
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinction of electron pathways at titanium oxide/liquid interfaces in photocatalytic processes and co-catalyst effects†
Shota
Kuwahara
a and
Kenji
Katayama
 *b
*b
aDepartment of Chemistry, Faculty of Science, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan. E-mail: syouta.kuwahara@sci.toho-u.ac.jp
bDepartment of Applied Chemistry, Chuo University, 1-13-27, Kasuga, Bunkyo, Tokyo 112-8551, Japan. E-mail: kkata@kc.chuo-u.ac.jp; Fax: +81-3-3817-1913; Tel: +81-3-3817-1913
First published on 12th August 2016
Abstract
Photocatalytic reactions include several different steps and routes for photoexcited carriers, and each dynamic is closely related to the reaction efficiency. Although commonly used time-resolved techniques can reveal the kinetics of photoexcited carriers, the reaction pathways are difficult to distinguish due to decay kinetics extending over many temporal orders and various contributions from the carriers and species involved. Herein, we report the distinction of the electron dynamics in the photocatalytic processes of titanium oxide through the combination of the transient grating method and maximum entropy analysis for the estimation of time constants. We were able to confirm three different carrier responses corresponding to an intrinsic recombination, an interfacial transfer or the decay of surface-trapped electrons, and the decay of polarons. Based on the responses, it appears that both gold and platinum work as good electron acceptors, but that only platinum shortened the lifetime of the polaron state due to the acceleration in the adsorption/desorption exchange of ions, which explains the shorter cycles of the photocatalytic reactions for platinum.
Introduction
Inorganic photocatalytic materials have been utilized for the decomposition of environmental pollution, in particular they are expected to work under sunlight.1 Recently, such materials have been intensively applied to artificial photosynthesis such as hydrogen or oxygen generation and CO2 reduction.2–7 Furthermore, various organic syntheses have been proposed using photocatalytic reactions.8 In these applications, both photo-excited electrons and holes are effectively utilized for reduction and oxidation. For the development of efficient photocatalytic materials, doping of various atoms has been applied to expand the absorption wavelength region,9–14 and deliberately controlled deposition of metal nanoparticles has also been utilized for the enhancement of photocatalytic activity.15,16 Recently, active reaction sites were studied using a single-molecule imaging technique, and the surface activities of the crystals and the deposited metal nanoparticles were revealed.17,18For material researchers, the activity of photocatalytic materials has been typically evaluated from the yield of products. However, from a mechanistic point of view, photocatalytic reactions include various processes; it is necessary to understand the photo-excitation of electrons and holes, diffusion, interfacial charge transfer and trapping of these carriers, formation of radical and intermediate species, etc., and these processes must be well understood to control the total reaction. It has been clarified that the fundamental processes, such as the interfacial charge transfer, charge trapping, and recombination, occur in broad time scales from picoseconds to seconds.19–21 The key point is to specify which process affects the photocatalytic reactions and how they do so.22,23 Time-resolved spectroscopic techniques, such as transient absorption/reflectance spectroscopy, have been utilized16,20,21,24–34 to reveal the dynamics of photogenerated electrons and holes by using a specific absorption band for each carrier from the ultraviolet (UV) region to the infrared (IR) region, and electron–hole recombination and trapping processes have been studied.
With regard to titanium oxide (TiO2), one of the most well-known photocatalysts, it has been clarified that there are some specific absorption bands for photo-excited electrons and holes in the visible to the near-infrared region, and the trapped holes, trapped electrons, and free electrons were assigned to the bands in 400–600 nm, 500–1000 nm, and >500 nm, respectively.20,24,34–36 Mostly, the corresponding transient responses cannot be fitted to a simple exponential decay and they extend over many orders of time; an earlier report by Yamakata et al. showed that the decay kinetics were composed of multiple exponential components with six different lifetimes in the range of sub-nanoseconds to seconds.30 For photogenerated electrons, the responses are typically composed of different lifetimes and have been often observed in the time regions of (i) <1 μs, (ii) 1–10 μs, and (iii) milliseconds to seconds. However, it is still unclear what the differences between each process are and which response is actually responsible for the photocatalytic reaction, meaning which types of electrons are transferred to the reactants. Although electron/hole scavengers or reactive vapors could cause changes in the responses, the reasons have not been understood well. In many cases, the absorption of the target carriers overlapped with those for other carriers and co-catalysts, and the extraction of sufficient information about the target carriers from the obtained transient responses has been one of the issues. Additionally, IR transmission/reflectance spectroscopy is a powerful tool to distinguish carriers, but usually it cannot be applied for photocatalysts in contact with liquid reactants.
