
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid acquisition of wideline MAS solid-state NMR spectra with fast MAS, proton detection, and dipolar HMQC pulse sequences†
Aaron J.
Rossini
*ab,
Michael P.
Hanrahan
ab and
Martin
Thuo
bc
aIowa State University, Department of Chemistry, Ames, IA 50011, USA. E-mail: arossini@iastate.edu; Tel: +1-515-294-8952
bUS DOE Ames Laboratory, Ames, Iowa 50011, USA
cIowa State University, Materials Science and Engineering Department, Ames, IA 50011, USA
First published on 22nd August 2016
Abstract
The solid-state NMR spectra of many NMR active elements are often extremely broad due to the presence of chemical shift anisotropy (CSA) and/or the quadrupolar interaction (for nuclei with spin I > 1/2). These NMR interactions often give rise to wideline solid-state NMR spectra which can span hundreds of kHz or several MHz. Here we demonstrate that by using fast MAS, proton detection and dipolar hetero-nuclear multiple-quantum (D-HMQC) pulse sequences, it is possible to rapidly acquire 2D spectra which correlate 1H chemical shifts to the indirectly detected wideline MAS powder patterns of dipolar coupled hetero-nuclei. The D-HMQC pulse sequence enables broadband excitation of the wideline hetero-nuclear NMR spectrum and provides higher sensitivity by detecting the narrower and more sensitive 1H NMR signal. This approach is demonstrated for the rapid acquisition of 2D 1H detected 195Pt solid-state NMR spectra of cisplatin and transplatin and the 71Ga solid-state NMR spectrum of a self-assembled Ga coordination polymer of unconfirmed structure. This approach should be broadly applicable for the rapid acquisition of wideline MAS solid-state NMR spectra of moderately abundant NMR nuclei.
Introduction
Solid-state nuclear magnetic resonance (NMR) spectroscopy is an extremely powerful probe of structure and dynamics for both crystalline and partially ordered/amorphous solid materials.1–7 Many of the elements of the periodic table possess NMR active nuclei that can potentially be studied by solid-state NMR spectroscopy. The solid-state NMR spectra of heavier spin-1/2 nuclei (e.g., 77Se, 113Cd, 109Ag, 183W, 195Pt, 207Pb, etc.) are frequently significantly broadened by chemical shift anisotropy (CSA).6,8 With the increasing availability of high field NMR spectrometers the effects of CSA are becoming more important even for lighter spin 1/2 nuclei such as 13C, 15N, 19F and 31P since the broadening due to CSA scales with the strength of the applied field. Similarly, many integer and half-integer quadrupolar nuclei give rise to very broad solid-state NMR spectra due to inhomogeneous broadening by the quadrupolar interaction.8–12 Here we refer to a solid-state NMR spectrum covering a frequency range greater than 200 kHz as a wideline spectrum. Wideline solid-state NMR spectra of spin-1/2 and quadrupolar nuclei have been used to obtain valuable information about structure, bonding and dynamics for a variety of organic, inorganic and biological systems.13–33Unfortunately, the broadening of solid-state NMR spectra often results in a drastic loss in sensitivity. This is because the sensitivity of an NMR experiment is inversely proportional to the square root of the observed linewidth of the signal when a standard Bloch decay or spin echo pulse sequence is used for acquisition [sensitivity ∝ (FWHH)−1/2 = (πT2*)1/2]. For this reason special wideline solid-state NMR techniques based upon the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence are typically used to reduce experiment times on static samples.8,34–36 CPMG experiments exploit the fact that in solids with inhomogeneously broadened NMR spectra, the effective refocused transverse relaxation time (T2′, measured with a spin echo or CPMG experiment) is usually much longer than the apparent transverse relaxation time (T2*) determined by the observed linewidth. With CPMG detection, multiple spin echoes are acquired in each scan and sensitivity is proportional to the square root of T2′, rather than the typically much shorter T2*. CPMG pulse sequences can provide a dramatic increase in sensitivity.8,34–36
Wideline solid-state NMR spectra are also challenging to acquire due to their broad frequency ranges and the limited excitation bandwidths of conventional pulses. This can be addressed by incorporating frequency swept WURST pulses into static CPMG experiments to improve the bandwidth of excitation and refocusing pulses (WCPMG)37,38 and/or cross-polarization steps (BRAIN-CP).8,39 In many cases the excitation/refocusing bandwidth is insufficient, even with frequency swept pulses, and a frequency stepped, piece-wise acquisition of the wideline NMR spectrum must be performed (i.e., using the VOCS procedure).8,14,16 Static CPMG experiments incorporating frequency swept pulses often provide the best absolute sensitivity since large sample volumes can be used and large excitation bandwidths can be realized. However, the analysis of a solid-state NMR spectrum of a stationary powdered sample will be challenging if there are overlapping powder patterns arising from multiple sites, therefore, MAS experiments could be preferable since they can provide higher resolution.
Application of MAS to ordered/crystalline systems results in narrowing of the peaks and substantially improves the resolution of solid-state NMR spectra of spin-1/2 nuclei. However, in disordered materials or nuclei with other broadening mechanisms the peaks may only be partially narrowed under MAS and often remain broad due to combinations of chemical shift distributions from disorder, susceptibility broadening, second-order quadrupolar interactions, paramagnetic interactions, scalar/dipolar coupling to quadrupolar nuclei, temperature gradients due to MAS, etc. When the peaks are inhomogeneously broadened under MAS, rotor-synchronized CPMG experiments can be applied to improve sensitivity.35,40–42 However, MAS solid-state NMR spectra of heavy spin-1/2 nuclei such as 195Pt and 207Pb often possess isotropic and spinning sideband peaks that are several hundred Hz or several kHz broad, even in highly crystalline materials.15,24,43–49 This is an unfavorable regime where T2* ≈ T2′ and CPMG techniques will provide negligible gains in sensitivity.
An additional complication of MAS experiments arises when the CSA is large. The MAS sideband manifold may cover hundreds of kHz and the signal will be dispersed into numerous sidebands, making it difficult to uniformly excite or refocus the spectrum. For example, square planar Pt complexes usually give rise to wideline 195Pt solid-state NMR spectra covering >800 kHz since the span (Ω) is often greater than 7000 ppm.27,43 Conventional ramped CPMAS experiments are typically not applicable in such cases since they suffer from very poor bandwidth due to the narrowband spin lock pulse in the CP step.50 Subsequently, direct excitation experiments, possibly incorporating frequency swept shaped pulses, are often used to acquire wideline MAS NMR spectra.51–53 Similar to static wideline NMR experiments, if the MAS sideband manifold is not uniformly excited, frequency stepped acquisition of the MAS pattern may be performed.49,51,54 However, in diamagnetic materials direct excitation experiments often give poor sensitivity due to the long longitudinal relaxation times of the heavy spin-1/2 nuclei in solids.27,43,51 We note that broadband polarization transfer from wideline MAS 1H NMR signals has been demonstrated with dipolar TEDOR and INEPT pulse sequences in paramagnetic materials, but, they have not been demonstrated on diamagnetic materials.52,53,55,56
Here we demonstrate that many of the challenges associated with the acquisition of MAS wideline solid-state NMR spectra can be alleviated by fast MAS 1H-detected dipolar hetero-nuclear multiple quantum coherence (D-HMQC) 2D correlation experiments. The dipolar-HMQC pulse sequence is depicted in Fig. 1A and is based upon the classic solution NMR pulse sequence. Gan and Bodenhausen separately demonstrated the use of HMQC pulse sequences in the solid state for the indirect detection of isotropic 14N solid-state NMR spectra by 13C.57–59 Further development and refinement of D-HMQC pulse sequences included optimization of dipolar recoupling schemes and addition of homo-nuclear decoupling during the indirect dimension evolution time (t1).60–65 The advantages of this pulse sequence for obtaining dipolar correlation NMR spectra with half-integer and integer quadrupolar spins are clear;60–66 the only manipulation of the hetero-nucleus involves two relatively broadband π/2 pulses for excitation and reconversion. Importantly, the D-HMQC pulse sequence also incorporates efficient proton detection, which can provide a large gain in sensitivity.67–70
 | ||
| Fig. 1 (A) Constant time D-HMQC pulse sequence used to acquire 2D 1H-detected dipolar correlation spectra. The spin echo block in the middle of the sequence is fixed to a constant duration (2n × τr > t1,max) and the θ pulses are stepped outwards to enable arbitrary, rotor asynchronous t1 increments. (B) The 2D 1H–195Pt D-HMQC correlation spectrum of cisplatin (Alfa Aesar) acquired with a 50 kHz MAS frequency, 32 scans per increment, a 0.5 s recycle delay, m = 27 (2m × τr = 1.08 ms), n = 6 (n × τr = 120 μs), 512 individual t1 increments (256 hyper-complex points) and t1 was incremented in steps of 0.8 μs (1.25 MHz indirect dimension spectral width). θ pulses 0.6 μs in duration with a 278 kHz rf field (60° tip angle) were used. The total experiment time was 2.3 hours. (C) The positive projection of the indirectly detected 195Pt dimension (black trace) is compared to an analytical simulation with the previously reported values of Ω and κ (green trace δiso = −1834 ppm; Ω = 8975 ppm, κ = −0.96).51 An analytical fit of the sideband manifold yielded similar CS tensor parameters (red trace, δiso = −1834 ppm; Ω = 8561 ppm, κ = −0.96). The simulated sideband manifolds were offset from the experimental one to allow better comparison of sideband intensities. | ||
The D-HMQC pulse sequence is applied here for the rapid indirect detection of MAS wideline solid-state NMR spectra of 195Pt (I = 1/2) and the quadrupolar 71Ga (I = 3/2) isotopes. It was very recently suggested that D-HMQC could be beneficial for obtaining solid-state NMR spectra of spin-1/2 nuclei with large CSA such as 195Pt.71 However, to the best of our knowledge, the D-HMQC pulse sequence has never been demonstrated for indirect detection of a wideline MAS solid-state NMR spectrum. This is because in 2D D-HMQC spectra the spectral width of the indirect dimension is normally set equal to the MAS frequency or half of the MAS frequency (i.e., by using a t1-increment of one or two rotor periods, respectively). Here we show that it is possible to use an arbitrary indirect spectral width in the D-HMQC pulse sequence and rapidly indirectly detect wideline MAS solid-state NMR spectra. The high rf powers delivered by the small diameter rf coil of a fast MAS probe provide large excitation bandwidths, enabling the uniform excitation of the indirectly detected wideline solid-state NMR spectra.
