
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Planarity and multiple components promote organic photovoltaic efficiency by improving electronic transport†
Matthew B.
Goldey
 ,
Daniel
Reid
,
Juan
de Pablo
and
Giulia
Galli
*
,
Daniel
Reid
,
Juan
de Pablo
and
Giulia
Galli
*
Institute for Molecular Engineering, The University of Chicago, Chicago, Illinois, USA. E-mail: gagalli@uchicago.edu
First published on 9th September 2016
Abstract
Establishing how the conformation of organic photovoltaic (OPV) polymers affects their electronic and transport properties is critical in order to determine design rules for new OPV materials and in particular to understand the performance enhancements recently reported for ternary blends. We report coupled classical and ab initio molecular dynamics simulations showing that polymer linkage twisting significantly reduces optical absorption efficiency, as well as hole transport rates in donor polymers. We predict that blends with components favoring planar geometries contribute to the enhancement of the overall efficiency of ternary OPVs. Furthermore, our electronic structure calculations for the PTB7–PID2–PC71BM system show that hole transfer rates are enhanced in ternary blends with respect to their binary counterpart. Finally, our results point at thermal disorder in the blend as a key reason responsible for device voltage losses and at the need to carry out electronic structure calculations at finite temperature to reliably compare with experiments.
1 Introduction
Organic photovoltaic (OPV) materials possess a moderate yet rapidly increasing efficiency.1–3 A relative abundance of semiconducting polymers, the ease of solution-based processing, and the relatively high optical absorption coefficients of OPV materials are all promising properties for grid-scale solar energy harvesting, together with the low production cost.2,4,5 Typical OPV cells consist of a π-conjugated polymeric donor material, commonly poly-3-hexyl-thiophene (P3HT), along with a fullerene acceptor functionalized to enhance solubility, such as phenyl-C61-butyric acid methyl ester (PC61BM) or phenyl-C71-butyric acid methyl ester (PC71BM).Solar cells constructed from these materials are highly nonuniform by design. They contain a bulk heterojunction (BHJ) in order to maximize interfacial contact between components, which is necessary to promote exciton dissociation and hence harvest electrons and holes; the latter are transported through the acceptor (fullerene) and donor (polymer) regions, respectively. The spatial segregation of transport pathways is accomplished through microphase separation between acceptors and donors and by their relative energy levels6,7—this separation of pathways putatively serves to reduce recombination events which decrease current density.8–10 The open-circuit voltage, in principle proportional to the energy difference between the donor highest occupied molecular orbital (HOMO) and acceptor lowest unoccupied molecular orbital (LUMO), is in part controlled by adjusting the position and spacing of these energy levels,11–13 including their onset;14 however, voltage losses are also determined by other factors, e.g. the energy required to separate electrons and holes.12,15–21
One traditional approach to organic solar cell optimization has involved tuning the HOMO energy level and optical gaps of donor materials, in addition to thin film morphology optimization. This approach has led to developing new donor materials consisting of copolymers made from two different building blocks,22 which resulted in added flexibility in tuning band positions and exciton binding energies, and thus hole and electron separation.23 Recently, more complex systems have been proposed, consisting of ternary blends of two donor materials (polythieno[3,4-b]-thiophene/benzodithiophene (PTB7), and poly-3-oxothieno[3,4-d]isothiazole-1,1-dioxide/benzodithiophene (PID2)) and one fullerene acceptor (PC71BM), resulting in photoconversion efficiencies which exceeded those of binary solar cells. These improvements were hypothesized to come from changes in charge separation and transport with respect to binary blends and from changes in the blend morphology.24
In order to provide design rules for new OPV materials, as well as understand the performance enhancement reported for ternary blends, it is important to establish how the conformation of the OPV polymers affects the electronic properties of the blend, particularly level alignments, and how the overall morphology of the blend affects hole mobilities in donor regions.
The rational design of OPV materials using computational methods is a challenging task, due to the size and morphology of the blends composing OPV devices and the numerous interdependent properties affecting the photoconversion efficiency. Typical molecular weights of OPV polymers suggest polymer sizes of 10–1000 repeating units, composed of 1000–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, which extend way beyond the regime of accessibility of first principles calculations. Hence, isolated oligomers with truncated side-chains are typically utilized to model the electronic properties of OPV materials using ab initio methods.25–27 Although useful starting points, isolated oligomers, often represented by their zero temperature optimized geometry, do not account for the inherent disorder present in OPV blends,28 or for the presence of polydispersity, and of many different stereoisomers resulting from either variation in polymeric linkages or side-chain conformations.29 Changes in polymers (including side-chains, bridging units, and atomic substitutions) may result in surprisingly varied morphologies, electronic properties, and ultimate photoconversion efficiency.30 Morphologies of thin OPV films used in solar cells are indeed rather complex, encompassing glassy and crystalline domains, whose structures and sizes are believed to strongly affect the overall device performance.7,31–38
000 atoms, which extend way beyond the regime of accessibility of first principles calculations. Hence, isolated oligomers with truncated side-chains are typically utilized to model the electronic properties of OPV materials using ab initio methods.25–27 Although useful starting points, isolated oligomers, often represented by their zero temperature optimized geometry, do not account for the inherent disorder present in OPV blends,28 or for the presence of polydispersity, and of many different stereoisomers resulting from either variation in polymeric linkages or side-chain conformations.29 Changes in polymers (including side-chains, bridging units, and atomic substitutions) may result in surprisingly varied morphologies, electronic properties, and ultimate photoconversion efficiency.30 Morphologies of thin OPV films used in solar cells are indeed rather complex, encompassing glassy and crystalline domains, whose structures and sizes are believed to strongly affect the overall device performance.7,31–38
In this work, we used classical molecular dynamics (MD) to obtain realistic disordered structures of ternary polymer blends, whose electronic and transport properties were then investigated using first principles methods. In particular, we considered the ternary blend of PTB7, PID2, and PC71BM, recently proposed experimentally24 and shown in Fig. 1, and we determined the relative energy level alignment of the three components and the transport properties of the composite system, using Marcus theory to describe the hopping transport39–41 of holes.
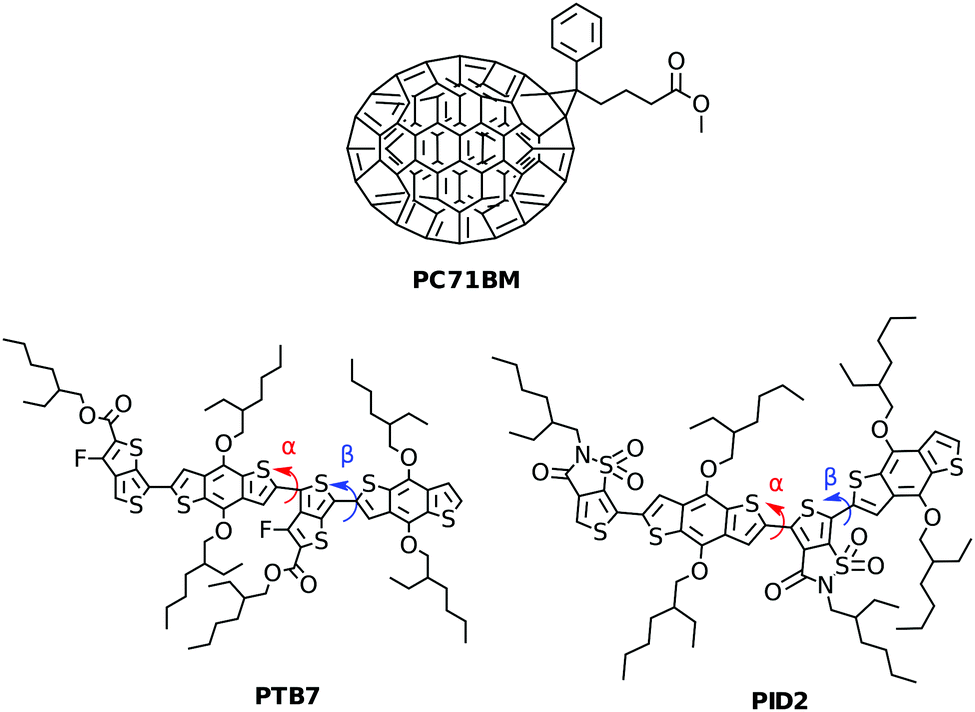 | ||
| Fig. 1 Structures of compounds included in the ternary blend examined in this paper. Two different dihedral angles α and β (S–C–C–S) between subunits are labeled for use in distinguishing configurations. | ||
Our focus on hopping transport builds on the work of Brédas et al.,42 Coropceanu, et al.,43 and Bässler and Köhler,44 who proposed that hole transport occurs primarily via hopping in disordered conjugated polymers, after charge transfer to fullerene acceptors has occurred.6 Measured current versus time curves, obtained by time of flight experiments, are consistent with the proposed picture of transport occurring primarily by hopping.45 In addition measured optical absorption profiles support the commonly accepted picture that hole and electron transport occur in the donor and acceptor regions, respectively.46–48
Our coupled classical and ab initio molecular dynamics simulations showed that polymer linkage twisting significantly reduces optical absorption efficiency, as well as hole transport rates in donor polymers compared to those of polymer blends with planar, ordered geometries. Hence, we predicted that blends with components favoring planar geometries contribute to the enhancement of the overall efficiency of ternary OPVs. Furthermore, our electronic structure calculations showed that hole transfer rates from PC71BM → PID2 are much larger than those from PC71BM → PTB7, contributing to an improved charge current density in ternary blends with respect to their binary counterpart.
