and solid state computations. It allows tracking of molecular transformations in the chemical reaction pathways taking into account the local atomic environment.23,24 Herein, we present a comparative study of possible pathways leading to the release of the first molecule of H2 from tetrameric clusters [M(NH2BH3)2]4, where M = Mg (1), Ca (2), and Sr (3).
Results and discussion
A. Structure of [M(NH2BH3)2]4 tetramers
In the present work, a tetrameric cluster, [Ca(NH2BH3)2]4 (2), was obtained by optimization of a cutout from crystalline CaAB. CaAB adopts a monoclinic structure with the C2 space group.12 Each Ca atom is coordinated with two NH2 and four BH3 groups (Fig. 1a). The minimal distance between H+ and H− of adjacent layers is about 2.4 Å, i.e. no dihydrogen bonds occur. This observation points out that dehydrogenation proceeds within a layer. A chosen piece of crystalline structure is shown in Fig. 1a and the optimized geometry of the resultant C2h symmetric tetrameric cluster is presented in Fig. 1b (projection onto the XY plane). The z-axis is chosen to be perpendicular to the layer. NH2BH3 groups in the cluster can be classified according to their positions as shown in Fig. 1b. Expectedly, the Ca–N–B angles are the most affected by optimization, which is reflected in the formation of additional Ca–H(BH3) bonds with x- and y-NH2BH3 groups. The environment of BH3 groups in z-NH2BH3 is less affected upon the optimization of the cutout from the crystal tetramer. Overall, Ca–H(BH3) distances in the resultant tetramer are in the range 2.30–2.45 Å. In general, optimization leads to bond shortening with respect to the experimental structure. While all experimental B–N bond lengths in the crystal are 1.575 Å, the optimized values in the tetramer are 1.570, 1.543, and 1.527 Å in x-, y-, and z-NH2BH3, respectively. The respective values of Ca–N distances are 2.352, 2.400, and 2.461 Å, while in the CaAB crystal the Ca–N bond lengths are 2.383 Å.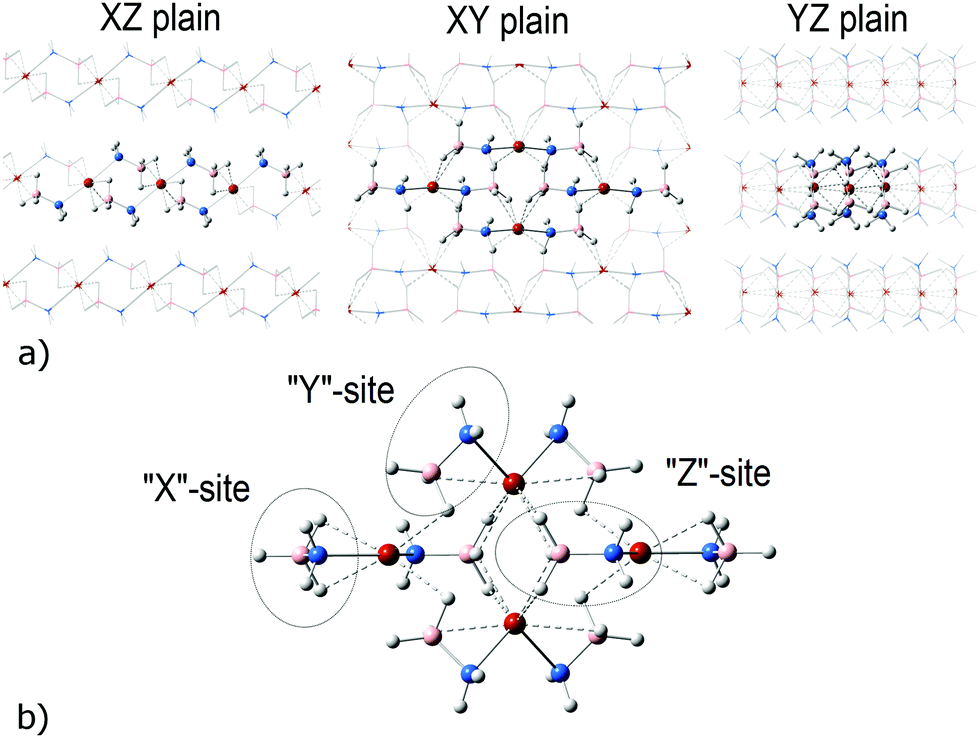 | ||
