
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the stability of alanine-based helices by conformer-selective IR spectroscopy†
Waldemar
Hoffmann
ab,
Mateusz
Marianski
b,
Stephan
Warnke
b,
Jongcheol
Seo
b,
Carsten
Baldauf
b,
Gert
von Helden
b and
Kevin
Pagel
*ab
aFreie Universität Berlin, Takustraße 3, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
First published on 5th July 2016
Abstract
Polyalanine based peptides that carry a lysine at the C-terminus ([Ac-AlanLys + H]+) are known to form α-helices in the gas phase. Three factors contribute to the stability of these helices: (i) the interaction between the helix macro dipole and the charge, (ii) the capping of dangling C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups by lysine and (iii) the cooperative hydrogen bond network. In previous studies, the influence of the interaction between the helix dipole and the charge as well as the impact of the capping was studied intensively. Here, we complement these findings by systematically assessing the third parameter, the H-bond network. In order to selectively remove one H-bond along the backbone, we use amide-to-ester substitutions. The resulting depsi peptides were analyzed by ion-mobility and m/z-selective infrared spectroscopy as well as theoretical calculations. Our results indicate that peptides which contain only one ester bond still maintain the helical conformation. We conclude that the interaction between the charge and the helix macro-dipole is most crucial for the formation of the α-helical conformation and a single backbone H-bond has only little influence on the overall stability.
O groups by lysine and (iii) the cooperative hydrogen bond network. In previous studies, the influence of the interaction between the helix dipole and the charge as well as the impact of the capping was studied intensively. Here, we complement these findings by systematically assessing the third parameter, the H-bond network. In order to selectively remove one H-bond along the backbone, we use amide-to-ester substitutions. The resulting depsi peptides were analyzed by ion-mobility and m/z-selective infrared spectroscopy as well as theoretical calculations. Our results indicate that peptides which contain only one ester bond still maintain the helical conformation. We conclude that the interaction between the charge and the helix macro-dipole is most crucial for the formation of the α-helical conformation and a single backbone H-bond has only little influence on the overall stability.
Introduction
Several decades of protein-structure investigation have led to an in-depth understanding of the effects that stabilize α-helices in the condensed phase.1–4 Also in the “cleanroom” environment of the gas phase helical peptides have been studied extensively.5–7 For these investigations, especially two techniques are widely used. Ion mobility-mass spectrometry (IM-MS) is a powerful tool that separates ions according to their size, shape, charge and mass and therefore provides information about the overall shape of an ion.8,9 Complementary to this approach is mid-IR spectroscopy, which probes intramolecular vibrations and therefore is highly diagnostic for the underlying H-bond network.10,11Due to their simplicity and strong tendency to adopt helical structures, especially polyalanine based peptides turned out to be highly suitable systems. It has been shown that if the Alan peptide chain is C-terminated by a lysine, peptides as short as [Ac-AlanLys + H]+ (n > 7) are able to form stable α-helices in the gas phase.12 Furthermore, water adsorption measurements,13 IM-MS,14 gas-phase infrared multiple photon dissociation (IRMPD) spectroscopy15 as well as theoretical methods12,15,16 showed that the protonated peptide [Ac-Ala10Lys + H]+ exists solely as an α-helix in the gas phase.
Three factors stabilize [Ac-Ala10Lys + H]+ in the α-helical conformation (Fig. 1): (i) the attractive interaction between protonated lysine and the helix macro-dipole, (ii) the capping to dangling carbonyls at the C-terminal end of the helix by protonated lysine, and (iii) the cooperative network of H-bonds in the helix. Multiple studies have been designed to quantify these12–14,16–18 and other15,19–22 factors. In particular, Jarrold and co-workers showed that simply by shifting the lysine residue from C- to N-terminus eliminates the attractive interaction between the charge and the helix macro-dipole, which substantially destabilizes the α-helix.14,17,18 As a result, these peptides adopt more compact structures, which exhibit significantly shorter drift times in IM-MS experiments. In another study, Kim et al. coordinated the protonated lysine side chain with 18-crown-6 ether in order to inhibit its H-bonding to the dangling carbonyl groups near the C-terminus.17 Surprisingly, it is observed that the isolation of the charge enhances the peptide helicity, presumably due to the elimination of geometry constraints posed by the H-bonds with the lysine amine group. The importance of the third factor, however, the network of backbone H-bonds on the stability of the helix, so far remains less understood.