On the other hand, the heterodyne transient grating (HD-TG) method is one of the time-resolved methods that monitors the refractive index change due to photochemical or photophysical processes.37 The dynamics of photo-generated electrons and holes in a particulate TiO2 film have been detected by this method.38–40 Furthermore, the generation of active oxygen species on spherical TiO2 surfaces with nanometer sizes in a sol solution and their adsorption/desorption were successfully observed.41 Recently ionic dynamics at the TiO2/electrolyte interface in dye-sensitized solar cells, such as the motion of the charged ion species, were monitored by this method.42,43 This is because the refractive index change is sensitive to the change in dipole at a solid/liquid interface, which is the reason why this method could exclusively detect such transparent dynamics at the interfaces.
In this study, a nanoporous TiO2 thin film in contact with various liquids was measured by the HD-TG technique after UV pump excitation, and the responses were compared in the presence and absence of the chemicals which were subject to the photocatalytic reactions. Photo-generated carriers were successfully detected in three different time regions. Each response was assigned as the electron–hole recombination in a bulk, the trapped electrons at the interface, and the polarons in TiO2, assigned not only by the HD-TG but also by the transient absorption responses, and further confirmed by comparing responses in the presence and absence of the radical and hole scavengers. Based on the results, the effect of the co-catalysts on the carrier dynamics was investigated.
Experimental
A titania nanoparticle paste with an anatase crystal structure (Ti-Nanooxide T, SOLARONIX) was used for the deposition of a transparent mesoporous TiO2 layer. It was coated on a glass slide with a fluorine doped tin oxide (transmittance: 80–82% (visible), Aldrich) using the doctor-blade technique, and it was sintered at 450 °C for 1 h. A cell for the measurement was prepared by putting another glass plate together with a silicon rubber spacer (thickness: 0.5 mm), and a solution was sandwiched within the gap.For the deposition of metal nanoparticles onto the sintered TiO2, a platinum nanoparticle dispersion (3 nm in diameter, Aldrich) was dropped onto the TiO2 substrate. Then, the dispersion was washed away with distilled water after 1 minute. Finally, the prepared substrate was sintered again at 450 °C for 1 h. Gold nanoparticles (5 nm in diameter, stabilized suspension in citrate buffer, Aldrich) were also deposited onto the TiO2 substrate in the same procedure. Since the absorption spectrum for the TiO2 substrate with metal nanoparticles did not change by deposition, the effect of the absorption of the pump and probe light by the nanoparticles is minimal. The dispersions of the gold and platinum nanoparticles were used as purchased. Distilled water, ethanol, and acetonitrile were used as purchased. (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl (TEMPO) was dissolved in acetonitrile at 0.1 M.
In the HD-TG measurements, the pump light source was the third harmonic of a Nd:YAG pulsed laser (pulse width: 4 ns, wavelength: 355 nm) with an intensity of 1–1.5 mJ per pulse, and the probe light source was a Nd:YVO4 laser (wavelength: 532 nm). The grating spacing used for the HD-TG measurement was 80 μm. The responses were detected by a photodiode (DET110, Thorlabs) connected to a voltage amplifier (DHPVA, Femto), the signal was sent to the oscilloscope (Wave Runner 6100A, Lecroy), and the data was stored.
For the lifetime analysis of the obtained HD-TG responses, a hybrid analysis that combines the maximum entropy method (MEM) developed by P. J. Steinbach, MemExp software, was utilized, which can give the lifetime distribution in responses and it is a powerful tool for the analysis for the responses extending over many temporal orders. All fitting data were normalized before the lifetime analysis. Thus, the quantitative information such as the number of carriers was not considered.
