
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcerted nitrogen inversion and hydrogen bonding to Glu451 are responsible for protein-controlled suppression of the reverse reaction in human DPP III†
A.
Tomić
a,
B.
Kovačević
*b and
S.
Tomić
*a
aDepartment of Organic Chemistry and Biochemistry, Rudjer Boskovic Institute, Bijenicka cesta 54, 10000, Zagreb, Croatia. E-mail: Antonija.Tomic@irb.hr; Sanja.Tomic@irb.hr
bDepartment of Physical Chemistry, Rudjer Boskovic Institute, Bijenicka cesta 54, 10000, Zagreb, Croatia. E-mail: Borislav.Kovacevic@irb.hr
First published on 2nd September 2016
Abstract
Human dipeptidyl-peptidase III (h.DPP III) is a zinc-exopeptidase that hydrolyses dipeptides from the N-terminus of its substrates. Its mechanism of action was assumed to be similar to that of thermolysin, but was never thoroughly investigated. This study presents the first insight into the reaction mechanism of h.DPP III, determined on the model and real (hydrated enzyme with Leu-enkephalin bound in the active site) systems. The Glu451-assisted water addition on amide carbon atoms and nitrogen inversion (i.e. change of pyramidalization on the leaving nitrogen) are shown to be the rate-determining steps with the activation energies in a good agreement with the experimental results for the Leu-enkephalin hydrolysis. The energy barrier for nucleophilic attack is about 28 kJ mol−1, while barriers for the N-inversion differ as a consequence of the number of hydrogen bonds that have to be changed, which is smaller in the model active site than in the solvated enzyme. Although precisely defined geometry of the enzyme binding site puts an additional restraint on the hydrogen bonding interactions, at the same time it stimulates the forward reaction towards the final hydrolytic product. Namely, different from the model, the N-inversion is in a concerted fashion followed by favourable hydrogen bonding with Glu451 that immediately “locks” the system into the configuration where reversion to the enzyme–substrate complex is hardly achievable. Therefore we propose that the functional significance of DPP III is dual: to lower the energy barrier of the peptide hydrolysis and to suppress the reverse reaction.
Introduction
Dipeptidyl-peptidase III (DPP III; EC 3.4.14.4) is a two-domain monozinc metalloexopeptidase from the peptidase family M49 (according to the MEROPS database, http://www.merops.ac.uk) that hydrolyzes dipeptides from the N-terminus of its substrates consisting of three or more amino acids.1 The motifs HEXXGH and E*EXR(K)AE(D) are considered as its trademark,1,2 wherein histidines from the first motif and Glu* from the second motif take part in the zinc ion coordination (i.e. His450, His455 and Glu508 in the h.DPP III). The Glu from the first motif (Glu451 in the h.DPP III) is crucial for the catalytic activity of DPP III.In mammalian tissues, DPP III is broadly distributed and thought to contribute to the final steps of normal intracellular protein catabolism. There are strong indications of its role in the endogenous pain-modulation system,3,4 as well as in the endogenous defense against oxidative stress.5 Pathophysiological roles of DPP III are indicated in cataractogenesis,6 malignant growth7 and influenza virus infection.8 The most recent studies showed that DPP III is a member of a six-gene signature that accurately predicts human breast cancer patient survival.9 Hast et al. revealed that DPP III contributes to the activation of transcription factor Nrf2 by binding to its negative regulator, KEAP1, a member of the Keap1 functional E3 ubiquitin ligase complex.10 The Nrf2-Keap1 signalling pathway is a major regulator of the cytoprotective response to oxidative and electrophilic stress. All these findings characterize DPP III as a valuable drug target.
There are several crystal structures of h.DPP III available. The structure of the ligand-free enzyme (pdb codes: 3FVY and 3EGY) was determined in the so-called open and closed form, while that of the liganded enzyme only in a more compact, closed, form (pdb codes: 3T6B, 3T6J, 5E33, 5E3A, 5E2Q, 5EHH and 5E3C). The structure of the open ligand-free enzyme has a wide cleft (∼40 Å wide and ∼40 Å high) between the two domains (so-called upper and lower domain) which can be easily accessed by peptide substrates. The increased protein globularity, which is a result of protein closure, has also been observed during molecular dynamic (MD) simulations of the ligand-free h.DPP III.11 During the simulations the enzyme transformed from an extended (open) form to the compact (closed) form while the conformation of each protein domain remained conserved.
In the closed (chemically active) enzyme structure the catalytic zinc ion, bound to the upper domain at the “roof” of the cleft, interacts with the carbonyl oxygen from the scissile amide bond of the substrate molecule bound to the lower protein domain.12–14 The substrate is bound in the form of a β-strand to the five-stranded β-core from a lower protein domain (of an enzyme) in an antiparallel fashion.
Although the DPP III structure represents a novel fold, the mode of zinc binding closely resembles that observed in other structurally unrelated metallopeptidases, such as neprilysin and thermolysin. These two enzymes also coordinate with the metal ion through two histidines and one glutamate, and superposition of the zinc-binding residues reveals very similar coordination geometry.15 Neprilysin and thermolysin have the structurally equivalent residues to evolutionarily 100% conserved His568 and Glu451 from the h.DPP III. It was shown that in the thermolysin catalyzed reaction the glutamic acid residue acts as a general base during the deprotonation of the water molecule that attacks the scissile amide bond, while the role of histidine is to stabilize the tetrahedral intermediate.16,17 The kinetic data on h.DPP III showed that the His568 to Asn mutation significantly decreases enzyme activity, for example the kcat value for hydrolysis of synthetic substrate Arg–Arg–2-naphthylamide (RR–2-NA) is three orders of magnitude lower in the His568Asn mutant than in the wild type protein, however the Km values are similar.18 In addition, the Tyr318 to Phe substitution (Tyr318 is also 100% conserved residue) resulted in a significant reduction of the enzymatic activity, the kcat for RR–2-NA hydrolysis decreased two orders of magnitude while the ligand binding affinity was not affected.19 These results indicate that these highly conserved residues could be involved in transition state stabilization during the catalytic action of h.DPP III.
We approached the study of human DPP III hydrolysis progressively. First we performed a quantum mechanical (QM) study on a small model system shown in Fig. 1 that was followed by a quantum mechanics/molecular mechanics (QM/MM) study of the reaction in the solvated enzyme – Leu-enkephalin (Tyr–Gly–Gly–Phe–Leu) complex. Using DFT calculations we elucidated the role of the individual enzyme residues in the catalytic reaction and determined the protonation state of the 100% conserved His568 residue.