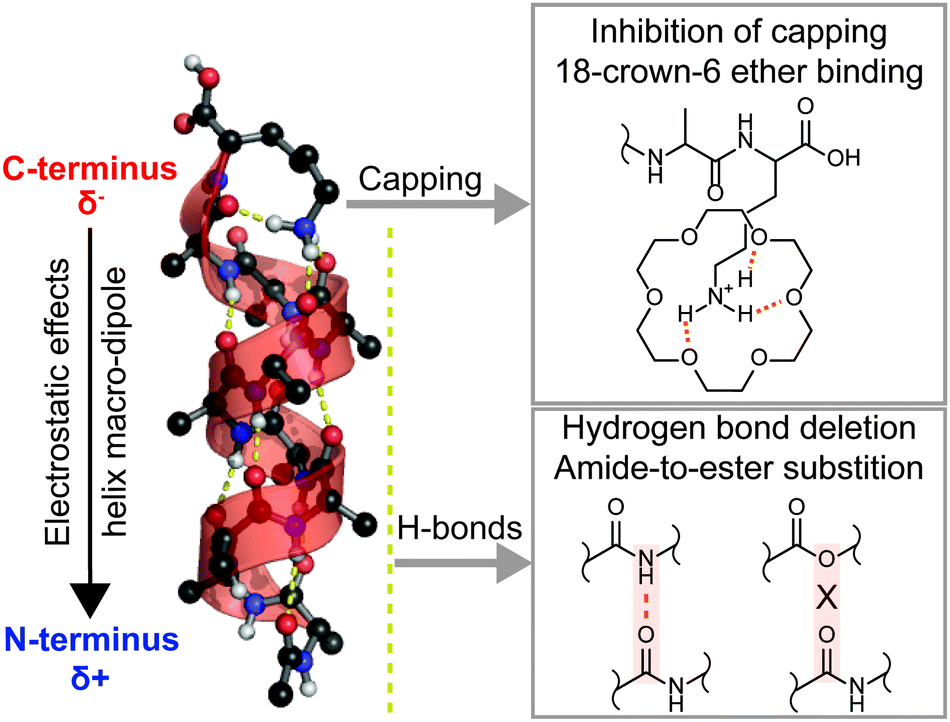 | ||
| Fig. 1 Three factors that stabilize [Ac-Ala10Lys + H]+ in the α-helical conformation in the gas phase. Capping of the lysine side-chain residue to carbonyl groups can be turned off by attaching 18-crown-6 ethers. H-bonding can be selectively deleted via amide-to-ester substitutions. | ||
In this work, we study the impact of individual H-bonds in [Ac-Ala10Lys + H]+ (AK00) on the conformational stability by introducing amide-to-ester substitutions at specific positions within the sequence.20,23–25 The replacement of an alanine building block by an isostructural lactic acid unit results in formation of an ester bond (often referred to as a depsi peptide bond, Fig. 1). This replacement of an N–H donor group by an oxygen atom selectively turns off one specific hydrogen bond while the dipole moment associated with the peptide bond remains largely unaltered (1.7 D vs. 3.5 D for the ester and regular amide bonds, respectively).26 The position of the depsi peptide bond is systematically varied from C- (AK01) to N-terminus (AK10), with the exception of the peptide bond between the Lys and the Ala residue. The resulting peptides are characterized by a combination of experimental (IM-MS coupled to IRMPD spectroscopy) and theoretical techniques (enhanced sampling molecular dynamics, MD). This approach allows probing the molecular structure on complementary levels, ranging from the overall molecular shape via IM-MS, through secondary-structure sensitive gas-phase IR spectroscopy, up to atomistic resolution MD simulations.
