
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of LiH on the rehydrogenation behavior of halide free rare earth (RE) borohydrides (RE = Pr, Er)†
Michael
Heere
a,
Seyed Hosein
Payandeh GharibDoust
b,
Christoph
Frommen
a,
Terry D.
Humphries
ac,
Morten B.
Ley
bd,
Magnus H.
Sørby
a,
Torben R.
Jensen
 b and
Bjørn C.
Hauback
*a
b and
Bjørn C.
Hauback
*a
aPhysics Department, Institute for Energy Technology, NO-2027 Kjeller, Norway. E-mail: Bjorn.Hauback@ife.no
bInterdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, University of Århus, Langelandsgade 140, DK-8000 Århus C, Denmark
cDepartment of Physics and Astronomy, Fuels and Energy Technology Institute, Curtin University, GPO Box U1987, Perth, 6845 WA, Australia
dMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm Platz 1, DE-45470 Mülheim/Ruhr, Germany
First published on 12th August 2016
Abstract
Rare earth (RE) metal borohydrides are receiving immense consideration as possible hydrogen storage materials and solid-state Li-ion conductors. In this study, halide free Er(BH4)3 and Pr(BH4)3 have been successfully synthesized for the first time by the combination of mechanochemical milling and/or wet chemistry. Rietveld refinement of Er(BH4)3 confirmed the formation of two different Er(BH4)3 polymorphs: α-Er(BH4)3 with space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , a = 10.76796(5) Å, and β-Er(BH4)3 in Pmm with a = 5.4664(1) Å. A variety of Pr(BH4)3 phases were found after extraction with diethyl ether: α-Pr(BH4)3 in Pa with a = 11.2465(1) Å, β-Pr(BH4)3 in Pmm with a = 5.716(2) Å and LiPr(BH4)3Cl in I
, a = 10.76796(5) Å, and β-Er(BH4)3 in Pmm with a = 5.4664(1) Å. A variety of Pr(BH4)3 phases were found after extraction with diethyl ether: α-Pr(BH4)3 in Pa with a = 11.2465(1) Å, β-Pr(BH4)3 in Pmm with a = 5.716(2) Å and LiPr(BH4)3Cl in I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, a = 11.5468(3) Å. Almost phase pure α-Pr(BH4)3 in Pa with a = 11.2473(2) Å was also synthesized. The thermal decomposition of Er(BH4)3 and Pr(BH4)3 proceeded without the formation of crystalline products. Rehydrogenation, as such, was not successful. However, addition of LiH promoted the rehydrogenation of RE hydride phases and LiBH4 from the decomposed RE(BH4)3 samples.
3m, a = 11.5468(3) Å. Almost phase pure α-Pr(BH4)3 in Pa with a = 11.2473(2) Å was also synthesized. The thermal decomposition of Er(BH4)3 and Pr(BH4)3 proceeded without the formation of crystalline products. Rehydrogenation, as such, was not successful. However, addition of LiH promoted the rehydrogenation of RE hydride phases and LiBH4 from the decomposed RE(BH4)3 samples.
Introduction
In recent years, rare earth (RE) metal borohydrides have received increasing interest on a variety of research frontiers. Firstly, the hydrogen capacities of these materials, which vary between 9.0 wt% H2 for Y(BH4)3 to 5.5 wt% H2 for Yb(BH4)3, are highly comparable to the more established hydrogen storage materials, such as MgH2 (7.6 wt% H2) or NaAlH4 (5.4 wt% H2).1–15 Secondly, RE borohydrides are also being pursued for other technological applications. For instance, their electrochemical properties have been investigated for solid state electrolytes in new battery applications,16–19 novel optical and magnetic properties have also been identified.20–22The established procedure for synthesizing RE borohydrides is the ‘solvent-free’ approach, where a RE-chloride and an alkali metal borohydride (Li, Na, K) are reacted in a one-step mechanochemical synthesis. LiBH4 has, so far, proven to be the most efficient precursor during these reactions,1–12,16,17,23 while attempts with other metal borohydrides, e.g. NaBH4, generally offer poor yields at the most. This is perfectly illustrated in the recently reported synthesis of Er(BH4)3.24 Meanwhile, the solvent based synthesis of metal borohydrides has been employed for over 50 years.25 The advantage of this method is the ability to remove the byproducts (e.g. LiCl) to yield almost pure RE borohydride products, and thus allowing for the accurate determination of their physical properties.1,14,16,26 The detriment of the solvent based synthesis is the possible decomposition of the RE borohydride upon removal of the solvent.27 On the contrary, mechanochemical synthesis does allow for the facile synthesis of these materials without complicated in vacuo manipulations and possible decomposition of the desired product.25
A number of RE borohydrides (RE = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Er, Yb, Lu) have been extensively characterized by in situ synchrotron radiation powder X-ray diffraction (SR-PXD), thermal analysis and vibrational spectroscopy.2,13,14,24 In general, the materials can be categorized into several groups. RE = La, Ce and Nd form LiRE(BH4)3Cl complexes crystallizing in space group I3m. In a recent study, Olsen et al.13 demonstrated that mechanochemical manipulation of PrCl3 and LiBH4 only yields the LiPr(BH4)3Cl complex. RE = Sm, Gd, Tb, Er and Yb form the corresponding RE(BH4)3, crystallizing in the space group Pa, which at high temperatures undergo a phase transition to form polymorphs in the space group Fmc. RE = Yb and Lu form [RE(BH4)4]− complex anions, which are stabilized by Li+ cations and are determined to crystallize in the space group P2c. Incidentally, the structures of the [RE(BH4)4]− complex anions are analogous to ASc(BH4)4 (A = Li, Na, K).15,28–30 Furthermore, RE borohydrides (RE = Sm, Gd) react with LiCl to form LiRE(BH4)3Cl complexes upon heating,13 while trivalent RE = Sm and Eu in LiRE(BH4)3Cl are reduced upon heating to divalent Sm and Eu (RE(BH4)2 (space group Pbcn)).14
The majority of the previous studies have been conducted on product mixtures containing both metal borohydrides and alkali metal chlorides, in most cases LiCl, making it virtually impossible to determine the specific effect of the halide salt on the pure metal borohydride. This is particularly related to reversible hydrogen sorption. For example, Er(BH4)3 was first synthesized from ErCl3 and LiBH4 by mechanochemical synthesis.24 Thermal decomposition of the Er(BH4)3/LiCl product exhibited a hydrogen desorption of 3.4 wt%.24 Rehydrogenation was achieved during absorption experiments applying p(H2) = 60 bar and T = 400 °C. Although the hydrogenation products were not determined in detail, it was stated that “Er(BH4)3 shows partial reversibility and reabsorbs about 20% of its original hydrogen content”.24 The present study illustrates that it is, surprisingly, the influence of LiH rather than LiCl or Li-ion which is responsible for reversible hydrogenation under the employed conditions.
In this study, Er(BH4)3 and Pr(BH4)3 have been synthesized halide free for the first time by mechanochemical and/or solvent techniques. The decomposition of both materials has been studied by in situ SR-PXD and the products and intermediates identified and their crystal structures determined by the Rietveld method. Moreover, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), as well as mass spectrometry (MS) have been conducted. Finally, we have investigated the rehydrogenation behavior of RE(BH4)3 and decomposed RE(BH4)3 with addition of LiCl and/or LiH.
Experimental
Sample preparation
LiBH4 (95%), ErCl3 (99.9%), PrCl3 (99.99%), dimethyl sulfide (S(CH3)2, anhydrous, 99.9%), diethyl ether ((C2H5)2O, anhydrous, 99.7%) and toluene (C6H5CH3, anhydrous, 99.8%) were purchased from Sigma Aldrich and used as received. Generally, a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 RECl3:LiBH4 were reacted. The individual sample descriptions of the RE borohydrides analyzed in this study are presented in Table 1, including their reaction products, composition and lattice parameters determined by Rietveld refinements.