 | ||
| Fig. 1 Minimal model of the human DPP III active site. The substrate (CH3CO–Gly–NHCH3) and water are shown in red and blue, respectively. His568 is shown in its protonated state with the Hδ atom shown in green. Electrostatic interactions are shown with black dashed lines. Atom labels used further in the text are indicated. | ||
Computational methods
QM calculations on the model system
The experimentally determined crystal structure of the closed h.DPP III in complex with tynorphine (pdb code: 3T6B) was used as a starting structure for QM calculations. Since the closed DPP III structure obtained by X-ray diffraction represents the E451A mutant of h.DPP III and lacks the zinc ion, we mutated Ala451 back to Glu before the simulations and added the zinc ion using the crystal structure of the open h.DPP III (pdb code 3FVY) as a template. It should be emphasised that the protonation states of the histidines, and thereby His568, were determined based on their ability to form hydrogen bonds with neighbouring amino acid residues or to coordinate to the metal ion.QM calculations were carried out for a minimal model of the active site consisting of the zinc ion ligated by one water molecule, two 4-methylimidazoles, and one propanoate group, replacing the enzyme residues His450, His455 and Glu508, respectively. The conserved residues Tyr318 and Glu451 were modelled by 1-methly-4-hydroxyphenyl and butyrate groups, while His568 by 4-methylimidazol or 4-methylimidazolium cations, in the case of the neutral (histidine with protons on the Nε atom) or protonated histidine residue, respectively (Fig. 1). The substrate was modelled by CH3CO–Gly–NHCH3, i.e. glycine was capped with acetyl (Ace) at the N-terminal and methylamide (Mam) at the C-terminal (Ace–Gly–Mam). The initial position of the water molecule coordinating the zinc ion was chosen based on our previous QM/MM calculations on the zinc ion coordination.12 Since the previous theoretical studies of the peptide hydrolysis by thermolysin had revealed the Glu143-assisted water addition as a preferable mechanism, the water molecule attached to Zn2+ was hydrogen bonded to Glu451 in our model system as well. This is in line with our QM/MM calculations on the Zn2+ ion coordination12 which clearly indicated that one water molecule is always hydrogen bonded to Glu451 in the closed structure of h.DPP III. Another water molecule found close to the metal ion12 coincides with the substrate oxygen location and was deleted.
In the crystal structures of DPP III–peptide complexes, the amide group of the second amino acid from the substrate N-terminus makes a hydrogen bond with Tyr318, while its hydroxyl group is hydrogen bonded to His568. The particular hydrogen-bond pattern was preserved in our model structure (see Fig. S1, ESI†).
The B97D20 density functional method as implemented in GAUSSIAN 0921 program package was used for all the present optimization calculations. Stationary points were determined using the 6-31G(d) basis set for the H, N, C and O atom and the LANL2DZ-ECP basis set for the Zn2+ ion.22 During the optimization of the stationary points (ES, INT and P) the terminal methyl group carbon atoms of the model enzyme residues (Y318, H450, E451, H455, E508 and H568) were fixed. However, to search for the minima and transition states, the unconstrained relaxed potential energy surface scan was used. To guess the transition state structure the Ow–Cs and Cs–Ns distances were scanned since these bond lengths most closely describe the imaginary frequencies of the first and second transition states proposed in the study of thermolysin hydrolysis.17 No constraints were applied on the substrate during the optimization. The transition state structures were obtained either using the Synchronous Transit-Guided Quasi-Newton (STQN) method or the transition state optimization method.23–25
Each transition state was confirmed by the existence of one negative eigenvalue (one imaginary frequency), with the vibrational vector leading to the desired neighbouring minima. In addition, transition state geometries were connected to the ground state geometries using intrinsic reaction coordinate (IRC) calculations that allow one to follow the path downhill from the transition state in both forward and reverse directions.26
Calculations were performed in the presence of the low dielectric media (dielectric constant 4) utilizing the polarizable continuum model (CPCM).27,28
QM/MM calculations on the hydrated enzyme–substrate complex
The optimized structure of the closed h.DPP III in complex with peptide substrate Leu-enkephalin (Tyr–Gly–Gly–Phe–Leu) obtained after 30 ns of MD simulation (as described in our previous publication)12 was used as a starting structure for QM/MM optimization of the hydrated enzyme–substrate system. Only 3502 water molecules closest to the protein, i.e. constituting enzyme first and second solvation spheres, were retained. Besides the structure taken directly from MD simulations, a reaction mechanism was also investigated in the “hybrid” enzyme structure where positions of Tyr318, Glu451 and His568, as well as those of the zinc-coordinating residues, were adjusted using the closed h.DPP III structure (pdb code: 3T6B) as a template. It was accomplished by aligning the simulated and the experimental structure backbones. The water molecule coordinated to the zinc ion was added manually based on its position in the QM model system. The constructed system closely resembles the crystal structure of the human DPP III–Leu-enkephalin complex (pdb code: 5E3A) only recently available (see Fig. S1, ESI†). Two differently charged systems were considered, one with neutral and the other with protonated His568.QM/MM geometry optimization of the enzyme binding site was performed using the 2-layer ONIOM (Our own N-layered Integrated molecular Orbital and molecular Mechanics) methodology implemented in program GAUSSIAN 09:
| EONIOM = EMM(S) + EQM(SM) − EMM(SM) | (1) |
For the purpose of 2-layer ONIOM structure optimization the system was divided into two layers which were handled at different levels of theory. At the places where borders “cut” covalent bonds (Cα–Cβ), the link atoms were placed. The QM layer, consisting of Y318, H450, E451, H455, E508 and H568 side chains, Leu-enkephalin, the zinc ion and a water molecule, was treated by the B97D DFT method and two different basis sets: 6-31G(d) for the H, N, C and O atoms, and LANL2DZ-ECP for the Zn2+ atom. The MM part (the rest of the protein) was treated using the AMBER force field (parm96).31 To take into account solvation, water molecules from the enzyme first and second solvatation sphere (additional 3501 water molecules closest to the protein molecule) were considered as a part of the MM layer as well. The initial cycle of geometry optimizations was performed using mechanical embedding (ME), and the final geometry optimization was performed using electronic embedding (EE).30 The net charge of the whole system was either −24 e or −23 e, while the QM region had a zero or plus one charge depending on whether the H568 is neutral or protonated, respectively. During QM/MM geometry optimization and potential energy surface scan, protein residues and water molecules found within 8 Å from the substrate molecule were allowed to move. Minima and saddle points were designated according to the vibrational analysis. Transition states were confirmed by the existence of one negative eigenvalue (one imaginary frequency), with the vibrational vector leading to the desired neighbouring minima. In addition, each transition state was connected to the neighbouring ground states by IRC calculations.
Results and discussion
The reaction mechanism was determined for two systems of different complexities: (a) model (CH3CO–Gly–NHCH3 bound into the model of the h.DPP III active site), and (b) real (the hydrated enzyme–Leu-enkephalin complex) system.Reaction mechanism determined for the model systems
The reaction mechanism was investigated using two model systems, with His568 in its neutral (proton on Nε atom) and protonated state. Present calculations revealed that 100% conserved His568 residue is in its protonated state during the hydrolysis. Namely, the amide bond hydrolysis was shown to be energetically more feasible in the model system with protonated His568. Therefore this mechanism is discussed in detail further in the text, while the reaction mechanism determined with neutral His568 is given in the ESI† (Fig. S5–S7, Tables S1 and S2).Details of the amide bond hydrolysis mechanism determined using the model system with protonated His568 are summarized in Fig. 2, with the energy profile for individual reaction steps illustrated in Fig. 3a and Table 2. The three-dimensional structures of stationary points are shown in Fig. S2 (ESI†). The selected bond lengths in the optimized structures are given in Table 1.
