
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of solvent polarity on the structure of drop-cast electroactive tetra(aniline)-surfactant thin films†
Thomas G.
Dane
a,
Julia E.
Bartenstein
a,
Beatrice
Sironi
a,
Benjamin M.
Mills
a,
O.
Alexander Bell
a,
J.
Emyr Macdonald
b,
Thomas
Arnold
c,
Charl F. J.
Faul
*a and
Wuge H.
Briscoe
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: charl.faul@bris.ac.uk; wuge.briscoe@bris.ac.uk
bSchool of Physics and Astronomy, Cardiff University, Cardiff CF24 3AA, UK
cDiamond Light Source Ltd., Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, UK
First published on 12th August 2016
Abstract
The influence of processing conditions on the thin film microstructure is a fundamental question that must be understood to improve the performance of solution-processed organic electronic materials. Using grazing-incidence X-ray diffraction, we have studied the structure of thin films of a tetra(aniline)-surfactant complex prepared by drop-casting from five solvents (hexane, chloroform, tetrahydrofuran, dichloromethane and ethanol), selected to cover a range of polarities. We found that the structure, level of order and degree of orientation relative to the substrate were extremely sensitive to the solvent used. We have attempted to correlate such solvent sensitivity with a variety of solvent physical parameters. Of particular significance is the observation of a sharp structural transition in the thin films cast from more polar solvents; such films presented significantly greater crystallinity as measured by the coherence length and paracrystalline disorder parameter. We attribute this higher structural order to enhanced dissociation of the acid surfactant in the more polar solvents, which in turn promotes complex formation. Furthermore, the more polar solvents provide more effective screening of (i) the attractive ionic interaction between oppositely charged molecules, providing greater opportunity for dynamic reorganisation of the supramolecular aggregates into more perfect structures; and (ii) the repulsive interaction between the positively charged blocks permitting a solvophobic-driven aggregation of the aromatic surfaces during solvent evaporation.
Introduction
A key advantage of organic electronic materials over their inorganic counterparts is their high solubility in a broad range of common organic solvents. This attractive property enables high-throughput, low-cost deposition on cheap, flexible substrates using techniques such as spin coating, dip coating, spray coating, roll-to-roll processing, drop casting and inkjet printing.1 The optoelectronic properties of these materials depend strongly on their supramolecular structures as well as the presence of amorphous regions, disorder, polymorphism and structural anisotropy.2–4 Understanding the effect of processing conditions on the resulting film microstructure and physical properties therefore is of both fundamental and practical importance.One important factor in the self-assembly behaviour of such organic molecules is the processing solvent. The cost, toxicity and properties of the solvent are also relevant commercial and environmental considerations. The processing solvent has been shown to influence the microstructure and charge carrier mobility in devices based on the well-studied semiconducting polymer poly(3-hexylthiophene) (P3HT).5–11 Some investigations, for example, suggest that films spin-cast from high boiling point solvents demonstrate a higher degree of structural order and orientation than from lower boiling point solvents.8,9 This observation was ascribed to the longer evaporation time, which permitted the formation of thermodynamically stable structures. It was also found that the P3HT aromatic planes were parallel to the substrate when drop-cast from chloroform, but perpendicular when cast from dichloromethane (DCM); such induced differences would have obvious consequences on the direction of charge transport through the film.10
The bulk-phase supramolecular structure of poly(aniline)—a conducting polymer offering environmental stability and switchable conductivity and optical transitions—is strongly dependent on the processing conditions;12–19 however, there have been very few studies on such dependencies for thin films. The use of oligomers of poly(aniline) offers a means to overcome the polydispersity and structural disorder inherent to such polymers,20 and can induce high levels of supramolecular order via an ionic self-assembly (ISA) route.21,22 Such model compounds therefore provide opportunities to understand physical mechanisms and control subtle factors that govern structure20 and functionality.21
