
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insight into the catalytic implication of head/tail-first orientation of arachidonic acid in human 5-lipoxygenase: consequences for the positional specificity of oxygenation†
Patricia
Saura
ab,
Jean-Didier
Maréchal
*a,
Laura
Masgrau
 b,
José M.
Lluch
ab and
Àngels
González-Lafont
*ab
b,
José M.
Lluch
ab and
Àngels
González-Lafont
*ab
aDepartament de Química, Universitat Autonòma de Barcelona, 08193 Bellaterra, Barcelona, Spain. E-mail: Jeandidier.Marechal@uab.cat; Angels.Gonzalez@uab.cat
bInstitut de Biotecnologia i de Biomedicina (IBB), Universitat Autonòma de Barcelona, 08193 Bellaterra, Barcelona, Spain
First published on 25th July 2016
Abstract
In the present work we have combined homology modeling, protein–ligand dockings, quantum mechanics/molecular mechanics calculations and molecular dynamics simulations to generate human 5-lipoxygenase (5-LOX):arachidonic acid (AA) complexes consistent with the 5-lipoxygenating activity (which implies hydrogen abstraction at the C7 position). Our results suggest that both the holo and the apo forms of human Stable 5-LOX could accommodate AA in a productive form for 5-lipoxygenation. The former, in a tail-first orientation, with the AA carboxylate end interacting with Lys409, gives the desired structures with C7 close to the Fe–OH− cofactor and suitable barrier heights for H7 abstraction. Only when using the apo form structure, a head-first orientation with the AA carboxylate close to His600 (a residue recently proposed as essential for AA positioning) is obtained in the docking calculations. However, the calculated barrier heights for this head-first orientation are in principle consistent with 5-LOX specificity, but also with 12/8 regioselectivity. Finally, long MD simulations give support to the recent hypothesis that the Phe177 + Tyr181 pair needs to close the active site access during the chemical reaction, and suggest that in the case of a head-first orientation Phe177 may be the residue interacting with the AA carboxylate.
1 Introduction
Lipoxygenases (LOXs) constitute a family of non-heme iron dioxygenases that catalyze the regio- and stereospecific peroxidation of polyunsaturated fatty acids containing 1,4-Z,Z-pentadiene units to give the corresponding conjugated hydroperoxide derivatives. Mammalian LOXs are classified according to the specific position of oxygenation of their main substrate, arachidonic acid (AA), in such a way that they are named 5-, 8-, 12-, and 15-LOX.1–7 AA is the precursor of a large number of functional molecules. Amongst them are leukotrienes and lipoxins which are signaling compounds with pro- and anti-inflammatory effects, respectively, and are involved in a range of inflammatory disorders such as asthma, allergic rhinitis, and have a role in atherosclerosis and some types of cancer.3,8–10 Importantly, while the isolated action of human 5-LOX (h5-LOX) converts AA into leukotrienes, its action in combination with 15-LOX leads to the synthesis of lipoxins.11,12 These two LOX isoforms are not present in the same cell type and lipoxin synthesis has been reported to be transcellular.13 The conversion of 5-LOX into a 15-lipoxygenating form has been proposed as an appealing strategy for the induction of lipoxin synthesis and consequently to turn an inflammatory activity into an anti-inflammatory one. Along this line, mutagenesis experiments on 5-LOX involving residues F359, A424 and N425 allowed one to reduce the volume of its active site cavity and led to an increase of 15-LOX products and a decrease of 5-LOX ones.5,14It is generally accepted that the rate-determining step of the global hydroperoxidation process is the initial hydrogen abstraction from a bisallylic group of AA by the Fe(III)–OH− cofactor, yielding Fe(II)–OH2 and a pentadienyl π radical (see Scheme 1). An attack of molecular oxygen followed by a retro-hydrogen transfer to the peroxy radical produces the final hydroperoxy product and restores the Fe(III)–OH− moiety of the LOX active form.2,10 The positional specificity depends on which is the methylene group that transfers a hydrogen in the first step of the reaction. In fact, there are three bisallylic methylene groups in AA that can transfer a hydrogen atom, and it is assumed7,15,16 that the proximity of these groups to the Fe(III)–OH− cofactor determines the hydrogen atom to be abstracted.
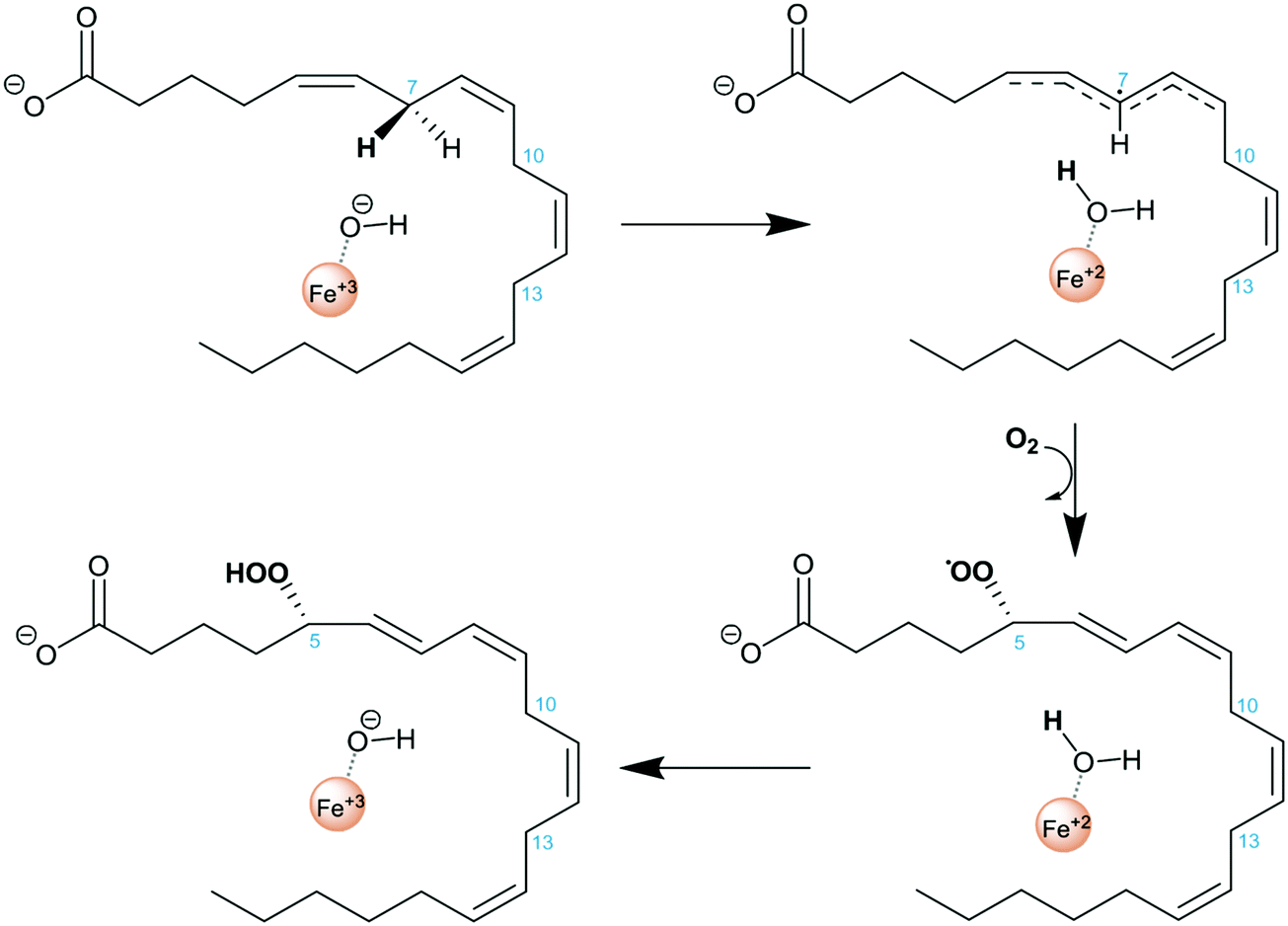 | ||
| Scheme 1 Reaction mechanism of AA hydroperoxidation by 5-LOX. | ||
Several structures of human 5-LOX have been recently released including a substrate bound form.17,18 All were obtained from the so-called human Stable 5-LOX form of the enzyme,17 which consists of a stabilized, soluble (as h5-LOX is a membrane bound protein) and active mutant of h5-LOX. The Stable 5-LOX mutant was solubilized by deletion of putative membrane-insertion amino acids (Δ40 to 44GS, W13E, F14H, W75G and L76S) as well as a pair of cysteines (C240A and C561A). In addition, the enzyme was stabilized by replacing a KKK sequence by an ENL one for residues from 653 to 655. Most of the available structures correspond to a substrate free system (pdb code 3O8Y)17 for which additional mutation could lead to a phosphorylated mimic Ser663Asp (pdb codes 3V98 and 3V99)18 or a non-phosphorylated one Ser663Ala (pdb code 3V92).18 Only one of those structures (pdb code 3V99) represents a substrate bound form of 5-LOX (the AA:Stable 5-LOX-Ser663Asp mutant). While all the substrate free structures (with and without the S663D mutation) are highly similar (rmsd = 0.3 Å), the comparison with the holo form shows an enzyme with substantial plasticity. Between the unbound and bound system the enzyme has suffered a major conformational change, mainly in the region of α2 helix (its end part) and the arched helix. In addition, displacement of the α18 helix is observed, and other regions (some affecting α2 helix) are disordered in the holoenzyme (substrate-bound) crystal structure but not in the apo (substrate-free) one.
Unfortunately, the X-ray coordinates of some residues in crystal structure 3V99 are missing (flexible loops from residues 172 to 176, 190 to 198, 294 to 299, 611 to 613, α helix from residues 414 to 429, and terminal Ile673), and the partial density does not permit18 one to unambiguously determine the orientation of the substrate in the binding site.
Based on the X-ray structures of 5-LOX, two major hypotheses have been raised on how the substrate gains access to the binding cavity: (i) access from the residues Phe177–Tyr181 (FY-cork) or (ii) access from Trp147 which is at the opposite end (Fig. 1).17 The way the substrate enters the binding site will not be discussed here, but the way it is placed once it has entered. So, from here on head/tail-first orientation refers to the relative position of the AA carboxylate group with respect to Lys409, which is supposed to play a similar role as Arg403 in rabbit 15-LOX,19,20 in such a way that tail-first orientation means that AA carboxylate points to the cavity end where Lys409 is placed, whereas head first means the opposite orientation.
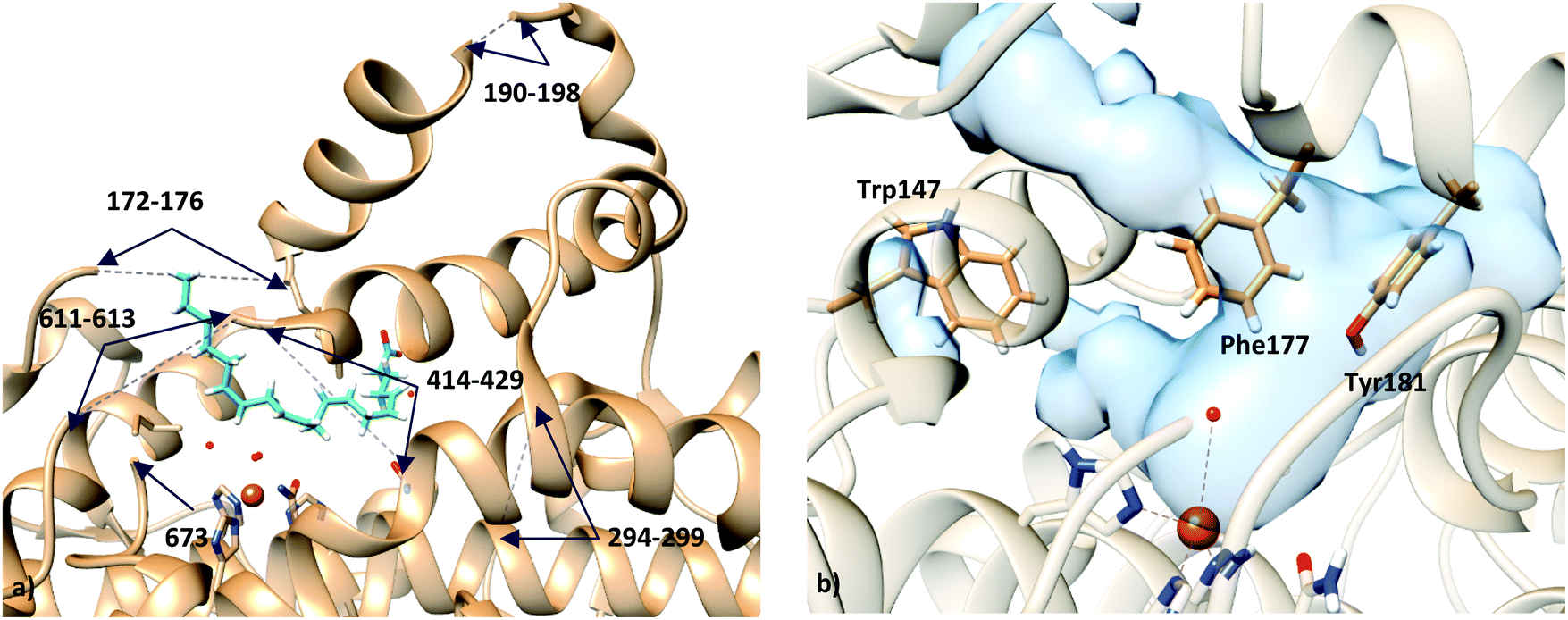 | ||
| Fig. 1 (a) Representation of holo structure 3V99 (subunit A), showing the missing fragments with dashed lines; (b) active site of apo structure 3O8Y, showing the residues proposed for substrate access to the binding site: residue Trp147 in one end, and residues Phe177 and Tyr181 (FY-cork) at the opposite end. | ||
Two hypotheses have been proposed to explain the different positional specificities of lipoxygenases. According to the “space hypothesis”, AA alignment would be more or less conserved (head first) among the LOX isoenzymes, and the volume of the substrate-binding pocket would ultimately tune the positional specificity.2,21 Conversely, assuming the “orientation hypothesis”, tail first would be required for 15-lipoxygenation (which involves an initial hydrogen abstraction from C13),2,21 while only a head-first orientation would make possible the 5-lipoxygenation2,22 (which requires an initial hydrogen abstraction from C7). Very recently, mutagenesis (H600A and H600V) experiments have suggested that His600 (located at the bottom of the cavity on the opposite end of Lys409) is required for productive alignment of the substrate in the active site, presumably because its polar/charged character favors the head-first orientation of AA.23 In the present study, we try to shed some light on the 5-LOX case, for which, to date, no experiment has been able to conclusively determine whether AA is placed head or tail first in 5-LOX.
