
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near edge X-ray absorption mass spectrometry of gas phase proteins: the influence of protein size
D.
Egorov
 *a,
L.
Schwob
b,
M.
Lalande
b,
R.
Hoekstra
a and
T.
Schlathölter
*a
*a,
L.
Schwob
b,
M.
Lalande
b,
R.
Hoekstra
a and
T.
Schlathölter
*a
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, Netherlands. E-mail: d.egorov@rug.nl; t.a.schlatholter@rug.nl
bCIMAP, UMR 6252 (CEA/CNRS/ENSICAEN/Université de Caen Normandie), Caen, France
First published on 5th September 2016
Abstract
Multiply protonated peptides and proteins in the gas phase can respond to near edge X-ray absorption in three different ways: (i) non dissociative ionization and ionization accompanied by loss of small neutrals, both known to dominate for proteins with masses in the 10 kDa range. (ii) Formation of immonium ions, dominating for peptides in the 1 kDa range. (iii) Backbone scission leading to sequence ions which is typically weaker and has mainly been observed for peptides in the 1 kDa range. We have studied carbon 1s photoexcitation and photoionization for a series of peptides and proteins with masses covering the range from 0.5 kDa to more than 10 kDa. The gas phase protonated molecules were trapped in a radiofrequency ion trap and exposed to synchrotron radiation. Time of flight mass spectrometry was employed for investigation of the photoionization and photofragmentation processes. A smooth transition from the photofragmentation regime to the non-dissociative photoionization regime is observed. Mass spectra are most complex in the few kDa regime, where non-dissociative ionization, backbone scission and immonium ion formation coexist. The observed correlation between protein size and fragmentation, i.e. radiation damage, is of relevance for soft X-ray microscopy.
Introduction
Mass spectrometry is the key technology for protein sequence determination and protein identification. The two key aspects of protein mass spectrometry are ionization and activation. For the initial soft ionization process, most mass spectrometers rely either on electrospray ionization or on matrix assisted laser desorption. For the subsequent activation that causes the desired breakup of the protein ions on the other hand, a large collection of techniques is available. The most commonly used method of collision induced dissociation (CID) for instance involves stepwise energy deposition, favoring the rupture of weaker bonds. The resulting mass spectrum is dominated by sequence specific fragments due to backbone scission and by loss of small neutral groups.1 Electron capture induced dissociation and related electron based activation techniques imply the formation of an odd-electron radical. Here, fragmentation is faster and although the resulting mass spectrum is typically dominated by sequence specific fragments, different bonds are broken as for the case of CID. Both methods are complementary. A third class of activation techniques is based on photoabsorption.2 For low energy infrared photons, similar to CID, energy deposition is stepwise and predominantly weak bonds are ruptured. Single photon induced dissociation becomes possible in the UV range. Here, dissociation can occur either after intramolecular vibrational energy redistribution, leading to similar fragmentation patterns as CID, or on a much shorter timescale directly from the excited state. The latter process crucially depends on the photon energy and can open up new fragmentation channels.3 Simultaneous IR and UV exposure of ubiquitin has been found to combine advantages from both approaches and particularly rich mass spectra were obtained.4Only very recently, the interaction of energetic photons with gas phase protonated proteins and peptides was studied for the first time. Using synchrotron radiation Milosavljevic et al.5 and Bari et al.6 investigated VUV photoionization of protonated cytochrome c (m = 12.4 kDa) and leucine enkephalin (m = 555 Da), respectively. The resulting mass spectra for cytochrome c were found to be dominated by non-dissociative single ionization, accompanied by loss of small neutral groups. For the much smaller leucine enkephalin, non-dissociative ionization was found to be suppressed and fragmentation into small fragments was observed. In contrast to the findings from conventional activation methods, sidechain loss leading to formation of immonium ions was identified as the main process.
A straightforward next step was near K-edge photoionization of gas phase peptides and proteins, using soft X-rays. The appeal of such experiments is the possibility of element specific excitation of a 1s electron into an unoccupied molecular orbital. For instance, resonant K-shell excitation of the peptide model system N-methylacetamide, a relatively simple molecule containing a single peptide bond, can strongly favor the selective rupture of this bond.7,8
First soft X-ray inner shell photoionization studies on protonated cytochrome c and leucine enkephalin have been performed by Milosavljevic et al.9 and Gonzalez-Magaña et al.,10 respectively. Qualitatively similar dynamics were found as for VUV photoionization, with fragmentation patterns dominated by non-dissociative single and double ionization for cytochrome c and with disintegration into small fragments for leucine enkephalin. Also for protonated ubitiquin (m = 8.5 kDa), inner shell photoionization turns out to be predominantly non-dissociative. Furthermore, with increasing protonation state, the inner shell ionization energy was found to increase but fragmentation stayed negligible.11 In summary for larger proteins, fast dissociation channels involving the photoexcited state are thus efficiently quenched whereas for small peptides, fast dissociation channels dominate. It has to be noted, however, that larger proteins such as ubiquitin respond like an ensemble of small peptides, when subjected to simultaneous absorption of multiple EUV photons in femtosecond free electron laser pulses.12
These findings entail two fundamental questions: how are the fragmentation dynamics changing with increasing protein size and what are the underlying fragmentation mechanisms? The answers to these questions have implications beyond mass spectrometric applications. Radiation damage is a limiting factor for the entire field of soft X-ray microscopies (e.g. soft X-ray photoemission electron microscopy or scanning transmission X-ray microscopy).13,14
In the following, we therefore compare near C K-edge X-ray photoabsorption data for a series of protonated peptides and proteins, with masses ranging from 0.5 to over 12 kDa: leucine enkephalin (555.6 Da), YG10F (899 Da), gramicidin A (1.9 kDa), the PK26-P fragment from collagen (2.3 kDa), melittin from honey bee venom (2.8 kDa), porcine insulin (5.8 kDa), bovine ubiquitin (8.6 kDa) and equine cytochrome c (12.4 kDa).
Experiment
The experiments were performed by interfacing a home built tandem mass spectrometer with the U49/2-PGM1 soft X-ray beamline at Helmholtz-Zentrum Berlin.15 The setup is displayed in Fig. 1. Briefly, protonated peptide or protein cations were produced by means of electrospray ionization. | ||
| Fig. 1 Sketch of the experimental setup. The length of the time of flight (TOF) system between the 3D RF trap and the MCP detector at the end of the TOF tube is not to scale. | ||
Solutions of the 8 samples under study were prepared using mixtures of HPLC grade methanol and water with formic acid added as an ionization agent as listed in Table 1. The mixtures were selected based on protein solubility. All chemicals and target molecules except the synthetic PK26-P (ProteoGenix, France) and YG10F (JPT Peptides, Berlin, Germany) were purchased from Sigma-Aldrich, the Netherlands. The protonation state was chosen to increase with peptide/protein size, to keep the mass over charge ratio close to 1000 with the exception of the smallest peptide under study (leucine enkephalin). Gramicidin A was a mixture of the sequences HCO-L-X-Gly-L-Ala-D-Leu-L-Ala-D-Val-L-Val-D-Val-L-Trp-D-Leu-L-Trp-D-Leu-L-Trp-D-Leu-L-Trp-NHCH2CH2OH, containing L- and D-amino acids with X being either V or L. Experiments were done with the X = V type. PK26-P has the sequence PGGPPGPKGNSGEPGAPGSKGDTGAK.