We have previously investigated the thin self-assembly behaviour of aniline oligomers when deposited by drop casting. This process, of relevance to widely used thin film deposition processes, proved to be a facile route to highly ordered thin films. We found that the oligomer thin film self-assembly behaviour depends strongly on the molecular architecture and thermal treatment.25,26
Of particular interest is the TANI(BEHP)0.5 oligomer–surfactant complex, shown in Fig. 1(a), which is formed from phenyl/phenyl end-capped tetra(aniline) (Ph/Ph TANI) and two equivalents of the acid surfactant dopant bis(ethyl hexyl) hydrogen phosphate (BEHP). This system forms a highly ordered bilayer-type structure as represented in Fig. 1(b). In the bulk phase, TANI(BEHP)0.5 forms a rhombohedral 2D unit cell with lattice parameters a = 2.35 nm, b = 2.7 nm and γ = 80°.21 Thin films drop-cast from tetrahydrofuran (THF) adopt a hexagonal lattice (a = b = 2.53 nm, γ = 120°), whereby the bilayers preferentially orient parallel to the substrate.25,26
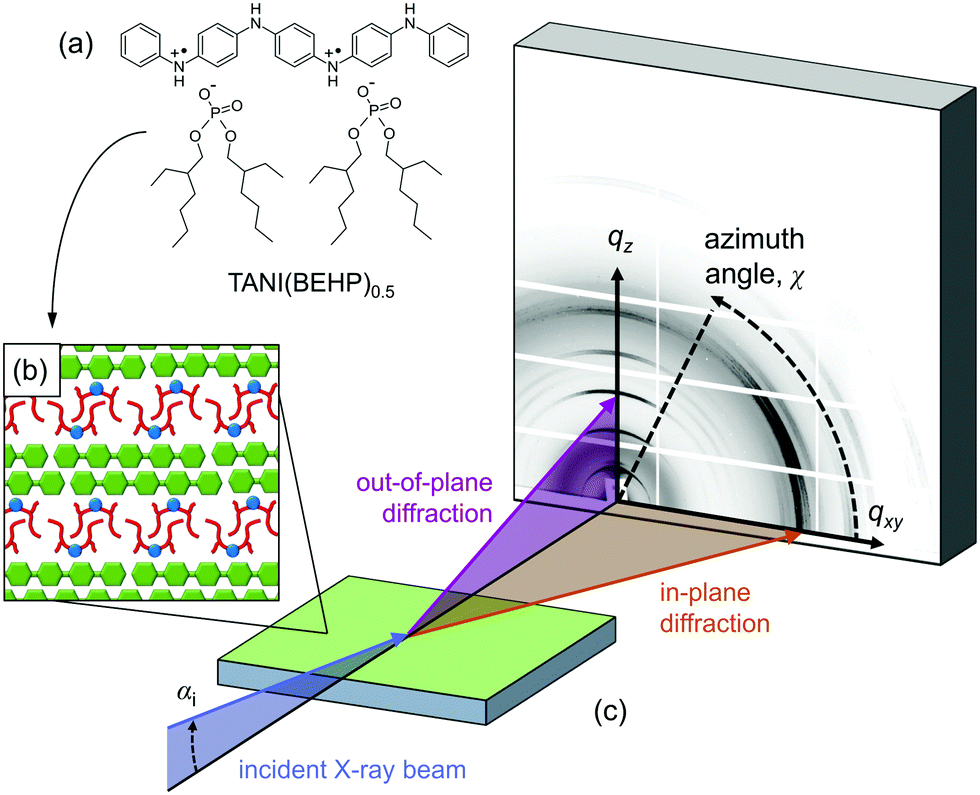 | ||
| Fig. 1 (a) Chemical structure of the TANI(BEHP)0.5 complex, which self-assembles into a bilayer-type arrangement (b) when drop-cast onto silicon, with a typical d-spacing of ∼2.2 nm. These films have been studied using GIXD (c). Synchrotron X-rays are incident on the sample at an angle of αi = 0.36°. The GIXD pattern collected on a 2D detector reveals structural information about the out-of-plane ordering (vertical direction, qz) and in-plane ordering (horizontal direction, qxy). | ||
Grazing-incidence X-ray diffraction (GIXD) is a valuable tool in ascertaining structural information of soft matter thin films.27 In combination with a 2D detector, the structure normal to the surface (out-of-plane, qz) and within the surface plane (in-plane, qxy) can be simultaneously probed (as represented pictorially in Fig. 1(c)). Here we investigate the effect of solvent used for drop-casting on the resulting structure and degree of order in TANI(BEHP)0.5 thin films using GIXD. We determine the level of order within the films by considering the broadening of diffraction peaks and relate these structural characteristics to physical properties of the solvents (such as boiling point, vapour pressure and polarity). We have considered five solvents (including the previously studied THF as a control), and have summarised their relevant physical properties in Table 1. It is expected that the results from this initial study will provide fundamental insight into the role of the solvent, and thus stimulate further investigations into the influence of solvent properties on structure formation, self-assembly and function.
| Solvent | ε 23 | Hansen solubility parameters (Pa1/2)24 | b.p.23 (°C) | v.p.23 (kPa) | d 23 (g mL−1) | |||
|---|---|---|---|---|---|---|---|---|
| δ Total | δ D | δ P | δ H | |||||
| Hexane | 1.89 | 14.9 | 14.9 | 0.0 | 0.0 | 68.7 | 20.2 | 0.661 |
| Chloroform | 4.81 | 19.0 | 17.8 | 3.1 | 5.7 | 61.2 | 26.2 | 1.479 |
| THF | 7.52 | 19.4 | 16.8 | 5.7 | 8.0 | 65.0 | 21.6 | 0.883 |
| DCM | 8.93 | 20.3 | 18.2 | 6.3 | 6.1 | 40.0 | 58.2 | 1.327 |
| Ethanol | 25.3 | 26.5 | 15.8 | 8.8 | 19.4 | 78.3 | 7.87 | 0.789 |
Experimental
Ph/Ph-TANI was synthesised according to a published method.28 Bis(ethyl hexyl)phosphate (BEHP), hexane, chloroform, THF, dichloromethane (DCM) and ethanol were obtained from Sigma Aldrich and used as received. Silicon wafers (Si(100) crystal face with a native SiO2 surface layer) obtained from University Wafers were cut into 10 × 10 mm pieces. The substrates were cleaned by sonication for 5 min each in acetone, ethanol and MilliQ water, and dried under nitrogen flow. Solutions of TANI(BEHP)0.5 (3.3 mg mL−1) were prepared by dissolving emeraldine base TANI (∼6.7 mg) and BEHP (∼9.8 mg) in each of the chosen solvents (5 mL). Given the reduced solubility of TANI in some of the solvents compared with THF (e.g., hexane), the solutions were stirred for one week in sealed vials prior to casting to ensure complete dissolution.Films were produced by drop-casting as follows: a droplet of solution (40 μL) was placed on the substrate and allowed to evaporate for one hour in a closed atmosphere saturated with vapour from the solvent in question (by exposure to an open vial of the solvent (∼5 mL)). The films were then examined by GIXD within 24 hours of film preparation.