In the last decade, docking simulations of potential inhibitors of 5-LOX have been widely performed but using the apoenzyme (substrate-free)24,25 structure or the incomplete coordinates of the 3V99 protein structure as a receptor,26 but a complete model of a holoenzyme (substrate-bound) structure has never been proposed. Moreover, previous docking simulations of the AA substrate in the 3O8Y structure have been performed with constraints to keep the carboxylate end close to Lys409/Arg411 and C7 close to the catalytic iron.14 However, other ways of positioning the substrate in the active site, while maintaining its consistency with 5-lipoxygenating catalysis, have not been explored.
In this work, a molecular modeling study is presented, aimed at identifying binding orientations that would determine the catalytic specificity of 5-LOX. A multi-scale approach has been set up and applied. A homology modeling study has first been performed to generate a model of the holo form of human Stable 5-LOX that completes the missing regions of the crystallographic structure (pdb code 3V99). Then a series of intensive protein–ligand docking experiments have been carried out to generate AA:Stable 5-LOX complexes both in the holo model and in the apo structure (pdb code 3O8Y). Different docking solutions, corresponding to tail-first orientation in the holo structure and head-first orientation in both holo and apo structures, have been selected to pursue the reactivity studies. First, as AA is a very flexible ligand, configurations of the complex have been generated by molecular dynamics simulations and a selection of snapshots have been used to perform QM/MM calculations to check the catalytic reliability (energy barrier for hydrogen abstraction consistent with the regio- and stereo-selectivity of the enzyme:AA reaction) of the generated models. Finally, additional 100 ns molecular dynamics simulations have been carried out for each Michaelis complex to gain insight into some structural hypotheses recently proposed in the literature.
2 Methodology
2.1 Homology modeling of the holoenzyme structure
The first step of this study consisted of building a valid and complete 3D model of the holo form of human Stable 5-LOX. As X-ray data indicate that the apo and holo forms present significantly different conformations, we decided to build the holo model based on the only crystal structure of holo 5-LOX available (pdb code 3V99), even though it lacks the coordinates of some residues. Our solution consisted of a mixed strategy that implied both loop modeling and structural overlapping. All the calculations were carried out using the program Modeller (version 9.12).27 We think that our choice of template is justified by the fact that this is actually the only available holoenzyme structure. Nevertheless, molecular dynamics simulations were carried out to structurally relax the generated complexes.Because the objective of this part of the study is to complete the missing regions of the crystallographic structure of the holo form of the enzyme (Table 1), no templates from analogous enzymes were used to construct the model. The structure of subunit A of the pdb 3V99 was used as our template, as it is the only one displaying electron density associated with the presence of the substrate.
| Subunit | Missing segment | Characteristics |
|---|---|---|
| 3V99-A | 172–176 | Loop |
| 190–198 | Loop | |
| 294–299 | Loop | |
| 414–429 | α helix | |
| 611–613 | Loop | |
| 673 | C-terminal Ile |
All the loop regions that lack less than 9 residues were modeled with the loop refinement procedure built in Modeller, using the non-default option slow (to ensure an exhaustive level of conformational sampling and maintain an affordable time of calculation). The region composed of residues 414 to 429 represents a far wider gap and overcomes the limitation in the accuracy of the loop refinement algorithm provided by Modeller.27 Therefore, this region was built using structural matching with the apo structure of the same protein (pdb code 3V92). Last but not least, the initial structure misses the iron-coordinating terminal Ile673 residue. Therefore, a complete coordination sphere of the metal was modeled by applying distance restraints (2.04 Å between the Fe and its coordinating atoms) during the modeling. The Asn554 residue was included in the coordination sphere. In total, the Modeller runs were constituted by the generation of 20 models followed by 20 steps of loop refinement. The full models were generated with a degree of refinement of the Modeller (option slow) that allows a thorough molecular dynamics and simulated annealing procedure. Then, the 20 runs of the loop modeling process used a more extensive refinement procedure (option slow large).
The most encouraging structures were minimized with a full atom force field of CHARMM,28,29 and the quality of the resulting models was assessed with ERRAT,30 VERIFY-3D,31 and the PROCHECK Ramachandran plot32 (web server: http://nihserver.mbi.ucla.edu/SAVES/).
2.2 Docking simulations
Docking simulations were run with the program GOLD5.2.33 Two different receptors were used, the final model of the holoenzyme generated by homology modeling as described above, and subunit A of the structure 3O8Y, corresponding to the apoenzyme. The option of GOLD to deal with the interaction of metal ions of metalloenzymes with organic ligands was active during the docking although limited at the search of hexacoordinated geometries of the metal. Prior to docking, the protonation states of the titrable residues were determined by the PROPKA server.34 Simulations were carried out for both rigid and flexible side chain receptors in all cases except for the mutants of the holoenzyme, for which only rigid receptors were used.To ensure a complete exploration of the conformational space for AA, the maximum efficiency of the genetic algorithm was selected in the docking setup. The binding site cavity of the receptor consisted of a 19 Å radius sphere centered in the Fe atom. The fitness function used was the Chemscore function.33 The parameters of the genetic algorithm were set to the default values of the program. In the case of considering local rearrangements of the LOX binding site, different rotameric schemes were tested selecting the rotamers from the GOLD library.35
Finally, the solutions were analyzed in order to be consistent with catalytic processes. As such, the full benches of docking solutions of each experiment were analyzed throughout a clustering process based on rmsd calculations. After that, in order to identify possible pre-reactive structures, the docking solutions were filtered as a function of the distances between the reactive oxygen and the different unsaturated carbons that could be involved in the mechanism.
2.3 In silico mutants
In silico triple mutant F359W/A424I/N425M has been generated by changing the corresponding residues with the Mutator Plugin of the VMD program.36 However, the side-chain of tryptophan is too large to be put just by simple substitution of the side-chain of phenylalanine. So, for mutation F359W, the position of the side chain of W359 was optimized by CHARMM molecular mechanics (MM) optimization.28,292.4 Reactivity studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, with about 10600 of them belonging to the protein.
000 atoms, with about 10600 of them belonging to the protein.
The system was submitted to some MM minimization steps, and then, MD simulations under periodic boundary conditions (PBC) were started. The system was gradually heated from 25 K to 300 K during 80 ps, followed by an equilibration step of 400 ps at 300 K. During the heating steps, harmonic restraints were applied to the protein, which were gradually released, while the ligand (and water molecules) was kept free of restraints. Finally, a production step of 20 ns was carried out in order to relax the system and to generate a set of snapshots ready for some hydrogen abstraction reactions.
The MD simulations were done at constant pressure and temperature (CPT) using the extended system constant pressure and the Hoover constant temperature algorithms.37 A time step of 2 fs was used for the production steps. All of the bonds and angles involving hydrogen atoms were constrained by the SHAKE algorithm.38 PBCs were built with the CRYSTAL facility of CHARMM. For long-range electrostatics, the particle mesh Ewald method was used. MD simulations were run with CHARMM version c35-b1. The force field topology and parameters for AA were derived from the CHARMM27 ones.39,40 The CHARMM2228,29 force field was used for protein atoms except Fe and its first coordination sphere, for which the parameters specifically derived by Saam et al.41 were used, updating the parameters for the coordination with Asn554. Structural analysis of the simulations was performed with the CORREL module of CHARMM.
All QM/MM calculations have been performed with the modular program package ChemShell,42 using TURBOMOLE43 for all of the DFT calculations. The MM calculations were carried out by the DL_POLY44 module in ChemShell, using the CHARMM2228,29 and CHARMM2739,40 (for the lipid moiety included in the MM region) force fields. An electrostatic embedding scheme45 has been adopted in all calculations, and a hydrogen link atom scheme has been employed to treat the QM/MM boundary using the charge shift model. No cutoffs were introduced for the nonbonding MM and QM/MM interactions.46
QM/MM optimizations were carried out by employing the limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm47,48 in the case of energy minimizations, and the microiterative optimizer, combining both the partitioned rational function optimizer (P-RFO)49,50 and the L-BFGS algorithm, was used during the transition state structure searches. All of these algorithms are implemented in the HDLCopt (Hybrid Delocalized Internal Coordinate Scheme)51 ChemShell module. A small core containing six atoms (C, pro-S and pro-R hydrogens, and Fe–OH− cofactor) was selected for the P-RFO algorithm, which treats these atoms with an explicit Hessian matrix. In every case a transition state structure was located and characterized by a frequency calculation that ensured a single imaginary frequency describing the reactive process under study.
For the QM subsystem the hybrid functional B3LYP was used with the 6-31G(d) Pople basis set52 for all atoms except Fe, which was described by the LANL2DZ basis set.53 The QM region includes 64 atoms (link atoms not included): as for the enzyme, there are 11 atoms of each of the 3 His residues in the Fe coordination sphere (His367, His372, His550), 8 atoms of Asn554, 3 atoms of the terminal Ile673 in the coordination sphere, and the Fe(III)–OH− cofactor, able to accept the hydrogen from AA; the QM region also includes the pentadienyl group around the methylene of interest, and the adjacent C atoms (and its corresponding hydrogens), containing a total of 17 QM atoms of the lipid substrate. Seven link atoms were used, five along the bonds Cα-QM atom of the five residues in the Fe coordination sphere and two in the AA chain. Scheme 2 shows the QM/MM partition.
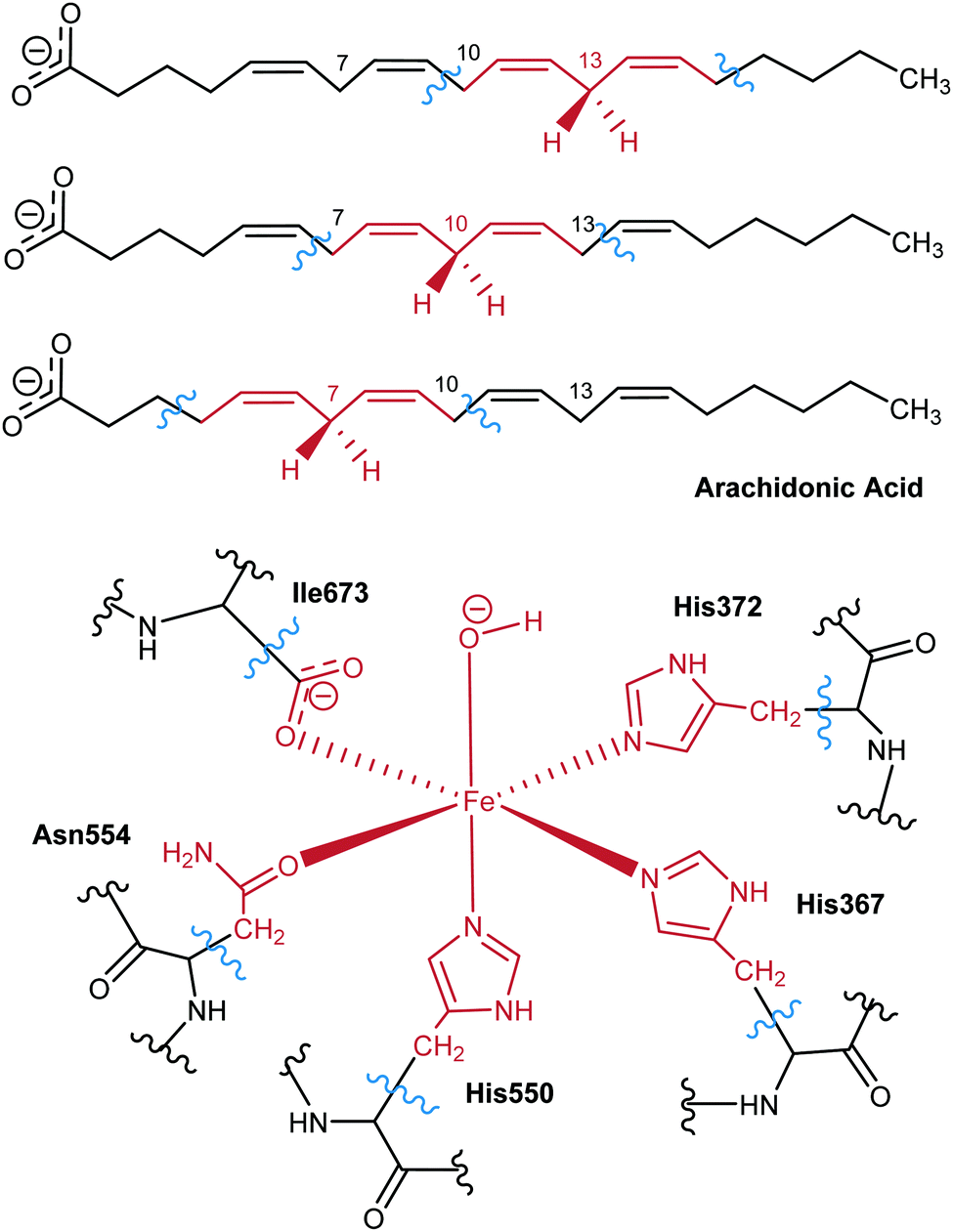 | ||
| Scheme 2 QM region used for each of the hydrogen abstractions considered in this work. | ||
2.5 Molecular dynamics simulations
Long molecular dynamics simulations with production runs of 100 ns were run for each of the three AA:Stable 5-LOX complexes considered in this work. The details of the calculations are as described in Section 2.4 but with box sizes of 118 × 93/80 × 73/71 Å using also PBC for the holo/apo complexes.3 Results and discussion
3.1 Homology modeling of the holoenzyme structure
The use of the only available X-ray structure of holoenzyme to generate a binding mode of AA into human-5-LOX compatible with hydrogen atom abstraction at C7 (the one leading to 5-lipoxygenation) presented the problem that this structure lacks the coordinates of some parts. Thus, we employed homology modeling techniques to build them. A total of 400 3D models of the holo form of the human-Stable 5-LOX have been produced in this part of the study accounting for both modeling and loop refinement processes. The best models were first analyzed based on the energies provided by Modeller and the 3 lowest energy solutions were finally selected, as those values were not discriminative enough. For each of these models, the 3 lowest energy loop models were also selected which led to a total of 9 model candidates that were further refined and analyzed. First, a CHARMM MM optimization of the side chains was performed in order to reduce possible constraints remaining from the modeling process. Then, the quality of the 9 minimized models was evaluated by ERRAT, VERIFY-3D, and the PROCHECK Ramachandran plot. In all cases, the final values of each geometry checker were higher than 90%, which suggests that all candidates are potentially good models for human-Stable 5-LOX (Table 2). The final model was selected using a consensus between both energetic and structural criteria.| Model | ERRAT | VERIFY-3D | PROCHECK |
|---|---|---|---|
| 1 | 90.348 | 92.79 | 91.5 |
| 2 | 89.984 | 93.71 | 91.7 |
| 3 | 90.317 | 92.18 | 91.5 |
| 4 | 93.228 | 93.56 | 92.0 |
| 5 | 93.196 | 93.87 | 92.0 |
| 6 | 93.207 | 93.71 | 92.0 |
| 7 | 94.462 | 94.48 | 91.5 |
| 8 | 93.987 | 92.79 | 92.2 |
| 9 | 94.453 | 91.41 | 92.2 |
According to these results, model 7 is the best solution and the one selected to proceed with the study. Fig. 2 shows the final model.