Results and discussion
MAS wideline 1H–195Pt D-HMQC experiments
The square planar platinum complexes cisplatin (cis-(NH3)2PtCl2) and transplatin (trans-(NH3)2PtCl2) were chosen as a test case since these complexes possess some of largest known CSA (Ω > 8000 ppm in both cases).43 The large CSA gives rise to 195Pt solid-state NMR spectra which cover >800 kHz at the moderate magnetic field strength of 9.4 T used here (ν0(1H) = 400 MHz, ν0(195Pt) = 86 MHz).43195Pt is a spin-1/2 nucleus which possesses good NMR receptivity [natural abundance = 33.8% and R(195Pt)/R(13C) = 20.7].72 However, 195Pt solid-state NMR is often challenging due to the tendency of 195Pt to give rise to very broad solid-state NMR spectra because of large CSA.Fig. 1B shows the 2D 1H–195Pt D-HMQC spectrum of cisplatin acquired at 9.4 T with a 1.3 mm diameter rotor and an MAS frequency of 50 kHz. The SR421 symmetry based dipolar recoupling sequence73 was used in the D-HMQC pulse sequence here since it is insensitive to rf field inhomogeneity and offsets and subsequently yields better efficiency than rotary resonance recoupling (R3).61 A second 1H–195Pt D-HMQC spectrum of cisplatin was also acquired with an MAS frequency of 40 kHz to confirm the position of the isotropic chemical shift (Fig. S1, ESI†). The 1.3 mm diameter rotor enables fast MAS which provides reasonable 1H NMR resolution and improved sensitivity by lengthening the 1H T2′ and narrowing the 1H NMR lines. As expected, the 2D D-HMQC spectrum of cisplatin correlates the amine protons with a 1H chemical shift of 4.3 ppm to the wideline MAS 195Pt NMR sideband manifold. This 2D spectrum was acquired in an experiment time of only 2.3 hours! This is despite the fact that cisplatin was found to possess an unfavorable 11.7 s 1H longitudinal relaxation time (T1). The 2D 1H–195Pt D-HMQC experiment time is much shorter than those reported by Lucier et al. for acquisition of static 195Pt solid-state NMR spectra of cisplatin with direct excitation WCPMG experiments.43 Their frequency-stepped, piece-wise acquisition of the static 195Pt NMR spectrum required acquisition of 11-subspectra and 9 hours total experiment time (using large sample volumes with 5 mm glass tubes).43 Here, the entire MAS sideband manifold could be uniformly excited with a single transmitter offset. This provides considerable time savings and simplifies acquisition. See below for a comparison of direct and indirectly detected 195Pt solid-state NMR spectra.
There are several clear advantages of the fast MAS D-HMQC approach for acquisition of MAS wideline NMR spectra: (i) the pulse sequence enables broadband excitation, which is sufficient to acquire the wideline MAS 195Pt solid-state NMR spectrum of cisplatin, (ii) a high resolution MAS solid-state NMR spectrum is obtained permitting accurate measurement of the isotropic chemical shift, CS tensor parameters and possibly enabling overlapping powder patterns to be resolved either in the indirectly detected high resolution 195Pt dimension or by correlations to different 1H chemical shifts, (iii) 1H detection provides a substantial absolute gain in signal to noise ratio and significantly reduces experiment times, (iv) the small diameter rotors required for fast MAS solid-state NMR typically only require ca. 5 to 10 μL of sample.
Constant time D-HMQC
In order to indirectly detect the wideline MAS 195Pt sideband manifold a D-HMQC pulse sequence with a constant echo time was used (i.e., constant n, Fig. 1A). In the previous implementations of HMQC in solid-state NMR the indirect dimension evolution period (t1) was incremented in a rotor-synchronized manner and the indirect spectral width was set to the MAS frequency.57,60,63,64 This was accomplished by simultaneously incrementing the separation of the θ pulses by one rotor cycle and increasing the duration of the central spin echo block (by simultaneously incrementing n and t1, Fig. 1A).57,60,63,64 Setting the t1-increment to the rotor period (τr) eliminates sidebands in the indirect dimension and provides maximum sensitivity since the indirect dimension signal will always sample the top of the rotary echoes. Here a constant echo time version of D-HMQC was used because the t1-increment and the spectral width of the indirect dimension can easily be set to any value. This is realized by fixing n to a constant value for all t1-increments within the 2D data set, and then incrementing the θ pulses outwards from the central π-pulse. Note that the constant time pulse sequence also has the advantage that broadening in the indirect dimension due to 1H T2′ relaxation is eliminated.However, there are several minor disadvantages arising from the use of a rotor asynchronous t1-increment: (i) the total duration of the central spin echo block must be set so that it is greater than the maximum t1 evolution time (2n × τr > t1,max). Therefore, depending upon the T2′ of the 1H nuclei a reduction in signal to noise ratio will occur because there is transverse relaxation during the long spin echo element used for all of the increments in the 2D experiment. Note that it is possible to incorporate decoupling schemes into the spin echo block to extend 1H T2′ and partially address this issue.63,64 (ii) When the indirect dimension spectral width is very large (i.e., the t1-increment is very small), then a larger number of points in the indirect dimension are required to capture the decay of the indirectly detected signal. However, in practice truncation of the indirect dimension will broaden the peaks and does not substantially affect the relative intensities of the sidebands. (iii) When the t1-increment is rotor asynchronous, many of the increments composing the 2D spectrum contain very little signal. This is because when the CSA is large signal is only observed when the t1 evolution period approaches integer multiples of the rotor cycle (Fig. S2, ESI†). A larger indirect dimension spectral width therefore decreases the signal to noise ratio and increases the experiment time compared to rotor synchronized indirect dimension spectral widths. Despite these drawbacks, constant time D-HMQC experiments with large indirect dimension spectral widths enables the rapid indirect detection of MAS wideline NMR spectra as evidenced by the short experiment times required for 1H–195Pt D-HMQC experiments on cisplatin and transplatin (ca. 2 hours each). Below we directly quantify the sensitivity gains provided by indirect detection 1H–195Pt D-HMQC as compared to direct detection 195Pt NMR experiments.
Quantifying the sensitivity gains provided by indirect detection
The rapid acquisition of the wideline MAS 195Pt solid-state NMR spectra is enabled by the large gain in sensitivity provided by 1H detection. Here we quantitatively compare the sensitivity of the indirectly detected MAS 1H–195Pt D-HMQC spectrum of cisplatin to (i) a direct excitation MAS 195Pt Bloch decay (pulse-acquire) NMR spectrum, (ii) a 1H–195Pt RESPIRATION-CPMAS74 spectrum and (iii) a direct excitation 195Pt WCPMG static solid-state NMR spectrum. By measuring the factors that restrict the efficiency of the HMQC experiments it is possible to obtain a better understanding of detection limits and identify the factors that control efficiency.The gain in sensitivity (ξ) provided by indirect 1H detection as compared to direct excitation and detection of 195Pt can be estimated by the following modified expression:67,75
 | (1) |
The gain in sensitivity provided by indirect detection can be illustrated and partly quantified by examining the SNR of various 1H solid-state NMR spectra of cisplatin. Fig. 2 compares a 50 kHz MAS 1H solid-state NMR spectra of cisplatin acquired with a standard rotor synchronized spin echo pulse sequence (Fig. 2A), a spin echo pulse sequence with SR421 recoupling applied for a total duration of 2.16 ms (Fig. 2B) and a 1H–195Pt D-HMQC spectrum acquired with the empirically optimized 2.16 ms total SR421 recoupling (Fig. 2C). All spectra were acquired with 16 scans and a 14.8 s recycle delay (which corresponds to 1.3 × T1(1H)) and processed with 200 Hz exponential line broadening. The 1H spin echo NMR spectrum has a SNR of ca. 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600. The 1H spin echo spectrum acquired with SR421 recoupling applied for 2.16 ms has a signal which is reduced by a factor of ca. 6 (0.09/0.015). This is due to relaxation during the 2.16 ms recoupling period, however, the recoupled spin echo spectrum still has a signal to noise ratio of ca. 2100. In the 1H–195Pt D-HMQC spectrum the 1H NMR signal is reduced by a factor 0.015 and 0.09 compared to the spin echo and the recoupled spin echo, respectively. However, despite the low efficiency and losses due to relaxation the 1H–195Pt D-HMQC spectrum still has a SNR of ca. 166 in only 16 scans, corresponding to a sensitivity [SNR/(time)−1/2] of 84 min−1/2.
600. The 1H spin echo spectrum acquired with SR421 recoupling applied for 2.16 ms has a signal which is reduced by a factor of ca. 6 (0.09/0.015). This is due to relaxation during the 2.16 ms recoupling period, however, the recoupled spin echo spectrum still has a signal to noise ratio of ca. 2100. In the 1H–195Pt D-HMQC spectrum the 1H NMR signal is reduced by a factor 0.015 and 0.09 compared to the spin echo and the recoupled spin echo, respectively. However, despite the low efficiency and losses due to relaxation the 1H–195Pt D-HMQC spectrum still has a SNR of ca. 166 in only 16 scans, corresponding to a sensitivity [SNR/(time)−1/2] of 84 min−1/2.