GIXD measurements were performed on beamline I07, Diamond Light Source, UK. The X-ray beam energy was E = 10 keV (wavelength λ = 1.24 Å) and the beam size was ∼300 × 300 μm (FWHM) with an approximately Gaussian intensity profile. An incident angle of αi = 0.36° (∼2 × αc, the critical angle) was used, as this allowed for complete illumination of the film with minimal substrate-reflected beam which could complicate analysis (see Section S3 in the ESI† for further discussion). Data were collected on a PILATUS 2M detector (DECTRIS) at a distance of 340.02 mm from the sample, which was calibrated by silicon powder in transmission mode. Samples were enclosed in a helium-filled chamber,29 mounted on a hexapod attached to a 2+3 circle Huber diffractometer.
The GIXD scattering geometry is shown in Fig. 1(c). GIXD data were reduced using a custom-made software package written in IgorPro (Wavemetrics®).26 The diffraction patterns were transformed from the raw images into reciprocal space maps, i.e., scattered intensity as a function of the in-plane (qxy) and out-of-plane (qz) scattering vectors. Diffraction features along the out-of-plane direction reveals information about structuring normal to the surface, whilst in-plane features are related to the ordering within the plane of the film. Out-of-plane line profiles (intensity vs. qz) were extracted by integrating a sector of data at χ = 90° (where χ is the azimuthal angle relative to the surface plane, cf.Fig. 1), with an angular width of Δχ = 25°. In-plane line profiles (intensity vs. qz) were extracted similarly at χ = 5°, with an angular width of Δχ = 5°. The larger angular integration width for the out-of-plane profiles was chosen to account for the splitting of q-space at higher qz values. Azimuthal line profiles (intensity vs. χ) were assessed to determine the degree of crystallite orientation relative to the surface and were extracted at a fixed q ∼ 0.55 Å−1 with a radial width of Δq = 0.05 Å−1. The integration regions used to extract out-of-plane, in-plane and azimuthal line profiles were the same for each film and are shown in Fig. S2 in the ESI.†
Results and discussion
Film morphology
The five films were imaged with atomic force microscopy (AFM) in PeakForce Tapping mode, the results of which are presented in Fig. S1 of the ESI.† The hexane-, chloroform-, and THF-cast films present a homogenous roughness with a height variation on the order of 1 μm. By contrast, crystalline structures are present in the DCM- and ethanol cast films, which is particularly evident in the PeakForce Error images (Fig. S1(i) and (j) in the ESI†). The 2D GIXD patterns (recorded at room temperature and incident angle αi = 0.36°) for TANI(BEHP)0.5 films prepared from different solvents are shown in the left hand column of Fig. 2. The corresponding integrated line-profiles in the out-of-plane direction (normal to the sample surface along qz at qxy ∼ 0 Å−1) and the in-plane direction (parallel to the surface along qxy at qz ∼ 0 Å−1) are shown in the right-hand column of Fig. 2 (purple and orange curves, respectively).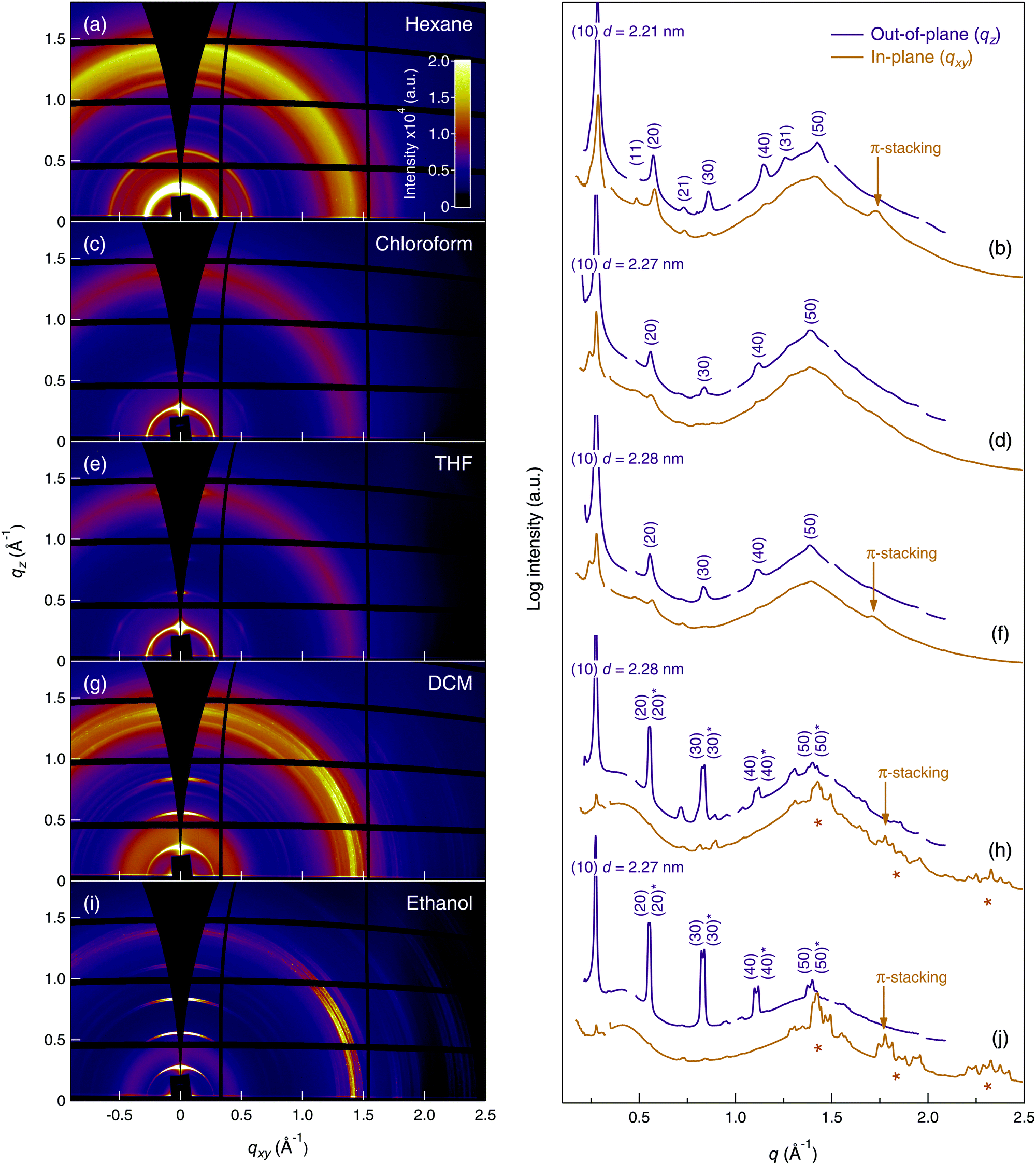 | ||
