
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying short surface ligands on metal phosphide quantum dots†
Edwin A.
Baquero‡
a,
Wilfried-Solo
Ojo‡
a,
Yannick
Coppel
b,
Bruno
Chaudret
a,
Bernhard
Urbaszek
a,
Céline
Nayral
*a and
Fabien
Delpech
*a
aLPCNO (Laboratoire de Physique et Chimie des Nano-Objets), Université de Toulouse, INSA, UPS, CNRS, 135, avenue de Rangueil, F-31077 Toulouse, France. E-mail: fabien.delpech@insa-toulouse.fr; celine.nayral@insa-toulouse.fr
bLaboratoire de Chimie de Coordination, UPR-CNRS 8241, 205 route de Narbonne, 31077 Toulouse Cedex, France
First published on 13th June 2016
Abstract
The control and understanding of the chemical and physical properties of quantum dots (QDs) demands detailed surface characterization. However, probing the immediate interface between the inorganic core and the ligands is still a major challenge. Here we show that using cross-polarization magic angle spinning (MAS) NMR, unprecedented information can be obtained on the surface ligands of Cd3P2 and InP QDs. The resonances of fragments which are usually challenging to detect like methylene or methyl near the surface, can be observed with our approach. Moreover, ligands such as hydroxyl and ethoxide which have so far never been detected at the surface can be unambiguously identified. This NMR approach is versatile, applicable to any phosphides and highly sensitive since it remains effective for identifying quantities as low as a few percent of surface atoms.
Over the last decade, quantum dots –QDs– have emerged as an important class of materials for applications ranging from electronics to biomedicine.1 These significant achievements have been made possible thanks to the development of procedures leading to nano-objects of controlled-shape, -structure, -composition and surface chemistry. This latter feature is obviously of key importance for the synthesis but it is also central for the chemical and physical properties of individual QDs as well as their ordered-assemblies. As an important example, the switching from insulating organic ligands to inorganic ones allows the establishment of conductive bridges between QDs. This has paved the way for applications such as LEDs, solar cells, electronic circuits, and photodetectors.2 While the concepts and the principles that have been established during these works could in principle be applied to a wide variety of materials, the vast majority of advances have been achieved for II–VI and IV–VI semiconductors. In contrast to the success story of these chalcogenides QDs, the chemistry of phosphides QDs has achieved only limited success to date.2,3 For example, InP QDs whose chemistry is the most developed, still suffer from comparably large size distributions (>15%) and broad emission line widths (40–60 nm at FWHM), limited range of diameters and hence emission wavelengths (mainly 500–600 nm) and lower fluorescence quantum yields (generally <60%).3
Aiming at overcoming these limitations, significant efforts have been invested over the last decade into the clarifications of the mechanisms involved in the synthesis and the stabilization of the phosphide QDs.3–6 The point of convergence of these works is the surface of the QDs. In contrast to the now well-known surface chemistry of the lead and cadmium chalcogenides nanocrystals (NCs),7 that of phosphide QDs is much less understood and displays a diversity that does not seem to exist for selenides or sulfides. For instance, we have previously shown for InP QDs that POx (with x = 3 and predominantly 4), In2O3, and InOy(OH)3−2y (y = 0, 1) species exist at the surface.8 The formation of these species is the direct consequence of the high oxophilicity of these QDs which react with water existing as a residual impurity in reactants or solvents or coming from side-reactions of ligands at high temperature. Given the extraordinary complexity of these phosphide QDs surface, the full characterization of the surface ligand is not an easy task. Among the various spectroscopic tools probing QDs surface at the molecular scale (as for instance, infrared, and X-ray photoelectron spectroscopies), nuclear magnetic resonance (NMR) stands out.7c,9,10 This commonly available technique is probably the most effective because (i) many ligands display characteristic spectral features that allow their identification, (ii) it is nondestructive, easy to set up and it allows an analysis directly in the native environment, (iii) the free and bound species can be distinguished, and (iv) the ligand dynamics can be monitored.9,10 Nevertheless, an important and still open challenge for the characterization of NC/ligand systems, is to obtain a detailed picture of the QD surface for short ligands11 as could be the case of hydroxyl8b,12 or alkoxide.10c,11b,13 The observation of moieties close to the surface of QDs is extremely difficult for two main reasons: first, the NMR resonances are broadened due to dipolar couplings, chemical shift anisotropies, structural disorder and potential electronic effects of the nanoparticles –NPs– (including the Knight shift).7c,10,14 Second, the interface represents only a small proportion of the sample. In this communication, we describe our solution to this blocking point by using conventional solid state NMR. Using Cd3P2 and InP QDs as model systems, we show far-reaching capability for identifying surface fragments or ligands and in particular, the so far unobserved P–OH fragment. Moreover, we demonstrate that concentrations as low as a few percent of surface atoms can be identified using this straightforward and easy to set-up method. The generality of this approach to other phosphide QDs is also provided and allow demonstrating the presence of hydroxyl and ethoxide at the surface of InP QDs.