Results and discussions
Fig. 1 shows the HD-TG responses for nanoporous TiO2 thin films in contact with liquids. The HD-TG signal intensity decreased initially with an oscillation in the time region of 10−7 to 10−5.5 s, then rose up and back to the baseline in the time region of 10−5–10−2.5 s, which was similar to the responses for a TiO2 thin film in air as previously reported.38 The oscillating signal up to several microseconds was due to an acoustic response induced by the generated heat, and the rising component corresponds to the thermal diffusion in the TiO2/liquid system, and these two responses are typically observed in the HD-TG method. Since a fringe pattern of the pump pulse is irradiated in this technique, photo-excitation is induced by the same pattern. The generated heat causes thermal expansion to occur inhomogeneously according to the fringe pattern, and an acoustic standing wave with a wavelength equal to the fringe spacing is induced, causing oscillation of the signal. Similarly, thermal diffusion occurs in the sample with an inhomogeneous temperature distribution according to the fringe pattern. Since the sample is a nanoporous TiO2 substrate including liquid in the pores and the fringe spacing is much larger than the nanostructure, the thermal diffusion reflects the thermal property of the TiO2/liquid as a whole. The dropping response in the microsecond region is related to the dynamics of the photo-excited carriers.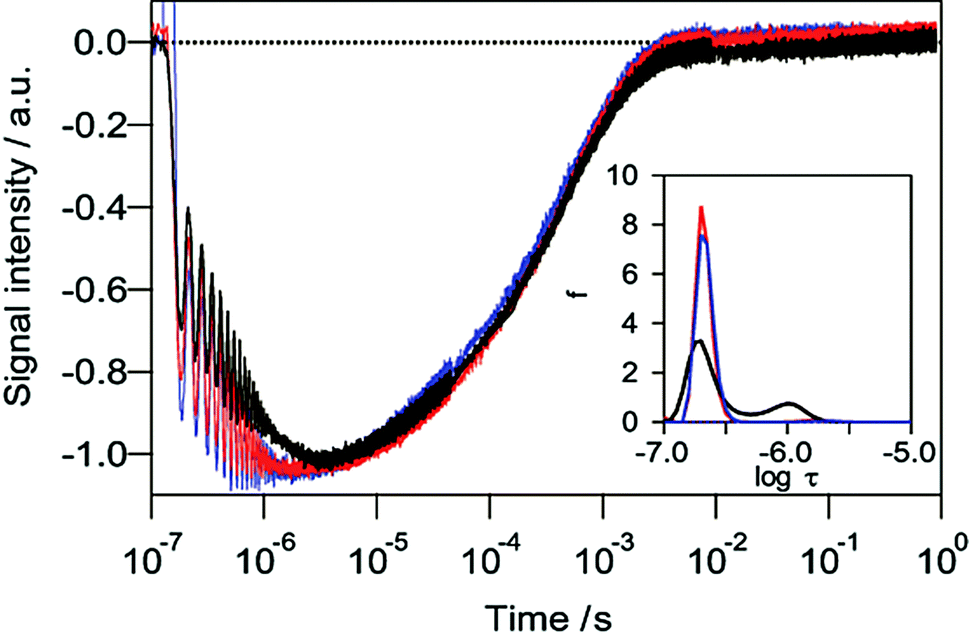 | ||
| Fig. 1 HD-TG responses for TiO2 films in acetonitrile (black) and 0.1 M TEMPO/acetonitrile (blue). HD-TG response in a Pt/TiO2 film in acetonitrile is also shown (red). The time constant distributions were analysed by using the MEM/NLS method in the range of 10−7 to 10−5 s (inset). | ||
The time constant of the dropping response in the time range of 10−7–10−5.5 was analysed using MEM analysis,44 which gives a continuous distribution of the lifetimes of discrete exponential responses by maximizing the entropy of the error between the experimental data and the fitting. As shown in the inset in Fig. 1, the decreasing part consisted of two components with the average time constants of 0.21 μs and 0.96 μs as shown in Table 1 (see the ESI† for the fitting curves). It is thought that the two components obtained in the HD-TG response were caused by trapped electrons or holes in the TiO2 since trapping of the carriers occurs at less than 1 ps.21,22,27
| Photocatalyst | Solvent | τ 1st/μs | τ 2nd /μs |
|---|---|---|---|
| a The time constants for the components which had the amplitudes <0.05 were ignored. | |||
| TiO2 | ACN | 0.21 | 0.96 |
| TiO2 | TEMPO/ACN | 0.21 | — |
| Pt/TiO2 | ACN | 0.21 | — |
| TiO2 | EtOH | 0.21 | 2.4 |
| Pt/TiO2 | EtOH | 0.21 | 4.2 |
To confirm the origin of each component, a commonly used radical scavenger, TEMPO, was added to the solution. In the presence of the radical scavenger, the second component with a time constant of 0.96 μs disappeared, and correspondingly the dropping response became faster than the response in acetonitrile only. It can be understood that the electrons trapped at the surface defects were immediately transferred to TEMPO because the process was faster than the recombination with holes. Similarly the HD-TG response for a platinum deposited TiO2 film also lost the second component (Fig. 1). This result is consistent with the report that photo-generated electrons were transferred to platinum within a few picoseconds for platinum deposited TiO2.27 Based on these results, the second component was assigned as surface-trapped electrons at the TiO2/solution interface (τ = 0.96 μs), while the first component was assumed to be due to intrinsic decay due to the electron–hole recombination inside TiO2 (τ = 0.21 μs). The distinction between electrons and holes was not possible at the moment, and a better time resolution measurements are necessary.
