
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The reactivity of platinum microelectrodes
Leon
Jacobse
,
Stefan J.
Raaijman
and
Marc T. M.
Koper
*
Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: m.koper@lic.leidenuniv.nl
First published on 20th September 2016
Abstract
Despite the widespread application of ultramicroelectrodes (UMEs), the customary method of their electrochemical characterization via outer sphere redox probing has serious limitations. In this study we provide additional insights into this subject by measuring not only outer sphere redox couples, but also blank voltammetries and the reactivity towards various catalytic reactions of Pt UMEs. The data for the UMEs are compared to those for macroscale Pt electrodes that can be flame-annealed. Although the reactivity is similar for reactions that are rather insensitive to the surface structure (and/or composition), UMEs perform much worse for more surface sensitive catalytic reactions. This effect can be explained by the UMEs being contaminated much faster, though it remains to be established if the origin of this contamination lies in the preparation method or in the impurities in the (high-purity grade) chemicals used. Our study recommends that catalytic reactivity measurements on ultramicroelectrodes should always be accompanied by blank voltammetries and the results have to be interpreted extremely carefully.
1 Introduction
A wealth of information concerning ‘conventional’ and newer types of (electro-)catalysts has been gained in decades of catalysis research. Due to improved catalytic properties, experimental approaches have to be increasingly specialized for the reaction studied, to be not (kinetically) limited by the setup used. Mass transport and resistance can easily become issues while studying electrochemical reactions with large kinetic rate constants. In such cases, decreasing the geometrical dimensions of the electrode to the micrometer scale or smaller can help remedy these inconveniences. This strategy is based on the fact that miniaturization improves mass transport and decreases absolute currents, both beneficial for situations where high kinetic rates are concerned.1So long as the electrode is not reduced to the nanometer scale, it should be possible to directly compare their results to the macro-scale world. Upon further size reduction, e.g. as in the case of nanoparticles, there are plenty of examples of size dependent reactivity due to the presence of specific nanoscale effects.2–12
On a different note, it is known from the literature that the selectivity of Pt for the reduction of O2 to H2O (with H2O2 as an intermediate/sideproduct) decreases by increasing the mass transport rate.13,14 These observations were attributed to the competition between the adsorption/desorption of H2O2 and trace contaminants that are present even in the highest purity grade chemicals.13–15 Furthermore, a recent publication by Chen et al. indicates, unexpectedly, that already on the micrometer scale differences in catalytic reactivity can be observed for the oxidation of hydrazine on Pt electrodes in HClO4.16 This effect cannot be explained by competitive adsorption/desorption as no stable intermediates are known for hydrazine oxidation to N2.17,18
These results call for a more in-depth comparison between the reactivities of platinum macroscale electrodes and ultramicroelectrodes (UMEs). UMEs come in many varieties, but have in common at least one dimension that is smaller than the diffusion layer thickness (≃10 μm) although still much larger than the double layer (10–100 Å).1,19
As reviewed by Climent and Feliu, the characterization of platinum surfaces in electrochemistry is, for historical reasons, most often performed in dilute sulfuric acid.20 In this medium, the defining features in cyclic voltammograms (CVs) are clearly visible and well understood.21 With the introduction of flame annealing, these so-called blank CVs (blanks) have enjoyed excellent reproducibility between different labs, making it the default method for electrode characterization. From such blanks, one can discern the presence and/or absence of contaminating species and specific reaction sites.20,22
Even though such practices are now widespread within the community studying Pt electrochemistry, blank CVs of (Pt) UMEs are rarely published and if at all only for high potential scan rates23–26 which make them rather insensitive towards slow reactions. Reactions with slow kinetics or reactions which are diffusion limited (e.g. trace contamination) will be suppressed under these conditions. Instead, surface probes such as the [Fe(CN)6]4−/3− or [FcCH2OH]0/1+ redox couple enjoy high popularity for electrode characterization.27–30 The problem is that such outer sphere redox couples are, although some complications are known,31,32 in general, insensitive to the surface structure, composition, and cleanliness.33 The main benefits of these experiments are determination of electrical contact and gain information regarding the geometrical dimensions of the electrode,1 but their suitability for the assessment of surface reactivity is highly debatable.
Outer sphere redox couples, blank voltammetry, and catalytic (inner sphere) reactions were measured in this study to make a solid comparison between the catalytic reactivity of macroelectrodes and UMEs. The redox couples studied were the oxidation of ferrocenemethanol ([FcCH2OH]0/1+) and the reduction of hexaammineruthenium ([Ru(NH3)6]3+/2+). Similarly, catalytic reduction and oxidation reactions were also studied. For catalytic reactions, a further distinction was made: reactions that are well catalyzed by Pt in general (largely surface insensitive) and reactions that require specific surface ‘defects’ (surface sensitive). The chosen reactions are oxidation of hydrazine (N2H4),16,17,34,35 oxidation of methanol (CH3OH),11,36–40 and reduction of nitrate (NO3−).41–43 For each of these reactions, the Tafel slope, onset potential, and specific activity (where applicable) were compared to the values obtained on flame-annealed platinum electrodes under similar experimental conditions.
