
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of mass-transport conditions on the ethanol oxidation reaction (EOR) mechanism of Pt/C electrocatalysts
Antoine
Bach Delpeuch†
*abc,
Marjorie
Jacquot
ab,
Marian
Chatenet‡
bc and
Carsten
Cremers
a
aFraunhofer Institut für Chemische Technologie ICT, Joseph-von-Fraunhofer-Straße 7, 76327 Pfinztal, Germany. E-mail: antoine.bachdelpeuch@gmail.com
bUniv. Grenoble Alpes, LEPMI, F-38000 Grenoble, France
cCNRS, LEPMI, F-38000 Grenoble, France
First published on 12th August 2016
Abstract
This study aims to provide further understanding of the influence of different parameters that control mass-transport (the revolution rate of the rotating disk electrode and the potential scan rate) on the ethanol oxidation reaction (EOR). The experiments were conducted on a home-made carbon-supported 20 wt% Pt/C electrocatalyst, synthesized using a modified polyol method, and characterized in terms of physicochemical properties by thermogravimetric analysis (TGA), powder X-ray diffraction (XRD) and transmission electron microscopy (TEM). The EOR at the thin active layer of this electrocatalyst was characterized using both differential electrochemical mass spectrometry (DEMS) in a flow cell configuration and the rotating disc electrode (RDE). The results demonstrate that operating under stationary conditions (low scan rate and high RDE speed) hinders complete ethanol electrooxidation into CO2 and favors the poisoning of the electrocatalyst surface by hydroxide and strong ethanol adsorbates. As such, the EOR appears to be more efficient and faster under dynamic conditions than in near steady-state.
1. Introduction
Direct ethanol fuel cells (DEFCs) hold promise as an alternative power source for portable applications, owing to their theoretical high energy density1 and non-toxicity of their fuel: ethanol. Moreover, ethanol presents additional advantages compared to the classical hydrogen fuel of low-temperature fuel cells: its production in a pre-existing infrastructure, transportation and storage are rather straightforward. Yet, the DEFC performance is impeded by the sluggish kinetics of the ethanol oxidation reaction (EOR) (acceptable currents are only obtained at large anode overpotential values) and its poor selectivity toward the formation of CO2, the desired product. Indeed, the complete ethanol electrooxidation into CO2 (12e− per ethanol molecule – pathway (3) in Fig. 1) is hindered by the difficult cleavage of the ethanol C–C bond into COad and CHx,ad which leads, as a consequence, to the formation of acetaldehyde (2e− – pathway (1)) and acetic acid (4e− – pathway (2)) by-products2–6 and thereby severely decreases the faradaic efficiency of practical DEFCs. To summarize, present DEFC systems fail to complete their theoretical promises in terms of practical cell voltage and energy density. To overcome these hurdles, more fundamental insights into the EOR phenomenon are required, especially at the best single-metal EOR electrocatalyst, platinum.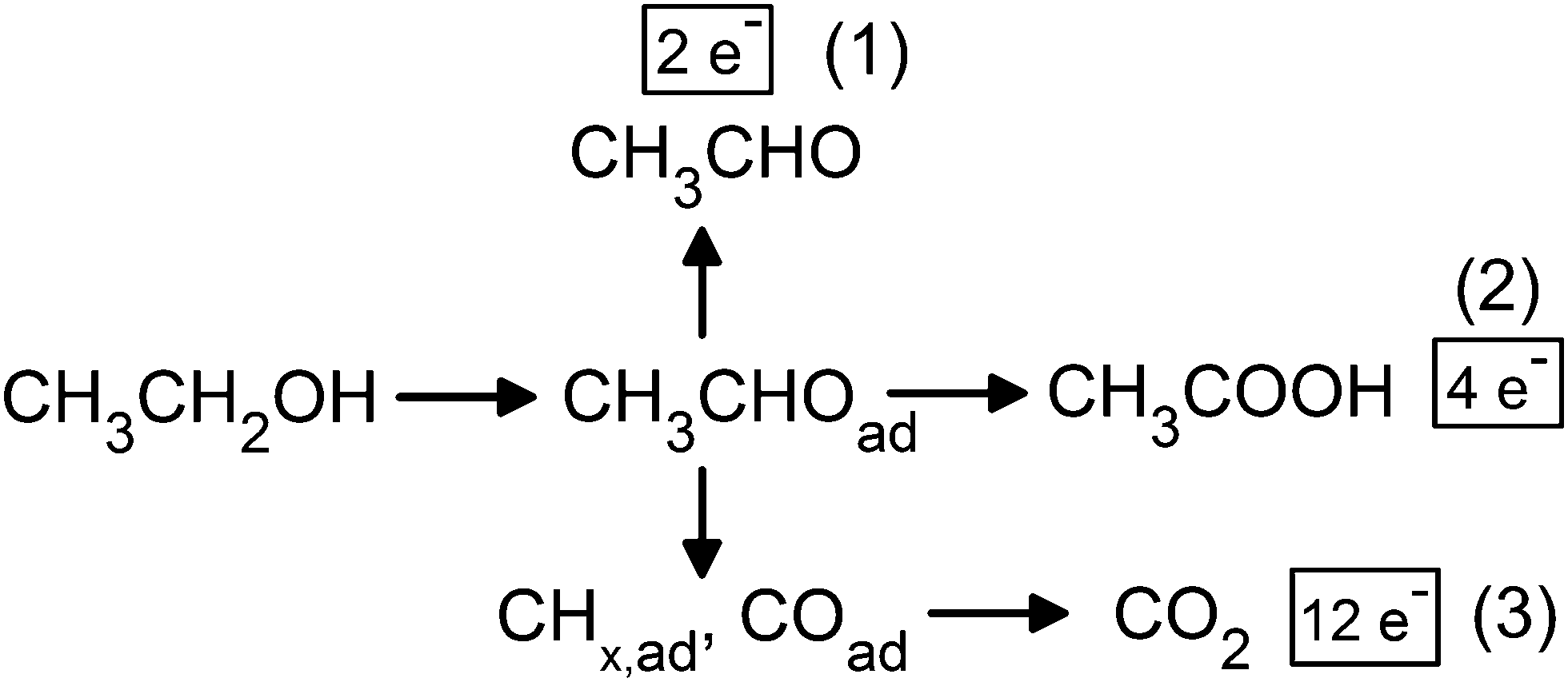 | ||
| Fig. 1 Scheme of the EOR mechanism on Pt. | ||