:3 RECl3:LiBH4 were reacted. The individual sample descriptions of the RE borohydrides analyzed in this study are presented in Table 1, including their reaction products, composition and lattice parameters determined by Rietveld refinements.
| Sample | Reactants | Method | Products | Amount (wt%) | Unit cell (Å) | Space group |
|---|---|---|---|---|---|---|
| S1 | ErCl3–3LiBH4 | BM + S(CH3)2 + heat treatment | α-Er(BH4)3 | 98.4(1) | a = 10.78517(7) |
Pa |
| β-Er(BH4)3 | 1.6(1) | a = 5.478(1) |
Pmm |
|||
| S2 | ErCl3–3LiBH4 | BM + S(CH3)2 + heat treatment | α-Er(BH4)3 | 87.8(1) | a = 10.76796(5) |
Pa |
| β-Er(BH4)3 | 12.2(1) | a = 5.4664(1) |
Pmm |
|||
| S3 | PrCl3–3LiBH4 | (C2H5)2O | α-Pr(BH4)3 | 32.7(5) | a = 11.2465(1) |
Pa |
| LiPr(BH4)3Cl | 35.8(5) | a = 11. 5468(3) |
I3m |
|||
| LiBH4 | 28.4(4) | a = 7.144(1) |
I3m |
|||
| b = 4.421(1) | ||||||
| c = 6.779(2) | ||||||
| β-Pr(BH4)3 | 3.1(6) | a = 5.716(2) |
Pmm |
|||
| S4 | PrCl3–3LiBH4 | C6H5CH3 + S(CH3)2 | α-Pr(BH4)3 | a = 11.2473(2) |
Pa |
|
Mechanochemical synthesis (ball milling, BM) was executed using a Fritsch Pulverisette 6 planetary mill, employing an 80 ml tungsten carbide coated steel vial and balls with a ball to powder mass ratio of 40:1. Initially for samples S1 and S2, the powders were milled for 5 min followed by a break of 2 min at 400 rpm. After 24 repetitions, the reaction progress was determined by powder X-ray diffraction (PXD) followed by another 24 repetitions. The products were then dissolved in 20 ml S(CH3)2 per 1 g sample and stirred for three days. The LiCl was filtered from the solution and the Er(BH4)3 precipitated in vacuo using a rotary evaporator at 55 °C. To remove the coordinated S(CH3)2, the powder was heat treated at 145 °C in an oil bath, ground and heat treated once more. S2 was left under dynamic vacuum at 150 °C for 24 h. S1 was a light pink colored powder, whereas S2 was light orange.
S3 was synthesized by dissolving three molar equivalents of LiBH4 per PrCl3 in (C2H5)2O (60 ml per 1 g sample). After one day of stirring, the solution was filtered and the (C2H5)2O removed in vacuo using a rotary evaporator at 70 °C, yielding in a light green powder.
For S4, PrCl3 was activated by ball milling using 20 repetitions of 5 min milling and 2 min pause at 400 rpm. LiBH4 was recrystallized in (C2H5)2O and dried in vacuo at 90 °C. PrCl3 and LiBH4 were mixed in C6H5CH3 and stirred for five days. The suspension was refluxed at 90 °C for 3 h. The C6H5CH3 was then removed in vacuo at room temperature (RT) for 4 h. S(CH3)2 was added and the solution stirred for two days before removal of the S(CH3)2in vacuo at 120 °C for 2 h.
Characterization and thermal decomposition studies
Decomposition experiments were conducted in an in-house manufactured Sieverts type apparatus.31 Decomposition of S2 and S4 was executed using a temperature ramp of 5 °C min−1 from RT to 400 °C under 3 bar hydrogen pressure, followed by a 21 h isothermal step. Hydrogen reabsorption for S2 was carried out at 340 °C, under 100 bar H2, for 21 h. Furthermore, the decomposed S2 and S4 were physically mixed with 100 wt% LiCl and/or 50 wt% LiH with a mortar and pestle. The reabsorption was repeated at 400 °C under 100 bar H2 followed by an 18 h isotherm.Powder X-ray Diffraction (PXD) data were collected using a Rigaku SmartLab diffractometer. The data were collected in Debye–Scherrer transmission geometry using monochromatic CuKα1 radiation, (λ = 1.54056 Å), a curved position-sensitive detector and scattering angle (2θ) 10–70°. The samples were contained within rotating glass capillaries with an inner diameter of 0.5 mm, sealed with silicone grease after filling.
In situ SR-PXD data were collected at the Swiss Norwegian Beam Line (SNBL), BM01A at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Sapphire capillaries were connected to an in-house manufactured remote controlled gas rig and high pressure manifold cell. The sapphire capillary was rotated by 10° to improve powder averaging. The sample-to-detector distance was 146 or 245.8 mm, respectively, and the wavelength was 0.69733 or 0.77787 Å, calibrated from a NIST LaB6 standard. The in situ cycling experiments were conducted using a heating/cooling rate of 5 °C min−1 with a typical hydrogen pressure of ∼100 bar for absorption and ∼3 bar for desorption. Data were collected using a Pilatus 2M detector. The exposure time was set to 30 s giving a temperature resolution of 2.5 °C per pattern. Single crystal reflections from the sapphire tube were masked manually in Fit2D and Bubble.32,33 The Rietveld refinements were performed using GSAS and Expgui software.34,35 Three Gaussian and one Lorentzian parameter were modeled with a Thompson–Cox–Hastings pseudo-Voigt function.36 The background was fitted with a shifted Chebyshev polynomial with up to 36 terms. Atomic positions for α- and β-RE(BH4)3 (RE = Er, Pr) and LiPr(BH4)3Cl were taken from Y(BH4)3 (ref. 1 and 6) and LiCe(BH4)3Cl (ref. 18), respectively, and not refined.
Simultaneous thermogravimetric and differential scanning calorimetry (TG-DSC) was conducted on S1–S4 using a Netzsch STA 449 F3 Jupiter analyzer. Samples were loaded in aluminum crucibles (∼5–10 mg) and heated from RT to 400 °C at a heating rate of 5 °C min−1. An argon flow of 20 ml min−1 was set as a protection gas flow with a purge gas rate of 50 ml min−1.
S2 and S4 were measured by temperature programmed desorption (TPD-MS) in an in-house manufactured apparatus connected to a MKS Microvision-IP Rest Gas Analyser. The powders (∼50 mg) were loaded in a steel sample holder and heated from RT to 400 °C with a heating rate of 5 °C min−1 under dynamic vacuum. S3 was measured by mass spectrometry (MS) using a Netzsch STA449C analyzer connected to a Hiden Analytical HPR-20 QMS. Samples (∼5–10 mg) were loaded in aluminum-oxide crucibles and heated to 500 °C at a heating rate of 5 °C min−1. An argon flow of 20 ml min−1 was set as a protection gas flow, while purge gas rate was 30 ml min−1.
Results
Investigation of erbium borohydride
Fig. 1 shows Rietveld refinements at RT for the SR-PXD data of S1 and S2. Two phases are present in the samples which were identified as α-Er(BH4)3 and β-Er(BH4)3, respectively.1,6 α-Er(BH4)3 crystallizes in the space group Pa, while β-Er(BH4)3 crystallizes in Pmm. Neither starting materials (LiBH4, ErCl3) nor by-products (LiCl) are observed. For S1, the phase fractions were refined to 98.4(1) wt% for α-Er(BH4)3 and 1.6(1) wt% for β-Er(BH4)3. For S2, the phase fractions were determined to 87.8(1) and 12.2(1) wt% for α- and β-Er(BH4)3, respectively.
