Unravelling the fundamentals of thermal and chemical expansion of BaCeO3 from first principles phonon calculations
Received
17th August 2016
, Accepted 19th October 2016
First published on 19th October 2016
Abstract
Differentiating chemical and thermal expansion is virtually impossible to achieve experimentally. While thermal expansion stems from a softening of the phonon spectra, chemical expansion depends on the chemical composition of the material. In the present contribution, we, for the first time, completely decouple thermal and chemical expansion through first principles phonon calculations on BaCeO3, providing new fundamental insights to lattice expansion. We assess the influence of defects on thermal expansion, and how this in turn affects the interpretation of chemical expansion and defect thermodynamics. The calculations reveal that the linear thermal expansion coefficient is lowered by the introduction of oxygen vacancies being 10.6 × 10−6 K−1 at 300 K relative to 12.2 × 10−6 K−1 for both the protonated and defect-free bulk lattice. We further demonstrate that the chemical expansion coefficient upon hydration varies with temperature, ranging from 0.070 to 0.115 per mole oxygen vacancy. Ultimately, we find that, due to differences in the thermal expansion coefficients under dry and wet conditions, the chemical expansion coefficients determined experimentally are grossly underestimated – around 55% lower in the case of 10 mol% acceptor doped BaCeO3. Lastly, we evaluate the effect of these volume changes on the vibrational thermodynamics.
1. Introduction
Mismatch between the functional components of a device as a consequence of thermal and chemical expansion can be detrimental for the overall performance and durability, and a detailed understanding of the origins of these expansion mechanisms is thus essential. The linear chemical expansion coefficient, εi, of a crystalline material along the a-, b- or c-axis can, for a given defect i, be described by| | 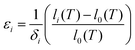 | (1) |
where δi is the defect concentration given as the number of defects per formula unit, whereas li(T) and l0(T) are the dimensions of the defective and perfect supercells, respectively. For proton conductors such as acceptor doped BaZrO3 and BaCeO3, these defects consist of protons (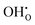 ) and oxygen vacancies (
) and oxygen vacancies (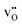 ), where the hydration equilibrium
), where the hydration equilibrium| | 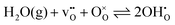 | (2) |
describes their relative dominance. In addition to a chemical expansion stemming from variations in the defect concentrations, materials are also subject to thermal expansion;| | 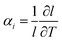 | (3) |
where αi is the linear thermal expansion coefficient along one of the unit cell axes. While chemical expansion depends on the chemical composition of the material, thermal expansion originates from a softening of the phonon spectra of the lattice. Together, these two expansion processes, (1) and (3), comprise the thermochemical expansion.
The two expansion coefficients (εi and αi) are usually determined simultaneously from structural measurements, e.g. X-ray powder diffraction or dilatometry, followed by subsequent measurements to determine the specific defect chemical concentrations, e.g. thermogravimetry or coulometric titration. This often entails assuming that αi is constant throughout the temperature interval investigated, i.e. the thermal volume expansion is independent of any chemical reactions occurring (hydration in the case of proton conductors). Such an assumption can obviously lead to an under- or overestimation of the chemical expansion coefficient (εi) if αi itself is changing. Recent experimental work on proton conducting BaCeY0.2O3−δ1 and BaCe0.2Zr0.7Y0.1O3−δ2 showed that the thermal expansion coefficient is higher when the material is hydrated under wet relative to dry conditions, changing from 11.1 × 10−6 K to 9.9 × 10−6 K and 15.0 × 10−6 K to 12.7 × 10−6 K, respectively. However, in both cases, these differences were neglected, and chemical expansion coefficients were extracted assuming a constant thermal expansion coefficient. Such an assumption has also been employed for the chemical expansion due to oxidation of several oxide ion conductors in recent work by Marrocchelli et al.3–7 In the present contribution, we will through quasiharmonic first principles phonon calculations, address the validity and consequences of this assumption by decoupling the thermal and chemical expansion mechanisms, ultimately providing new fundamental insights to lattice expansion. This will include assessing the effects of defects on the thermal expansion and how this in turn may affect the interpretation of chemical expansion and defect thermodynamics. We have chosen to study the state of the art proton conducting perovskite BaCeO3, which has already been studied extensively in terms of hydration,8–14 thus serving as a good comparative basis for the present work.
2. Methodology
Thermodynamic formalism
Vibrational thermodynamics and thermal properties of solids can be calculated by evaluating their phonon density of states. In the harmonic approximation and under constant volume, the phonon contributions to the Helmholtz energy, Fvib, are given by;| | 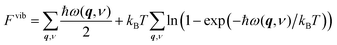 | (4) |
where q and ν are the wave vector and band index, respectively while ω(q,ν) is the phonon frequency at the specific q and ν.
As an approach to include anharmonic effects, we here adopt the quasiharmonic approximation,15,16 hereafter referred to as QHA. The Gibbs free energy, G(T,p), is taken as the unique minimum of the sum of the total electronic energy, Eel(V), the Helmholtz vibrational energy, Fvib, and pV work with respect to volume given by:
| | 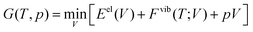 | (5) |
It should be noted that for each
p and
V, the function inside the square brackets of
(5) is minimised with respect to volume.
Fvib is assumed to only be a function of temperature at each specific volume. By fitting the thermodynamic functions on the right hand side of
(5) to the third order Birch–Murnaghan isothermal equation of state (B–M EoS),
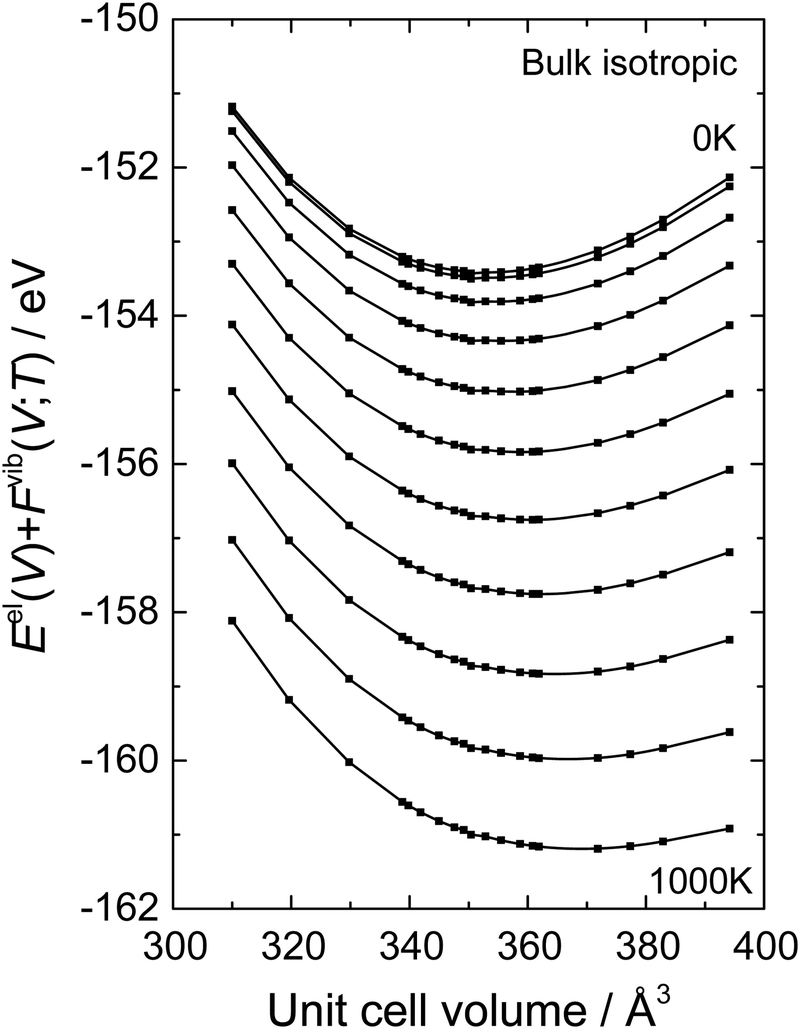
17 Gibbs free energies at finite temperatures are obtained allowing the evaluation of all thermal properties as a function of temperature and pressure. The fitting procedure is demonstrated in
Fig. 1 for bulk BaCeO
3 where a fully isotropic expansion has been chosen. The calculated values of
Eel(
V) +
Fvib(
T;
V) are plotted as a function of the unit cell volume at 100 K intervals between 0 and 1000 K. The solid lines represent the fits from the B–M EoS. Corresponding equilibrium volumes and isothermal bulk moduli are obtained simultaneously from this fitting procedure. The entropy (
S) and heat capacity at constant pressure (
Cp) are then given by the first and second order partial derivatives of
G(
T,
p) with respect to
T, respectively.