The impact of the morphology of bulk platinum (Pt) and carbon-supported Pt electrocatalysts (Pt/C) on the EOR has already been investigated to some extent in the literature. On the one hand, studies on monocrystalline electrocatalysts revealed that the C–C bond cleavage occurs mainly on surface steps,7–9 whereas no influence of the terrace was observed.7 Colmati et al. found out using in situ FTIR that both C–C bond breaking and COad electrooxidation are enhanced at low potential in steps, whereas a competitive formation of acetaldehyde and acetic acid takes place at high potential on the same sites.7 On the other hand, investigations on Pt/C showed contradictory results on the influence of the nanoparticle size on the EOR mechanism. Perez et al. obtained an optimal EOR activity on ca. 2.5 nm diameter nanoparticles and explained their performance by a compromise between a large electrochemical surface area and enhanced oxophilicity of small nanoparticles compared to the bigger ones (with a diameter comprised between ca. 3.3 and 3.6 nm).10 In contrast, Li et al. found the highest EOR specific activity on their smallest Pt nanoparticles (ca. 1.7 nm) against nanoparticles of ca. 2.4 and 4.0 nm.11
The mass-transport of the reactant/soluble intermediates/products plays an important role in electrochemical processes in general, with special emphasis on complex electrocatalytic reactions.12–18 This is not less true for the EOR, a very complex electrocatalytic reaction, being admitted that DEFCs will likely be fed using pumped ethanol and that electroactive soluble intermediates/by-products are formed in the course of the reaction. To some extent, it is possible that difference in mass-transport conditions from one study to the other can explain some divergences in the EOR literature. Unfortunately, the influence of mass-transport on the EOR mechanism has hardly been reviewed. To the authors' knowledge, there is only a single paper truly dedicated to that matter, by Rao et al.19 They demonstrated the decrease of the CO2 current efficiency (CCE) under enhanced flow rate conditions on Pt/C, which was attributed to the lower residence time of ethanol inside the catalytic layer thickness.19 A similar study on the methanol oxidation reaction (MOR) at the smooth Pt electrode20 highlighted the hindered MOR activity at a faster revolution rate of the rotating disc electrode (RDE), and ascribed it to the larger poisoning of the electrocatalyst surface by COad. This phenomenon was however not confirmed for Pt/C electrodes, where no influence of the diffusion–convection conditions was reported on the MOR activity.21
In this paper, the influence of the mass-transport on the EOR has been studied by varying the diffusion and convection conditions on state-of-the-art Pt/C electrocatalysts. The impact of the diffusion conditions has been studied by means of DEMS using the m/z = 22 and m/z = 29 mass-to-charge signals attributed to CO2 and acetaldehyde via modification of the scan rate from ν = 2 to 10 mV s−1. The role of the electrolyte convection in the EOR mechanism has been examined by varying the revolution rate of the RDE between 0 and 2500 rpm.