In our analysis of the ternary blend solar cell, we first characterized the equilibrium geometries of PTB7 and PID2 using density functional theory. Computational details are given in Section 2. We started by investigating the twisting of links between subunits in an oligomer picture, followed by the analysis of ground state periodic configurations, as described in Section 3.1. As validation of our methodology and optimized structures, we compared the computed energy levels of the lowest lying configurations to the experimental measurements in Section 3.2. Next, we used molecular dynamics to sample the effects of structural disorder on the properties of PC71BM and PTB7 in Section 3.3. Then, we used classical molecular dynamics simulations to assess the effect of PID2 on the morphology of PTB7 in Section 3.4. Using umbrella sampling49,50 combined with the weighted histogram analysis method,51 we find that the presence of PID2 forces PTB7 to adopt a more planar geometry, thereby increasing charge transfer rates. Finally, we computed the charge transport rates at interfaces between donor polymers and PC71BM and these are discussed in Section 3.5.
2 Computational methods
The characterization of isolated monomers of PTB7 and PID2 was performed within the Gaussian basis set code ORCA 352 using the def2-TZVP53 basis set for density functional calculations (Perdew–Burke–Ernzerhof54 (PBE), the hybrid PBE0,55 and the hybrid B3LYP56–58) and the Dunning basis cc-pVTZ59 for resolution of the identity second order Møller–Plesser perturbation theory60 (MP2) calculations. Unless otherwise noted, all density functional approximations used for Gaussian basis set calculations included the Grimme dispersion correction (-D3)61 with Becke–Johnson (BJ) damping62,63 and the geometric counterpoise correction (gCP) of Kruse, et al.64 Gaussian basis set calculations used the chain-of-spheres approximation65,66 and density fitting for Coulomb integrals.67,68For periodic systems, plane wave density functional theory (DFT) calculations were performed using Quantum Espresso,69 with Troullier–Martins and Hartwigsen–Goedecker–Hutter pseudopotentials70,71 and energy cutoffs of 85 and 340 Ry for the wavefunction and charge density, respectively. First principles molecular dynamics simulations were performed with the PBE density functional approximation using Qbox72 and norm-conserving pseudopotentials of the HSCV type73,74 and a cutoff of 85 Ry. van der Waals complexes of PTB7 and PID2 with PC71BM were optimized using the rVV10 van der Waals density functional.75,76
Various conformations of the polymers representing structural disorder were obtained from constant volume and temperature (NVT) classical molecular dynamics (MD) simulations performed with the LAMMPS package77 and a modified OPLS-AA force field.78,79 A Nose–Hoover80,81 thermostat was used with a time step of 1 femtosecond and a temperature of 300 K.
To examine the structural effects of PID2 on PTB7, we performed bulk simulations of PTB7 and PID2, which were prepared by initializing systems at low density, then equilibrating at 1 atm and 500 K for 1 ns until condensed. Systems were then equilibrated at 500 K for 1 ns. Sampling was performed over 2.5 ns at 500 K and 1 atm. We find that polymer chains are within the diffusive regime in these timescales, as shown in ESI.† We used PTB7 and PID2 chains with 5 monomer units, allowing for rapid relaxation. Umbrella restraints were applied to the appropriate PTB7 dihedral during the entire simulation. We performed umbrella sampling using 50 evenly space umbrellas with a restraint strength of 200 kcal mol−1.
Given that the major differences between OPV binary and ternary blends come from variation in the donor material composition, we focused our investigation on hole transport, hypothesized to primarily occur in the donor region of OPVs8 by hopping. Charge transfer parameters for hopping transport in semiconducting systems are commonly computed using either constrained DFT (CDFT),82–84 excited state wavefunction methods,85 or model Hamiltonians,86–88 and then subsequently used to estimate transfer rate using, e.g. Marcus theory.
In our calculations of transport parameters, we assumed that holes were localized either on a copolymer unit (consisting of both a thienothiophene and a benzodithiophene building block) or a PC71BM molecule. Indeed, ab initio calculations of hole wavefunctions showed that holes do localize within one repeating unit of copolymers45 or within four monomer units of the much smaller homopolymer P3HT,89 where a P3HT tetramer is approximately the same size as one monomer of PTB7 or PID2. Localization of charge carriers in organic polymer semiconductors is further supported by terahertz spectroscopy experiments.90 Previous studies using drift–diffusion models hypothesized that current in these materials is largely determined by the variability in 1-dimensional conductivity along polymer chains;91 to that end, we calculated charge transport parameters for extended chain configurations.
We computed hole transfer rates hole transfer rates (kAB) at the interfaces of the donor polymers PTB7 and PID2 with PC71BM using Marcus theory:
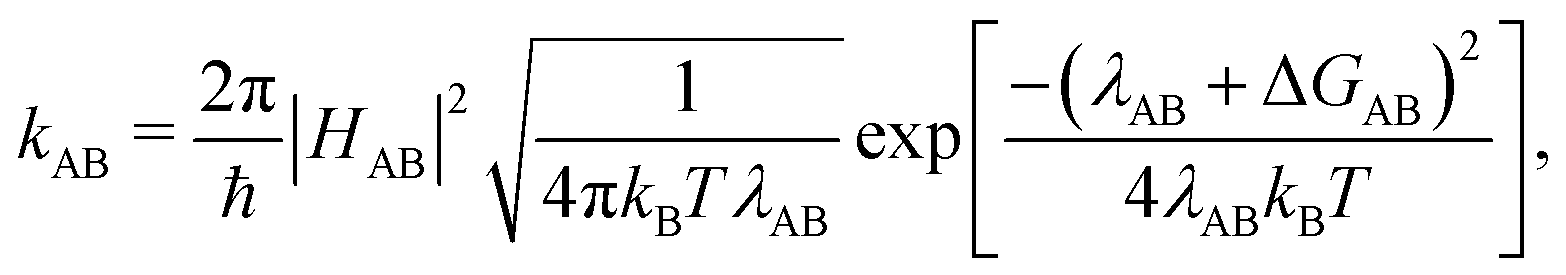 | (1) |
In order to understand how conformation changes the electronic properties of OPV polymers, we computed the energy levels and hole transport couplings of 200 thermally disordered configurations of extended polymers, which were generated by classical MD. To determine specific structure–property relationships, we performed a clustering analysis of dihedral angles in these configurations; we identified distinct structural motifs solely from the dihedral angles between polymer subunits, and we computed averaged properties per cluster. First, we used principal component analysis99 to perform a dimensionality reduction to two principal components. We then identified clusters (i.e. structural motifs) in this reduced space with the affinity propagation method,100 relying upon a Euclidean distance metric. Our identification of clusters was performed using the scikit-learn python library.101
3 Results and discussion
In the following, we present our results for structural optimizations and electronic structure calculations. As mentioned in the introduction, we first computed optimized geometries of monomers and periodic conformers using density functional theory, and we compared computed energy levels with experimental electron affinities and ionization potentials. Then, we established the effect of structural disorder on electronic properties using configurations generated by classical molecular dynamics simulations, focusing on hole transport parameters. Finally, we calculated charge-transfer rates for specific interfacial geometries to address the increased charge current density observed experimentally in ternary blends,24 and we found that that hole transfers from PC71BM → PID2 proceed orders of magnitude faster than from PC71BM → PTB7; these results indicate that ternary blends will have significantly improved charge carrier collection resulting from photoexcitation of PC71BM. Our results are discussed in detail below.3.1 Conformer identification