Fig. 2 shows the HD-TG response of a TiO2 film in ethanol, which is well-known to work as a hole scavenger, and the time constant distribution in the microsecond region was analyzed by MEM analysis. The time constant of the second component in ethanol increased compared with that in acetonitrile. It is understood that the probability of recombination with the photo-generated holes was reduced in the presence of the hole scavenger, causing the survival of the surface-trapped electrons. On the contrary, the hole scavenger did not affect the time constant for the first component, probably because the scavenger removed only the surface-trapped holes.
 | ||
| Fig. 2 HD-TG responses in a TiO2 film in acetonitrile (black) and ethanol (green). The time constant distributions were analysed by the MEM analysis in the range of 10−7 to 10−5 s (inset). | ||
Interestingly, a new component appeared in the time range of 10−3–1 s in the presence of ethanol. In this time region, the diffusion of the photo-generated chemical species is often observed. To distinguish whether it is due to the diffusion of species, the grating spacing dependence for the HD-TG response was checked (Fig. S2, ESI†),38 but no dependence was observed, which indicated that this component was not due to the diffusion of species. The HD-TG method also provides the distinction between whether it is due to the refractive index or the absorption change, by changing the phase difference between the signal and the reference probe lights.37 The response up to 10−3 s was reversed by changing the phase, which indicated the signal was due to the refractive index change, while the slower component (10−3 to 1 s) did not, indicating that it was caused by the absorption change. Furthermore, it was confirmed that this slow component was also observed in the transient absorption response (Fig. S2 in ESI†).
Alcohol molecules are known to adsorb onto TiO2 in the form of alkoxide, and it is predicted that photo-generated holes are transferred to the adsorbed alkoxide on the order of femtoseconds, while the photo-generated electrons are trapped and stabilized in the d-orbital of the Ti site, representing the polaron state.45–47 Polarons have been observed by the transient absorption in the near-infrared region only in the presence of water or methanol vapor as hole scavengers,16,30 which are similar conditions to this case. Furthermore, it was confirmed by the naked eye that the TiO2 film turned blue at the pump spot in ethanol when it was irradiated with high excitation light intensity >3 mJ. It is known that this color change is caused by the change in the valence number of Ti (Ti(IV) to Ti(III)).15 The wavelength of the probe light (532 nm) is in the wavelength range for the color change. Thus this slower component (10−3–1 s) was assigned to electrons self-trapped in the form of polarons, localized at the individual Ti site.
The long lifetime of the polaron state is considered. Photo-generated electrons are trapped either in the bulk state or the surface state. In the presence of hole scavengers, a lack of holes in TiO2 reduced the electron–hole recombination, meaning that the transfer of electrons to acceptors on the liquid side was the only pathway for the decay of the remaining electrons. However, the electron acceptors, possibly protons released from alcohol at the interface,48 would be consumed by the surface-trapped electrons; further electron acceptors are necessary for the decay of the excess electrons, which could be transported from the bulk liquid. It was reported that adsorption/desorption for the active oxygen species at the TiO2 nanoparticles/water interface occurred on the order of milliseconds to seconds,41 and it is understood that the lifetime of polarons was controlled by the transport of the electron acceptors; namely the rate of the adsorption/desorption of species on the liquid side (Scheme 1). With regard to the acceptor, we could not detect hydrogen until now, possibly because of the small amount of sample in the cell. Since the response did not depend on the pump intensity, namely the electron number, the trap states were filled with electrons, and the decay was controlled by the acceptors on the liquid side under our experimental conditions.