2. Experimental
2.1. Material preparation
A modified polyol method was used to synthesize the 20 wt% Pt/C electrocatalyst. An appropriate amount of the metal precursor (H2PtCl6, Alfa Aesar) was dissolved in a solution containing Milli-Q water and ethylene glycol (EG) (volumetric ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A carbon black suspension in ethylene glycol, prepared separately and dispersed by sonication, was then added into the first solution. The pH of the resulting suspension was then adjusted with 0.5 M NaOH solution (diluted in EG) to pH = 11.7 and the suspension was allowed to stir for one hour in argon, before being heated up to T = 160 °C and was maintained at this temperature for three hours. After this step, the suspension was cooled down and was allowed to stir overnight. Its pH was thereafter decreased to pH = 3 with a 0.5 M H2SO4 aqueous solution and stirred for an additional 24 h. The electrocatalyst powder was finally filtered, washed abundantly with Milli-Q water and dried overnight in a drying oven at T = 80 °C.
:1). A carbon black suspension in ethylene glycol, prepared separately and dispersed by sonication, was then added into the first solution. The pH of the resulting suspension was then adjusted with 0.5 M NaOH solution (diluted in EG) to pH = 11.7 and the suspension was allowed to stir for one hour in argon, before being heated up to T = 160 °C and was maintained at this temperature for three hours. After this step, the suspension was cooled down and was allowed to stir overnight. Its pH was thereafter decreased to pH = 3 with a 0.5 M H2SO4 aqueous solution and stirred for an additional 24 h. The electrocatalyst powder was finally filtered, washed abundantly with Milli-Q water and dried overnight in a drying oven at T = 80 °C.
2.2. Physical characterization
The metal–carbon ratio of the Pt/C electrocatalyst was evaluated by thermogravimetric analysis (TGA, Q 5000 from TA Instruments). Information about the crystalline structure and the associated lattice parameter was obtained by powder X-ray diffraction (XRD, Bruker AXS D8) in the 2θ range from 15° to 90° using Cu Kα radiation with a scan rate of 0.74° min−1. The dispersion of the nanoparticles on the carbon support was observed using transmission electron microscopy (TEM, Jeol 2010). The associated nanoparticle size distribution (PSD) histograms were constructed after examination of the diameter of 400 metal nanoparticles.2.3. Electrochemical measurements
For the DEMS investigation, the working electrode was a uniform electrocatalyst film deposited on top of an Au-sputtered (55 nm) Gore-Tex PTFE membrane (60 μm thickness, 0.02 μm mean pore size, 50% porosity). The electrocatalytic ink was made of 3 mg of electrocatalyst dispersed by sonication in a 400 μL mixture of isopropanol and water (volumetric ratio 3:1). 9 μL of this ink was then deposited onto the sputtered Au/PTFE membrane (i.e. 9.9 μg Pt). The amount of Nafion® ionomer deposited onto the film was always equal to 50 wt% of the electrocatalyst loading on the working electrode. The counter-electrode was a platinum foil. The DEMS measurements were carried out in a flow cell described elsewhere.22
The RDE analysis was achieved in a standard three-electrode electrochemical cell, on a thin Pt/C active layer deposited on a Ø 0.4 mm glassy carbon substrate/current collector (0.13 cm2 geometric area) working electrode connected to a rotator (Pine Research Instrumentation). The counter-electrode was a platinum wire. The reference electrode was a reversible hydrogen electrode (RHE) for both the RDE and the DEMS studies. In any case, the electrochemical results presented hereafter are normalized by the electrochemical active surface area (ECSA) of the material of the Pt/C working electrode.
