
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new insight into the photochemistry of avobenzone in gas phase and acetonitrile from ab initio calculations†
Marko
Kojić
,
Milena
Petković
and
Mihajlo
Etinski
*
Faculty of Physical Chemistry, University of Belgrade, Studentski trg 12-16, 11000 Belgrade, Serbia. E-mail: etinski@ffh.bg.ac.rs; Fax: +381112187133; Tel: +381113336632
First published on 12th July 2016
Abstract
Avobenzone (4-tert-butyl-4′-methoxydibenzoylmethane, AB) is one of the most widely used filters in sunscreens for skin photoprotection in the UVA band. The photochemistry of AB includes keto–enol tautomerization, cis–trans isomerization, rotation about the single bond and α bond cleavages of carbonyl groups. In this contribution we study chelated and non-chelated enol, rotamers Z and E, and keto tautomers of AB in the ground and excited states in gas phase and acetonitrile by means of a coupled cluster method. Our findings suggest that torsion around the double C2–C3 bond of photoexcited chelated enol leads to internal conversion to the ground state and formation of rotamer E. In addition, opening of the chelated hydrogen ring by torsion of the hydroxyl group creates non-chelated enol. The possible mechanisms of rotamer Z formation are discussed. The solvent dependent photolability is related to the relative order of the lowest triplet ππ* and nπ* states of the keto tautomer.
1 Introduction
The Sun emits ultraviolet radiation in the UVA (320–380 nm), UVB (290–320 nm), and UVC (100–290 nm) bands. Nevertheless, on the Earth surface only radiation in UVA and UVB bands is relevant since ozone strongly absorbs radiation below 290 nm. As a result of the toxic, mutagenic and carcinogenic effects of UVA and UVB radiation on skin tissue,1,2 there is a necessity for producing safe and efficient filters that will provide photoprotection.3 Photostability of a UV filter is the principal condition for its application since potential photoproducts generally less efficiently absorb UVA and UVB light. In addition, the photoproducts could be deleterious to human skin because they are usually highly reactive radical species.In contrast to the broad range of UVB filters, a suitable UVA filter is lacking. One of the main classes of UVA filters in cosmetic sunscreens consists of dibenzoylmethane (DBM) derivatives. Particularly, 4-tert-butyl-4′-methoxydibenzoylmethane (trade name avobenzone (AB)) is one of the most widely used UVA filters.3 Its photochemistry is similar to that of the parent compound DBM, although AB is known to photodegrade via the Norrish type I mechanism in nonpolar solvents.4 The absence of fluorescence at room temperature indicates an efficient nonradiative relaxation mechanism in the first excited state or a fast photochemical reaction. Despite the fact that AB has practical importance as an UVA filter, complete understanding of its photophysics and photochemistry is still missing due to solvent-dependent multiple relaxation pathways.4–22
AB is a β-dicarbonyl compound liable to keto–enol tautomerization. Many theoretical and experimental studies provided evidence that in the ground state the chelated enol form is favored due to the intramolecular hydrogen bond.12,21,23,24 However, particularly in solutions the keto tautomer is present since the equilibrium constant is not large.23 The chelated enol absorbs around 355 nm, while the keto tautomer displays an absorption band in the UVC range around 265 nm. The keto tautomer can be produced upon irradiation of chelated enol in polar nonprotic solvents such as dimethylsulfoxide12 and acetonitrile15 although with low quantum yield (0.014 in acetonitrile15). Tautomerization was not observed in nonpolar and polar protic solvents. Particularly, the absence of tautomerization and the increase of the recovery rate to chelated enol in polar protic solvents have been attributed to a fast proton exchange mechanism via intermolecular hydrogen bonding with the solvent molecules.8 Since the keto form is thermodynamically unstable, it recovers to the more stable chelated enol tautomer within approximately 5 hours in the dark.15 No reverse tautomerization to the enol form has been observed upon irradiation of keto AB in acetonitrile.15 On the other hand, the photoexcitation of the keto tautomer leads to the formation of a triplet state which is believed to be a precursor for photodegradation.10,12 AB is found to be essentially photostable in polar protic solvents, while it photodegrades appreciably in nonpolar solvents.8,12 The main products of photodegradation are benzoyl or phenacyl radicals resulting from α-cleavage reactions.4,12