| Fig. 2 GIXD patterns (left-hand column) and line profiles (right-hand column) for TANI(BEHP)0.5 thin films cast from: hexane, (a) and (b); chloroform, (c) and (d); THF, (e) and (f); DCM, (g) and (h); ethanol (i) and (j). Purple curves in the line profiles are extracted from the out-of-plane direction and orange from the in-plane direction. The out-of-plane line profiles (purple curves) have been offset from the in-plane (orange curves) for clarity. Out-of-plane lamellar reflections are indexed (purple numbers), and those for in-plane π-stacking are indicated with orange arrows. Additional in-plane ordering present for the DCM- and ethanol-cast films are identified with orange * markers. | ||
There are a number of features in these data to be highlighted: firstly, for each of the films examined, five orders of the (h0) family of lamellar reflections were present in the GIXD patterns (and out-of-plane line profiles, which are indexed with purple labels), suggesting the formation of the TANI(BEHP)0.5 lamellar structure with the (10) planes preferentially oriented parallel to the substrate. For all systems, this lattice spacing was 2.27–2.28 nm, which is in good agreement with one of the three polymorphs found in the previous study (2.29 nm),26 except for the hexane-cast film, which presented d(10) = 2.21 nm. There is a diffuse isotropic ring in the GIXD patterns (and present as very broad peaks in the line profiles) at q ∼ 1.4 Å−1, which arises from the disordered alkyl chains of the BEHP dopant. The scattering intensity of this feature relative to that of the lamellar reflections varies between samples. Lastly, an important feature in the diffraction patterns is the reflection arising from the π-stacking interaction of the TANI units. When present, this feature occurs at q ∼ 1.75 Å−1 (lattice spacing d ∼ 0.36 nm) and is most apparent in the in-plane line profiles, which indicates that these planes preferentially orient normal to the surface. This observation is consistent with the (10) preferential orientation relative to the surface. These features in Fig. 2 are labelled with orange arrows on the in-plane line profiles. Fig. 2(e) shows the GIXD data from the THF-cast thin film, which is consistent with our previous studies.26 Despite the presence of common features, there were a number of crucial differences between the data for each of the five solvents used for film casting.
In contrast to the other four films, the GIXD pattern of the hexane-cast film showed isotropic Debye–Scherrer diffraction rings with little azimuthal intensity variation (cf.Fig. 2(a)), indicating that the crystallites were randomly oriented relative to the substrate plane. A reflection at d = 0.36 nm, present only in the in-plane line profile, indicates that some π-stacking was present in the film, with these domains preferentially oriented perpendicular to the substrate. Furthermore, the (11), (21) and (31) reflections, absent from the films cast from other solvents, were observed here for the hexane-cast film.