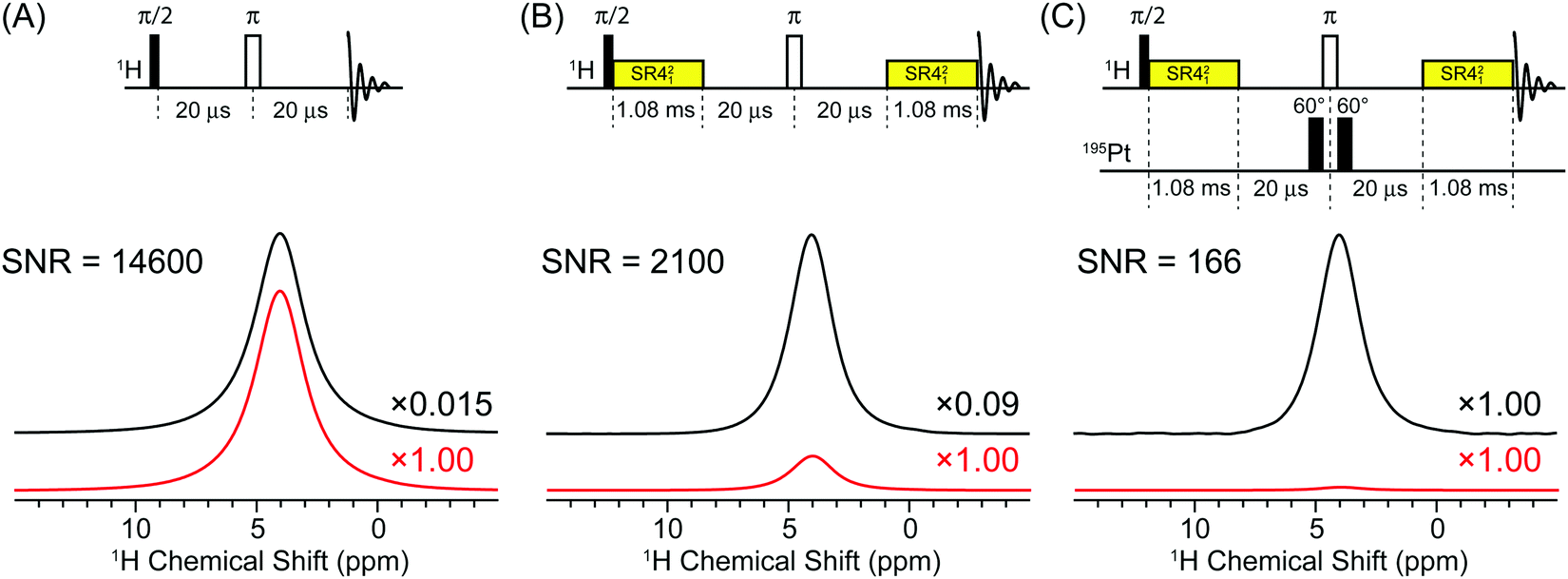 | ||
| Fig. 2 Comparison of 50 kHz MAS 1H solid-state NMR spectra of cisplatin acquired with (A) a rotor synchronized spin-echo pulse sequence, (B) a rotor synchronized spin echo with 2.16 ms of SR421 recoupling and (C) a 1D 1H–195Pt D-HMQC experiment with 2.16 ms of SR421 recoupling. All spectra were acquired with 16 scans and a 14.8 s recycle delay which corresponds to 1.3 × T1. The upper black traces shows all 1H NMR spectra scaled to the same height, with the scaling factors relative to 1H–195Pt D-HMQC spectrum indicated to the right of each spectrum. The lower red traces show the 1H NMR spectra plotted on the same absolute scale. The signal to noise is indicated to the left of each spectrum. | ||
Comparison of the signal intensities in Fig. 2 suggests fHMQC2 ≈ 0.015 and fHMQC ≈ 0.12. However, taking into account that 195Pt is only 33.8% abundant, this suggests fHMQC ≈ 0.36 and fHMQC2 ≈ 0.13. Using eqn (1) with (γ1H/γ195Pt)5/2 = 47, (Ω/νrot) = 15, fHMQC2 ≈ 0.13, (W195Pt/W1H) = 1.2 and assuming [T1(195Pt)/T1(1H)]1/2 ≈ 3 and that all other factors/ratios in eqn (1) equal 1, we calculate ξ = 330. This is for comparison of the sensitivity of the first increment of a 1H–195Pt D-HMQC experiment and a 195Pt NMR spectrum obtained with direct excitation and detection.
In order to estimate the sensitivity gain for a 2D 1H–195Pt D-HMQC experiment, α1/2 was calculated by comparing the SNR of the first increment of the 2D D-HMQC spectrum to the SNR of the indirect dimension of the 2D spectrum. The SNR of the first increment is 15 (with 32 scans × 0.5 s), and this corresponds to a sensitivity of 29 min−1/2. In the 2D spectrum the SNR of the most intense peak in the 195Pt dimension is 22 (with 32 scans × 0.5 s × 512 increments), corresponding to a sensitivity of 1.9 min−1/2 (Fig. 3B). However, the positive projection of the 2D D-HMQC spectrum had a SNR of ca. 83 and sensitivity of 7.2 min−1/2 (Fig. 3C). This analysis indicates α1/2 ≈ 15 (for a single column of the 2D) and α1/2 ≈ 4 (for the positive projection). The measured values of α directly include additional losses in sensitivity from t1-noise since the SNR of the indirectly detected 195Pt spectrum was measured. Therefore, for the full 2D D-HMQC experiment with eqn (1) we estimate ξ is between 22 and 82.
 | ||
| Fig. 3 Comparison of 1H detected and 195Pt detected solid-state NMR spectra of cisplatin. The sensitivity [S = SNR/(time1/2)] is indicated above each spectrum. All MAS experiments were performed with a 50 kHz MAS frequency. (A) Analytical simulation of the MAS 195Pt solid-state NMR spectrum with δiso = −1834 ppm, Ω = 8975 ppm and κ = −0.96. (B and C) MAS 195Pt NMR spectra obtained from the most intense column and the positive projection of the 2D D-HMQC spectrum presented in Fig. 1B (acquired in 2.3 hours). (D) MAS direct excitation 195Pt Bloch decay NMR spectrum obtained with 1024 scans and a 120 s recycle delay (34.1 hours). (E) 1H–195Pt RESPIRATION-CPMAS spectrum obtained with 2000 scans and a 14.8 s recycle delay (8.2 hours). (F) Direct excitation 195Pt WCPMG static NMR spectrum obtained with 680 scans and a 120 s delay (22.7 hours). | ||
Fig. 3 shows direct comparisons of indirectly detected MAS 195Pt spectra obtained from the 2D D-HMQC spectrum (Fig. 1B) and 1D direct detection MAS 195Pt solid-state NMR spectra of cisplatin obtained with Bloch decay and RESPIRATION-CP pulse sequences. After 34.1 hours of acquisition (1024 scans × 120 s) the Bloch decay spectrum has a SNR of 8, corresponding to a sensitivity of 0.18 min−1/2 (Fig. 3D). Comparison of the sensitivity of the Bloch decay spectrum and the 1D 1H–195Pt D-HMQC spectrum (Fig. 2C) obtained with an optimal 14.8 s recycle delay results in a ξ of ca. 467. Comparison to the 2D 1H–195Pt D-HMQC spectrum results in ξ of 10 and 40 if the most intense column or the positive projection of the 2D spectrum are used, respectively. Note that the 0.5 s recycle delay that was used for acquisition of the 2D spectrum is far less than the optimal value of 14.8 s. The sensitivity of the 2D D-HMQC spectrum and ξ could potentially be increased by a factor of ca. 3 with use of the much longer optimal recycle delay (at the expense of a much longer experiment time). Therefore, the measured values of ξ are consistent with those estimated using eqn (1).
A wideline CPMAS 1H–195Pt solid-state NMR spectrum of cisplatin was also obtained with RESPIRATION-CP (Fig. 3E).74 The RESPIRATION-CP pulse sequence has previously been applied to obtain wideline CPMAS 1H–14N and 1H–2H solid-state NMR spectra.76,77 Note that application of the RESPIRATION-CP pulse sequence requires prior knowledge of the peak positions since the carrier frequency for the θ pulses on the hetero-nucleus must be within a few kHz of an isotropic or sideband peak.74,76,77 After 8.2 hours (2000 scans × 14.8 s) the RESPIRATION-CP spectrum had a SNR of 25, corresponding to a sensitivity of 1.1 min−1/2. Comparison of the sensitivity of RESPIRATION-CP to that of the 1D 1H–195Pt D-HMQC gives ξ ≈ 75 and comparison to the 2D 1H–195Pt D-HMQC gives ξ ≈ 1.7 or 6.5 depending if a single column or the positive projection is considered. If 1H detected 1H–195Pt D-HMQC and 195Pt detected 1H–195Pt CPMAS NMR spectra are compared then eqn (1) should be modified so that the ratio of γ term is raised to the power 3/2 (instead of 5/2) and the T1 term should be eliminated since both experiments use the 1H T1. This reduces the maximum estimated ξ by factors of 14 and this is consistent with the reduced ξ observed when comparing the D-HMQC and RESPIRATION-CP spectra.