| Fig. 1 Three projections of the experimental crystalline structure of CaAB12 (a) and the optimized structure of [Ca(NH2BH3)2]4 (b). Molecules extracted from the crystal structure are shown in ball & bond format, other molecules are shown as a wireframe. Red refers to Ca atoms, blue to N, orange to B and white to H atoms. The Ca–H(BH3) bond network is shown by dashed lines. “x”-, “y”- and “z” positions of NH2BH3 units are marked with ovals. | ||
Several initial experimental attempts of the synthesis of pure MgAB failed,14,25 which led to the conclusion that the condensed charge on Mg2+ cations leads to structural instability. However, pure MgAB was finally obtained,11 but unfortunately, the crystal structure of the compound has not been determined. A computational modelling15 predicts that MgAB adopts a structure in the same space group as experimentally known CaAB. In the present study, we assumed the structure of [Mg(NH2BH3)2]4 (1) to be similar to that of 2. The optimized structure of 1 (Fig. S1 in the ESI†) indeed resembles that of 2 though the pattern of the M–H(BH3) contacts is slightly different. Mg–H(BH3) distances are in the range 2.02–2.25 Å. Mg–N bond lengths are 2.026, 2.052 and 2.136 Å, and B–N bond lengths are 1.589, 1.592 and 1.522 Å in x-, y-, and z-NH2BH3, respectively.
SrAB adopts a similar structural type in the crystal as CaAB with different atomic coordinates. Positions of hydrogen atoms in the unit cell of SrAB were not determined.13 To be consistent, we assumed the geometry of the tetrameric cluster [Sr(NH2BH3)2]4 (3) to be similar to that of 2. In the optimized structure of 3, Sr–H(BH3) distances are in the range 2.47–2.60 Å. Sr–N bond lengths are 2.518, 2.586, and 2.632Å, and B–N bond lengths are 1.571, 1.539, and 1.526 Å in x-, y-, and z-NH2BH3, respectively.
In the following discussion the energy values are reported with respect to the optimized tetrameric clusters 1, 2, and 3.
B. Cleavage of B–H bonds and M3H moiety formation