 | ||
| Scheme 1 Schematic illustrations of the carrier dynamics in photocatalytic processes in the time range of microseconds to seconds; (top) in acetonitrile, (middle) in ethanol, and (bottom) in ethanol in the presence of a co-catalyst. | ||
The effect of the metal nanoparticles deposited on TiO2 was studied from the responses. The component corresponding to the surface-trapped electrons disappeared in the presence of metal nanoparticles, either gold or platinum in acetonitrile, as shown in Fig. 3, which indicated that both types of metal nanoparticle worked as electron acceptors. In ethanol, however, the component for the surface-trapped electrons was observed even in the presence of metal nanoparticles (Fig. 4a). It is understood that the removal of photo-generated holes gave a pathway not only to the metal nanoparticles, but also to the surface-trapped states. The time constants for the surface-trapped electrons were increased for Pt/TiO2 compared with those for TiO2 alone or Au/TiO2 (Fig. 4b), probably because of the better electron/hole separation for Pt/TiO2.
 | ||
| Fig. 3 HD-TG responses in acetonitrile with TiO2 (black), Pt/TiO2 (red) and Au/TiO2 thin film (blue). (inset) The time constant distributions were analysed by MEM analysis in the range of 10−7 to 10−5 s. | ||
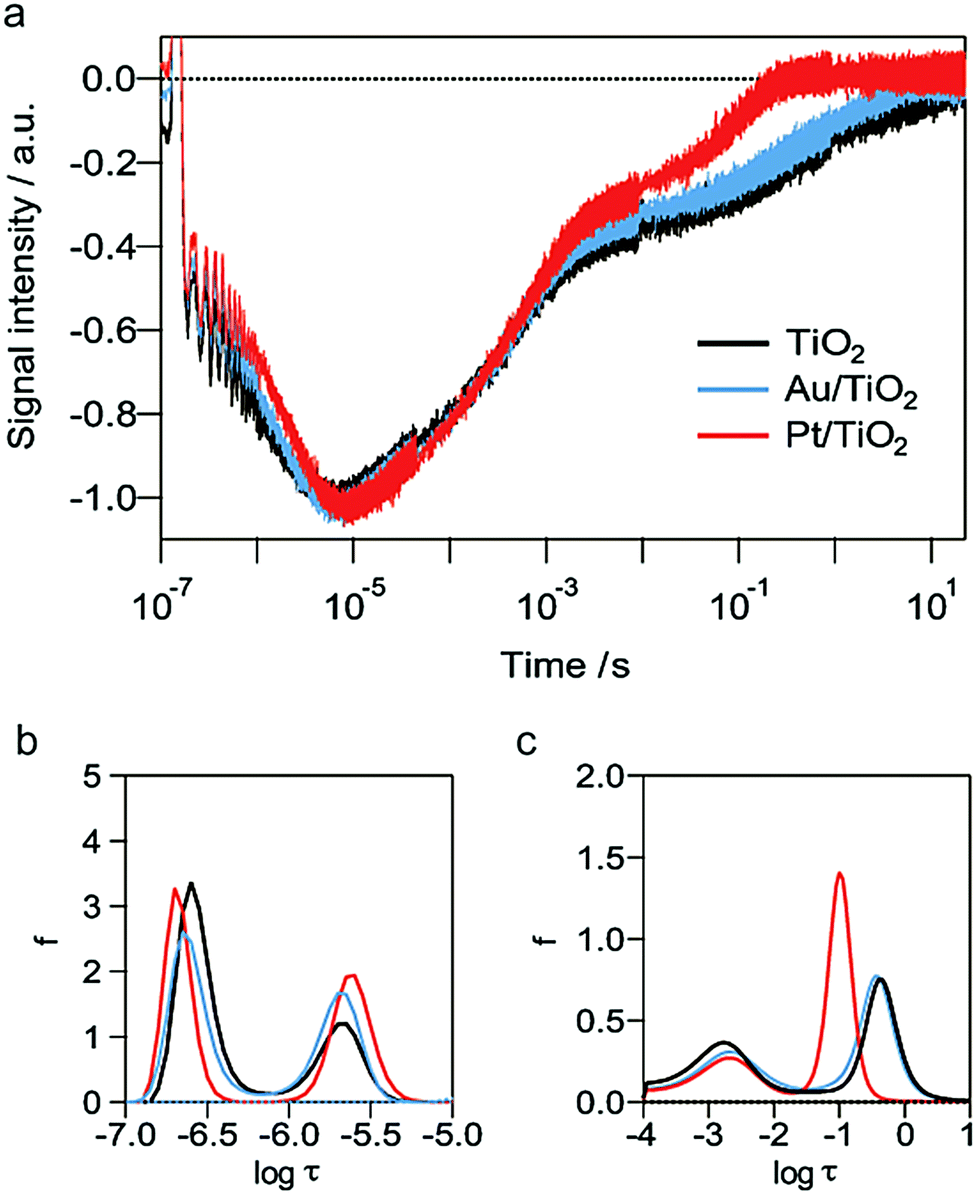 | ||
| Fig. 4 (a) HD-TG responses in ethanol with TiO2 (black), Pt/TiO2 (red) and a dispersion of Au/TiO2 thin film (blue). (b and c) The time constant distributions analysed by the MEM method (b) in the range of 10−7 to 10−5 s and (c) 10−4 to 101 s. | ||
The lifetime of the polarons in ethanol was decreased in the presence of platinum nanoparticles compared with those in TiO2 alone and Au/TiO2. (Fig. 4a, c and Table 1, Scheme 1). The result indicated that the rate of the adsorption/desorption of protons on platinum nanoparticles was faster than those for TiO2 alone and Au/TiO2, as a result of the acceleration of the transport of protons. Muhich et al. reported that the binding energy of the electron scavengers on the surface depended on the defect states or the deposited metals on TiO2 and also the charge stored in TiO2.48 It is assumed that the adsorption/desorption of protons on the platinum nanoparticles was accelerated due to such reasons, and this is in consensus with the high affinity between protons and platinum.
Conclusions