2 Experimental
UMEs were prepared from platinum wire (50 μm diameter, Goodfellow, 99.99%) which was electrochemically etched to the desired thickness.1,19 Next, the wire was dipped in freshly prepared piranha (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v H2SO4 (Sigma-Aldrich, Puriss. p.a.) and H2O2 (Merck)) for a few seconds and rinsed with ultrapure (>18.2 MΩ cm−1, Millipore Milli-Q) water. The electrode was made by sealing the wire into a soda lime glass capillary using a butane torch. The absence of bubbles surrounding the seal was confirmed using an optical microscope. A microdisk was exposed by polishing with increasingly fine-grained sandpaper, after which alumina (Kemet α-alumina powder solution) or diamond paste (Kemet diamond emulsion) was used for a final polish on a microcloth (Buehler). To finish, the electrode was rinsed thoroughly with ultrapure water, acetone (Sigma-Aldrich, 99% Puriss.) and ethanol (Sigma-Aldrich, 99.8% Puriss. p.a.) after which it was sonicated (Bransonic 2510, Branson) in ultrapure water to remove any residue of the polishing process. When the UME was used for experiments, mechanical polishing and sonication were applied only when blank voltammetry indicated this to be necessary, to increase the reproducibility and the ‘lifetime’ of the electrode (e.g. Pt/glass seal, surface conditions).
:1 v/v H2SO4 (Sigma-Aldrich, Puriss. p.a.) and H2O2 (Merck)) for a few seconds and rinsed with ultrapure (>18.2 MΩ cm−1, Millipore Milli-Q) water. The electrode was made by sealing the wire into a soda lime glass capillary using a butane torch. The absence of bubbles surrounding the seal was confirmed using an optical microscope. A microdisk was exposed by polishing with increasingly fine-grained sandpaper, after which alumina (Kemet α-alumina powder solution) or diamond paste (Kemet diamond emulsion) was used for a final polish on a microcloth (Buehler). To finish, the electrode was rinsed thoroughly with ultrapure water, acetone (Sigma-Aldrich, 99% Puriss.) and ethanol (Sigma-Aldrich, 99.8% Puriss. p.a.) after which it was sonicated (Bransonic 2510, Branson) in ultrapure water to remove any residue of the polishing process. When the UME was used for experiments, mechanical polishing and sonication were applied only when blank voltammetry indicated this to be necessary, to increase the reproducibility and the ‘lifetime’ of the electrode (e.g. Pt/glass seal, surface conditions).
Different previously reported (see e.g.ref. 19, 26, 44 and 45) UME cleaning methods were used to prepare the UME for daily use of which the following routine gave the best and most reproducible results. At the start of each day, the UME was subsequently rinsed with ultrapure water; sonicated in ultrapure water (5 min); and 500 times cycled between 0.06 and 1.60 V vs. RHE (0.5 M H2SO4 at 1 V s−1) to yield a blank voltammogram that was reproducible over multiple days when the same pretreatment was applied. In between experiments on the same day, the electrode was rinsed with ultrapure water and cycled (same conditions as before) 100 times. At the end of the day, the UME was once more cycled 500 times, rinsed with ultrapure water and stored in a desiccator until the next use. Adsorption of CO by bubbling through the electrolyte or from methanol decomposition followed by CO stripping, commonly used to clean Pt nanoparticles,46,47 was also tested to clean the UMEs. However, this did not improve the blank voltammetries while increasing the risk of introducing contamination. The platinum electrodes used for comparison (referred to as macroelectrode) have a much larger geometric surface area (tens of mm2) than the UMEs and were cleaned by annealing in a butane flame.
All glassware was cleaned by initially boiling in a 3:1 v/v mixture of sulfuric acid (Sigma-Aldrich, Puriss. p.a.) and nitric acid (Sigma-Aldrich, ACS reagent grade) and stored in permanganate solution (1 g L−1 KMnO4 dissolved in 0.5 M H2SO4) in-between experiments. Prior to each experiment, trace amounts of residual permanganate and manganese oxide were removed with dilute piranha, after which the glassware was cleaned by repeated boiling in ultrapure water. Electrolyte solutions were prepared by dissolving the appropriate amount of chemicals in 25 or 100 mL ultrapure water. The electrolyte was purged of oxygen by bubbling argon for 20 minutes.
Two different set-ups were used. The macroelectrode experiments were carried out in a two-compartment electrochemical cell with the reference separated by a Luggin capillary. Microelectrode measurements were carried out in a one compartment cell in a Faraday cage situated on a weighed table. A Pt wire was used as a counter electrode (CE). A reversible hydrogen electrode (RHE) (blank CVs and catalytic reactions) and a Ag|AgCl|KClsat electrode (outer sphere redox reactions) were employed as reference electrodes. All potentials are reported versus the reversible hydrogen electrode scale. Potentials were controlled using a potentiostat/galvanostat from either Autolab (PGSTAT12) or Bio-Logic (SP-300). [Ru(NH3)6]·3Cl (Acros organics, 98%); FcCH2OH (Aldrich, 97%), methanol (Fluka, 99.9% LC-MS CHROMASOLV), N2H4·H2O (Sigma-Aldrich, 98%), NaNO3 (Sigma-Aldrich), KCl (Sigma-Aldrich, ACS reagent grade), H2SO4 solution (Fluka, for trace analysis), and hydrogen and argon (Linde 6.0) were used as received.
All data shown here (except the green curves in Fig. 1b and 2) result from a single UME. Other Pt UMEs always showed a similar behavior. The geometrical surface area of the electrode was determined via the limiting current of the outer sphere redox couples to be discussed in Section 3.1.1 assuming a microdisk geometry. Both redox couples were measured in triplicates and yielded very similar values for the disk radius (4.2 ± 0.01 and 4.5 ± 0.05 μm for [Ru(NH3)6]3+ and FcCH2OH respectively). The average radius of 4.4 μm is in line with observations of optical microscopy.