 | ||
| Fig. 2 Reaction mechanism for the amide bond breakdown determined using the model system (CH3CO–Gly–NHCH3 bound into the model of the h.DPP III active site) with His568 in its protonated state (for clarity His568 and Tyr318 are not shown). Calculations were performed at the B97D/[6-31G(d)+LanL2DZ-ECP] level of theory. ES denotes the enzyme–substrate complex, TS1-4 denote the transition states, INT1-3 denote the intermediates and P and P′ denote the products. Hydrogen bonds and bonds that are either broken or formed during hydrolysis are depicted as dashed or dotted black lines, respectively. Covalent and coordinative bonds are depicted as solid black lines. | ||
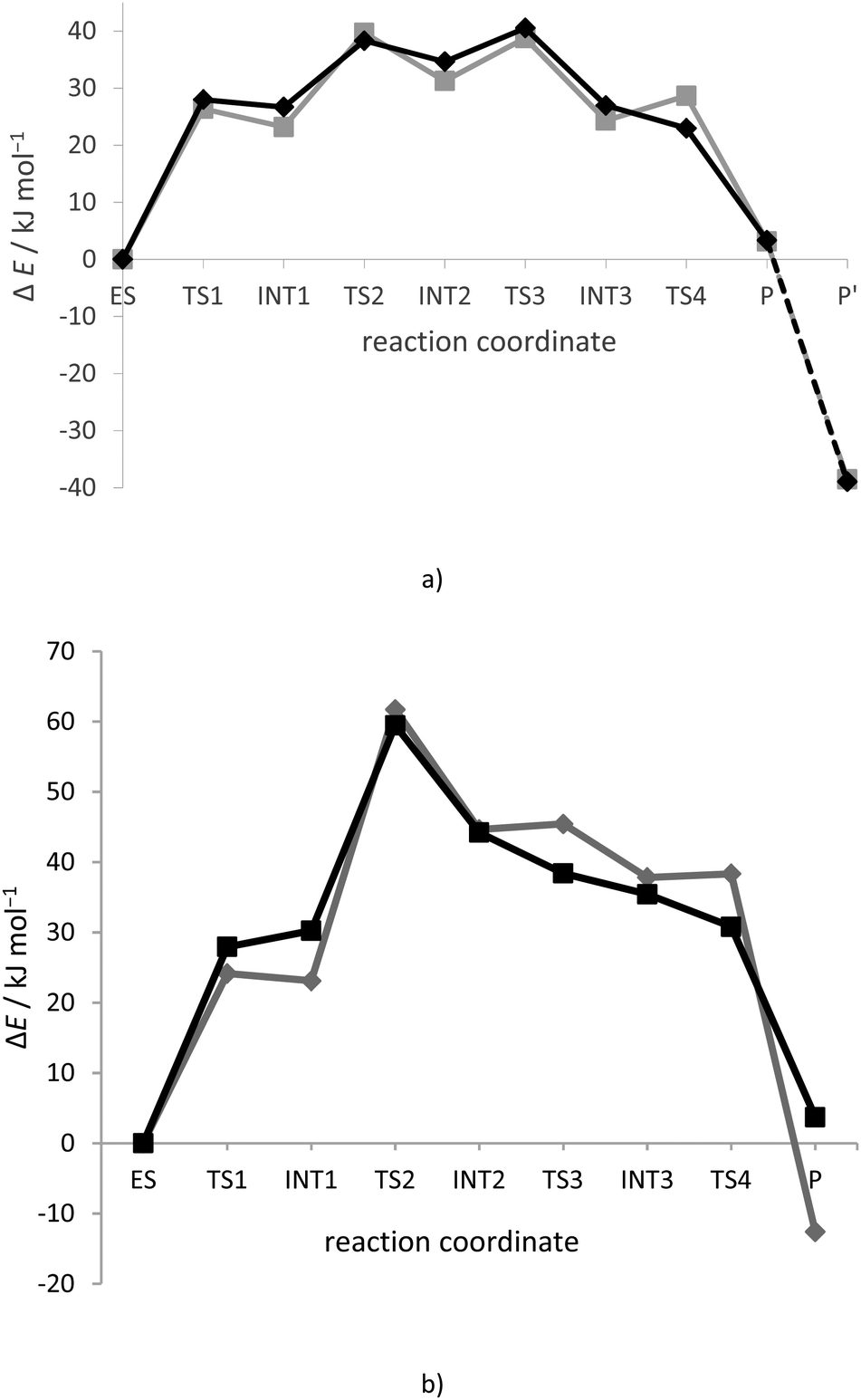 | ||
| Fig. 3 Energy profile for: (a) breakdown of the amide bond in CH3CO–Gly–NHCH3 bound into the model of the h.DPP III active site (in the presence of a solvent with ε = 4), and (b) Leu-enkephalin hydrolysis in the active site of h.DPP III. Calculations were performed at: B97D/[6-31G(d)+LanL2DZ-ECP] (gray rhombus and line) and B97D/[6-31G(d)+LanL2DZ-ECP] + ZPVEB97D/[6-31G(d)+LanL2DZ-ECP] (black squares and line) levels of theories, with His568 in its protonated form. | ||
| ES | TS1 | INT1 | TS2 | INT2 | TS3 | INT3 | TS4 | P | P′ | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Distances between the zinc ion and either nitrogen (Nδ) or oxygen (carboxyl) atoms from the histidine or glutamate amino acid residue, respectively. b Distance between the Tyr318 hydroxyl oxygen and amide hydrogen of the second amino acid residue from the substrate (sub) N terminus. c C and C′ are carbon atoms adjacent to Ns and Cs, respectively, while Hs is a hydrogen bonded to Ns, see Fig. S3 (ESI). | ||||||||||
| d(Os–Zn)/Å | 2.42 | 2.09 | 2.03 | 2.04 | 2.02 | 2.05 | 2.06 | 2.07 | 2.12 | 4.17 |
| d(Ow–Zn)/Å | 2.06 | 2.18 | 2.36 | 2.26 | 2.40 | 2.26 | 2.25 | 2.26 | 2.36 | 2.01 |
| d(Cs–Ns)/Å | 1.34 | 1.39 | 1.43 | 1.40 | 1.45 | 1.46 | 1.50 | 1.55 | 2.76 | 4.15 |
| d(Ow–Cs)/Å | 3.23 | 1.79 | 1.55 | 1.60 | 1.49 | 1.48 | 1.44 | 1.42 | 1.27 | 1.28 |
| d(Os–Cs)/Å | 1.26 | 1.32 | 1.36 | 1.36 | 1.38 | 1.38 | 1.37 | 1.35 | 1.30 | 1.27 |
| d(Ow–Hw2)/Å | 0.98 | 0.99 | 1.00 | 1.00 | 1.00 | 0.99 | 1.00 | 1.02 | 1.67 | 3.66 |
| d(Ow–Hw1)/Å | 1.09 | 1.58 | 1.67 | 1.65 | 1.71 | 2.11 | 2.60 | 2.49 | 2.69 | 5.05 |
| d(Hw2–Oe2)/Å | 2.82 | 2.83 | 2.84 | 2.76 | 2.74 | 2.50 | 1.71 | 1.60 | 1.01 | 1.06 |
| d(Hw1–Oe1)/Å | 1.41 | 1.03 | 1.01 | 1.02 | 1.01 | 0.99 | 1.06 | 1.24 | 2.22 | 2.82 |
| d(Ns–Hw1)/Å | 5.07 | 2.95 | 2.96 | 2.72 | 2.76 | 2.19 | 1.61 | 1.30 | 1.02 | 1.02 |
| d(Ns–Hw2)/Å | 4.66 | 3.19 | 3.10 | 3.07 | 3.01 | 2.92 | 2.59 | 2.60 | 3.59 | 1.59 |
| d(H450–Zn)a/Å | 2.21 | 2.17 | 2.15 | 2.15 | 2.14 | 2.14 | 2.16 | 2.15 | 2.11 | 2.13 |
| d(H455–Zn)a/Å | 2.15 | 2.11 | 2.11 | 2.11 | 2.11 | 2.11 | 2.12 | 2.12 | 2.11 | 2.11 |
| d(E508–Zn)a/Å | 2.11 | 2.09 | 2.08 | 2.08 | 2.07 | 2.06 | 2.05 | 2.05 | 2.04 | 2.05 |
| d(H568[Hε]–Os)/Å | 1.68 | 1.53 | 1.45 | 1.46 | 1.42 | 1.43 | 1.49 | 1.53 | 1.66 | 1.60 |
| d(H568[Nε–Hε])/Å | 1.05 | 1.08 | 1.10 | 1.10 | 1.12 | 1.11 | 1.09 | 1.08 | 1.05 | 1.08 |
| d(Y318–sup)b/Å | 1.82 | 1.84 | 1.83 | 1.84 | 1.84 | 1.87 | 1.95 | 1.95 | 1.94 | 1.96 |
| ω 1(C–Ns–Cs–Hs)c/° | 175.6 | −133.5 | −125.6 | 163.9 | 122.1 | 124.9 | 122.7 | 121.5 | ||
| ω 2(C′–Cs–Ns–Os)c/° | −178.6 | 137.6 | 126.5 | 131.1 | 126.3 | 127.1 | 125.9 | 125.0 | ||
Model of the enzyme–substrate complex
In the optimized structure of the model of the enzyme–substrate complex (ES) five ligands (His450, His455 and Glu508, water and the substrate) coordinate with the zinc ion; however, the 2.42 Å long Os–Zn distance indicates the reduced contribution of the Os atom to the zinc coordination.The zinc-coordinated water is polarized by a negatively charged Glu451 (strong Oe1–Hw1–Ow hydrogen bond), which is a strong base, and by the zinc cation (2.06 Å long Zn–Ow coordination bond). Consequently, the Hw1 proton is partially shared between Glu451 and water as indicated by Oe1–Hw1 and Ow–Hw1 distances of 1.41 Å and 1.09 Å, respectively. Apparently the pKa value of the metal bound water molecule is lower than that of the one in solution. It should be noticed that the water Hw2 proton makes an additional hydrogen bond with the substrate terminal acetyl group. Besides by its coordination to the metal ion, the substrate is further stabilized by hydrogen bonds with evolutionally conserved His568 and Tyr318. Tyr318 is additionally hydrogen bonded to Glu508 and this hydrogen bond is preserved throughout the reaction. A similar substrate-stabilization pattern is observed in the active site of thermolysin.16
Glu451-assisted water addition