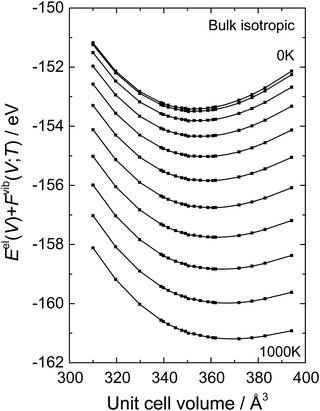 |
| | Fig. 1
E
el(V) + Fvib(T;V) as a function of the unit cell volume for 0–1000 K with temperature intervals of 100 K of bulk BaCeO3 where a fully isotropic expansion has been chosen. The points denote calculated values of Eel(V) + Fvib(T;V) while the solid lines are fitted by the Birch–Murnaghan third order equation of state. | |
The free energy of defect formation (ΔFFdefect or ΔFGdefect) is given by;
| | 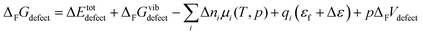 | (6) |
where Δ
Etotdefect denotes the total electronic energy difference between the defective and pristine supercell, while Δ
FGvibdefect (or Δ
FFvibdefect) is the vibrational (phonon) contribution to the free energy of formation. Δ
ni is the change in the number of defects
i with chemical potential,
μi, while
qi is the effective charge of the defect and
εf is the Fermi level. Δ
ε is the shift in the core potentials of the perfect and defective supercell to correct for shifts in the band edges due to the jellium background charge while
pΔ
FVdefect represents the contributions from the pressure and defect formation volume under zero/constant pressure conditions. Further, the chemical potential of gaseous species,
i, is defined by:
| | 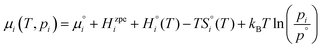 | (7) |
where
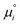
is set to the total electronic energy of the isolated
i-molecule as determined by DFT.
Hzpei is the zero-point energy obtained from experimental vibrational frequencies, while
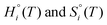
are taken from the HSC 8.2.0 database.
18pi is the partial pressure of
i while
p° is the standard pressure (1 bar).
Computational methodology
All first principles calculations in this work have been performed within the Density Functional Theory (DFT) formalism as implemented in the VASP code.19 Both the LDA20 and GGA-PBE21 functionals have been used and the core potentials were treated using the projector-augmented wave (PAW) method.22 The electronic wave functions were expanded using a set of plane waves with a constant cut-off energy of 500 eV. The defect calculations were performed using both 2 × 1 × 2 (80 atoms) and 2 × 2 × 2 (160 atoms) supercell expansions of the experimentally determined orthorhombic Pnma BaCeO3 20 atom unit cell.23 Electronic integrations over the Brillouin zone were performed using a 4 × 4 × 4 Monkhorst–Pack k-mesh for the unit cell,24 and reduced accordingly for larger supercells. To ensure sufficient convergence of the calculated forces, all calculations were performed with ionic and electronic convergence criteria of 10−4 eV Å−1 and 10−8 eV, respectively.
Orthorhombic BaCeO3 exhibits two structurally inequivalent oxygen sites; an apical oxygen site occupying Wyckoff position 4c and an equatorial site occupying the Wyckoff position 8d. Previous work8,14 has demonstrated that both protons and oxygen vacancies are most stable at the 4c site, and we have therefore only considered this site for both defects. For all charged defects, the total charge of the supercells was adjusted to simulate the desired charge state, by the standard means of charge-compensation with a homogenous jellium charge. Note that we have explicitly not included any acceptors in the supercells due to the large computational demand that would impose. While this is perfectly reasonable in a dilute limit with few defects, thermodynamics and thermal properties may be influenced by the type and amount of acceptor at higher doping levels (≥10 mol%).8,25–27 Further work is needed to assess such dopant effects.
The phonon frequencies were obtained from the Hellmann–Feynman forces acting on all ions within the harmonic (HA) and quasiharmonic approximations (QHA) using finite displacements of ±0.01 Å as implemented in the Phonopy code.15,16,28 For the QHA calculations, lattice parameters were varied isotropically up to ±4% using up to 20 volume points. For the defective cells, the phonon spectra are only evaluated at the Γ-point due to the large size of the supercells.
3. Results and discussion
Thermal and chemical expansion of orthorhombic BaCeO3
The heat capacities at constant pressure, Cp, and constant volume, CV, for isotropic defect-free bulk BaCeO3 as a function of temperature are displayed in Fig. 2 along with experimental values for Cp.29–31 The heat capacities for bulk BaCeO3 are in good agreement with the experimental values from literature obeying the classical Debye law at the lowest temperatures where the heat capacities exhibit a T3-dependency.32 At higher temperatures (above 400 K), CV approaches a constant value of 3N × R (∼125 J K−1 mol−1) as per the Dulong–Petit law, where N is the number of atoms per formula unit (5 for BaCeO3). The calculated Cp maintains a positive slope and starts deviating from the literature values at T ∼ 600 K with values being higher than those reported from experiments. The difference between the calculated and experimentally determined Cp-values could stem from the fact that BaCeO3 undergoes several phase changes in the temperature range measured,9,33,34 and may as such display different Cp values than the orthorhombic Pnma phase used for the DFT calculations. This is also in accordance with work by Ligny and Richet35 on SrZrO3 in which the high temperature cubic polymorph (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) was shown to exhibit an apparent lower heat capacity than its low temperature orthorhombic ground state structure.
m) was shown to exhibit an apparent lower heat capacity than its low temperature orthorhombic ground state structure.