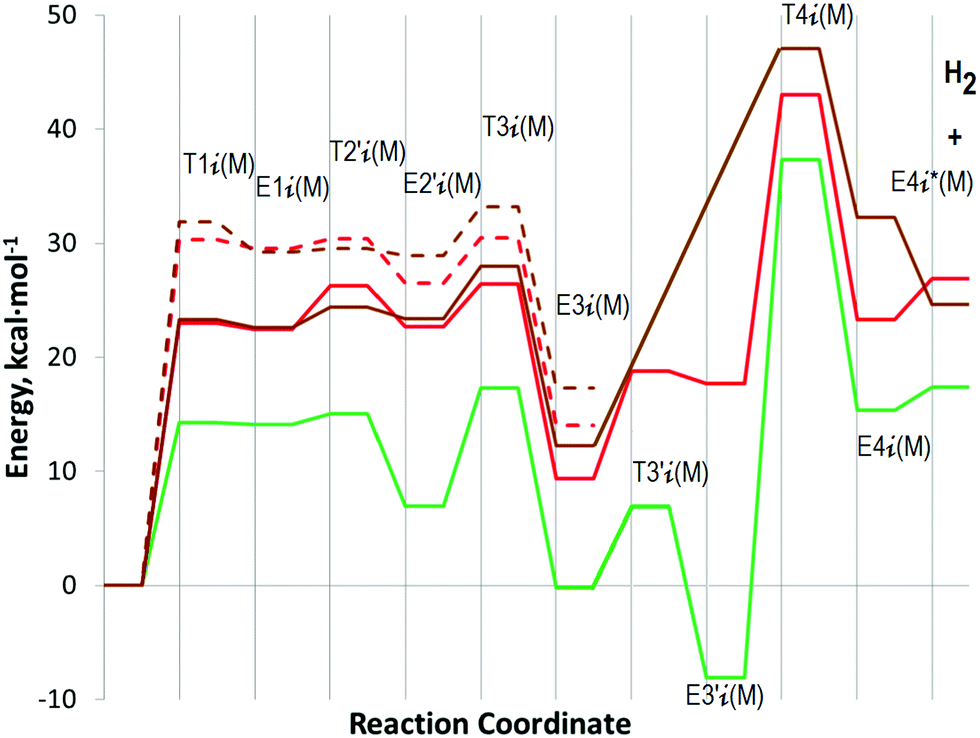
B–H dissociation energies in MAB are lower than N–H dissociation energies,18 thus, the first obvious step in the dehydrogenation mechanism is the cleavage of the B–H bonds. Metal atoms in the considered tetramers form a diamond-shaped arrangement in the XY plane (Fig. 1b). Atomic charges of Mg from the natural population analysis are 1.398 for atoms arranged parallel to the X-axis and 1.500 ē for atoms arranged parallel to the Y-axis. The respective charges of Sr are 1.157 and 1.260 ē. The NBO analysis provides the respective charges for Ca in 2, 0.857 and 0.930 ē, to be somewhat smaller than those for Sr in 3 because of the higher natural population of valence orbitals. B–H bonds of z-NH2BH3 directed toward the XY plane are the longest (for example, 1.259 Å compared to 1.229 Å for other B–H bonds in this BH3 group in 2) with the largest negative charge at H atoms (NBO charges are −0.100 ē; −0.070 ē; and −0.114 ē for 1, 2, and 3, respectively).Cleavage of these B–H bonds leads to the hydrogen atom takeover by three neighboring metal cations in E1z(M) local minima (see the energy diagram in Fig. 2). It was shown earlier that the formation of a Li3H moiety is a key feature of dehydrogenation pathways in LiAB.23,24 The analogous pyramidal M3H moiety is also formed upon activation of alkaline-earth amidoboranes. The highest bonding orbital of E1z(Ca) is delocalized over the Ca3H moiety (Fig. 3). Geometrical parameters of the M3H moieties in E1z(M) are provided in the ESI† (Table S1). The removed hydrogen atom gains significant charge in the environment of the three metal cations. It has a natural charge of −0.580, −0.358 and −0.484 ē for M = Mg, Ca, and Sr, respectively. The transition state leading to the cleavage of the B–H bond T1z(M) is only slightly higher than E1z(M). The energy barriers that should be crossed to split the B–H bond are 23.0 and 23.3 kcal mol−1 for 2 and 3, respectively. It is much lower for 1, only 14.3 kcal mol−1.