| Sample | Water (%) | Methanol (%) | Formic acid (%) | Conc. (μM) |
|---|---|---|---|---|
| Leucine enkephalin | 0 | 99 | 1 | 40 |
| YG10F | 0 | 99 | 1 | 40 |
| Gramicidin A | 0 | 99.8 | 0.2 | 40 |
| PK26-P | 50 | 49.99 | 0.01 | 50 |
| Melittin | 0 | 99.995 | 0.005 | 40 |
| Insulin (porcine) | 0 | 97 | 3 | 40 |
| Ubiquitin (bovine) | 39.98 | 60 | 0.024 | 40 |
| Cytochrome c (equine) | 99.94 | 0 | 0.062 | 40 |
The ions entered the first vacuum chamber (p ∼ 5 × 10−1 mbar) through a heated capillary (T ∼ 350 K, inner diameter: 0.76 mm; length: 20 cm). A radiofrequency (RF) ion funnel was used for phase-space compression of the ions into a well-defined beam. The ions were then guided into the next vacuum chamber (p ∼ 5 × 10−4 mbar) housing an RF-octopole ion guide. Here, ions were accumulated by biasing a diaphragm at the end of the ion guide. The ions were extracted from the octopole, by lowering the diaphragm voltage for 50–100 ms. The extracted ions passed an RF quadrupole ion guide, operated in mass selective mode, where the desired m/z ratio was isolated (p ∼ 1 × 10−6 mbar). Eventually, the ions entered a 3 dimensional RF trap (Paul trap) through one of the hyperbolic end-caps. A He buffer gas pulse of 100 ms duration was injected into the trap to facilitate the dissipation of the molecular ions kinetic energy necessary for efficient ion trapping. The diameter of the trapped ion cloud was measured to be about 400 μm for [ubi + 10H]10+ and for all the other molecules under study similar diameters are expected.
The photon beam crossed the RF-trap through two opposite bores in the ring electrode and was focused to about 200 μm in the trap center. The exposure of the trap content to the soft X-ray photons was controlled by means of a mechanical shutter. The photon flux through the trap was monitored using a silicon p–n junction photodiode (SXUV type, Opto Diode, Camarillo, US).
Photoabsorption was investigated around the carbon K-edge. For each peptide/protein, photon energies around 288.5 eV, leading to 1s excitation and around 300 eV, leading predominantly to 1s ionization, were used. Typically the photon fluxes were between 5 × 1012 and 1.5 × 1013 photons per s. To limit the contribution of multiphoton absorption processes to the recorded mass spectra, exposure times were chosen in a way that guaranteed single photon absorption conditions for more than 90% of the ionized peptides/proteins.
The trapped ions and in particular their fragments were then cooled again by application of a second He buffer gas pulse of 100 ms duration, to dissipate the kinetic energy released in photofragmentation processes. Thereafter the ions were extracted into the TOF system (resolution M/ΔM ∼ 300–400) by applying a high voltage pulse to the hyperbolic end-caps of the RF-trap. The ions were detected using a microchannel plate (MCP) detector and the induced signal was recorded by a 1 GHz digitizer.
To account for fluctuations in electrospray ion source performance, from each photoionization mass spectrum a subsequently recorded reference spectrum obtained with a closed shutter was subtracted. The resulting difference spectrum was cleaned from peaks due to ionization of residual gas, by subtracting a mass spectrum recorded with an empty trap.
The limited capacity of the RF trap required a large number of acquisition cycles (typically 500–1000) to obtain a mass spectrum with sufficient statistics.
One of the critical issues for a quantitative analysis of the mass spectra is the strong dependence of the MCP sensitivity on the velocity of the ions and thus on their m/z. Earlier it was established16 that the MCP detection efficiency P can be well approximated by the equation

Results and discussion
Photoabsorption and Auger ionization
Near edge X-ray absorption spectroscopy is based on inner shell excitation or ionization. In the following, we focus on the C K-edge, i.e. excitation of the C 1s electron into an unoccupied bound state or into the continuum. In general, a core level vacancy can lead to radiative or Auger-type de-excitation processes. For light (low Z) atoms such as C, O and N, radiative decay is inefficient and Auger decay dominates. Here, an electron from a higher orbital de-excites to fill the core level vacancy. The excess energy facilitates the simultaneous transfer of a second electron into the continuum. Typically, the net result of C 1s excitation to a bound excited state is single ionization via the subsequent Auger electron emission, while for C 1s ionization subsequent Auger decay leads to double ionization. Depending on the molecular orbitals from which the two electrons originate, the molecule can be left in an excited state. The role of this excitation energy in peptide/protein photofragmentation will be discussed later.As an example, Fig. 2 displays two immonium ion yields for photoabsorption of [YG10F + H]+ as a function of photon energy. The most distinct features of these spectra are peaks at 285.5 eV and 288.5 eV.10 Both features are due to C 1s-π* transitions. The low energy peak at 285.5 eV is due to such transitions in the rings of the aromatic amino acids (phenylalanine (F), tyrosine (Y), and tryptophan (W)).20 Accordingly, the relative intensity of the peak reflects the relative content of aromatic residues. The peak at 288.5 eV is a feature commonly observed in all proteins and peptides and arises from C 1s-π* transitions in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of the amide bonds.
O groups of the amide bonds.
 | ||
| Fig. 2 Partial ion yields for the protonated synthetic peptide [YG10F + H]+ as a function of photon energy around the C K-edge. The data for the two strongest fragment ions (see Fig. 4) are shown. Red symbols: immonium ions from phenylalanine (F), m/z = 91; blue symbols: immonium ions from tyrosine (Y), m/z = 136. The peak at 285.5 eV is due to 1s-π* transitions in the aromatic rings. The main resonance at 288.5 eV is due to 1s-π*CO transitions in the amide groups. The grey bars indicate the two photon energies for which mass spectra are presented. At 300 eV, 1s photoionization is dominating. | ||
The broad feature starting at about 290 eV is driven by C 1s excitation to Rydberg and σ states and to ionization into the continuum. At 300 eV ionization is dominating, which subsequently leads to protein double ionization. We have therefore chosen 288.5 eV and 300 eV as exemplary photon energies for the excitation and ionization regimes to be discussed in the following section. The respective transitions are independent of amino acid composition and predominantly lead to single and double ionization, respectively.
Mass spectra
In the following figures the transition of photoionization and photofragmentation from the small molecule regime to the large molecule regime is studied. All spectra are normalized to the total photoabsorption yield. Because immonium (and related) ions dominate the small m/z region of the spectra, Table 2 shows the relevant ion masses.| Amino acid | 1-letter code | Immonium ion mass (Da) | Related ion masses (Da) (from CID18,19) |
|---|---|---|---|
| Lysine | K | 101 | 70, 84, 112, 129 |
| Glutamine | Q | 101 | 56, 84, 129 |
| Methionine | M | 104 | 61 |
| Histidine | H | 110 | 82, 121, 123, 138, 166 |
| Arginine | R | 129 | 59, 70, 73, 87, 100, 112 |
| Isoleucine | I | 86 | 44, 72 |
| Leucine | L | 86 | 44, 72 |
| Tryptophane | W | 159 | 77, 117, 130, 132, 170, 171 |
| Tyrosine | Y | 136 | 91, 107 |
| Phenylalanine | F | 120 | 91 |
Leucine enkephalin (M = 0.56 kDa, 5 residues) is the smallest peptide we studied. Photoionization of [leucine-enkephalin + H]+ leads to mass spectra dominated by small fragments with m/z = 80–140.