Methods
Materials
The peptide Ac-Ala10Lys-OH (AK00) and the corresponding depsi-modified (AK01–AK10) peptides were purchased from KareBay Biochemicals (Rochester, New York) and used without further purification. Samples were dissolved at 50 μM in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:MeCN solution with 1% trifluoroacetic acid (TFA).
:1 H2O:MeCN solution with 1% trifluoroacetic acid (TFA).
Experimental details
Ion mobility-mass spectrometry (IM-MS) experiments were performed using an in-house built drift-tube instrument, which is partially based on a previously described design.27 A detailed description of the instrument can be found elsewhere.28 Briefly, ions are generated using a nano-electrospray ionization source (nESI) and transferred into the gas phase. An electrodynamic ion funnel collects and pulses ions into the drift region where they travel through helium buffer gas (∼5 mbar) under the influence of a weak and homogenous electric field (10–20 V cm−1). At the end of the drift-tube a second electrodynamic ion funnel collects and guides ions into a quadrupole mass filter, where mass-to-charge ratio (m/z) separation occurs. By measuring the time-dependent ion current of m/z selected ions, characteristic arrival time distributions (ATDs) can be obtained. From these ATDs, absolute collision cross sections (CCSs) of a particular ion species can be determined using the Mason Shamp equation.29 The CCS is a unique molecular property, representing the overall shape of a particular ion. Absolute CCS values are not dependent on instrumental parameters and can therefore be compared to theoretically calculated values.29 For the acquisition of conformer- and m/z-selected infrared (IR) spectra of ions in the gas phase, the drift-tube instrument is coupled to the Fritz-Haber-Institute's Free Electron Laser (FHI-FEL), described in detail elsewhere.30 First, a narrow drift-time window (100 μs, corresponding to a narrow CCS distribution) is selected by electrostatic deflection of unwanted ions. After traversing the quadrupole mass filter, the conformer and m/z-selected ions are irradiated by an intense IR laser pulse of 10 μs length (macropulse energy up to 50 mJ). When the IR wavelength is resonant with an IR-allowed molecular vibration, the absorption of many photons leads to an increase in internal ion energy and can induce fragmentation. This process is known as IR multiple photon dissociation (IRMPD, Fig. S1, ESI†). Subsequently, fragment analysis is performed by means of time-of-flight (ToF) mass spectrometry. An IR action spectrum is recorded by monitoring the intensity of the precursor ion signal and the corresponding fragment signals as a function of the laser wavelength (Fig. S2, ESI†).31,32Simulation details
Molecular dynamics (MD) simulations were performed using the gromacs-4.5.5 package.33 The OPLS-AA force field34,35 was augmented with parameters for the depsi peptide bond taken from a previous work on polylactic acids.36,37 The AK00–AK10 peptides were first thermalized at 300 K for 1 ns, followed by 100 ns production runs with 1 fs time step. The temperature of 300 K was maintained during the simulations using stochastic velocity rescaling thermostat38 with a coupling constant of 0.05 ps. To enhance the sampling of the conformational space, replica-exchange MD (REMD) simulations were performed for AK00 (2.0 μs accumulated time of 4 replicas at 300–380 K) and AK03–AK06 (8.0 μs accumulated time of 16 replicas at 300–915 K). For each system, snapshots in 25 ps intervals were extracted from the 300 K trajectory. The recorded geometries were clustered with a single-linkage algorithm and the cutoff of 0.1 nm. Clustering yielded up to two different clusters.The central structures of most abundant clusters were optimized using the non-local hybrid B3LYP39 density-functional and the cc-pVDZ basis set using the Gaussian09 package.40 B3LYP shows an excellent performance in electrostatic-dominated systems like peptides studied here.41 The geometry relaxations at the B3LYP level introduced only minor structural changes. The calculated RMSD values between the initial (from MD) and optimized structures are typically smaller than 1 Å. Only for AK00, AK03, AK04 and AK08 somewhat larger RMSD values are observed, which essentially reflect rearrangements in the lysine side chain. Nevertheless, these changes should be attributed to the different meaning of the arbitrary chosen of the central structure, which represents a cluster, and the 0 K DFT optimized energetic minimum. The overlays of each pair also show that there is no significant rearrangement (see Fig. S6, ESI†). Harmonic vibrational frequencies were calculated for the relaxed structures. Predicted vibrations were then scaled by 0.971 and convoluted with Gaussian functions (bandwidth 10 cm−1).