The hydrolysis reaction is initiated via deprotonation of the water molecule by Glu451 whilst the emerging hydroxyl group approaches the amide carbon (Cs) atom. The strong hydrogen bond between His568 and substrate Os oxygen in the model of the enzyme–substrate complex (ES) facilitates nucleophile attack of the activated water molecule by dragging the negative charge away from the Cs atom and by positioning the carbonyl group for the attack (i.e. the plane of the peptide bond is perpendicular to the direction of OH− attack). This reaction step has the highest activation energy of 28 kJ mol−1. Already in the TS1 structure Glu451 is protonated and the planarity of the peptide bond is distorted as indicated by the carbon and nitrogen pyramidalization (see improper dihedral angles ω2 and ω1 in Table 1, respectively, and Fig. S3, ESI†). The advancing formation of the tetrahedral oxy-anion in the TS1 and INT1 structures is stabilized by substrate coordination to the metal dication (the Os–Zn distance is reduced by ∼0.4 Å as compared to the ES structure) and the strong hydrogen bond with His568 (Os–His568 distances of 1.53 and 1.45 Å, in TS1 and INT1 structures, respectively). The Ow–Cs bond formation system relaxes in the first intermediate structure, INT1 (distance Ow–Cs reduces from 3.23, 1.79 to 1.55 Å, as the system evolves from the ES and TS1 to INT1 structure, respectively).Once the tetrahedral intermediate is formed (INT1), it can either lose the –NH2CH3 group (giving the hydrolytic product) or the –OH group (reverting to the starting compound). Deslongchamps32,33 has proposed that one of the factors affecting this choice is the conformation of the intermediate; more specifically, the position of the lone pairs. In this view, a leaving group –NH2CH3 or –OH can depart only if each of the remaining heteroatoms bound to the carbon (Cs) atom has an orbital antiperiplanar to the breaking bond, either Cs–Ns or Cs–Ow. This factor was called the stereoelectronic control or effect. From the principle of microscopic reversibility it follows that the lone-pair orbitals on Os and Ns atoms upon OH− nucleophilic attack must be oriented antiperiplanar to the new Cs–Ow bond, i.e. initial pyramidalization on the Ns atom (INT1 structure) with the lone pair pointing away from the Glu451 is influenced by the direction of OH− attack.
N-inversion
To become a good leaving group, the nitrogen (Ns) of –NH2CH3 has to accept protons from Glu451 prior to or at least during the Cs–Ns bond cleavage. Inspection of the tetrahedral intermediate (INT1) structure reveals that the configuration of the leaving nitrogen is not suitable for this proton transfer since its lone pair orbital, which should accept the proton, points away from Glu451. So, the inversion of the absolute configuration of the Ns atom (so-called N-inversion) that would orient its lone pair towards protonated Glu451 is necessary. Inversion of the pyramidal nitrogen atoms carrying only hydrogen or carbon substituents is normally a very rapid process.34 For example the inversion energies determined for ammonia and trimethylamine at 300 K are about 24 kJ mol−1 and 34.7 kJ mol−1, respectively.34,35The N-inversion from the (R) to (S) isomer through the near planar transition state (ω1 = 163.9° in TS2 structure) occurs in the second reaction step with an activation barrier of 11.6 kJ mol−1. The improper torsion angle ω1, defining the degree of nitrogen pyramidalization, changes by more than 110° as the system evolves from the INT1 to INT2 structure. It should be noticed that the structures INT1 and INT2 differ only in the lone pair position at Ns, namely, the N-inversion (the change in the Ns configuration from (R) to (S)) leads to the new tetrahedral intermediate in which the positions of the N–H bond and the lone pair orbital are interchanged. According to the energy profile, the (R) isomer (INT1) is about 8 kJ mol−1 more stable than the (S) isomer (INT2). Apparently the nitrogen in the INT2 structure can easily surmount the low energy barrier (3.7 kJ mol−1) and interconvert back to the (R) isomer (INT2 → INT1 transition). Instead, the formation of the hydrogen bond between the Ns lone pair and the Glu451 Ow–Hw1 group “locks” Ns in (S) configuration (INT2 → INT3 transition). The energy barrier for this transition is 5.9 kJ mol−1. Once the Ns–Glu451 hydrogen bond is formed, the reverse reaction is hindered. Namely, the energy barrier of 13.5 kJ mol−1 is much higher for the reverse (INT3 → INT2 transition) than for the forward (INT3 → P transition) reaction which is barrierless (Fig. 3a). Furthermore, the lone pair orbitals at Ns and Os are not antiperiplanar to the Cs–Ow bond, and therefore, according to the stereoelectronic effect, this reaction is not feasible. In summary, for the reaction determined using the model system, TS3 is the point from which the reaction can proceed only in the forward direction, i.e. from which the reverse reaction becomes unfavourable. The first three reaction steps can be considered as rate-determining steps with a total activation energy of 45.5 kJ mol−1 (obtained as a sum of 28 kJ mol−1 + 11.6 kJ mol−1 + 5.9 kJ mol−1). During the third reaction step (INT2 → INT3 transition) the hydroxyl group (Ow–Hw2) bound to the substrate reoriented in a way to form a hydrogen bond with the Glu451 (Ow–Hw2–Oe2 hydrogen bond in the INT3 structure). Only at this point, after establishing strong Ow–Hw2–Oe2 and Ns–Hw1–Oe1 hydrogen bonds, does the (S) isomer in the INT3 structure become equally stable as the (R) isomer in the INT1 structure.