 | ||
| Fig. 2 Energy diagram for dehydrogenation pathways in [M(NH2BH3)2]4, M = Mg (green), Ca (red) and Sr (brown). Lines (i = z) refer to the z-path and dashed lines (i = y) refer to the y-path. | ||
 | ||
| Fig. 3 The highest bonding orbital of E1z(Ca) local minimum. Red refers to Ca atoms, blue to N, orange to B and white to H atoms. | ||
Alternatively, the H atom could be removed from y-NH2BH3. Transition states T1y(M) leading to the cleavage of the B–H bonds in such a case are higher in energy (Fig. 2, dashed lines). Activation energies are 27.8, 30.3 and 31.9 kcal mol−1 for 1, 2, and 3, respectively. The pathways go through the formation of a kite-shaped M3H arrangement in T1y(M) and the subsequent E1y(M). Similar kite-shaped Li3H moieties were found by GRRM26 scanning of B–H bond cleavage pathways for LiAB tetramers.23 While intermediates E1y(Ca) and E1y(Sr) are similar, E1y(Mg) is quite different. The local minima E1y(M) are shown in Fig. 4. In the case of Ca and Sr, the highest bonding orbitals are delocalized over the removed H atom and two neighboring metal atoms, and the NH2BH2 intermediate is released (Fig. 4a). As in the case of the z-pathway, E1y(Ca) and E1y(Sr) are only slightly lower in energy than the respective transition states T1y(M). In the case of Mg, the release of the NH2BH2 intermediate is not observed. Instead, [BH3NH2CaNH2BH2]+ is formed, where one H bridges two BH2 groups (Fig. 4b). This significantly stabilizes the E1y(Mg) state  .
.
 | ||
| Fig. 4 Optimized structures and the highest bonding orbitals of E1y(M), M = Sr (a) and Mg (b). Brown/green refers to Sr/Mg atoms, blue to N, orange to B and white to H atoms. | ||
C. Dehydrogenation from the same NH2BH3 group
Dehydrogenation implies the cleavage of N–H bonds in the NH2BH2 intermediate with the subsequent release of H2 molecules. This pathway is analogous to the D-path considered by Kim et al.21 for M(NH2BH3)2 (M = Mg and Ca). While the first step involves the cleavage of the B–H bonds in the NH2BH3 group, H2 is released in the next step via the M–H⋯H–N transition state. According to Kim et al.,21 for Ca(NH2BH3)2 these steps are nearly equal in energy (∼36 kcal mol−1 at the MP2/6-311++G** level of theory). The dehydrogenation step for Mg(NH2BH3)2 has about the same energy barrier (∼36 kcal mol−1) but the cleavage of the B–H bonds proceeds more easily (∼24 kcal mol−1).The dehydrogenation steps for the z- and y-pathways are denoted as T2z(M) and T2y(M), respectively (Fig. 2); optimized structures for M = Ca are shown in Fig. 5. IRC defines local minima, E1′z(M)/E1′y(M), which are connected to E1z(M)/E1y(M)via additional steps, T1′z(M)/T1′y(M), with low energy barriers between them (Fig. 2). Transition states leading to the formation of H2 are the key states for the z- and y-pathways with activation energies ranging from 40.9 to 44.4 kcal mol−1 (Fig. 2). The M3H pyramidal moiety in T2z(M) and the M3H kite-shaped arrangement in T2y(M) are destroyed (Fig. 5a). The hydrogen atoms in the transition states are located between two M atoms. The destruction of the M3H pyramidal moiety requires somewhat higher energy barriers. The T2z(M) energies increase in the order Mg < Ca < Sr. It should be noted that the IRC scan leads to the intermediate E2z(Mg), which is somewhat different from E2z(Ca) and E2z(Sr) (see the ESI,† Fig. S2).
 | ||
| Fig. 5 Transition states and the final products for z- (a) and y- (b) pathways for [Ca(NH2BH3)2]4. | ||
While E2z(Ca) and E2z(Sr) are lower in energy with respect to E2z*(M) and free H2 molecules, E2z(Mg) is higher in energy (Fig. 2).
For the sake of comparison, we also explored a direct dehydrogenation pathway which proceeds through hydrogen evolution via N–Hδ+⋯Hδ−–B interaction in the same NH2BH3 group. This pathway was found to be the most favorable for the CaAB dimer in the theoretical study of Yuan et al.22 (TS could be as low as 21.7 kcal mol−1 computed within an extended unit cell PBE/PAW method). The direct intramolecular dehydrogenation pathway was also assumed for CaAB and MgAB thermal decomposition in the theoretical study of Wang et al.16 The release of the first H2 molecule requires to overcome a barrier of 69.0 kcal mol−1 for MgAB and 73.5 kcal mol−1 for CaAB at the CCSD(T)/6-311++G(3d,2p) level of theory.16 In the present study on the example of 2 we found this pathway to be the most energetically demanding. The energy of the N–H⋯H–B transition state (Fig. S3 in the ESI†) leading to the release of the first H2 molecule from x-NH2BH3 is 59.7 kcal mol−1. Optimization of the corresponding transition states for y- and z-NH2BH3 converged to the alternative transition states T2z(M)/T2y(M) considered above.