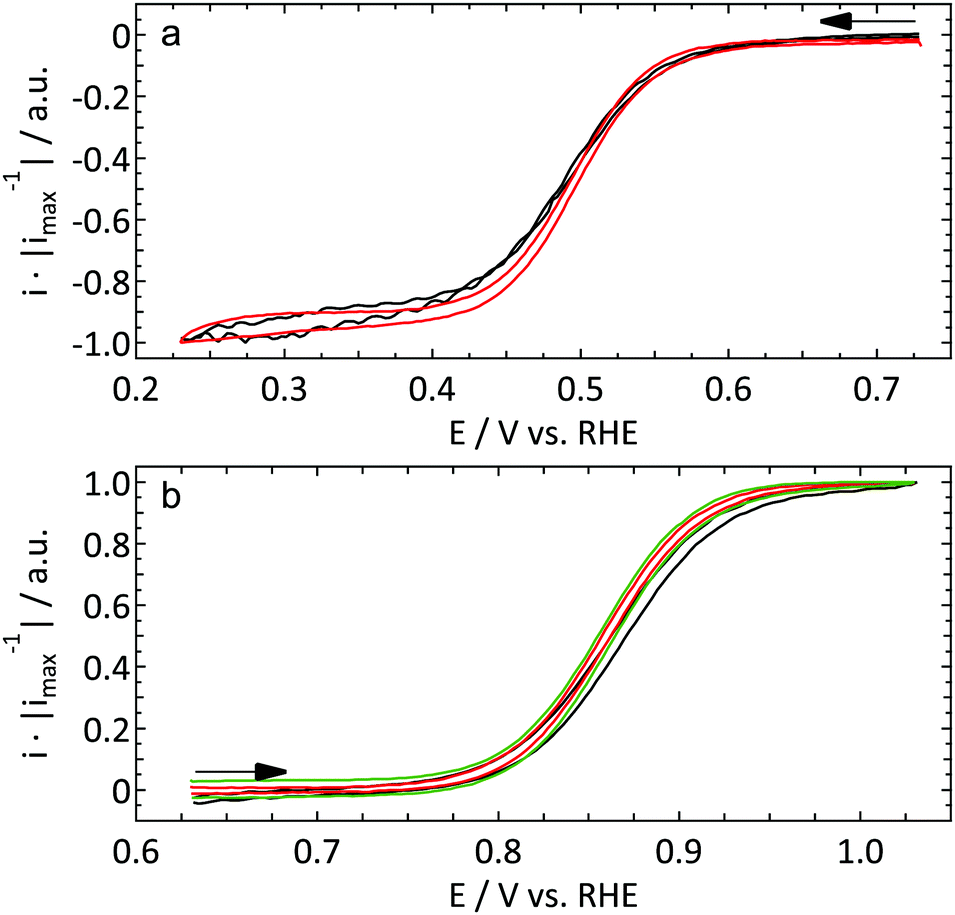 | ||
| Fig. 1 First cycle of the reduction of 1 mM [Ru(NH3)6]3+ (a) and oxidation of 1 mM FcCH2OH (b) in 0.1 KCl on a Pt UME (r = 4.4 μm, ν = 10 mV s−1, red) and a macroelectrode (hanging meniscus configuration, f = 2800 r.p.m., ν = 50 mV s−1, black). The response of a different UME (r = 8.5 μm) in a separate experiment under identical conditions is shown in green in (b). | ||
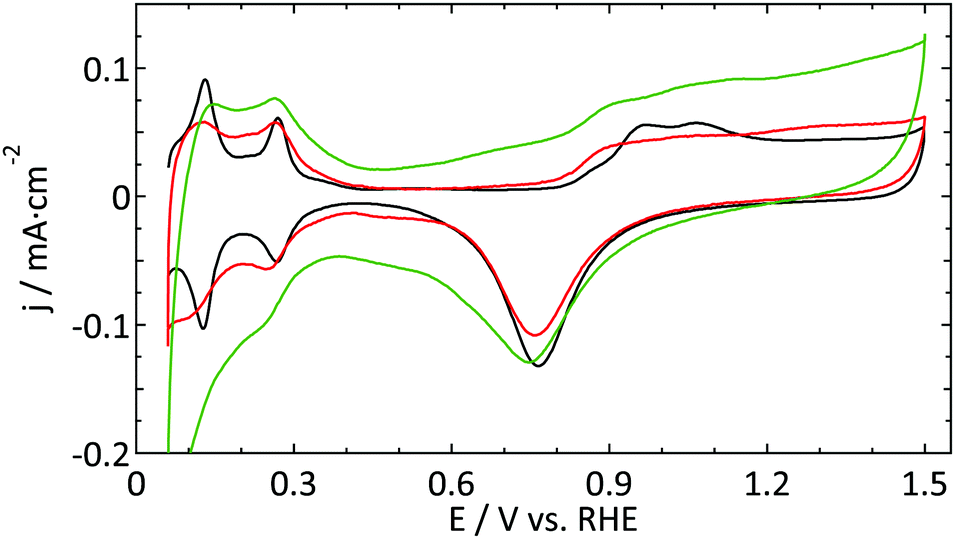 | ||
| Fig. 2 Blank cyclic voltammogram (ν = 50 mV s−1) of a platinum ultramicroelectrode (r = 4.4 μm, red and r = 8 μm, green) and a flame-annealed platinum spiral (black) in 0.5 M H2SO4. | ||
The electrochemically active surface area was calculated from the hydrogen desorption integral in the region 0.06 < E < 0.6 V after subtraction of the double layer, using the recently revisited value of 230 μC cm−2 for a polycrystalline Pt surface in sulfuric acid.46 The ratio between the electrochemically active and geometrical surface areas yielded roughness factors that were typically in the 10–12 range, which is reasonable compared to the size of the the UME with the polishing material.
