
Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals
 a
and
a
and
Abstract
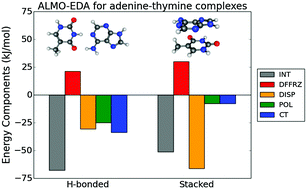
An energy decomposition analysis (EDA) separates a calculated interaction energy into as many interpretable contributions as possible; for instance, permanent and induced electrostatics, Pauli repulsions, dispersion and charge transfer. The challenge is to construct satisfactory definitions of all terms in the chemically relevant regime where fragment densities overlap, rendering unique definitions impossible. Towards this goal, we present an improved EDA for Kohn–Sham density functional theory (DFT) with properties that have previously not been simultaneously attained. Building on the absolutely localized molecular orbital (ALMO)-EDA, this second generation ALMO-EDA is variational and employs valid antisymmetric electronic wavefunctions to produce all five contributions listed above. These contributions moreover all have non-trivial complete basis set limits. We apply the EDA to the water dimer, the T-shaped and parallel-displaced benzene dimer, the p-biphthalate dimer “anti-electrostatic” hydrogen bonding complex, the biologically relevant binding of adenine and thymine in stacked and hydrogen-bonded configurations, the triply hydrogen-bonded guanine–cytosine complex, the interaction of Cl− with s-triazine and with the 1,3-dimethyl imidazolium cation, which is relevant to the study of ionic liquids, and the water–formaldehyde–vinyl alcohol ter-molecular radical cationic complex formed in the dissociative photoionization of glycerol.
 Please wait while we load your content...
Please wait while we load your content...