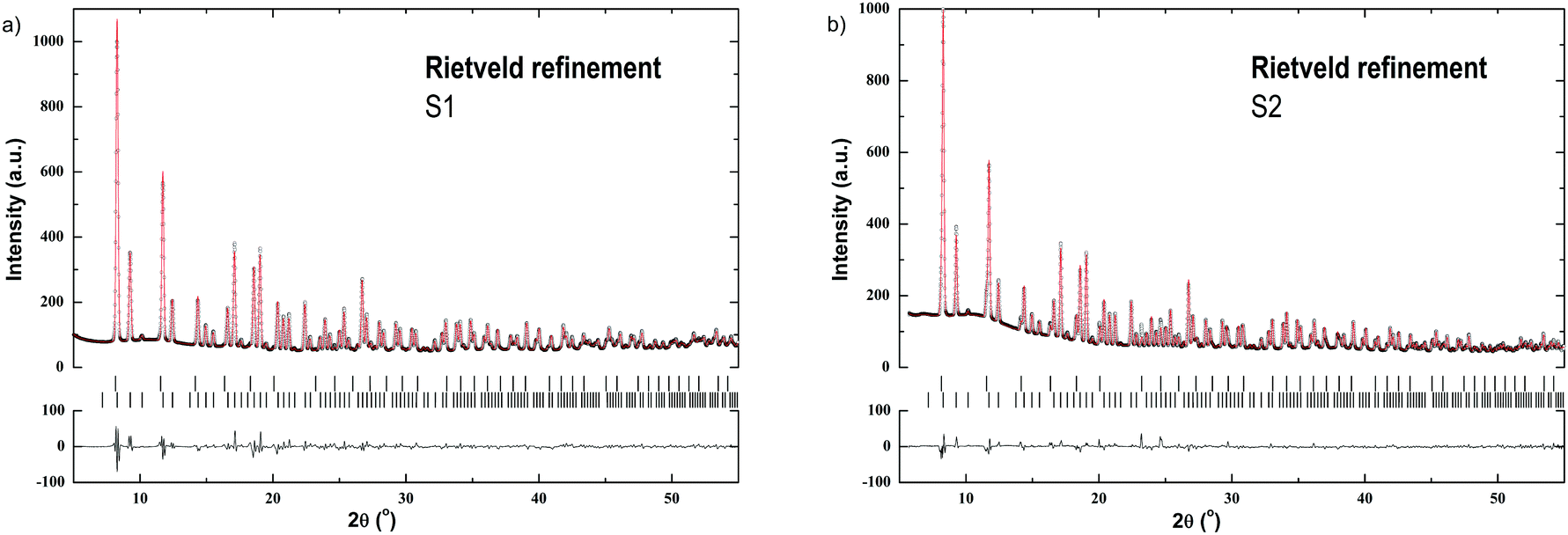 | ||
| Fig. 1 (a) Rietveld refinement with the SR-PXD data of S1 at RT showing experimental (circles) and calculated (red line) patterns and a difference plot (below). Vertical ticks mark the Bragg peak positions for (from top): β-Er(BH4)3, α-Er(BH4)3. Rwp = 4.01%. λ = 0.77787 Å; (b) Rietveld refinement with the SR-PXD data of S2 at RT showing experimental (circles) and calculated (red line) patterns and a difference plot (below). Vertical ticks mark the Bragg peak positions for (from top): β-Er(BH4)3, α-Er(BH4)3. Rwp = 3.53%. λ = 0.77787 Å. | ||
The combined TPD-MS and TG-DSC data of S2 is shown in Fig. 2a. S2 releases 4.7 wt% of gas between RT and 400 °C. The slope of the TG measurement indicates a smooth decomposition event; starting at 184 °C with a moderate slope which gradually becomes steeper. The endothermic event observed by DSC has a peak maximum at 281 °C which correspond to the steepest slope of the TG curve. A possible decomposition pathway is:37
| Er(BH4)3 → 0.75ErB4 + 0.25ErH2 + 5.75H2 (5.4 wt% H2) | (1) |
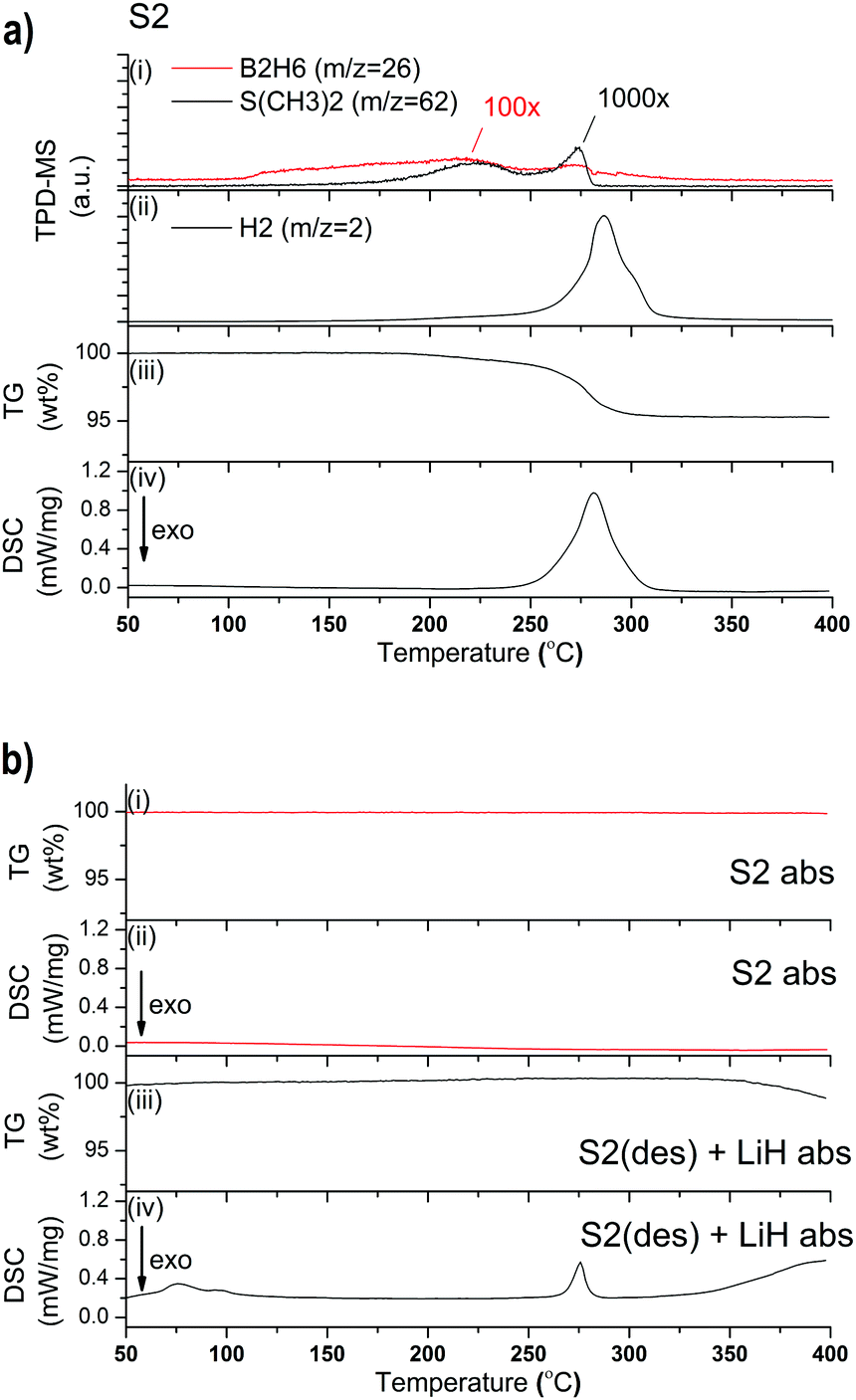 | ||
| Fig. 2 (a) TPD-MS-TG-DSC analysis from RT to 400 °C of S2 showing release of S(CH3)2 and B2H6 (i), H2 (ii), TG data (iii) and DSC signal (iv); (b) TG data (i) and DSC signal (ii) of S2 absorbed (red) and TG data (iii) and DSC signal (iv) of S2(des) + LiH absorbed (black) (temperature ramp of 5 °C min−1). | ||
For S2, one desorption–absorption-cycle was performed in the Sieverts-type apparatus. The desorption was performed at a heating rate of 5 °C min−1 from RT to 400 °C, followed by a 21 h isothermal step. A 3 bar H2 pressure was employed during desorption which is reported to increase reversibility compared to samples desorbed in vacuum.23,37,38 Absorption was performed at 340 °C under 100 bar H2 for 21 h. However, the TG-DSC measurement of the absorbed sample shown in Fig. 2b (S2 abs) indicates that rehydrogenation has not occurred during this experiment.
Fig. 3 illustrates the in situ SR-PXD desorption data of pristine S2 from RT to 300 °C, under 1 bar H2. This measurement confirms that all Bragg peaks of Er(BH4)3 have disappeared by 300 °C, which is consistent with the TG-DSC data, showing that decomposition is complete by ∼300 °C. After cooling to RT, no crystalline reaction product is observed (not shown). At the end of the measurement, the background is slightly modulated, which may be indicative of nanocrystalline or amorphous phases. The Bragg peak intensities in the in situ SR-PXD measurement of S2 fluctuate significantly from exposure to exposure. The reason for which remains undetermined.