Finally, a static 195Pt solid-state NMR spectrum of cisplatin was acquired with direct excitation and the WCPMG pulse sequence (Fig. 3F). With WCPMG the entire 195Pt powder pattern was obtained in a single transmitter offset since the 1.3 mm HX probe had a large excitation/receiving bandwidth. The static 195Pt WCPMG NMR spectrum of cisplatin had a signal to noise ratio (SNR) of 27 after a total experiment time of 22.7 hours (680 scans × 120 s delay), corresponding to a sensitivity of 0.7 min−1/2. Comparison of the static WCPMG spectrum and the indirectly detected 1H–195Pt D-HMQC illustrates that D-HMQC simultaneously gives the best sensitivity and provides informative, high resolution 2D NMR spectra.
Measuring 195Pt CS tensor parameters
The 195Pt MAS NMR sideband manifold of cisplatin extracted from the projection of the indirect dimension of the 2D D-HMQC spectrum is shown in Fig. 1C along with an analytical simulation which used the previously published 195Pt CS tensors for cisplatin (green trace (isotropic chemical shift) δiso = −1834 ppm; Ω = 8975 ppm, (skew) κ = −0.96).43 Comparison of the analytical simulations and experimentally measured, indirectly detected sideband manifold shows all of the expected sidebands are present, without a significant reduction in the relative intensity of the outer sidebands. An analytical fit of the observed sideband manifold yields very similar CS tensor parameters with a slightly lower value of Ω obtained (red trace, δiso = −1834 ppm; Ω = 8561 ppm, κ = −0.96). This illustrates that the broad sideband manifold which covers ca. 800 kHz can be uniformly excited. This is possible because the 1.3 mm probe used for the experiments is equipped with a small diameter solenoid coil that provides very high rf fields. A 195Pt rf field of ca. 278 kHz corresponding to a 195Pt π/2 pulse width of 0.9 μs could be obtained with 160 W of input power.Numerical simulations indicate that with a 278 kHz rf field it is possible to excite and detect a broad range of isotropic chemical shifts (Fig. S3A, ESI†). Excitation bandwidths can be significantly increased at the expense of efficiency if smaller tip angle pulses are used. Numerical simulation of the D-HMQC experiment for 195Pt sites with different Ω indicate that θ pulses with tip angles less than 90° provide the largest D-HMQC signal for 195Pt sites with large Ω (Fig. S3B, ESI†). Note that sites with different Ω also show different D-HMQC efficiencies and different oscillation/nutation with the θ pulse tip angle. For sites with large Ω, the there is good agreement between the simulated and experimentally observed variation in the D-HMQC signal of transplatin with the θ pulse tip angle (Fig. S3C, ESI†). Finally, comparison of simulations of the experimental, indirectly detected wideline MAS 195Pt sideband manifold of cisplatin to a numerical simulation confirms that with these conditions the wideline spinning sideband manifold is uniformly excited (Fig. S3D, ESI†). While conventional pulses worked well here, alternative excitation pulses and schemes can possibly further improve excitation bandwidths in HMQC pulse sequences.71,78
2D 1H–195Pt D-HMQC spectra of transplatin were acquired with total experiment times of 1.7 hours and 3.0 hours with MAS frequencies of 50 kHz and 40 kHz, respectively (Fig. 4). Note that these short total acquisition times were possible despite the fact that an unfavorable 1H T1 of 17 s was measured for transplatin. For both MAS frequencies, a high SNR, uniformly excited wideline MAS 195Pt solid-state NMR spectrum was once again obtained. We note that Lucier et al. previously acquired a Bloch decay MAS 195Pt NMR spectrum of transplatin with a similar or lower signal to noise ratios compared to the indirectly detected spectra presented here. However, an experiment time of ca. 20 hours was required with a MAS frequency of 26 kHz and a larger 2.5 mm rotor.43 This again highlights the large gains in absolute sensitivity that can be obtained with 1H detected D-HMQC experiments. Analytical fits of the indirectly detected MAS wideline sideband manifolds once again yield good agreement with the previously published CS tensor parameters and demonstrate uniform excitation of the sideband manifold (Fig. 4).
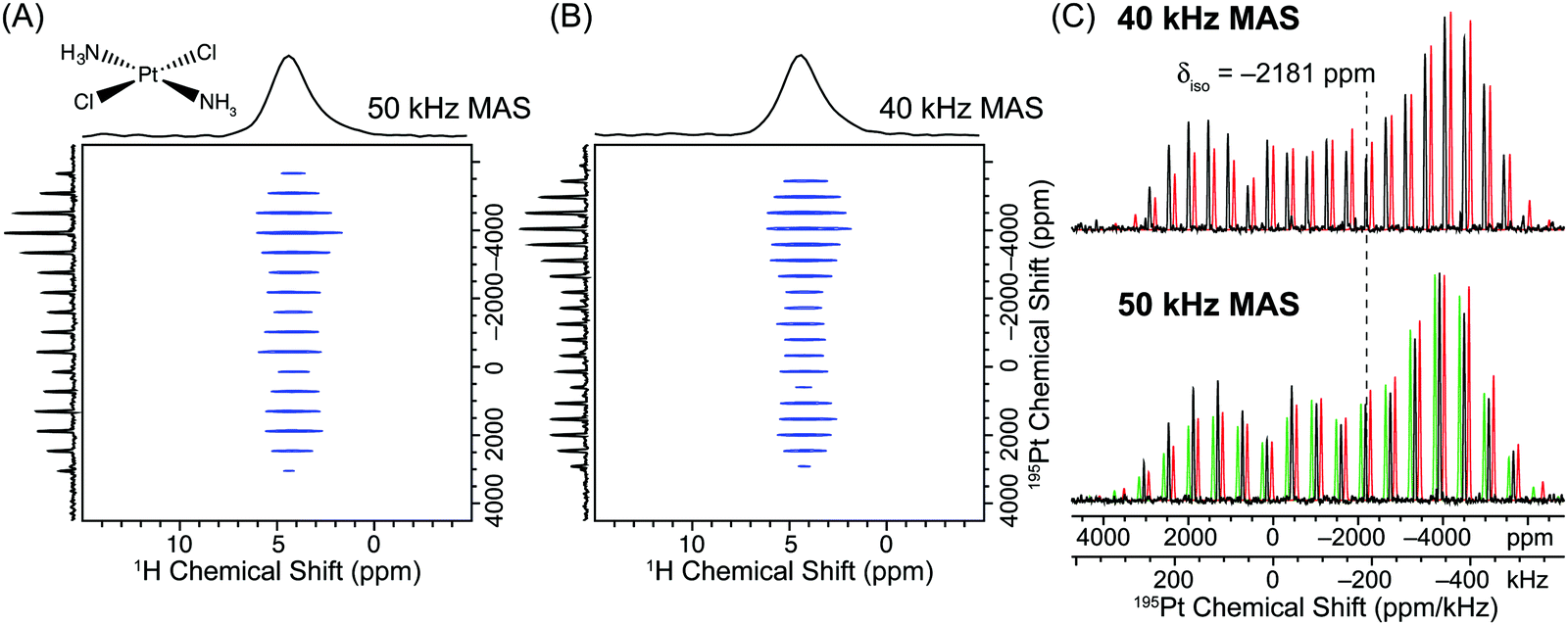 | ||
| Fig. 4 (A) 2D 1H–195Pt D-HMQC correlation spectrum of transplatin (Alfa Aesar) acquired with a 50 kHz MAS frequency, 32 scans per increment, a 0.5 s recycle delay, m = 27 (2m × τr = 1.08 ms), n = 6 (n × τr = 120 μs), 380 individual t1 increments and t1 was incremented in steps of 0.8 μs (1.25 MHz indirect dimension spectral width). The total experiment time was 1.7 hours. (B) 2D 1H–195Pt D-HMQC correlation spectrum acquired with an MAS frequency of 40 kHz, 32 scans per increment, a 1.0 s recycle delay, m = 27 (2m × τr = 1.35 ms), n = 5 (n × τr = 125 μs), 336 individual t1 increments and t1 was incremented in steps of 0.8 μs (1.25 MHz indirect dimension spectral width). The total experiment time was 3.0 hours. In both (A) and (B) θ pulses 0.6 μs in duration with a 278 kHz rf field (60° tip angle) were used for excitation/reconversion of the 195Pt signal. (C) The positive projections of the indirectly detected 195Pt dimension from the 40 kHz (upper set) and 50 kHz MAS frequency (lower set) D-HMQC spectra (black traces) are compared to analytical simulations/fits. For the 50 kHz MAS D-HMQC spectrum an analytical simulation was performed with the previously reported values of Ω and κ (green trace δiso = −2181 ppm; Ω = 9100 ppm, κ = −0.60).28 An analytical fit of the 50 kHz MAS sideband manifold yielded similar CS tensor parameters (lower red trace, δiso = −2181 ppm; Ω = 8850 ppm, κ = −0.64). The analytical fit of the 40 kHz MAS sideband manifold yielded similar CS tensor parameters to those determined from the 50 kHz MAS sideband manifold (upper red trace, δiso = −2185 ppm; Ω = 9001 ppm, κ = −0.63). The simulated sideband manifolds were offset from the experimental one to allow better comparison of sideband intensities. | ||
Detecting Pt containing impurities with 1H–195Pt D-HMQC
During the course of this work 1H–195Pt D-HMQC experiments were also conducted on different samples of cisplatin and transplatin obtained from Sigma Aldrich (Fig. S4, ESI†). This second cisplatin sample showed the presence of several other platinum sites/sideband manifolds with relatively low intensities in the 2D 1H–195Pt D-HMQC spectrum (Fig. S4, ESI†). Note that these other sites also give rise to very broad MAS sideband manifolds which cover a similar frequency range to the cisplatin manifold. These other impurity sites would therefore be challenging or impossible to detect in static experiments. The sample of transplatin from Sigma Aldrich showed the presence of an impurity which must possess an octahedral Pt coordination environment given the much smaller Ω and δiso closer to 0 ppm for this site (Fig. S4, ESI†). The intensity of the signals from the octahedral Pt impurity are strongly amplified in the 2D 1H–195Pt D-HMQC spectrum since the site has a small CSA and all intensity is focused into the isotropic peak. We also note that there were several different spinning sideband manifolds and isotropic chemical shifts which were similar in appearance and position, but not the same as that observed in the Alfa Aesar sample of transplatin. This could indicate that there are polymorphs of transplatin or other impurities present in the second sample of transplatin, however, additional experiments beyond the scope of this work are needed to definitively test this hypothesis. These examples demonstrate the advantages of 2D 1H–195Pt D-HQMC experiments for resolving different Pt sites and detecting dilute Pt containing impurities/phases.Indirect detection of MAS wideline solid-state NMR spectra of quadrupolar nuclei
Quadrupolar nuclei (nuclear spin I > 1/2) frequently give rise to solid-state NMR spectra which are significantly broadened by the quadrupolar interaction (QI), and in some cases by the combined effects of the QI and CSA.9–11 By fitting static and MAS solid-state NMR spectra of the central or satellite transitions of quadrupolar nuclei it is possible to measure the EFG tensor parameters: the quadrupolar coupling constant (CQ) and the EFG tensor asymmetry parameter (ηQ). CQ depends upon the magnitude of the EFG at the nuclear site which is determined by the degree of spherical symmetry at the nuclear site; highly spherically symmetric coordination environments give rise to negligible CQ and minimal broadening, while asymmetric sites will possess large CQ and give rise to very broad solid-state NMR spectra. The presence or absence of rotational symmetry axes at the nuclear site will give rise to extreme values of ηQ of 0 and 1. Measurement of the EFG tensor parameters serves as a powerful probe of bonding, structure and dynamics.10,11,79–85Application of MAS to integer spin quadrupolar nuclei (e.g., 2H and 14N) typically results in relatively narrow peaks and a broad manifold of spinning sidebands, which is reminiscent of the MAS NMR spectrum of a spin-1/2 nucleus subjected to large CSA. However, the intensity of the sidebands and symmetry of the sideband manifold are determined by the EFG tensor parameters, CQ and ηQ. HMQC pulse sequences with rotor synchronized indirect dimensions have been extensively applied to indirectly detect single quantum, double quantum and overtone MAS 14N solid-state NMR spectra.57–59,86–88 Constant time HMQC with large indirect spectral widths could potentially be used to indirectly detect the broad manifolds of spinning sidebands seen in the MAS NMR spectra of integer spin quadrupolar nuclei. However, here we focus on indirect detection of MAS wideline NMR spectra of half-integer quadrupolar nuclei.