In order to build realistic model geometries of OPV polymers, we first investigated the total energy of polymer twisting as a function of the dihedral angle between copolymer subunits. We considered isolated monomer models of the polymers, similar to previous work.102–104 As noted by Bhatta et al.,102 the dependence of the energy of these polymers on structural parameters is well-represented using only one-dimensional dihedral scans since the inter-mode vibrational couplings for dihedral fluctuations are small (0.11 kcal mol−1), on the order of the errors expected when using finite basis density functional approximations for conformational energies. In Fig. 2, we show potential energy surfaces as a function of dihedrals (denoted in Fig. 1) for PBE-D3(BJ)-gCP/def2-TZVP geometries of PTB7 and PID2 constrained at each dihedral value. To remove the effect of side-chains, the 2-ethyl-hexyl groups were replaced by hydrogens. The MP2, PBE, and B3LYP surfaces present minima in correspondence to approximately aligned sulfurs (hereafter denoted as a cis geometry) or anti-aligned (trans geometry), with an offset of 10–30 degrees for PTB7 and 0–20 degrees for PID2. This preference for near-planarity is consistent with the conjugated, or quinoidal, nature of these polymers along the backbone. The barriers to twisting the linkage between the subunits are appreciably higher in PID2 (∼6 kcal mol−1 in Fig. 2, top) than in PTB7 (∼3 kcal mol−1 in Fig. 2, bottom), and these barriers, as well as the relative positions of minima, are within chemical accuracy for the three methods used. In addition to a higher barrier, the energy surface of PID2 presents local minima with a higher energetic separation of 3–4 kcal mol−1. From these results, we expect the conformations accessed by PID2 at finite temperature to be significantly less diverse than those of PTB7.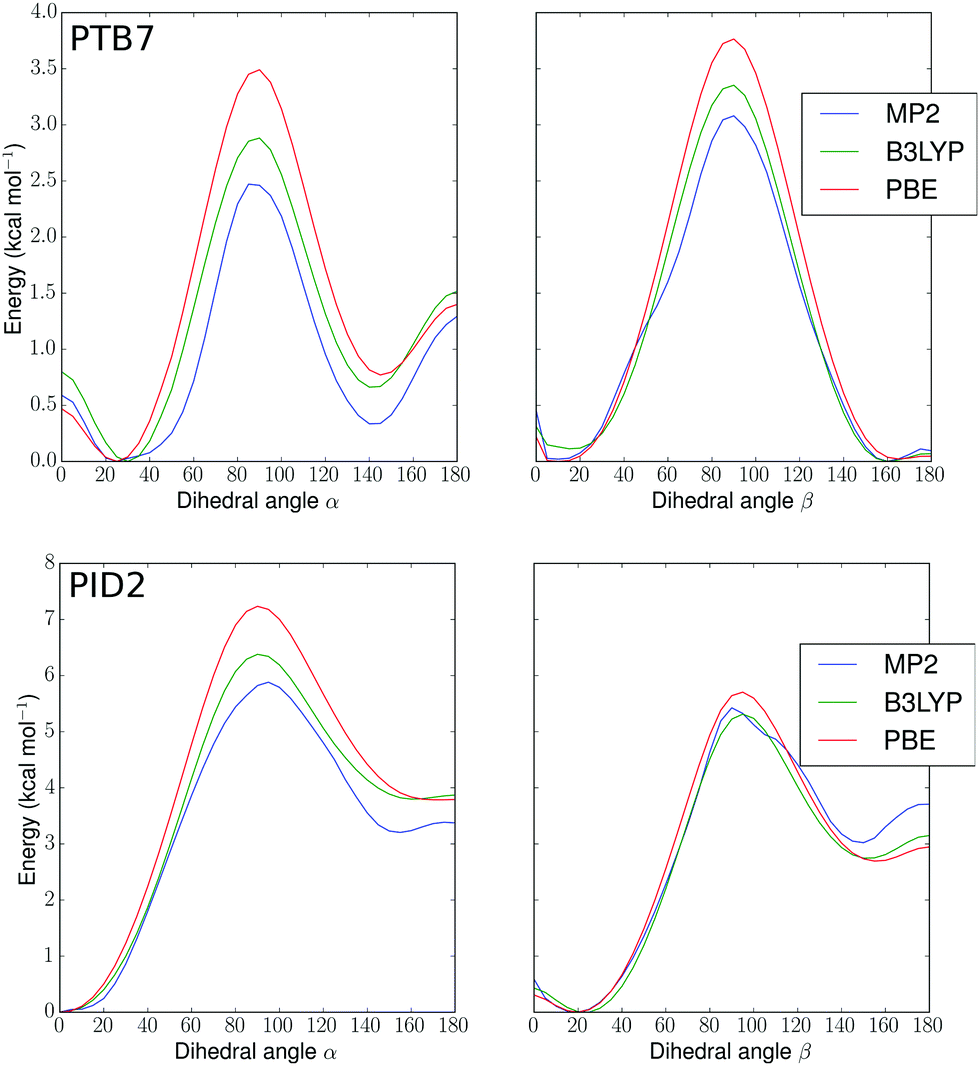 | ||
| Fig. 2 Potential energy surfaces (kcal mol−1) for PTB7 (top) and PID2 (bottom) monomers versus α and β dihedral angles in degrees (see Fig. 1). MP2, B3LYP, and PBE denote different levels of theory (see text). | ||
From the configurations with minimal energy determined above, we constructed periodic configurations for use in plane-wave density functional theory (DFT) models in order to determine the energy levels of extended chains. Since each dihedral potential energy surface has two minima, we examined a total of four conformers for extended chains. Each of the four conformers for PTB7 and PID2 were optimized using the PBE density functional approximation with 5 k-points in the direction of periodicity and 20 Å separation between images. Geometry optimizations were performed with hydrogens and methyl groups in place of the full side chains. We report structural parameters (dihedral angles), fundamental gaps, and relative energies in Tables 1 and 2. The lowest energy conformation for PTB7 is cis–cis aligned for the dihedrals, as denoted in Table 1. Similarly, the cis–cis conformer of PID2 is also lowest lying, with the other conformers much higher in energy. We note that our calculations of PTB7 conformer energies are in agreement with those of Bhatta, Perry, and Tsige,102 while we found different minimum structures from those reported by Khoshkholgh, et al.103 and Li,104 and Freeman,104 possibly due to a very limited configurational sampling in previous works. The variability of our calculated band gaps as a function of the structural motif indicates that models using a single, possibly high energy configuration are inadequate to describe the electronic properties of the blends considered here.
| PTB7 conformer | α 1 | α 2 | β 1 | β 2 | E g | Energy |
|---|---|---|---|---|---|---|
| cis–trans | −57 | 64 | 165 | −173 | 2.3 | 20.9 |
| cis–cis | 23 | 19 | −22 | −21 | 1.9 | 0.0 |
| trans–cis | 117 | −120 | −16 | 15 | 2.4 | 17.9 |
| trans–trans | −141 | −141 | 144 | 148 | 2.0 | 2.2 |
| PID2 conformer | α 1 | α 2 | β 1 | β 2 | E g | Energy |
|---|---|---|---|---|---|---|
| cis–trans | −49 | −58 | −130 | −138 | 2.5 | 24.5 |
| cis–cis | 30 | 9 | −11 | −33 | 2.1 | 0.0 |
| trans–cis | −160 | 137 | 63 | −39 | 2.5 | 23.8 |
| trans–trans | 160 | 161 | −162 | −157 | 2.0 | 4.0 |
3.2 Energy levels of ternary blend systems
Fig. 3 presents the calculated (PBE, PBE0, and B3LYP) and experimental (cyclic voltammetry and ultraviolet photoelectron spectroscopy) HOMO and LUMO energy levels for the ternary blend examined by Lu, et al.,24 using our lowest energy configurations for extended polymer chains and the equilibrium geometry of PC71BM. Our calculations are in agreement105 with the proposed cascade of energy levels measured by cyclic voltammetry (CV),24,106 as well as vertical ionization potential (IP) and electron affinity (EA) onsets measured with ultraviolet photoelectron spectroscopy (UPS) and inverse photoemission spectroscopy (both marked as UPS in Fig. 3).107–117 Both UPS and CV measurements reported here (corresponding to adiabatic and vertical ionization potentials, respectively) agree on the trends in electronic energy levels, as discussed in literature.118,119 The high variability in UPS threshold ionization potentials and electron affinities may stem from the effects of contaminants and differing surface orientations of materials.111,120,121 The DFT HOMO energy levels are clearly separated for each component, with spacings similar to experiment regardless of the density functional chosen. The dielectric constant ε∞ for thin films of PTB7 and PC71BM is ∼4;122 hence, PBE0 (25% exact exchange) or B3LYP (20% exact exchange) are expected to yield good trends in ionization potentials and electron affinities since the optimal percent of exact exchange is inversely proportional to ε∞ in condensed phase materials.123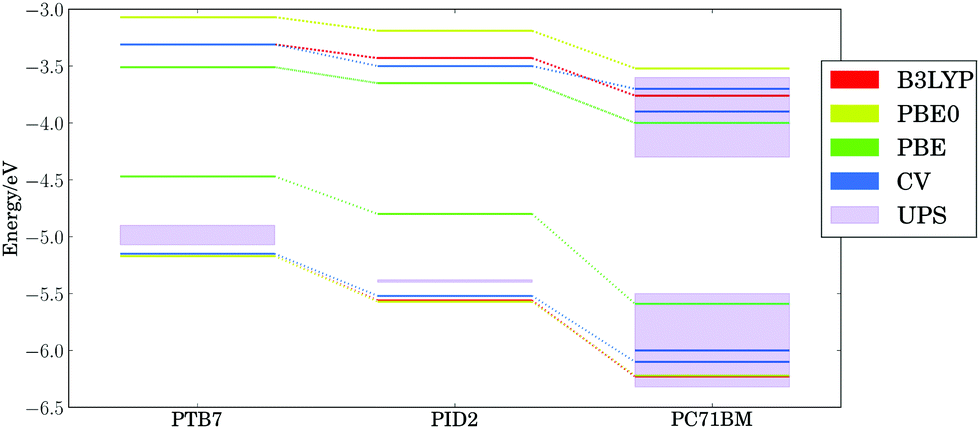 | ||
| Fig. 3 Calculated (PBE, PBE0, and B3LYP levels of theory) and experimental (cyclic voltammetry/CV and ultraviolet photoelectron spectroscopy/UPS) HOMO and LUMO energy levels for ternary system of PTB7, PID2, and PC71BM. | ||
3.3 Thermal configurational disorder
As a first step toward assessing the effect of thermal disorder on polymer energy levels, we performed a first principles molecular dynamics simulation of benzodithiophene. This simulation allowed us to isolate the influence of the thermal disorder of side-chains on the electronic properties of organic polymers, and our results provided qualitative validation of classical MD simulations of the full PTB7 polymer. Fig. 4 presents the PBE HOMO and LUMO energy levels for periodic benzodithiophene configurations with varying side-chain lengths. Geometries were obtained from a first principles NVT molecular dynamics trajectory at 300 K using the Bussi–Donadio–Parrinello thermostat,124 with 80 snapshots taken every 1.2 femtoseconds over a trajectory of 400 steps, sampled with a timestep of 0.242 femtoseconds. Increasing side-chain length shifts the average position of the HOMO energy level up and broadens the energy level positions. The presence of highly branched side-chains induces significantly less broadening, attributable to a loss of flexibility and steric hindrances. From these calculations, we concluded that the different thermal configurations of side chains induce significant band broadening, and hence they need to be taken into account in calculations of electronic energy levels.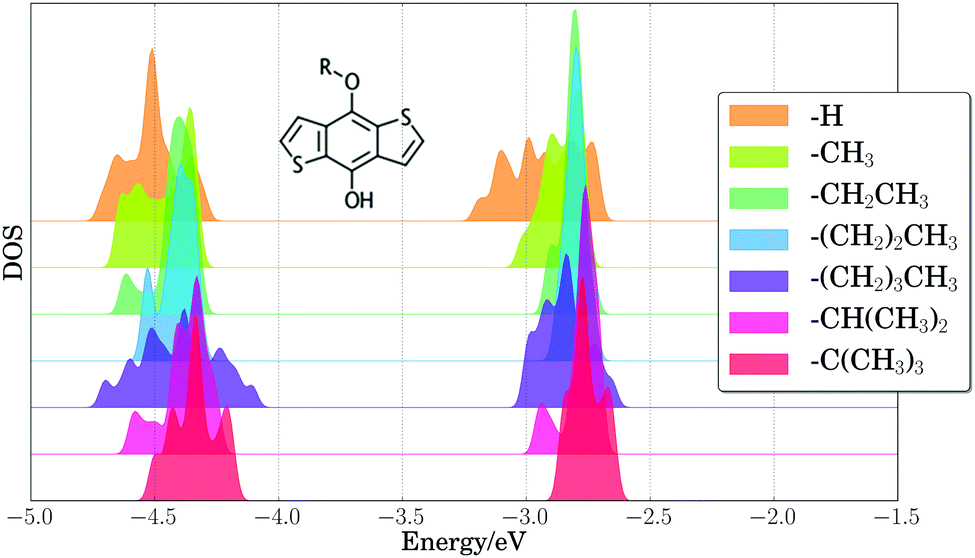 | ||
| Fig. 4 Density of states (DOS) of calculated PBE HOMO and LUMO energy levels (eV) for 80 benzodithiophene configurations with increasing R group complexity. Plotted densities of states have been vertically shifted as a visual aide. A Gaussian broadening of 0.02 eV was applied to computed energy levels. | ||
To estimate the energy level broadening of PC71BM, we performed first principles molecular dynamics simulations of PC71BM, whose B3LYP HOMO−2 through LUMO+2 energy levels (calculated using B3LYP/def2-TZVP) are presented in Fig. 5. Eighty configurations were obtained in the same manner as for the benzodithiophene configurations above. The distribution of HOMO energy levels (−5.86 ± 0.06 eV) displays two peaks and a shoulder, with a dispersion of ca. 0.25 eV. The distribution of LUMO energy levels (−3.47 ± 0.10 eV) displays four peaks, with a dispersion of ca. 0.45 eV. The apparent width of these distributions exceeds two standard deviations, indicating that different atomic configurations accessible due to thermal disorder exhibit different electronic properties. The energy onset of the LUMO (ca. −3.7 eV) is 0.2 eV lower than the mean energy level for these configurations, which suggests that disorder of PC71BM geometries may result in a voltage loss of 0.2 V for the entire device.