In INT1-3 structures, the amide bond has a single bond character as indicated by the Cs–Ns distance (1.43, 1.45 and 1.50 Å). Oxyanion intermediates (INT1-3) and transition states (TS2-3) are stabilized by the strong hydrogen bonding to His568 and Glu451 (Ow–Hw1–Oe1 hydrogen bond in INT1 and INT2 structures, and Ns–Hw1–Oe1 hydrogen bond in the INT3 structure), as well as by bidentate coordination to the zinc cation. Strong hydrogen bonding between the substrate and His568 is indicated by short Hε–Os distances (1.45, 1.42 and 1.49 Å in INT1, INT2 and INT3 structures, respectively), and consequently slightly extended His568 Nε to Hε atom distances (1.10, 1.12 and 1.09 Å in INT1, INT2 and INT3 structures, respectively).
The amide bond cleavage
Collapse of the INT3 structure into the product occurs relatively easily since the structural conditions are fulfilled. Firstly, the lone pair orbital on the Ns atom should accept the proton to make nitrogen a good leaving group oriented toward the Oe1–Hw1 group of the Glu451. Secondly, the substrate is transformed into a conformation that according to Deslongchamps allows the cleavage of the Cs–Ns bond (Os and Ow bound to Cs have lone pair orbitals antiperiplanar to the scissile Cs–Ns bond). Namely, one of the three lone pair orbitals on Os oxygen always occupies the required position, while for the Ow oxygen atom this was enabled after the Ow–Hw2 group reorientation in a way to form a hydrogen bond with Glu451.Therefore, the fourth reaction step, during which the system relaxes into the final product (P), consists of: transfer of the proton Hw1 from Glu451 to leaving nitrogen (Ns), followed by cleavage of the amide bond and regeneration of the protonated state of the carboxyl of Glu451 by the second Hw2 proton from the Ow atom. One can notice that upon making a zero-point correction to electronic energy, the energy of TS4 structures becomes lower than the energy of the INT3 structure suggesting that the system more or less continuously, without energy barrier, relaxes from the INT3 structure into the product P.
The final optimized product complex P has a bidentate bond between the zinc and the acetate part of the product of the hydrolysis (although the Ow–Zn coordination bond of 2.36 Å is slightly long). In addition, the acetate part of the product is stabilized by the strong hydrogen bond with His568. The methylamine part of the product is only weakly bound to the model of the enzyme active site via a single hydrogen bond to the Glu451 Oe1 oxygen (with the Oe1–Hw1 distance equal to 2.22 Å) and weak van der Waals interactions with His450 and His568.
As suggested for thermolysin17 the second proton (Hw2) transfer by Glu451 towards the Ns nitrogen could probably be considered as a next step of the product conversion, although this step is not crucial for peptide bond cleavage. We were not able to locate protonated amine as a final product. Instead, the product P′, where the Hw1 proton is placed on Glu451, and where Zn2+ is tetrahedrally coordinated (with one coordination to acetate part of the product), was found to be the most stable product structure.
Enzymatic hydrolysis
The mechanism for peptide (Leu-enkephalin, Fig. 4) hydrolysis by h.DPP III was investigated considering two slightly different initial ES geometries, one obtained directly from MD simulations and the other constructed (as described above). We succeeded to obtain a reaction mechanism for both these geometries; however, the reaction profiles are slightly different. The energy barriers determined for the reaction mechanism in the latter are lower than those determined for the reaction obtained using the MD snapshot. Hereinafter we focus on the more efficient reaction mechanism while the results obtained using the system taken from MD simulations are given in the ESI.†The mechanism for the peptide bond hydrolysis obtained for the hydrated enzyme is summarized in Fig. 5, with the energy profile for individual reaction steps given in Fig. 3b and Table 2. Three-dimensional structures of the energy optimized stationary points are shown in Fig. S4 (ESI†), and the selected bond lengths in these structures are given in Table 3.
| ΔE (model)/kJ mol−1 | ΔE (real)/kJ mol−1 | ||||
|---|---|---|---|---|---|
| A | B | A | B | ||
| ES | 0.00 | 0.00 | ES | 0.00 | 0.00 |
| TS1 | 26.38 | 27.99 | TS1 | 24.16 | 27.94 |
| INT1 | 23.19 | 26.68 | INT1 | 23.11 | 30.25 |
| TS2 | 39.66 | 38.30 | TS2 | 61.72 | 59.48 |
| INT2 | 31.24 | 34.62 | INT2 | 44.65 | 44.22 |
| TS3 | 38.74 | 40.52 | TS3 | 45.44 | 38.39 |
| INT3 | 24.32 | 26.96 | INT3 | 37.82 | 35.44 |
| TS4 | 28.64 | 22.97 | TS4 | 38.35 | 30.80 |
| P | 3.13 | 3.32 | P | −12.61 | 3.71 |
| P′ | −38.51 | −38.94 | |||
| ES | TS1 | INT1 | TS2 | INT2 | TS3 | INT3 | TS4 | Pd | P′d | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Distances between the zinc ion and either nitrogen (Nδ) or oxygen (carboxyl) atoms from the histidine or glutamate amino acid residue, respectively. b Distance between the Tyr318 hydroxyl oxygen and amide hydrogen of the second amino acid residue from the substrate (sub) N terminus. c C and C′ are carbon atoms adjacent to Ns and Cs, respectively, while Hs is a hydrogen bonded to Ns, see Fig. S3 (ESI). d Corresponds to the enzyme-product complex. | ||||||||||
| d(Os–Zn)/Å | 2.89 | 2.17 | 2.09 | 2.09 | 2.10 | 2.11 | 2.15 | 2.13 | 2.85 | |
| d(Ow–Zn)/Å | 2.02 | 2.15 | 2.26 | 2.17 | 2.16 | 2.16 | 2.14 | 2.15 | 2.03 | |
| d(Cs–Ns)/Å | 1.36 | 1.42 | 1.44 | 1.45 | 1.53 | 1.57 | 1.69 | 1.74 | 3.18 | |
| d(Ow–Cs)/Å | 2.56 | 1.74 | 1.58 | 1.52 | 1.46 | 1.45 | 1.41 | 1.39 | 1.31 | |
| d(Os–Cs)/Å | 1.24 | 1.31 | 1.33 | 1.34 | 1.34 | 1.33 | 1.32 | 1.31 | 1.24 | |
| d(Ow–Hw2)/Å | 0.99 | 0.99 | 0.99 | 0.98 | 1.00 | 1.01 | 1.07 | 1.14 | 1.69 | |
| d(Ow–Hw1)/Å | 1.07 | 1.56 | 1.61 | 2.10 | 2.53 | 2.48 | 2.41 | 2.42 | 3.58 | |
| d(Hw2–Oe2)/Å | 1.88 | 2.58 | 2.63 | 2.45 | 1.71 | 1.64 | 1.45 | 1.31 | 1.01 | |
| d(Hw1–Oe1)/Å | 1.45 | 1.03 | 1.02 | 1.00 | 1.08 | 1.18 | 1.57 | 1.64 | 3.56 | |