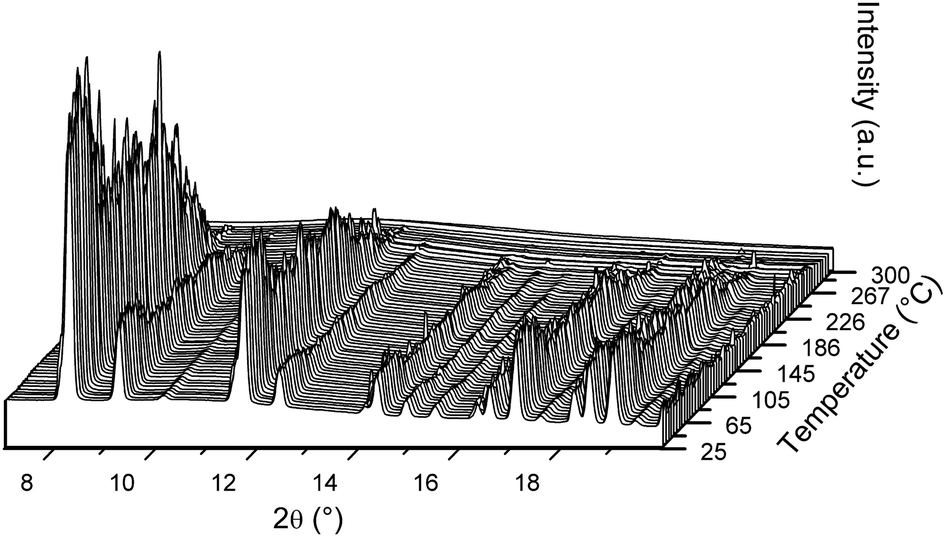 | ||
| Fig. 3 In situ SR-PXD data from the desorption process of S2 between RT and 300 °C under 1 bar H2 (temperature ramp of 5 °C min−1). Rietveld refinements with phase composition for initial state (front) are given in Fig. 1b. See text for discussion. λ = 0.77787 Å. | ||
Investigation of praseodymium borohydride
S3 was synthesized in (C2H5)2O, the LiCl by-product was removed by filtration and the solvent was then evaporated in vacuo (see Table 1). S4 was synthesized in C6H5CH3 and extracted with S(CH3)2. The Rietveld refinement plots of S3 and S4 are shown in Fig. 4. The refinement of S3 reveals a mixture of α-Pr(BH4)3 crystallized in space group Pa1 and β-Pr(BH4)3 in Pmm,39 as well as LiBH4 and LiPr(BH4)3Cl in space groups Pnma and I3m, respectively.5,18 No traces of the PrCl3 starting material is observed in S3. The residual powder, after filtration, was also analyzed by PXD, indicating the presence of PrCl3 and LiCl and thus suggesting that the reaction did not go to completion (Fig. A1, ESI†).
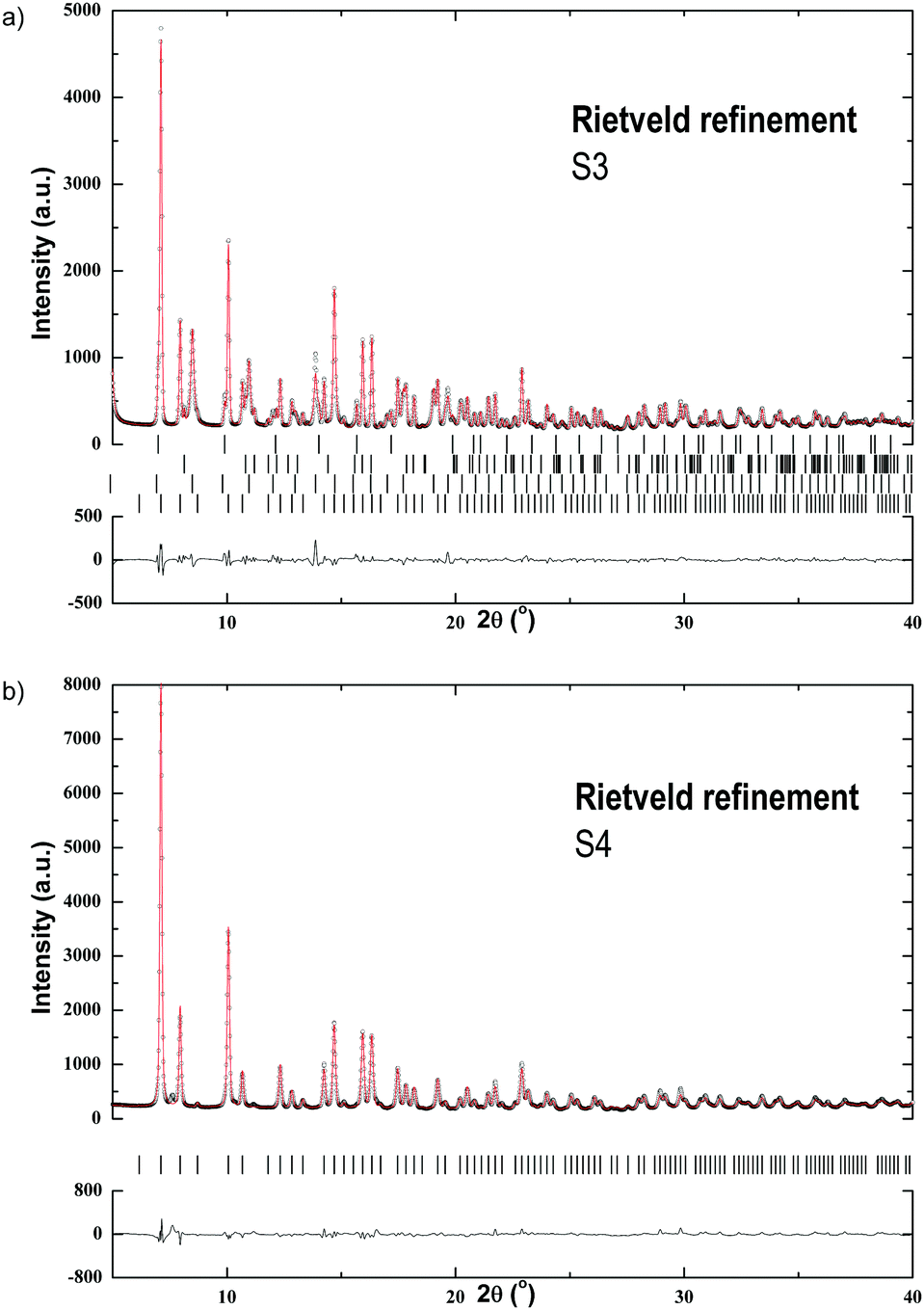 | ||
| Fig. 4 (a) Rietveld refinement with the SR-PXD data of S3 at RT showing experimental (circles) and calculated (red line) patterns and a difference plot (below). Vertical ticks mark the Bragg peak positions for (from top): β-Pr(BH4)3, o-LiBH4, LiPr(BH4)3Cl and α-Pr(BH4)3. Rwp = 4.78%. λ = 0.69733 Å; (b) Rietveld refinement with the SR-PXD data of S4 at RT showing experimental (circles) and calculated (red line) patterns and a difference plot (below). Vertical ticks mark the Bragg peak positions for: α-Pr(BH4)3. Rwp = 6.25%. λ = 0.69733 Å. | ||
MS analysis of S3 reveals that H2 release begins at 175 °C and continues through multiple desorption events until 375 °C (Fig. 5a(ii)). (C2H5)2O release is observed between 75 and 150 °C, while minor quantities of B2H6 are observed in a rather narrow range between 175 and 200 °C (Fig. 5a(i)). The TG data in Fig. 5a(iii) shows a release of 11.7 wt% and the DSC data reveals that it is a multistep process, with at least six major endothermic events. The first DSC peak can be assigned to the phase transition of orthorhombic-LiBH4 (o-LiBH4) to hexagonal-LiBH4 (h-LiBH4). The second peak at 176 °C can be attributed to the decomposition of LiPr(BH4)3Cl as it coincides with mass loss observed by TG. The same decomposition process is observed at 191 °C during in situ SR-PXD, however, that reaction occurred under hydrogen pressure which shifts the reaction to higher temperature (Fig. 6a). A third, minor peak at 191 °C, which is also observed for S4 (Fig. 5b), could not be assigned to any known process, but is most likely correlated to a structural phase transition, as discussed for S4 below. The endothermic peak at 260 °C is probably the decomposition of the parent borohydride. Further peaks at 284 and 345 °C have not yet been assigned, but are probably related to the multistep decomposition of the borohydride species and unknown formation, as observed in the in situ SR-PXD (Fig. 6a), which coincides again with the mass loss of 8.6 wt% between 200 and 360 °C.