3 Results and discussion
3.1 Electrode characterization
000 r.p.m.). This explains the slightly less steep current increase for the macroelectrode. Apart from that no significant differences are observed between the different electrodes.
Fig. 2 shows a decent blank voltammogram of a Pt UME (in red) compared to a flame annealed macroelectrode (in black). Although one would expect these CVs to be the same, there are some obvious differences. The hydrogen adsorption and desorption peaks (around 0.12 and 0.27 V) of the UME are not as well defined as and less reversible than in the case of the macroelectrode. Also, the most negative adsorption/desorption (ca. 0.12 V) peak should be larger than the second peak (ca. 0.27 V), which it is not. The latter could in principle be explained by the production process of these thin Pt wires which is known to lead to a preferential grain orientation.48 Due to the small size of a UME, it is likely that the surface consists of a single grain and could thus be considered to be a high-index single crystal. However, this mainly influences the ratio between different hydrogen-related peaks and to a much smaller extent their sharpness.22 Furthermore, the microelectrodes typically show a larger surface normalized double layer (DL) current. One could explain this by the underestimation of the surface area of UMEs due to less well-defined hydrogen desorption. However, this seems in contradiction with the observation that the oxide reduction peak (ca. 0.78 V) is typically smaller for the UME than for the macroelectrode. Besides the larger DL current, one could argue that there is an additional peak present around 0.45 V in the negative-going scan. Finally, the shape of the platinum oxidation region usually does not clearly show two bumps, in fact showing very few identifying features at all.
The second UME blank voltammogram (green) that is shown in Fig. 2 demonstrates the superiority of electrode characterization using blank voltammetry instead of an outer sphere redox couple. This blank was measured prior to the oxidation of FcCH2OH shown in Fig. 1b (green curve). Whereas the FcCH2OH data suggest that this electrode performs well, this does not follow from the blank voltammetry. In this case our normalization procedure (using the hydrogen desorption area) clearly leads to an underestimation of the active surface area leading to much too high hydrogen adsorption, oxide formation, and DL current densities. Obviously, the shape also deviates significantly from what it should be. This implies that there is a process inhibiting the hydrogen desorption and oxide reduction reactions without a significant impact on outer sphere redox chemistry.