Prior to the measurements, the investigated Pt/C electrocatalyst was subjected to potentiodynamic cycling in the supporting electrolyte (0.5 M H2SO4) until stabilization of its performance (overlay of successive CVs). CO-stripping CVs were recorded in 0.5 M H2SO4 subsequently to CO potentiostatic adsorption (8 min) at Ead = 0.15 V vs. RHE and the flush of the solution in Ar (20 min) under potential control. The EOR investigation was then carried out in a 0.5 M H2SO4 + 0.1 M EtOH electrolyte solution.
2.4. Calibration of the mass spectrometer
The calibration of the m/z = 22 mass-to-charge signal was carried out according to the protocol detailed in ref. 23 and 24.3. Results and discussion
3.1. Physical characterization
The XRD profile of the home-made 20 wt% Pt/C electrocatalyst is shown in Fig. 2. The sharpness of the diffraction peaks, was localized at ca. 40°, 46.5°, 67.8°, 82°, and 86° and assigned to the (111), (200), (220), (311), and (222) planes, respectively, which is characteristic of the expected face-centered cubic (fcc) structure of platinum, demonstrate that the sample is well crystallized. The average size of the crystalline domains was determined from Scherrer’s equation; the (220) diffraction peak was used for that purpose, owing to its isolated character on the diffractogram (it does not overlap with other peaks). The lattice parameters and the average size of the Pt crystalline domains are summarized in Table 1. | ||
| Fig. 2 XRD profile of the investigated Pt/C electrocatalyst prepared by a polyol method at pH = 11.7. | ||
| Electrocatalyst | Effective Pt loading by TGA (wt%) | a/Å | d XRD/nm | d Elec/nm | d N/nm | d S/nm | d V/nm |
|---|---|---|---|---|---|---|---|
| a: lattice parameter; dXRD: mean nanoparticle size (XRD); dElec: electrochemical mean particle size; dN: number-averaged diameter (TEM); dS: surface-averaged diameter (TEM); dV: volume-averaged diameter (TEM). | |||||||
| Pt/C | 22.1 | 3.84 | 2.0 | 6.8 | 1.8 | 5.2 | 9.7 |
Fig. 3 shows a typical TEM image of the Pt/C electrocatalyst and the associated particle size distribution (PSD) histogram of the sample. Pt nanoparticles that were small and homogeneous in size were uniformly disseminated on their carbon support. Some large nanoparticles/agglomerates (which account for ca. 1.9% of the total number of nanoparticles) were, however, also occasionally detected during the TEM observations, which explains the deviance between the number-, surface- and volume-averaged diameters, dN, dS and dV, respectively (see Table 1), calculated as in ref. 25. A quite good agreement is nevertheless found between dS and dElec. The difference between the dV and dXRD values is attributed to the fact that the large Pt ensemble detected in TEM consists of agglomerates of small nanoparticles rather than large monocrystalline nanoparticles; indeed, the large Pt feature at the extreme left of the carbon grain in Fig. 3 exhibits a very rough surface and complex contrast, which would agree with its polycrystalline (and therefore agglomerated) nature.
 | ||
| Fig. 3 (a) Typical TEM images and (b) associated particle size distribution (PSD) of the investigated Pt/C electrocatalysts; inset: magnification at low particle percentage. | ||
3.2. Effect of the scan rate
 | ||
| Fig. 4 Subtracted (a) raw and (b) amplified CO-stripping CVs in 0.5 M H2SO4 on Pt/C (metal loading: 9.9 μg) and corresponding (c) non-amplified and (d) amplified MSCVs for the m/z = 22 mass-to-charge signals multiplied by a number indicated in brackets; T = 25 °C. | ||
Moreover, a similar trend was found in Fig. 4c by the use of the m/z = 22 mass-to-charge signal (ascribed to CO2). According to Maillard et al., the onset potential of the CO-stripping at ν = 2 mV s−1 is close to the equilibrium potential of the OHad formation.26 However, as the scan rate increases and the reactions take place deeper under non-stationary conditions, the onset potential, which is believed to be controlled by COad + OHad interactions, shifts positively due to the low mobility of the COad on Pt surface, and more particularly Pt terraces.