Atomic partial charges (ESP charges) for the equilibrium structures were derived by fitting to the electrostatic potential with the Merz–Singh–Kollman scheme.42 These ESP charges are used in the calculation of theoretical CCSs using the trajectory method43 as implemented in the MOBCAL program.44 In the trajectory method, each atom of the molecule is represented by a 12-6-4 potential (Lennard-Jones term plus ion-induced dipole interactions) and the effective potential of an ion is built by summation of the potentials of all atoms. The trajectories of the incoming buffer gas (He) are simulated under the effective potential, then the scattering angle is determined as a function of the impact parameter for a given collision geometry. The scattering angle is related to the CCS and an average CCS is calculated by integrating over all possible collision geometries.
Results and discussion
The IM-MS arrival time distributions (ATDs) of AK00 and its depsi-peptide analogues (AK01–AK10) exhibit narrow single peaks, having a width that indicates the presence of a single conformer (Fig. S3a and S4, ESI†). The drift time of AK00 corresponds to an overall collision cross section (CCS) of 237 Å2. This is in good agreement with the previously reported 235 Å2, which has been assigned to an α-helical structure.12–14The NH groups close to the N-terminus are not involved in H-bonds in helical structures and typically point towards the N-terminus. As a result, amide-to-ester substitution at these dangling N–H groups (peptides AK08–AK10) does not delete any H-bonds and induces only minor structural changes (AK08 = 234 Å2, AK09 = 236 Å2, AK10 = 237 Å2, see Fig. S3a–S5, ESI†). Intuitively, the deletion of an NH donor group close to the C-terminus or in the middle of the helix may have a larger impact on the H-bond network and therefore on the conformational stability, since these residues are involved in the formation of stabilizing H-bonds.20 However, IM-MS analysis of peptides AK01–AK07 reveals drift times and CCSs that correspond to conformations of similar (AK07 = 236 Å2 and AK0n = 237 Å2 with n = 0, 2, 4, and 6) or slightly extended size (AK01 = 240 Å2, AK03 = 242 Å2 and AK05 = 241 Å2). Remarkably, none of these peptides adopts a more compact, globular structure, as observed for protonated [Ac-LysAla10 + H]+ (reported CCS 219 Å2).12,14,17 This observation therefore indicates that for the here-investigated molecules the helical scaffold remains largely unaltered.
To further investigate the structural details of the depsi-peptide analogues we probed the shape and m/z-selected ions using IRMPD spectroscopy. The resulting gas-phase IR spectra shown in Fig. 2a and Fig. S3b (ESI†), exhibit two characteristic bands at 1600–1700 cm−1 (amide I band) and 1450–1550 cm−1 (amide II band). The IR features above 1700 cm−1 originate from carbonyl groups of free or only weakly-interacting carboxylic acids (∼1780 cm−1) and the decoupled carbonyl stretching mode of the introduced ester group (1720–1750 cm−1). The dominant amide I band of the α-helical AK00 appears at 1675 cm−1 and the position of the respective band in the amide-to-ester substituted peptides shows little variation. The amide I bands of AK01–AK07 are somewhat broader than the respective band in the unsubstituted AK00 and AK08–AK10 peptides. This may be due to the deletion of hydrogen bonds positioned in the middle of the H-bond chain, which decouples in-phase stretching modes of carbonyl groups and effectively broadens the amide I band.19 Theoretical spectra of the α-helical peptides (red line in Fig. 2a) confirm that the substitution in the middle of the helix leads to such a broadening of the amide I band.