The chloroform- and THF-cast films presented very similar features in their GIXD patterns (cf.Fig. 2(c) and (e)), and line profiles ((d) and (f)). Both films displayed hexagonal symmetry, evident from the presence of the (02) reflections at 60° to the (20) reflection (cf. azimuthal profile in Fig. 3). The main difference between these two films was the presence of a π-stacking reflection (d = 0.37 nm) for the THF-cast film, which was not present for the chloroform-cast film.
 | ||
| Fig. 3 Azimuthal integration about the (20) reflection for each of the TANI(BEHP)0.5 films. Dotted lines represent Lorentzian fits to the intensity maxima at χ = 90°. | ||
Both the DCM- and ethanol-cast films also displayed the typical lamellar reflections with a relatively high degree of preferential orientation of crystallites parallel to the substrate. The GIXD patterns for both films showed many additional reflections, evident from the peaks in the range 1.25 Å−1 < qxy < 2.5 Å−1 in the in-plane line profiles (highlighted with orange stars in Fig. 2(h) and (j)). These sharp reflections extended to the limit of observable reciprocal space, suggesting highly crystalline structures. This observation is consistent with the AFM images showing well-defined crystallites (Fig. S1(i) and (j) in the ESI†). It has not yet been possible to interpret the detailed structure of this phase, though our investigations are ongoing. No hexagonal symmetry was observed in either the DCM- or ethanol-cast films. For both samples, we observed π-stacking reflections of lattice spacing d = 0.35 nm.
Note that for the DCM- and ethanol-cast films, the reflections were split into two sets of closely overlapping peaks, visible in both the 2D patterns and the line profiles, with expanded views shown in Fig. S3 in the ESI.† We attribute it to two possible explanations.‡ First, polymorphism could be present (two structures with differing lattice parameters, well-known for organic thin films). Alternatively, the large footprint of the X-ray beam caused smearing of the scattering pattern from isolated thicker regions of the film. These two out-of-plane reflection series are referred to below with the notation (I) and (II).
Orientation of the crystallite domains in the film
The degree to which the ordered domains are oriented relative to the surface can be further ascertained by examining the intensity as a function of the azimuthal angle (χ) about a Bragg reflection.27 As a consequence of the grazing-incidence geometry, data at qxy = 0 Å−1 cannot be observed, thus the azimuthal profile cannot produce a complete pole figure to represent the full texture of the film.§ Nevertheless, the GIXD azimuthal profile can be used to compare the relative degree of orientation between different samples. Azimuthal integration was performed about the (20) reflection for each film (as shown in Fig. S2 in the ESI†), the results of which are shown in Fig. 3. Lorentzian profiles were fitted to the intensity maxima (at χ = 90°, normal to the substrate, a constant background was also fitted) to quantify the relative degree of orientation of the lamellae relative to the underlying substrate. The numerical results of the fitting are summarised in Table 2. The hexane-cast sample was almost completely isotropic as evidenced by the lack of a clear intensity maximum in the azimuthal profile. The films cast from chloroform and THF had extremely sharp intensity maxima at χ = 90°; the full-width at half-maximum (FWHM) of their Lorentzian azimuthal profiles was Δχ(20) ∼ 4°, indicating that the crystalline domains in the thin film were preferentially aligned parallel to the substrate. The DCM- and ethanol-cast films also exhibited a high degree of orientation relative to the substrate, although less than those of the chloroform- and THF-cast films, with a broader FWHM Δχ(20) ∼ 19° for both samples.| Solvent | Lamellar spacing d (nm) | Coherence length La (nm) | Disorder parameter g (%) | Azimuthal FWHM Δχ(20) (°) |
|---|---|---|---|---|
| Hexane | 2.21 | 22.4 ± 0.3 | 2.4 ± 0.3 | — |
| Chloroform | 2.27 | 23.4 ± 0.7 | 2.4 ± 0.3 | 4.1 ± 0.3 |
| THF | 2.28 | 23.7 ± 0.3 | 2.6 ± 0.4 | 3.5 ± 0.2 |
| DCM | ||||
| (I) | 2.30 | 69.3 ± 2.6 | 2.0 ± 0.6 | — |
| (II) | 2.26 | 66.5 ± 1.7 | 1.6 ± 0.6 | — |
| Average | 2.28 | 67.9 ± 2.2 | 1.8 ± 0.6 | 18.8 ± 0.4 |
| Ethanol | ||||
| (I) | 2.29 | 66.6 ± 1.1 | 1.7 ± 0.4 | — |
| (II) | 2.25 | 86.6 ± 1.6 | 1.7 ± 0.3 | — |
| Average | 2.27 | 76.6 ± 1.4 | 1.7 ± 0.4 | 19.4 ± 0.2 |
Coherence length and paracrystalline disorder in the thin films
The broadening of a Bragg reflection is related to a number of factors, including (1) the finite domain size, as characterized by the coherence length, La, (2) the paracrystalline disorder parameter, g, which is due to the fluctuations in the lattice spacing about the mean value (d),29–32 and (3) structural defects and disorder on a nanoscopic scale. These parameters (La and g) can be determined by examining the FWHM (Δq) of a series of reflections along a reciprocal lattice vector, in this case the (h00) family. For an ideal crystal, in which the lattice spacing (d) does not fluctuate, Δq(h00) should be constant for all values of h (= 1, 2, 3, etc., the Bragg reflection order). For a paracrystalline sample, Δq(h00) increases with increasing h, and the g parameter is proportional to the slope of Δq2vs. h4. By fitting a line to a plot of Δq(h00)2/(2π)2 as a function of h4, the La and g parameters can be determined. The coherence length is given by La = K/c1/2, where K is the shape factor (K = 0.9) and c is the intercept on the y-axis. The paracrystalline disorder parameter is given by g = (md2)1/4/π, where m is the linear gradient and d is the lattice spacing. A more detailed explanation of this theory, as well as derivations of the error propagation are given in Section S5 in the ESI.†The FWHM Δq and d-spacing values for each of the TANI(BEHP)0.5 samples cast from different solvents were determined from fitting the out-of-plane line profiles (Fig. 2) using the MultiPeak fitting routines in IgorPro (Wavemetrics) (fitted data are shown in Fig. S5 in the ESI†). The paracrystalline disorder plots are shown in Fig. 4, with the data points shown as open symbols and the fits by dashed lines.¶ Note that the data for the second phases (II) of the DCM- and ethanol-cast films are shown as filled symbols and the fits as solid lines. These data are also plotted separately for clarity in Fig. S6 in the ESI.† The full numerical fitting results are summarised in Table 2.