The aim of the current study is not to obtain the perfect blank CV of a Pt UME, but merely to provide a better insight into the reliability of the currently available methods to study UMEs. Within this framework, the blank shown in Fig. 2 (red curve) was among the best, in terms of quality and reproducibility, that we obtained. Therefore this was considered as a good starting point to study catalytic reactions. It is expected that the underestimation of the active surface area due to a blocking process as demonstrated in Fig. 2 also occurs, to some extent, for the other electrodes. To prove that this has only a minor effect on the observed differences; the blank CVs measured prior to hydrazine and methanol oxidation are shown in Fig. 3. The blank CV measured prior to the nitrate reduction is shown in Fig. 6. From these blanks it is clear that the error in the determination of the active surface area is always smaller than a factor of 2.
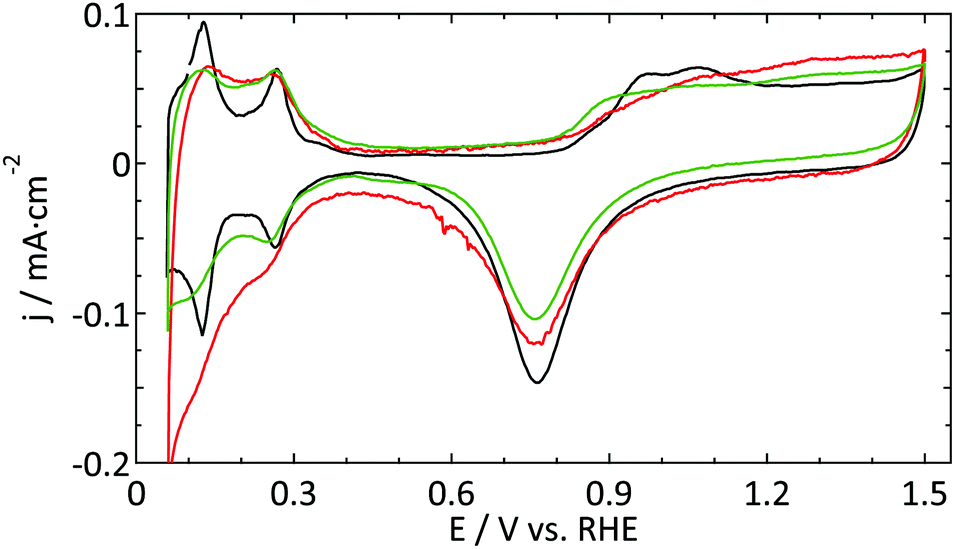 | ||
| Fig. 3 Blank CVs (ν = 50 mV s−1) of a platinum UME (r = 4.4 μm) prior to the oxidation of 1 mM hydrazine (red) and 0.5 M methanol (green) in 0.5 M H2SO4. As the macroelectrode blanks were virtually identical, only one CV is shown (black). | ||
3.2 Catalytic reactivity
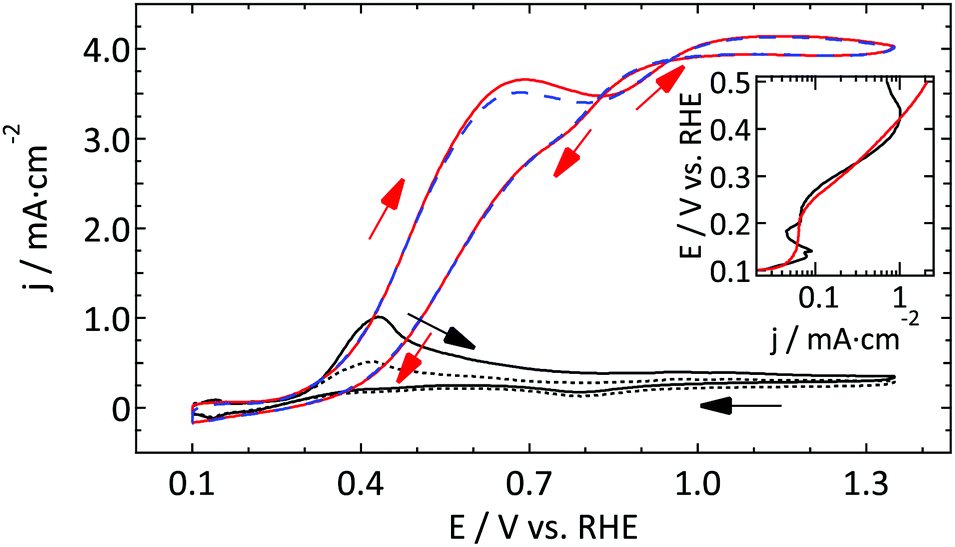 | ||
| Fig. 4 First and fourth CV (ν = 50 mV s−1) of the oxidation of 1 mM N2H4 in 0.5 M H2SO4 on a Pt UME (r = 4.4 μm, red and dashed blue respectively) and a macroelectrode (full and dotted black line respectively). The inset shows a Tafel plot of the first anodic scans. | ||
As Pt is a very good catalyst for the oxidation of hydrazine, diffusion limited current is observed for both electrodes. Due to different diffusion geometries the CVs seem to be very different at first sight. However, if one takes a closer look at the characteristic features such as the onset potential and the Tafel slope, it can be concluded that both electrodes show almost the same reactivity. The only (minor) difference regarding catalytic activity is the fact that the change in the Tafel slope is slightly higher for the UME than for the macroelectrode. However, as the current density is limited by the diffusion of hydrazine to the surface and not by the catalytic properties of the working electrode for most of the potential range studied, it is to be expected that both electrodes show a similar behavior. Actually, this result is more or less similar to the outer sphere reactions discussed in Section 3.1.1.
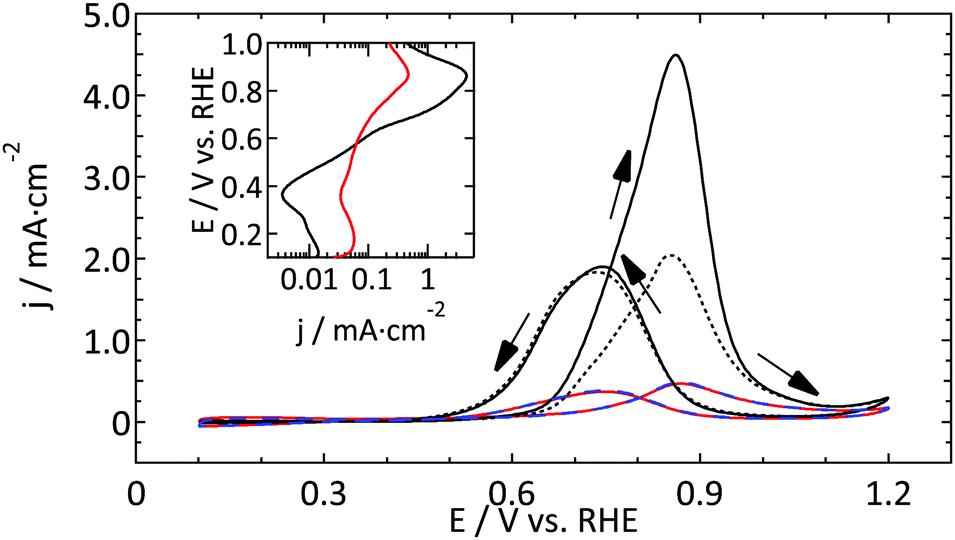 | ||
| Fig. 5 First and fourth CV (ν = 50 mV s−1) of the oxidation of 0.5 M methanol in 0.5 M H2SO4 on a Pt UME (r = 4.4 μm, red and dashed blue respectively) and a macroelectrode (full and dotted black line respectively). The inset shows a Tafel plot of the first anodic scans. | ||
Comparing the absolute reactivity, it is very clear that the UME is much less active than the macroelectrode. From the good agreement between the normalized blank CVs (Fig. 3) it is confirmed that this large difference (about a factor 10) cannot be explained by an error in the calculated surface areas. Furthermore, although the onset potential seems similar, the higher Tafel slope in the case of the microelectrode masks the precise onset potential.