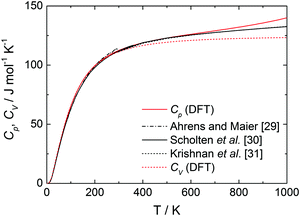 |
| | Fig. 2 Heat capacity at constant pressure, Cp (red solid), and volume, CV (red dotted), as a function of temperature along with experimental values for Cp29–31 (black). | |
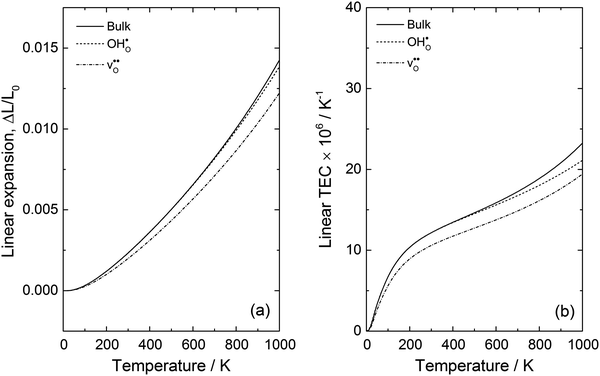
The linear thermal expansion of BaCeO3 and the resulting linear thermal expansion coefficients, αi, given by (3), are shown in Fig. 3(a) and (b), respectively. Note that all linear thermal expansion coefficients are given by 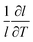 where
where 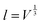 , i.e. we only evaluate a single average coefficient for each expansion. We also assess the effect of an oxygen vacancy and a proton on the thermal expansion. As the figure demonstrates, the linear thermal expansion coefficient, αi, for the pristine and defective BaCeO3 cells becomes increasingly positive with temperature. All coefficients at lower temperatures (300–600 K) are in reasonable agreement with literature values on undoped and acceptor doped BaCeO3 determined by dilatometry and XRD; 8–14 × 10−6 K−1.1,2,36–40 At higher temperatures (600–1000 K), the calculated thermal expansion coefficients vary from 13 to 27 × 10−6 K−1, and are somewhat higher than corresponding values from literature. This difference is likely to stem from phase transitions to other high temperature polymorphs as for instance dilatometry measurements on BaCe0.8−xZrxY0.2O3−δ (x ≤ 0.4) have been shown to yield different αi-values above and below ∼600 °C.39,40 Computational studies on other oxides such as CeO2 and SrTiO3 have also shown that the GGA functional tends to overestimate the thermal expansion coefficient.35,41,42
, i.e. we only evaluate a single average coefficient for each expansion. We also assess the effect of an oxygen vacancy and a proton on the thermal expansion. As the figure demonstrates, the linear thermal expansion coefficient, αi, for the pristine and defective BaCeO3 cells becomes increasingly positive with temperature. All coefficients at lower temperatures (300–600 K) are in reasonable agreement with literature values on undoped and acceptor doped BaCeO3 determined by dilatometry and XRD; 8–14 × 10−6 K−1.1,2,36–40 At higher temperatures (600–1000 K), the calculated thermal expansion coefficients vary from 13 to 27 × 10−6 K−1, and are somewhat higher than corresponding values from literature. This difference is likely to stem from phase transitions to other high temperature polymorphs as for instance dilatometry measurements on BaCe0.8−xZrxY0.2O3−δ (x ≤ 0.4) have been shown to yield different αi-values above and below ∼600 °C.39,40 Computational studies on other oxides such as CeO2 and SrTiO3 have also shown that the GGA functional tends to overestimate the thermal expansion coefficient.35,41,42
 |
| | Fig. 3 (a) Linear thermal expansion of bulk BaCeO3 with or without any defects and (b) the resulting thermal expansion coefficients determined by quasiharmonic first principles phonon calculations. For the defective systems, a single oxygen vacancy, 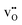 , or proton, , or proton, 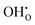 has been investigated. All lattice parameters have been varied isotropically and linear thermal expansion coefficients are given by has been investigated. All lattice parameters have been varied isotropically and linear thermal expansion coefficients are given by 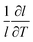 where where 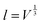 . . | |
Interestingly, the supercells containing defects both display lower α-values compared to bulk BaCeO3, which arguably stems from a blue shift of the phonon spectra due to their negative formation volumes (−20.4 and −6.8 Å3 for 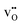 and
and 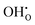 , respectively). There is also a significant difference in the thermal expansion coefficients between the cells containing
, respectively). There is also a significant difference in the thermal expansion coefficients between the cells containing 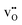 and
and 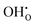 demonstrating that BaCeO3 may thermally expand differently depending on the water vapour partial pressure employed. This is also in good correspondence with recent work on similar compositions.1,2,43 Drawing any overall conclusions regarding the effect of ionic defects on thermal expansion is difficult based on the present work alone, but there appears to be a correlation between the defect formation volume (or defect chemical expansion coefficient) and the change in the thermal expansion coefficient. For instance, the cell containing a proton or an oxygen vacancy, which both display negative formation volumes, also exhibit lower thermal expansion coefficients than that of the pristine system. This could suggest that defects with positive formation volumes, such as large acceptor dopants44 or neutral oxygen vacancies,45 may increase the thermal expansion coefficient. This opens up for the possibility of altering the thermal expansion of a material system simply by changing its defect structure, and may help in limiting thermal mismatch in device fabrication.
demonstrating that BaCeO3 may thermally expand differently depending on the water vapour partial pressure employed. This is also in good correspondence with recent work on similar compositions.1,2,43 Drawing any overall conclusions regarding the effect of ionic defects on thermal expansion is difficult based on the present work alone, but there appears to be a correlation between the defect formation volume (or defect chemical expansion coefficient) and the change in the thermal expansion coefficient. For instance, the cell containing a proton or an oxygen vacancy, which both display negative formation volumes, also exhibit lower thermal expansion coefficients than that of the pristine system. This could suggest that defects with positive formation volumes, such as large acceptor dopants44 or neutral oxygen vacancies,45 may increase the thermal expansion coefficient. This opens up for the possibility of altering the thermal expansion of a material system simply by changing its defect structure, and may help in limiting thermal mismatch in device fabrication.
The chemical expansion coefficient due to hydration per mole oxygen vacancy, εhydr, is given by the relative difference of the chemical expansion coefficients for the formation of an oxygen vacancy and a proton, 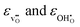 , respectively:
, respectively:
| | 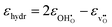 | (8) |
Thus,
εhydr is calculated with respect to the defect-free lattice, while experimental determination of
εhydr uses the volume differences between the hydrated and dry lattice
i.e. relative to the defective lattice. To assess the effect of such differences, we calculate
εhydr in three separate ways;
| | 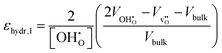 | (9) |
| | 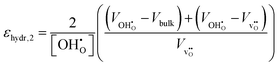 | (10) |
| | 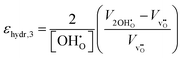 | (11) |
where
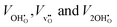
denote the equilibrium supercell volumes with a proton, oxygen vacancy, and 2 protons in the same supercell, respectively, while
Vbulk represents the volume of the pristine lattice. In all three cases,
εhydr was determined to be virtually identical yielding a maximum difference of a mere 2% suggesting that all calculated chemical expansion coefficients, regardless of calculation method, should be comparable to experiments.