It has been shown,8,15 that the analysis of surface QDs using the basic cross-polarization (CP) sequence MAS NMR is of high interest because it brings complementary information to those given by solution NMR: the low motion of the surface ligand can be advantageously used through the CP experiment to enhance the signals which are typically too broad and too weak to be not observable in solution. The CP experiment where magnetization is transferred from a proton nucleus to a spin nucleus of lower sensitivity (e.g.13C, 29Si, 31P) relies on heteronuclear dipolar couplings. This effect is favored by the low motion and, it decreases fast with distance (inversely proportional to the cube of the distance). Thus, it can be used as a filter to probe neighboring fragments. On this basis, we have chosen to implement a double CP16 also named Forth and Back CP (FBCP)17 which has been recently used for nanoscale silica-based system involving 1H and 29Si nuclei.18 In our case, we utilize two consecutive CP transfers from the 1H of the ligands to 31P located at the surface of QDs, followed by CP from 31P to 1H close to the surface, allowing to probe distance less than 0.5 nm. In addition, we have adapted the sequence for the general and challenging case of the NPs bearing mobile alkyl chain ligands, where the end groups of the ligands display sharp and intense NMR signal. It often prevents the detection of the broad and weak resonances of groups close to their surface. The case of phosphide QDs is well-suited for using the FBCP sequence for the three following reasons: (i) the NMR active 31P nucleus is a 100% naturally abundant and a sensitive probe that can be used as a relay for the polarization transfer, (ii) the 31P nucleus in phosphide QDs possesses long T1 relaxation time (usually T1(31P) is between 20 and 200 s)19 that is an important property for a good elimination of the residual 1H magnetization, (iii) 31P NMR is very sensitive to its chemical environment and it allows in this work to cross-check the validity of the observations made with the FBCP sequence.
In a first step, the potentialities of the FBCP sequence were tested using Cd3P2 QDs as a reference because they have important advantages as a model system. These QDs can be synthesized at RT and thus, side-reactions which are known to occur at high (300 °C) or moderately high (150 °C) temperature are avoided.8,20 In addition, the surface chemistry of these QDs is versatile and can be modified in order to introduce various ligands and to test the limits of this method.
First, we studied Cd3P2 QDs stabilized by a mixture of classical amine and carboxylate ligands namely hexadecylamine (HDA) and acetate (OAc). The QDs were prepared following a procedure previously described with octylamine instead of HDA that is the addition at room temperature of the phosphorus source P(SiMe3)3 into a solution containing 3 equiv. of Cd(OAc)2 and 7 equiv. of HDA.20 After 2 h and the subsequent work-up, spheroidal Cd3P2 QDs of 2.4(0.3) nm are obtained. The UV-Vis, PL spectra, XRD data and TEM picture are provided in Fig. S1 (ESI†). Using the established NMR spectroscopy techniques available for the QDs analysis (solution and solid-state NMR studies), a first insight into both the inorganic and organic components can be gained.8,20 The 31P CP-MAS showed only one sharp signal around −250 ppm corresponding to 31P in the immediate proximity of the Cd3P2 NC surface (Fig. S2, ESI†). In the 31P Hahn-echo MAS NMR spectrum, a broad resonance was observed and shifted at low frequency (around −270 ppm) because of the presence of a second component at −286 ppm as shown by the deconvolution in the Fig. 1a. This latter signal can be assigned to the inner 31P atoms of the core of Cd3P2 QDs. Both 31P resonances lie in the high-field range typically found for NPs of metal phosphides8,20 and are consistent with the formation of Cd3P2 as the only phosphorus-based material. Concerning the composition of the coordination sphere, 13C CP MAS NMR experiments (Fig. 1b and Fig. S3, ESI†) show signals corresponding to carbon containing moieties that are in restricted motions as the ligand on NPs.