Immonium ions and related species are the main contributors, in particular tyrosine (Y) at m/z = 91, 107, and 136 and phenylalanine (F) at m/z = 91 and 120 (see Fig. 3). A number of weaker fragments due to backbone scission (b2+: m/z = 221, b3,y3: m/z = 278) and N-terminal fragments associated with tyrosine sidechain loss ((a2-107)+: m/z = 86, (b2-107)+: m/z = 114, (c2-107)+: m/z = 131, (b3-107)+: m/z = 171) can be identified as well. Non-dissociative ionization, i.e. formation of [leucine enkephalin + H]2+, is not observed. Very similar spectra are observed below (hν = 288.5 eV) and above (hν = 300 eV) the C K-edge. The latter spectrum exhibits slightly reduced peak intensities for m/z > 110 and slightly increased intensities for m/z < 110. A detailed discussion of soft X-ray induced photofragmentation of [leucine-enkephalin + H]+ has been published previously.10 For VUV photon energies (8–40 eV), previous studies6 found higher relative yields of fragments due to backbone scission and lower intensities in the low m/z range, but qualitatively similar overall mass spectra were found.
 | ||
| Fig. 3 C 1s photofragmentation mass spectra of [leucine enkephalin + H]+ at photon energies of 288.5 eV and 300 eV. | ||
YG10F (M = 0.9 kDa, 12 residues) is a synthetic peptide, previously investigated in the VUV regime.21 Despite its substantially higher mass, photofragmentation of [YG10F + H]+ is qualitatively similar to that of [leucine-enkephalin + H]+. The mass spectra remain dominated by fragments with m/z < 170 accompanied by larger fragments extending up to m/z ∼ 250 albeit with much lower yields (see Fig. 4). The main features are once again Y and F immonium and related ions, but small fragments due to backbone scission ((y1-18)+: m/z = 148, y1+: m/z = 166) are relatively more intense, as compared to the case of [leucine-enkephalin + H]+. Larger fragments due to backbone scission remain absent.
 | ||
| Fig. 4 C 1s photofragmentation mass spectra of [YG10F + H]+ at photon energies of 288.5 eV and 300 eV. | ||
Gramicidin A is an antibiotic compound (M = 1.88 kDa, 15 residues) commonly investigated in mass spectrometric studies. The soft X-ray photofragmentation spectra for doubly protonated [gramicidin A + 2H]2+ exhibit the strongest peaks in the low-mass region (see Fig. 5), which are due to immonium ions related to tryptophan (W) (m/z = 117, 130, 159, 170 and 171) and leucine (L) (m/z = 86). The high abundance of immonium ions related to tryptophan reflects the high tryptophan content of the molecule (4 out of 15 residues) and the low local ionization potential of the tryptophan sidechain. Sequence ions due to backbone scission become significant. The most intense sequence ions have N-terminal character (a1+: m/z = 100, b3+: m/z = 185, a3+: m/z = 228, b3+,a62+: m/z = 256, b4+: m/z = 369, b5+: m/z = 441, b6+: m/z = 540) but also C-terminal fragments are observed (z1+: m/z = 231). Below the C K-edge (hν = 288.3 eV), traces of non-dissociative ionization can be seen ([gramicidin A + 2H]3+, m/z = 628), whereas for core ionization (hν = 300 eV) double and single non-dissociative ionization channels are absent. Besides this, the step from 1s excitation to ionization has only very weak effects on photofragmentation.
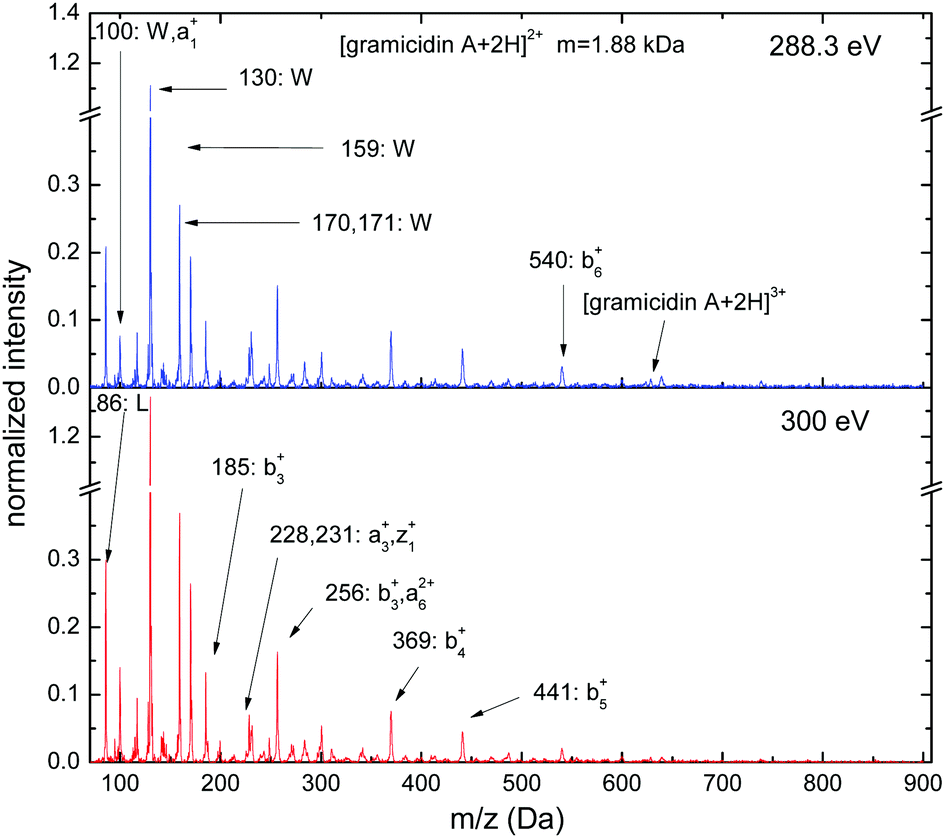 | ||
| Fig. 5 C 1s photofragmentation mass spectra of [gramicidin A + 2H]2+ at photon energies of 288.3 eV and 300 eV. | ||
PK26-P is a collagen fragment rich in proline. The non-dissociative ionization yield at 288.3 eV is surprisingly high for such a small molecule and the peak is accompanied by an equally intense peak due to loss of a m = 44 fragment, probably CO2 (see Fig. 6). Immonium ions are represented only by lysine (m/z = 129). The lower diversity of peaks related to immonium fragments might in part be due to the fact that some of the amino acids constituting PK26-P can produce immonium ions with masses below the Paul trap cutoff (m/z = 80). Backbone scission fragments dominate the spectra: both N-terminal (c1+: m/z = 115, a2+: m/z = 127, b2+: m/z = 155, b3+: m/z = 212, b4+: m/z = 309) and C-terminal (z1+: m/z = 130, x2+-H2O: m/z = 226, z4+: m/z = 360, y4+: m/z = 377) fragments are observed. Above the ionization threshold (hν = 300 eV) the fragmentation intensity is higher and the non-dissociative ionization yields are strongly reduced.