Fig. 4 displays
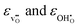
, and the resulting chemical expansion coefficient upon hydration calculated by
(9),
εhydr, as a function of temperature. Note that we have chosen to plot the individual contributions to
εhydr,
i.e.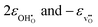
. As the figure demonstrates, the chemical expansion coefficient for the supercells containing
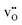
and
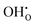
becomes increasingly negative with increasing temperature. The resulting chemical expansion coefficient due to hydration varies to a much smaller extent, ranging from 0.070 to 0.115 in the temperature range presented. This is in reasonable agreement with other experimental and computational work.
1,12,38
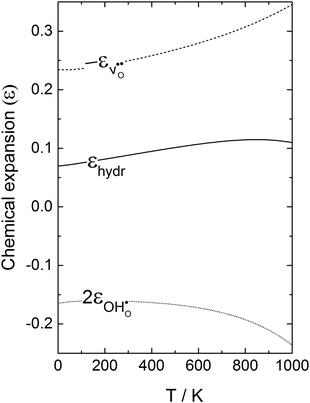 |
| | Fig. 4 Chemical expansion coefficient due to hydration, εhydr, and its individual contributions 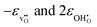 , as calculated by quasiharmonic phonon calculations as a function of temperature. , as calculated by quasiharmonic phonon calculations as a function of temperature. | |
Given that εhydr varies with temperature, distinctly different volume expansions are expected when hydrating acceptor doped BaCeO3 isothermally at different temperatures; 0.35 to 0.58% for 10 mol%. The results presented here can thus partially explain the large scatter in chemical expansion coefficients due to hydration found experimentally. For instance, the difference in εhydr of 20 mol% acceptor doped BaCeO3 determined by Yamaguchi and Yamada38 and Andersson et al.1 (0.096 and 0.151, respectively) could stem from the differences in the water vapour partial pressures (pH2O) used during heating and cooling. While Yamaguchi and Yamada38 humidified the gas with a constant pH2O of ∼0.03 atm, Andersson et al.1 were not able to monitor or control pH2O but estimated it to be around 10−4 atm based on the volume expansion/contraction onset and the material's hydration thermodynamics. Such a large difference in pH2O means that the material will hydrate (or dehydrate) in different temperature intervals, and the corresponding difference in lattice expansion will thus be affected due to the temperature dependency of εhydr. Consequently, careful attention to the experimental conditions is needed when comparing specific chemical expansion coefficients extracted.
By combining the expressions for the thermal and chemical expansion coefficient, (3) and (1), respectively, a full description of the thermochemical lattice expansion of BaCeO3 as a function of temperature can be obtained. This requires an expression for the proton concentration as a function of temperature. For that purpose, we will firstly assume that the effectively negative charged acceptors, Acc′, are fully charge compensated by oxygen vacancies and/or protons i.e. all other defects are considered to have negligible concentrations:
| | 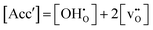 | (12) |
Secondly, the oxygen site restriction for perovskites is imposed:
| | 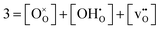 | (13) |
The proton concentration is then determined by a combination of
(12) and (13) along with the hydration equilibrium constant,
Khydr, for
reaction (2), for undoped BaCeO
3. For any further information regarding the hydration thermodynamics, the reader is referred to work by Bjørheim
et al.
12 The volumetric thermal expansion coefficient upon hydration,
α, is assumed to change linearly from the thermal expansion coefficient of the oxygen vacancy,
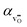
, to that of the proton,
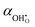
:
| | 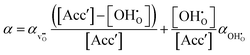 | (14) |
The linear relationship in
(14) is from the simple rule of mixtures, often used in predicting thermal and mechanical properties of composite materials. It assumes that the physical properties of each phase are not influenced by each other's presence in the mixture.
46,47 By combining the expressions for the thermal and chemical expansion coefficient upon hydration (
cf.Fig. 4), the thermochemical expansion of the BaCeO
3 unit cell can now be determined where the unit cell volume at temperature
Tf,
Vf, is given by:
| | 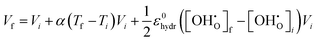 | (15) |
Vi represents the unit cell volume of the dehydrated system at
Ti, while
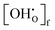
and
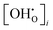
denote the proton concentration at
Tf and
Ti, respectively. Note that we have chosen to use a constant value for the chemical expansion coefficient,
ε0hydr, which differs from
εhydr, given in
(8), in that it is entirely temperature independent stemming from the equilibrium volumes of all supercells calculated at 0 K and excludes all zero-point energy contributions (0.078). This is intentionally done to avoid double counting the temperature dependencies of the equilibrium volumes, which is already implemented in the thermal expansion coefficient in
(14).
Fig. 5a displays the change in the unit cell volume as a function of temperature for different acceptor dopant concentrations (5–20 mol%) under humid conditions,
pH2O = 0.025 atm, while
Fig. 5b gives the unit cell volume for 20 mol% doped BaCeO
3 where
pH2O is varied from 0.025 to 1 atm.
Fig. 5a demonstrates that the volume increases to a larger extent upon hydration with increasing doping concentrations reflecting higher proton concentrations. While the unit cell volume only changes gradually upon hydration (or dehydration) for the lower doping concentrations (5–15 mol%), the volume change for 20 mol% doped BaCeO
3 is more abrupt, and the volume is almost constant in a small temperature interval for 20 mol% doped BaCeO
3. In
Fig. 5b, protons are retained to higher temperatures for
pH2O = 1 atm and further heating is therefore needed for the volume to decrease compared to the corresponding unit cell volume at
pH2O = 0.025 atm.
Fig. 5b shows an additional interesting feature where the volume of the unit cell is not the same at lower temperatures (<600 K) when the water vapour partial pressure is changed. This stems from the difference in the thermal expansion coefficients upon hydration (
cf.Fig. 3) where the unit cell at
pH2O = 1 atm is thermally contracting to a larger extent than the corresponding unit cell at 0.025 atm.