D. Oligomerization pathways
An oligomerization pathway going through B–N bond formation between NH2BH3 and NH2BH2 units was found to be the most favorable in the case of LiAB tetramers.23 Compared with the D-path, the O-path was found to be more favorable for LiAB, KAB, and NaAB dimers and less favorable for CaAB and MgAB monomers in Kim's study.21For tetramers, considered in the present work, B–N bond formation between NH2BH3 and NH2BH2 units can proceed along both z- and y- pathways. Oligomerization pathways are structurally similar for M = Mg, Ca and Sr. As an example, structural transformation leading to the B–N bond formation along the z-pathway is shown in Fig. 6 for M = Ca. The Ca3H moiety is retained along the oligomerization pathway. Structural transformations along the y-pathway are analogous (ESI,† Fig. S4). The energy diagram is presented in Fig. 7. T3y(M) energies in the oligomerization step (30.5 and 33.2 kcal mol−1 for M = Ca and Sr, respectively) are similar to those leading to the cleavage of the B–H bonds. For the z-pathway the oligomerization step is more energetically demanding than the first T1z(M) step by 3–5 kcal mol−1, but somewhat lower in energy than the oligomerization step for the y-pathway. Energies of T3z(M) are 17.3, 26.4, and 28.0 kcal mol−1 for M = Mg, Ca and Sr, respectively. Formation of the B–N bond significantly stabilizes E3z(M) though it is still notably endothermic for M = Ca and Sr. In contrast, formation of E3z(Mg) is slightly exothermic (−0.2 kcal mol−1), and after an isomerization step viaT3′z(Mg), formation of the E3′z(Mg) product is exothermic by −8.1 kcal mol−1 (see Fig. 7).
 | ||
| Fig. 6 Oligomerization z-pathway for [Ca(NH2BH3)2]4. Dashed lines show the Ca3H pyramidal moiety. For E3z(Ca), the number of sites of NH2 groups is shown. | ||
 | ||
| Fig. 7 Energy diagram of oligomerization pathways for [M(NH2BH3)2]4, M = Mg (green), Ca (red) and Sr (brown). Lines (i = z) refer to the z-path and dashed lines (i = y) refer to the y-path. | ||
The dehydrogenation step implies further cleavage of N–H bonds. Possible sites of involved NH2 groups are shown in Fig. 6 for the product E3z(M). Five transition states found for the dehydrogenation of E3z(Ca) are given in Fig. S5, ESI.† Energies of T4z(Ca) vary from 43 to 53 kcal mol−1. The dehydrogenation transition states T4z(M) in the oligomerization pathway are higher in energy than T2z(M) in the pathway avoiding the oligomerization step, similar to previous findings for the alkaline-earth amidoborane monomers.21
The lowest energy barrier corresponds to the transition state leading to the hydrogen release from site number 3 where the NH2 group is associated with two B atoms. The transition state, T4z(Ca), and its product E4z*(Ca) are shown in Fig. 8. Similar transition states also exist in the case of Mg and Sr. The energies of the states are 37.3, 43.0 and 47.1 kcal mol−1 for M = Mg, Ca and Sr, respectively. The final product of the step is notably endothermic for all considered tetramers; 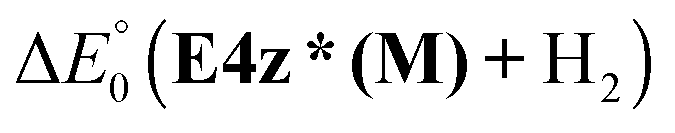 is 17.4, 26.9, and 24.6 for M = Mg, Ca, and Sr, respectively.