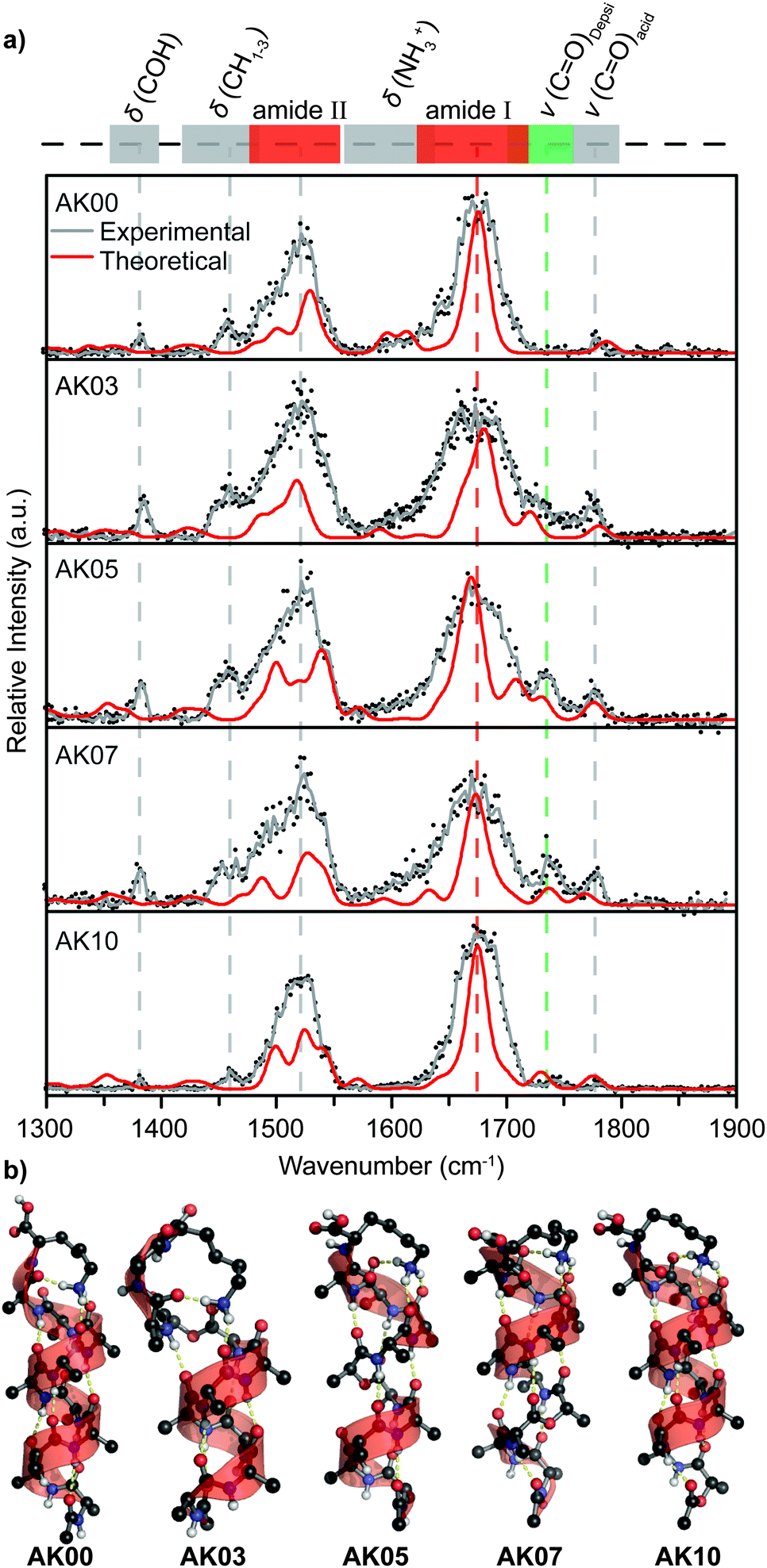 | ||
| Fig. 2 Gas-phase infrared spectra of ion-mobility and m/z-selected conformers (a) and representative theoretical gas-phase structures (b). Data from two individual scans per species are depicted as black dots. The solid gray lines represent an average of these two individual scans. A qualitative band assignment is schematically shown in the upper panel. Theoretical vibrational spectra of geometry-optimized structures (b) are displayed as solid red lines in (a). | ||
In order to better understand the structure of the peptides on the atomistic level, we performed a conformational search with enhanced sampling MD or plain MD techniques (see simulations details). While the starting geometry determines the fraction of the phase space that is sampled and insufficient sampling might lead to trapping of a system in a meta-stable-state,45 we believe that an idealized α-helix is a reasonable choice starting point for MD simulations. Previous theoretical studies used extensive sampling techniques to show that lysine C-terminated polyalanine peptides form α-helices in the gas phase16 while N-terminated they form other motives.15 Moreover, Jarrold et al. have shown that the protonated AK00 peptide exists solely as an α-helix in the gas phase.12 Our results show that the amide-to-ester substitution in AK08–AK10 does not alter the overall α-helical structure (Fig. 2b and Fig. S5, ESI†) whereas the deletion of a hydrogen bond in AK06 and AK07 is compensated by minor rearrangements between 310 and α-helices in the vicinity of the lactic acid. Such transitions have been observed before in amide-to-ester substituted helices in crystallized peptides.46,47 Substitution in the middle of the helix (AK05) yields two abundant conformers. While one maintains the α-helical character (calculated CCSth = 238 Å2), the other conformer adopts a rather compact structure (calculated CCSth = 230 Å2). In the latter, amide-to-ester substitution terminates the helix (Fig. S5, ESI†), which renders part of the peptide unstructured. The theoretical CCS of this more compact structure, however, does not agree with the experimental