| d(Ns–Hw1)/Å | 2.78 | 2.83 | 2.80 | 1.95 | 1.53 | 1.34 | 1.08 | 1.06 | 1.03 | |
| d(Ns–Hw2)/Å | 3.87 | 3.21 | 3.12 | 2.92 | 2.54 | 2.55 | 2.64 | 2.72 | 4.46 | |
| d(H450–Zn)a/Å | 2.12 | 2.15 | 2.14 | 2.13 | 2.12 | 2.12 | 2.11 | 2.12 | 2.10 | |
| d(H455–Zn)a/Å | 2.12 | 2.15 | 2.15 | 2.14 | 2.14 | 2.14 | 2.13 | 2.14 | 2.14 | |
| d(E508–Zn)a/Å | 2.04 | 2.08 | 2.08 | 2.07 | 2.07 | 2.07 | 2.07 | 2.08 | 2.04 | |
| d(H568[Hε]–Os)/Å | 1.78 | 1.60 | 1.56 | 1.50 | 1.55 | 1.57 | 1.61 | 1.61 | 1.81 | |
| d(H568[Nε–Hε])/Å | 1.04 | 1.07 | 1.08 | 1.10 | 1.09 | 1.08 | 1.07 | 1.07 | 1.04 | |
| d(Y318–sub)b/Å | 2.30 | 2.29 | 2.30 | 2.27 | 2.15 | 2.15 | 2.20 | 2.21 | 2.47 | |
| ω 1(C–Ns–Cs–Hs)c/° | −151.5 | −132.0 | −128.3 | 158.1 | 130.1 | 127.9 | 128.0 | 127.9 | ||
| ω 2(C′–Cs–Ns–Os)c/° | 173.7 | 138.6 | 131.2 | 132.0 | 129.8 | 128.9 | 126.4 | 125.0 | ||
The Michaelis complex
In the active site of the optimized h.DPP III–Leu-enkephalin complex structure (ES) the zinc ion is tetrahedrally coordinated with His450, His455, Glu508 and the water molecule. The substrate carbonyl oxygen (Os), located 2.89 Å from the catalytic ion, interacts with it electrostatically (the same zinc-coordination geometry is found in the h.DPP III–Leu-enkephalin crystal structure, see Fig. S1, ESI†). The hydrogen bond network that bounds the substrate in a form of a β-strand in the crystal structure remained preserved during geometry optimization (see Fig. 4 and Fig. S1, ESI†). The completely buried N terminus of the bound peptide makes hydrogen bonds with Glu316, Asn394 and Ans391, while the carbonyl group of the substrate tyrosine residue is hydrogen bonded to Asn391. The third substrate residue from the N-terminus, Gly, makes hydrogen bonds with Ala388 and Gly389. Besides these hydrogen bonds, mostly with the protein β-core, the second substrate residue, Gly, makes hydrogen bonds with His568 (the His568–Os distance of 1.78 Å indicates that this is quite a strong hydrogen bond) and Tyr318 which also makes a hydrogen bond with Glu508. The zinc-bound water molecule (Ow–Zn distance of 2.02 Å) is additionally polarized by strong hydrogen bonding to Glu451 (distances Oe1–Hw1 and Ow–Hw1 of 1.45 Å and 1.07 Å indicate the partially delocalized nature of proton Hw1). | ||
| Fig. 4 The Michaelis complex of the h.DPP III and Leu-enkephalin (with carbon atoms coloured yellow) and amino acid residues that electrostatically interact with it (for clarity only either backbone or side chain atoms is shown with carbon atoms coloured green) are shown as sticks. The zinc ion is shown as a black sphere. Hydrogen bonds, as well as the electrostatic and substrate–Zn2+ interactions, are indicated by magenta dashed lines. | ||
 | ||
| Fig. 5 Reaction mechanism for the Glu451-assisted peptide bond hydrolysis in h.DPP III calculated at the B97D/[6-31G(d)+LANL2DZ-ECP] level of theory (for clarity His568 and Tyr318 are not shown). ES denotes the enzyme–substrate complex, TS1-4 denote the transition states, INT1-3 denote the intermediate structures and P denotes the product. Hydrogen bonds and bonds that are either broken or formed during the hydrolysis are depicted as dashed and dotted black lines, respectively. Covalent and coordinative bonds are depicted as solid lines. | ||
Antiparallel binding of the β-stranded substrate to the protein β-core influences mild initial pyramidalization on the Ns atom (ω1 = −151.5°) hydrogen bonded to Gly389.
Glu451-assisted water addition and change in the zinc coordination
Like in the reaction obtained using the model system, the concerted deprotonation of the water molecule by Glu451 and nucleophilic attack of the emerging hydroxyl group on the amide carbon (Cs) is the first reaction step with an equally high energy barrier (28 kJ mol−1). The Ow–Cs distance of 2.56 Å in ES gradually decreases to 1.74 Å in TS1, and to 1.58 Å in INT1 resulting in the formation of a new chemical bond. By establishing the zinc-substrate coordination (distance Zn–Os decreases from 2.89 Å in the ES structure, to 2.17 Å and 2.09 Å in the TS1 and INT1 structures, respectively), the zinc coordination number increases from 4 to 5 however, the zinc–water interaction weakens (the Zn–Ow distance increased from 2.02 Å in ES, to 2.15 Å and 2.26 Å in TS1 and INT1 structures, respectively). Such variability in zinc coordination was also reported for thermolysin16 and arises from its flexibility in bonding.36 Besides by coordination to the metal ion, the oxy-anion is stabilized by strong hydrogen bonding to protonated His568 in TS1 and INT1 structures. By system evolution from ES to INT1 the Hε(His568)–Os distance reduces from 1.78 Å, to 1.60 Å and 1.56 Å in ES, TS1 and INT1 structures, respectively, while the distance between His568 Nε and Hε increases from 1.04 Å to 1.08 Å and 1.09 Å, respectively, indicating the partially delocalized nature of the Hε proton between Nε and Os. Apparently, the transition state, TS1, structure closely resembles that of the intermediate, INT1, which is more tightly bound to the enzyme than the substrate. Similar findings were determined for thermolysin.16Nucleophilic attack of OH− on Cs is accompanied by pyramidalization on the carbon (Cs), see Table 2, resulting not only in the formation of the tetrahedral intermediate INT1, but also in the further pyramidalization of Ns. Namely, during the system evolution from ES to INT1, ω2 and ω1 dihedral angles reduce more than 40° and 20°, respectively. Due to the nitrogen Ns atom pyramidalization its lone pair electrons orient away from Glu451 in ES, TS1 and INT1 structures. As already emphasized in the discussion about the reaction obtained using the model system, the (R) isomer of Ns is stereoelectronically controlled by the direction of OH− attack. In the enzyme this control is further supported by the favourable hydrogen bonding to the protein, namely the Gly389 carbonyl group.
By adding the zero-point correction to electronic energy, TS1 becomes lower in energy than INT1 (Fig. 3b) suggesting that at this level of theory the TS1 structure cannot be considered as a real transition state.