 | ||
| Fig. 4 Paracrystalline disorder plot of FWHM of Bragg reflections as a function of h4 (Miller index h = 2 to 5) for the out-of-plane lamellar reflections in TANI(BEHP)0.5 films, along with linear fits to the data. Plots for both phases of types I and II for the DCM and ethanol films are shown. | ||
The paracrystalline disorder plots reveal a clear difference between the level of order in the films cast from more polar solvents (DCM and ethanol) and those cast from less polar solvents (hexane, chloroform and THF). The fitting for the polar solvents gave a lower intercept on the y-axis and a smaller gradient, indicating both larger domain sizes (∼70 nm vs. ∼23 nm) and less paracrystalline disorder (∼1.7% vs. ∼2.4%) in these films compared with those of the less polar solvents. We now proceed to relate the origin of these structural differences with the physical properties of the solvents.
Relationship between solvent properties and film structure
Although it is well recognized that the thin film self-assembly behaviour is sensitive to the processing solvent, our understanding of the mechanism remains on an empirical level.It is desirable to correlate structural characteristics with certain solvent physical parameters, which would offer clear guidance for solvent choice and processing conditions. Previously, the solvent boiling point, vapour pressure and dielectric constant have been used to explain observed variations in the thin film structure.5–11 Hildebrand and Scott proposed a solubility parameter δ defined as the square root of the cohesive energy density given by33
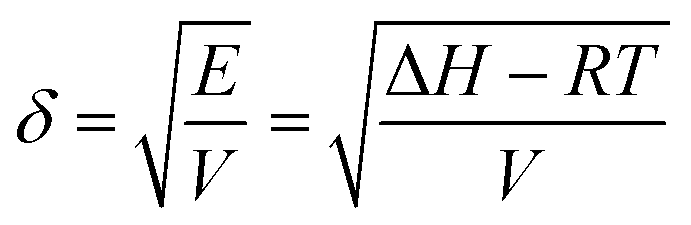 | (1) |
 | (2) |
There appears to be no correlation between the level of structural order and the boiling point of the solvent (cf. Fig. S7(a) in the ESI†). Whilst previous studies on P3HT have reported correlations between the boiling point and thin film structure,8,9 the similarity between the diffraction patterns for the DCM- and ethanol-cast films (boiling points 40 °C and 77 °C, respectively) suggests that, as a parameter reflecting the total intermolecular attractions between the solvent molecules, the solvent boiling point cannot explain the structural differences as a result of TANI(BEHP)0.5 self-assembly in different solvents.
It is well known that the residual patterns from the evaporative drying process of a sessile drop on a surface depend on the evaporation rate.35,36 However, the vapour pressure (which is a measure of the evaporation rate of the solvents) does not show any correlation with the thin film structure (cf. Fig. S7(b) in the ESI†). This is most apparent in the DCM and ethanol cast films, which have significantly different vapour pressures (58.2 and 7.87 kPa, respectively) yet similar thin film structures. The microscopic morphology and topography of the residual patterns depend on the capillary and Marangoni flows which are steered by evaporation and thus depend on the evaporation rate. Indeed, our AFM imaging (Fig. S1 in the ESI†) has revealed different topographies of the films drop cast from different solvents. However, the GIXS results examine the structure on a different length scale (<5 nm) reflecting the intermolecular packing. Thus, the nanoscopic structural order we observe is not affected by the different evaporation rates, whist the residual patterns and topography on a larger length scale are.
The dispersion and hydrogen-bonding HSPs also do not correlate with the film structure (cf. Fig. S9 in the ESI†). However, the polarity of the solvent measured both by the dielectric constant (cf. Fig. S8(a) in the ESI†) and the polar HSP correlate more closely with the resulting film order as shown Fig. 5. Note that the average values of La and g have been plotted for the (I) and (II) phases of the DCM- and ethanol-cast films. The structural order within the film also correlates with the total solubility parameter (cf. Fig. S8(b), though not δ–δP as shown in Fig. S10, ESI†). However, given the dispersion and hydrogen-bonding HSPs do not seem to play a significant role, it can be inferred that the polarity of the solvent is here the dominating factor.
 | ||
| Fig. 5 Coherence length (purple circles) and paracrystalline disorder parameter (orange triangles) of the lamellar planes parallel to the substrate as a function of the polar HSP of the casting solvent for TANI(BEHP)0.5 films. The dashed lines are sigmoidal fits to the data and are meant solely as a guide for the eye. | ||
The plot of coherence length and paracrystalline disorder parameter as a function of the polar HSP shown in Fig. 5 indicates that the ionic self-assembly of TANI(BEHP)0.5 is promoted in a more polar environment and that there exists a critical polarity, above which the resulting level of structural order in the films is significantly greater than below this polarity. This critical polarity occurs at around 6 Pa1/2, and we should note that this value is specific to the chemical system investigated.