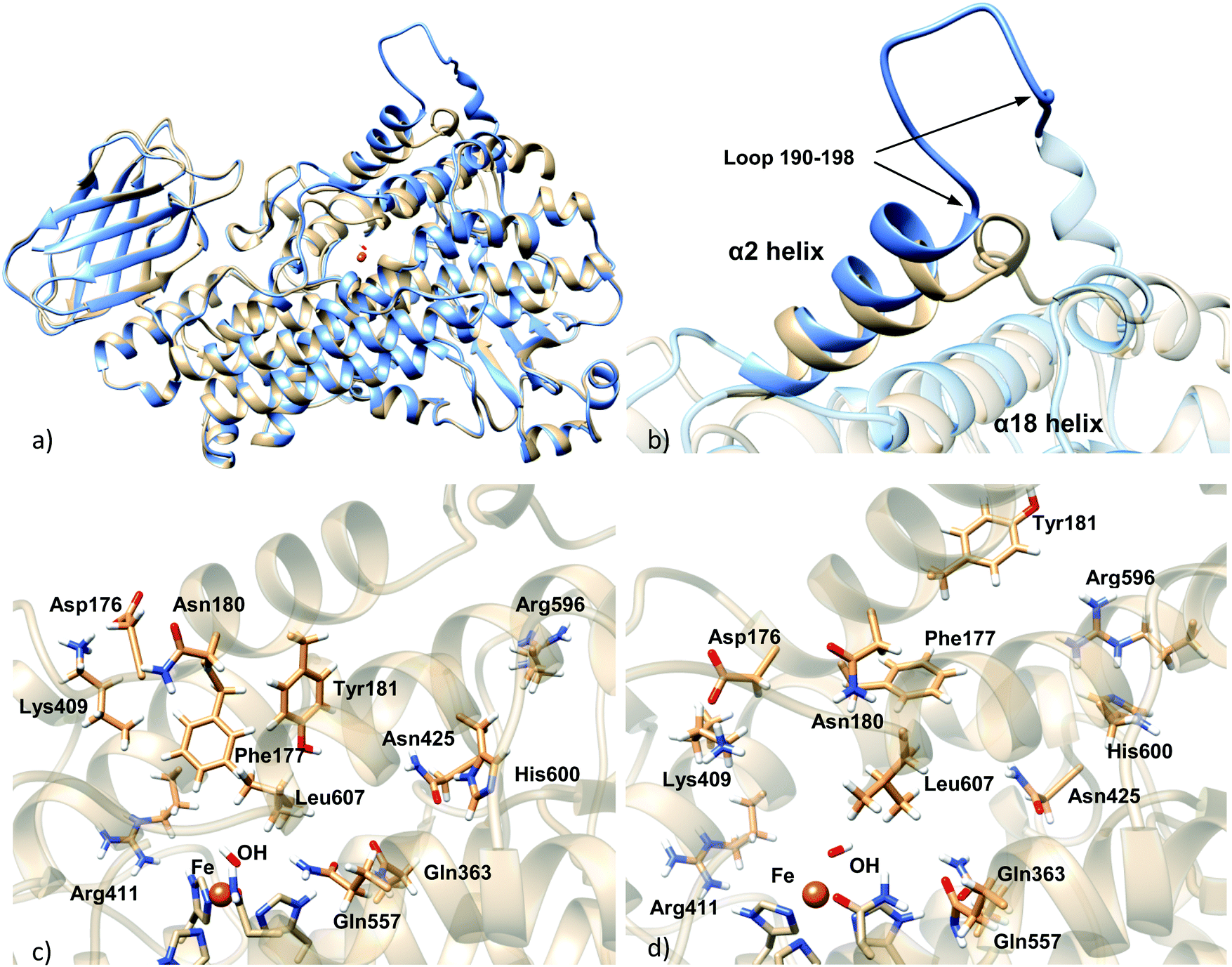 | ||
| Fig. 2 (a) Superposition of the homology model holoenzyme (blue) and the apoenzyme crystal structure (light brown); (b) details of α2 helix, showing the modeled loop 190–198; (c) active site of the apoenzyme structure; (d) active site of the homology model holoenzyme. | ||
3.2 Comparison between apoenzyme and holoenzyme
The main difference between the Stable 5-LOX-apoenzyme crystal structure and the Stable 5-LOX-holoenzyme model generated by homology modeling is the position of the α2 helix (Fig. 2). In the structure of the 5-LOX apoenzyme, α2 helix comprises residues 176–193 and presents a geometry that is unique in 5-LOX with respect to other LOX isoforms such as 15-LOX and 8-LOX. In the structure of the 5-LOX holoenzyme, α2 connects with an unfolded loop (residues 190–198) that displays a very wide solvent-exposed conformation (see Fig. S1 in the ESI†). In the corresponding apo form of the enzyme, this loop is packed and is folded toward the cavity.On the other hand, clear displacement of the segment 414–431 occurs in the substrate-bound enzyme with respect to its position in the substrate-free enzyme, this way letting more free space for the substrate (see Fig. S2, ESI†).
There are also differences in the side chain orientation of some residues of the active site, principally in Phe177, Tyr181, Leu607, Lys409, and Gln363. The cases of Lys409 and Phe177 stand out. Lys409, which forms a salt bridge with Asp176, is placed away from the active site in the apoenzyme, and therefore, it cannot tie the carboxylate end of AA inside the binding pocket. In the holoenzyme this salt bridge is placed toward the active site and Lys409 could bind the AA inside the cavity. Phe177 and Tyr181, which belong to α2 helix, form a cork which blocks the cavity entrance in the apoenzyme, but in the holoenzyme their positioning is in a different orientation that would allow the entrance of the substrate to the binding pocket. Another important difference between apo and holo forms is an apparently slight repositioning of α18 helix, which can slightly affect the shape and volume of the active site. Arg596 and His600, which belong to this helix, present different side chain conformations in apo and holo forms (Fig. 2c and d).
3.3 Generation of AA:Stable 5-LOX complexes
As commented, in order to generate AA:Stable 5-LOX complexes compatible with catalysis the use of the holoenzyme structure (with the missing parts modeled as explained above) seemed the most reliable choice and the first approach we followed. Docking calculations of AA into in silico mutants of this holo form were also carried out in order to better test our model. On the other side, and despite the substantially packed binding site of the apoenzyme structure, the possibility of this apo conformation to bear some activity cannot easily be ruled out. Moreover, as will be presented below, our docking calculations on the holoenzyme receptor did not predict solutions with the AA carboxylate interacting with His600 (a residue that has very recently been proposed in the literature as required for positioning the substrate for catalysis, most probably in head-first orientations).23 Thus, docking calculations into the apoenzyme structure were also performed in order to investigate this hypothesis.Taking both approaches together, the scoring values of the resulting solutions ranged from 34.30 to 26.41 Chemscore units (Table 3). No improvement of the predicted binding energies is obtained here by the incorporation of side chain flexibility. Given the wide geometric variability obtained, and in order to facilitate the analysis of binding modes, the solutions were clustered. An rmsd of 5.88 (10.30) Å was used, which led to 7 (8) clusters, for the rigid (flexible side chain) approaches. The scoring function value of each representative model of the clusters is shown in Table 3. The wide conformational variability obtained for the 5-LOX:AA complexes using the holoenzyme model structure can be observed in Fig. 3, which corresponds to the results obtained with the flexible side chain approach. In this approach, using such a number of rotamer residues provokes the generation of non-productive solutions in which the substrate falls away from the binding site (as shown in Fig. 3), but allows better exploration of productive configurations inside the cavity.
| Cluster number | Rigid | Flexible | ||
|---|---|---|---|---|
| Score | Population | Score | Population | |
| 1 | 34.30 | 16 | 33.03 | 23 |
| 2 | 34.26 | 53 | 31.47 | 349 |
| 3 | 33.97 | 107 | 31.32 | 34 |
| 4 | 33.25 | 3 | 31.15 | 20 |
| 5 | 32.34 | 18 | 30.99 | 50 |
| 6 | 30.41 | 2 | 30.17 | 7 |
| 7 | 30.32 | 1 | 29.73 | 16 |
| 8 | — | — | 26.41 | 1 |
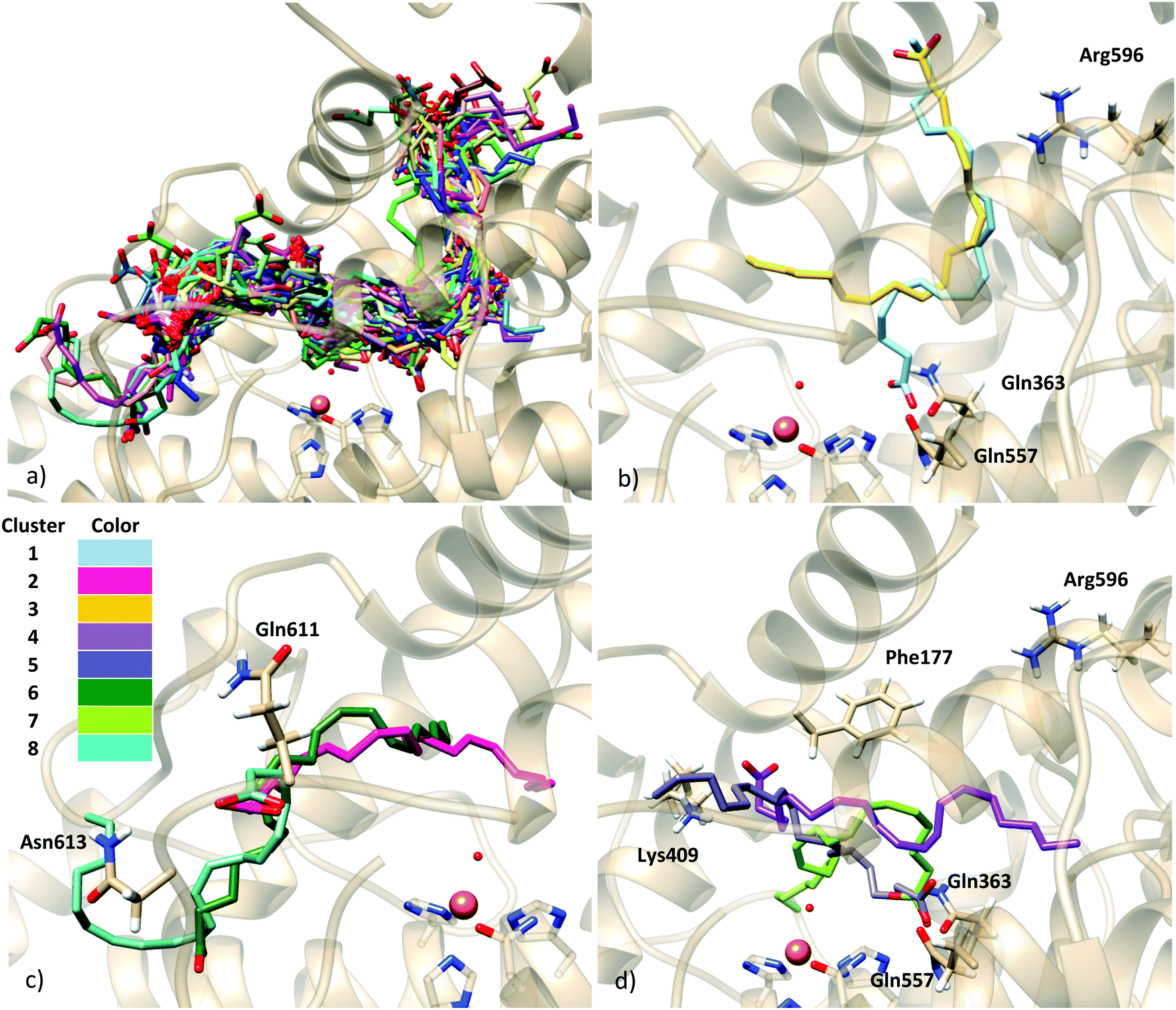 | ||
| Fig. 3 (a) Solutions of the docking simulation of the holoenzyme model with flexible side chains; (b)–(d) representative models of the eight clusters obtained in the clustering analysis with rmsd = 10.30 Å. A representative structure from each cluster is depicted, from cluster 1 in blue (top) to cluster 8 in cyan (bottom). | ||
As can be observed in Fig. 3, H-bond donor residues close to the AA carboxylate group are Lys409 and the backbone of Phe177, Gln363, Gln557, Asn425, Gln611, Asn613 and Arg596. There are about 300 solutions in the vicinity of Gln611, but these solutions are non-productive since the substrate is very far from the catalytic iron and they are not considered in further analyses. Solutions bonded to Lys409 (one of the proposed anchoring residues) and with hydrophobic contacts with Phe177 (15 solutions) exhibit a tail-first type orientation, while solutions interacting with Gln363 and Gln557 (which is spatially close to Gln363) (12 solutions) present a head-first type orientation. Flexible side chains allow the AA methyl end to be located inside the binding cavity when the carboxylate end is bound to Gln363 (Gln557), allowing head-first orientation positioning (see Fig. 3d), in contrast to the case of the rigid holoenzyme (see Fig. S4, ESI†), in which case solutions H-bonded to Gln363 had the AA tail pointing outward from the cavity.