Application of MAS to half-integer quadrupolar nuclei results in only a partial narrowing of the NMR spectrum since MAS cannot fully average the broadening of the central transition (CT) or satellite transitions (STs) by the second-order QI. It is important to note that for half-integer quadrupolar nuclei, usually only the central transition (CT) is observed since STs are substantially broadened by the first-order QI. Since the CT is usually inhomogeneously broadened by the second-order QI, CPMG pulse sequences are often applied for signal enhancement of both static and MAS solid-state NMR spectra.34,35,40 Here we demonstrate that D-HMQC can also be applied to indirectly detect wideline MAS solid-state NMR spectra of half-integer quadrupolar nuclei.
Fig. 5 shows MAS and static 71Ga solid-state NMR spectra of a gallium acetate hydroxide coordination complex with an empirical molecular formula of Ga(OH)2(CH3CO2) (1). The absolute solid-state structure of 1 is yet to be confirmed, however, this complex is likely a self-assembled coordination polymer, i.e., [Ga(OH)2(CH3CO2)]m. All Ga ions are likely equivalent and reside in distorted octahedral coordination environments which are formed by bridging hydroxide and acetate ligands. For the purposes of this work the structural details are unimportant, but a more complete investigation of the solid-state structure will be described in a forthcoming publication.89 The static 71Ga solid-state NMR spectrum of 1 is substantially broadened by the second-order QI, resulting in a 71Ga CT powder pattern which covers a frequency range of ca. 380 kHz (Fig. 5A). This indicates that there is a large EFG at the Ga site, which is consistent with a distorted octahedral Ga coordination environment in 1. A static 71Ga solid-state NMR spectrum with a SNR of 41 (sensitivity = 14.1 min−1/2) was acquired with the WCPMG pulse sequence38 and a single transmitter offset. The static 71Ga WCPMG NMR spectrum was obtained in only 8.5 minutes due to the favorable combination of a short 71Ga T1 (a 0.5 s recycle delay was used) and long 71Ga T2 (60 spin echoes were acquired over 14 ms). The static 71Ga NMR spectrum can readily be simulated to determine the 71Ga EFG tensor parameters (CQ and ηQ) and estimate the CS tensor parameters (Fig. 5A). These parameters are given in the ESI† (Table S1).
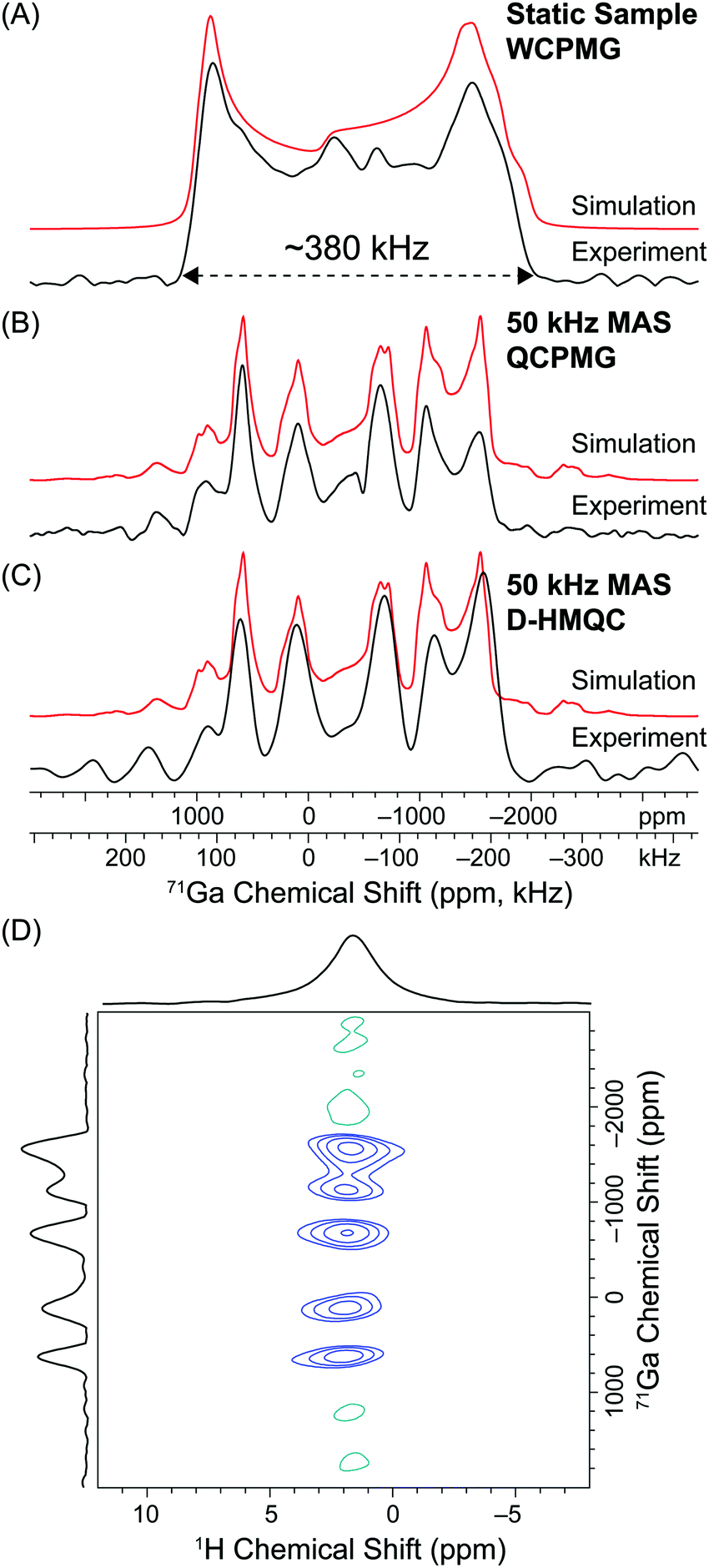 | ||
| Fig. 5 (A) Static 71Ga solid-state NMR spectrum of 1 obtained with the WCPMG pulse sequence. Total experiment time was 8.5 minutes (1024 scans, 0.5 s recycle delay). (B) 50 kHz MAS 71Ga solid-state NMR spectrum of 1 acquired with the QCPMG pulse sequence. The total experiment time was 9.4 minutes (1128 scans, 0.5 s recycle delay). (C) 50 kHz MAS 71Ga solid-state NMR spectrum obtained from the indirect dimension of the 2D 1H–71Ga D-HMQC spectrum at a 1H chemical shift of 1.8 ppm. (D) 2D 1H–71Ga D-HMQC spectrum acquired with an MAS frequency of 50 kHz, 32 scans per increment, a 1.0 s recycle delay, m = 16 (2m × τr = 640 μs), n = 3 (n × τr = 60 μs), 80 individual t1 increments and t1 was incremented in steps of 1.25 μs (800 kHz indirect dimension spectral width). 0.61 μs π/2 CT selective pulses were used for excitation/reconversion of the 71Ga signal. The total experiment time was 43 minutes. Analytical simulations are shown above the experimental spectra (red traces) and EFG and CS tensor parameters are given in the ESI.† | ||
A 50 kHz MAS 71Ga solid-state NMR spectrum with a SNR of 45 was acquired in 9.4 minutes (sensitivity = 14.7 min−1/2) with a rotor-synchronized QCPMG pulse sequence (Fig. 5B). For MAS experiments on half-integer quadrupolar nuclei, the MAS frequency must be greater than the width of the isotropic MAS NMR CT powder pattern to avoid spinning sideband overlap. Here the MAS frequency is not fast enough to avoid overlap of the broad isotropic 71Ga CT powder pattern with its own broad spinning sidebands. At this applied magnetic field a MAS frequency >120 kHz would be required to obtain a “clean” isotropic MAS CT 71Ga powder pattern free of sideband overlap. Subsequently, the 50 kHz MAS 71Ga solid-state NMR spectrum of 1 consists of an isotropic MAS CT powder pattern overlapping with its own sidebands. The resulting spectrum is referred to as an “intermediate MAS NMR spectrum”. At first sight the resulting intermediate MAS NMR spectrum of 1 appears to be much more complicated than the corresponding static spectrum since there are now several discontinuities. However, note that the breadth of the intermediate MAS NMR spectrum is essentially the same as that of the static NMR spectrum. It is therefore possible to easily estimate the magnitude of CQ simply by comparing the breadths of the intermediate MAS NMR spectrum to that of a simulated static solid-state NMR spectrum. Furthermore, the intermediate MAS NMR spectrum can be readily fit with established simulation software packages in order to directly determine CQ and ηQ with reasonable accuracy (Fig. 5B).