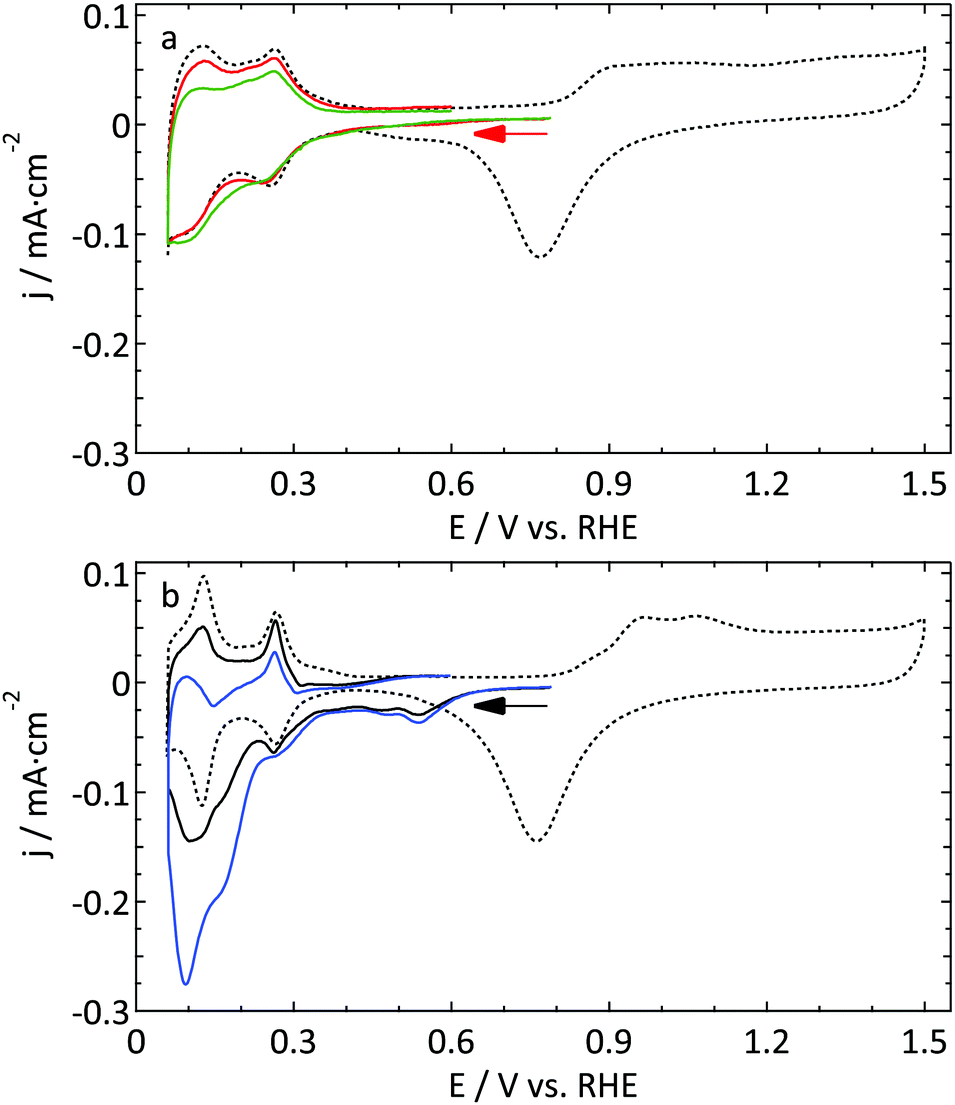 | ||
| Fig. 6 Cyclic voltammograms (ν = 50 mV s−1) of the reduction of 0.1 and 0.5 M nitrate in 0.5 M H2SO4 on (a) a Pt UME (r = 4.4 μm, red and green respectively) and (b) a macroelectrode (black and blue respectively). The dotted lines show the blank voltammograms measured right before the nitrate reduction experiment. The same scale is used for both graphs. | ||
3.3 UME vs. macroelectrode
In general, one could summarize the data for the catalytic reactions as exhibiting the same features as the outer sphere and blank results. Reactions that are relatively insensitive to the arrangement of the surface atoms (or their very nature) show the same reactivity for UMEs and macroelectrodes. However, for slow, more structure-sensitive reactions the UMEs are outperformed by macroelectrodes. The data suggest that the difference does not lie in the onset potential, but in the absolute current density. Also, from the blanks there is no reason to conclude that a preferred grain orientation causes these differences. The most likely explanation is that part of the surface is blocked by contaminant species. As the most reactive sites will also bind the contaminants in the strongest manner, it makes sense that the largest effect is observed for slow, strong catalytic reactions that occur preferentially at these sites.A crucial distinction lies in the source of the contamination: Is it introduced during the preparation/cleaning of the UME, or are the UMEs that much more sensitive due to faster diffusion and a smaller surface area as compared to macroelectrodes? Significantly, no improvement was observed after cleaning the UME with O2 plasma. Thus, if UMEs are inherently contaminated to a larger degree because flame-annealing is not an option, the source of contamination is probably not organic in nature.
Even though the cleanest chemicals available in our lab were used, the level of contamination might be relevant on this scale. For example, the sulfuric acid used (Fluka, for trace analysis) contains, according to the manufacturer's specifications, among others, ≤0.1 mg kg−1 Cl− ions. Assuming the maximum concentration, this means that our 0.5 M H2SO4 electrolyte contains 1.41 × 10−7 M chloride. Using a diffusion coefficient for chloride of 2.0 × 10−5 cm2 s−1, the surface of our UME (roughness factor, ≃10) can be fully covered with Cl− within approximately 20 minutes. This suggests that a significant fraction of the surface could be poisoned by chloride within the experimental timeframe.
A similar argument was made by Katsounaros et al. who studied the effect of chloride ions, catalyst loading, and electrode rotation speed on the reduction of H2O2.14 On the other hand, the cell used for the UME experiments has a rather small electrolyte volume (15 mL), so that the total number of chloride ions present is of the same order of magnitude as the amount needed to generate a single adlayer. Finally, we consider the concentrations of other contaminants, e.g. organic carbon from ultrapure water (<5 ppb) or nitrates from glassware cleaning, to be too low to play a role here. Thus, the most plausible explanation is that the contamination originates from the (UME) preparation. It is noteworthy to mention that another often used supporting electrolyte, HClO4, typically contains a 100-fold higher concentration of Cl− than H2SO4. Although it proved to be more difficult to obtain good blank voltammograms in perchloric acid electrolytes, the best ones we obtained (Fig. 7) are not inferior to the ones in sulfuric acid.