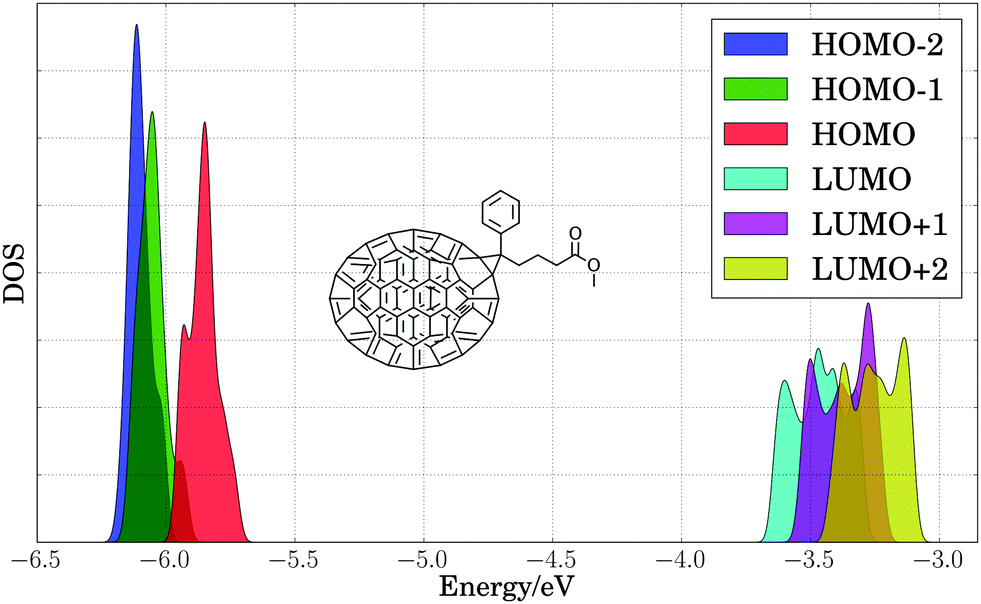 | ||
| Fig. 5 Density of states (DOS) of calculated B3LYP HOMO−2, HOMO−1, HOMO, LUMO, LUMO+1, and LUMO+2 energy levels (eV) for 80 PC71BM configurations, with each energy level marked by color. A Gaussian broadening of 0.02 eV was applied to computed energy levels. | ||
Our results indicated that side chain dynamics must be included to describe PTB7, as well as changes in torsions and out-of-plane bending angles, whose timescales extend beyond those sampled by first principles molecular dynamics. Hence, we performed classical NVT molecular dynamics calculations on periodic cells with two polymer repeating units of PTB7, and we considered 200 snapshots taken every 0.5 nanoseconds after 100 nanoseconds of equilibration. Fig. 6 shows the broadening of the HOMO and LUMO energy levels at the PBE level of theory for those 200 configurations. We note the large spread for the HOMO and LUMO energy levels, with band onsets of 0.2–0.3 eV from the average values of −4.95 eV (HOMO) and −3.65 eV (LUMO), possibly accounting for 0.2 V in operating voltage losses for these materials. The voltage losses due to thermal disorder will be cumulative, adding up to 0.4 V for the PTB7/PC71BM binary mixture from the band widths of the PTB7 HOMO and the PC71BM LUMO. Since the theoretical maximum voltage for that binary mixture is 1.45 V, and the experimental open-circuit voltage is 0.72 V,24, voltage losses of 1.45 − 0.72 V = 0.73 V must be explained. We attribute part of these losses (0.40 V) to thermal broadening of energy levels. The remainder of the voltage losses (∼0.33 V) can be reasonably attributed to the energy required to separate charges (bound by large excitonic binding energies, ca. 0.3 eV, in organic materials125).
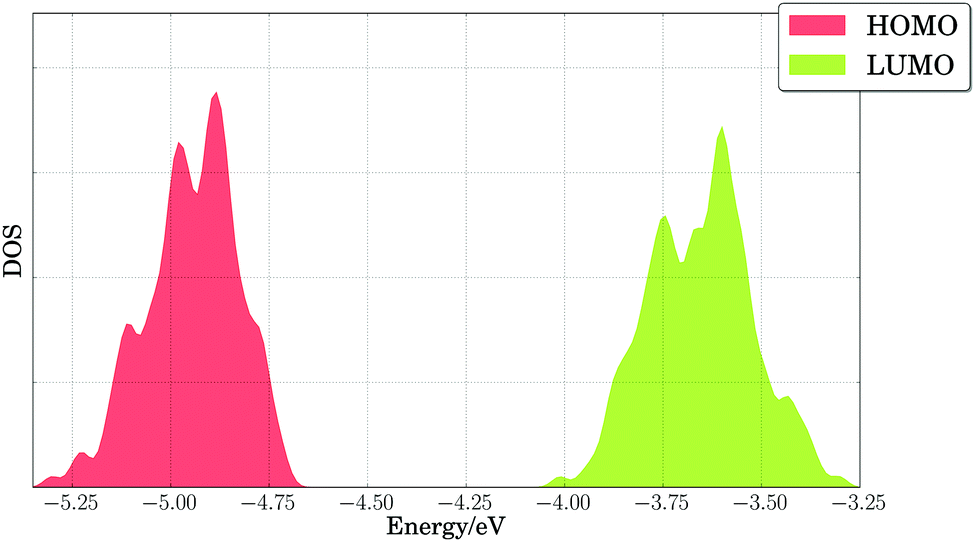 | ||
| Fig. 6 Density of states (DOS) of calculated PBE HOMO and LUMO energy levels for PTB7 extended chains. A Gaussian broadening of 0.02 eV was used in smoothing the results. | ||
In order to establish correlations between electronic and transport properties and specific structural motifs, we performed a cluster analysis (principal component analysis,99 followed by clustering with the affinity propagation method100) of the inter-unit dihedral angles of these configurations. From the 200 configurations, we identified 11 clusters. We present our results for the electronic properties (PBE HOMO, LUMO, band gap, and HAB nonadiabatic coupling for hole transfer) of each cluster within Table 3.