Fig. 5C shows the indirectly detected intermediate MAS 71Ga solid-state NMR spectrum of 1 (SNR = 15, sensitivity = 2.3 min−1/2) obtained from the 2D 1H–71Ga D-HMQC correlation spectrum acquired with a MAS frequency of 50 kHz (Fig. 5D). The 2D 1H–71Ga D-HMQC spectrum correlates the 1H nuclei of the acetate/hydroxide ligands to the wideline intermediate MAS 71Ga solid-state NMR spectrum. The 2D 1H–71Ga D-HMQC spectrum was acquired in a total experiment time of 43 minutes which is ca. 4–5 times longer than the time required for the directly detected static or MAS 71Ga QCPMG NMR experiments. 71Ga has a relatively high gyromagnetic ratio (γ1H/γ71Ga = 3.28) and a nuclear spin of I = 3/2. Subsequently the magnetic moment (μ) of 1H is only ca. 1.5 times larger than that of 71Ga. Indirect 1H detection can provide a sensitivity gain of at most ca. 8.7 considering only γ and μ. Furthermore, direct 71Ga WCPMG and QCPMG experiments are very efficient for 1 because the 71Ga T1 is short and the 71Ga T2′ is long. For quadrupolar nuclei with relatively high γ and/or high nuclear spin, proton detection with D-HMQC is unlikely to provide a net gain in sensitivity compared to CPMG techniques. Indeed, here we obtain approximately 7 times higher sensitivity with direct detection CPMG for 71Ga solid-state NMR experiments on 1.
However, the indirectly detected MAS 71Ga solid-state NMR spectrum is very similar in appearance to the directly detected intermediate MAS spectrum. The 2D 1H–71Ga D-HMQC spectrum has the advantage that dispersion of the signals into two dimensions can potentially resolve overlapping intermediate MAS sideband patterns by obtaining correlations to different peaks in the higher resolution 1H dimension. The 2D spectrum also enables the spatial proximities of the metal center and the 1H nuclei of the ligands to be confirmed.
Conclusions
In conclusion we have demonstrated that fast MAS 1H-detected D-HMQC experiments enables the rapid acquisition of wideline MAS solid-state NMR spectra of both spin-1/2 and quadrupolar nuclei. D-HMQC is a promising alternative to CPMG techniques for the acquisition of wideline solid-state NMR spectra of spin-1/2 and quadrupolar nuclei. By employing a simple constant time D-HMQC pulse sequence, arbitrary indirect dimension spectral widths can be easily used. This enabled the wideline MAS solid-state NMR spectrum to be detected in the indirect dimension of the 2D correlation spectrum. The D-HMQC pulse sequence is ideally suited for indirect detection of wideline MAS solid-state NMR spectra because it provides efficient, broadband coherence transfer between 1H and the hetero-nucleus and a large gain in sensitivity from proton detection. D-HMQC enabled rapid acquisition of 2D 1H–195Pt correlation sold-state NMR spectra of cisplatin and transplatin in experiment times of ca. 2 hours. Comparison of 195Pt solid-state NMR spectra obtained with direct detection and the 1H detected 2D D-HMQC spectrum demonstrate that indirect detection provides sensitivity gains between 1 to 2 orders of magnitude. 2D 1H–71Ga D-HMQC experiments required slightly longer experiment time than direct detection MAS and static CPMG experiments. However, the 2D NMR spectrum offers the advantage that overlapping sites could potentially be resolved by correlation to different 1H chemical shifts. This would be similar to the way in which overlapping MAS quadrupolar NMR powder patterns correlated to isotropic chemical shifts are resolved in an MQMAS experiment. In this regard high magnetic fields would obviously be very advantageous for experiments with half-integer quadrupolar nuclei since the 1H chemical shift dispersion would increase and simultaneously the breadth of the quadrupolar NMR powder pattern would decrease.One key drawback of the D-HMQC pulse sequence is that both the MAS frequency and spectrometer must be very stable to avoid excessive t1-noise because the signals from uncoupled 1H spins are imperfectly suppressed by phase cycling alone.62,65,90,91 Therefore, this method may not be applicable to isotopes with very low abundance, when the hetero-nuclei are dilute or when there are multiple overlapping 1H signals. However, this method should be generally applicable for the rapid indirect detection of 2D solid-state NMR dipolar correlation spectra of a wide variety of moderately abundant spin-1/2 and quadrupolar nuclei that give rise to wideline solid-state NMR spectra or in cases where there are multiple isotropic spin-1/2 signals with large frequency differences. These experiments will likely benefit from the development of probe technology that provide faster MAS rates and larger rf fields.
Experimental
Samples of cisplatin and transplatin were purchased from Alfa Aesar (solid-state NMR spectra shown in Fig. 1–4) and Sigma Aldrich (solid-state NMR spectra shown in Fig. S4, ESI†) and used without further purification. The Ga coordination polymer 1 was prepared in the following way: eutectic gallium–indium metal particles were prepared by the SLICE technique.92 Treatment of the eutectic gallium–indium metal particles with acetic acid under ambient conditions lead to the etching of Ga from the passivating oxide layer. 1 was then observed to precipitate in situ as a white powder which consisted of high aspect ratio nano-beams. The synthesis, composition, structure and properties of 1 will be described in detail in a forthcoming publication.89All solid-state NMR experiments were performed on a Bruker wide-bore 9.4 T (ν0(1H) = 400 MHz) NMR spectrometer equipped with a Bruker Avance III HD console and a Bruker 1.3 mm broadband HX fast MAS probe. 195Pt rf pulses were calibrated on the 127I resonance of potassium iodide. 71Ga rf pulses were calibrated on an aqueous solution of GaCl3 and for solid-state NMR experiments the solution NMR pulse widths were divided by a factor 2 to obtain CT selective pulse widths. 1H rf pulses were directly calibrated on the samples of interest. 1H chemical shifts were referenced to neat tetramethylsilane by the use of adamantane (δiso(1H) = 1.82 ppm) as a secondary chemical shift standard. 195Pt and 71Ga chemical shifts were indirectly referenced to the established chemical shift standards using the previously published relative NMR frequencies.72
D-HMQC experiments were performed with the previously described pulse sequence.60–65 However, as described in the main text, rather than simultaneously incrementing the central spin echo block and t1 evolution period, the central spin echo block was fixed to a constant time period and the θ pulses were incremented outwards during the 2D experiments. In all D-HMQC experiments the symmetry based recoupling sequence supercycled (S)R421,73 was applied to the 1H nuclei for a fixed duration in order to build-up and re-convert anti-phase coherence (IxSz) via hetero-nuclear dipolar couplings.60,61 We note that other recoupling schemes are also available.61 In all cases the rf field for SR421 was set to two times the sample spinning rate to fulfill the second order rotary resonance recoupling (R3) condition. The second order R3 condition was precisely calibrated by performing a 1H 90° pulse-spin lock pulse sequence with the spin lock pulse power varied around the value previously calibrated for a 100 kHz rf field.93 The precise spin lock pulse power leading to the lowest observable 1H NMR signal was then utilized.93 For all D-HMQC experiments the length of the recoupling sequence was empirically optimized on the sample of interest and the recoupling time providing the largest signal was used. For experiments on 195Pt the θ pulse width was directly optimized on the samples and for both cisplatin and transplatin 0.6 μs 60° pulses (corresponding to a 278 kHz rf field) were found to provide optimal signal (Fig. S3, ESI†). Details on the exact experimental settings used for D-HMQC experiments (number of scans, pulse widths, number of t1 increments, recycle delay, etc.) are provided in the figure captions and main text.
The 50 kHz MAS Bloch decay (pulse-acquire) 195Pt NMR spectrum of cisplatin was acquired with a 0.6 μs 60° excitation pulse, 1024 scans and a 120 s delay. Prior to Fourier transform the Bloch decay FID was shifted to the first rotational echo to simplify phasing of the spectrum and eliminate a broad probe background signal which was likely from the 207Pb NMR signal of lead metal. The 50 kHz 1H–195Pt RESPIRATION-CPMAS74,76,77 spectrum was obtained with simultaneous rotor-synchronized 0.3 μs θ pulses with a ca. 160 kHz rf field applied on both 1H and 195Pt. The rf field of the phase alternated spin lock pulses was ca. 100 kHz which corresponds to the second order R3 condition. The contact time was 600 μs which corresponds to 30 repetitions of the RESPIRATION-CP element. The static 195Pt solid-state NMR spectrum of cisplatin was obtained with the previously described WCPMG pulse sequence,38 680 scans and a 120 s recycle delay. WURST-80 pulses 25 μs in duration and sweeping across a spectral width of 1.6 MHz were used for excitation and refocusing. The 195Pt rf field was 70 kHz for the WURST pulses.