is 17.4, 26.9, and 24.6 for M = Mg, Ca, and Sr, respectively.
 | ||
| Fig. 8 The lowest transition state, T4z(Ca), and the product of H2 release, E4z*(Ca), from [Ca(NH2BH3)2]4via an oligomerization pathway. | ||
It should be noted that the energy of the lowest dehydrogenation transition state from the [LiNH2BH3]4 cluster is 31.2 kcal mol−1 at the M06/6-311(d,p) computational level. The energy of the lowest transition state leading to the oligomerization is 25.5 kcal mol−1.23 Analogous values for tetramer 2 at the same level of theory are 42.6 T4z(Ca) and 26.0 T3z(Ca) kcal mol−1. While oligomerization in the case of [Ca(NH2BH3)2]4 needs to overcome nearly the same energy barrier as that in the case of [LiNH2BH3]4, the dehydrogenation step is significantly more energy consuming. Presumably the reason is the better mobility of the closed shell LiH unit formed after the cleavage of the B–H bond. In contrast to LiAB tetramers, the diamond-shaped arrangement of metal atoms in 2 is maintained in the cluster during all transformations in the reaction direction.
While H2 elimination is a major result of thermal decomposition of MAB, some amount of ammonia release is observed experimentally.6,7,13 According to the mechanisms of the thermal decomposition of the alkali-metal amidoboranes suggested by Fijalkowski et al.,8 this secondary NH3 release proceeds via the B–N bond cleavage accompanied by intermolecular transfer of Hδ+ and intermediate formation of weakly bonded [MNH3]+ cations. To estimate whether the cleavage of B–N bonds in alkaline-earth metal amidoboranes is a competitive process with the H2 elimination mechanisms suggested above, we considered several B–N bond breaking pathways in 2. Three different transition states leading to the cleavage of B–N bonds of z-NH2BH3 are provided in the ESI† (Fig. S6). One of the possibilities, T5z(Ca), leads to the formation of [CaNH3]+ cations in E5z(Ca). However, the activation energy is 81.1 kcal mol−1, i.e. 3.5 times larger than the activation energy of B–H bond cleavage viaT1z(Ca), and about twice as large as that of the key state for H2 release via the oligomerization pathway, T4z(Ca). Transfer of BH3 to one of the y-NH2BH3 units is accompanied by the occupation of the [NH2]− residue at the position between three Ca atoms forming a pyramidal Ca3NH2 moiety analogously to the Ca3H moiety discussed above. One of the transition states, T6z(Ca) with an energy of 50.8 kcal mol−1, leads to bridging of the released BH3 with the BH3 group of y-NH2BH3 by the H atom. The other transition state, T7z(Ca) with an energy of 51.3 kcal mol−1, leads to head-to-tale dimerization with the formation of a [BH3NH2BH3]− unit. Thus, it follows that B–N bond breaking pathways have larger activation energies than H2 elimination pathways. This is consistent with the minor role of mechanisms leading to the release of ammonia.