In conclusion, by using the HD-TG method, the three different electron dynamics, the intrinsic recombination with holes, a transfer or the decay of the surface-trapped electrons, and the decay of polarons were successfully distinguished in different time regions, respectively. In the presence of metal nanoparticles on the TiO2 films, the surface-trapped electrons were captured at the nanoparticles in acetonitrile, while a part of the excess electrons could survive in the surface-trap state in the presence of ethanol. The polaron state with a lifetime of 0.1–0.5 s was observed in ethanol solutions, and its lifetime was shortened in the presence of platinum nanoparticles because the adsorption/desorption of electron acceptors was accelerated. The shorter lifetime of polarons in the presence of platinum nanoparticles indicates that the valence number of Ti was regenerated smoothly, and it would provide faster catalytic cycles. The HD-TG method will pave the way to explore the carrier dynamics of various inorganic films by combination with the transient grating method.Acknowledgements
The research was financially supported by the Sasakawa Scientific Research Grant from The Japan Science Society, by JSPS KAKENHI Grant (#15K05549, and #15K17879), and by the Institute of Science and Engineering, Chuo University.Notes and references
- O. M. Alfano, D. Bahnemann, A. E. Cassano, R. Dillert and R. Goslich, Catal. Today, 2000, 58, 199–230 CrossRef CAS.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- N. Hoffmann, Aust. J. Chem., 2015, 68, 1621–1639 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- A. M. Czoska, S. Livraghi, M. C. Paganini, E. Giamello, C. Di Valentin and G. Pacchioni, Phys. Chem. Chem. Phys., 2011, 13, 136–143 RSC.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- R. Jaiswal, N. Patel, D. C. Kothari and A. Miotello, Appl. Catal., B, 2012, 126, 47–54 CrossRef CAS.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454–456 CrossRef CAS.
- M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353–358 CrossRef CAS.
- A. Yamakata, T. Ishibashi and H. Onishi, J. Phys. Chem. B, 2001, 105, 7258–7262 CrossRef CAS.
- T. Tachikawa, S. Yamashita and T. Majima, J. Am. Chem. Soc., 2011, 133, 7197–7204 CrossRef CAS PubMed.
- T. Tachikawa, T. Yonezawa and T. Majima, ACS Nano, 2013, 7, 263–275 CrossRef CAS PubMed.
- T. Tachikawa, S. Tojo, K. Kawai, M. Endo, M. Fujitsuka, T. Ohno, K. Nishijima, Z. Miyamoto and T. Majima, J. Phys. Chem. B, 2004, 108, 19299–19306 CrossRef CAS.