| Cluster | α 1 | β 1 | α 2 | β 2 | γ | HOMO | LUMO | E g | H AB | Frequency |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 35 ± 16 | 47 ± 21 | 61 ± 24 | 77 ± 22 | 52 ± 7 | −5.01 ± 0.10 | −3.62 ± 0.11 | 1.39 ± 0.13 | 298 ± 124 | 19 |
| B | 127 ± 15 | 122 ± 27 | 105 ± 18 | 95 ± 24 | 61 ± 6 | −5.05 ± 0.10 | −3.57 ± 0.07 | 1.48 ± 0.13 | 243 ± 98 | 17 |
| C | 115 ± 23 | 112 ± 21 | 33 ± 20 | 42 ± 21 | 50 ± 8 | −4.97 ± 0.10 | −3.62 ± 0.11 | 1.35 ± 0.17 | 336 ± 168 | 26 |
| D | 153 ± 16 | 140 ± 19 | 33 ± 17 | 31 ± 12 | 33 ± 6 | −4.85 ± 0.07 | −3.79 ± 0.12 | 1.06 ± 0.12 | 552 ± 108 | 22 |
| E | 64 ± 20 | 61 ± 17 | 40 ± 22 | 51 ± 14 | 53 ± 7 | −4.94 ± 0.07 | −3.60 ± 0.09 | 1.34 ± 0.13 | 368 ± 78 | 17 |
| F | 20 ± 17 | 42 ± 18 | 28 ± 18 | 45 ± 20 | 34 ± 7 | −4.82 ± 0.09 | −3.74 ± 0.08 | 1.08 ± 0.13 | 523 ± 120 | 18 |
| G | 67 ± 19 | 67 ± 20 | 113 ± 18 | 94 ± 24 | 67 ± 8 | −5.07 ± 0.10 | −3.49 ± 0.11 | 1.59 ± 0.14 | 220 ± 110 | 18 |
| H | 56 ± 16 | 54 ± 16 | 150 ± 14 | 153 ± 15 | 41 ± 7 | −4.91 ± 0.05 | −3.71 ± 0.08 | 1.19 ± 0.10 | 470 ± 70 | 16 |
| I | 141 ± 18 | 137 ± 17 | 136 ± 18 | 133 ± 16 | 43 ± 7 | −4.88 ± 0.05 | −3.69 ± 0.11 | 1.19 ± 0.11 | 447 ± 72 | 12 |
| J | 107 ± 19 | 76 ± 27 | 137 ± 15 | 141 ± 19 | 53 ± 8 | −5.05 ± 0.06 | −3.61 ± 0.08 | 1.44 ± 0.04 | 207 ± 125 | 11 |
| K | 34 ± 13 | 33 ± 17 | 136 ± 16 | 129 ± 15 | 40 ± 8 | −4.90 ± 0.08 | −3.72 ± 0.11 | 1.18 ± 0.15 | 485 ± 113 | 24 |
| Overall | 48 ± 13 | −4.95 ± 0.12 | −3.65 ± 0.13 | 1.29 ± 0.21 | 386 ± 164 | 200 | ||||
The calculated electronic properties for clustered PTB7 configurations in Table 3 are statistically distinguishable, as determined by Kruskal–Wallis126 and one-way127 analysis of variance p-values (p < 1 × 10−12 for all calculated values). The clusters with the highest hole nonadiabatic couplings have average dihedral angles approximately 30–40 degrees out of plane, with sequential dihedrals typically either both aligned (cis) or anti-aligned (trans), corresponding to the ground state models examined in Section 5.1. By comparison, clusters with a lower degree of planarity (e.g. B and G) exhibit much lower hole transport couplings and higher band gaps. Cluster I includes all-trans model geometries, and cluster F all-cis geometries; however, several motifs with low band gap and high hole transfer couplings have a cis–cis–trans–trans character (e.g. clusters D and K). These results suggest that the local pairwise alignment of dihedrals and overall planarity are key descriptors to optimize opto-electronic properties of semiconducting polymers.
As a further simplification, we reduced the number of structural parameters to 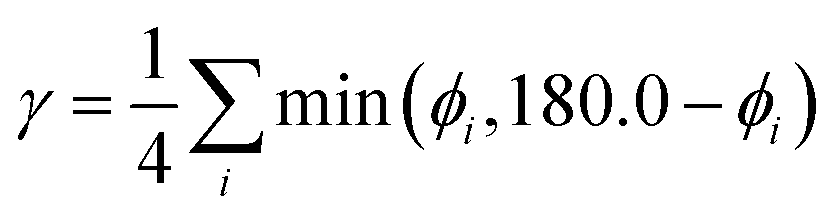 , i.e. the variation of dihedrals from 0 or 180 deg. (the planar configurations). Fig. 7 shows the dependence of the HOMO–LUMO gap and the along-chain hole transfer nonadiabatic couplings on γ. As the chains deform, conjugation is broken, raising the HOMO–LUMO gap linearly as a function of γ, and thus shifting the absorption threshold towards values less optimal for solar energy absorption. This breaking of conjugation also interrupts the charge transport along the chain, reducing the hole transfer integrals by a factor of four. Since rates depend quadratically on the transfer integrals (within Marcus theory), such a reduction translates into a decrease of rates by a factor of 16. These results highlight the key role of microphase structural changes in determining optical absorption and charge transport in OPV blends. In particular, structural aggregation motifs (induced by solvent, additives, or ternary components) are expected to change the planarity of polymer chains and thus hole conductivities and the efficiency of light harvesting. Our conclusion from this analysis is that the planarity of the backbone is highly correlated with optimal electronic gaps and efficient hole transport.
, i.e. the variation of dihedrals from 0 or 180 deg. (the planar configurations). Fig. 7 shows the dependence of the HOMO–LUMO gap and the along-chain hole transfer nonadiabatic couplings on γ. As the chains deform, conjugation is broken, raising the HOMO–LUMO gap linearly as a function of γ, and thus shifting the absorption threshold towards values less optimal for solar energy absorption. This breaking of conjugation also interrupts the charge transport along the chain, reducing the hole transfer integrals by a factor of four. Since rates depend quadratically on the transfer integrals (within Marcus theory), such a reduction translates into a decrease of rates by a factor of 16. These results highlight the key role of microphase structural changes in determining optical absorption and charge transport in OPV blends. In particular, structural aggregation motifs (induced by solvent, additives, or ternary components) are expected to change the planarity of polymer chains and thus hole conductivities and the efficiency of light harvesting. Our conclusion from this analysis is that the planarity of the backbone is highly correlated with optimal electronic gaps and efficient hole transport.
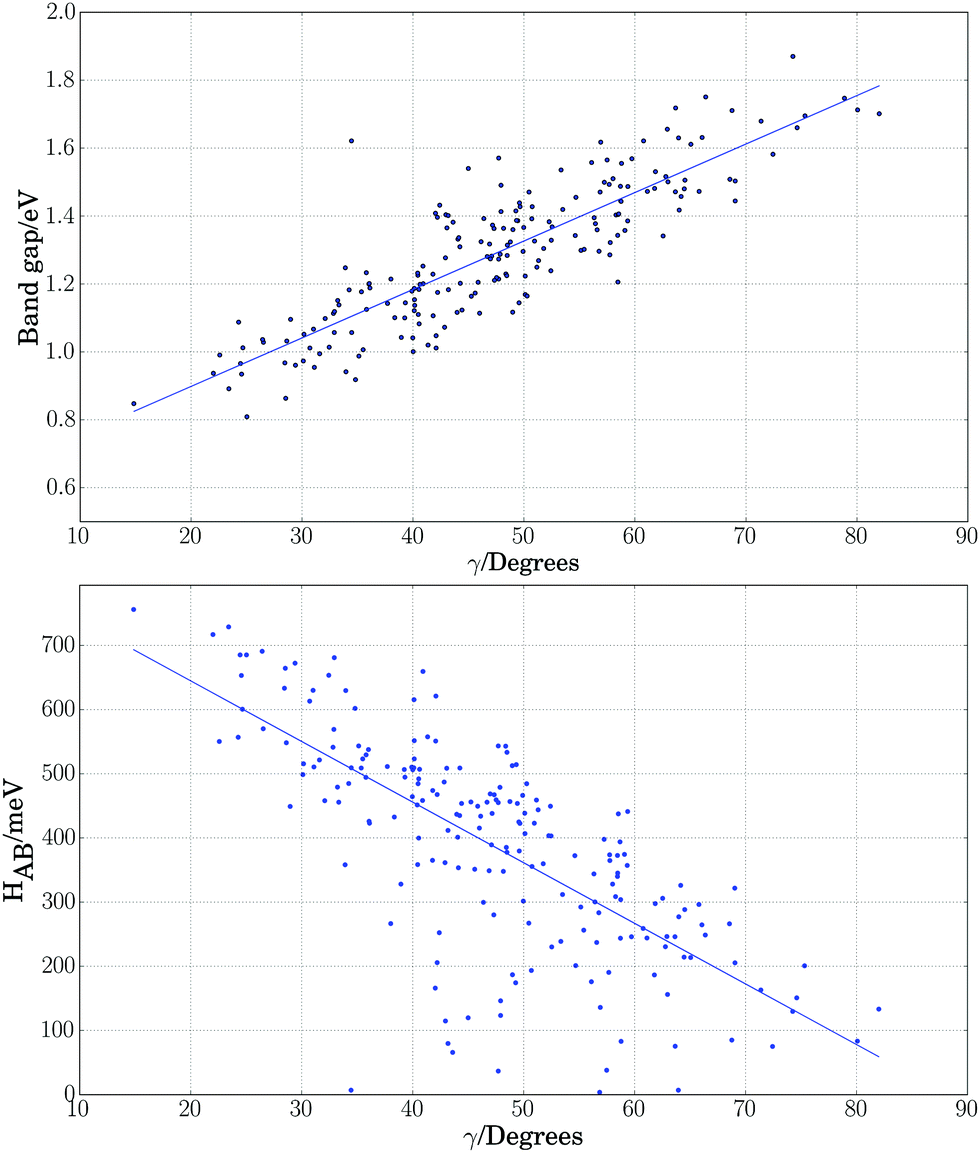 | ||
Fig. 7 Electronic band gap obtained at the PBE level of theory (top) and nonadiabatic coupling (bottom) versus the variation of dihedrals from planarity 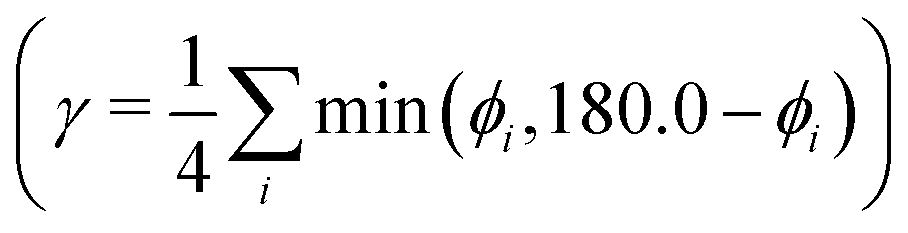 for PTB7 chains. for PTB7 chains. | ||
3.4 Templating of PTB7 planarity by PID2
To assess the effect of PID2 on the morphology of PTB7, we used the weighted histogram analysis method combined with umbrella sampling to determine the free energy of rotation for the middle dihedral in a PTB7 5-mer, which is the β dihedral (see Fig. 1). We determined the free energy of this rotation in two different solvents: a solvent of PTB7, and a solvent of PID2. The solvent consisted of 25 PTB7 or PID2 5-mers, depending on the sample—a system large enough to prevent self-interaction of the PTB7 5-mer of interest. We sampled the free energy of the entire dihedral rotation using 50 uniformly spaced umbrellas, each with a restraint strength of 200 kcal mol−1. Sampling was performed at 500 K, well above the glass transition temperature of PTB7. Samples were prepared by first initializing molecules on a low-density lattice, then evolving the system with a barostat set to 1 atmosphere at 500 K until volume reached equilibrium (5 ns). A temperature above the glass transition