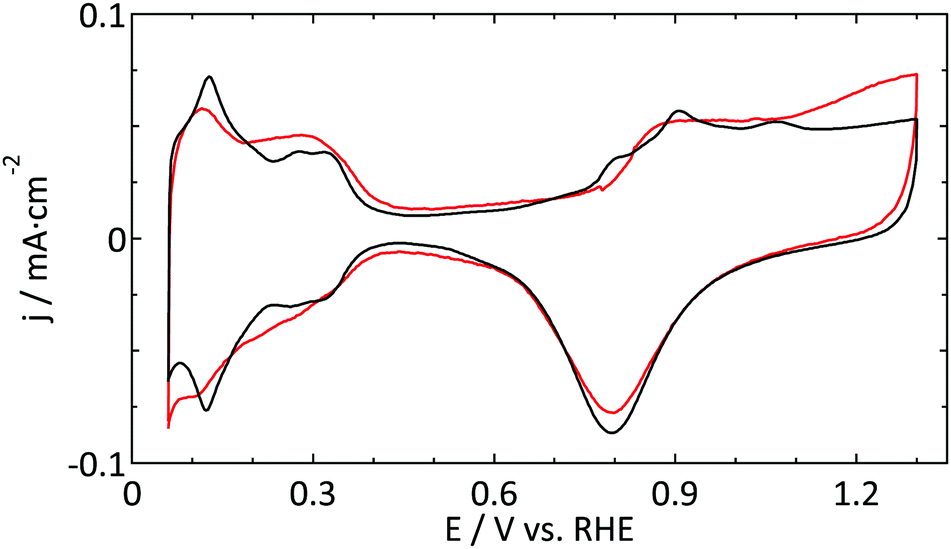 | ||
| Fig. 7 Blank CVs (ν = 50 mV s−1) of a platinum UME (r = 4.4 μm, red) and a macroelectrode (black) in 0.1 M HClO4. | ||
4 Conclusion
Despite the widespread application of UMEs, the customary method of electrochemical characterization via outer sphere redox probing is insufficient if the reactivity of the electrode is studied. In this study we provided a better insight into this subject by measuring not only outer sphere redox couples, but also blank CVs and the catalytic reactivity of Pt UMEs. Reactions were chosen for which plenty of data on macroelectrodes is available from the literature. To validate the UME results, the data for macroelectrodes were measured as well for comparison.Activity for the reduction of [Ru(NH3)6]3+ and the oxidation of FcCH2OH were shown to agree very well when comparing UME responses to annealed electrodes. The onset potential was virtually identical, and a minor difference in the Tafel slope was explained by the diffusion rates of the UME being faster than those experimentally possible for a macroelectrode. Also in the case of the reaction for which Pt is known to be a very good catalyst, the oxidation of N2H4, no significant differences between the UME and the macroelectrode were observed.
However, for reactions that strongly depend on the surface structure, significant differences are observed. Blank CVs of the UME do not show well-defined hydrogen and oxide related peaks that are observed for a polycrystalline electrode. Furthermore the normalized current in the DL region is too high, while simultaneously the oxide reduction current is too low. In the case of methanol oxidation on Pt the CVs are similarly shaped, but the current density of the UME is about an order of magnitude lower than expected based on the electrochemical surface area. For the reduction of nitrate the effect is even more drastic, with the UME exhibiting hardly any catalytic activity. The voltammograms do not provide a means to identify a specific contaminant as no clear additional peaks were observed, although this does not imply that none were present. Concluding, the most active sites on the UME surface seem to be blocked, with drastic consequences for reactions which depend heavily on the presence of these sites. Although the responsible process is not yet fully elucidated, there seem to be two reasonable explanations; either there is something bound so strongly to the surface (e.g. residual from the UME preparation) that it is very difficult to obtain a clean sample without flame-annealing, or the diffusion is so fast that the surface becomes easily covered by trace contaminants in the electrolyte within the experimental timeframe. The amount of contaminants in the used chemicals is too low to result in a significant coverage of the UME. Thus, it seems most plausible that the limited possibilities to clean the UMEs prior to use are the source of the observed effects.
Although it is uncommon to publish blank voltammograms for UMEs, our comparison to macroelectrodes shows that there is a crucial mismatch between the catalytic reactivities of these electrodes. Unfortunately none of the widely used cleaning methods for UMEs provide electrode cleanliness similar to flame annealing. Thus, in many cases the reactivity of UMEs will be underestimated if no proper electrochemical characterization is performed. Obviously this may have severe consequences for the interpretation and reproducibility of the data.
References
- R. J. Forster, Encycl. Electrochem., 2002, 3, 160–195 Search PubMed.
- M. T. M. Koper, Nanoscale, 2011, 3, 2054–2073 RSC.
- R. J. Gilliam, D. W. Kirk and S. J. Thorpe, Electrochem. Commun., 2007, 9, 875–878 CrossRef CAS.
- R. Gilliam, D. Kirk and S. Thorpe, Electrochem. Commun., 2007, 9, 2276–2279 CrossRef CAS.
- W. P. Zhou, M. Li, C. Koenigsmann, C. Ma, S. S. Wong and R. R. Adzic, Electrochim. Acta, 2011, 56, 9824–9830 CrossRef CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- S. Chumillas, C. Busó-Rogero, J. Solla-Gullón, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, Electrochem. Commun., 2011, 13, 1194–1197 CrossRef CAS.
- T. L. Tan, L.-L. Wang, J. Zhang, D. D. Johnson and K. Bai, ACS Catal., 2015, 5, 2376–2383 CrossRef CAS.