To better analyze the dockings in the context of pre-reactive orientations, the results were filtered as a function of the distances between the reactive groups of the substrate and the Fe–OH− moiety. Based on our actual knowledge on the reaction of LOX, only solutions with the C7 atom below the distance threshold of 5 Å would lead to the correct product of the reaction. In the flexible side chain docking calculations, 47 solutions are obtained with C7 close to the oxygen of the Fe–OH− cofactor. Thus, the holoenzyme structure could correspond to a productive form of 5-LOX. In this group there are both solutions with head-first and tail-first orientation with good values of the scoring function. Fig. 4 shows one representative example of each type of orientation. Solution 34, which belongs to cluster 1, has the AA carboxylate interacting with Lys409 (tail first), a d(C7–OH) of 3.74 Å, and carbon atoms of its hydrophobic tail (from C12) at contact distances (<4.4 Å) from carbon atoms of His600, Phe359 or Ala424. Solution 12, with the opposite orientation (head first), has the AA carboxylate close to Gln363 (Gln557), a d(C7–OH) of 4.25 Å, and the methyl end in the region between the terminal Ile673, Lys409 and Phe177. These two structures will be used to study the regiospecificity of the products expected for these two different binding orientations (tail/head first) in the holoenzyme structure.
 | ||
| Fig. 4 Representation of solutions 34 (tail first, green) and 12 (head first, gray) of docking simulations of the flexible side-chain holoenzyme model. | ||
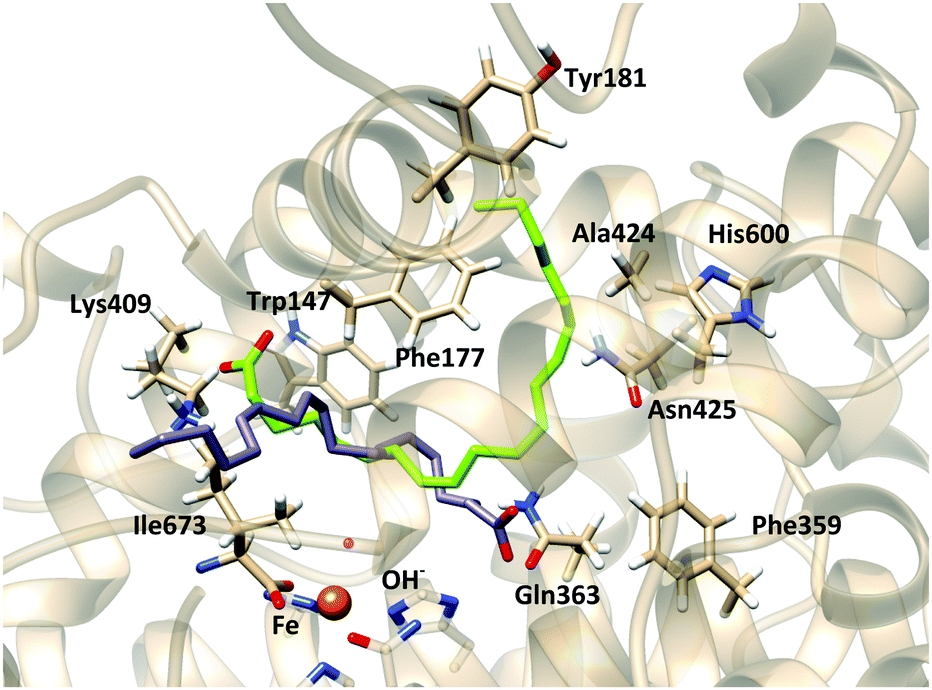
In the F359W/A424I/N425M triple mutant (reduced cavity), the number of docking solutions in which AA interacts with Lys409 decreases from 116 (out of 200 runs) in the WT enzyme to 5 in the triple mutant (Table S1, ESI†); solutions bound to Lys409 seem to need a larger volume to accommodate the AA tail. In Stable 5-LOX the channel where Arg596 is placed can do so while the AA carboxylate is interacting with Lys409 (Fig. 5). However, in the triple mutant, the three residues (359, 424 and 425) together block this channel and the AA tail is twisted inside the binding cavity. This provokes that some solutions appear close to Arg411. This residue which is spatially close to Lys409 (Fig. 5b) does not present any interaction with AA carboxylate in the docking solutions of the Stable 5-LOX. On the other hand, the number of docking solutions in which the AA carboxylate interacts with Gln363 increases (from 34 to 170, Table S1, ESI†) when the cavity volume is decreased by the effect of the triple mutation. An increase in the number of solutions with d(C7–OH) < 5 Å (from 29 to 126, Table S2, ESI†), as well as in the number of solutions with more than one methylene close to the OH− group, is also obtained for this triple mutant. Thus, the more twisted poses that AA adopts around the OH− group may also favor that several AA methylenes can have access to this group for reaction. This is in qualitative agreement with the experimental observation that the F359W/A424I/N425M triple mutant produces more than one regioproduct.5,14 Our results support the “space hypothesis” mentioned in the Introduction2,21 (with the tail-first orientation being responsible for 5-lipoxygenation), although the reason why a reduction of the 5-LOX active-site volume leads to 15-lipoxygenation is that the head-first orientation becomes the favored one in the mutant. As expected, the docking solutions interacting with Lys409 or Gln363 disappear in mutants K409L or Q363L, respectively, which also leads to more variability in the carbon atom suitably positioned for H abstraction.
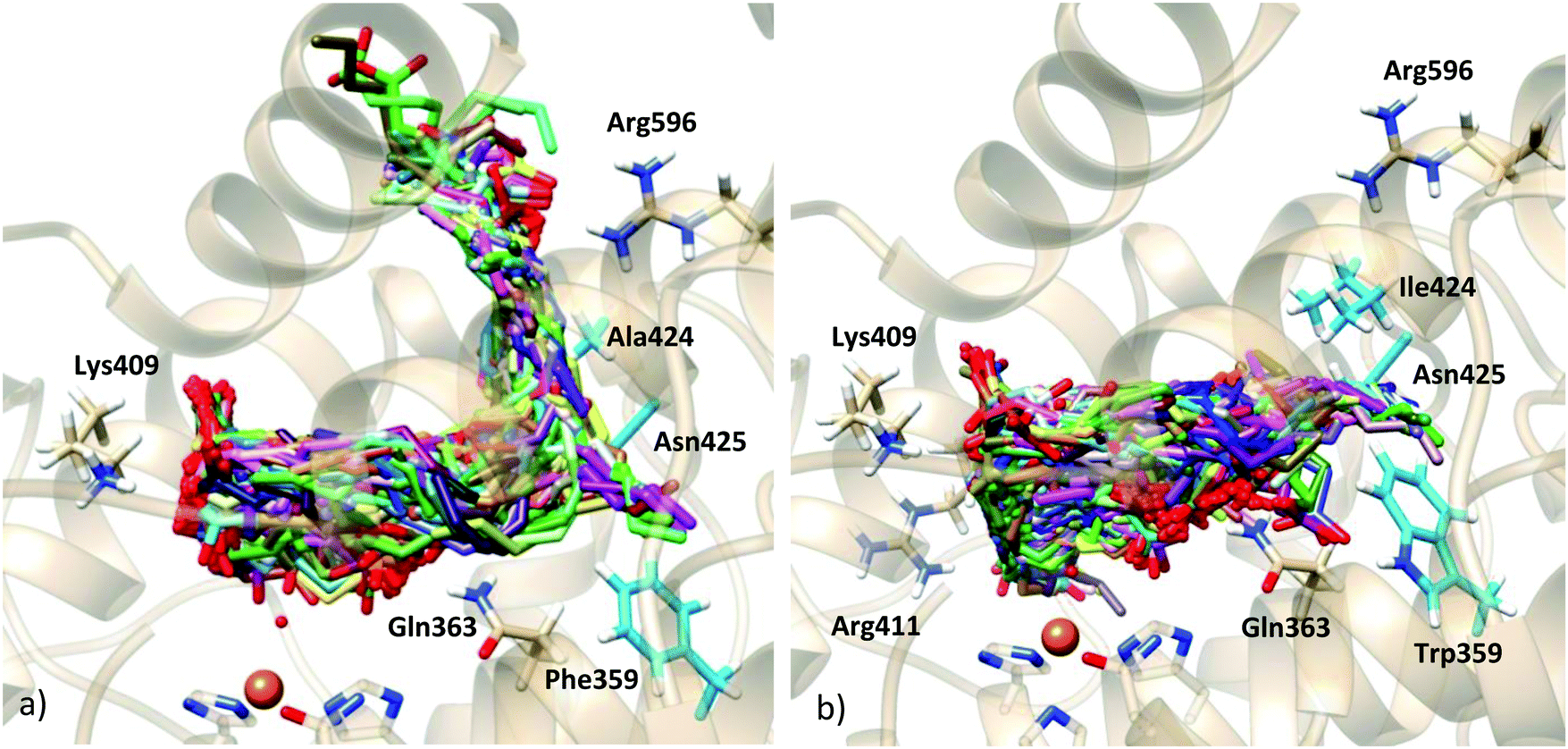 | ||
| Fig. 5 Docking solutions of (a) stable 5-LOX holoenzyme and (b) F359W/A424I/N425M-Stable 5-LOX mutant holoenzyme. | ||
 | ||
| Fig. 6 (a) Solutions of the docking simulation of the apoenzyme structure with rigid side chains; (b) representative models of clustering with rmsd = 2.96 Å, showing residues of interest (dashed line pointing towards Asn425 indicates that this residue is behind the substrate as it cannot be shown in this picture). | ||
| Cluster number | Rigid | Flexible | ||
|---|---|---|---|---|
| Score | Population | Score | Population | |
| 1 | 32.29 | 35 | 33.53 | 39 |
| 2 | 32.18 | 29 | 30.56 | 60 |
| 3 | 31.83 | 22 | 30.38 | 48 |
| 4 | 31.25 | 15 | 29.45 | 21 |
| 5 | 30.78 | 7 | 27.32 | 28 |
| 6 | 30.15 | 87 | 22.86 | 1 |
| 7 | 28.24 | 5 | 21.94 | 3 |
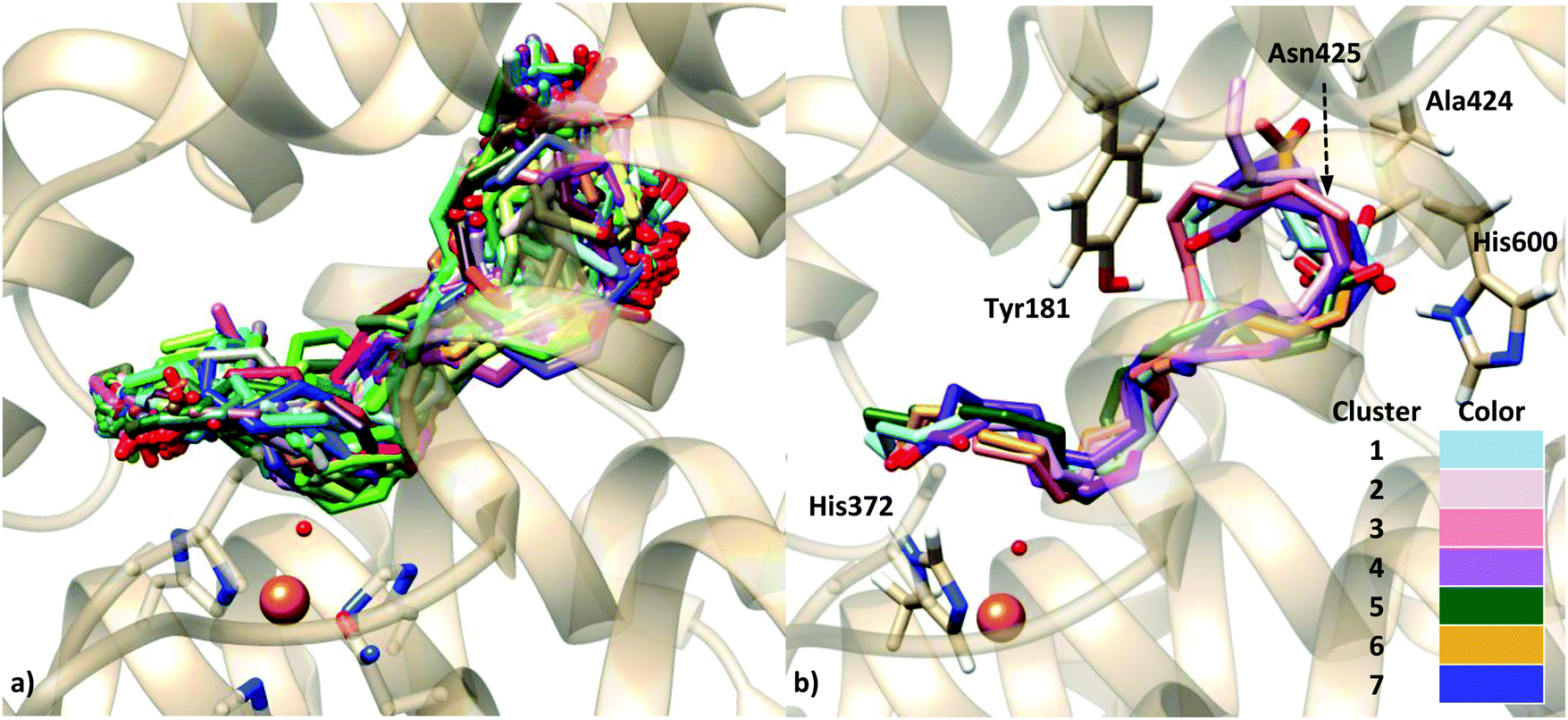
The binding modes of AA were characterized by the presence of polar and H-bond donor residues of the protein in H-bond distances to the oxygen atoms of the AA carboxylate. Five residues fulfill this condition: Ala424 (the backbone), Asn425, His600, Tyr181 and His372.