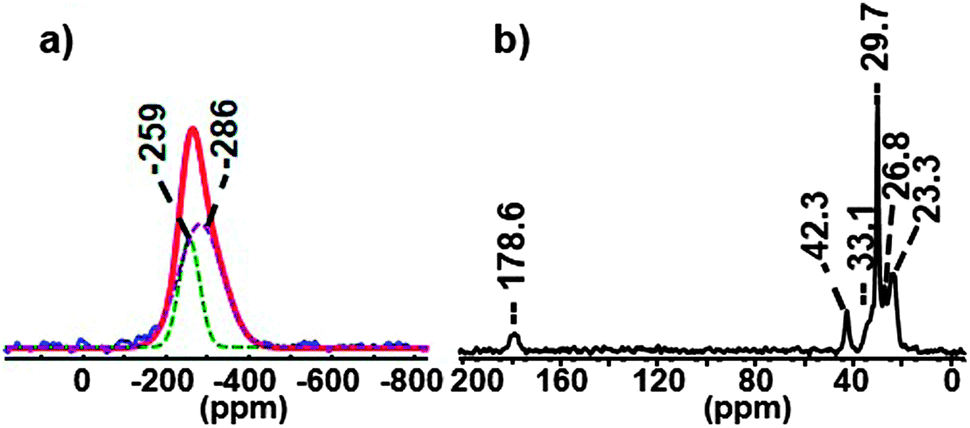 | ||
| Fig. 1 31P Hahn-echo MAS (blue) NMR spectrum (spinning speed 20 kHz) compared to simulated spectrum (red) (a) and 13C CP MAS NMR spectrum (spinning speed 16 kHz) of Cd3P2 QDs stabilized by a mixture of HDA and OAc (b). | ||
The presence of HDA and OAc at the surface is attested by the resonances of the methylene group located α to the amine functional group (δ = 42.3 ppm) and of the acyl (δ = 178.6 ppm) together with its methyl moiety (δ = 23.3 ppm). While being much more sensitive than the NMR of all other nuclei, the up-to-date 1H NMR methods remain not well-suited for the identification of the ligands. In the 1H MAS NMR spectrum of the Cd3P2 QDs (bottom spectrum of Fig. 2), the only resonances clearly visible are located at δ 1.28 and 0.89 ppm and can be assigned respectively to those of the 14 methylenes (i.e. all the CH2 except the closest to the nitrogen atom) and the methyl moieties of the alkyl chain of HDA.
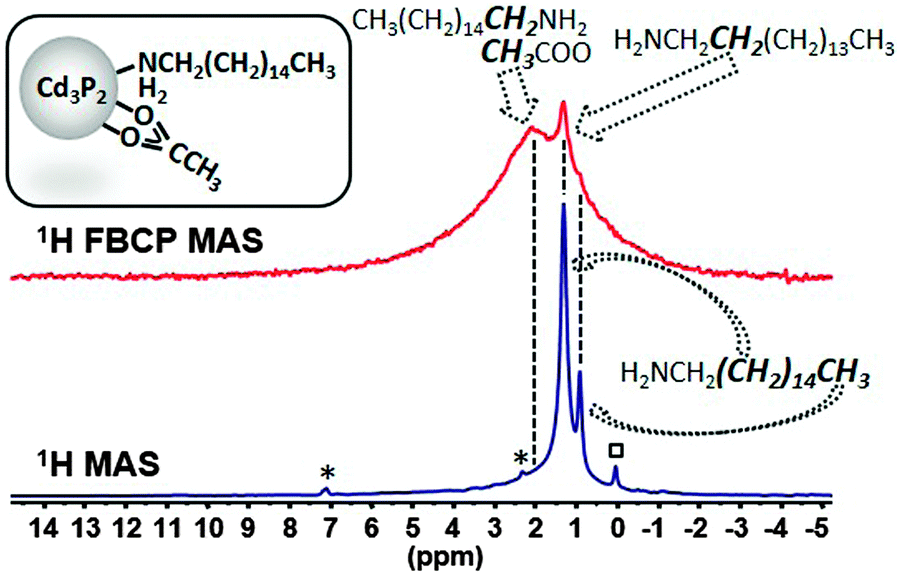 | ||
| Fig. 2 Comparison of the 1H MAS NMR and FBCP 1H MAS NMR spectra (spinning speed 20 kHz) of Cd3P2 QDs stabilized by a mixture of HDA and OAc, (*: residual toluene, □: grease). | ||
These resonances are sharp because the high mobility of the HDA alkyl chain at the methyl terminus strongly reduces the 1H–1H dipolar coupling that usually broaden the 1H NMR signal. In contrast, the resonances which allow the identification of specific groups near the surface (methylene α to the amine functional group and the methyl group of acetate) are too weak to be observable. Similarly, in solution the 1H NMR spectrum in CDCl3 (Fig. S4, ESI†) shows mostly sharp signals for the mobile groups of HDA (central methylenes at 1.28 ppm and methyl groups at 0.90 ppm). However for these small NPs (2.4(0.3) nm), very broad and weak resonances can be observed for the methylene α to NH2 (2.71 ppm) and the OAc methyl group (2.01 ppm). Additional information can be gained with NOESY and DOSY experiments that evidenced strongly surface-bound OAc species and a mixture of strongly and weakly-bound HDA species (Fig. S5 and S6, ESI† respectively). Applying the upgraded FBCP sequence (see ESI†) allows the enhancement of the protons close to the surface at the expense of the intensity of the peaks belonging to mobile fragments (Fig. 2). A new broad signal due to a resonance distribution centered around 2.1 ppm can indeed be observed and assigned to the acetate and the methylene α to the amine functional group. This method, thus, addresses the current failure existing for 1H MAS NMR, concerning the characterization of restricted motion fragments in the presence of mobile ones which is the most frequent situation in ligand-capped metal NC systems.