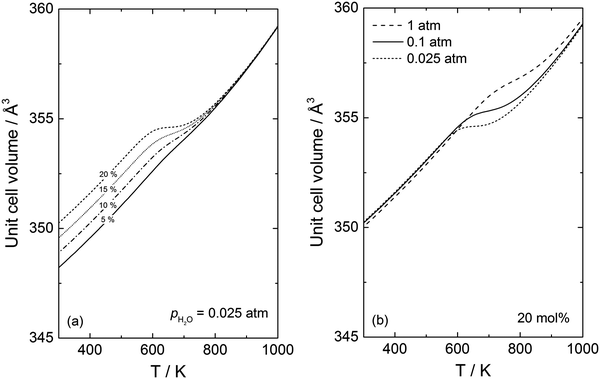 |
| | Fig. 5 Unit cell volume of BaCeO3 calculated using the quasiharmonic approximation as a function of temperature for (a) different acceptor concentrations (5–20 mol%) for pH2O = 0.025 atm and (b) 20 mol% acceptor doped BaCeO3 under different water vapour partial pressures (0.025–1 atm). | |
As both the chemical and thermal expansion coefficients change as a function of temperature, and the two processes are interrelated, their accurate determination through experiments may be difficult. Through the present DFT calculations, we are able to decouple the expansion mechanisms, demonstrating that experimental work may easily over- or underestimate the chemical expansion coefficient. Since α for a protonated cell is higher than the corresponding cell with an oxygen vacancy (Fig. 3), the lattice will in fact thermally contract to a larger extent upon cooling for a hydrated material. This, in turn, means that the chemical expansion coefficient determined experimentally will be grossly underestimated, as it is often assumed that the difference in the volume under wet and dry conditions at lower temperatures is only given by chemical expansion. For 20 mol% acceptor doped BaCeO3, this results in an apparent chemical expansion coefficient of 0.056 at 300 K whereas the actual coefficient is far higher (0.078) i.e. an underestimation of 27%. This discrepancy increases with decreasing acceptor concentrations yielding an underestimation of 55% at 300 K for 10 mol%. This underestimation can for instance help explain the large scatter in the chemical expansion coefficients determined experimentally, which for acceptor doped BaCeO3, vary between 0.030 and 0.151.1,38
Defect thermodynamics of bulk BaCeO3
The thermochemical expansion of BaCeO3 will undoubtedly affect the defect thermodynamics due to changes it will impose on the phonon spectra. While most of the defect thermodynamics have already been treated in our previous contribution using the harmonic approximation (HA),12 this work assumed that the defects themselves did not affect thermal expansion. We are, thus, only determining the effect of anharmonicity on the thermodynamics by using the QHA, and the reader is referred to ref. 12 for further details on the thermodynamic formalism and defect thermodynamics of BaCeO3 and other perovskite oxides. As the results herein were obtained using 2 × 1 × 2 supercells (80 atoms), we first assess the effect of the supercell size on the thermodynamics. Fig. 6 shows (a) the calculated vibrational formation entropies of an oxygen vacancy and proton while (b) shows the resulting hydration entropies using the LDA and PBE functional and 2 × 1 × 2 and 2 × 2 × 2 supercells from phonon calculations within the harmonic approximation as a function of temperature. Both functionals and supercells yield similar values with the entropies generally being slightly more negative for larger supercells and with the PBE functional.
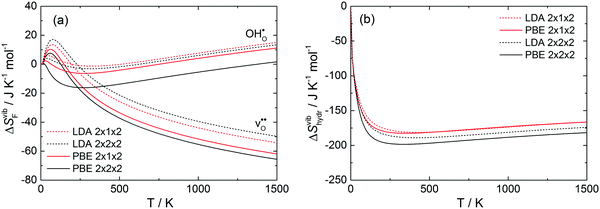 |
| | Fig. 6 (a) Vibrational formation entropy for an oxygen vacancy and proton and (b) the resulting hydration entropy determined by harmonic phonon calculations for BaCeO3 with different supercell sizes and functionals. | |
Fig. 7 displays the vibrational formation entropy for an oxygen vacancy and proton, along with the resulting hydration entropy for BaCeO3, as a function of temperature calculated by the HA and the QHA where the latter accounts for the volume dependency of the phonons. While there is virtually no difference in the proton formation entropy calculated by the two approximations, the oxygen vacancy formation entropy differs by almost 35 J K−1 mol−1 at 1000 K. This difference could among other things stem from assuming, in the HA, that the partial derivative of the entropy with respect to volume, 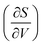 , does not change upon defect formation;12,41,48–51
, does not change upon defect formation;12,41,48–51
| | 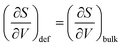 | (16) |
Under constant pressure,
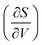
is further given by
| | 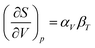 | (17) |
where
αV and
βT are the volumetric thermal expansion coefficient and isothermal bulk modulus, respectively. Thus, for this assumption to be valid, the product
αVβT should not change upon defect formation. However, through the present work using the QHA, the thermal expansion coefficient has been shown to change significantly when defects are introduced, especially in the case of oxygen vacancies, relative to the pristine system (
Fig. 3). This could thus explain the difference in the oxygen vacancy formation entropies calculated using the two approximations. Whether one could alleviate the differences between the approximations by using larger supercells in the calculations,
i.e. more dilute defect concentrations, remains unkown but more work is needed to address these aspects further. Interestingly, for the hydration entropy, which is calculated by subtracting the defect formation entropies through
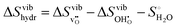
, the difference between the approximations again only stems from the oxygen vacancy formation entropy reaching a maximum difference of about 35 J K
−1 mol
−1 at higher temperatures. Ultimately, we find that most of the vibrational thermodynamics are appropriately described by the HA as anharmonic effects appear to be of a minor concern.
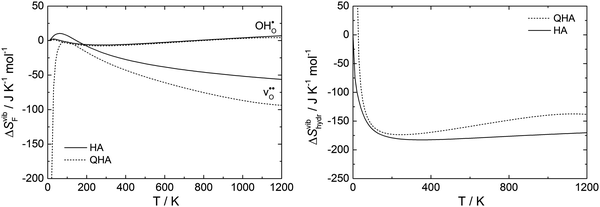 |
| | Fig. 7 Vibrational formation entropy for an oxygen vacancy and a proton, and the entropy of hydration for BaCeO3, as a function of temperature calculated by the harmonic and quasiharmonic approximations, HA and QHA, respectively. The latter also accounts for the volume dependency of the phonon spectra. | |
4. Conclusions
In the present contribution we have investigated the thermal and chemical expansion of BaCeO3 from first principles phonon calculations. This has allowed us, for the first time, to decouple these expansion processes providing fundamental insights to lattice expansion. The calculations reveal that the formation of defects such as oxygen vacancies or protons can play a significant role in thermal expansion displaying lower thermal expansion coefficients compared to that of the defect-free bulk. Further, due to the differences in the thermal expansion coefficients between the dry and protonated unit cell, we demonstrate that experimentally determined chemical expansion coefficients due to hydration can be grossly underestimated being around 55% lower in the case of 10 mol% acceptor doped BaCeO3. The chemical expansion coefficient is also found to change with temperature ranging between 0.070 and 0.115 per mole oxygen vacancy thus demonstrating that different experimental conditions can significantly change the chemical expansion of the lattice. Overall, our results rationalise the large scatter in the chemical expansion coefficients determined experimentally, which for acceptor doped BaCeO3 varies from 0.030 to 0.151 per mole oxygen vacancy. Lastly, we have assessed how thermochemical expansion may influence the defect thermodynamics by including the volume dependency of the phonon spectra.