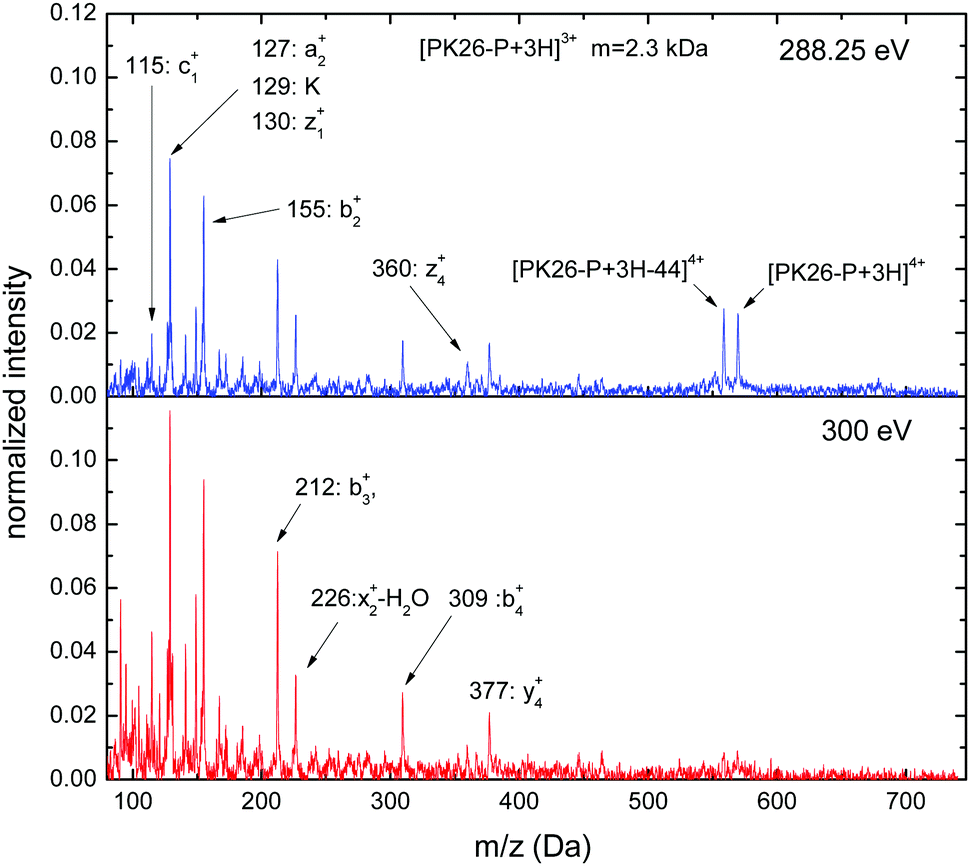 | ||
| Fig. 6 C 1s photofragmentation mass spectra of [PK26-P + 3H]3+ at 288.3 eV and at 300 eV. | ||
Melittin (M = 2.85 kDa, 26 residues), the active component of honey bee venom, was studied triply protonated [melittin + 3H]3+. Near C K-edge photoabsorption (see Fig. 7) still features strongest peaks due to immonium and related ions stemming from lysine (K) (m/z = 84, 101, 112), glutamine (Q) (m/z = 84, 101, 129), isoleucine (I) (m/z = 86), leucine (L) (m/z = 86), arginine (R) (m/z = 112, 129) and tryptophan (W) (m/z = 130). Sequence ions due to backbone scission are of comparable relative intensity as observed for [gramicidin A + 2H]2+ and the most intense peaks also have N-terminal character (z1+: m/z = 129, a2+: m/z = 143, b2+: m/z = 171, b3+: m/z = 228, a4+: m/z = 271, a5+: m/z = 370, a122+: m/z = 541). Sequence ions with C-terminal character are observed as well (x52+: m/z = 370, z4+, y133+: m/z = 541). The most striking aspect of the photofragmentation mass spectrum of [melittin + 3H]3+ at hν = 288.3 eV is the occurrence of the [melittin + 3H]4+ peak due to non-dissociative Auger ionization, accompanied by loss of a m = 43 fragment which most likely is a CONH group from the amidated C-terminal. On increasing the photon energy above the K-edge (hν = 298 eV), the single-ionization peak shows the expected strong decrease. Non-dissociative double photoionization, however, is not observed yet. Instead, additional fragmentation is observed, leading to increased relative intensities of most of the fragments with m/z < 700. It is also notable that in the mass-range of m/z > 300 the peak density is so high that it proves difficult to assign all individual peaks.
 | ||
| Fig. 7 C 1s photofragmentation mass spectra of [melittin + 3H]3+ at photon energies of 288.3 eV and 298 eV. | ||
Insulin (from porcine pancreas, M = 5.78 kDa, 51 residues) is a two-chain polypeptide hormone that for instance regulates carbohydrate metabolism. Chain A consists of 21 residues and chain B consists of 30 residues. Both chains are linked by disulfide bridges between cysteine residues.
For [insulin + 5H]5+ C 1s excitation (hν = 288.3 eV), non-dissociative single ionization into [insulin + 5H]6+ is the strongest channel and also non-dissociative double ionization is clearly observed (see Fig. 8), while the intensity of the CO2 loss peak is low. Unambiguous assignment of the sequence ion peaks with m/z > 300 is difficult. The low m/z range is dominated by immonium and related ions stemming from leucine (L) (m/z = 86), tyrosine (Y) (m/z = 107, 136), histidine (H) (m/z = 110), phenylalanine (F) (m/z = 120), arginine (R) (m/z = 129) and glutamine (Q) (m/z = 129). Sequence ions from both the N-terminals of both chains (A: a2+: m/z = 143, b2+: m/z = 171, b4+: m/z = 399; B: a1+: m/z = 148, c1+: m/z = 165, a2+: m/z = 219, b102+: m/z = 561) and from the C-terminals (A: y1+: m/z = 133, y3+: m/z = 315, y82+: m/z = 416; B: z4+: m/z = 399, z103+: m/z = 400, y4+: m/z = 416, z5+: m/z = 562) can be observed. The number of sequence peaks is much smaller than what is observed for gramicidin A or melittin, which might be due to the tertiary gas-phase structure of insulin.
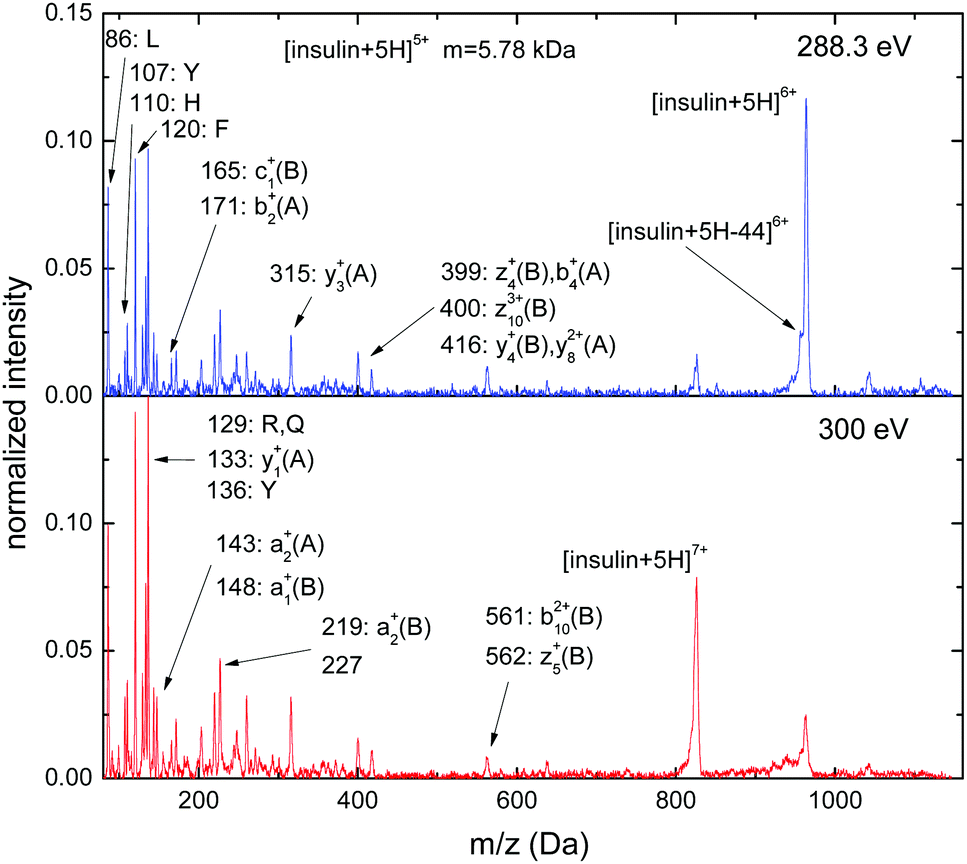 | ||
| Fig. 8 C 1s photofragmentation mass spectra of [insulin + 5H]5+ at photon energies of 288.3 eV and 300 eV. | ||
Upon increase of the photon energy to hν = 300 eV, non-dissociative single ionization strongly decreases as expected, whereas the corresponding double ionization channel strongly increases. Furthermore, the yields of immonium ions and sequence ions with m/z < 300 increase.