Besides the positive effect on the onset potential, the ascending current soars faster at slow scan rates (Fig. 4a and b) giving the CO-stripping ascending and descending parts of the peak a more symmetric and narrower profile. The more moderate current increase at ν = 10 mV s−1versus ν = 2 mV s−1 likely results from the slow diffusion of the COad species on the Pt surface; this surface-diffusion is believed to limit the reaction kinetics of the CO-stripping reaction,26 even though the slow water dissociation was also put forth to account for the reaction sluggishness.29
 | ||
| Fig. 5 (a) CVs of the EOR in 0.5 M H2SO4 + 0.1 M EtOH on Pt/C and corresponding MSCVs for the (b) m/z = 29 and (c) m/z = 22 mass-to-charge signals; at ν = 2, 5 and 10 mV s−1; T = 25 °C. | ||
The m/z = 22 mass-to-charge signal demonstrates that the behavior of the CO2 production during the EOR contrasts with the electrooxidation of COad. Indeed, although the peak potential of the CO2 generation during the EOR depends on the scan rate similarly to the CO-stripping, the extent of the shift is much lower in the case of the EOR. This difference finds its origin in the distinct mechanisms that yield CO2 in the two cases: while the CO-stripping peak current is reached at intermediate consumption of the COad layer (and therefore attributed to a lack of COad-species at the electrocatalyst surface), the CO2 peak of the EOR in Fig. 5c originates from the surface poisoning by ethanol and hydroxide adsorbates. Besides, the onset potential of the CO2 formation during the EOR (Fig. 5c) remains constant with the rise of the potential scan rate, a behavior that differs from the positive shift of the CO-stripping initiation, attributed to the slow mobility of COad-species.26 This independence of the initiation of the CO2 formation against the scan rate shows that the reaction is not limited by COad mobility (like in a CO-stripping), but to other phenomena, like the C–C bond cleavage or the supply in hydroxide species.
It can further be remarked that the current of the oxidation reaction at 0.4 < E < 1.1 V vs. RHE does not increase as much as that of the reaction at E > 1.1 V vs. RHE, as the potential scan rate rises from ν = 2 to 10 mV s−1. This phenomenon is ascribed to the slow kinetics of the EOR in the medium potential region which involves the production of acetaldehyde (2e−) and acetic acid (4e−) and CO2 (12e−). Moreover, the complete ethanol electrooxidation into CO2 represents a larger part of the total current at ν = 10 mV s−1 than at ν = 2 mV s−1 (see CCE values in Fig. 6). Even if ethanol electrooxidation to CO2 yields to 12e− against 2 or 4e− for the acetaldehyde and acetic acid production, respectively, the reaction kinetics are much lower and may induce the generation of lower currents due to the very low turnover frequency of the Pt sites. The reaction at E > 1.1 V vs. RHE may correspond to the oxidation of strong ethanol adsorbates, such as CHx,ad or/and C2-adsorbates.24,30,31
 | ||
| Fig. 6 CO2 current efficiency (CCE) values calculated from cyclic voltammetries run in 0.5 M H2SO4 + 0.1 M EtOH (represented in Fig. 5a) at ν = 2, 5 and 10 mV s−1 and corresponding MSCVs (Fig. 5b) on Pt/C. | ||
The evolution of the CCE against the potential is represented in Fig. 6. The drop of the CCE versus the rise of the applied potential is attributed to the growing contamination of the electrocatalyst surface by ethanol and hydroxide adsorbates, which hinders the dissociative adsorption of ethanol. The identity of these ethanol adsorbates has been still under speculation to date,30 but may correspond to CHx,ad or C2-adsorbates, which only oxidize at high potential (typically at E > 0.9 V vs. RHE).24,31 The maximal value obtained at E = 0.6 V vs. RHE is very similar for each scan rate, which rules out the influence of the potential scan rate (at least between ν = 2, 5 and 10 mV s−1) on the CCE. This result contrasts with what was expected. Indeed, ethanol adsorbate stripping studies have shown that the C–C bond breaking can occur at potentials as low as E = 0.05 V vs. RHE on Pt-based carbon-supported electrocatalysts.24,31 As such, spending a longer time between E = 0.07 (CV initial potential) and E = 0.6 V vs. RHE (CO2 generation initiation) during a slow-scan CV was expected to enhance ethanol dissociative adsorption and boost the amount of CO-like adsorbates at the electrocatalyst surface at the beginning of the CO2 generation. At E ≥ 0.7 V vs. RHE, the degree at which the CCE decreases against the potential is rather different depending on the sweep rate: the CCE at E = 0.7 V vs. RHE is twice lower at ν = 2 mV s−1 (CCE = 0.09) than at ν = 10 mV s−1 (CCE = 0.18). This is explained by the low-potential blockage of the electrocatalyst surface by the mentioned poisoning adsorbates (enhanced at lower potential scan rate), which gives more time to the poisoning process to operate.