Acknowledgements
The authors gratefully acknowledge the Research Council of Norway (RCN) through the project “HydraThermPro” (#214252) for financial support. The calculations were performed on resources provided by UNINETT Sigma2 – the National Infrastructure for High Performance Computing and Data Storage in Norway through project NN4604k.
References
- A. K. E. Andersson, S. M. Selbach, C. S. Knee and T. Grande, J. Am. Ceram. Soc., 2014, 97, 2654–2661 CrossRef CAS.
- G. C. Mather, G. Heras-Juaristi, C. Ritter, R. O. Fuentes, A. L. Chinelatto, D. Pérez-Coll and U. Amador, Chem. Mater., 2016, 28, 4292–4299 CrossRef CAS.
- D. Marrocchelli, S. R. Bishop and J. Kilner, J. Mater. Chem. A, 2013, 1, 7673–7680 CAS.
- D. Marrocchelli, N. H. Perry and S. R. Bishop, Phys. Chem. Chem. Phys., 2015, 17, 10028–10039 RSC.
- D. Marrocchelli, S. R. Bishop, H. L. Tuller and B. Yildiz, Adv. Funct. Mater., 2012, 22, 1958–1965 CrossRef CAS.
- S. R. Bishop, D. Marrocchelli, C. Chatzichristodoulou, N. H. Perry, M. B. Mogensen, H. L. Tuller and E. D. Wachsman, Annu. Rev. Mater. Res., 2014, 44, 205–239 CrossRef CAS.
- S. R. Bishop, D. Marrocchelli, W. Fang, K. Amezawa, K. Yashiro and G. W. Watson, Energy Environ. Sci., 2013, 6, 1142–1146 CAS.
- A. Løken, T. S. Bjørheim and R. Haugsrud, J. Mater. Chem. A, 2015, 3, 23289–23298 Search PubMed.
- K. S. Knight, Solid State Ionics, 1994, 74, 109–117 CrossRef CAS.
- M. Oishi, K. Yashiro, K. Sato, J. Mizusaki, N. Kitamura, K. Amezawa, T. Kawada and Y. Uchimoto, Solid State Ionics, 2008, 179, 529–535 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1999, 125, 285–302 CrossRef CAS.
- T. S. Bjørheim, A. Løken and R. Haugsrud, J. Mater. Chem. A, 2016, 4, 5917–5924 Search PubMed.
- M. Oishi, S. Akoshima, K. Yashiro, K. Sato, J. Mizusaki and T. Kawada, Solid State Ionics, 2009, 180, 127–131 CrossRef CAS.
- J. Hermet, F. Bottin, G. Dezanneau and G. Geneste, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 205137 CrossRef.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- A. Togo, L. Chaput, I. Tanaka and G. Hug, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174301 CrossRef.
- F. Birch, Phys. Rev., 1947, 71, 809–824 CrossRef CAS.
- HSC Chemistry 8.2.0, Outetec Technologies, 2015.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- K. S. Knight and N. Bonanos, J. Mater. Chem., 1994, 4, 899–901 RSC.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- A. Løken, S. W. Saeed, M. N. Getz, X. Liu and T. S. Bjørheim, J. Mater. Chem. A, 2016, 4, 9229–9235 Search PubMed.
- A. Løken, C. Kjølseth and R. Haugsrud, Solid State Ionics, 2014, 267, 61–67 CrossRef.
- K. D. Kreuer, S. Adams, W. Münch, A. Fuchs, U. Klock and J. Maier, Solid State Ionics, 2001, 145, 295–306 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- M. Ahrens and J. Maier, Thermochim. Acta, 2006, 443, 189–196 CrossRef CAS.
- M. J. Scholten, J. Schoonman, J. C. v. Miltenburg and E. H. P. Cordfunke, Thermochim. Acta, 1995, 268, 161–168 CrossRef.
- R. Venkata Krishnan, K. Nagarajan and P. R. Vasudeva Rao, J. Nucl. Mater., 2001, 299, 28–31 CrossRef CAS.
- P. Debye, Ann. Phys., 1912, 344, 789–839 CrossRef.
- V. M. Egorov, Y. M. Baikov, N. F. Kartenko, B. T. Melekh and Y. N. Filin, Phys. Solid State, 1998, 40, 1911–1914 CrossRef CAS.
- T. Ohzeki, S. Hasegawa, M. Shimizu and T. Hashimoto, Solid State Ionics, 2009, 180, 1034–1039 CrossRef CAS.
- D. de Ligny and P. Richet, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 3013–3022 CrossRef CAS.
- J.-X. Wang, L.-P. Li, B. J. Campbell, Z. Lv, Y. Ji, Y.-F. Xue and W.-H. Su, Mater. Chem. Phys., 2004, 86, 150–155 CrossRef CAS.
- S. Yamanaka, M. Fujikane, T. Hamaguchi, H. Muta, T. Oyama, T. Matsuda, S.-i. Kobayashi and K. Kurosaki, J. Alloys Compd., 2003, 359, 109–113 CrossRef CAS.
- S. Yamaguchi and N. Yamada, Solid State Ionics, 2003, 162–163, 23–29 CrossRef CAS.
- Y. G. Lyagaeva, D. A. Medvedev, A. K. Demin, P. Tsiakaras and O. G. Reznitskikh, Phys. Solid State, 2015, 57, 285–289 CrossRef CAS.
- J. Lagaeva, D. Medvedev, A. Demin and P. Tsiakaras, J. Power Sources, 2015, 278, 436–444 CrossRef CAS.
- S. Grieshammer, T. Zacherle and M. Martin, Phys. Chem. Chem. Phys., 2013, 15, 15935–15942 RSC.
- Y. Lu, D. Jia, F. Gao, T. Hu and Z. Chen, Solid State Commun., 2015, 201, 25–30 CrossRef CAS.
- C. Hiraiwa, D. Han, A. Kuramitsu, A. Kuwabara, H. Takeuchi, M. Majima and T. Uda, J. Am. Ceram. Soc., 2013, 96, 879–884 CrossRef CAS.
- E. Jedvik, A. Lindman, M. Þ. Benediktsson and G. Wahnström, Solid State Ionics, 2015, 275, 2–8 CrossRef CAS.
- T. S. Bjørheim, M. Arrigoni, D. Gryaznov, E. Kotomin and J. Maier, Phys. Chem. Chem. Phys., 2015, 17, 20765–20774 RSC.
- H. A. Bruck and B. H. Rabin, J. Am. Ceram. Soc., 1999, 82, 2927–2930 CrossRef CAS.
- H. S. Kim, S. I. Hong and S. J. Kim, J. Mater. Process. Technol., 2001, 112, 109–113 CrossRef.
- T. S. Bjørheim, E. A. Kotomin and J. Maier, J. Mater. Chem. A, 2015, 3, 7639–7648 Search PubMed.
- P. Ágoston and K. Albe, Phys. Chem. Chem. Phys., 2009, 11, 3226–3232 RSC.