A variety of dynamic processes and interactions are present in solutions of TANI and BEHP. These can be summarized as follows: (a) formation of the charged species via dissociation of the acid surfactant (and protonation of the basic TANI molecule): TANI + 2BEHP ⇌ TANI2+ + 2BEHP−; (b) ionic interactions between the charged species: TANI2+ + 2BEHP− ⇌ TANI(BEHP)2; (c) solubility governed by solvent–molecule interactions for the three equilibrium states (uncharged TANI and BEHP, charged TANI2+ and BEHP− and charge-neutralized TANI(BEHP)2); (d) aggregation driven by van der Waals, π-stacking and hydrogen bonding interactions; (e) entropy of aggregation. The relative importance of these processes (in addition to being dependent on the solvent properties) is also dependent on concentration and are thus temporally dynamic during the evaporation process.
Whilst we have only considered five solvents, we hypothesize that the clear correlation between the degree of structural order and solvent polarity (and lack of correlation with any other solvent properties) is due to an increased driving force for self-assembly in more polar solvents, which can be explained as follows. In more polar solvents (higher dielectric constant), the degree of dissociation of the acid surfactant is greater. Whilst the strength of the ionic interaction between the charged species is weaker in more polar solvents (due to greater charge-screening of this interaction), the increased population of the charged species promotes the formation of the TANI(BEHP)0.5 complex. In the more polar solvents, the weaker ionic interactions between the ionic species means that there is a greater opportunity for dynamic reorganisation of the charged species into more thermodynamically stable structures during aggregation in the final stages of evaporation, leading to an increase in the overall structural order.
Furthermore, the charge screening also reduces repulsive interactions between charged nitrogen atoms on the TANI units, which facilitates aggregation through π-stacking. This solvophobic interaction is particularly favourable in more polar solvents so as to minimise contact of the hydrophobic aromatic planes with the polar medium, and may serve to direct further self-assembly. This hypothesis is supported by the observation of both enhanced crystallinity in the π-stacking direction and larger bilayer stacking domains for films cast from the more polar solvents, DCM and ethanol.
Conclusions
The results presented here demonstrate that the thin film self-assembly behaviour of TANI(BEHP)0.5 is extremely sensitive to the solvent used for film casting. In addition to observing variations in the overall morphology of the film, the degree of order within the film was strongly correlated with the polarity of the solvent. It appears that there is a critical solvent polarity above which the oligomer–surfactant complex self-organises into much larger crystalline domains, with less disorder within the bilayers. For example, the domain size along this direction when cast from hexane was only 22 nm, yet this could be increased to more than 76 nm when using ethanol as the solvent. We ascribe this to charge screening by the polar medium, which reduces the strength of ionic interactions and permits reorganisation into larger domains. Furthermore, the solvophobic aromatic interaction drives greater organisation along the self-assembly along the π-stacking direction. We are undertaking experiments to explore this self-assembly in situ to gain a better understanding of the physical mechanism. Furthermore, the degree of orientation could be tuned through solvent selection, with chloroform and THF yielding the most highly oriented films. These results provide the first fundamental insight into the role of the solvent on the thin film self-assembly behaviour and the resulting structure of oligo(aniline) materials. Such insights can be used to optimise fabrication of solution-processed devices, where certain structural motifs are crucial to maximising device performance and functionalities, and stimulate further investigations into the role of solvent on thin film self-assembly.Acknowledgements
This work was supported by the EPSRC (EP/K502996/1 and EP/J500379/1). We gratefully acknowledge Diamond Light Source for access to synchrotron radiation (proposal number SI8702). We thank Dr Robert Harniman and the EMU (School of Chemistry, University of Bristol) for assistance with the AFM imaging. PeakForce atomic force microscopy carried out in the Chemical Imaging Facility, University of Bristol, purchased under EPSRC Grant “Atoms to Applications” Grant ref. EP/K035746/1. MS analysis was performed on instrumentation bought through the Core Capability for Chemistry Research – Strategic Investment in Mass Spectrometry EPSRC grant (EP/K03927X/1). We thank Dr Samuele Lilliu (Department of Physics and Astronomy, University of Sheffield) for helpful discussions. WHB acknowledges funding from the Engineering and Physical Science Research Council (EPSRC), the Royal Society and the European Research Council (ERC), Taiho Kogyo Tribology Research Foundation (TTRF); the European for Cooperation in Science and Technology (CMST COST) Action CM1101, and the Marie Curie Initial Training Network (MC-ITN) NanoS3 (Grant No. 290251).Notes and references
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- A. Salleo, R. J. Kline, D. M. DeLongchamp and M. L. Chabinyc, Adv. Mater., 2010, 22, 3812–3838 CrossRef CAS PubMed.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS.
- K. E. Aasmundtveit, E. J. Samuelsen, M. Guldstein, C. Steinsland, O. Flornes, C. Fagermo, T. M. Seeberg, L. A. A. Pettersson, O. Inganäs, R. Feidenhans'l and S. Ferrer, Macromolecules, 2000, 33, 3120–3127 CrossRef CAS.