CCS of 241 Å2. Finally, the replacement of an amide bond by an ester bond close to the C-terminus of the helix (AK01–AK04) results in the formation of a π-turn, combined with a partial unfolding of the helix induced by binding of the charged lysine to the dangling carbonyls. More importantly, however, also here the helical structure largely prevails. Such unfolding at the C-terminus justifies structures with larger CCSs in IM-MS and the broadening of the amide I absorption band within the IRMPD spectra in Fig. 2a (and Fig. S3b, ESI†).
Conclusions
The protonated peptide [Ac-Ala10Lys + H]+ (AK00) is known to adopt an α-helical structure in the gas phase. This structure is stabilized by three factors: (i) the attraction between the helix macro-dipole and the charge, (ii) the capping to dangling carbonyls by the protonated lysine side chain, and (iii) the cooperative H-bonding along the helix (Fig. 1).12–14 In this work, we complement previous studies focused on the first two factors by evaluating the impact of individual backbone H-bonds on the stability of the α-helix scaffold.IM-MS experiments reveal that regardless of the position within the sequence, amide-to-ester substitution of one peptide bond only results in minor structural changes and conformations that, if altered at all, are slightly more extended than the unmodified peptide. This is confirmed by IRMPD spectroscopy experiments, which indicate that the predominantly helical conformation of the unmodified peptide AK00 is still present in the depsi analogues. Molecular modelling shows that the observed subtle changes in structure are largely dependent on the sequence position at which the H-bond is deleted. Close to the N-terminus (AK08, AK09, and AK10), the removal of a single H-bond leaves the α-helix essentially unaffected. Further towards the helix center (AK06 and AK07), H-bond deletion introduces small structural rearrangements to bifurcated 310/α-helices. A substitution close to the C-terminus (AK01–AK05) leads to unfolding of the helix and formation of a π-turn induced by the protonated lysine. Nevertheless, the α-helix scaffold remains the defining structural motif.