71Ga WCPMG experiments were performed with the previously described pulse sequence.38 Continuous wave 1H hetero-nuclear decoupling with an rf field of 50 kHz was applied for the duration of the static WCPMG experiments. The 71Ga solid-state NMR spectrum of 1 was acquired with 25 μs WURST pulses with a total frequency sweep width of 600 kHz and a single transmitter offset. Two static 71Ga NMR spectra were acquired with WURST pulses of opposite sweep direction and were co-added. The two 71Ga WCPMG NMR spectra were acquired with 512 scans, a 0.5 s recycle delay and 60 echoes were acquired, each of which was 100 μs duration each. The intermediate MAS 71Ga solid-state NMR spectrum was acquired with an MAS frequency of 50 kHz with a rotor synchronized QCPMG pulse sequence34,35 with 1128 scans and a 0.5 s recycle delay. The MAS QCPMG spectrum was acquired with 0.94 μs and 1.88 μs central transition selective π/2 and π pulses and 40 echoes were acquired, each of which was 200 μs in duration. Echo reconstructed MAS and static 71Ga SSNMR spectra were formed from the CPMG spectra by co-adding each of the spin echoes in the CPMG echo-train together in the time domain, then Fourier transforming the resulting whole spin echo.94,95
Analytical simulations of static and MAS 71Ga and 195Pt solid-state NMR spectra were performed in the solid lineshape analysis (SOLA) module v2.2.4 included in the Bruker Topspin v3.2 software. Numerical simulations of the 195Pt D-HMQC experiments (Fig. S3, ESI†) were performed with the SIMPSON v.4.1 software package running on a personal computer.96–98
Acknowledgements
A. J. R. thanks Iowa State University and the Ames Laboratory (Royalty Account) for support. The Ames Laboratory is operated for the U.S. DOE by Iowa State University under contract no. DE-AC02-07CH11358. M. T. acknowledges support from Iowa State University through startup funds and a Black & Veatch Faculty Fellowship. We thank Dr Simge Çınar (Iowa State University, Materials Science and Engineering Department) for synthesizing the gallium coordination polymer. We thank Dr Luke A. O'Dell (Deakin University) for helpful comments and reading of the manuscript. We thank Dr Julien Trebosc (Université Lille) for providing initial D-HMQC pulse programs.References
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- Special Issue “Frontiers in Solid State NMR Technology”, Acc. Chem. Res., 2013, 46.
- Special Issue on Solid-State NMR Spectroscopy, Phys. Chem. Chem. Phys., 2009, 11.
- D. D. Laws, H. M. L. Bitter and A. Jerschow, Angew. Chem., Int. Ed., 2002, 41, 3096–3129 CrossRef CAS.
- C. Dybowski and S. Bal, Anal. Chem., 2008, 80, 4295–4300 CrossRef CAS PubMed.
- K. J. D. Mackenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, Oxford, 1st edn, 2002 Search PubMed.
- M. J. Duer, Solid-State NMR Spectroscopy Principles and Applications, Blackwell Science, Oxford, UK, 2002 Search PubMed.
- R. W. Schurko, Acc. Chem. Res., 2013, 46, 1985–1995 CrossRef CAS PubMed.
- A. P. M. Kentgens, Geoderma, 1997, 80, 271–306 CrossRef CAS.
- S. E. Ashbrook and M. J. Duer, Concepts Magn. Reson., 2006, 28A, 183–248 CrossRef CAS.
- S. E. Ashbrook, Phys. Chem. Chem. Phys., 2009, 11, 6892–6905 RSC.
- J. V. Hanna and M. E. Smith, Solid State Nucl. Magn. Reson., 2010, 38, 1–18 CrossRef CAS PubMed.
- H. E. Rhodes, P.-K. Wang, H. T. Stokes, C. P. Slichter and J. H. Sinfelt, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 3559–3568 CrossRef CAS.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. P. Coutures, Solid State Nucl. Magn. Reson., 1995, 4, 241–248 CrossRef CAS PubMed.
- F. Fayon, I. Farnan, C. Bessada, J. Coutures, D. Massiot and J. P. Coutures, J. Am. Chem. Soc., 1997, 119, 6837–6843 CrossRef CAS.
- A. Medek, V. Frydman and L. Frydman, J. Phys. Chem. A, 1999, 103, 4830–4835 CrossRef CAS.
- A. S. Lipton, G. W. Buchko, J. A. Sears, M. A. Kennedy and P. D. Ellis, J. Am. Chem. Soc., 2001, 123, 992–993 CrossRef CAS PubMed.
- N. Pooransingh-Margolis, R. Renirie, Z. Hasan, R. Wever, A. J. Vega and T. Polenova, J. Am. Chem. Soc., 2006, 128, 5190–5208 CrossRef CAS PubMed.
- K. J. Ooms, V. V. Terskikh and R. E. Wasylishen, J. Am. Chem. Soc., 2007, 129, 6704–6705 CrossRef CAS PubMed.
- A. S. Lipton, R. W. Heck, W. A. de Jong, A. R. Gao, X. J. Wu, A. Roehrich, G. S. Harbison and P. D. Ellis, J. Am. Chem. Soc., 2009, 131, 13992–13999 CrossRef CAS PubMed.
- A. J. Rossini, R. W. Mills, G. A. Briscoe, E. L. Norton, S. J. Geier, I. Hung, S. Zheng, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2009, 131, 3317–3330 CrossRef CAS PubMed.
- P. J. Knijn, P. J. M. van Bentum, E. R. H. van Eck, C. Fang, D. L. A. G. Grimminck, R. A. de Groot, R. W. A. Havenith, M. Marsman, W. L. Meerts, G. A. de Wijs and A. P. M. Kentgens, Phys. Chem. Chem. Phys., 2010, 12, 11517–11535 RSC.
- L. A. O'Dell, R. W. Schurko, K. J. Harris, J. Autschbach and C. I. Ratcliffe, J. Am. Chem. Soc., 2011, 133, 527–546 CrossRef PubMed.
- B. J. Greer, V. K. Michaelis, M. J. Katz, D. B. Leznoff, G. Schreckenbach and S. Kroeker, Chem. – Eur. J., 2011, 17, 3609–3618 CrossRef CAS PubMed.
- R. Hajjar, C. Volkringer, T. Loiseau, N. Guillou, J. Marrot, G. Ferey, I. Margiolaki, G. Fink, C. Morais and F. Taulelle, Chem. Mater., 2011, 23, 39–47 CrossRef CAS.
- F. A. Perras and D. L. Bryce, Angew. Chem., Int. Ed., 2012, 51, 4227–4230 CrossRef CAS PubMed.
- B. E. G. Lucier, K. E. Johnston, W. Q. Xu, J. C. Hanson, S. D. Senanayake, S. Y. Yao, M. W. Bourassa, M. Srebro, J. Autschbach and R. W. Schurko, J. Am. Chem. Soc., 2014, 136, 1333–1351 CrossRef CAS PubMed.
- C. A. O'Keefe, K. E. Johnston, K. Sutter, J. Autschbach, R. Gauvin, J. Trébosc, L. Delevoye, N. Popoff, M. Taoufik, K. Oudatchin and R. W. Schurko, Inorg. Chem., 2014, 53, 9581–9597 CrossRef PubMed.
- R. Gupta, G. Hou, R. Renirie, R. Wever and T. Polenova, J. Am. Chem. Soc., 2015, 137, 5618–5628 CrossRef CAS PubMed.
- J. Catalano, A. Murphy, Y. Yao, G. P. A. Yap, N. Zumbulyadis, S. A. Centeno and C. Dybowski, Dalton Trans., 2015, 44, 2340–2347 RSC.
- I. D. Seymour, D. S. Middlemiss, D. M. Halat, N. M. Trease, A. J. Pell and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 9405–9408 CrossRef PubMed.
- M. Soorholtz, L. C. Jones, D. Samuelis, C. Weidenthaler, R. J. White, M.-M. Titirici, D. A. Cullen, T. Zimmermann, M. Antonietti, J. Maier, R. Palkovits, B. F. Chmelka and F. Schüth, ACS Catal., 2016, 6, 2332–2340 CrossRef CAS.
- T. Kobayashi, F. A. Perras, T. W. Goh, W. Huang and M. Pruski, J. Phys. Chem. Lett., 2016, 7, 2322–2327 CrossRef CAS PubMed.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, J. Phys. Chem. A, 1997, 101, 8597–8606 CrossRef CAS.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, J. Magn. Reson., 1998, 131, 144–147 CrossRef CAS PubMed.
- I. Hung and Z. H. Gan, J. Magn. Reson., 2010, 204, 256–265 CrossRef CAS PubMed.
- R. Bhattacharyya and L. Frydman, J. Chem. Phys., 2007, 127, 194503 CrossRef PubMed.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- K. J. Harris, A. Lupulescu, B. E. G. Lucier, L. Frydman and R. W. Schurko, J. Magn. Reson., 2012, 224, 38–47 CrossRef CAS PubMed.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, Mol. Phys., 1998, 95, 1185–1195 CrossRef CAS.
- J. Trebosc, J. W. Wiench, S. Huh, V. S. Y. Lin and M. Pruski, J. Am. Chem. Soc., 2005, 127, 7587–7593 CrossRef CAS PubMed.