- D. W. Breiby and E. J. Samuelsen, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2375–2393 CrossRef CAS.
- J. F. Chang, B. Sun, D. W. Breiby, M. M. Nielsen, T. I. Sölling, M. Giles, I. McCulloch and H. Sirringhaus, Chem. Mater., 2004, 16, 4772–4776 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Fréchet and M. F. Toney, Macromolecules, 2005, 38, 3312–3319 CrossRef CAS.
- H. Yang, S. W. LeFevre, C. Y. Ryu and Z. Bao, Appl. Phys. Lett., 2007, 90, 172116 CrossRef.
- K. Sethuraman, S. Ochiai, K. Kojima and T. Mizutani, Appl. Phys. Lett., 2008, 92, 183302 CrossRef.
- A. MacDiarmid and A. Epstein, Synth. Met., 1994, 65, 103–116 CrossRef CAS.
- J. P. Pouget, Z. Oblakowski, Y. Nogami, P. A. Albouy, M. Laridjani, E. J. Oh, Y. Min, A. G. Macdiarmid, J. Tsukamoto, T. Ishiguro and A. J. Epstein, Synth. Met., 1994, 65, 131–140 CrossRef CAS.
- Y. Xia, A. G. MacDiarmid and A. J. Epstein, Macromolecules, 1994, 27, 7212–7214 CrossRef CAS.
- Y. Cao and P. Smith, Synth. Met., 1995, 69, 191–192 CrossRef CAS.
- Y. Cao, J. Qiu and P. Smith, Synth. Met., 1995, 69, 187–190 CrossRef CAS.
- Y. Xia, J. M. Wiesinger, A. G. MacDiarmid and A. J. Epstein, Chem. Mater., 1995, 7, 443–445 CrossRef CAS.
- O. T. Ikkala, L. O. Pietila, L. Ahjopalo, H. Osterholm and P. J. Passiniemi, J. Chem. Phys., 1995, 103, 9855–9863 CrossRef CAS.
- T. Vikki, L. O. Pietila, H. Osterholm, L. Ahjopalo, A. Takala, A. Toivo, K. Levon, P. Passiniemi and O. Ikkala, Macromolecules, 1996, 29, 2945–2953 CrossRef CAS.
- Z. Wei and C. F. J. Faul, Macromol. Rapid Commun., 2008, 29, 280–292 CrossRef CAS.
- Z. X. Wei, T. Laitinen, B. Smarsly, O. Ikkala and C. F. J. Faul, Angew. Chem., Int. Ed., 2005, 44, 751–756 CrossRef CAS PubMed.
- C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed.
- Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, Florida, USA, 85th edn, 2004 Search PubMed.
- A. F. M. Barton, Handbook of Solubility Parameters and Other Cohesion Parameters, CRC Press, Boca Raton, Florida, USA, 2nd edn, 1991 Search PubMed.
- T. G. Dane, P. T. Cresswell, O. Bikondoa, G. E. Newby, T. Arnold, C. F. J. Faul and W. H. Briscoe, Soft Matter, 2012, 8, 2824–2832 RSC.
- T. G. Dane, P. T. Cresswell, G. A. Pilkington, S. Lilliu, J. E. Macdonald, S. W. Prescott, O. Bikondoa, C. F. J. Faul and W. H. Briscoe, Soft Matter, 2013, 9, 10501–10511 RSC.
- M. L. Chabinyc, Polym. Rev., 2008, 48, 463–492 CrossRef CAS.
- Z. Shao, P. Rannou, S. Sadki, N. Fey, D. M. Lindsay and C. F. J. Faul, Chem. – Eur. J., 2011, 17, 12512–12521 CrossRef CAS PubMed.
- S. Lilliu, T. Agostinelli, E. Pires, M. Hampton, J. Nelson and J. E. Macdonald, Macromolecules, 2011, 44, 2725–2734 CrossRef CAS.
- R. Hosemann and A. M. Hindeleh, J. Macromol. Sci., Part B: Phys., 1995, B34, 327–356 CrossRef CAS.
- T.-M. Wu, J. Blackwell and S. N. Chvalun, Macromolecules, 1995, 28, 7349–7354 CrossRef CAS.
- L. Jimison, A. Salleo, M. Chabinyc, D. Bernstein and M. Toney, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 125319 CrossRef.
- L. H. Sperling, Introduction to Polymer Physical Science, John Wiley & Sons, Hoboken, New Jersey, USA, 4th edn, 2006 Search PubMed.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, Taylor & Francis Group, Boca Ranton, Florida, USA, 2nd edn, 2007 Search PubMed.
- T. P. Bigioni, X.-M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed.
- H. Wu, L. X. Chen, X. Q. Zeng, T. H. Ren and W. H. Briscoe, Soft Matter, 2014, 10, 5243–5248 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05221e |
| ‡ We rule out the possibility of the incoming X-ray beam reflecting from the underlying substrate and acting as a second illumination source (see ESI† for full discussion). |
| § The “missing” data can be obtained by setting the incident angle such that the specular condition coincides with the Bragg reflection,7 which was not possible due to limitations in the experimental configuration. |
| ¶ Note that we plot only the orders h = 2 to 5 because at the first order reflection, the I and II phases for the DCM- and ethanol-cast films are too closely overlapping to separately resolve. |
| This journal is © the Owner Societies 2016 |