Finally, it would have been of interest to lead this study at faster scan rates, to see whether the trend observed under quasi-stationary conditions were confirmed under non-stationary ones. However, higher scan rates (i) would have led to longer delays between the CVs and MSCVs thereby corrupting the reliability of the corresponding results, and (ii) would consist of experimental conditions that substantially differ in the practical operation of a DEFC, which is not desirable.
3.3. Effect of the mass-transport rate at rotating disc electrodes
The influence of the mass-transport on the potentiodynamic ethanol electrooxidation was investigated by means of RDE. Fig. 7 displays cyclic voltammetries acquired on Pt/C in 0.5 M H2SO4 + 0.1 M EtOH at different revolution rates of the RDE: ω = 0, 100, 225, 400, 900, 1600, 2500 rpm. Three separate features of oxidation current are attributed to the EOR: the first one between E = 0.3 and 1 V vs. RHE, a second one at E > 1.1 V vs. RHE (both in the anodic scan), and a last one located between E = 0.8 and 0.4 V vs. RHE during the cathodic scan. | ||
| Fig. 7 (a) CV in 0.5 M H2SO4 + 0.1 M EtOH on Pt/C (metal loading: 9.9 μg) on a RDE at different rotation speeds: 0, 100, 225, 400, 900, 1600, and 2500 rpm; evolution of the peak current between (b) E = 0.3 and 1 V vs. RHE (Ip,a1) and between (c) E = 0.8 and 0.4 V vs. RHE (Ip,c) against the rotation speed; ν = 10 mV s−1; T = 25 °C. | ||
It can be observed that the current between E = 0.3 and 0.85 V vs. RHE increases together with the rise of the revolution rate. The reactions occurring in this potential range are therefore to some extent mass-transport limited. Besides, the potential of the peak current (Ip,a1) shifts toward lower values with the rise of the revolution rate. This phenomenon, also observed for formic acid and formaldehyde oxidation reactions, is however opposite to that observed for the methanol oxidation reaction (MOR):20 the (different) behavior of methanol electrooxidation is attributed to an enhancement of the formaldehyde formation over the CO2 production under increased mass-transport conditions, which thus induce a reduction of the number of exchanged electrons20 and thus a decrease of the generated current. In the present case, the increase of the EOR current at large revolution rates is ascribed to the fast ethanol supply to the electrocatalytic sites free of the ethanol adsorbates, which formerly oxidized into acetaldehyde (the kinetics of this reaction being fast). However, the enhanced mass-transport conditions also lead to the faster poisoning of the Pt sites by bulk contaminants (hydroxide, sulfate and strong ethanol adsorbates), which thus results in the shift toward lower potential of the EOR oxidation peak (referred as “a1” in Fig. 7a). During the cathodic scan, the amplified rise of the current at E < 0.8 V vs. RHE (potential at which hydroxide species start to reduce and to free the Pt sites) at fast revolution rates is also attributed to a faster supply in the bulk reactant to the electrocatalyst sites free of ethanol adsorbates, which were oxidized in the meantime.
Interestingly, a non-linear dependence of the peak current between E = 0.3 and 1 V vs. RHE (Ip,a1) and between E = 0.8 and 0.4 V vs. RHE (Ip,c) against the square root of the revolution rate is observed in Fig. 7b. This result was expected and illustrates the complexity of ethanol electrooxidation (inducing slow kinetics), which is a multi-step reaction yielding up to 12 electrons. In particular, the reaction rate may not only depend on the access to the bulk reactant (linear behavior of the “limiting current” vs. ω1/2, Levich equation), but also to the formation/desorption/poisoning of adsorbed species, therefore giving this peculiar behavior (quasi-absence of limiting current, presence of multiple peaks with activation/inhibition regions).