Our findings are in good agreement to previous work of Schultz and co-workers who investigated the effect of amide-to-ester substitution in protein secondary structures in the condensed phase. They found that a single ester group destabilizes α-helices by a mere 0.7–1.7 kcal mol−1.48 This surprisingly small energetic penalty might be an effect of increased solvent accessibility to the backbone and/or be an electrostatic effect associated with the backbone.49 In the gas phase the solvent is absent and electrostatic effects are amplified substantially. As a result, the energetic penalty of the amide-to-ester substitution in the depsi-peptides studied in this work might even be smaller than 0.7–1.7 kcal mol−1, and therefore too small to significantly disturb the α-helix.
Previous studies have shown that the attractive interaction between the helix macro-dipole and the charge shapes the energy landscape of polyalanine peptides, whereas capping to dangling carbonyl groups by the protonated lysine side-chain only has a minor influence on the stability of the α-helix.15,17,18 We show here that also the removal of a single backbone H-bond only has a small impact on the stability of the helix in the gas phase. Therefore, we conclude that the interplay of the charge and the helix macro-dipole is by far the most crucial parameter for the formation of the α-helical conformation in AK00. Capping of the protonated lysine side-chain to dangling carbonyls can slightly disturb the helicity, while individual backbone H-bonds only have a very minor influence on the overall stability.
Acknowledgements
The authors thank Sandy Gewinner and Wieland Schöllkopf for technical support at the FHI FEL facility.References
- K. R. Shoemaker, P. S. Kim, E. J. York, J. M. Stewart and R. L. Baldwin, Nature, 1987, 326, 563–567 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- A. Chakrabartty, T. Kortemme and R. L. Baldwin, Protein Sci., 1994, 3, 843–852 CrossRef CAS PubMed.
- S. Marqusee and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8898–8902 CrossRef CAS.
- A. E. Counterman and D. E. Clemmer, J. Phys. Chem. B, 2003, 107, 2111–2117 CrossRef CAS.
- A. E. Counterman and D. E. Clemmer, J. Phys. Chem. B, 2002, 106, 12045–12051 CrossRef CAS.
- A. E. Counterman and D. E. Clemmer, J. Am. Chem. Soc., 2001, 123, 1490–1498 CrossRef CAS PubMed.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- B. C. Bohrer, S. I. Merenbloom, S. L. Koeniger, A. E. Hilderbrand and D. E. Clemmer, Annu. Rev. Anal. Chem., 2008, 1, 293–327 CrossRef CAS PubMed.
- S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, J. Am. Chem. Soc., 2015, 137, 4236–4424 CrossRef CAS PubMed.
- A. I. González Flórez, E. Mucha, D.-S. Ahn, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, Angew. Chem., Int. Ed., 2016, 55, 3295–3299 CrossRef PubMed.
- R. R. Hudgins, M. A. Ratner and M. F. Jarrold, J. Am. Chem. Soc., 1998, 120, 12974 CrossRef CAS.
- M. Kohtani and M. F. Jarrold, J. Am. Chem. Soc., 2004, 126, 8454–8458 CrossRef CAS PubMed.
- R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1999, 121, 3494–3501 CrossRef CAS.
- F. Schubert, M. Rossi, C. Baldauf, K. Pagel, S. Warnke, G. von Helden, F. Filsinger, P. Kupser, G. Meijer, M. Salwiczek, B. Koksch, M. Scheffler and V. Blum, Phys. Chem. Chem. Phys., 2015, 17, 7373–7385 RSC.
- M. Rossi, V. Blum, P. Kupser, G. von Helden, F. Bierau, K. Pagel, G. Meijer and M. Scheffler, J. Phys. Chem. Lett., 2010, 1, 3465–3470 CrossRef CAS.
- J. Y. Ko, S. W. Heo, J. H. Lee, H. B. Oh, H. Kim and H. I. Kim, J. Phys. Chem. A, 2011, 115, 14215–14220 CrossRef CAS PubMed.