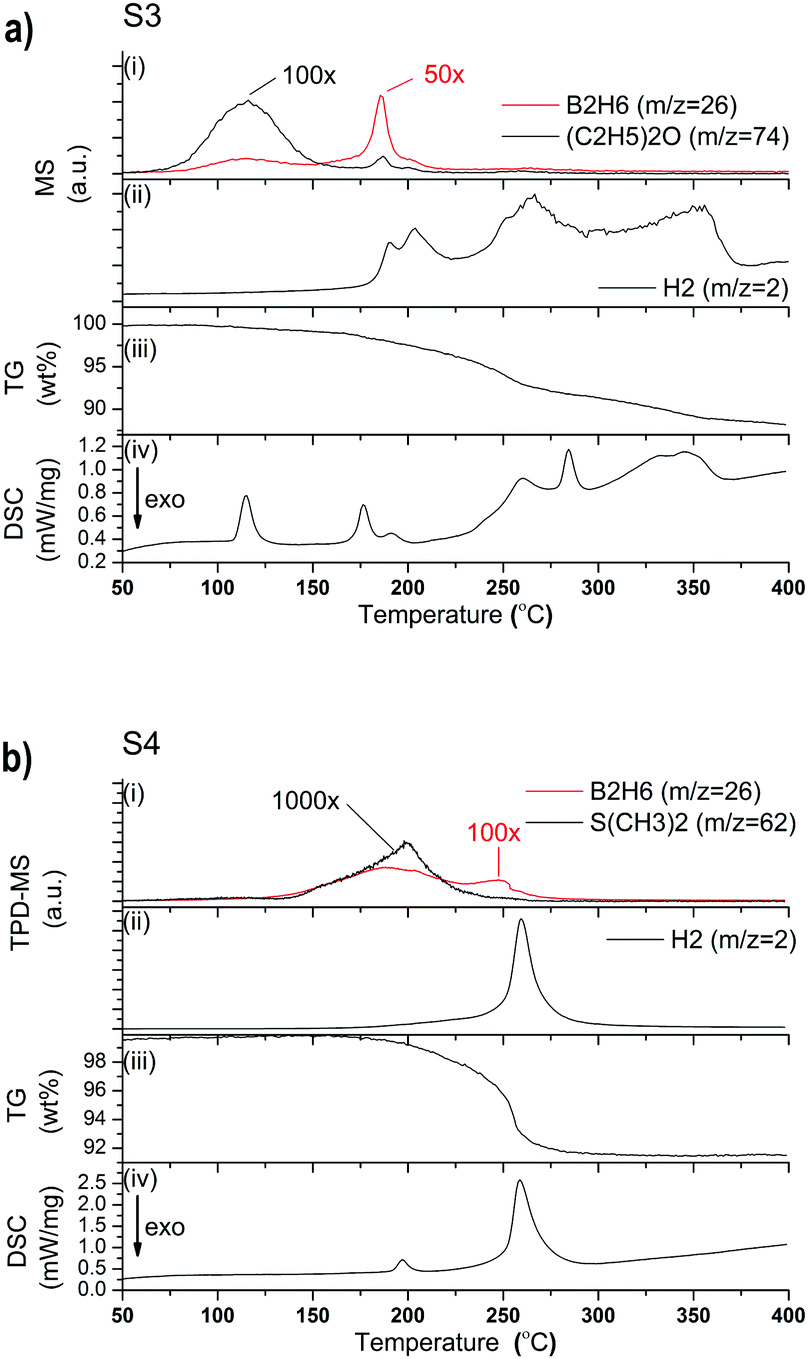 | ||
| Fig. 5 (a) MS-TG-DSC analysis from RT to 400 °C of S3 showing release of (C2H5)2O and B2H6 (i), H2 (ii), TG data (iii) and DSC signal (iv); (b) (TPD-MS)-TG-DSC analysis from RT to 400 °C of S4 showing release of S(CH3)2 and B2H6 (i), H2 (ii), TG data (iii) and DSC signal (iv). | ||
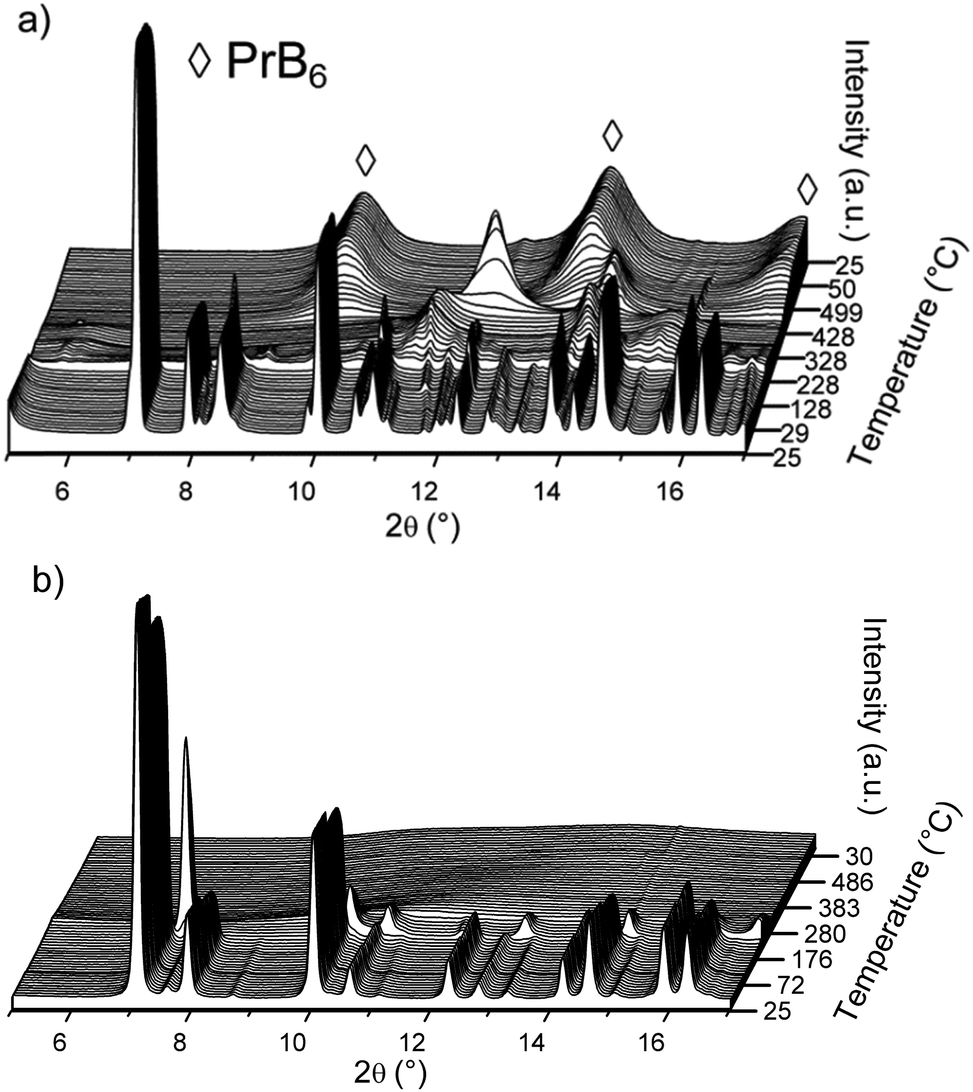 | ||
| Fig. 6 In situ SR-PXD of decomposition reaction of S3 (a) and S4 (b) heated from RT to 500 °C (temperature ramp of 5 °C min−1) and cooling to RT. Rietveld refinements with phase composition for initial state (front) are given in Fig. 4. See text for discussion. λ = 0.69733 Å. | ||
The in situ decomposition of S3 studied by SR-PXD is shown in Fig. 6a and supports the multistep decomposition pathway suggested from the DSC data. The sample was heated from RT to 500 °C at a ramp of 5 °C min−1, followed by cooling to RT. At 103 °C, the phase transition of o-LiBH4 to h-LiBH4 is observed. The Bragg peaks of LiPr(BH4)3Cl and α-Pr(BH4)3 disappear at 191 °C and 230 °C, respectively. A third phase, possibly a alternative polymorph of Pr(BH4)3, disappears at 282 °C. New Bragg peaks appear at 214 °C and vanish at 318 °C, which could originate from PrH3. From 346 °C, a second new phase appears and is the only crystalline phase in the pattern until 413 °C, when it starts to decrease to a minimum at 506 °C. PrB6 is observed from 399 °C, becomes the major phase until 507 °C and remains after cooling to RT.