Ubiquitin (M = 8.6 kDa, 76 residues) is a regulatory protein, functioning for instance as a signal in protein degradation.
Near C K-edge photoabsorption of [ubiquitin + 9H]9+ (see Fig. 9) gives rise to a mass spectrum that is totally different from what is observed for smaller systems. For hν = 289 eV non-dissociative single ionization leading to [ubiquitin + 9H]10+ is by far the strongest feature in the spectrum. As for many previous spectra the non-dissociative ionization peak is accompanied by a CO2 loss peak. The pedestal of the peak is most likely due to various small-neutral loss channels.
 | ||
| Fig. 9 C 1s photofragmentation mass spectra for [ubiquitin + 9H]9+ at photon energies of 289 eV and 298 eV. | ||
Fragmentation is almost negligible and leads mainly to formation of immonium and related ions stemming from lysine (L) (m/z = 101, 112, 129), methionine (M) (m/z = 104), histidine (H) (m/z = 110), arginine (R) (m/z = 112, 129) and glutamine (Q) (m/z = 101, 129). Sequence ions due to backbone scissions are barely observed with the exception of m/z = 467 and m/z = 479, which may be assigned to a82+ and a133+.
Increasing the photon energy to the core ionization regime (hν = 298 eV) shows the expected transition to non-dissociative double ionization into [ubiquitin + 9H]11+. However, the intensity of the [ubiquitin + 9H]10+ peak remains high and amounts to more than 50% of the [ubiquitin + 9H]11+ yield. Fragment yields remain minute but are slightly higher as compared to the case of hν = 289 eV.
It is remarkable, that the dramatic change in molecular response upon photoionization occurs upon a moderate increase in size from insulin (M = 5.78 kDa) to ubiquitin (M = 8.6 kDa).
The largest protein under study, is the cytochrome complex (cytochrome c) (from equine heart, M = 12.4 kDa, 104 residues). In this protein, the polypeptide chain is linked to an iron coordinating heme group.
Near C K-edge photoabsorption of [cytochrome c + 16H]16+ (see Fig. 10) leads to a mass spectrum with very similar features as observed already for ubiquitin. For C 1s excitation at h = 288.3 eV, non-dissociative single ionization into [cytochrome c + 16H]17+ is the main feature of the spectrum. The peak is accompanied by the respective double ionization feature [cytochrome c + 16H]18+, both located on a broad pedestal that is most likely due to losses of small neutral fragments. In contrast to the previous spectra, here CO2 loss does not lead to a clearly distinguishable peak, as the mass resolution is insufficient. The pedestal is more pronounced than for ubiquitin. The fragmentation channels are even weaker than for ubiquitin. The only peaks clearly exceeding the background are immonium fragments related to arginine (R), glutamine (Q) and lysine (K). No sequence ions are observed.
 | ||
| Fig. 10 C 1s photofragmentation mass spectra for [cytochrome c + 16H]16+ at photon energies of 288.3 eV and 300 eV. | ||
Electronic excitation and internal temperature
An important direct conclusion from our data is the almost total disappearance of small and medium size fragment ions for the largest proteins (ubiquitin and cytochrome c) – an observation that is in line with a statistical fragmentation scenario. In this scenario, the energy deposited by photoabsorption and subsequent Auger decay is assumed to thermalize by internal vibrational redistribution (IVR), before fragmentation sets in.The Auger process is a two-electron process, where the initial 1s hole induced by soft X-ray absorption is filled by a molecular valence electron, and simultaneously a second valence electron is ejected into the continuum. The distribution of the kinetic energies of the Auger electrons (and thus of molecular excitation energies) depends on the valence electronic structure and on the C 1s binding energy. Both can be different for different C 1s ionization sites. However, as all proteins under study are composed from a wide range of amino acids, it is assumed that differences in the soft X-ray absorption induced excitation energy distributions average out. Sanchez-Gonzalez et al.22 have measured the kinetic energy distribution of Auger electrons emitted after C 1s ionization of gas phase glycine, the simplest amino acid, using 560 eV photons (Fig. 11).
 | ||
| Fig. 11 Auger-electron spectrum for core ionization of gas-phase glycine molecules using hν = 560 eV photons.22 The top axis gives the excitation energy, as determined from the cutoff energy (here estimated as 269 eV). The inset sketches Auger electron emission involving the two most loosely bound electrons. | ||
Highest Auger electron kinetic energies correspond to de-excitation and emission of the two most loosely bound valence electrons (the inset in Fig. 11 sketches this scenario). The resulting molecule is a ground state dication. In all cases, where lower lying valence electrons are involved in the Auger process, the glycine dication is left in a singly or multiply excited state.
The Auger electron spectrum in Fig. 11 exhibits two clear peaks at excitation energies of 6 eV and 18.5 eV, respectively. The average excitation energy (∼21 eV) is indicated with a dashed line.
In order to relate the yields for fragmentation and non-dissociative ionization to excitation energy and peptide/protein size, it is required to determine internal temperatures of the molecular ions. Assuming a complete conversion of electronic excitation into vibrational excitation by IVR, the absolute internal temperature T depends on the excitation energy Eexc as follows23

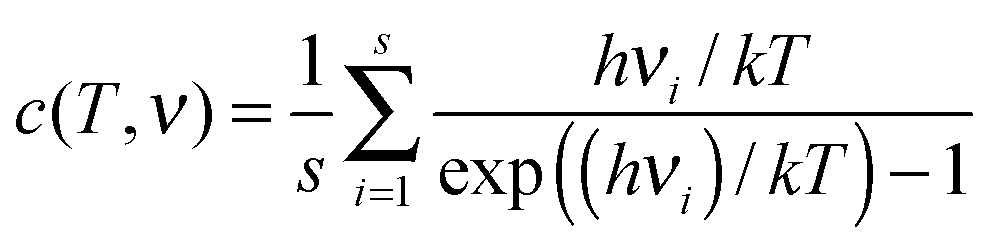
The exact choice of the array of frequencies νi depends on the “frequency model” used. For molecules of the same class, such as peptides, the various existing frequency models are similar,25–27 and accordingly the function c(T,ν) is fairly model independent. Therefore this function can be considered as only temperature-dependent. The T-dependency of this function as published by Drahos and Vekey24 is used by us to relate the energy per degree of freedom and internal temperature.