- A. Kabbabi, F. Gloaguen, F. Andolfatto and R. Durand, J. Electroanal. Chem., 1994, 373, 251–254 CrossRef CAS.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792–5798 CrossRef CAS.
- C. K. Rhee, B.-J. Kim, C. Ham, Y.-J. Kim, K. Song and K. Kwon, Langmuir, 2009, 25, 7140–7147 CrossRef CAS PubMed.
- T. Frelink, W. Visscher and J. A. R. van Veen, J. Electroanal. Chem., 1995, 382, 65–72 CrossRef.
- S. Chen and A. Kucernak, J. Phys. Chem. B, 2004, 108, 3262–3276 CrossRef CAS.
- I. Katsounaros, J. C. Meier and K. J. J. Mayrhofer, Electrochim. Acta, 2013, 110, 790–795 CrossRef CAS.
- D. A. Scherson and Y. V. Tolmachev, Electrochem. Solid-State Lett., 2010, 13, F1 CrossRef CAS.
- C. H. Chen, L. Jacobse, K. McKelvey, S. C. S. Lai, M. T. M. Koper and P. R. Unwin, Anal. Chem., 2015, 87, 5782–5789 CrossRef CAS PubMed.
- M. D. García, M. L. Marcos and J. G. Velasco, Electroanalysis, 1996, 8, 267–273 CrossRef.
- V. Rosca and M. T. M. Koper, Electrochim. Acta, 2008, 53, 5199–5205 CrossRef CAS.
- C. G. Zoski, Electroanalysis, 2002, 14, 1041–1051 CrossRef CAS.
- V. Climent and J. M. Feliu, J. Solid State Electrochem., 2011, 15, 1297–1315 CrossRef CAS.
- F. J. Vidal-Iglesias, R. M. Arán-Ais, J. Solla-Gullón, E. Herrero and J. M. Feliu, ACS Catal., 2012, 2, 901–910 CrossRef CAS.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- M. C. Tavares, S. A. S. Machado and L. H. Mazo, Electrochim. Acta, 2001, 46, 4359–4369 CrossRef CAS.
- D. A. Cook, PhD thesis, University of Southampton, 2005.
- D. Zhan, J. Velmurugan and M. V. Mirkin, J. Am. Chem. Soc., 2009, 131, 14756–14760 CrossRef CAS PubMed.
- I. Fromondi, P. Shi, A. Mineshige and D. A. Scherson, J. Phys. Chem. B, 2005, 109, 36–39 CrossRef CAS PubMed.
- K. Aoki, C. Zhang, J. Chen and T. Nishiumi, Electrochim. Acta, 2010, 55, 7328–7333 CrossRef CAS.
- S. Chen and A. Kucernak, J. Phys. Chem. B, 2002, 106, 9396–9404 CrossRef CAS.
- J. Guo, C.-N. Ho and P. Sun, Electroanalysis, 2011, 23, 481–486 CrossRef CAS.
- U. K. Sur, A. Dhason and V. Lakshminarayanan, J. Chem. Educ., 2012, 89, 168–172 CrossRef CAS.
- L. Peter, W. Dürr, P. Bindra and H. Gerischer, J. Electroanal. Chem., 1976, 71, 31–50 CrossRef CAS.
- C. Beriet and D. Pletcher, J. Electroanal. Chem., 1993, 361, 93–101 CrossRef CAS.
- X. Xiao, S. Pan, J. S. Jang, F.-R. F. Fan and A. J. Bard, J. Phys. Chem. C, 2009, 113, 14978–14982 CAS.
- L. Aldous and R. G. Compton, Phys. Chem. Chem. Phys., 2011, 13, 5279–5287 RSC.
- B. Álvarez Ruiz, R. Gómez, J. M. Orts and J. M. Feliu, J. Electrochem. Soc., 2002, 149, D35 CrossRef.
- E. Herrero, K. Franaszczuk and A. Wieckowski, J. Phys. Chem., 1994, 98, 5074–5083 CrossRef CAS.
- S. C. S. Lai, N. P. Lebedeva, T. H. M. Housmans and M. T. M. Koper, Top. Catal., 2007, 46, 320–333 CrossRef CAS.
- T. H. M. Housmans, A. H. Wonders and M. T. M. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS PubMed.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663–3674 CrossRef CAS.
- J. Jiang and A. Kucernak, J. Electroanal. Chem., 2005, 576, 223–236 CrossRef CAS.
- M. T. De Groot and M. T. M. Koper, J. Electroanal. Chem., 2004, 562, 81–94 CrossRef CAS.
- G. E. Dima, G. L. Beltramo and M. T. M. Koper, Electrochim. Acta, 2005, 50, 4318–4326 CrossRef CAS.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, J. Electroanal. Chem., 2003, 554–555, 15–23 CrossRef CAS.
- T. Sun, P.-Y. Blanchard and M. V. Mirkin, Anal. Chem., 2015, 87, 4092–4095 CrossRef CAS PubMed.
- S. A. G. Evans, J. M. Elliott, L. M. Andrews, P. N. Bartlett, P. J. Doyle and G. Denuault, Anal. Chem., 2002, 74, 1322–1326 CrossRef CAS PubMed.
- Q.-S. Chen, J. Solla-Gullón, S.-G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982–7994 CrossRef CAS.
- J. Solla-Gullon, V. Montiel, a. Aldaz and J. Clavilier, J. Electrochem. Soc., 2003, 150, E104–E109 CrossRef CAS.
- G. Greenwood, Z. Kristallogr. – Cryst. Mater., 1931, 78, 242–250 CAS.
| This journal is © the Owner Societies 2016 |