However, by segmenting in Fig. 7b the x-axis in two regions, at 0 < ω1/2 < 20 rpm1/2 (I) and 20 < ω1/2 < 50 rpm1/2 (II), two straight lines (the slope of which equals pI = 2.78 × 10−3 mV rpm−1/2 and pII = 1.37 × 10−3 mV rpm−1/2 respectively) can be used to model the peak current evolution against the speed rotation. The ratio between the two distinct slopes, which equals pI/pII = 2.03, indicates by the Levich equation that a loss of two electrons occurs during the EOR mechanism at high rotation speed (20 < ω1/2 < 50 rpm1/2) versus that at low rotation speed (0 < ω1/2 < 20 rpm1/2). This loss of two electrons is attributed to a change of the EOR dominating pathway from that leading to the formation of acetic acid (and generating 4e− per ethanol molecule) at low rotation speed to that yielding acetaldehyde as the final product (generating only 2e− per ethanol molecule) at high rotation speed. This result is attributed to: (i) the ejection of freshly formed acetaldehyde (from ethanol dehydrogenation) from the inside of the Pt/C film, which prevents its re-adsorption and further oxidation to acetic acid; (ii) the faster poisoning of the Pt/C surface by ethanol adsorbates at high rotation speed, which impedes OH− adsorption necessary for the formation of acetic acid: and (iii) a combination of both. This matches closely with the results obtained in another similar work on the influence of the flow rate on the EOR mechanism in alkaline medium.32
The oxidation current at E > 1.1 V vs. RHE does not follow the same tendency. Besides a rise of the peak current between ω = 0 and 100 rpm, the current remains quasi-constant independently of the electrode rotation. Actually, it reaches a threshold at 100 rpm, which does not vary with increasing revolution rates. This trend shows that the reaction occurring in this potential region is rather independent of the electrolyte mass-transport and likely corresponds to the electrooxidation of ethanol adsorbates, which form between E = 0.3 and 0.9 V vs. RHE and can only oxidize at E > 0.9 V vs. RHE.31,33,34 Indeed, it is hardly believable that some new species can manage to adsorb at E = 1.1 V vs. RHE on the electrocatalyst surface that is already fully blocked by OH-adsorbates and ethanol adsorbates (formed at lower potential values).
4. Conclusions
The influence of the mass-transport (both diffusion and convection) on the EOR was investigated by means of RDE and DEMS on a Pt/C electrocatalyst. The latter was physically characterized by TGA, XRD and TEM.The comparison between the EOR and the CO-stripping revealed the quasi-non-dependence of the CO2 production rate of the EOR with the scan rate, which contrasts with the dependence of that of the CO-stripping. A different rate determining step, which is the COad mobility on the electrocatalyst surface in the case of the CO-stripping and the C–C bond cleavage for the EOR, may explain this divergent behavior.
The investigation also revealed an enhanced contamination of the electrocatalyst surface by hydroxide and sulfate species and strong ethanol adsorbates (CHx,ad or/and C2-adsorbates) under quasi-stationary conditions (at low scan rate and high RDE speed), which hinders dramatically the complete ethanol electrooxidation into CO2.
These results illustrate the difficulty to completely oxidize ethanol into CO2 under fuel cell stationary conditions. Instead, a 2e− mechanism pathway leading to the formation of acetaldehyde seems to be favored in the near steady-state.
Acknowledgements
Financial support of this work by the German Federal Ministry of Research (BMBF) and the French National Research Agency (ANR) through the “Program Inter Carnot Fraunhofer” part project “Efficient use of Bio-Ethanol in Fuel Cells (EUBECELL)” is gratefully acknowledged. M. Chatenet thanks the Institut Universitaire de France (IUF) for its support.References
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J.-M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS
.