The number of vibrational degrees of freedom is 3N−6, where N is the number of atoms in the molecule, which ranges from 228 for leucine-enkephalin to 5268 for cytochrome c (see Table 3). Typically, protein ions get collisionally cooled to ambient temperature by a buffer gas in less than 10 ms.28 As we apply buffer gas pulses of about 100 ms duration, the protein internal temperature prior to photoabsorption can be assumed to be 300 K. From this temperature, the initial thermal excitation energy E0exc can be determined. Values between 0.92 eV (leucine enkephalin) and 21.28 eV (cytochrome c) are obtained (see Table 3). The protein internal energy after photoabsorption can then be determined by adding to E0exc the photoexcitation energy estimated from the Auger-electron spectrum. We will discuss our data assuming 18.5 eV as an excitation energy, as it corresponds to one of the maxima in the excitation energy spectrum and as it is close to the average excitation energy (21 eV). The final internal temperatures vary from 1678 (leucine enkephalin) to 413 K (cytochrome c).
| Protein/peptide | DOF | E 0exc (eV) | T 1 (K) | T 2 (K) |
|---|---|---|---|---|
| Leucine enkephalin | 228 | 0.92 | 878 | 1678 |
| YG10F | 339 | 1.37 | 726 | 1312 |
| Gramicidin A (V) | 828 | 3.34 | 510 | 810 |
| PK26-P | 939 | 3.79 | 489 | 763 |
| Melittin | 1293 | 5.22 | 444 | 662 |
| Insulin | 2361 | 9.54 | 384 | 523 |
| Ubiquitin | 3714 | 15.00 | 356 | 454 |
| Cytochrome c | 5268 | 21.28 | 340 | 413 |
Dissociation timescales are the last issue to be discussed, here. For conventional heating, e.g. in a heated capillary flow reactor, for the example of [leucine-enkephalin + H]+ thermal dissociation rates k = 81 s−1 for T = 608 K and k = 730 s−1 for T = 653 K were obtained. Characteristic dissociation times thus were of the order of few ms.29 In our experiment, trapping periods are typically two orders of magnitude longer. On such timescales and particularly at low T, thermal dissociation possibly competes with radiative cooling by infrared photon emission. For leucine enkephalin, typical values for the radiative cooling constant are 7.5 ± 0.5 s−1 at T = 298 K30 and 30 s−1 at T = 438 K.28 This implies, that for relatively low internal temperatures below 600 K, where dissociation times exceed few ms, infrared cooling likely is the dominating process and the experimentally observed fragmentation pattern still is due to dissociation processes occurring in the very first ms after photoabsorption.
Partial ion yields
It is now possible to directly relate yields of various ionization and fragmentation channels to the temperature of the peptide/protein cation after photoabsorption.Fig. 12 displays the relative fragmentation yield (YF), defined as the ratio of the integral intensity of all fragment peaks and the total loss integral, determined from the loss of the parent peak. For all but the two heaviest molecules YF exceeds 1, implying that more than one charged fragments are produced per molecule. For the large systems (ubiquitin and cytochrome c), the yield of small fragments is much lower than 1.
 | ||
| Fig. 12 Relative fragmentation yields as a function of internal temperature under assumption that soft X ray photoabsorption followed by Auger decay deposits 18.5 eV of external energy into the molecules. The data points are labelled with the first letter of the corresponding molecules. | ||
The fragmentation yield reaches a maximum for gramicidin A and subsequently drops for YG10F and leucine-enkephalin. One of the reasons for this drop is the limited number of charges initially present on the YG10F and leucine-enkephalin molecule cations after photoabsorption and subsequent Auger decay. YG10F and leucine-enkephalin are initially both singly protonated. Their charge state is raised to 2+ in the excitation-Auger ionization regime (288.5 eV) and 3+ in the ionization-Auger ionization regime (300 eV). Therefore for 288.5 eV photons, the maximum number of charged fragments is limited to 2 for [YG10F + H]+ and [leucine-enkephalin + H]+ (see Fig. 12). This implies that for the lighter molecules the charged-fragment yields are not solely determined by the internal energies but that they are capped by the available number of charges on the molecules after photoabsorption and subsequent Auger decay.
We can use this idea by defining the maximum number of charged fragments nmax per cation as the number of positive charges on the intermediate protein ion directly after photoabsorption and subsequent Auger decay. For photon energies below the C 1s ionization threshold (photoexcitation), nmax equals the initial protonation state +1, while above the threshold (photoionization) +2 has to be added. For instance, for [melittin + 3H]3+nmax = 4 for 288.3 eV photons and nmax = 5 for 300 eV photons. The relative fragmentation yields divided by nmax (YcorrF) are displayed in Fig. 13 as a function of internal temperature.
 | ||
| Fig. 13 Charge-state corrected fragmentation yields as a function of internal temperature under assumption that soft X ray photoabsorption followed by Auger decay deposits 18.5 eV of external energy into the molecules. The data points are labelled with the first letter of the corresponding molecules. | ||
From Fig. 13 it is seen that fragmentation increases rapidly as a function of internal temperature, i.e., going to smaller molecular sizes, until saturation is reached for gramicidin A and lighter systems. At this point we want to point out that the data for PK26-P appear to drop below the generic trend line. This will be further discussed in the framework of the non-dissociative channels, which are complementary to the fragmentation channels.
For 300 eV photon energy the fragmentation yields show a drop in normalized fragmentation for YG10F and leucine-enkephalin. Most likely, this is an artefact due to the low mass cutoff of the ion trap: photofragmentation patterns for these two molecules show strongest peaks close to the low mass cutoff of the trap already at 288.5 eV (see Fig. 3 and 4) and the increase to 300 eV clearly shifts a sizeable fraction of fragment ions below the trap cutoff.
It is clear from both figures, that fragmentation becomes weak for the two largest systems (ubiquitin and cytochrome c). However, it should be noted that the peaks forming the pedestal near the non-dissociative peaks (cf.Fig. 9 and 10) are not included in the fragmentation yields.
From Fig. 12 and 13 an estimate can be made of the internal temperature threshold region where significant fragmentation sets in. The onset temperature for fragmentation can be estimated to be about 500 K. This temperature corresponds to a molecule with about 2700 DOF (∼900 atoms) (see also Table 3). This would correspond to a threshold protein mass of about 6500 Da. Above this threshold, non-dissociative ionization clearly dominates the mass spectra.
It should be taken into account that in comparison to mass-spectra obtained after heating in a capillary flow reactor, by collision induced dissociation (CID) or by surface induced dissociation (SID) clear differences are obvious: in all three conventional cases, highest yields are observed for sequence ions due to backbone scission, whereas immonium ions are low.29,31
Why does extensive fragmentation set in at much lower T for the case of soft X-ray absorption and why are fragmentation dynamics so different? There are two aspects to be discussed:
(i) soft X-ray absorption involves ionization, i.e. besides the excitation, the peptide/protein charge state increases by 1 or 2. In particular for a small peptide such as leucine enkephalin, this is expected to dramatically lower stability against fragmentation. However, the charge state is not likely to play a role for the larger systems under study and these are the systems, where the onset of fragmentation is observed.