Computational details
The conventional transition state theory was used to predict the optimized structures and transition states of [M(NH2BH3)2]4 tetramers, where M = Mg, Ca, and Sr. The intrinsic reaction coordinate (IRC) scans confirmed the connectivity of all the transition states to reactants and products of a given step. It was noted in previous theoretical investigations that taking into account van der Waals interactions is important to reproduce experimental parameters of SrAB.20 All computations were performed within DFT using the M0627 functional, which takes dispersion interaction into account. The TZVP28 basis set was used throughout. The same computation level was used for the Natural Bond Orbital (NBO) analysis.29 The Gaussian 09 code30 was utilized in all computations.Conclusion
A cluster approximation was used to explore the release of the first H2 molecule from alkaline-earth metal amidoboranes. To form the cluster, geometries of four neighboring molecules were extracted from a layer of experimental crystal structure of CaAB. The cleavage of B–H bonds in the NH2BH3 unit in the environment of alkaline-earth metal atoms leads to the “capture” of the released H atom by neighboring metal atoms with the formation of a M3H moiety. Such a moiety was found to be a key feature of the dehydrogenation process in small (trimeric and tetrameric) LiAB clusters.23,24 The formation of this Li3H moiety was ascribed to the existence of stable Li3H clusters.31,32 Despite the fact that similar free M3H clusters are not found in alkaline-earth metal hydrides (unlike Li3H, Ca3H is an open shell construct), the M3H moiety (M = Mg, Ca, and Sr) plays an important role in the structural transformations of alkaline-earth metal amidoboranes. The M3H moiety has a delocalized bonding orbital and the M–H bond lengths are only slightly larger than the sum of the respective covalent radii.While the cleavage of the B–H bonds and further oligomerization of amidoboranes require moderate energy, the cleavage of N–H bonds accompanied by destruction of the M3H moiety in favor of H2 formation is much more energy consuming. This step requires significant energy uptake both for oligomerization and non-oligomerization pathways.
Kinetically, after the cleavage of the first B–H bond, the cleavage of other B–H bonds or/and oligomerization with B–N bond formation require much less energy uptake than a direct H2 release. This indicates that compounds featuring MNH2BH2NH2BH3 units are potential intermediates in the dehydrogenation process. The large number of possible isomers of such compounds makes the use of a convenient transition state method ineffective for the exploration of pathways for the release of second and subsequent hydrogen molecules. In such a situation, use of the GRRM26 method is recommended for further studies.
The cleavage of B–N bonds is found to be significantly less favorable than the cleavage of B–H bonds. If local overheating leads to the B–N bond cleavage, then the formation of the M3NH2 moiety is more favorable than the formation of the MNH3 complex. This may be accompanied by the formation of an intermediate [BH3NH2BH3]− or the release of diborane. The following NH3 release is likely to be a multistep side process which requires additional studies which are outside the scope of the present research.
Tetramers of alkaline-earth metal (Mg, Ca and Sr) amidoboranes show similar tendencies along the reaction pathways with energy barriers increasing in the order Mg < Ca ≤ Sr. However, according to experiments,11 a higher temperature is required for the start of the dehydrogenation in the case of MgAB, which suggests a larger barrier for H2 release from MgAB compared to CaAB and SrAB. One of the reasons for this disagreement could be our assumption of a similar crystal structure for MaAB and CaAB, and, consequently, a similar local environment of the atoms in tetramer clusters 1 and 2. Note that previous computational studies based on such an assumption also resulted in lower energy barriers for the thermal decomposition of MgAB compared to CaAB.18 In the present study, we demonstrate that unlike Ca and Sr, in the case of Mg, oligomerization of NH2BH3 and NH2BH2 units in 1 makes the formation of the E3′z (Mg) intermediate exothermic and significantly increases the activation energy for the subsequent dehydrogenation steps. We suppose that similar transformations occur in the crystal structure of MgAB prior to the dehydrogenation which increases the overall activation energy for MgAB dehydrogenation compared to CaAB and SrAB.
Acknowledgements
This work was financially supported by the Russian Science Foundation grant 14-13-00151. This research was carried out using computational resources provided by the Resource Center “Computer Center of SPbU”.References
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS PubMed.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS PubMed.
- Y. S. Chua, P. Chen, G. Wua and Z. Xionga, Chem. Commun., 2011, 47, 5116 RSC.
- R. Owarzany, P. J. Leszczyński, K. J. Fijalkowski and W. Grochala, Crystals, 2016, 6, 88 CrossRef.
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS PubMed.