temperature was used to ensure that the system was equilibrated. The dihedral restraint for each umbrella was active during the entire simulation. Free energies of rotation for PTB7 in a solvent of PTB7 and PID2 solvent at 500 K are shown in Fig. 8. The free energies of rotation reproduce the distinguishing characteristics of the dihedral potential energy surfaces shown in Fig. 2, in that planar configurations are strongly favored. Comparing the free energies of rotation in a solvent of PTB7 to that in a solvent of PID2, the planar configurations centered around 0 and 180 degrees are stabilized by PID2 solvent. While the energy maxima at 90 and 270 degrees are of nearly identical magnitude in a solvent of PTB7, PID2 imparts an asymmetry, with the energy maxima centered around 90 degrees 0.7 kcal mol−1 lower than that centered around 270 degrees. We calculated the average deviation from planarity to be 36.4 and 34.6 degrees for PTB7 in a solvent of PTB7 and PID2, respectively, showing that PID2 imparts a mild degree of planarity onto PTB7. Interestingly, we performed the same experiment at 400 K and calculated the average deviation from planarity to be 32.3 and 29.2 degrees. These free energies are shown in the ESI.† Planarity increases significantly as temperature drops, demonstrating that planarity is the enthalpically favorable state. Note also that PTB7 at 500 K is on average 1.8 degrees more planar in a solvent of PID2 than in PTB7. At 400 K, this difference increases to 3.1 degrees. Our results show that PID2 leads to a higher degree of planarity of PTB7 in bulk heterojunctions, increasing charge transfer rates in donor regions and thus device efficiency.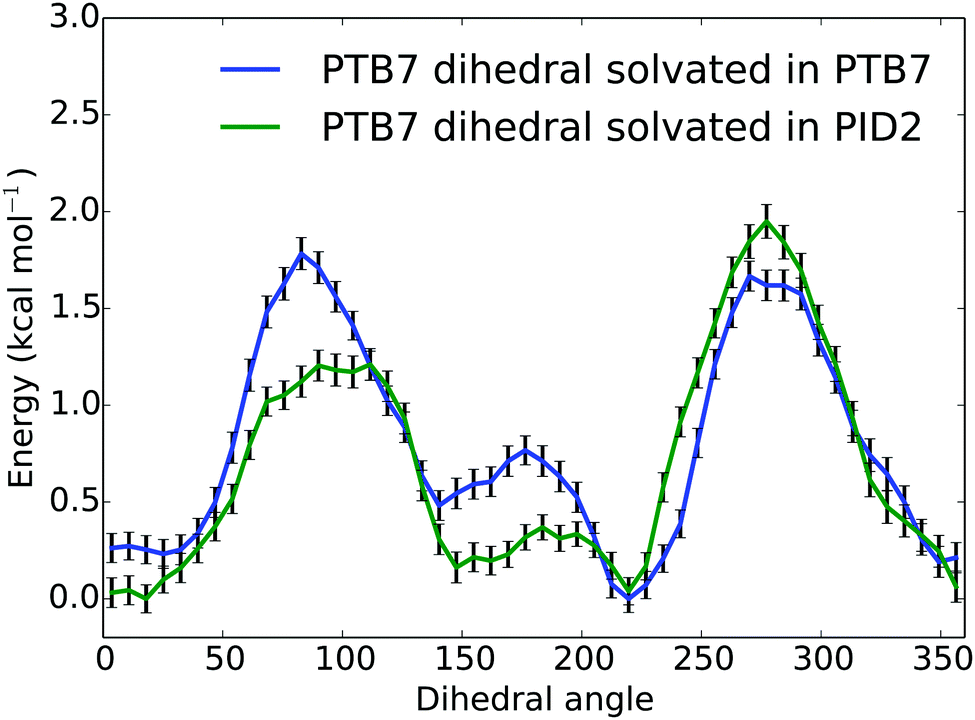 | ||
| Fig. 8 Free energies (kcal mol−1) of rotation for a central PTB7 β dihedral angle in degrees (see Fig. 1) in two different solvents (PTB7 and PID2) at 500 K. | ||
3.5 Charge transfer at interfaces
Differences in hole transfer properties at interfaces between components in organic solar cell systems have been proposed as one reason for improved efficiency of the PTB7, PID2, and PC71BM system.24 Upon photo-excitation of PC71BM, electron hole pairs may be harvested if holes can successfully be extracted from the PC71BM microphases. To this end, we examined the hole transfer rates from PC71BM → PTB7 and →PID2 using constrained DFT for interfacial structures containing two repeating units of a polymer and one PC71BM molecule. Fig. 9 shows isosurfaces of the difference between the charged and neutral densities for a positively ionized interfacial structure of the PTB7/PC71BM interface. Our constrained DFT implementation provided us with direct access to the nonadiabatic couplings (HAB), as well as with differences in energy between diabatic states (ΔGAB). We approximated our reorganization energy (λAB = λint.AB + λext.AB) using the internal reorganization energy (λint.AB) of the isolated, periodic polymer and the isolated PC71BM, with a 0.11 eV estimate of the external reorganization energy (λext.AB) in these systems (corresponding to the response of the bulk environment), which was taken from a literature study on similar materials.128 Our total reorganization energies (the sum of λint.AB and λext.AB) for these systems are of the order 0.2–0.3 eV. The resulting rates for hole transfer at the PTB7/ PC71BM interface are presented in Table 4. In addition to Marcus theory, we also present results using a different rate equation129 proposed by Troisi93 for reactions with large nonadiabatic coupling (HAB ≥ λAB/2). A comparison of the magnitudes of our calculated nonadiabatic couplings and reorganization energies, showing HAB ≥ λAB/2, suggests that the use of the Troisi rate equation may be appropriate. However, we computed HAB using semi-local functionals and based on calculations reported in the literature for small molecules,130,131 we may expect our values to be overestimated compared to hybrid functional results. Hence, which rate equation is applicable may depend upon the level of theory used to compute HAB. Therefore, we calculated hole transfer rates using both rate equations (Marcus and Troisi) and examined trends. The different rate equations yielded substantially different results for along-chain hole transfer rates; however, the qualitative trends for hole transfer rates are similar. The calculated hole transfer rates suggest extremely efficient hole transfer along the polymer backbone (k ∼ 1014–1015 second−1 within Marcus theory), while transfer of holes from PC71BM → PTB7 is calculated to occur 6–8 orders of magnitude more slowly; this suggests that few charge carrier collection events will occur resulting from photoexcitation of the PC71BM in binary PTB7/PC71BM solar cells. | ||
| Fig. 9 Isosurfaces of the difference between charged and neutral densities (ρ = ρneutral − ρcharged) corresponding to constrained DFT diabatic states with a hole localized on two PTB7 subunits (1-red, 2-blue) and PC71BM (green). | ||
| Hole transfer | H AB (eV) | ΔGAB (eV) | λ AB (eV) | k Marcus (s−1) | k Troisi (s−1) |
|---|---|---|---|---|---|
| PTB7-1 → PTB7-2 | 0.60 | −0.07 | 0.23 | 3.9 × 10+15 | 1.2 × 10+23 |
| PTB7-1 → PC71BM | 0.12 | 0.76 | 0.27 | 1.3 × 10−2 | 8.4 × 10−2 |
| PTB7-2 → PC71BM | 0.14 | 0.82 | 0.27 | 1.3 × 10−4 | 1.6 × 10−3 |
| PTB7-2 → PTB7-1 | 0.60 | 0.07 | 0.23 | 3.0 × 10+14 | 9.1 × 10+21 |
| PC71BM → PTB7-1 | 0.12 | −0.76 | 0.22 | 8.8 × 10+8 | 5.1 × 10+9 |
| PC71BM → PTB7-2 | 0.14 | −0.82 | 0.22 | 4.4 × 10+7 | 4.8 × 10+8 |
The hole transfer dynamics at the PID2/PC71BM interface are presented in Table 5. Here, hole transfer rates along the backbone of PID2 are calculated to occur at similar rates to within PTB7 (k ∼ 1013–1015 s−1 within Marcus theory), while the hole transfer from the PC71BM can occur at comparatively fast rates (k ∼ 1013 s−1) to the nearest PID2 subunit. As suggested by Lu, et al.,24 a lower driving force, ΔGAB, of the reaction is the primary difference for this hole extraction process, indicating this reaction is within the inverted Marcus regime.132 Thus the difference in HOMO energy level between PTB7 and PID2 is responsible for different hole transport rates at interfaces with PC71BM. Hence, our results predict that only a small portion of PID2 at interfaces with PC71BM would be necessary for significant increases in charge collection following PC71BM excitation.