The SR-PXD pattern for S4 at RT is shown in Fig. 4b. The diffraction pattern is distinctively different to that of S3, which is due to removal of S(CH3)2, LiCl and LiBH4. The refined parameters are given in Table 1. Fig. 5b(i–iv) shows TPD-MS and TG-DSC data of S4. A phase transition is observed in the DSC data at 192 °C. S4 loses ∼8.3 wt% when heated from RT to 400 °C.
| Pr(BH4)3 → 0.5PrB6 + 0.5PrH2 + 5.5H2 (5.7 wt% H2) | (2) |
The in situ SR-PXD decomposition of S4 between RT and 500 °C is shown in Fig. 6b. During heating from RT to 204 °C, the pattern is dominated by α-Pr(BH4)3 crystallized in space group Pa1 with a = 11.2473(2) Å at RT. At 204 °C, a structural phase transition occurs, which is consistent with our DSC data. Bragg peaks from the high temperature Pr(BH4)3 phase increase in intensity until 217 °C and vanish at 275 °C, after which no more crystalline products are observed. After reaching 500 °C, the sample is cooled to RT without appearance of any crystalline compounds.
The influence of LiH
Both the ex and in situ SR-PXD measurements show that hydrogen is not reversibly stored in S2. This is in disagreement with Gennari who reported that the Er(BH4)3, synthesized by ball milling, had a reversibility of 20% at 400 °C, under 60 bar H2.24 However, this may be attributed to the presence of Li-ions in the sample, with a formation of LiBH4 instead of regenerating Er(BH4)3. In our in situ SR-PXD absorption study, 100 bar H2 was applied to the decomposed sample at the same temperature and this time crystalline metal borohydride products were not observed. This is in agreement with the absorption experiment carried out in the Sieverts-type apparatus at 340 °C, under 100 bar H2. Furthermore, the lack of reversibility was confirmed by our TG-DSC measurements (Fig. 2b). Overall, this suggests an altered decomposition pathway in our halide free Er(BH4)3. In the absence of LiCl, the formation of amorphous and/or nanocrystalline phases occurs during decomposition without formation of crystalline phases upon rehydrogenation.In order to corroborate this assumption, the decomposition product of S2 was physically mixed in a 1:1 mass ratio with LiCl. The hydrogenation experiment was carried out in and ex situ without the resultant appearance of any crystalline rehydrogenation products (in situ SR-PXD in Fig. A2; TG-DSC in Fig. A3, ESI†). The experiment was repeated with addition of 50 wt% of LiH to desorbed S2 instead of LiCl, as LiH is less stable than LiCl. The hydrogenation process was followed in situ at 400 °C, under 100 bar H2. The products of this reaction are ErH3 and LiBH4, as shown in Fig. 7. ErH3 is formed at 400 °C (* in Fig. 7). During cooling to RT the h-LiBH4 appears at 268 °C. The transition to o-LiBH4 is also detected, which is in agreement with our DSC data, shown in Fig. 2b(iv), in which the presence of LiBH4 is suggested by the phase transition of o- to h-LiBH4 at 97 °C. The process behind the endothermic event at 74 °C is undetermined. In our sample, melting is observed at 272 °C, which is strongly indicative of the presence of LiBH4. The TG data in Fig. 2b shows that 1 wt% H2 is released during heating to 400 °C, probably linked to the decomposition of LiBH4.
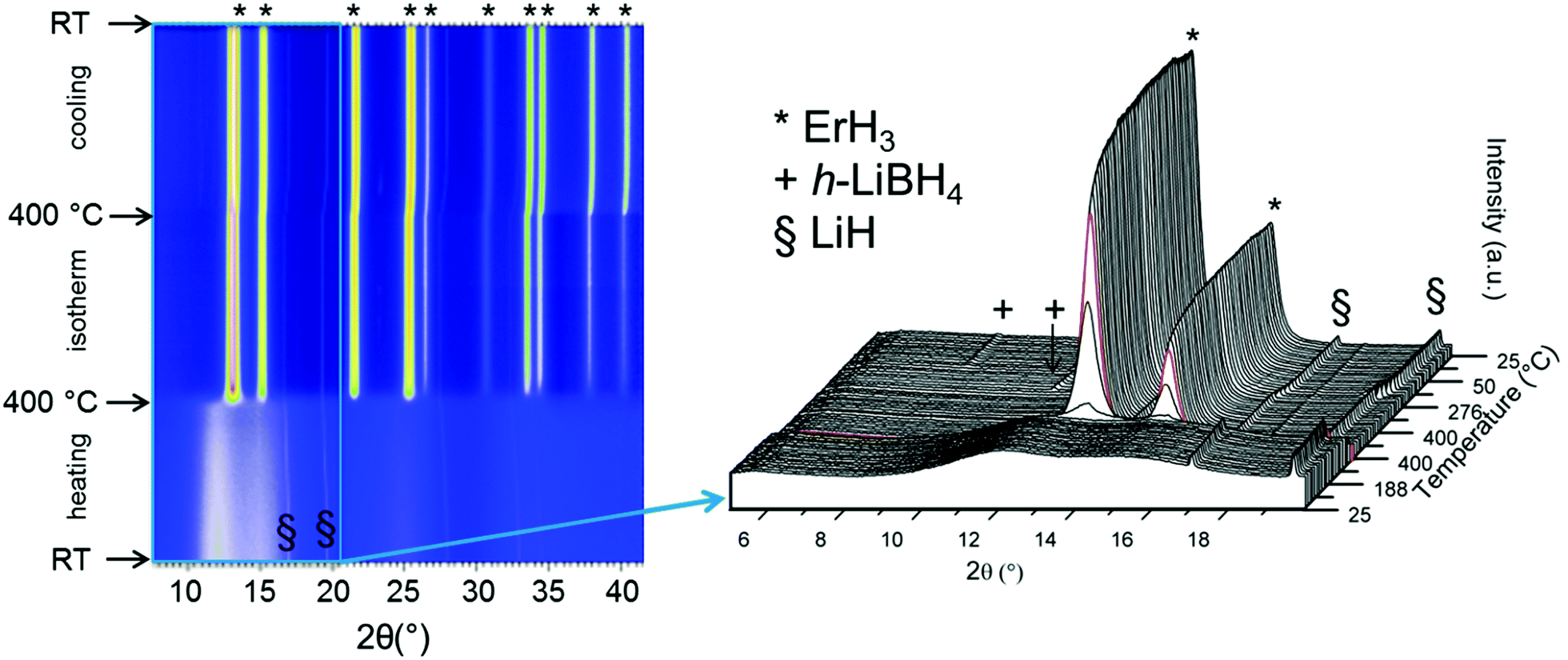 | ||
| Fig. 7 In situ SR-PXD data of a powder mixture containing desorbed S2 + 50 wt% LiH. The composite was heated from RT to 400 °C with a 1 h isothermal stage and cooling to RT in p(H2) = 100 bar (temperature ramp of 5 °C min−1). Inset showing magnification of the 2θ range 5 to 20° to enhance the visibility of the appearance of Bragg peaks for h-LiBH4 (+). Red curve in magnification marks the temperature of 400 °C. λ = 0.69733 Å. | ||
For the ex situ desorbed S4, the PXD pattern (not shown) did not show any crystalline phases. Thus, we assume that the reaction mechanism is the same as for S2. Therefore, 50 wt% of LiH was mixed with desorbed S4 (Fig. A4, ESI†) and rehydrogenation was carried out ex situ using the same conditions. PXD data (Fig. A5, ESI†) indicates the product PrH2 while TG-DSC data (Fig. A6, ESI†) indicates the formation of LiBH4. A weight loss of 1.7 wt% H2 is observed by TG. The observation of LiBH4 can often be very difficult in diffraction experiments since the scattering of LiBH4 is weak.