Concerted N-inversion and hydrogen bonding to Glu451
To obtain a good leaving group in the h.DPP III peptide bond hydrolysis, the nitrogen atom (Ns) should accept the proton from Glu451. Since in the model system, the configuration of the leaving nitrogen in the tetrahedral intermediate (INT1) structure is not suitable for this proton transfer since the nitrogen lone pair orbital which should accept the proton points away from Glu451. Again, the “inside out” turnover of the amide nitrogen (N-inversion shown in Fig. 6) that enables the Hw1 proton transfer to Ns is the next reaction step with the activation energy of 29.2 kJ mol−1. During a system evolution from INT1 to INT2 the dihedral angle ω1 changes more than 100°. Simultaneously, the hydrogen bond between the substrate amide and the Gly389 carbonyl group weakens as the hydrogen bond angle decreases from 160° in INT1 to 145° and 135° in structures TS2 and INT2, respectively. Unlike in the model system, N-inversion is simultaneously followed by the formation of the hydrogen bond between the nitrogen (Ns) lone pair and the Glu451 Hw1–Oe1 group (TS2 in Fig. 6). Final relaxation of the system into the INT2 structure is accompanied by the Ow–Hw2 group reorientation and formation of the Ow–Hw2–Oe2 hydrogen bond (Fig. 6). | ||
| Fig. 6 Pyramidalization on the Ns nitrogen atom (blue opaque sphere) in INT1, TS2, to INT2 structures, wherein the first structure corresponds to the (R) and the latter two to (S) isomer on the nitrogen Ns atom. Edges of the triangular pyramid topped by the Ns atom are shown with dashed black lines. Selected hydrogen bond interactions are shown with yellow dashed lines. Distances are in Å. | ||
The energy cost for the system to have a favourable (S) isomer on the leaving nitrogen atom is higher in the enzyme (59.5 kJ mol−1) than in the model system (45.5 kJ mol−1). This is a result of the N-inversion step (as energies for the water addition step are equal) that costs 29.2 kJ mol−1 in the enzyme active site, while 17.5 kJ mol−1 in the model system. A difference of ∼12 kJ mol−1 is mostly an outcome of more hydrogen bonds being broken in the enzyme active site than in the model during transformation of the (R to S) isomer. In both real and model systems, by interconversion to the favourable (S) isomer on the Ns atom, two strong, Ow–Hw2–Oe2 and Ns–Hw1–Oe1, hydrogen bonds are being formed, while the one between the added Ow–Hw2 group and the substrate (carbonyl group from the first amino acid or the terminal acetyl group) disappears. Meanwhile in the enzyme active site transformation from the INT1 to INT2 structure leads to weakening of the hydrogen bond between the leaving amide group and Gly389, as well as the total loss of the hydrogen bond between the lone pair orbital on the leaving Ns atom and the water molecule found in its vicinity (see Fig. 6).
Hydrogen transfers and peptide bond cleavage
In the next reaction step proton Hw1 is transferred from Glu451 to the leaving nitrogen (Ns) atom. This leads to stretching of the Cs–Ns bond from 1.53 Å in the INT2 structure to 1.69 Å in the INT3 structure indicating the ionic character of the peptide bond in the latter structure. Afterwards, the proton Hw2 is transferred from the substrate molecule to the Glu451 carboxylate group and the peptide bond is broken (distance Cs–Ns increased to 3.81 Å in P). In the product P the carboxylate ion monodentately coordinates with the zinc ion, and the tetrahedral coordination of the zinc ion present in the ES structure is restored. It is interesting to notice that as in the ES complex, the metal ion is coordinated by Ow and not by the Os atom. The amine part of the product forms a hydrogen bond with Gly389 from the enzyme β-core and the water molecule found in its vicinity (the same one that made a hydrogen bond with it in the INT1 structure), while the carboxylate ion is stabilized via monodentate coordination to the metal ion and strong hydrogen bonds with His568 and Glu451. It should be noticed that the product carboxylate group is planar and that Glu451 is protonated.From the energetic point of view we can conclude that once the system passes the TS2 transition state it more or less continuously develops towards the final product P, i.e. once the configuration of the leaving nitrogen is suitable for proton transfer, the system relaxes in the final product without passing any energy barrier. This shows that forward reaction, leading to a final product, is even more preferred in real than in model systems, i.e. that reversible reaction is more difficult to appear in the enzyme active site than in a model system.
Calculations have shown that emerging negative charge on the Os atom in the rate-determine steps is mostly stabilized by coordination to the metal dication and by the strong hydrogen bonding with protonated His568 as indicated by the short His568–Os distances. This is in agreement with the experimental measurement that has shown a 1296-fold decrease in the kcat value after mutation of His568 to Asn. Kinetic data also suggested the importance of Tyr318 in enzymatic hydrolysis, i.e. Tyr318 to Phe mutation resulted in a 100-fold decrease in the kcat value. The presented results clearly indicate stabilization of the transition state by His568, and participation of Tyr318 in positioning of both the substrate and Glu508.
Conclusions
The mechanism of peptide hydrolysis by human DPP III was investigated in several systems of different complexities and protonation states of His568. Using the reduced model system reaction profiles were obtained with His568 in both protonation states, while using the hydrated enzyme–Leu-enkephalin complex two slightly different reaction profiles were obtained with this 100% conserved histidine in its protonated form.According to the calculations the reaction rate is determined by two consecutive steps, the nucleophilic attack and the nitrogen (Ns) inversion. While the nucleophilic attack (by Glu451-assisted deprotonated water molecule) energy barriers determined on the systems of different complexities are of the same height (28 kJ mol−1), the energy barriers for the N-inversion differ by 12 kJ mol−1. The reason for this difference is the larger number of hydrogen bonds that need to be broken during the N-inversion in the real system than in the model system. In order to properly position toward the Glu451 hydroxyl (Oe1–Hw1) and to enable the proton (Hw1) transfer, the scissile peptide bond nitrogen (Ns) has to interconvert. While unconstrained in the model system, this interconversion is hindered by two hydrogen bonds, Ns–Hs to Gly389 and Ns to H2O, in the enzyme. This is probably the reason why in a real system, the N-inversion step occurs in a concerted fashion followed by the hydrogen bonding to Glu451, and consequently why the interconversion immediately “locks” the system in configuration from which the backward reaction in more difficult. From the energetic point of view, once the system passes the TS2 transition state it almost barrierlessly develops towards the final product P. On the other hand, in the model system, one can observe equilibrium between the (R) and (S) isomers on the Ns atom, i.e. the substrate is more free to explore different conformations. To be more precise, from the INT2 structure, the model system can equally easy revert to ES or go forward to P passing over energy barriers of 3.7 and 5.9 kJ mol−1, respectively. Therefore, a greater possibility of reversible reaction leading to a starting compound is observed in the model system than in the real system.
These findings can also be explained by stereoelectronic theory. In the model system, in the INT2 structure, the NS atom has a favourable configuration for a Hw1 proton transfer (the lone pair on the Ns atom is pointing toward Glu451, although the Ns–Hw1 distance of 2.76 Å is still quite long for proton transfer to occur) but there is no lone pair orbital on the Ow atom antiperiplanar to the C–N bond to allow its cleavage. So, from this point the system can easily transform into an ES complex. Only after the Ow–Hw2–Oe2 hydrogen bond is formed, orientation of the lone pair on the Ow atom becomes antiperiplanar to the C–N bond, and the Ns–Hw1 distance reduces to 1.61 Å. This transformation “locks” the system in a configuration where the reversion to the enzyme–substrate complex is not favourable. Moreover, the lone pair orbitals antiperiplanar to the newly formed Cs–Ow bond (needed for the reversible reaction) do not exist at Ns and Os atoms. Therefore only after the favourable hydrogen bond interactions between the substrate and Glu451 are established, the equilibrium between two tetrahedral intermediates is likely to be largely on the side of the (S) isomer than the (R) isomer in the model system.