(ii) The initial energy deposition mechanism is different: soft X-ray absorption initially leads to a localized electronic excitation, rather than an unspecific heating of the entire molecule. The influence of the localization of an excitation on the fragmentation can be seen in soft X-ray absorption in leucine-enkephaline10 where fragmentation spectra depend on whether photon energy corresponds to unspecific 1s-π*CO transition or to 1s-π* transitions in the aromatic rings. In such a case, it is clear that IVR has to compete with fast (and local) fragmentation processes. This could be tested in the future by investigation of soft X-ray photoabsorption of much larger proteins (M > 30 kDa) for which fragmentation due to the resulting temperature increase can be ruled out. In our recent study on fs EUV multiphoton ionization of [ubiquitin + 10H]10+ we already found strong indications for such localization effects.12
The quantity complementary to the fragmentation yield, is the relative yield for non-dissociative ionization to which we have added the yield for the respective CO2 loss channel. YNDl is defined as ratio of these peak integrals and the total photoabsorption yield. YNDI characterizes channels which do not result in extensive protein fragmentation. This yield is displayed in Fig. 14. For 288.5 eV, the data is for single ionization and for 300 eV, the data is for double ionization. In both cases, the yields decrease dramatically with E/DOF, with a steeper slope for double ionization. The data from Fig. 14 is plotted on a log scale as a function of excitation energy per degree of freedom in Fig. 15. There is a clear exponential dependence for 288.3 eV and 300 eV with the exception of the data points for PK26-P, for which extraordinarily high non-dissociative yields are observed. This is in line with the lower fragmentation yields for this peptide. It should be noted that the significant drop in the fragmentation yield for PK26-P might be also partly affected by the fact that the many immonium fragments for PK26-P have masses below the Paul Trap cutoff. However, judging from the fragmentation pattern in Fig. 6 formation of very small m/z fragments is unlikely a strong channel. The reason for higher non-dissociative yield and lower fragmentation yield most likely lies in the fact, that PK26-P is a model system for the proline-rich collagen, which is the most common structural protein in the human body (for PK26-P, 6 out of 26 amino acids (23%) are proline). Proline has a much higher conformational rigidity as compared to other amino acids, as the side-chain is cyclic. It seems logic that the enhanced conformational rigidity increases non-dissociative ionization in favor of fragmentation. The other proteins and peptides studied here do not have high proline content (melittin: 4%, insulin: 2% ubiquitin: 4%, cytochrome c: 4%, gramicidin, YG10F and leucine-enkephalin: 0%).
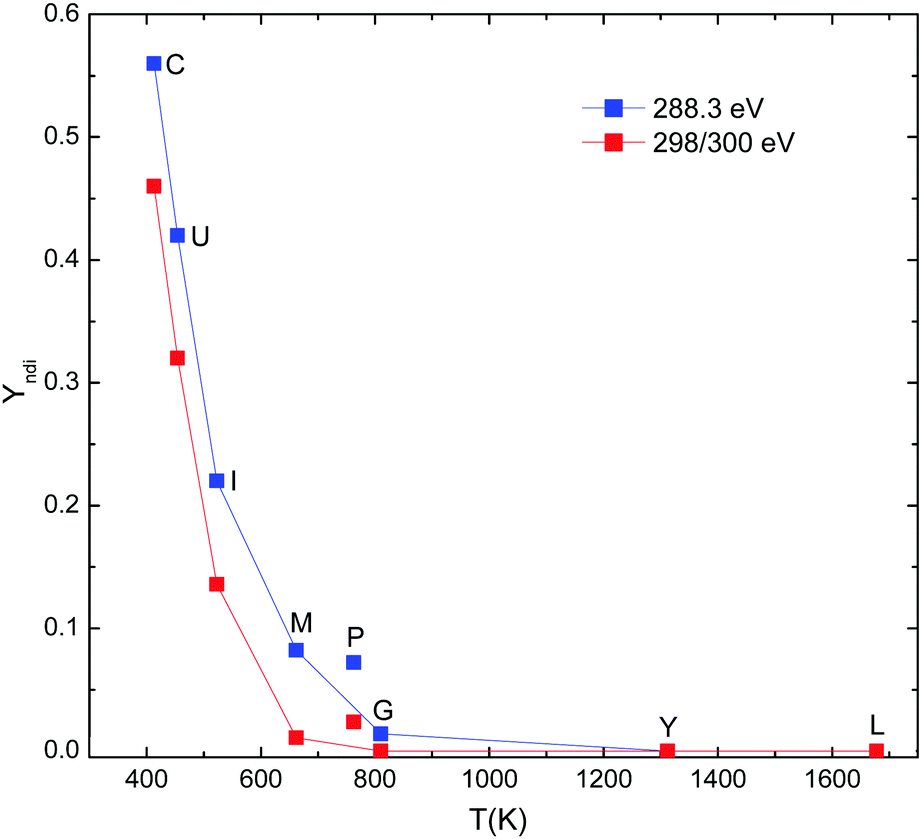 | ||
| Fig. 14 Log-scale plot of the data from figure as a function of excitation energy per DOF. | ||
 | ||
| Fig. 15 Non-dissociative ionization yield as a function of protein/peptide temperature for E = 18.5 eV. | ||
It is also notable that the slope of the 288.3 eV curve in Fig. 15 is smaller than the slope for the 300 eV data. The reason for this difference lies in weakening of the intramolecular bonds by Coulomb repulsion. At 300 K [leucine enkephalin + H]+ is barely able to stabilize a second charge. For [gramicidin A + 2H]2+, a stable [gramicidin A + 2H]3+ ion is observed but [gramicidin A + 2H]4+ is not observed.
Conclusions
We have presented results of near-edge X-ray absorption mass-spectrometry of peptides and proteins of various masses for photon energies corresponding to the 1s-π*CO transition in the amide bonds and for a photon energy above the ionization threshold. We have observed the transition from the small-molecule mode where non-dissociative ionization is absent and predominantly small fragments are formed, to the large molecule regime, dominated by non-dissociative ionization accompanied by a low yield of small fragments. In the framework of a simplified harmonic oscillator model, the photoexcitation energy is transformed into a peptide/protein internal temperature. Fragmentation was observed to increase strongly with internal temperature, with the onset being at lower temperature than what is commonly observed with conventional vibrational excitation of the system (CID, SID, direct heating). Fragmentation patterns obtained upon soft X-ray absorption are markedly different from what is observed with conventional methods. Most likely, the reason for the differences lies in the competition of IVR and fast local fragmentation, once the Auger process is completed.
The obtained relations between fragmentation and non-dissociative ionization yields and protein temperature are in agreement with earlier research of protein laser desorption and thermal desorption, though the qualitive picture of thermal desorption spectra differs from soft X-ray absorption spectra mainly by higher abundance of backbone scission fragments and lower abundance of immonium ions.
Acknowledgements
The authors would like to acknowledge the contribution of the COST Action CM1204 “XUV/X-ray light and fast ions for ultrafast chemistry” (XLIC). We thank HZB for the allocation of synchrotron radiation beamtime. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 312284. We thank the LCLS LF62 collaboration and in particular Alvaro Sanchez-Gonzalez, Thomas Barillot and Přemysl Kolorenč, for providing the experimental Auger electron spectrum for gas-phase glycine.Notes and references
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548, DOI:10.1002/mas.20024.
- J. S. Brodbelt, Chem. Soc. Rev., 2014, 43, 2757–2783, 10.1039/c3cs60444f.