Discussion
For sample S2, two Er(BH4)3 polymorphs are detected at RT by SR-PXD. Earlier reports have shown a slight deviation in products. Olsen et al. observed only the β-Er(BH4)3 polymorph after 5 h of mechanochemical synthesis from ErCl3 and 3LiBH4.13 After annealing at 100 °C for 50 h, the α-Er(BH4)3 crystallized and replaced the metastable β-Er(BH4)3 polymorph.13 In contrast, Gennari reported that only the α-Er(BH4)3 polymorph was formed by mechanochemical synthesis.24 The differences in reports may be a result of the different pause time used during mechanochemical synthesis. Gennari describes the synthesis with 10 min milling followed by 10 min pause,24 opposed to Olsen et al. who milled for 5 h without breaks.13 Gennari also reported a reversible phase transition from α-Er(BH4)3 to β-Er(BH4)3 at 220 °C, which was neither determined in our DSC measurements, nor during our in situ SR-PXD analysis. Olsen et al. only observed a decrease of the parent borohydride Bragg peaks, and an increasing background during in situ SR-PXD while heating from 170–210 °C.13 However, the polymorphic transition is not observed in our study. To summarize, it seems that the reaction conditions have a major influence on the formation of the different Er(BH4)3 polymorphs. S1 primarily consists of α-Er(BH4) (98.4(1) wt%), along with β-Er(BH4)3 (1.6(1) wt%) (Fig. 1a), while in S2, it is approximately a 9:1 ratio of α-Er(BH4)3 to β-Er(BH4)3 (Fig. 1b). Thus, milder reaction conditions appear to favor the formation of α-Er(BH4)3.
The modulated background and the poor crystallinity of S2 during in situ SR-PXD can be compared with a recent in situ decomposition study, where solvent extracted Gd(BH4)3 was investigated.16 The intensity of Bragg peaks belonging to Gd(BH4)3 decreases between 180 and 270 °C, where after poor crystallinity and a modulated background is observed between 270 and 400 °C. This temperature region is the same as where pristine S2 becomes amorphous and a modulated background is observed.
The theoretical hydrogen capacity for the decomposition reaction of S2, according to eqn (1) is 5.4 wt%. Thus, the 4.7 wt% measured is 13% lower than expected. This deviation could be attributed to incomplete decomposition and the formation of a combination of different ErHx phases and/or the combination of nanocrystalline ErH2+δ and ErH3 as reported recently.37,40,41 The peak temperature of decomposition in our DSC measurement is 10–20 °C higher compared to earlier reports.24 However, the Ar flow rate was about 50% lower compared to former reports, while heating rates were consistent at 5 °C min−1.24 Olsen et al. used the same conditions for Ar flow, and heating rates, compared to the present experiments, but the decomposition temperature of Er(BH4)3, in the presence of six molar equivalents of LiBH4, was 240 °C,13,37 and thus lower compared to the decomposition temperature of S2 at 281 °C. As reported recently for Y(BH4)3 + LiBH4, this decrease is probably an effect of the co-melt/decomposition of Er(BH4)3 and LiBH4.19,26,42 A similar result was found when pure RE(BH4)2 with RE = Eu, Sm14 were compared to the impure compounds containing residual LiCl in the powder.13,14 It was determined that the latter had a decreased decomposition temperature, probably caused by residual LiBH4.14 LiCl or LiBH4 were not present in S2, which causes the higher decomposition temperature in our study.
The appearance of LiBH4 during rehydrogenation with addition of 50 wt% LiH to both S2 and S4 was not simply a recrystallization effect. Our rehydrogenation of S2, without additional LiH, showed no endothermic peaks in the DSC data. Thus, it is strongly suggested, that there was no LiBH4 in the sample. The reaction conditions are much milder compared to reported values of 155 bar H2 and 600 °C for rehydrogenation of pure LiBH4.43 A control experiment with the addition of LiCl was not successful, which again suggests that the influence of LiH on rehydrogenation goes beyond being a source of Li-ions. In analogy, the formation of LiBH4 from hydrogenation of reactive hydride composites with LiH, such as LiH + MgB2, are already known from literature, again under much harsher conditions or usage of catalysts.44,45
The effect of LiH is not fully understood, but one explanation as to why this effect has not been discovered earlier in metal borohydrides could be that LiH is a decomposition product of LiBH4, which is a likely impurity phase in all metal borohydrides synthesized by mechanochemical synthesis. Unreacted LiBH4 thus acts as a source for LiH during the thermal decomposition of such metal borohydrides. This can be very difficult to observe in diffraction experiments since the scattering of LiH is very weak and the Bragg peaks are often overlapped by other Bragg peaks, thus making LiH difficult to identify.
The first direct synthesis of Pr(BH4)3 has been shown in S3 by the solvation method. By itself, (C2H5)2O facilitates the production of α-Pr(BH4)3, LiPr(BH4)3Cl and minor amounts of β-Pr(BH4)3. α-Pr(BH4)3 crystallizes in space group Pa, which is analogous to α-Y(BH4)3.6 S4 on the other hand shows that extraction with S(CH3)2 removes LiCl and forms α-Pr(BH4)3 (see Fig. 4b).
The attempt to synthesize Pr(BH4)3 halide free for the first time was partly successful. Unwanted LiCl impurities could not be removed entirely, which led to formation of LiPr(BH4)3Cl in S3. The decomposition pathways of S3 and S4 are completely different, which is assumed to be an effect of the remaining LiCl. It seems to play a crucial role in the decomposition pathway of S3, as decomposition is rather complex with at least six major endothermic events. Decomposition of S4 without LiCl is rather simple. This opposed behavior suggests that the LiCl has a role beyond being an inert “dead mass” compound.
Conclusions
Two new halide free RE borohydrides, Er(BH4)3 and Pr(BH4)3, have been synthesized. Er(BH4)3, synthesized via mechanochemical synthesis followed by solvent extraction shows a deviation in behavior compared to previous mechanochemical synthesis containing the reaction product LiCl.24 In the absence of any LiCl, the reaction pathway during in situ SR-PXD decomposition of Er(BH4)3 indicates that the reaction products are completely amorphous. Rehydrogenation of the decomposition products at 400 °C under 100 bar H2 was unsuccessful.To prove the assumption that the absence of LiCl is responsible for the amorphous decomposition product(s) of S2 and negligible rehydrogenation, the desorbed sample was mixed with LiCl, without any effect upon rehydrogenation. The physical mixture of the desorbed Er(BH4)3 with 50 wt% of LiH was also investigated upon rehydrogenation, as LiH is less stable than LiCl. At 400 °C, under 100 bar H2, the crystalline reaction products, ErH3 and LiBH4, appeared during cooling. The same reaction mechanism occurred for the desorbed Pr(BH4)3 + 50 wt% LiH sample. PrH2 and LiBH4 were formed upon rehydrogenation at the same conditions. Thus, the presence of LiH improves the crystallinity and rehydrogenation properties.
An attempt to increase the total hydrogen storage capacity by the investigation of a 4LiBH4 + ErH3 composite was unsuccessful. PXD analyses (Fig. A7, ESI†) reveals, that the composite cannot be cycled between 4LiBH4 + ErH3 and ErB4 + 4LiH + 7.5H2 at the conditions described above. This observation reveals that erbium hydride needs to be formed in situ to have a destabilization effect on LiBH4. This is also in agreement with Gennari et al. who investigated a 4LiBH4 + YH3 composite and also showed the superior destabilization of LiBH4 by in situ formed YH3.23 Anyhow, ErH3 synthesized directly from the elements is not able to destabilize LiBH4 (for further information see Fig. A7 and A8 in the ESI†).
The Pr(BH4)3 synthesized in (C2H5)2O alone, shows that LiPr(BH4)3Cl is also formed in tandem with Pr(BH4)3, while recrystallization from S(CH3)2 and further heat treatment produced almost pure α-Pr(BH4)3. This material undergoes a phase transition during heating at 192 °C, although the nature of this phase transition will be studied in a follow up investigation.
In summary, we have synthesized pure RE(BH4)3, making it possible to investigate properties on the pure RE borohydrides. Additionally, the full gravimetric hydrogen content of these materials is now available. The supporting role of LiH was found to be essential for reversibility towards the formation of RE hydrides and LiBH4.