More interestingly, this method can be used to identify ligands that have been so far invisible. For this purpose, we have prepared Cd3P2 QDs displaying few hydroxyl ligands by adding small amounts of water (see Experimental part). For sake of clarity, these QDs will be noted Cd3P2–OH QDs. The UV-Vis, PL spectra, XRD data and TEM picture of Cd3P2–OH NCs are provided in Fig. S7 (ESI†). Besides the resonances of the phosphides, the 31P Hahn-echo MAS NMR spectrum displays a small peak which is barely visible at ca. 3 ppm (Fig. 3). The existence of this peak was confirmed using the 31P CP MAS NMR experiment (Fig. 3). This resonance is assigned to oxidized phosphorus.8
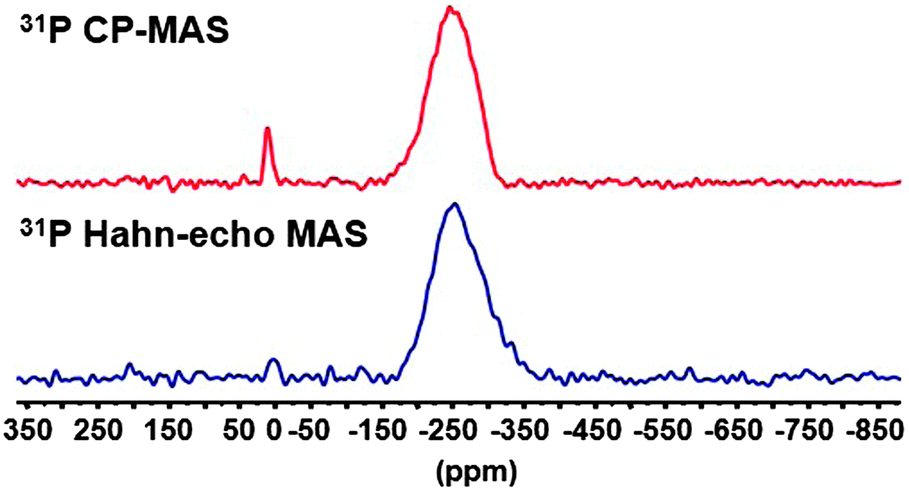 | ||
| Fig. 3 31P Hahn-echo MAS NMR and 31P CP MAS NMR spectra of Cd3P2–OH QDs (spinning speed 20 kHz). | ||
The 1H MAS NMR spectrum performed at 20 kHz of the Cd3P2–OH QDs shows no clear difference with that of Cd3P2 QDs (Fig. S8, ESI†). However, when applying the FBCP sequence, the resulting 1H NMR spectrum displays on one hand a broad resonance around δ 2.1 ppm similar to the one observed for the previous Cd3P2 QDs (methyl of acetate and methylenes located α and β to the amine functional group). On the other hand, an additional broad resonance at higher frequencies and in particular, around 8 ppm is clearly evidenced (Fig. 4). This latter signal is consistent with the oxidized phosphorus species identified in the 31P CP MAS NMR spectra of Cd3P2–OH QDs. It can be assigned to the acidic phosphate protons P–O–H of HPO42− (or H2PO4−) moiety as previously observed with calcium phosphates.21 The broadness of this resonance can be interpreted as the consequence of different hydrogen bonded environments and water content. The chemical shift of hydroxyl is dependent on hydrogen bonding (the stronger the hydrogen-bond, the higher the chemical shift) and chemical exchange with surface-adsorbed water.21 Moreover, an evaluation of the amount of OH which is detected can be undertaken: the size of the QDs (2.3(0.3) nm) implies that around 30% of the P atoms are located at the surface, this value being also supported by the 31P Hahn-echo MAS NMR spectrum shown in Fig. 3. Given the percentage of P–O–H (2% of the total P atoms) which is determined by integrating the surface of the P–O–H resonance in the 31P Hahn-echo MAS NMR spectrum, we can estimate that 2/30 i.e. ca. only 7% of the P surface atoms are P–O–H. Thus, this shows that besides being effective for identifying moieties close to the surface, this method is also highly sensitive. The implications of the presence of hydroxyl ligand at the surface go beyond the mere issue of the identification of new ligands: hydroxyl has been recently shown to play a key role in stabilizing the (111) facets of PbS NCs. Moreover, it is suspected to be at the origin of (i) the deterioration of the optical and electrical properties of PbSe22 and PbS12b QDs-based films and (ii) the InP QDs growth inhibition.8,5e,6b Last, as small-sized anionic ligand, it plays a central role to establish charge neutrality that could not be achieved by sterically hindering fatty carboxylate.12a Thus, this tool is highly relevant in view of controlling the cation/anion stoichiometry in QDs as a tool to further manipulate QD material properties.23