- G. A. Camara and T. Iwasita, J. Electroanal. Chem., 2005, 578, 315–321 CrossRef CAS
- T. Iwasita, B. Rasch, E. Cattaneo and W. Vielstich, Electrochim. Acta, 1989, 34, 1073–1079 CrossRef
- L.-W. H. Leung, S.-C. Chang and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1989, 266, 317–336 CrossRef CAS
- J. M. Perez, B. Beden, F. Hahn, A. Aldaz and C. Lamy, J. Electroanal. Chem. Interfacial Electrochem., 1989, 262, 251–261 CrossRef CAS
- P. Gao, S.-C. Chang, Z. Zhou and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1989, 272, 161–178 CrossRef CAS
- F. Colmati, G. Tremiliosi-Filho, E. R. Gonzalez, A. Berná, E. Herrero and J. M. Feliu, Phys. Chem. Chem. Phys., 2009, 11, 9114–9123 RSC
- S. C. S. Lai and M. T. M. Koper, Faraday Discuss., 2009, 140, 399 RSC
- J. Shin, W. J. Tornquist, C. Korzeniewski and C. S. Hoaglund, Surf. Sci., 1996, 364, 122–130 CrossRef CAS
- J. Perez, V. A. Paganin and E. Antolini, J. Electroanal. Chem., 2011, 654, 108–115 CrossRef CAS
- X. Li, X. Qiu, H. Yuan, L. Chen and W. Zhu, J. Power Sources, 2008, 184, 353–360 CrossRef CAS
- A. Schneider, L. Colmenares, Y. E. Seidel, Z. Jusys, B. Wickman, B. Kasemo and R. J. Behm, Phys. Chem. Chem. Phys., 2008, 10, 1931–1943 RSC
- P. S. Ruvinskiy, A. Bonnefont, M. Bayati and E. R. Savinova, Phys. Chem. Chem. Phys., 2010, 12, 15207–15216 RSC
- P. S. Ruvinskiy, A. Bonnefont, C. Pham-Huu and E. R. Savinova, Langmuir, 2011, 27, 9018–9027 CrossRef CAS PubMed
- K. S. Freitas, B. M. Concha, E. A. Ticianelli and M. Chatenet, Catal. Today, 2011, 170, 110–119 CrossRef CAS
- P.-Y. Olu, C. R. Barros, N. Job and M. Chatenet, Electrocatalysis, 2014, 5, 288–300 CrossRef CAS
- P.-Y. Olu, A. Bonnefont, M. Rouhet, S. Bozdech, N. Job, M. Chatenet and E. Savinova, Electrochim. Acta, 2015, 179, 637–646 CrossRef CAS
- P.-Y. Olu, F. Deschamps, G. Caldarella, M. Chatenet and N. Job, J. Power Sources, 2015, 297, 492–503 CrossRef CAS
- V. Rao, C. Cremers, U. Stimming, L. Cao, S. Sun, S. Yan, G. Sun and Q. Xin, J. Electrochem. Soc., 2007, 154, B1138–B1147 CrossRef CAS
- F. Seland, R. Tunold and D. A. Harrington, Electrochim. Acta, 2010, 55, 3384–3391 CrossRef CAS
- S. L. Gojković, J. Electroanal. Chem., 2004, 573, 271–276 CrossRef
- C. Cremers, D. Bayer, B. Kintzel, M. Joos, F. Jung, M. Krausa and J. Tübke, ECS Trans., 2008, 16, 1263–1273 CAS
- A. B. Delpeuch, M. Chatenet, M. S. Rau and C. Cremers, Phys. Chem. Chem. Phys., 2015, 17, 10881–10893 RSC
- A. Bach Delpeuch, M. Chatenet, C. Cremers and T. Tübke, Electrochim. Acta, 2014, 141, 102–112 CrossRef CAS
- L. Dubau, J. Durst, F. Maillard, L. Guétaz, M. Chatenet, J. André and E. Rossinot, Electrochim. Acta, 2011, 56, 10658–10667 CrossRef CAS
- F. Maillard, E. R. Savinova and U. Stimming, J. Electroanal. Chem., 2007, 599, 221–232 CrossRef CAS
- L. Palaikis, D. Zurawski, M. Hourani and A. Wieckowski, Surf. Sci., 1988, 199, 183–198 CrossRef CAS
- A. Couto, M. C. Pérez, A. Rincón and C. Gutiérrez, J. Phys. Chem., 1996, 100, 19538–19544 CrossRef CAS
- Z. Jusys, J. Kaiser and R. J. Behm, Phys. Chem. Chem. Phys., 2001, 3, 4650–4660 RSC
- A. Bach Delpeuch, F. Maillard, M. Chatenet, P. Soudant and C. Cremers, Appl. Catal., B, 2016, 181, 672–680 CrossRef CAS
- H. Wang, Z. Jusys and R. j. Behm, Fuel Cells, 2004, 4, 113–125 CrossRef CAS
- J. O. Meier, C. Cremers, U. Stimming and J. Tübke, ECS Trans., 2013, 50, 2019–2030 CrossRef
- U. Schmiemann, U. Müller and H. Baltruschat, Electrochim. Acta, 1995, 40, 99–107 CrossRef CAS
- V. M. Schmidt, R. Ianniello, E. Pastor and S. González, J. Phys. Chem., 1996, 100, 17901–17908 CrossRef CAS
Footnotes |
| † Current address: Department of Industrial Engineering, Section of Chemistry for Technologies, University of Padova, Via Marzolo 1, I-35131 Padova (PD), Italy. |
| ‡ Member of the French University Institute (IUF). |
| This journal is © the Owner Societies 2016 |