The distances between the carbon C7, C10 and C13 of AA and the oxygen atom of the OH− group were measured for all the docking solutions (200 solutions for each approach, see Table S3, ESI†). For the rigid-protein dockings, only 3 solutions display d(C7–OH) < 5 Å (a distance threshold that could be consistent with the 5-lipoxygenating action of the enzyme); 2 solutions have d(C10–OH) < 5 Å; and 39 solutions present d(C13–OH) < 5 Å, and the AA carboxylate end close to His600. The introduction of flexible side chains in the docking leads to an increase of the number of solutions with d(C7–OH) < 5 Å (up to 17). It is worth mentioning that this is apparently a consequence of very subtle relaxations of the active site upon binding, since the final position of the residues that have been allowed with flexibility remains nearly unchanged with respect to the X-ray position (Fig. S6, ESI†). The highest number of catalytically consistent solutions are still those with d(C13–OH) < 5 Å (26 solutions).
Interestingly, and contrary to what is obtained in the dockings to the holoenzyme, there are numerous solutions with the substrate carboxylate end close to His600 in the case of using the apoenzyme structure for the receptor. This may be in part a consequence of the different orientations displayed by His600 in the apoenzyme (Fig. 2c) and the holoenzyme (Fig. 2d). In the holoenzyme crystal structure, the His600 side chain points away (outward) from the active site, whereas in the apoenzyme crystal structure it is directed towards (inward) the catalytic iron. Notice that this result is not a consequence of our homology modeling simulations since this residue does not belong to the missing regions (Table 1), so that this different orientation is originally found in the crystallographic structures. Also, the conformation of His600 is not correlated with the S663D mutation mentioned in the Introduction, a mutation found in the holoenzyme but also in some of the apo form structures (see Fig. S7, ESI† and comments therein).
Analysis of the distance between the bisallylic methylene groups and the OH− shows that in these solutions C13 is closer to the OH− group, whereas C7 is far away. Still, as in the AA dockings to the holoenzyme structure no solutions show this interaction, we have selected one of these AA:apoenzyme solutions to study this AA–His600 interaction. Solution 11 has been selected from the docking calculations into the rigid apoenzyme receptor (Fig. 7); this solution has a similar score to the most stable solution (score 30.78 in solution 11 and 32.29 in solution 1), but its C13–OH, C10–OH and C7–OH distances are smaller than the corresponding ones in the most stable solution; actually, it corresponds to the most stable solution with d(C13–OH) < 5 Å. The distance d(C7–OH) is 7.39 Å.
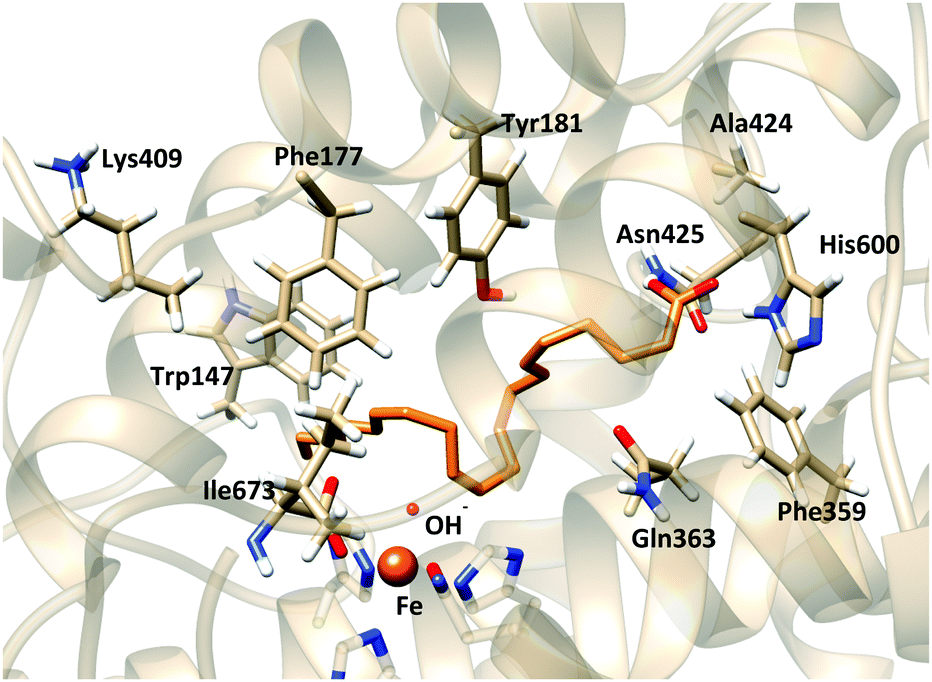 | ||
| Fig. 7 Representation of solution 11 (orange) of docking simulation of the rigid side-chain apoenzyme model. | ||
3.4 Reactivity evaluation of the different binding modes
Solutions 12 and 34 of the flexible side-chain holoenzyme docking simulation, representative of head-first and tail-first orientations, respectively, and solution 11 of the rigid apoenzyme dockings, representative of head-first orientation presenting the AA–His600 interaction, were selected to pursue the study. Our goal was to evaluate if any of these AA:Stable 5-LOX complexes and their corresponding binding modes were consistent with 5-lipoxygenation (regarded here as hydrogen abstraction occurring at C7). As we have shown in previous works,19,54 the distance criteria alone may not be enough to extract conclusions about the reactivity, and calculation of the energy barriers for hydrogen transfer is required. Basically, although the initial distance of the atom that is going to be abstracted has a crucial influence on the potential energy barrier, it is not the unique factor and other considerations have to be taken into account (for example, how difficult is the evolution from a non-planar structure in the reactant to a planar structure in the abstraction product). Nevertheless, when one hydrogen is significantly further away than another one, it can be expected that the latter will be preferably abstracted.Thus, we have first run molecular dynamics simulations of the three complexes, analyzed the distance distributions of the methylene groups along these simulations for each binding mode, and then selected representative snapshots to calculate the hydrogen abstraction reaction paths and energy barriers. Once each set was generated, we were able to analyze the feasibility of the different hydrogen abstractions for each of the three possible AA binding modes studied.
On the other hand, the pose that AA exhibits in crystal structure 3V99-A was also studied by performing a molecular dynamics simulation. The crystal structure 3V99-A and the holoenzyme model were superimposed, and AA coordinates were placed into the holoenzyme model to give a complete structure of the Michaelis complex.
Initially, AA in the crystal structure pose is not interacting with any positive residue of the protein, and the carboxylate end is placed at the bottom of the active site cavity (head-first orientation). During the MD simulation the carboxylate end moved towards the region where Arg596 and Asn425 are, and remained there for the rest of the 10 ns production run trajectory that was carried out for this complex. The average distance (standard deviation) from C7 to the oxygen of the OH− group was 13.44 (1.10) Å, whereas d(C10–OH) and d(C13–OH) were 10.92 (1.29) Å and 8.78 (1.33) Å, respectively. Thus, it is clear that starting from this complex it will be difficult to obtain structures with a bisallylic methylene close enough to the Fe–OH− cofactor to consider this model a catalytically productive one. Thus, the QM/MM calculations have been carried out on the other three complexes and the results are presented next.
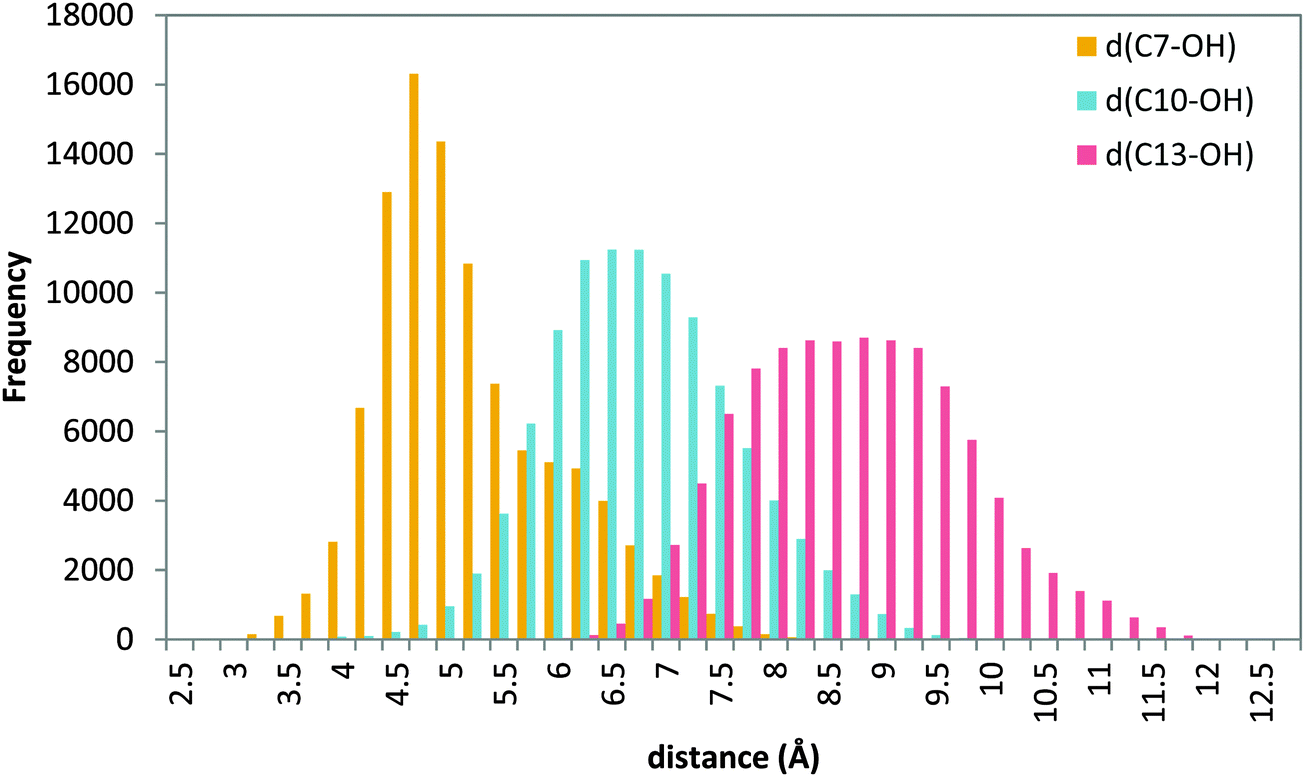 | ||
| Fig. 8 Histogram of distances C7–OH (yellow), C10–OH (blue) and C13–OH (red) during the 20 ns MD production step of solution 34 (tail first). | ||
The figure shows that d(C7–OH) presents a narrow distribution centered at 4.5 Å, whereas the distribution of distances d(C10–OH) and d(C13–OH) is broader and centered around 7.0 Å and 9.0 Å, respectively. These results suggest that a tail-first binding mode in which the AA carboxylate interacts with Lys409 gives conformations in which the C7 methylene is the only one close to the hydroxyl group. In Fig. S8 (ESI†) the cavity-lining residues for a representative head-tail AA orientation are indicated. Several invariant amino acids that have been shown to interact with AA in other LOXs are used in 5-LOX, although there are also some specific 5-LOX residues defining this binding site.
In order to further check if the produced models represent pre-reactive structures of the AA:Stable 5-LOX complexes (i.e. consistent with the 5-lipoxygenating activity), the reaction mechanism of the hydrogen abstraction step was studied.54 Snapshots from the latter simulations were selected and optimized at the QM/MM level (see Section 2.4) in order to obtain the initial reactant structures that initiate the reactive process. Table 5 shows the main geometric parameters of the different pre-reactive minima obtained. Snapshots I and II correspond to this tail-first orientation. Snapshot I is also depicted in Fig. 9a. In both cases, C7 is the closest methylene carbon atom to the OH− group. Moreover, H7 is not only close enough to be transferred but in a good orientation as d(C7–OH) is greater than d(H7–OH).