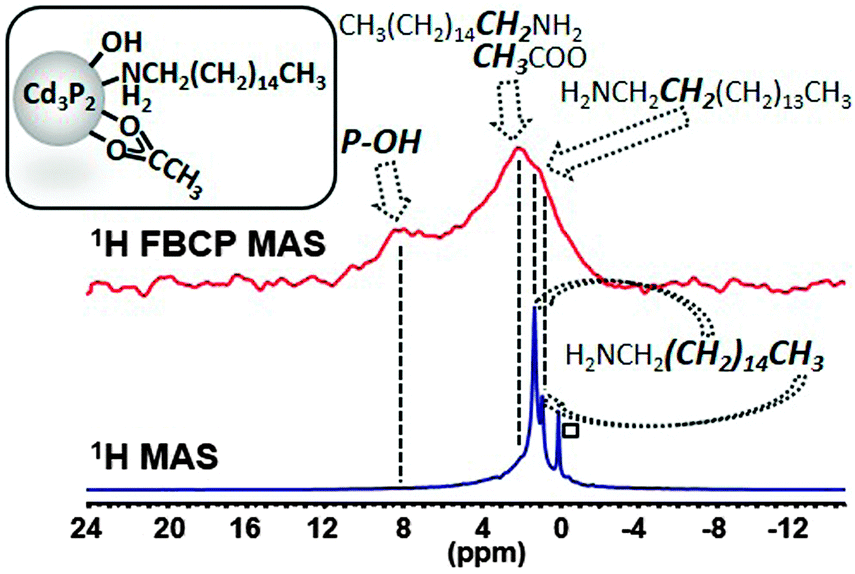 | ||
| Fig. 4 Comparison of the 1H MAS NMR and FBCP 1H MAS NMR spectra (spinning speed 20 kHz) of Cd3P2–OH QDs (□: grease). | ||
The FBCP sequence used here can be directly applied to other phosphides QDs like the highly valuable InP QDs.3 In this case, it allows unraveling so far unknown ligand at the surface of QDs. These QDs were prepared at 230 °C by adding P(SiMe3)3 into a solution containing indium(III) tris(amidinate) as indium precursor, and a mixture of HDA and palmitic acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. Similarly to the previous reports on the synthesis of InP QDs,8,17a the 31P MAS NMR spectra display several environments for the phosphorus atoms as evidenced by the resonances at δ 28, 2 and −185 ppm in a 8:7:85 ratio (Fig. S9a, ESI†). The major one is broad and intense and lies in the high-field range typically found for nanoparticulate metal phosphides. The two other resonances arise from surface oxidation, as evidenced by the significant increase in intensity in the 31P CP MAS NMR spectrum (Fig. S9b, ESI†). This phenomenon results from the reaction between the acid and the amine that yields to the amide formation and the concomitant in situ water formation.24 This side-reaction is clearly supported by the 13C CP MAS NMR spectrum (Fig. 5) which indicates the presence of amide. More importantly, this spectrum provides the first direct observation of the ethyl moiety of an ethoxide fragment (δ 60.0, 19.1 ppm for the methylene and the methyl groups respectively) at the QDs’ surface.10c,11b,13
:3 ratio. Similarly to the previous reports on the synthesis of InP QDs,8,17a the 31P MAS NMR spectra display several environments for the phosphorus atoms as evidenced by the resonances at δ 28, 2 and −185 ppm in a 8:7:85 ratio (Fig. S9a, ESI†). The major one is broad and intense and lies in the high-field range typically found for nanoparticulate metal phosphides. The two other resonances arise from surface oxidation, as evidenced by the significant increase in intensity in the 31P CP MAS NMR spectrum (Fig. S9b, ESI†). This phenomenon results from the reaction between the acid and the amine that yields to the amide formation and the concomitant in situ water formation.24 This side-reaction is clearly supported by the 13C CP MAS NMR spectrum (Fig. 5) which indicates the presence of amide. More importantly, this spectrum provides the first direct observation of the ethyl moiety of an ethoxide fragment (δ 60.0, 19.1 ppm for the methylene and the methyl groups respectively) at the QDs’ surface.10c,11b,13
 | ||
| Fig. 5 13C CP MAS NMR spectrum (spinning speed 16 kHz) of InP QDs. | ||