- J. P. Reilly, Mass Spectrom. Rev., 2009, 28, 425–447, DOI:10.1002/mas.20214.
- M. A. Halim, M. Girod, L. MacAleese, J. Lemoine, R. Antoine and P. Dugourd, J. Am. Soc. Mass Spectrom., 2016, 1–8, DOI:10.1007/s13361-016-1419-8.
- A. R. Milosavljevic, C. Nicolas, J. Lemaire, C. Dehon, R. Thissen, J. Bizau, M. Refregiers, L. Nahon and A. Giuliani, Phys. Chem. Chem. Phys., 2011, 13, 15432–15436, 10.1039/C1CP21211G.
- S. Bari, O. Gonzalez-Magaña, G. Reitsma, R. Hoekstra, J. Werner, S. Schippers and T. Schlathölter, J. Chem. Phys., 2011, 134, 024314, DOI:10.1063/1.3515301.
- P. Salen, M. Kaminska, R. J. Squibb, R. Richter, M. Alagia, S. Stranges, P. van der Meulen, J. H. D. Eland, R. Feifel and V. Zhaunerchyk, Phys. Chem. Chem. Phys., 2014, 16, 15231–15240, 10.1039/c4cp01067a.
- Y. Lin, C. Tsai, H. Lin, T. Hsieh, J. Chen, W. Hu, C. Ni and C. Liu, J. Phys. Chem. A, 2015, 119, 6195–6202, DOI:10.1021/acs.jpca.5b04415.
- A. R. Milosavljević, F. Canon, C. Nicolas, C. M. L. Nahon and a. A. Giuliani, J. Phys. Chem. Lett., 2012, 3, 1191–1196, DOI:10.1021/jz300324z.
- O. Gonzalez-Magaña, G. Reitsma, M. Tiemens, L. Boschman, R. Hoekstra and T. Schlathölter, J. Phys. Chem. A, 2012, 116, 10745, DOI:10.1021/jp307527b.
- A. R. Milosavljevic, C. Nicolas, M. L. J. Rankovic, F. Canon, C. Miron and A. Giuliani, J. Phys. Chem. Lett., 2015, 6, 3132–3138, DOI:10.1021/acs.jpclett.5b01288.
- T. Schlathölter, G. Reitsma, D. Egorov, O. Gonzalez–Magaña, S. Bari, L. Boschman, E. Bodewits, K. Schnorr, G. Schmid, C. D. Schröter, R. Moshammer and R. Hoekstra, Angew. Chem., Int. Ed., 2016, 55, 10741–10745, DOI:10.1002/anie.201605335.
- J. Wang, C. Morin, L. Li, A. P. Hitchcock, A. Scholl and A. Doran, J. Electron Spectrosc. Relat. Phenom., 2009, 170, 25–36, DOI:10.1016/j.elspec.2008.01.002.
- A. Gianoncelli, L. Vaccari, G. Kourousias, D. Cassese, D. E. Bedolla, S. Kenig, P. Storici, M. Lazzarino and M. Kiskinova, Sci. Rep., 2015, 5, 10250, DOI:10.1038/srep10250.
- Helmholtz-Zentrum Berlin für Materialien und Energie, Journal of Large-Scale Research Facilities, 2016, 2, A72, DOI:10.17815/jlsrf-2-75.
- T. Schlatholter, R. Hoekstra and R. Morgenstern, J. Phys. B: At., Mol. Opt. Phys., 1998, 31, 1321–1331, DOI:10.1088/0953-4075/31/6/017.
- H. Winter, M. Vana, G. Betz, F. Aumayr, H. Drexel, P. Scheier and T. D. Märk, Phys. Rev. A: At., Mol., Opt. Phys., 1997, 56, 3007, DOI:10.1103/PhysRevA.56.3007.
- A. M. Falick, W. M. Hines, K. F. Medzihradzky, M. A. Baldwin and B. W. Gibson, J. Am. Soc. Mass Spectrom., 1993, 4, 882–893, DOI:10.1016/1044-0305(93)87006-X.
- I. A. Papayannopoulos, Mass Spectrom. Rev., 1995, 14, 49–73, DOI:10.1002/mas.1280140104.
- W. Zhang, V. Carravetta, O. Plekan, V. Feyer, R. Richter, M. Coreno and K. C. Prince, J. Chem. Phys., 2009, 131, 035103, DOI:10.1063/1.3168393.
- O. Gonzalez-Magaña, G. Reitsma, S. Bari, R. Hoekstra and T. Schlathölter, Phys. Chem. Chem. Phys., 2012, 14, 4351–4354, 10.1039/C2CP23470J.
- A. Sanchez-Gonzalez, T. R. Barillot, R. J. Squibb, P. Kolorenc, M. Agaker, V. Averbukh, M. J. Bearpark, C. Bostedt, J. D. Bozek, S. Bruce, S. C. Montero, R. N. Coffee, B. Cooper, J. P. Cryan, M. Dong, J. H. D. Eland, L. Fang, H. Fukuzawa, M. Guehr, M. Ilchen, A. S. Johnsson, C. Liekhus-S, A. Marinelli, T. Maxwell, K. Motomura, M. Mucke, A. Natan, T. Osipov, C. Ostlin, M. Pernpointner, V. S. Petrovic, M. A. Robb, C. Sathe, E. R. Simpson, J. G. Underwood, M. Vacher, D. J. Walke, T. J. A. Wolf, V. Zhaunerchyk, J. Rubensson, N. Berrah, P. H. Bucksbaum, K. Ueda, R. Feifel, L. J. Frasinski and J. P. Marangos, J. Phys. B: At., Mol. Opt. Phys., 2015, 48, 234004, DOI:10.1088/0953-4075/48/23/234004.
- K. Vekey, J. Mass Spectrom., 1996, 31, 445–463, DOI:10.1002/(SICI)1096-9888(199605)31:5<445::AID-JMS354>3.0.CO;2-G.
- L. Drahos and K. Vekey, J. Am. Soc. Mass Spectrom., 1999, 10, 323–328, DOI:10.1016/S1044-0305(98)00156-1.
- K. Vekey, A. Somogyi and V. Wysocki, Rapid Commun. Mass Spectrom., 1996, 10, 911–918, DOI:10.1002/(SICI)1097-0231(19960610)10:8<911::AID-RCM593>3.0.CO;2-7.
- L. L. Griffin and D. J. Mcadoo, J. Am. Soc. Mass Spectrom., 1993, 4, 11–15, DOI:10.1016/1044-0305(93)85037-X.
- P. Derrick, P. Lloyd and J. Christie, Physical chemistry of ion reactions, 1995 Search PubMed.
- D. M. Black, A. H. Payne and G. L. Glish, J. Am. Soc. Mass Spectrom., 2006, 17, 932–938, DOI:10.1016/j.jasms.2006.01.001.
- M. Meotner, A. R. Dongre, A. Somogyi and V. H. Wysocki, Rapid Commun. Mass Spectrom., 1995, 9, 829–836, DOI:10.1002/rcm.1290090922.
- C. Peltz, L. Drahos and K. Vekey, J. Am. Soc. Mass Spectrom., 2007, 18, 2119–2126, DOI:10.1016/j.jasms.2007.09.011.
- J. Laskin and J. Futrell, Mass Spectrom. Rev., 2005, 24, 135–167, DOI:10.1002/mas.20012.
| This journal is © the Owner Societies 2016 |