| Snapshot | H-abstracted | Orientation | d(Cx–OH) (Å) | d(Hx,A–OH) (Å) | d(Hx,B–OH) (Å) |
|---|---|---|---|---|---|
| I | H7 proS | Tail first | 3.41 | 4.32 | 2.60 |
| I | H10B | Tail first | 5.42 | 5.89 | 5.08 |
| I | H13B | Tail first | 6.72 | 7.05 | 5.67 |
| II | H7 proS | Tail first | 3.49 | 4.32 | 2.55 |
| III | H7B | Head first | 5.31 | 5.15 | 4.85 |
| III | H13A | Head first | 5.26 | 4.16 | 4.18 |
| IV | H10A | Head first | 5.01 | 4.74 | 5.90 |
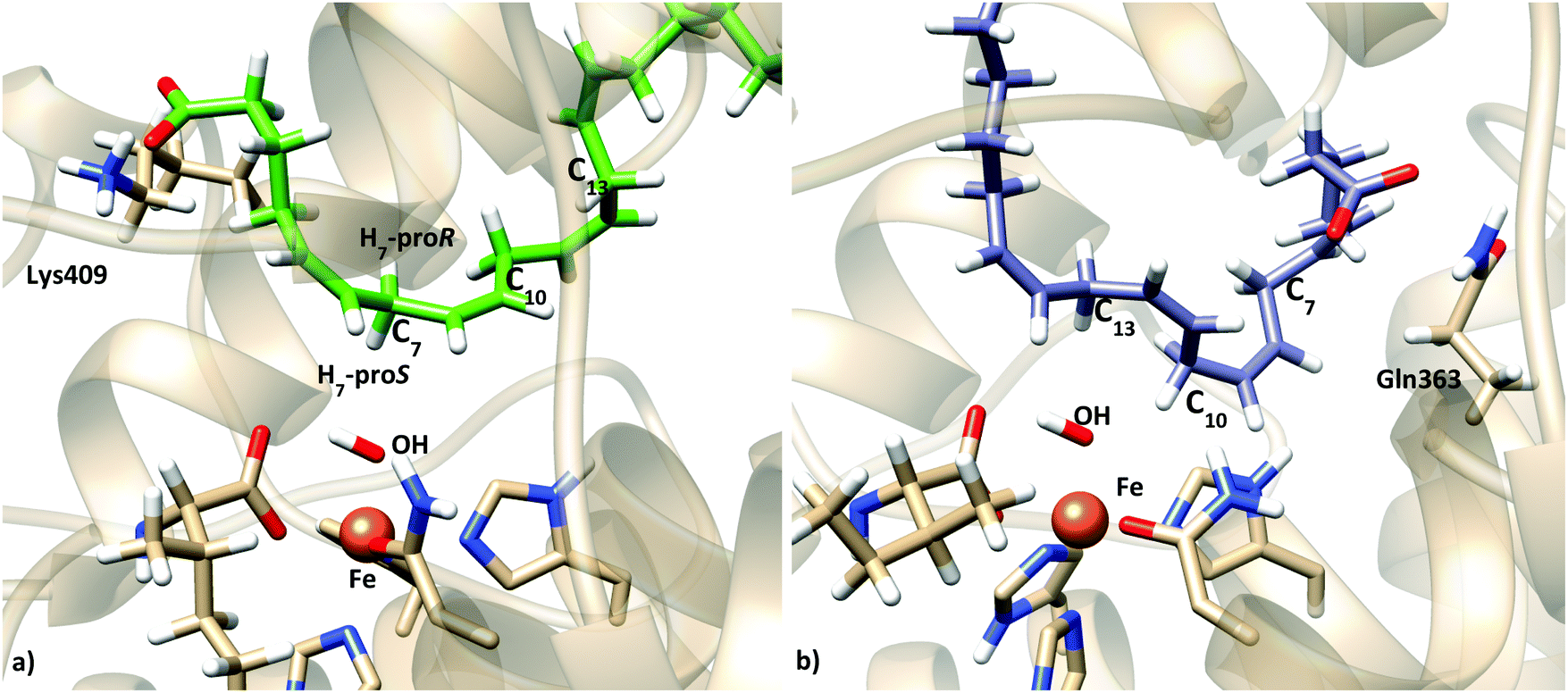 | ||
| Fig. 9 Representation of two characteristic reactant minima showing AA and the Fe coordination sphere: (a) structure obtained from optimization of snapshot I (tail first); (b) structure obtained from optimization of snapshot III (head first). | ||
The optimized geometry corresponding to each pre-reactive structure was used as a starting point to build the potential energy profile along the reaction coordinate for the hydrogen abstraction processes (see Section 2.4 for details). Note that for snapshot I abstraction of H10B and H13B was also considered in case they could still react at reasonable barrier heights even though they are significantly further away. The highest energy point of each profile was selected as the starting point for a direct transition state search, which was characterized by a frequency calculation. The potential energy barriers for all the processes are listed in Table 6 (snapshots I and II for this tail-first orientation). Values of additional geometric parameters corresponding to the pre-reactive minima and the transition state structures are given in Table S4 in the ESI.†
| Snapshot | H-abstracted | Orientation | Anchoring residue | d(Habs.–OH) (Å) | ΔE‡ (kcal mol−1) |
|---|---|---|---|---|---|
| I | H7 proS | Tail first | Lys409 | 2.60 | 18.1 |
| I | H10B | Tail first | Lys409 | 5.08 | 25.5 |
| I | H13B | Tail first | Lys409 | 5.67 | 50.7 |
| II | H7 proS | Tail first | Lys409 | 2.55 | 18.1 |
| III | H7B | Head first | Gln363 | 4.85 | 36.2 |
| III | H13A | Head first | Gln363 | 4.16 | 19.8 |
| IV | H10A | Head first | Gln363 | 4.74 | 25.0 |
We have to highlight the wide range of values obtained for the energy barriers, ranging from 18.1 to 50.7 kcal mol−1. This situation is directly related to the wide dispersion in the initial distance between the hydrogen that will be transferred and the accepting oxygen (from 2.55 to 5.67 Å), and the energy barriers are directly determined by how AA is placed in the active site of the enzyme. Importantly, the calculated potential energy barriers for H7 abstraction (18.1 kcal mol−1) are in qualitative agreement with experimental data on lipoxygenases. Contrary to this, the H10 and H13 abstraction barriers (7.4 and 32.6 kcal mol−1 higher than that for H7 abstraction, respectively) indicate that these are not plausible processes. Thus, taken together, our results show that this tail first binding mode considered is compatible with AA interacting with Lys409 and C7 being the closest methylene (and close enough to the OH− group) while C10 and C13 are further away, and the calculated energy barriers confirm that these pre-reactive minima would be favorable for H7 abstraction and unfavorable for H10 and H13 abstraction processes. In addition, the abstracted H7 turns out to be the proS one (see Tables 5 and 6, and Fig. 9a), in good agreement with the experimental stereoselectivity.
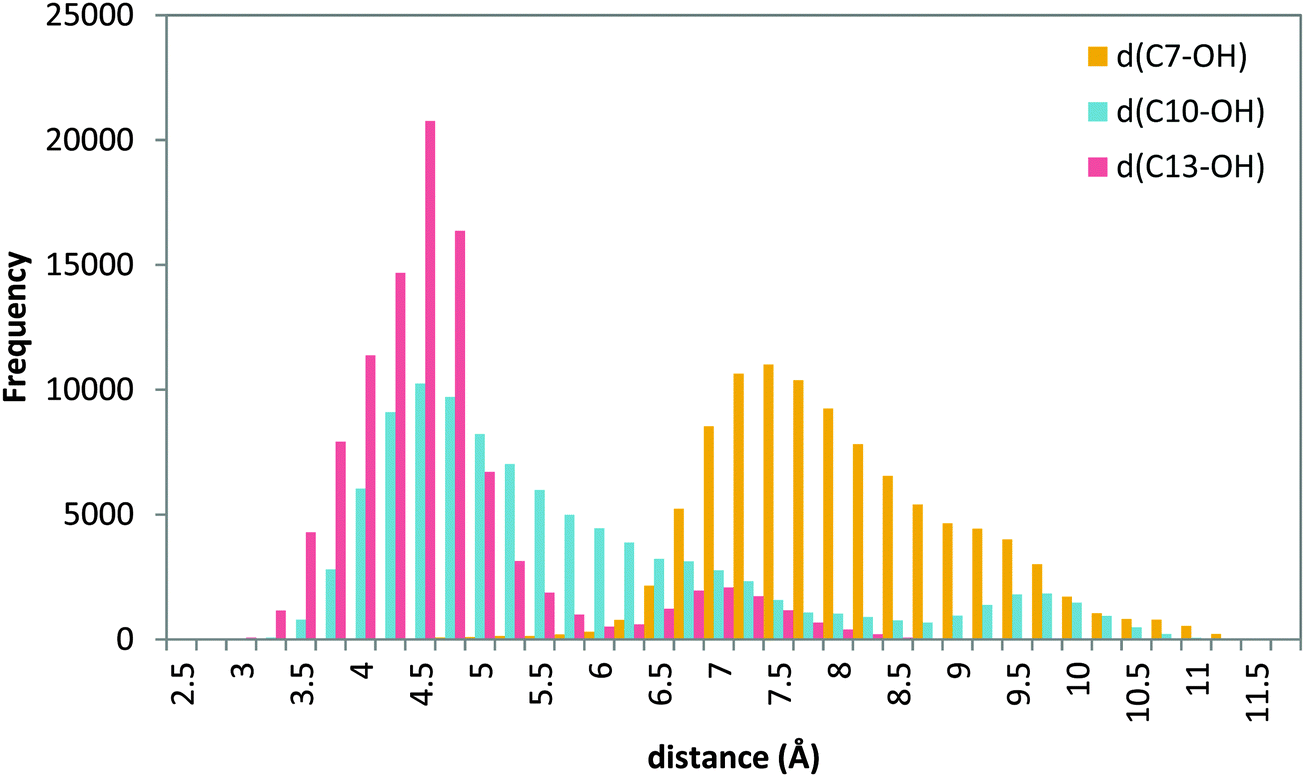 | ||
| Fig. 10 Histogram of distances C7–OH (yellow), C10–OH (blue) and C13–OH (red) during the 20 ns MD production step of solution 12 (head first). | ||
MD simulations of this solution 12 (head first) give broader distributions of all distances than those obtained for the tail-first orientation. d(C10–OH) and d(C13–OH) present lower values than d(C7–OH), which has its maximum population at ∼7.5 Å. AA remains with the carboxylate end bound to Gln363 (and Gln557), but its backbone adopts twisted conformations in which C10 and C13 can be simultaneously close to the hydroxyl group, while C7 is clearly placed away from the OH− group. Thus, these MD simulations suggest that this head-first model does not provide structures consistent with the 5-lipoxygenating function of this enzyme with the AA substrate. Still, two snapshots were selected to proceed with the QM/MM calculations. They are snapshots labeled as III and IV in Tables 5 and 6, and the molecular representation of the pre-reactive minima obtained for III is depicted in Fig. 9b.
The MD trajectories did not contain structures with H7 in a good position to initiate the reactive process, whereas H13 (and to a lesser extent H10) was close enough for the reaction to take place. The calculated potential energy barriers show that for this head-first orientation, the most favorable process is H13 abstraction, with a potential energy barrier of 19.8 kcal mol−1, whereas H10 abstraction is 5.2 kcal mol−1 higher and, most importantly, H7 abstraction is 16.4 kcal mol−1 higher. We have to underline at this point that, according to Fig. 10, the probability of having a snapshot in this head-first orientation with a C7–OH distance as short as in the pre-reactive structure optimized from snapshot III turns out to be extremely low. And, even in this case, the barrier for H7 abstraction would be over 36 kcal mol−1. Altogether our results strongly suggest that H7 abstraction is not occurring (or it would be extremely slow as compared to the experimental rate) when AA is in this head-first orientation.
However, very recent mutagenesis experiments showed that the H600A mutant displays less than 10% of Stable 5-LOX activity and has reduced affinity for the substrate, and that the H600V mutant has no detectable catalytic activity.23 These results led the authors to suggest that His600 is essential for proper AA positioning in the 5-LOX active site to give the 5-HPETE product. They also proposed that the presence of this amino acid deep in the cavity may be favoring the head-first substrate orientation.
The hypothesis of a head-first orientation in human 5-LOX is not new, and had been proposed in the literature in previous studies7 arguing that only with a head-first orientation could the stereochemistry of the hydroperoxidation product, 5S-HPETE, be justified, in contraposition with 15S-HPETE, the 15LOX product, which is obtained from an AA tail-first orientation. A head-first orientation has also been proposed for murine 8S-LOX.55 It is important to note that this interpretation of the experimental observations is not necessarily right, as it does not consider all the conformational changes that AA can undergo once placed inside the active site (methylene carbons consist of sp3 carbons that can rotate alternating the side of the plane that faces the Fe–OH− group),14 which in principle could yield both R- and S-HPETE products, even if starting from a given AA orientation. In fact, as mentioned above, the results obtained here with our model suggest that the proS-H7 can be transferred to the OH− group in a structure with a tail-first orientation (see Tables 5 and 6, and Fig. 9).
Nevertheless, as explained in Section 3.3, in order to study a possible head-first orientation bearing the AA–His600 interaction, the X-ray structure of apoenzyme has been used to generate the coordinates of complexes presenting such contact. The docking pose solution 11 (head first) was selected and prepared for an MD simulation following the same procedure as for the holoenzyme complexes. The position of the substrate is stable into the active site for the simulation time studied. The histogram of the distances from C7, C10 and C13 to the acceptor OH− is shown in Fig. 11.
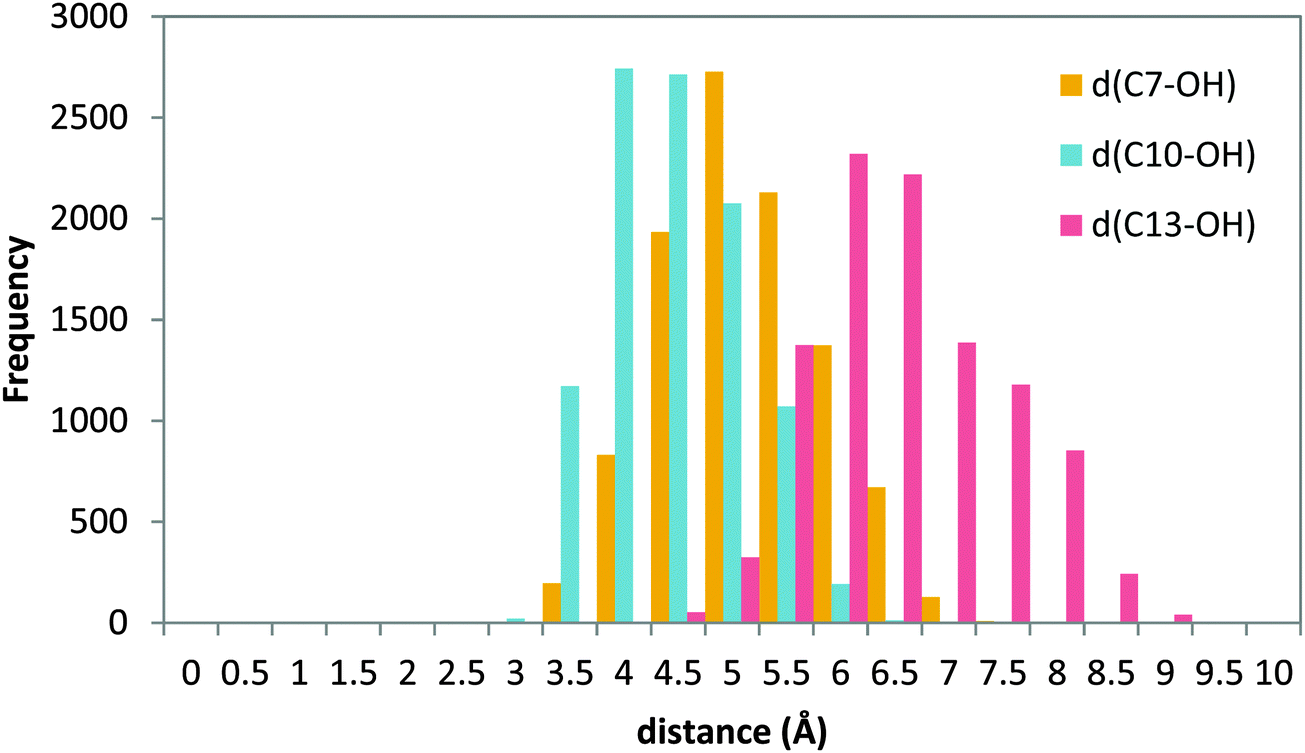 | ||
| Fig. 11 Histogram of distances C7–OH (yellow), C10–OH (blue), and C13–OH (red) during the 20 ns MD production step of solution 11 of the apoenzyme. | ||
A similar distance distribution can be observed for all the distances, with d(C10–OH) being the smallest of them, which is centered around 4 Å, followed by d(C7–OH) which is very similar and centered around 4.5 Å; d(C13–OH) is quite large with a distribution centered at 6.5 Å. In this situation the behavior is different from the case of the holoenzyme in which for a tail-first orientation (solution 34) the distance distribution is clearly more discriminating, with the d(C7–OH) distance being the shortest one (Fig. 8), whereas for the head-first orientation (solution 12), the distance distribution clearly differentiates d(C7–OH) (the greatest value) from d(C10–OH) and d(C13–OH), which are similar (Fig. 10). According to the distance criterion, in solution 11 of the apoenzyme both C7 and C10 could be suitable for transferring a hydrogen atom to the OH− group. It is also worth mentioning that along the MD simulation of solution 11, the aromatic side chains of the two FY-cork residues (Tyr181 and Phe177) remain as in the apoenzyme crystal structure. That is, once in the cavity the AA carboxylate end can interact with His600 without the need for ejecting or repositioning the FY-cork.23
The subsequent QM/MM study is aimed at determining if C7, C10 or even both positions are reactive for the hydrogen abstraction process, and then, which regiospecificity is consistent within this substrate binding mode. A series of snapshots from the MD simulation of solution 11, with a set of distances consistent with the distribution reported in the previous section, have been selected as the starting structures for the study of the first step of the reaction mechanism. The four selected snapshots were optimized giving the corresponding pre-reactive minima, whose characteristics are shown in Table 7. The values of additional geometric parameters corresponding to the pre-reactive minima and the transition state structures are given in Table S5 in the ESI† and Fig. 12 represents the structure of one of them.