It is important to stress that this so far unidentified ligand is suspected to play a key role for the isolation and purification processes of QDs.10c,11b,13 The high intensity in this CP MAS NMR spectrum clearly shows that this fragment is in restricted motion, presumably interacting through van der Waals interaction with others surface ligands. Given the absence of peaks in the OH region of the IR spectrum (Fig. S10, ESI†), it is probable that the ethoxide moiety belongs to ethoxide ligand rather than hydrogen-bonded or trapped ethyl alcohol.10c This ethoxyde ligand probably forms from the ligand exchange reaction with carboxylate as previously proposed by Hens et al.11b Last, palmitate completes the picture of the carbon-containing ligands shell as evidenced by its distinctive signal at δ 180.5 corresponding to the carboxylate resonance (Fig. 5). The direct 13C MAS NMR is well-suited for identifying mobile species and the corresponding spectrum shows residual mesitylene solvent molecules which remain in the sample (δ 136.2 and 126.1 ppm) (Fig. S11, ESI†). This is also evident from the 1H MAS NMR spectrum (δ 6.6 and 2.1 ppm) which displays in addition to the characteristic resonances of the long chain palmitate ligands clearly visible at δ 1.21 and 0.83 ppm (Fig. 6). However, when applying the FBCP sequence, the resulting 1H NMR spectrum shows the resonances of the fragments that are located close to the surface. Beyond the resonances of the methylenes and the methyl of palmitate, one can observe new broad peaks at 4.8 and 7.0 ppm (Fig. 6). The first one is consistent with the CH2 of ethoxide identified using 13C CP MAS NMR. The latter resonance belongs to more acidic protons and can be assigned, as for Cd3P2 QDs, to the hydroxyl fragments involved in different hydrogen-bonded environments with water molecules.21
 | ||
| Fig. 6 Comparison of the 1H MAS NMR and FBCP 1H MAS NMR spectra (spinning speed 16 kHz) of InP QDs (*: residual mesitylene, □: grease). | ||
In conclusion, we have described the potentialities of the conventional solid-state MAS NMR for providing a comprehensive overview of phosphides QDs. Remarkably, this allows the identification of new species that were so far invisible using IR and NMR spectroscopies because of the broadening of the surface resonances and of the strong signal of fragments located further from the surface. Moreover, the upgraded NMR-based approach called 1H Forth and Back Cross Polarization (FBCP) MAS enables the unprecedented visualization of 1H signals of rigid fragments or ligands at the surface of phosphides NCs. This is achieved thanks to selective enhancement of 1H nuclei at the inorganic surface. This experiment has been shown to be highly sensitive since very minor species can be identified (as low as a few percent) and thus, paves the way for re-examining in detail the surface of NPs and identifying the actual surface state. We have demonstrated the applicability of this approach to phosphide NCs and we believe that this approach can be extended to other NPs incorporating NMR active nuclei such as metals, metal chalcogenides, pnictogenides or halides.