- G. Kervern, S. Steuernagel, F. Engelke, G. Pintacuda and L. Emsley, J. Am. Chem. Soc., 2007, 129, 14118–14119 CrossRef CAS PubMed.
- B. E. G. Lucier, A. R. Reidel and R. W. Schurko, Can. J. Chem., 2011, 89, 919–937 CrossRef CAS.
- B. E. G. Lucier, K. E. Johnston, D. C. Arnold, J. L. Lemyre, A. Beaupre, M. Blanchette, A. M. Ritcey and R. W. Schurko, J. Phys. Chem. C, 2014, 118, 1213–1228 CAS.
- G. G. Briand, A. D. Smith, G. Schatte, A. J. Rossini and R. W. Schurko, Inorg. Chem., 2007, 46, 8625–8637 CrossRef CAS PubMed.
- R. K. Harris and A. Sebald, Magn. Reson. Chem., 1989, 27, 81–87 CrossRef CAS.
- S. P. Gabuda, S. G. Kozlova, V. V. Terskikh, C. Dybowski, G. Neue and D. L. Perry, Chem. Phys. Lett., 1999, 305, 353–358 CrossRef CAS.
- A. C. Larsson, A. V. Ivanov, K. J. Pike, W. Forsling and O. N. Antzutkin, J. Magn. Reson., 2005, 177, 56–66 CrossRef CAS PubMed.
- S. W. Sparks and P. D. Ellis, J. Am. Chem. Soc., 1986, 108, 3215–3218 CrossRef CAS.
- S. S. Wi, Z. H. Gan, R. Schurko and L. Frydman, J. Chem. Phys., 2015, 142, 064201 CrossRef PubMed.
- A. C. Poppler, J. P. Demers, M. Malon, A. P. Singh, H. W. Roesky, Y. Nishiyama and A. Lange, ChemPhysChem, 2016, 17, 812–816 CrossRef PubMed.
- A. J. Pell and G. Pintacuda, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 84, 33–72 CrossRef PubMed.
- G. Kervern, G. Pintacuda and L. Emsley, Chem. Phys. Lett., 2007, 435, 157–162 CrossRef CAS.
- A. J. Pell, R. J. Clement, C. P. Grey, L. Emsley and G. Pintacuda, J. Chem. Phys., 2013, 138, 114201 CrossRef PubMed.
- N. P. Wickramasinghe and Y. Ishii, J. Magn. Reson., 2006, 181, 233–243 CrossRef CAS PubMed.
- S. K. K. Swamy, A. Karczmarska, M. Makowska-Janusik, A. Kassiba and J. Dittmer, ChemPhysChem, 2013, 14, 1864–1870 CrossRef PubMed.
- Z. H. Gan, J. Am. Chem. Soc., 2006, 128, 6040–6041 CrossRef CAS PubMed.
- S. Cavadini, A. Lupulescu, S. Antonijevic and G. Bodenhausen, J. Am. Chem. Soc., 2006, 128, 7706–7707 CrossRef CAS PubMed.
- S. Cavadini, A. Abraham and G. Bodenhausen, Chem. Phys. Lett., 2007, 445, 1–5 CrossRef CAS.
- J. Trebosc, B. Hu, J. P. Amoureux and Z. Gan, J. Magn. Reson., 2007, 186, 220–227 CrossRef CAS PubMed.
- B. Hu, J. Trebosc and J. P. Amoureux, J. Magn. Reson., 2008, 192, 112–122 CrossRef CAS PubMed.
- O. Lafon, Q. Wang, B. W. Hu, F. Vasconcelos, J. Trebosc, S. Cristol, F. Deng and J. P. Amoureux, J. Phys. Chem. A, 2009, 113, 12864–12878 CrossRef CAS PubMed.
- S. Cavadini, V. Vitzthum, S. Ulzega, A. Abraham and G. Bodenhausen, J. Magn. Reson., 2010, 202, 57–63 CrossRef CAS PubMed.
- M. Shen, J. Trebosc, O. Lafon, F. Pourpoint, B. Hu, Q. Chen and J. P. Amoureux, J. Magn. Reson., 2014, 245, 38–49 CrossRef CAS PubMed.
- G. Tricot, J. Trebosc, F. Pourpoint, R. Gauvin and L. Delevoye, in Annual Reports on Nmr Spectroscopy, Vol 81, ed. G. A. Webb, 2014, vol. 81, pp. 145–184 Search PubMed.
- M. Deschamps and D. Massiot, in NMR of Quadrupolar Nuclei in Solid Materials, ed. R. E. Wasylishen, S. E. Ashbrook and S. Wimperis, John Wiley & Sons, 2012 Search PubMed.
- Y. Ishii and R. Tycko, J. Magn. Reson., 2000, 142, 199–204 CrossRef CAS PubMed.
- Y. Ishii, J. P. Yesinowski and R. Tycko, J. Am. Chem. Soc., 2001, 123, 2921–2922 CrossRef CAS PubMed.
- J. W. Wiench, C. E. Bronnimann, V. S. Y. Lin and M. Pruski, J. Am. Chem. Soc., 2007, 129, 12076–12077 CrossRef CAS PubMed.
- D. H. Zhou, G. Shah, M. Cormos, C. Mullen, D. Sandoz and C. M. Rienstra, J. Am. Chem. Soc., 2007, 129, 11791–11801 CrossRef CAS PubMed.
- M. Shen, J. Trebosc, O. Lafon, Z. H. Gan, F. Pourpoint, B. W. Hu, Q. Chen and J. P. Amoureux, Solid State Nucl. Magn. Reson., 2015, 72, 104–117 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- A. Brinkmann and A. P. M. Kentgens, J. Am. Chem. Soc., 2006, 128, 14758–14759 CrossRef CAS PubMed.
- S. Jain, M. Bjerring and N. C. Nielsen, J. Phys. Chem. Lett., 2012, 3, 703–708 CrossRef CAS PubMed.
- R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Oxford University Press, Oxford, 2nd edn, 1987 Search PubMed.
- S. K. Jain, A. B. Nielsen, M. Hiller, L. Handel, M. Ernst, H. Oschkinat, U. Akbey and N. C. Nielsen, Phys. Chem. Chem. Phys., 2014, 16, 2827–2830 RSC.
- K. Basse, S. K. Jain, O. Bakharev and N. C. Nielsen, J. Magn. Reson., 2014, 244, 85–89 CrossRef CAS PubMed.
- V. Vitzthum, M. A. Caporini, S. Ulzega and G. Bodenhausen, J. Magn. Reson., 2011, 212, 234–239 CrossRef CAS PubMed.
- F. Taulelle, Solid State Sci., 2004, 6, 1053–1057 CrossRef CAS.
- R. L. Vold and G. L. Hoatson, J. Magn. Reson., 2009, 198, 57–72 CrossRef CAS PubMed.
- C. Martineau, B. Bouchevreau, Z. J. Tian, S. J. Lohmeier, P. Behrens and F. Taulelle, Chem. Mater., 2011, 23, 4799–4809 CrossRef CAS.
- X. Q. Kong, L. A. O'Dell, V. Terskikh, E. Ye, R. Y. Wang and G. Wu, J. Am. Chem. Soc., 2012, 134, 14609–14617 CrossRef CAS PubMed.
- F. A. Perras and D. L. Bryce, J. Phys. Chem. C, 2012, 116, 19472–19482 CAS.
- S. E. Ashbrook and S. Sneddon, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef CAS PubMed.
- F. A. Perras, Pure Appl. Chem., 2016, 88, 95–111 CrossRef CAS.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, CrystEngComm, 2012, 14, 2654–2659 RSC.
- L. A. O'Dell, R. L. He and J. Pandohee, CrystEngComm, 2013, 15, 8657–8667 RSC.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, Mol. Pharmaceutics, 2013, 10, 999–1007 CrossRef CAS PubMed.
- S. Çınar, I. Tevis, A. J. Rossini, J. Chen, S. Thimmaiah, N. Bello, M. Foster, B. S. Chang and M. Thuo, 2016, manuscript in preparation.
- Y. Nishiyama, X. Y. Lu, J. Trebosc, O. Lafon, Z. H. Gan, P. K. Madhu and J. P. Amoureux, J. Magn. Reson., 2012, 214, 151–158 CrossRef CAS PubMed.
- M. K. Pandey, H. Kato, Y. Ishii and Y. Nishiyama, Phys. Chem. Chem. Phys., 2016, 18, 6209–6216 RSC.
- I. D. Tevis, L. B. Newcomb and M. Thuo, Langmuir, 2014, 30, 14308–14313 CrossRef CAS PubMed.
- Z. H. Gan, J. Magn. Reson., 2006, 183, 235–241 CrossRef CAS PubMed.
- F. H. Larsen, J. Skibsted, H. J. Jakobsen and N. C. Nielsen, J. Am. Chem. Soc., 2000, 122, 7080–7086 CrossRef CAS.
- R. Lefort, J. W. Wiench, M. Pruski and J. P. Amoureux, J. Chem. Phys., 2002, 116, 2493–2501 CrossRef CAS.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- Z. Tosner, T. Vosegaard, C. Kehlet, N. Khaneja, S. J. Glaser and N. C. Nielsen, J. Magn. Reson., 2009, 197, 120–134 CrossRef CAS PubMed.
- Z. Tosner, R. Andersen, B. Stevenss, M. Eden, N. C. Nielsen and T. Vosegaard, J. Magn. Reson., 2014, 246, 79–93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional solid-state NMR spectra and numerical simulations of solid-state NMR experiments is available. See DOI: 10.1039/c6cp04279a |
| This journal is © the Owner Societies 2016 |