- M. F. Jarrold, Phys. Chem. Chem. Phys., 2007, 9, 1659–1671 RSC.
- R. Wieczorek and J. J. Dannenberg, J. Phys. Chem. B, 2008, 112, 1320–1328 CrossRef CAS PubMed.
- J. A. Scheike, C. Baldauf, J. Spengler, F. Albericio, M. T. Pisabarro and B. Koksch, Angew. Chem., Int. Ed. Engl., 2007, 46, 7766–7769 CrossRef CAS PubMed.
- M. Kohtani, T. C. Jones, J. E. Schneider and M. F. Jarrold, J. Am. Chem. Soc., 2004, 126, 7420–7421 CrossRef CAS PubMed.
- A. Tkatchenko, M. Rossi, V. Blum, J. Ireta and M. Scheffler, Phys. Rev. Lett., 2011, 106, 118102 CrossRef PubMed.
- Y. Fu, J. Gao, J. Bieschke, M. A. Dendle and J. W. Kelly, J. Am. Chem. Soc., 2006, 128, 15948–15949 CrossRef CAS PubMed.
- S. Deechongkit, P. E. Dawson and J. W. Kelly, J. Am. Chem. Soc., 2004, 126, 16762–16771 CrossRef CAS PubMed.
- M. Wang, T. E. Wales and M. C. Fitzgerald, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2600–2604 CrossRef CAS PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, Boca Raton, 71st edn, 1990 Search PubMed.
- P. R. Kemper, N. F. Dupuis and M. T. Bowers, Int. J. Mass Spectrom., 2009, 287, 46–57 CrossRef CAS.
- S. Warnke, G. von Helden and K. Pagel, Proteomics, 2015, 15, 2804–2812 CrossRef CAS PubMed.
- E. Mason and W. McDaniel, Transport properties of ions in gases, Wiley, 1988 Search PubMed.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. M. Todd, Proc. SPIE, 9512, 2015 Search PubMed.
- J. Oomens, B. G. Sartakov, G. Meijer and G. von Helden, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef CAS.
- G. Papadopoulos, A. Svendsen, O. V. Boyarkin and T. R. Rizzo, Faraday Discuss., 2011, 150, 243–255 RSC.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- J. H. McAliley and D. A. Bruce, J. Chem. Theory Comput., 2011, 7, 3756–3767 CrossRef CAS PubMed.
- M. L. P. Price, D. Ostrovsky and W. L. Jorgensen, J. Comput. Chem., 2001, 22, 1340–1352 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Rev D.01, 2009, Wallingford, CT, USA Search PubMed.
- R. Wieczorek and J. J. Dannenberg, J. Am. Chem. Soc., 2005, 127, 14534–14535 CrossRef CAS PubMed.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- A. A. Shvartsburg and M. F. Jarrold, Chem. Phys. Lett., 1996, 261, 86–91 CrossRef CAS.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
- Y.-S. Lin, G. R. Bowman, K. A. Beauchamp and V. S. Pande, Biophys. J., 2012, 102, 315–324 CrossRef CAS PubMed.
- I. L. Karle, C. Das and P. Balaram, Biopolymers, 2001, 59, 276–289 CrossRef CAS PubMed.
- T. Ohyama, H. Oku, A. Hiroki, Y. Maekawa, M. Yoshida and R. Katakai, Biopolymers, 2000, 54, 375–378 CrossRef CAS PubMed.
- E. Chapman, J. S. Thorson and P. G. Schultz, J. Am. Chem. Soc., 1997, 119, 7151–7152 CrossRef CAS.
- J. T. Koh, V. W. Cornish and P. G. Schultz, Biochemistry, 1997, 36, 11314–11322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation details, ATDs, IRMPD spectra, CCSs, theoretical structures. See DOI: 10.1039/c6cp03827a |
| This journal is © the Owner Societies 2016 |