Acknowledgements
The research leading to these results has received funding from the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program FP7/2007-2013/under REA grant agreement no. 607040 (Marie Curie ITN ECOSTORE) and the Innovation Fund Denmark (project HyFill-Fast) and is thankfully acknowledged. We also acknowledge Prof. Radovan Černý for his scientific input and the Research Council of Norway for financial assistance. The authors acknowledge the skillful assistance from the staff of the Swiss-Norwegian Beamline, at the European Synchrotron Radiation Facility, Grenoble, France.Notes and references
- T. Sato, K. Miwa, Y. Nakamori, K. Ohoyama, H.-W. Li, T. Noritake, M. Aoki, S.-i. Towata and S.-i. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 104114 CrossRef.
- J. E. Olsen, C. Frommen, M. H. Sørby and B. C. Hauback, RSC Adv., 2013, 3, 10764–10774 RSC.
- T. Jaroń and W. Grochala, Dalton Trans., 2010, 39, 160–166 RSC.
- C. Frommen, N. Aliouane, S. Deledda, J. E. Fonneløp, H. Grove, K. Lieutenant, I. Llamas-Jansa, S. Sartori, M. H. Sørby and B. C. Hauback, J. Alloys Compd., 2010, 496, 710–716 CrossRef CAS.
- C. Frommen, M. H. Sørby, P. Ravindran, P. Vajeeston, H. Fjellvåg and B. C. Hauback, J. Phys. Chem. C, 2011, 115, 23591–23602 CAS.
- D. B. Ravnsbæk, Y. Filinchuk, R. Černý, M. B. Ley, D. r. Haase, H. J. Jakobsen, J. r. Skibsted and T. R. Jensen, Inorg. Chem., 2010, 49, 3801–3809 CrossRef PubMed.
- M. B. Ley, L. H. Jepsen, Y.-S. Lee, Y. W. Cho, J. M. B. von Colbe, M. Dornheim, M. Rokni, J. O. Jensen, M. Sloth and Y. Filinchuk, Mater. Today, 2014, 17, 122–128 CrossRef CAS.
- Y. Yan, H.-W. Li, T. Sato, N. Umeda, K. Miwa, S.-i. Towata and S.-i. Orimo, Int. J. Hydrogen Energy, 2009, 34, 5732–5736 CrossRef CAS.
- T. Jaron, W. Kozminski and W. Grochala, Phys. Chem. Chem. Phys., 2011, 13, 8847–8851 RSC.
- F. C. Gennari and M. R. Esquivel, J. Alloys Compd., 2009, 485, L47–L51 CrossRef CAS.
- H.-W. Li, Y. Yan, S.-i. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- B. J. Zhang, B. H. Liu and Z. P. Li, J. Alloys Compd., 2011, 509, 751–757 CrossRef CAS.
- J. E. Olsen, C. Frommen, T. R. Jensen, M. D. Riktor, M. H. Sørby and B. C. Hauback, RSC Adv., 2014, 4, 1570–1582 RSC.
- T. D. Humphries, M. B. Ley, C. Frommen, K. T. Munroe, T. R. Jensen and B. C. Hauback, J. Mater. Chem. A, 2015, 3, 691–698 CAS.
- R. Černý, D. B. Ravnsbæk, G. Severa, Y. Filinchuk, V. D'Anna, H. Hagemann, D. Haase, J. Skibsted, C. M. Jensen and T. R. Jensen, J. Phys. Chem. C, 2010, 114, 19540–19549 Search PubMed.
- M. B. Ley, S. Boulineau, R. Janot, Y. Filinchuk and T. R. Jensen, J. Phys. Chem. C, 2012, 116, 21267–21276 CAS.
- A. V. Skripov, A. V. Soloninin, M. B. Ley, T. R. Jensen and Y. Filinchuk, J. Phys. Chem. C, 2013, 117, 14965–14972 CAS.
- M. B. Ley, D. B. Ravnsbæk, Y. Filinchuk, Y.-S. Lee, R. l. Janot, Y. W. Cho, J. Skibsted and T. R. Jensen, Chem. Mater., 2012, 24, 1654–1663 CrossRef CAS.
- E. Roedern, Y.-S. Lee, M. B. Ley, K. Park, Y. W. Cho, J. Skibsted and T. R. Jensen, J. Mater. Chem. A, 2016, 4, 8793–8802 CAS.
- S. Marks, J. G. Heck, M. H. Habicht, P. Oña-Burgos, C. Feldmann and P. W. Roesky, J. Am. Chem. Soc., 2012, 134, 16983–16986 CrossRef CAS PubMed.
- P. Schouwink, M. B. Ley, A. Tissot, H. Hagemann, T. R. Jensen, L. Smrčok and R. Černý, Nat. Commun., 2014, 5, 5706 CrossRef CAS PubMed.
- P. Schouwink, E. Didelot, Y.-S. Lee, T. Mazet and R. Černý, J. Alloys Compd., 2016, 664, 378–384 CrossRef CAS.
- F. Gennari, L. F. Albanesi, J. Puszkiel and P. A. Larochette, Int. J. Hydrogen Energy, 2011, 36, 563–570 CrossRef CAS.
- F. C. Gennari, J. Alloys Compd., 2013, 581, 192–195 CrossRef CAS.
- B. James and M. Wallbridge, Prog. Inorg. Chem., 1970, 11, 99–231 CrossRef CAS.
- M. B. Ley, M. Paskevicius, P. Schouwink, B. Richter, D. A. Sheppard, C. E. Buckley and T. R. Jensen, Dalton Trans., 2014, 43, 13333–13342 RSC.
- M. Visseaux and F. Bonnet, Coord. Chem. Rev., 2011, 255, 374–420 CrossRef CAS.
- H. Hagemann, M. Longhini, J. W. Kaminski, T. A. Wesolowski, R. Černý, N. Penin, M. H. Sørby, B. C. Hauback, G. Severa and C. M. Jensen, J. Phys. Chem. A, 2008, 112, 7551–7555 CrossRef CAS PubMed.
- R. Černý, G. Severa, D. B. Ravnsbæk, Y. Filinchuk, V. D'Anna, H. Hagemann, D. Haase, C. M. Jensen and T. R. Jensen, J. Phys. Chem. C, 2009, 114, 1357–1364 Search PubMed.
- L. Huang, O. Elkedim and X. Li, J. Alloys Compd., 2012, 536, S546–S549 CrossRef CAS.
- H. Brinks, A. Fossdal, R. Bowman and B. C. Hauback, J. Alloys Compd., 2006, 417, 92–95 CrossRef CAS.
- A. Hammersley, Rep., ESRF97HA02T, 1997.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS PubMed.
- A. Larson and R. Von Dreele, General Structure Analysis System (GSAS); Report LAUR 86-748; Los Alamos National Laboratory, Los Alamos, NM, 2000.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- P. Thompson, D. Cox and J. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- C. Frommen, M. Heere, M. D. Riktor, M. H. Sørby and B. C. Hauback, J. Alloys Compd., 2015, 645(suppl 1), S155–S159 CrossRef CAS.
- J.-H. Shim, J.-H. Lim, S.-u. Rather, Y.-S. Lee, D. Reed, Y. Kim, D. Book and Y. W. Cho, J. Phys. Chem. Lett., 2010, 1, 59–63 CrossRef CAS.
- Y.-S. Lee, J.-H. Shim and Y. W. Cho, J. Phys. Chem. C, 2010, 114, 12833–12837 CAS.
- F. C. Gennari, Int. J. Hydrogen Energy, 2011, 36, 15231–15238 CrossRef CAS.
- M. Heere, S. H. Payandeh GharibDoust, C. Frommen, M. H. Sørby, T. R. Jensen and B. C. Hauback, 2016, to be submitted.
- K. Park, H.-S. Lee, A. Remhof, Y.-S. Lee, Y. Yan, M.-Y. Kim, S. J. Kim, A. Züttel and Y. W. Cho, Int. J. Hydrogen Energy, 2013, 38, 9263–9270 CrossRef CAS.
- P. Mauron, F. Buchter, O. Friedrichs, A. Remhof, M. Bielmann, C. N. Zwicky and A. Züttel, J. Phys. Chem. B, 2008, 112, 906–910 CrossRef CAS PubMed.
- G. Barkhordarian, T. Klassen, M. Dornheim and R. Bormann, J. Alloys Compd., 2007, 440, L18–L21 CrossRef CAS.
- J. J. Vajo, S. L. Skeith and F. Mertens, J. Phys. Chem. B, 2005, 109, 3719–3722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp04523e |
| This journal is © the Owner Societies 2016 |