| Snapshots | H-abstracted | d(Cx–OH) (Å) | d(Hx,A–OH) (Å) | d(Hx,B–OH) (Å) |
|---|---|---|---|---|
| V | H7 proS | 3.52 | 3.96 | 2.49 |
| VI | H10B | 3.50 | 4.15 | 2.44 |
| VII | H7 proS | 4.19 | 4.41 | 3.13 |
| VIII | H10A | 3.63 | 2.68 | 4.13 |
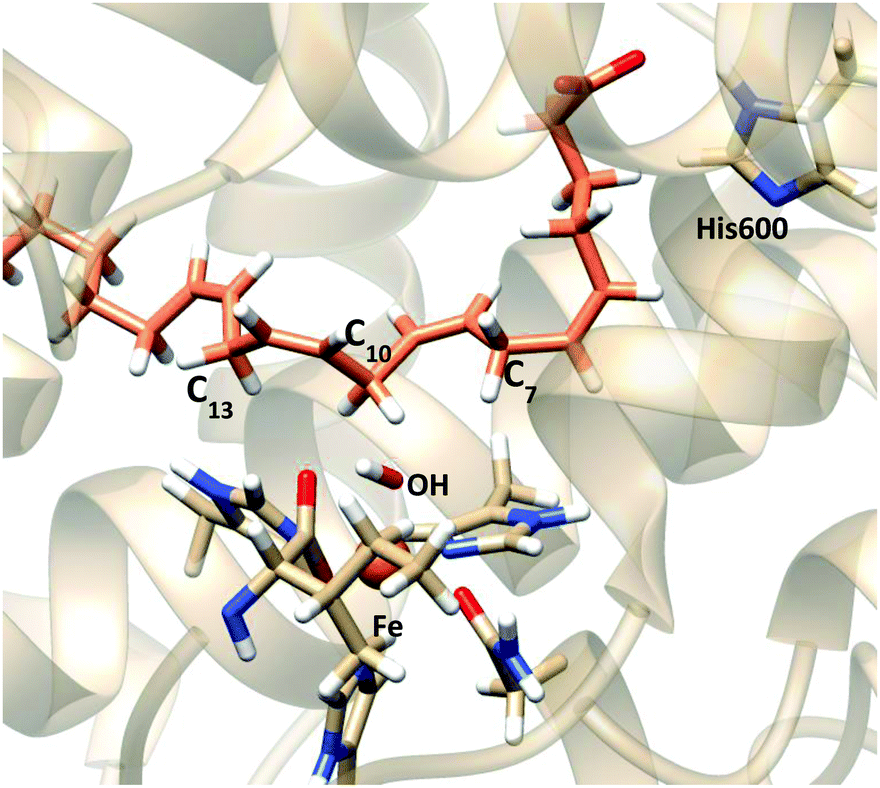 | ||
| Fig. 12 Representation of a characteristic reactant minimum showing AA and the Fe coordination obtained from optimization of snapshot VI. | ||
In this case the potential energy barriers range from 17.3 kcal mol−1 to 36.0 kcal mol−1 (Table 8), but the set of initial distances exhibit a narrower distribution than in the case of the holoenzyme structure, ranging from 2.44 to 3.13 Å. Thus, a wide dispersion of energy barriers comes from a narrow dispersion of initial geometries in terms of the set of atoms that constitute the reactive part of the system (the pentadienyl system defined by the bisallylic methylene and the Fe–OH− cofactor). Within these structures there are some that favor a H10 abstraction process, but there are also structures with an initial configuration favorable for proS-H7 abstraction. This binding mode of AA would be in principle consistent with 5-LOX specificity, but also with 12/8-specificity. In fact, 5-LOX is known to attack both carbon atoms, but for slightly different substrates: C7 is attacked in the first step to generate 5-HPETE and subsequently the H on C10 is abstracted from the first-oxidation product in the generation of leukotriene LTA4. As the substrates are different, though, it is not clear whether their binding orientation should be the same or not. Still, a model which puts both carbons proximal to the catalytic machinery, given the geometry of the bisallylic methylene groups, might indeed be plausible. Actually, in our previous studies on 15-LOX19,54 we have seen that AA can adopt conformations in the active site of the enzyme in which both C10 and C13 are at reactive distances of the OH− group. In that case, abstraction of H13 was favored by ∼4 kcal mol−1 (in agreement with the experimental ratio of 97:3 for 15/12 products in 15-LOX) probably due to steric hindrance for H10 abstraction, which is not necessarily the case here.
| Structure | H-abstracted | Orientation | Anchoring residue | d(Habs.–OH) (Å) | ΔE‡ (kcal mol−1) |
|---|---|---|---|---|---|
| V | H7 proS | Head first | His600 | 2.49 | 18.2 |
| VI | H10B | Head first | His600 | 2.44 | 17.3 |
| VII | H7 proS | Head first | His600 | 3.13 | 23.4 |
| VIII | H10A | Head first | His600 | 2.68 | 36.0 |
Our results suggest that both structures can accommodate AA in their active site while having C7 at a reactive distance. In particular, for the holoenzyme structure a tail-first binding mode has been found with C7 close to the OH− group, with the AA carboxylate interacting with Lys409 and the methyl tail occupying a region that would be consistent with mutagenic experiments;14 the corresponding calculated energy barriers confirm that these pre-reactive minima are consistent with the experimental 5-LOX regio- and stereoselectivity (favorable for H7 abstraction, and unfavorable for H10 and H13 abstraction processes). On the other hand, for the apo structure a head-first orientation has been identified with C7 close to the OH− group, with the AA carboxylate interacting with His600 and the methyl tail pointing towards Trp147; in this case the potential energy barriers for H10 and H7 abstraction indicate that both reactions (5-LOX or 12/8-LOX regioselectivity) are feasible. The latter is scarcely observed experimentally with AA as the substrate, which could be a reason to discard this alternative. However, the possibility exists that both H abstractions take place and that regiospecificity is determined by the later steps in the reaction (e.g. O2 attack). Thus, based on that we cannot completely discriminate between the two binding orientations, or conclude which crystal structure is the most suitable to model the reactive complexes that lead to 5-lipoxygenation.
The experimental observation that the only 5-LOX structure available with AA bound is different from the apo ones strongly suggests that a conformational change takes place upon substrate binding and indicates that the structure adopted by the enzyme in the pre-catalytic complex will probably be different from the apo structure. Unfortunately, in the X-ray structure of holo enzyme the density for AA does not allow one to unambiguously assign its orientation. More importantly, AA is located too far from the OH− group for reaction, so that this holo structure does not seem to correspond to a pre-reactive complex. In a very recent study,23 it has been proposed, based on the results of mutagenesis experiments, that the Phe177–Tyr181 pair does not act as a “cork” as had been proposed, but as a “twist-and-pour” closure. This implies that the conformation these residues present in the holo crystal structure would not be compatible with the pre-catalytic geometry (i.e. ready to catalyze the chemical reaction) of the enzyme.
This has prompted us to run long molecular dynamics simulations of the produced AA:Stable 5-LOX complexes in order to get new insights from their structural behaviour along the trajectory. Obviously, the way AA is positioned in the active site will affect the geometric changes that the enzyme experiences and vice versa.
We have performed 100 ns production runs for each of the complexes studied in the previous sections: solution 34 (holo, tail first), solution 12 (holo, head first) and solution 11 (apo, head first). The most relevant results of each simulation are commented here.
For solution 34 (tail first) of docking to holoenzyme, the complex remains relatively stable for 18 ns (Fig. S9–S11, ESI†) before AA comes out of the active site. During this 18 ns, the AA carboxylate interacts with Lys409 and the distance order (with respect to the Fe–OH− cofactor) C7 < C10 < C13 is maintained. Then, it remains at the protein surface and, later on, the AA carboxylate interacts with Lys423 and Arg596 (Fig. S12, ESI†), which are placed at the other end of the active site cavity. Finally, the AA tail enters the cavity through the opposite end, giving a reverse orientation to the starting one (Fig. S13, ESI†) and with the bisallylic carbons far from the OH− group (Fig. S10, ESI†). The Phe177 + Tyr181 pair stays in a similar conformation as in the crystallographic holoenzyme structure (Fig. S14a, ESI†), which gives AA access toward the solvent from inside the binding cavity.
For solution 12 (head first) of docking to holoenzyme, AA remains in the active site cavity for the whole 100 ns, and the bisallylic carbon C7 is quite far from the OH− group (Fig. S15–S17, ESI†). Initially, the AA carboxylate end is interacting with Gln363 and Gln557. At the end, it is positioned in the part of the cavity surrounded by Gln363, Gln557, Asn424, Ala425, Phe359, His600 and, notably, Tyr181 (Fig. S18, ESI†). Tyr181 has changed conformation and is now pointing into the cavity (as in the apo structure) and is making a hydrogen bond with the AA carboxylate (Fig. S14b, ESI†). Phe177 has also changed conformation and points into the cavity providing, altogether, a lid that seems to prevent AA from escaping. During the simulation, the hydrophobic tail moves from its initial position to the part of the active site delimited by Trp147, occupying the same zone as described for solution 11 of the apoenzyme dockings (head first).
For solution 11 (head first) of the docking to apoenzyme, the simulation shows less structural changes (Fig. S19–S21, ESI†). The distance distribution for C7, C10 and C13 remains qualitatively the same as the one shown in Fig. 11 (Fig. S19, ESI†), with both C7 and C10 being close to the OH− group (here C7 is more populated at shorter distances). The AA carboxylate starts interacting with His600 and Asn425 and, after 45 ns, a slight shift moves it to establish a hydrogen bond with Tyr181, as observed for solution 12 (Fig. S21 and S22, ESI†). His600 has rotated and it is now in the conformation observed for the holoenzyme (Fig. S23, ESI†). Phe177 and Tyr181 stay in a conformation similar to that observed in the crystal structure. Interestingly, during the 100 ns trajectories, solutions 12 and 11 (both head first) converge to be localized in the same region of the active site, although the central part of AA is bent down towards the Fe–OH− cofactor for solution 11, whereas it is bent towards the top (surface) of the cavity for solution 12, which brings the bisallylic carbons away from the Fe–OH− cofactor. Fig. 13 depicts the molecular representation of the last snapshot of the 100 ns simulation of each AA:Stable 5-LOX complex.