On the other hand in the enzyme active site, water addition and reorientation of the lone pair orbitals on Ns (N-inversion) and Ow atoms occur in a concerted fashion. Once the system is transformed into the favourable (S) configuration at the nitrogen, favourable hydrogen bonds with the Glu451 are formed that according to the stereoelectronic principle allow the C–N bond breakage. Thus one can say that the functional significance of the enzyme lies in the protein-controlled suppression of the reverse reaction.
Although most of the reaction steps that define the mechanism of action of thermolysin and neprilysin are similar to the ones determined for human DPP III, the importance of the nitrogen inversion was not reported in their case. However, we must emphasize that the importance of the N-inversion step was already found in catalytic pathways of aspartic peptidases.38
The QM/MM calculations on the enzyme–substrate complex carried out in this work indicate that the substrate carbonyl oxygen atom is not coordinated to the zinc ion in the Michaelis complex. Namely, the Zn2+ ion is tetrahedrally coordinated by His450, His455, Glu508 and water molecules. In the model system, the zinc coordination number is five since the substrate molecule also coordinates with the zinc ion, although rather weakly. Stabilization of the oxy-anion transition state is achieved through the formation of the strong coordination bond between the substrate carbonyl oxygen and the zinc ion as well as by its hydrogen bonding to the protonated His568. This suggests that both the transition state for nucleophilic attack and the oxy-anion intermediate are more tightly bound to the enzyme than the substrate itself.
Kinetic data obtained for the Leu-enkephalin hydrolysis by human DPP III are in a good agreement with our QM/MM calculations. Moreover, those obtained for His568 and Tyr318 DPP III mutants can be well explained by the results presented in this paper as well. A 1296-fold decrease in the kcat value after mutation of His568 to Asn is a consequence of direct stabilization of the transition state by histidine, while a 100-fold decrease in the kcat value after mutation of Tyr318 to Phe shows participation of tyrosine in positioning of both the substrate and Glu508.
Acknowledgements
This work has been supported by the Croatian Science Foundation under the project “7235 Flexibility, activity and structure correlations in the dipeptidyl peptidase III family”, the Croatian National Grid Infrastructure (CRO NGI, http:// www.cro-ngi.hr/) and computational cluster Isabella (http://www.srce.unizg.hr/en/isabella/).Notes and references
- J. M. Chen and A. J. Barrett, in Handbook of Proteolytic Enzymes, ed. A. J. Barrett, N. D. Rawlings and J. F. Woessner, Elsevier, Academic Press, London, 2nd edn, 2004, pp. 809–812 Search PubMed.
- K. Fukasawa, K. M. Fukasawa, H. Iwamoto, J. Hirose and M. Harada, Biochemistry, 1999, 38, 8299–8303 CrossRef CAS PubMed.
- T. Chiba, Y.-H. Li, T. Yamane, O. Ogikubo, M. Fukuoka, R. Arai, S. Takahashi, T. Ohtsuka, I. Ohkubo and N. Matsui, Peptides, 2003, 24, 773–778 CrossRef CAS.
- M. Barsun, N. Jajcanin, B. Vukelić, J. Spoljarić and M. Abramić, Biol. Chem., 2007, 388, 343–348 CrossRef CAS PubMed.
- Y. Liu, J. T. Kern, J. R. Walker, J. A. Johnson, P. G. Schultz and H. Luesch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5205–5210 CrossRef CAS PubMed.
- H. Zhan, Y. Yamamoto, S. Shumiya, M. Kunimatsu, K. Nishi, I. Ohkubo and K. Kani, Histochem. J., 2001, 33, 511–521 CrossRef CAS PubMed.
- S. Simaga, D. Babić, M. Osmak, M. Sprem and M. Abramić, Gynecol. Oncol., 2003, 91, 194–200 CrossRef CAS PubMed.
- V. A. Meliopoulos, L. E. Andersen, P. Brooks, X. Yan, A. Bakre, J. K. Coleman, S. M. Tompkins and R. A. Tripp, PLoS One, 2012, 7, e37169 CAS.
- M. He, D. P. Mangiameli, S. Kachala, K. Hunter, J. Gillespie, X. Bian, H.-C. J. Shen and S. K. Libutti, Clin. Cancer Res., 2010, 16, 249–259 CrossRef CAS PubMed.
- B. E. Hast, D. Goldfarb, K. M. Mulvaney, M. A. Hast, P. F. Siesser, F. Yan, D. N. Hayes and M. B. Major, Cancer Res., 2013, 73, 2199–2210 CrossRef CAS PubMed.
- A. Tomić, M. González and S. Tomić, J. Chem. Inf. Model., 2012, 52, 1583–1594 CrossRef PubMed.
- A. Tomić and S. Tomić, Dalton Trans., 2014, 43, 15503–15514 RSC.
- G. A. Bezerra, E. Dobrovetsky, R. Viertlmayr, A. Dong, A. Binter, M. Abramic, P. Macheroux, S. Dhe-Paganon and K. Gruber, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6525–6530 CrossRef CAS PubMed.
- P. Kumar, V. Reithofer, M. Reisinger, S. Wallner, T. Pavkov-Keller, P. Macheroux and K. Gruber, Sci. Rep., 2016, 6, 23787 CrossRef CAS PubMed.
- P. K. Baral, N. Jajcanin-Jozić, S. Deller, P. Macheroux, M. Abramić and K. Gruber, J. Biol. Chem., 2008, 283, 22316–22324 CrossRef CAS PubMed.
- J. Blumberger, G. Lamoureux and M. L. Klein, J. Chem. Theory Comput., 2007, 3, 1837–1850 CrossRef CAS PubMed.
- V. Pelmenschikov, M. R. A. Blomberg and P. E. M. Siegbahn, J. Biol. Inorg. Chem., 2002, 7, 284–298 CrossRef CAS PubMed.
- A. Tomić, M. Abramić, J. Spoljarić, D. Agić, D. M. Smith and S. Tomić, J. Mol. Recognit., 2011, 24, 804–814 CrossRef PubMed.
- B. Salopek-Sondi, B. Vukelić, J. Spoljarić, S. Simaga, D. Vujaklija, J. Makarević, N. Jajcanin and M. Abramić, Biol. Chem., 2008, 389, 163–167 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- C. Peng and H. Bernhard Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- H. Bernhard Schlegel, Theor. Chim. Acta, 1984, 66, 333–340 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- T. Vreven, K. Morokuma, O. Farkas, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 2003, 24, 760–769 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. a. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- P. Deslongchamps, Heterocycles, 1977, 7, 1271–1317 CrossRef CAS.
- P. Deslongchamps, Tetrahedron, 1975, 31, 2463–2490 CrossRef CAS.
- J. D. Swalen and J. A. Ibers, J. Chem. Phys., 1962, 36, 1914 CrossRef.
- A. M. Halpern, M. J. Ondrechen and L. D. Ziegler, J. Am. Chem. Soc., 1986, 108, 3907–3912 CrossRef CAS.
- A. G. Daniel and N. P. Farrell, Metallomics: integrated biometal science, 2014, 6, 2230–2241 RSC.
- S. Hay and N. S. Scrutton, Nat. Chem., 2012, 4, 161–168 CrossRef CAS PubMed.
- A. Barrett, N. Rawlings and J. Woessner, Handbook of Proteolytic Enzymes, Academic Press, 3rd edn, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: QM and QM/MM calculations of the systems with neutral His568, and QM/MM calculations of the system obtained directly from MD simulations. See DOI: 10.1039/c6cp04580d |
| This journal is © the Owner Societies 2016 |