Acknowledgements
This work was supported by the Université Paul Sabatier, the Région Midi-Pyrénées, the CNRS, the Institut National des Sciences Appliquées of Toulouse, the European Commission for ERC Grant No. 306719 and the Agence Nationale pour la Recherche (Project ANR-13-IS10-0004-01). W.-S. O. is grateful to the European Commission for a postdoctoral grant (ERC Grant No. 306719). E. A. B. is grateful to Marie Curie Actions and Campus France for a PRESTIGE post-doc fellowship (FP7/2007–2013) under REA grant agreement no. PCOFUND-GA-2013-609102.Notes and references
- (a) R. Costi, A. E. Saunders and U. Banin, Angew. Chem., Int. Ed., 2010, 49, 4878 CrossRef CAS PubMed; (b) D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012 CrossRef CAS PubMed.
- (a) S. Tamang, C. Lincheneau, Y. Hermans, S. Jeong and P. Reiss, Chem. Mater., 2016, 28, 2491 CrossRef CAS; (b) S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981 CrossRef CAS PubMed.
- (a) B. A. Glassy and B. M. Cossairt, Chem. Commun., 2015, 51, 5283 RSC; (b) M. H. Mobarok, E. J. Luber, G. M. Bernard, L. Peng, R. E. Wasylishen and J. M. Buriak, Chem. Mater., 2014, 26, 1925 CrossRef CAS; (c) S. Miao, T. Yang, S. G. Hickey, V. Lesnyak, B. Rellinghaus, J. Xu and A. Eychmüller, Small, 2013, 9, 3415 CrossRef CAS PubMed.
- (a) D. Franke, D. K. Harris, L. Xie, K. F. Jensen and M. G. Bawendi, Angew. Chem., Int. Ed., 2015, 54, 14507 CrossRef; (b) D. C. Gary, B. A. Glassy and B. M. Cossairt, Chem. Mater., 2014, 26, 1734 CrossRef CAS; (c) P. M. Allen, B. J. Walker and M. G. Bawendi, Angew. Chem., Int. Ed., 2010, 49, 760 CrossRef CAS PubMed; (d) D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432 CrossRef CAS; (e) L. Xie, D. K. Harris, M. G. Bawendi and K. F. Jensen, Chem. Mater., 2015, 27, 5058 CrossRef CAS; (f) D. C. Gary and B. M. Cossairt, Chem. Mater., 2013, 25, 2463 CrossRef CAS.
- (a) D. C. Gary, S. E. Flowers, W. Kaminsky, A. Petrone, X. Li and B. M. Cossairt, J. Am. Chem. Soc., 2016, 138, 1510 CrossRef CAS PubMed; (b) K. Kim, D. Yoo, H. Choi, S. Tamang, J.-H. Ko, S. Kim, Y.-H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714 CrossRef CAS PubMed.
- (a) M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141 CrossRef CAS PubMed; (b) J. S. Owen, Science, 2015, 347, 615 CrossRef CAS PubMed; (c) D. A. Hines and P. V. Kamat, ACS Appl. Mater. Interfaces, 2014, 6, 3041 CrossRef CAS PubMed; (d) A. J. Morris-Cohen, M. Malicki, M. D. Peterson, J. W. J. Slavin and E. A. Weiss, Chem. Mater., 2013, 25, 1155 CrossRef CAS; (e) Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664 CrossRef CAS PubMed.
- (a) A. Cros-Gagneux, F. Delpech, C. Nayral, A. Cornejo, Y. Coppel and B. Chaudret, J. Am. Chem. Soc., 2010, 132, 18147 CrossRef CAS PubMed; (b) H. Virieux, M. Le Troedec, A. Cros-Gagneux, W.-S. Ojo, F. Delpech, C. Nayral, H. Martinez and B. Chaudret, J. Am. Chem. Soc., 2012, 134, 19701 CrossRef CAS PubMed.
- (a) L. Marbella and J. Millstone, Chem. Mater., 2015, 27, 2721 CrossRef; (b) C. Bonhomme, C. Gervais and D. Laurencin, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 77, 1 CrossRef CAS PubMed.
- (a) Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211 CrossRef CAS; (b) J. De Roo, F. Van den Broeck, K. De Keukeleere, J. C. Martins, I. Van Driessche and Z. Hens, J. Am. Chem. Soc., 2014, 136, 9650 CrossRef CAS PubMed; (c) N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 18536 CrossRef CAS PubMed; (d) J. De Roo, Y. Justo, K. De Keukeleere, F. Van den Broeck, J. C. Martins, I. Van Driessche and Z. Hens, Angew. Chem., Int. Ed., 2015, 54, 6488 CrossRef CAS PubMed.
- (a) M. H. Mobarok and J. M. Buriak, Chem. Mater., 2014, 26, 4653 CrossRef CAS; (b) A. Hassinen, I. Moreels, K. De Nolf, P. F. Smet, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2012, 134, 20705 CrossRef CAS PubMed.