- Y. Mishin, M. R. Sørensen and A. F. Voter, Philos. Mag. A, 2001, 81, 2591–2612 CAS.
- T. Zacherle, P. C. Schmidt and M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235206 CrossRef.
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
 ) and oxygen vacancies (
) and oxygen vacancies ( ), where the hydration equilibrium
), where the hydration equilibrium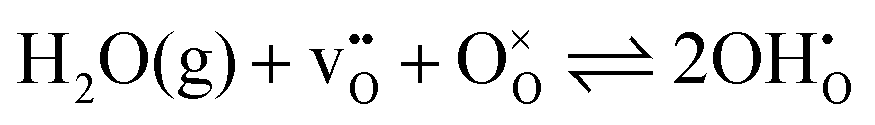



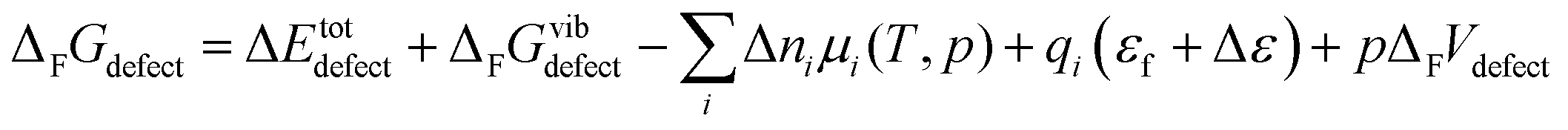

 is set to the total electronic energy of the isolated i-molecule as determined by DFT. Hzpei is the zero-point energy obtained from experimental vibrational frequencies, while
is set to the total electronic energy of the isolated i-molecule as determined by DFT. Hzpei is the zero-point energy obtained from experimental vibrational frequencies, while  are taken from the HSC 8.2.0 database.18pi is the partial pressure of i while p° is the standard pressure (1 bar).
are taken from the HSC 8.2.0 database.18pi is the partial pressure of i while p° is the standard pressure (1 bar).

 where
where  , i.e. we only evaluate a single average coefficient for each expansion. We also assess the effect of an oxygen vacancy and a proton on the thermal expansion. As the figure demonstrates, the linear thermal expansion coefficient, αi, for the pristine and defective BaCeO3 cells becomes increasingly positive with temperature. All coefficients at lower temperatures (300–600 K) are in reasonable agreement with literature values on undoped and acceptor doped BaCeO3 determined by dilatometry and XRD; 8–14 × 10−6 K−1.1,2,36–40 At higher temperatures (600–1000 K), the calculated thermal expansion coefficients vary from 13 to 27 × 10−6 K−1, and are somewhat higher than corresponding values from literature. This difference is likely to stem from phase transitions to other high temperature polymorphs as for instance dilatometry measurements on BaCe0.8−xZrxY0.2O3−δ (x ≤ 0.4) have been shown to yield different αi-values above and below ∼600 °C.39,40 Computational studies on other oxides such as CeO2 and SrTiO3 have also shown that the GGA functional tends to overestimate the thermal expansion coefficient.35,41,42
, i.e. we only evaluate a single average coefficient for each expansion. We also assess the effect of an oxygen vacancy and a proton on the thermal expansion. As the figure demonstrates, the linear thermal expansion coefficient, αi, for the pristine and defective BaCeO3 cells becomes increasingly positive with temperature. All coefficients at lower temperatures (300–600 K) are in reasonable agreement with literature values on undoped and acceptor doped BaCeO3 determined by dilatometry and XRD; 8–14 × 10−6 K−1.1,2,36–40 At higher temperatures (600–1000 K), the calculated thermal expansion coefficients vary from 13 to 27 × 10−6 K−1, and are somewhat higher than corresponding values from literature. This difference is likely to stem from phase transitions to other high temperature polymorphs as for instance dilatometry measurements on BaCe0.8−xZrxY0.2O3−δ (x ≤ 0.4) have been shown to yield different αi-values above and below ∼600 °C.39,40 Computational studies on other oxides such as CeO2 and SrTiO3 have also shown that the GGA functional tends to overestimate the thermal expansion coefficient.35,41,42
 , or proton,
, or proton,  has been investigated. All lattice parameters have been varied isotropically and linear thermal expansion coefficients are given by
has been investigated. All lattice parameters have been varied isotropically and linear thermal expansion coefficients are given by  where
where  .
.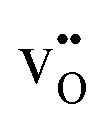 and
and  , respectively). There is also a significant difference in the thermal expansion coefficients between the cells containing
, respectively). There is also a significant difference in the thermal expansion coefficients between the cells containing  and
and  demonstrating that BaCeO3 may thermally expand differently depending on the water vapour partial pressure employed. This is also in good correspondence with recent work on similar compositions.1,2,43 Drawing any overall conclusions regarding the effect of ionic defects on thermal expansion is difficult based on the present work alone, but there appears to be a correlation between the defect formation volume (or defect chemical expansion coefficient) and the change in the thermal expansion coefficient. For instance, the cell containing a proton or an oxygen vacancy, which both display negative formation volumes, also exhibit lower thermal expansion coefficients than that of the pristine system. This could suggest that defects with positive formation volumes, such as large acceptor dopants44 or neutral oxygen vacancies,45 may increase the thermal expansion coefficient. This opens up for the possibility of altering the thermal expansion of a material system simply by changing its defect structure, and may help in limiting thermal mismatch in device fabrication.
demonstrating that BaCeO3 may thermally expand differently depending on the water vapour partial pressure employed. This is also in good correspondence with recent work on similar compositions.1,2,43 Drawing any overall conclusions regarding the effect of ionic defects on thermal expansion is difficult based on the present work alone, but there appears to be a correlation between the defect formation volume (or defect chemical expansion coefficient) and the change in the thermal expansion coefficient. For instance, the cell containing a proton or an oxygen vacancy, which both display negative formation volumes, also exhibit lower thermal expansion coefficients than that of the pristine system. This could suggest that defects with positive formation volumes, such as large acceptor dopants44 or neutral oxygen vacancies,45 may increase the thermal expansion coefficient. This opens up for the possibility of altering the thermal expansion of a material system simply by changing its defect structure, and may help in limiting thermal mismatch in device fabrication. , respectively:
, respectively:



 denote the equilibrium supercell volumes with a proton, oxygen vacancy, and 2 protons in the same supercell, respectively, while Vbulk represents the volume of the pristine lattice. In all three cases, εhydr was determined to be virtually identical yielding a maximum difference of a mere 2% suggesting that all calculated chemical expansion coefficients, regardless of calculation method, should be comparable to experiments. Fig. 4 displays
denote the equilibrium supercell volumes with a proton, oxygen vacancy, and 2 protons in the same supercell, respectively, while Vbulk represents the volume of the pristine lattice. In all three cases, εhydr was determined to be virtually identical yielding a maximum difference of a mere 2% suggesting that all calculated chemical expansion coefficients, regardless of calculation method, should be comparable to experiments. Fig. 4 displays  , and the resulting chemical expansion coefficient upon hydration calculated by (9), εhydr, as a function of temperature. Note that we have chosen to plot the individual contributions to εhydr, i.e.