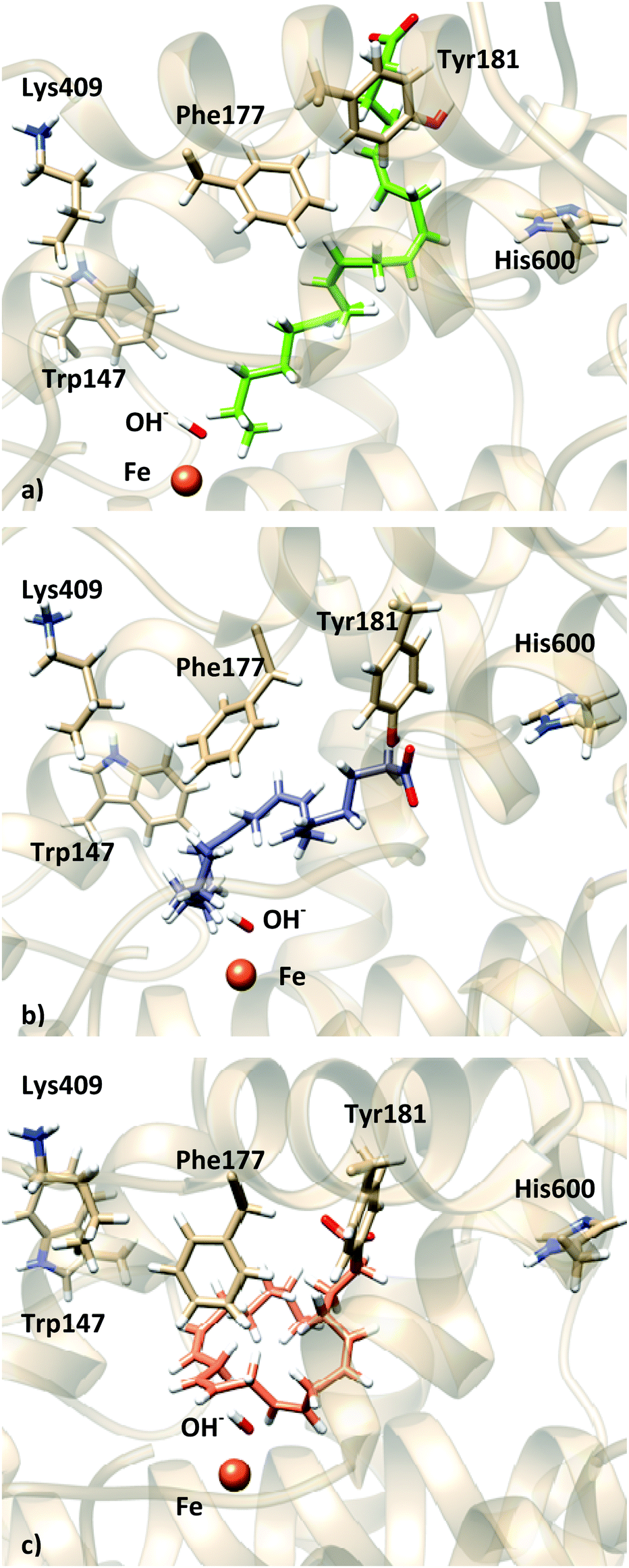 | ||
| Fig. 13 Representation of the last frame of the 100 ns MD simulations: (a) holo solution 34; (b) holo solution 12; (c) apo solution 11. | ||
Thus, these 100 ns simulations seem to support the idea proposed by Newcomer and coworkers that the Phe177 + Tyr181 pair would be “closing” the active site during the chemical reaction. Moreover, in a head-first orientation Tyr181 is the residue that interacts with the AA carboxylate. As for the tail-first orientation also studied in this work, we cannot conclude if the AA:Stable 5-LOX complex was not stable due to the binding mode or as a consequence of the FY cork being open. On the other hand, His600 seems to adopt the conformation observed in the holo crystal structure and it is not directly interacting with the AA carboxylate, which may be indicating that the effect observed experimentally upon mutation of this residue is due to a change in the shape and/or volume of the binding cavity (direct or by affecting other residues), as has been proposed by Kühn and coworkers for the F359W/A424I/N425M triple mutant.5,14
4 Conclusions
Human 5-LOX is a highly regio- and stereospecific enzyme. It catalyzes the oxygenation of its main substrate, arachidonic acid, at position 5. In spite of its huge pharmacological relevance, the way its catalytic mechanism works has not been well understood yet, mainly because no experiment has been able to determine the position of AA within the binding site so far. To shed light on this point, in this paper, we have employed a multiscale approach combining homology modeling, protein–ligand dockings, molecular dynamics simulations and quantum mechanics/molecular mechanics (QM/MM) calculations.We have started from the available crystallographic structures. All of them correspond to the human Stable 5-LOX form of the enzyme. In particular, we have used the unique existent AA bond form (holoenzyme, pdb code 3V99) and one of the essentially equivalent apo enzymes (with pdb code 3O8Y). For each one, we have successively determined the most probable AA binding modes, the regio- and stereoselectivity that could be expected for each binding mode, and the residence time of AA (that is, the AA stability) in each binding mode. Since the coordinates of some parts in the 3V99 structure are lacking, we have employed homology modeling techniques to complete the structure of the holoenzyme.
Our results show that two binding modes are possible in the holoenzyme: (a) a tail-first type orientation, with the AA carboxylate end interacting with Lys409, C7 being the closest methylene (and close enough to the OH− group) while C10 and C13 are further away, and with the H7 proS abstraction being mostly favored; and (b) a head-first orientation, with the AA carboxylate end bound to Gln363 (and Gln557), but with C10 and C13 much closer to the OH− group than C7, and with the H13 abstraction the most favorable process and the H7 abstraction not being possible. Thus, AA can be positioned in the holoenzyme active site with both tail-first and head-first orientations, but only tail-first orientation would yield structures reactive by C7, that is, with 5-lipoxygenating reactivity, whereas a head-first orientation would give 15/11-LOX or 12/8-LOX regioselectivity, in terms of hydrogen abstraction.
On the other hand, for the apoenzyme a head-first orientation has been identified with C7 close to the OH− group, with the AA carboxylate interacting with His600 and the methyl tail pointing towards Trp147. The His600 side chain points away (outward) in the active site of the holoenzyme, whereas in the apoenzyme it is directed towards (inward) the catalytic iron. As a consequence, AA cannot interact with His600 in the holoenzyme. Conversely, AA can interact with His600 in a head-first orientation inside the apoenzyme. If this were the case, our MD and QM/MM results predict that both the H7 proS and H10 abstractions would happen. Thus, we could suppose that the reactive form of AA inside the active site of human 5-LOX could be a head-first orientation interacting with His600 by its carboxylate end as proposed by an experimental model.23 However, at the current state of the experimental knowledge, it would seem somewhat unlikely for three reasons: firstly, AA should have entered the active site maintaining the inward His600 orientation corresponding to the apoenzyme, in contraposition to the outward His600 orientation of the available crystallographic structure of the holoenzyme. Second, that orientation would lead to both the H7 proS (5-LOX regioselectivity) and H10 (12/8-LOX regioselectivity) abstractions, which is scarcely observed experimentally. Third, in a recent study by Wu et al.,56 a large molecule known as a competitive inhibitor of 5-LOX was docked into the binding pocket of the apoenzyme crystal structure. The negative correlation coefficient obtained between the experimental and predicted IC50 made the authors conclude that this structure cannot be used for drug screening before the conformational flexibility of 5-LOX is further analyzed and included in the model.
A variety of fates have been found for the different AA binding modes at long times. In the case of the tail-first orientation in the holoenzyme, the complex remains stable, with its carboxylate interacting with Lys409, for 18 ns and then AA comes out of the active site. Later, the AA tail enters the cavity through the opposite end, leading to a reverse orientation to the starting one and with the bisallylic carbons (especially C7) far from the OH− group. On the other hand, the head-first orientation in the holoenzyme is maintained in the active site along the 100 ns, although the AA carboxylate initially interacts with Gln363 and Gln557, but ends up in the part of the cavity surrounded by Gln363, Gln557, Asn425, His600, and Tyr181, always with C7 quite far from the OH− group. Finally, the AA head-first orientation in the apoenzyme seems to remain quite stable, although the AA carboxylate evolves to form a hydrogen bond with Tyr181, while His600 rotates towards the outward orientation corresponding to the holoenzyme.
To summarize, based on the available crystallographic structures, we have found 3 possible binding modes for AA. Only one of them would clearly exhibit predominant 5-lipoxygenating reactivity. However, long MD simulations show that this binding mode is not stable, at least within the crystallographic holoenzyme structure used. It is evident that the reactive structure of human 5-LOX has to be a holoenzyme structure, although it is possible that the unique holoenzyme structure currently available is not good enough to reproduce the actual conformation of the enzyme when the chemical process occurs.
Acknowledgements
We thank the Spanish Ministerio de Economía y Competitividad (Grants CTQ2014-53144-P and CTQ2014-54071-P) and the Generalitat de Catalunya 2014SGR989 for financial support. L. M. thanks the “UAB-Banco Santander program”. We also acknowledge CSUC for computational facilities.References
- G. Coffa and A. R. Brash, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15579–15584 CrossRef CAS PubMed.
- I. Ivanov, D. Heydeck, K. Hofheinz, J. Roffeis, V. B. O'Donnell, H. Kühn and M. Walther, Arch. Biochem. Biophys., 2010, 503, 161–174 CrossRef CAS PubMed.
- Y. C. Joo and D. K. Oh, Biotechnol. Adv., 2012, 30, 1524–1532 CrossRef CAS PubMed.
- C. Schneider, D. A. Pratt, N. A. Porter and A. R. Brash, Chem. Biol., 2007, 14, 473–488 CrossRef CAS PubMed.
- K. Schwarz, M. Walther, M. Anton, C. Gerth, I. Feussner and H. Kühn, J. Biol. Chem., 2001, 276, 773–779 CrossRef CAS PubMed.
- R. Vogel, C. Jansen, J. Roffeis, P. Reddanna, P. Forsell, H. E. Claesson, H. Kühn and M. Walther, J. Biol. Chem., 2010, 285, 5369–5376 CrossRef CAS PubMed.
- M. E. Newcomer and A. R. Brash, Protein Sci., 2015, 24, 298–309 CrossRef CAS PubMed.
- A. Catalano and A. Procopio, Histol. Histopathol., 2005, 20, 969–975 CAS.
- A. D. Dobrian, D. C. Lieb, B. K. Cole, D. A. Taylor-Fishwick, S. K. Chakrabarti and J. L. Nadler, Prog. Lipid Res., 2011, 50, 115–131 CrossRef CAS PubMed.
- J. Z. Haeggstrom and C. D. Funk, Chem. Rev., 2011, 111, 5866–5898 CrossRef PubMed.
- O. Radmark and B. Samuelsson, Biochem. Biophys. Res. Commun., 2010, 396, 105–110 CrossRef PubMed.
- C. N. Serhan, N. Chiang and T. E. Van Dyke, Nat. Rev. Immunol., 2008, 8, 349–361 CrossRef CAS PubMed.
- G. Folco and R. C. Murphy, Pharmacol. Rev., 2006, 58, 375–388 CrossRef CAS PubMed.
- K. Hofheinz, K. R. Kakularam, S. Adel, M. Anton, A. Polymarasetty, P. Reddanna, H. Kühn and T. Horn, Arch. Biochem. Biophys., 2013, 530, 40–47 CrossRef CAS PubMed.
- I. Ivanov, H. Kühn and D. Heydeck, Gene, 2015, 573, 1–32 CrossRef CAS PubMed.
- H. Kühn, Prostaglandins Other Lipid Mediators, 2000, 62, 255–270 CrossRef.
- N. C. Gilbert, S. G. Bartlett, M. T. Waight, D. B. Neau, W. E. Boeglin, A. R. Brash and M. E. Newcomer, Science, 2011, 331, 217–219 CrossRef CAS PubMed.
- N. C. Gilbert, Z. Rui, D. B. Neau, M. T. Waight, S. G. Bartlett, W. E. Boeglin, A. R. Brash and M. E. Newcomer, FASEB J., 2012, 26, 3222–3229 CrossRef CAS PubMed.
- L. Toledo, L. Masgrau, J. D. Marechal, J. M. Lluch and À. González-Lafont, J. Phys. Chem. B, 2010, 114, 7037–7046 CrossRef CAS PubMed.
- A. Di Venere, T. Horn, S. Stehling, G. Mei, L. Masgrau, À. González-Lafont, H. Kühn and I. Ivanov, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 1079–1088 CrossRef CAS PubMed.
- M. F. Browner, S. A. Gillmor and R. Fletterick, Nat. Struct. Biol., 1998, 5, 179 CrossRef CAS.
- S. T. Prigge, B. J. Gaffney and L. M. Amzel, Nat. Struct. Biol., 1998, 5, 178–179 CrossRef CAS PubMed.
- S. Mitra, S. G. Bartlett and M. E. Newcomer, Biochemistry, 2015, 54, 6333–6342 CrossRef CAS PubMed.
- S. Ghatak, A. Vyas, S. Misra, P. O'Brien, A. Zambre, V. M. Fresco, R. R. Markwald, K. V. Swamy, Z. Afrasiabi, A. Choudhury, M. Khetmalas and S. Padhye, Bioorg. Med. Chem. Lett., 2014, 24, 317–324 CrossRef CAS PubMed.
- A. M. Schaible, R. Filosa, V. Temml, V. Krauth, M. Matteis, A. Peduto, F. Bruno, S. Luderer, F. Roviezzo, A. Di Mola, M. de Rosa, B. D'Agostino, C. Weinigel, D. Barz, A. Koeberle, C. Pergola, D. Schuster and O. Werz, Br. J. Pharmacol., 2014, 171, 2399–2412 CrossRef CAS PubMed.
- P. Prasher, Pooja and P. Singh, Bioorg. Med. Chem., 2014, 22, 1642–1648 CrossRef CAS PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinformatics, 2014, 47, 5.6.1–5.6.32 Search PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- A. D. Mackerell, M. Feig and C. L. Brooks, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- C. Colovos and T. O. Yeates, Protein Sci., 1993, 2, 1511–1519 CrossRef CAS PubMed.
- D. Eisenberg, R. Luthy and J. U. Bowie, in Macromolecular Crystallography, Pt B, ed. C. W. Carter and R. M. Sweet, Elsevier Academic Press Inc, San Diego, 1997, vol. 277, pp. 396–404 Search PubMed.
- R. A. Laskowski, M. W. Macarthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins: Struct., Funct., Genet., 2003, 52, 609–623 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins: Struct., Funct., Bioinf., 2005, 61, 704–721 CrossRef CAS PubMed.
- S. C. Lovell, J. M. Word, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Genet., 2000, 40, 389–408 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- S. E. Feller, K. Gawrisch and A. D. MacKerell, J. Am. Chem. Soc., 2001, 124, 318–326 CrossRef.
- S. E. Feller and A. D. MacKerell, J. Phys. Chem. B, 2000, 104, 7510–7515 CrossRef CAS.
- J. Saam, I. Ivanov, M. Walther, H. G. Holzhutter and H. Kühn, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13319–13324 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjovoll, A. Fahmi, A. Schafer and C. Lennartz, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- A. H. de Vries, P. Sherwood, S. J. Collins, A. M. Rigby, M. Rigutto and G. J. Kramer, J. Phys. Chem. B, 1999, 103, 6133–6141 CrossRef CAS.
- D. C. Liu and J. Nocedal, Mathematical Programming, 1989, 45, 503–528 CrossRef.
- J. Nocedal, Mathematics of Computation, 1980, 35, 773–782 CrossRef.
- J. Baker, J. Comput. Chem., 1986, 7, 385–395 CrossRef CAS.
- A. Banerjee, N. Adams, J. Simons and R. Shepard, J. Phys. Chem., 1985, 89, 52–57 CrossRef CAS.
- S. R. Billeter, A. J. Turner and W. Thiel, Phys. Chem. Chem. Phys., 2000, 2, 2177–2186 RSC.
- P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. Saura, R. Suardíaz, L. Masgrau, J. M. Lluch and À. González-Lafont, ACS Catal., 2014, 4351–4363 CrossRef CAS.
- M. Jisaka, R. B. Kim, W. E. Boeglin and A. R. Brash, J. Biol. Chem., 2000, 275, 1287–1293 CrossRef CAS PubMed.
- Y. R. Wu, C. He, Y. Gao, S. He, Y. Liu and L. H. Lai, J. Med. Chem., 2012, 55, 2597–2605 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Homology modeling; docking calculations; cavity lining residues; geometric parameters of pre-reactive minima and TSs; single point calculations; long MD simulation results. See DOI: 10.1039/c6cp03973a |
| This journal is © the Owner Societies 2016 |