- (a) D. Zherebetskyy, M. Scheele, Y. Zhang, N. Bronstein, C. Thompson, D. Britt, M. Salmeron, P. Alivisatos and L.-W. Wang, Science, 2014, 344(6190), 1380 CrossRef CAS PubMed; (b) V. Malgras, A. Nattestad, Y. Yamauchi, S. X. Doua and J. H. Kim, Nanoscale, 2015, 7, 5706 RSC.
- B. Shakeri and R. W. Meulenberg, Langmuir, 2015, 31, 13433 CrossRef CAS PubMed.
- J. P. Yesinowski, A. P. Purdy, H. Wu, M. G. Spencer, J. Hunting and F. J. Di Salvo, J. Am. Chem. Soc., 2006, 128, 4952 CrossRef CAS PubMed.
- (a) M. Tomaselli, J. L. Yarger, M. Bruchez, R. H. Havlin, D. de Graw, A. Pines and A. P. Alivisatos, J. Chem. Phys., 1999, 110, 8861 CrossRef CAS; (b) N. El Hawi, C. Nayral, F. Delpech, Y. Coppel, A. Cornejo, A. Castel and B. Chaudret, Langmuir, 2009, 7540 CrossRef CAS PubMed; (c) D. D. Lovingood, R. Achey, A. K. Paravastu and G. F. Strouse, J. Am. Chem. Soc., 2010, 132, 3344 CrossRef CAS PubMed; (d) F. Iacono, C. Palencia, L. de la Cueva, M. Meyns, L. Terracciano, A. Vollmer, M. J. de la Mata, C. Klinke, J. M. Gallego, B. H. Juarez and R. Otero, ACS Nano, 2013, 7, 2559 CrossRef CAS PubMed.
- J. Schaefer, R. A. Mc Kay and E. O. Stejskal, J. Magn. Reson., 1979, 34, 443 CAS.
- T. Tran, H. Bildsøe, H. Jakobsen and J. Skibsted, J. Magn. Reson., 2012, 221, 19 CrossRef CAS PubMed.
- (a) N. Baccile, G. Laurent, C. Bonhomme, P. Innocenzi and F. Babonneau, Chem. Mater., 2007, 19, 1343 CrossRef CAS; (b) N. Folliet, C. Roil, S. Bégu, A. Aubert, T. Mineva, A. Goursot, K. Selvaraj, L. Duma, F. Tielens, F. Mauri, G. Laurent, C. Bonhomme, C. Gervais, F. Babonneau and T. Azaïs, J. Am. Chem. Soc., 2011, 133, 16815 CrossRef CAS PubMed.
- (a) S. M. Holl, T. Kowalewski and J. Schaefer, Solid State Nucl. Magn. Reson., 1996, 6, 39 CrossRef CAS PubMed; (b) N. L. Adolphi, R. D. Stoddard, S. C. Goel, W. E. Buhro, P. C. Gibbons and M. S. Conradi, J. Phys. Chem. Solids, 1992, 53, 1275 CrossRef CAS.
- W.-S. Ojo, S. Xu, F. Delpech, C. Nayral and B. Chaudret, Angew. Chem., Int. Ed., 2012, 51, 738 CrossRef CAS PubMed.
- (a) J. P. Yesinowski and H. Eckert, J. Am. Chem. Soc., 1987, 109, 6274 CrossRef CAS; (b) J. Arends, J. Christoffersen, M. R. Christoffersen, H. Eckert, B. O. Fowler, J. C. Heughebaert, G. H. Nancollas, J. P. Yesinowski and S. J. Zawackiw, J. Cryst. Growth, 1987, 84, 515 CrossRef CAS.
- J. M. Luther, M. Law, Q. Song, C. L. Perkins, M. C. Beard and A. J. Nozik, ACS Nano, 2008, 2, 271 CrossRef CAS PubMed.
- (a) J. M. Luther and J. M. Pietryga, ACS Nano, 2013, 7, 1845 CrossRef CAS PubMed; (b) C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, ACS Nano, 2014, 8, 9463 CrossRef CAS PubMed.
- (a) H. Wu, Y. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522 CrossRef CAS PubMed; (b) M. Protière and P. Reiss, Chem. Commun., 2007, 2417 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental details and characterization data. See DOI: 10.1039/c6cp03564g |
| ‡ These authors have contributed equally to this work. |
| This journal is © the Owner Societies 2016 |