| Hole transfer | H AB (eV) | ΔGAB (eV) | λ AB (eV) | k Marcus (s−1) | k Troisi (s−1) |
|---|---|---|---|---|---|
| PID2-1 → PID2-2 | 0.58 | 0.14 | 0.23 | 3.4 × 10+13 | 5.7 × 10+2 |
| PID2-1 → PC71BM | 0.11 | 0.61 | 0.27 | 3.2 × 10+2 | 1.7 × 10+3 |
| PID2-2 → PC71BM | 0.14 | 0.47 | 0.27 | 1.8 × 10+6 | 1.8 × 10+7 |
| PID2-2 → PID2-1 | 0.58 | −0.14 | 0.23 | 8.1 × 10+15 | 1.4 × 10+23 |
| PC71BM → PID2-1 | 0.11 | −0.61 | 0.22 | 3.8 × 10+11 | 1.8 × 10+12 |
| PC71BM → PID2-2 | 0.14 | −0.47 | 0.22 | 3.7 × 10+13 | 3.2 × 10+14 |
4 Conclusions
In this paper, we addressed the question of increased efficiency of ternary OPV blends with respect to binary ones; in particular we focused on a promising system recently studied experimentally: the ternary blend of PTB7, PID2, and PC71BM, and we investigated the reasons underlying its measured absorption and transport efficiency. We used a combination of classical and ab initio MD and electronic structure calculations, and we found that sampling of conformational disorder is necessary for predictive computational design of the electronic and transport properties of OPV materials our results showed that fundamental band gaps and hole transfer nonadiabatic couplings are highly correlated with backbone dihedrals of PTB7, where planar configurations were found to exhibit superior hole transport rates and absorption onsets. Our simulations of different solvation environments found that the presence of PID2 resulted in more planar PTB7 geometries, which improves charge transport and overall device efficiency. We also found that structural disorder is a likely candidate for voltage losses of up to 0.4 V in the ternary blends investigated here, due to the dispersion of the HOMO energy level of PTB7 and the LUMO energy level of PC71BM as a function of temperature. These fundamental voltage losses are intrinsic to disordered organic polymer semiconductors and not only to the systems considered here. We found that improved current in ternary blends can be attributed to the superior hole transfer rates from PC71BM → PID2 relative to those from PC71BM → PTB7, in agreement with the experimental results and operational picture of Lu, et al.24In summary, our computational study showed that the higher planarity of PID2, coupled with the improved hole extraction from PC71BM when PID2 is present, explains the nonadditive improvements seen in ternary solar cell systems.24 As a direction forward in design of OPV materials, we recommend targeting planar structures to minimize voltage and current losses. These structures may be attainable through additives and ternary components or through computationally-aided design of more disorder-resistant (e.g. rigid or planar) OPV materials.
Acknowledgements
This research was supported by award 70NANB14H012 from the U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design (CHiMaD). This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. An award of computer time was provided by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research also used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357. Additionally, resources and computer time from the University of Chicago Research Computing Center were used in part of this research. Unpublished ultraviolet photoelectron spectroscopic data was provided by Donglin Zhao, Luping Yu, Sujitra Pookpanratana, and Lee Richter. Finally, we wish to acknowledge innumerable helpful discussions with Dr Márton Vörös, Dr Nicholas Jackson, and Mr Nicholas Brawand.References
- A. C. Mayer, S. R. Scully, B. E. Hardin, M. W. Rowell and M. D. McGehee, Mater. Today, 2007, 10, 28–33 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- R. F. Service, Science, 2011, 332, 293 CrossRef CAS PubMed.
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29–39 CrossRef CAS PubMed.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS.
- M. Williams, N. R. Tummala, S. G. Aziz, C. Risko and J.-L. Brédas, J. Phys. Chem. Lett., 2014, 5, 3427–3433 CrossRef CAS PubMed.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- S. Difley and T. V. Voorhis, J. Chem. Theory Comput., 2011, 7, 594–601 CrossRef CAS PubMed.
- G. Li, A. Nitzan and M. A. Ratner, Phys. Chem. Chem. Phys., 2012, 14, 14270 RSC.
- M. Scharber and N. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- M. Einax, M. Dierl, P. R. Schiff and A. Nitzan, Europhys. Lett., 2013, 104, 40002 CrossRef.
- B. M. Savoie, S. Dunaisky, T. J. Marks and M. A. Ratner, Adv. Energy Mater., 2015, 5, 1 Search PubMed.
- S. D. Collins, C. M. Proctor, N. A. Ran and T.-Q. Nguyen, Adv. Energy Mater., 2016, 6, 1501721 CrossRef.
- S. R. Yost, L.-P. Wang and T. V. Voorhis, J. Phys. Chem. C, 2011, 115, 14431–14436 CAS.
- M. Einax, M. Dierl and A. Nitzan, J. Phys. Chem. C, 2011, 115, 21396–21401 CAS.
- S. R. Yost and T. V. Voorhis, J. Phys. Chem. C, 2013, 117, 5617–5625 CAS.
- E. T. Hoke, K. Vandewal, J. A. Bartelt, W. R. Mateker, J. D. Douglas, R. Noriega, K. R. Graham, J. M. J. Fréchet, A. Salleo and M. D. McGehee, Adv. Energy Mater., 2013, 3, 220–230 CrossRef CAS.
- K. B. Whaley, A. A. Kocherzhenko and A. Nitzan, J. Phys. Chem. C, 2014, 118, 27235–27244 CAS.
- P. B. Deotare, W. Chang, E. Hontz, D. N. Congreve, L. Shi, P. D. Reusswig, B. Modtland, M. E. Bahlke, C. K. Lee, A. P. Willard, V. Bulović, T. Van Voorhis and M. A. Baldo, Nat. Mater., 2015, 14, 1130–1134 CrossRef CAS PubMed.
- Z. Zheng, J.-L. Brédas and V. Coropceanu, J. Phys. Chem. Lett., 2016, 7, 2616–2621 CrossRef CAS PubMed.
- H. J. Son, B. Carsten, I. H. Jung and L. Yu, Energy Environ. Sci., 2012, 5, 8158–8170 CAS.
- L. Lu and L. Yu, Adv. Mater., 2014, 26, 4413–4430 CrossRef CAS PubMed.
- L. Lu, T. Xu, W. Chen, E. S. Landry and L. Yu, Nat. Photonics, 2014, 8, 716–722 CrossRef CAS.
- R. Olivares-Amaya, C. Amador-Bedolla, J. Hachmann, S. Atahan-Evrenk, R. S. Sanchez-Carrera, L. Vogt and A. Aspuru-Guzik, Energy Environ. Sci., 2011, 4, 4849–4861 CAS.
- I. Y. Kanal, S. G. Owens, J. S. Bechtel and G. R. Hutchison, J. Phys. Chem. Lett., 2013, 4, 1613–1623 CrossRef CAS PubMed.
- C. Risko, M. D. McGehee and J.-L. Bredas, Chem. Sci., 2011, 2, 1200–1218 RSC.
- S. R. Yost, E. Hontz, D. P. McMahon and T. Van Voorhis, in Electronic and Optical Properties at Organic/Organic Interfaces in Organic Solar Cells, ed. D. Beljonne and J. Cornil, Springer, Berlin, Heidelberg, 2014, pp. 103–150 Search PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, T. J. Marks, L. X. Chen and M. A. Ratner, Macromolecules, 2014, 47, 987–992 CrossRef CAS.
- T. Wang, A. J. Pearson and D. G. Lidzey, J. Mater. Chem. C, 2013, 1, 7266–7293 RSC.
- N. Zhou, A. S. Dudnik, T. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. O. de la Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240–1251 CrossRef CAS PubMed.
- T. J. Fauvell, T. Zheng, N. E. Jackson, M. A. Ratner, L. Yu and L. X. Chen, Chem. Mater., 2016, 28, 2814–2822 CrossRef CAS.
- C. H. Woo, B. C. Thompson, B. J. Kim, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 16324–16329 CrossRef CAS PubMed.
- R. Mauer, M. Kastler and F. Laquai, Adv. Funct. Mater., 2010, 20, 2085–2092 CrossRef CAS.
- G. C. Welch, I. Ronald, C. Bakus, S. J. Teat and G. C. Bazan, J. Am. Chem. Soc., 2013, 135, 2298–2305 CrossRef CAS PubMed.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
- R. Noriega, A. Salleo and A. J. Spakowitz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16315–16320 CrossRef CAS PubMed.
- S. M. Ryno, C. Risko and J.-L. Brédas, ACS Appl. Mater. Interfaces, 2016, 8, 14053–14062 CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- R. Marcus and N. Sutin, Biochim. Biophys. Acta, Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- H. Bässler and A. Köhler, Top. Curr. Chem., 2012, 312, 1–65 CrossRef.
- J.-L. Brédas, B. David, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- H. Bässler and A. Köhler, Charge Transport in Organic Semiconductors, in Unimolecular and Supramolecular Electronics I: Chemistry and Physics Meet at Metal-Molecule Interfaces, ed. M. R. Metzger, Springer, Berlin, Heidelberg, 2012, pp. 1–65 Search PubMed.
- S. T. Hoffmann, F. Jaiser, A. Hayer, H. Bässler, T. Unger, S. Athanasopoulos, D. Neher and A. Köhler, J. Am. Chem. Soc., 2013, 135, 1772–1782 CrossRef CAS PubMed.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- R. Kline, M. McGehee, E. Kadnikova, J. Liu and J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- T. D. Anthopoulos, D. M. de Leeuw, E. Cantatore, P. van't Hof, J. Alma and J. C. Hummelen, J. Appl. Phys., 2005, 98, 054503 CrossRef.
- D. Chandler, Introduction to Modern Statistical Mechanics, Oxford University Press, 1987 Search PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. Thom, H. Dunning, T. H. Dunning, T. H. Dunning, Jr. and T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 24101 CrossRef PubMed.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- S. Kossmann and F. Neese, J. Chem. Theory Comput., 2010, 6, 2325–2338 CrossRef CAS PubMed.
- A. Sodt, J. E. Subotnik and M. Head-Gordon, J. Chem. Phys., 2006, 125, 194109 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- N. Troullier and J. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993 CrossRef CAS.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641–3662 CrossRef CAS.
- F. Gygi, IBM J. Res. Dev., 2008, 52, 137–144 CrossRef.
- D. R. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494–1497 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 8412–8415 CrossRef CAS.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 041108 CrossRef.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- D. Reid, M. Goldey, G. Galli and J. de Pablo, unpublished work.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- Q. Wu and T. Van Voorhis, Phys. Rev. A: At., Mol., Opt. Phys., 2005, 72, 7–10 Search PubMed.