- Y. Tamaki, A. Furube, M. Murai, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2006, 128, 416–417 CrossRef CAS PubMed.
- X. J. Yang and N. Tamai, Phys. Chem. Chem. Phys., 2001, 3, 3393–3398 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2007, 111, 5259–5275 CAS.
- D. W. Bahnemann, M. Hilgendorff and R. Memming, J. Phys. Chem. B, 1997, 101, 4265–4275 CrossRef CAS.
- G. Boschloo and D. Fitzmaurice, J. Phys. Chem. B, 1999, 103, 2228–2231 CrossRef CAS.
- A. Furube, T. Asahi, H. Masuhara, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1999, 103, 3120–3127 CrossRef CAS.
- K. Iwata, T. Takaya, H. Hamaguchi, A. Yamakata, T. A. Ishibashi, H. Onishi and H. Kuroda, J. Phys. Chem. B, 2004, 108, 20233–20239 CrossRef CAS.
- L. Q. Jing, J. Zhou, J. R. Durrant, J. W. Tang, D. N. Liu and H. G. Fu, Energy Environ. Sci., 2012, 5, 6552–6558 CAS.
- J. W. Tang, A. J. Cowan, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2011, 115, 3143–3150 CAS.
- A. Yamakata, T. Ishibashi and H. Onishi, Chem. Phys. Lett., 2001, 333, 271–277 CrossRef CAS.
- A. Yamakata, T. Ishibashi and H. Onishi, J. Phys. Chem. B, 2002, 106, 9122–9125 CrossRef CAS.
- A. Yamakata, M. Kawaguchi, N. Nishimura, T. Minegishi, J. Kubota and K. Domen, J. Phys. Chem. C, 2014, 118, 23897–23906 CAS.
- A. Yamakata, J. J. M. Vequizo and M. Kawaguchi, J. Phys. Chem. C, 2015, 119, 1880–1885 CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- Y. Tamaki, A. Furube, M. Murai, K. Hara, R. Katoh and M. Tachiya, Phys. Chem. Chem. Phys., 2007, 9, 1453–1460 RSC.
- J. W. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- M. Okuda and K. Katayama, Chem. Phys. Lett., 2007, 443, 158–162 CrossRef CAS.
- H. Hata, K. Katayama, Q. Shen and T. Toyoda, Jpn. J. Appl. Phys., 2012, 51, 042601 CrossRef.
- T. Morishita, A. Hibara, T. Sawada and I. Tsuyumoto, J. Phys. Chem. B, 1999, 103, 5984–5987 CrossRef CAS.
- Q. Shen, K. Katayama, T. Sawada, M. Yamaguchi, Y. Kumagai and T. Toyoda, Chem. Phys. Lett., 2006, 419, 464–468 CrossRef CAS.
- M. Okuda, T. Tsuruta and K. Katayama, Phys. Chem. Chem. Phys., 2009, 11, 2287–2292 RSC.
- S. Kuwahara, H. Hata, S. Taya, N. Maeda, Q. Shen, T. Toyoda and K. Katayama, Phys. Chem. Chem. Phys., 2013, 15, 5975–5981 RSC.
- N. Maeda, H. Hata, N. Osada, Q. Shen, T. Toyoda, S. Kuwahara and K. Katayama, Phys. Chem. Chem. Phys., 2013, 15, 11006–11013 RSC.
- P. J. Steinbach, R. Ionescu and C. R. Matthews, Biophys. J., 2002, 82, 2244–2255 CrossRef CAS PubMed.
- N. A. Deskins, R. Rousseau and M. Dupuis, J. Phys. Chem. C, 2009, 113, 14583–14586 CAS.
- G. Kolesov, D. Vinichenko, G. A. Tritsaris, C. M. Friend and E. Kaxiras, J. Phys. Chem. Lett., 2015, 6, 1624–1627 CrossRef CAS PubMed.
- P. M. Kowalski, M. F. Camellone, N. N. Nair, B. Meyer and D. Marx, Phys. Rev. Lett., 2010, 105, 146405 CrossRef PubMed.
- C. L. Muhich, Y. Zhou, A. M. Holder, A. W. Weimer and C. B. Musgrave, J. Phys. Chem. C, 2012, 116, 10138–10149 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fitting examples, TA responses, and grading spacing dependence. See DOI: 10.1039/c6cp04016k |
| This journal is © the Owner Societies 2016 |