- K. J. Fijalkowski and W. Grochala, J. Mater. Chem., 2009, 19, 2043 RSC.
- K. J. Fijalkowski, R. V. Genova, Y. Filinchuk, A. Budzianowski, M. Derzsi, T. Jaroń, P. J. Leszczyński and W. Grochala, Dalton Trans., 2011, 40, 4407 RSC.
- K. J. Fijalkowski, T. Jaroń, P. J. Leszczyński, E. Magos-Palasyuk, T. Palasyuk, M. K. Cyrańskic and W. Grochala, Phys. Chem. Chem. Phys., 2012, 14, 5778 RSC.
- K. J. Fijalkowski, R. Jurczakowski, W. Koźmińskia and W. Grochala, Phys. Chem. Chem. Phys., 2014, 16, 23340 RSC.
- R. Owarzany, K. J. Fijalkowski, T. Jaroń, P. J. Leszczyński, Ł. Dobrzycki, M. K. Cyrański and W. Grochala, Inorg. Chem., 2016, 55, 37 CrossRef CAS PubMed.
- J. Luo, X. Kang and P. Wang, Energy Environ. Sci., 2013, 6, 1018 CAS.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 14834 CrossRef CAS PubMed.
- Q. G. Zhang, C. X. Tang, C. H. Fang, F. Fang, D. Sun, L. Z. Ouyang and M. Zhu, J. Phys. Chem. C, 2010, 114, 1709 CAS.
- H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burrell, Angew. Chem., Int. Ed., 2007, 46, 8995 CrossRef PubMed.
- Y. Zhang and C. Wolverton, J. Phys. Chem. C, 2012, 116, 14224 CAS.
- K. Wang, V. Arcisauskaite, J.-S. Jiao, J.-G. Zhang, T.-L. Zhang and Z.-N. Zhou, RSC Adv., 2014, 4, 14624 RSC.
- Y. Zhang, T. Autrey and C. Wolverton, J. Phys. Chem. C, 2012, 116, 26728 CAS.
- W. Li, G. Wu, Y. Chua, Y. P. Feng and P. Chen, Inorg. Chem., 2012, 51, 76 CrossRef CAS PubMed.
- C. B. Lingam, K. R. Babu, S. P. Tewari, G. Vaitheeswaran and S. Lebègue, J. Phys. Chem. C, 2011, 115, 18795 Search PubMed.
- B. L. Chittari and S. P. Tewari, Mater. Chem. Phys., 2014, 148, 364 CrossRef CAS.
- D. Y. Kim, H. M. Lee, J. Seo, S. K. Shin and K. S. Kim, Phys. Chem. Chem. Phys., 2010, 12, 5446 RSC.
- P.-F. Yuan, F. Wang, Q. Sun, Y. Jia and Z. X. Guo, Int. J. Hydrogen Energy, 2013, 38, 11313 CrossRef CAS.
- A. V. Pomogaeva, K. Morokuma and A. Y. Timoshkin, J. Phys. Chem. A, 2016, 120, 145 CrossRef CAS PubMed.
- A. V. Pomogaeva, K. Morokuma and A. Y. Timoshkin, J. Comput. Chem., 2016, 37, 1259 CrossRef CAS PubMed.
- Y. S. Chua, G. Wu, Z. Xiong, A. Karkamkar, J. Guo, M. Jian, M. W. Wong, T. Autrey and P. Chen, Chem. Commun., 2010, 46, 5752 RSC.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision C.01, Wallingford, CT, 2009 Search PubMed.
- C. H. Wu and R. O. Jones, J. Chem. Phys., 2004, 120, 5128 CrossRef CAS PubMed.
- J. A. Jr. Montgomery, H. H. Michels, O. F. Guner and K. Lammertsma, Chem. Phys. Lett., 1989, 161, 291 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6, Table S1, and optimized structures of all clusters. See DOI: 10.1039/c6cp05835c |
| This journal is © the Owner Societies 2016 |