- Q. Wu and T. Van Voorhis, J. Chem. Phys., 2006, 125, 1–9 Search PubMed.
- Q. Wu and T. Van Voorhis, J. Chem. Theory Comput., 2006, 2, 765–774 CrossRef CAS PubMed.
- J. E. Subotnik, J. Vura-Weis, A. J. Sodt and M. A. Ratner, J. Phys. Chem. A, 2010, 114, 8665–8675 CrossRef CAS PubMed.
- F. Ortmann, F. Bechstedt and K. Hannewald, New J. Phys., 2010, 12, 23011 CrossRef.
- F. Ortmann, F. Bechstedt and K. Hannewald, Phys. Status Solidi, 2011, 248, 511–525 CrossRef CAS.
- S. K. Maiti and A. Nitzan, Phys. Lett. A, 2013, 377, 1205–1209 CrossRef CAS.
- D. L. Cheung, D. P. McMahon and A. Troisi, J. Am. Chem. Soc., 2009, 131, 11179–11186 CrossRef CAS PubMed.
- X. Ai, M. C. Beard, K. P. Knutsen, S. E. Shaheen, G. Rumbles and R. J. Ellingson, J. Phys. Chem. B, 2006, 110, 25462–25471 CrossRef CAS PubMed.
- B. Y. Finck and B. J. Schwartz, Phys. Rev. Appl., 2015, 4, 034006 CrossRef.
- K. Wynne and R. M. Hochstrasser, Coherence and Adiabaticity in Ultrafast Electron Transfer, John Wiley & Sons, Inc., 2007, pp. 263–309 Search PubMed.
- A. Troisi, Chem. Soc. Rev., 2011, 40, 2347–2358 RSC.
- In the case of the PC71BM → PID2-2 hole transfer (see Section 3.5), the activation energy is 0.07 eV, though calculations in Table 5 qualitatively distinguish this reaction from the others to/from PC71BM.
- H. Oberhofer and J. Blumberger, J. Chem. Phys., 2010, 133, 244105 CrossRef PubMed.
- M. Goldey, N. Brawand and G. Galli, unpublished work.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- J. Blumberger and K. P. McKenna, Phys. Chem. Chem. Phys., 2013, 15, 2184–2196 RSC.
- I. T. Jolliffe, Principal Component Analysis, Springer-Verlag, New York, 2002 Search PubMed.
- B. J. Frey and D. Dueck, Science, 2007, 315, 972–976 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- R. S. Bhatta, D. S. Perry and M. Tsige, J. Phys. Chem. A, 2013, 117, 12628–12634 CrossRef CAS PubMed.
- M. J. Khoshkholgh, F. Marsusi and M. R. Abolhassani, Spectrochim. Acta, Part A, 2015, 136(pt B), 373–980 CrossRef CAS PubMed.
- L.-H. Li, O. Y. Kontsevoi and A. J. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 195203 CrossRef.
- Our computed vertical ionization potentials are directly comparable with UPS data, not with CV ones, which represent adiabatic ionization potentials.
- L. Lu, W. Chen, T. Xu and L. Yu, Nat. Commun., 2015, 6, 7327 CrossRef CAS PubMed.
- D. Zhao and L. Yu, unpublished work.
- S. Pookpanratana and L. Richter, unpublished work.
- A. Nogimura, K. Akaike, R. Nakanishi, R. Eguchi and K. Kanai, Org. Electron., 2013, 14, 3222–3227 CrossRef CAS.
- R. Nakanishi, A. Nogimura, R. Eguchi and K. Kanai, Org. Electron., 2014, 15, 2912–2921 CrossRef CAS.
- D.-J. Yun, J. Chung, C. Jung, Y. Chung, S. Kim, S. Lee, K.-H. Kim, H. Han, G.-S. Park and S. Park, J. Appl. Phys., 2013, 114, 094510 CrossRef.
- E. L. Ratcliff, J. Meyer, K. X. Steirer, N. R. Armstrong, D. Olson and A. Kahn, Org. Electron., 2012, 13, 744–749 CrossRef CAS.
- H. Yoshida, J. Phys. Chem. C, 2014, 118, 24377–24382 CAS.
- J. Subbiah, C. M. Amb, I. Irfan, Y. Gao, J. R. Reynolds and F. So, ACS Appl. Mater. Interfaces, 2012, 4, 866–870 CAS.
- Q.-D. Yang, H.-W. Li, Y. Cheng, Z. Guan, T. Liu, T.-W. Ng, C.-S. Lee and S.-W. Tsang, ACS Appl. Mater. Interfaces, 2016, 8, 7283–7290 CAS.
- D. Shin, J. Lee, S. Park, J. Jeong, K.-W. Seo, H.-J. Kim, H.-K. Kim, M.-J. Choi, K.-B. Chung and Y. Yi, Appl. Phys. Express, 2015, 8, 095701 CrossRef.
- C. K. Song, B. J. Eckstein, T. L. D. Tam, L. Trahey and T. J. Marks, ACS Appl. Mater. Interfaces, 2014, 6, 19347–19354 CAS.
- B. M. Savoie, N. E. Jackson, T. J. Marks and M. A. Ratner, Phys. Chem. Chem. Phys., 2013, 15, 4538–4547 RSC.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17 RSC.
- H. Ishii and K. Seki, IEEE Trans. Electron Devices, 1997, 44, 1295–1301 CrossRef CAS.
- A. Kahn, Mater. Horiz., 2016, 3, 7–10 RSC.
- J. Singh and M. R. Narayan, Nanosci. Technol., 2013, 1, 8 Search PubMed.
- J. H. Skone, M. Govoni and G. Galli, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 195112 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Knupfer, Appl. Phys. A: Mater. Sci. Process., 2003, 77, 623–626 CrossRef CAS.
- W. H. Kruskal and W. A. Wallis, J. Am. Stat. Assoc., 1952, 47, 583–621 CrossRef.
- R. G. Lomax and D. L. Hahs-Vaughn, Statistical Concepts: A Second Course, Routledge Academic, 3rd edn, 2007 Search PubMed.
- T. Liu and A. Troisi, J. Phys. Chem. C, 2010, 115, 2406–2415 Search PubMed.
- In Tables 4 and 5, we calculated hole transfer rates using an adiabatic rate equation from Troisi's recent review,93 which has been modified to include the reaction driving force.
We employ a frequency ω = 1000 cm−1/ħ, corresponding to an optical phonon frequency which is representative for most organic semiconductors. Since ω enters primarily as a prefactor, it serves to rescale rates, and errors here will not change trends. - A. Kubas, F. Hoffmann, A. Heck, H. Oberhofer, M. Elstner and J. Blumberger, J. Chem. Phys., 2014, 140, 104105 CrossRef PubMed.
- A. Kubas, F. Hoffmann, A. Heck, H. Oberhofer, M. Elstner and J. Blumberger, Phys. Chem. Chem. Phys., 2014, 17, 14342–14354 RSC.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CAS.
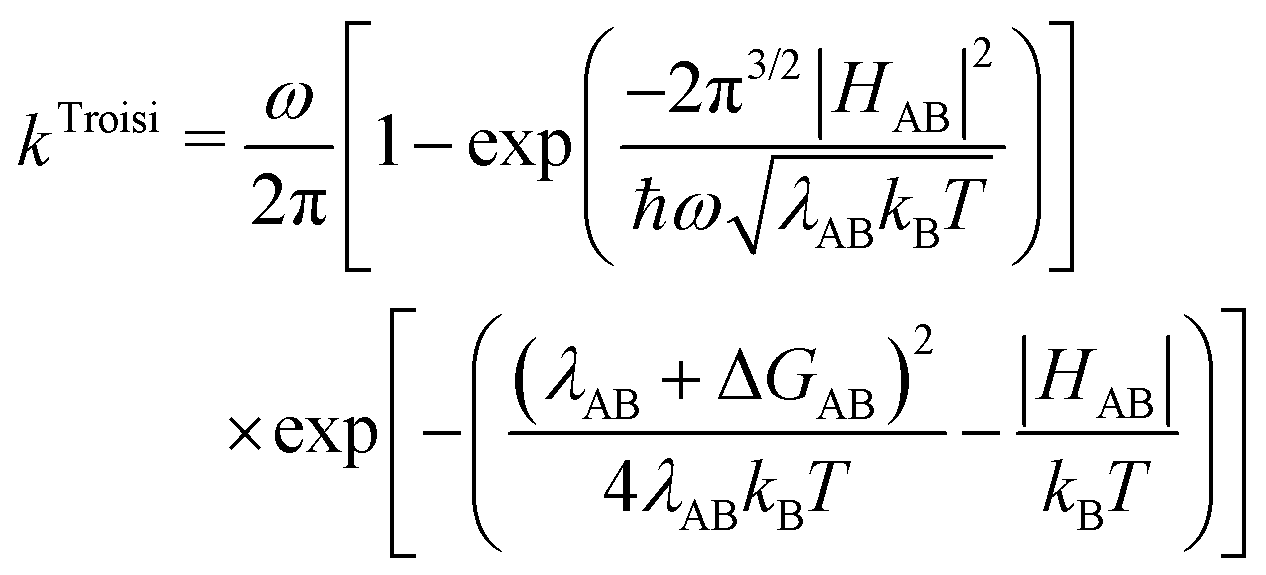
Footnote |
| † Electronic supplementary information (ESI) available: Ultraviolet photoelectron spectroscopy data for PTB7, PID2, and PC71BM which was used for Fig. 3. Free energies of rotation for PTB7 in PID2 and PTB7 at 400K, and mean-squared displacement versus time of PTB7 in our simulations. Histogram of linkage dihedrals for PTB7 snapshots taken from MD. Band gaps at the B3LYP level of theory for PTB7 and PID2 periodic configurations with hydrogen and methyl side chains. See DOI: 10.1039/c6cp04999k |
| This journal is © the Owner Societies 2016 |