, and the resulting chemical expansion coefficient upon hydration calculated by (9), εhydr, as a function of temperature. Note that we have chosen to plot the individual contributions to εhydr, i.e. . As the figure demonstrates, the chemical expansion coefficient for the supercells containing
. As the figure demonstrates, the chemical expansion coefficient for the supercells containing 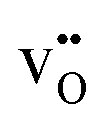 and
and  becomes increasingly negative with increasing temperature. The resulting chemical expansion coefficient due to hydration varies to a much smaller extent, ranging from 0.070 to 0.115 in the temperature range presented. This is in reasonable agreement with other experimental and computational work.1,12,38
becomes increasingly negative with increasing temperature. The resulting chemical expansion coefficient due to hydration varies to a much smaller extent, ranging from 0.070 to 0.115 in the temperature range presented. This is in reasonable agreement with other experimental and computational work.1,12,38

 , as calculated by quasiharmonic phonon calculations as a function of temperature.
, as calculated by quasiharmonic phonon calculations as a function of temperature.

 , to that of the proton,
, to that of the proton,  :
:
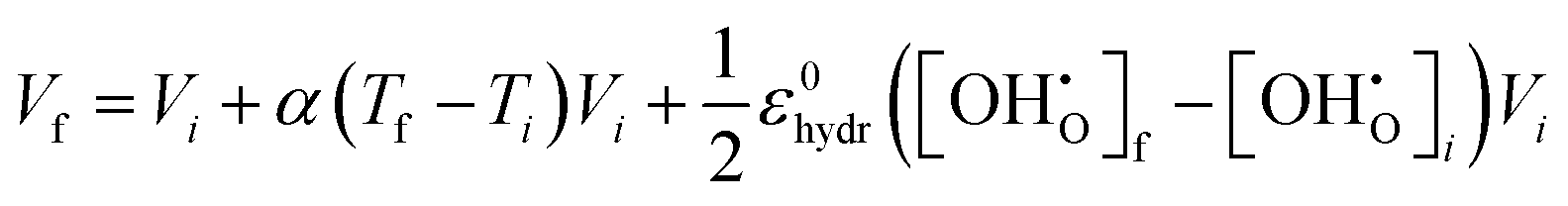
 and
and  denote the proton concentration at Tf and Ti, respectively. Note that we have chosen to use a constant value for the chemical expansion coefficient, ε0hydr, which differs from εhydr, given in (8), in that it is entirely temperature independent stemming from the equilibrium volumes of all supercells calculated at 0 K and excludes all zero-point energy contributions (0.078). This is intentionally done to avoid double counting the temperature dependencies of the equilibrium volumes, which is already implemented in the thermal expansion coefficient in (14). Fig. 5a displays the change in the unit cell volume as a function of temperature for different acceptor dopant concentrations (5–20 mol%) under humid conditions, pH2O = 0.025 atm, while Fig. 5b gives the unit cell volume for 20 mol% doped BaCeO3 where pH2O is varied from 0.025 to 1 atm. Fig. 5a demonstrates that the volume increases to a larger extent upon hydration with increasing doping concentrations reflecting higher proton concentrations. While the unit cell volume only changes gradually upon hydration (or dehydration) for the lower doping concentrations (5–15 mol%), the volume change for 20 mol% doped BaCeO3 is more abrupt, and the volume is almost constant in a small temperature interval for 20 mol% doped BaCeO3. In Fig. 5b, protons are retained to higher temperatures for pH2O = 1 atm and further heating is therefore needed for the volume to decrease compared to the corresponding unit cell volume at pH2O = 0.025 atm. Fig. 5b shows an additional interesting feature where the volume of the unit cell is not the same at lower temperatures (<600 K) when the water vapour partial pressure is changed. This stems from the difference in the thermal expansion coefficients upon hydration (cf.Fig. 3) where the unit cell at pH2O = 1 atm is thermally contracting to a larger extent than the corresponding unit cell at 0.025 atm.
denote the proton concentration at Tf and Ti, respectively. Note that we have chosen to use a constant value for the chemical expansion coefficient, ε0hydr, which differs from εhydr, given in (8), in that it is entirely temperature independent stemming from the equilibrium volumes of all supercells calculated at 0 K and excludes all zero-point energy contributions (0.078). This is intentionally done to avoid double counting the temperature dependencies of the equilibrium volumes, which is already implemented in the thermal expansion coefficient in (14). Fig. 5a displays the change in the unit cell volume as a function of temperature for different acceptor dopant concentrations (5–20 mol%) under humid conditions, pH2O = 0.025 atm, while Fig. 5b gives the unit cell volume for 20 mol% doped BaCeO3 where pH2O is varied from 0.025 to 1 atm. Fig. 5a demonstrates that the volume increases to a larger extent upon hydration with increasing doping concentrations reflecting higher proton concentrations. While the unit cell volume only changes gradually upon hydration (or dehydration) for the lower doping concentrations (5–15 mol%), the volume change for 20 mol% doped BaCeO3 is more abrupt, and the volume is almost constant in a small temperature interval for 20 mol% doped BaCeO3. In Fig. 5b, protons are retained to higher temperatures for pH2O = 1 atm and further heating is therefore needed for the volume to decrease compared to the corresponding unit cell volume at pH2O = 0.025 atm. Fig. 5b shows an additional interesting feature where the volume of the unit cell is not the same at lower temperatures (<600 K) when the water vapour partial pressure is changed. This stems from the difference in the thermal expansion coefficients upon hydration (cf.Fig. 3) where the unit cell at pH2O = 1 atm is thermally contracting to a larger extent than the corresponding unit cell at 0.025 atm.


 , does not change upon defect formation;12,41,48–51
, does not change upon defect formation;12,41,48–51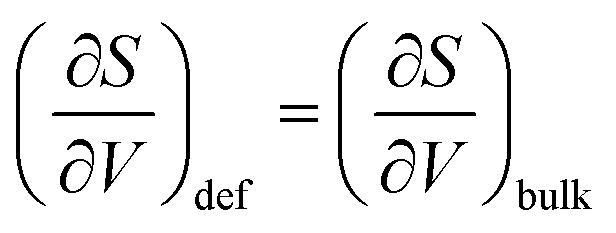
 is further given by
is further given by
 , the difference between the approximations again only stems from the oxygen vacancy formation entropy reaching a maximum difference of about 35 J K−1 mol−1 at higher temperatures. Ultimately, we find that most of the vibrational thermodynamics are appropriately described by the HA as anharmonic effects appear to be of a minor concern.
, the difference between the approximations again only stems from the oxygen vacancy formation entropy reaching a maximum difference of about 35 J K−1 mol−1 at higher temperatures. Ultimately, we find that most of the vibrational thermodynamics are appropriately described by the HA as anharmonic effects appear to be of a minor concern.