In addition to keto–enol tautomerization and photodegradation, photoexcited AB exhibits cis–trans isomerization and isomerization about single bonds.6,8,15,18–20 The produced enol tautomers (rotamers) are transient species in the photodynamics of AB. They absorb at higher wavelengths than the chelated enol form (≈300 nm) and recover to the chelated enol and to a minor extent to the keto form.9,10 Recent time-resolved spectroscopic experiments have provided additional insight into electronic and vibrational relaxation of the photoexcited chelated enol tautomer of DBM18,20 and AB.19 The findings from these studies revealed that the decay signal from the first bright state of chelated enol in acetonitrile exhibits a multiexponential behavior, which represents the kinetics of various relaxation channels. Brixner and coworkers revealed that the excited state absorption of DBM decays with a time constant of 147 fs. Based on global analysis of the transient signal, the authors also extracted time constants of 1.18 ps, 10.75 ps, 0.49 ns, and 10 ns. Furthermore, the absorption signal in deep UV related to the kinetics of rotamers remains present up to 3.2 ns whereas the absorption signal in near UV and visible decays within 14 ps.18 Intersystem crossing to a triplet state was observed on the time scale of a few picoseconds. Dunkelberger et al. measured the ultrafast transient absorption of AB.19 The authors concluded that photoexcited chelated enol electronically relaxes in hundreds of femtoseconds followed by vibrational relaxation in several picoseconds. Moreover, they argued that the rotamer species formed by rotation about the single bond is promptly created after the photoexcitation. The authors did not find evidence that this transient species was involved in the formation of the keto tautomer.19
Understanding the photochemistry of AB is an important issue because it can lead to the development of more efficient UV filters. Although recent time-resolved experiments have provided insight into the relaxation kinetics of DBM and AB,18–20 the mechanistic aspect of the relaxation is not yet fully understood and needs further investigation. The aim of the present study is to provide insight into the electronic relaxation mechanism upon photoexcitation to the bright state of AB. Moreover, we will characterize the experimentally observed transient species whose kinetics have previously been observed in time-resolved absorption experiments.18–20 In order to make this study computationally feasible, we decided to examine 4-methoxydibenzoylmethane, i.e. to exclude the tert-butyl group from AB. The electron-donating nature of this group decreases ππ* state energies13 and thus our computed energies of these states will be somewhat blueshifted compared to those of AB. In this work, we consider the following tautomers: chelated enol, non-chelated enol, rotamer E, rotamer Z and keto (cf.Fig. 1). The non-chelated enol tautomer is formed by rotation of the hydroxyl group in such a way that the intramolecular hydrogen bond is broken. Rotation of the non-chelated enol tautomer around the single C1–C2 and double C2–C3 bonds leads to rotamers Z and E, respectively.
 | ||
| Fig. 1 Atom labeling and the tautomers considered in this work: (a) chelated enol, (b) non-chelated enol, (c) keto, (d) rotamer E and (e) rotamer Z. | ||
The paper is organized as follows. In the next section we summarize computational methods and basis sets that were used in our calculations. In the subsequent section we present ground- and excited-state structures, and vertical and adiabatic electronic excitation energies. Furthermore, the formation of transient species that appear in the photodynamics of AB is discussed. Finally, we draw conclusions from our study.
2 Computational details
All calculations were performed using the TURBOMOLE program package.25 We employed the spin-component scaled26 coupled-cluster method with approximate treatment of doubles27 (SCS-CC2) implemented with the resolution-of-identity (RI) approximation28,29 for the ground state30 and in combination with linear response theory for excited-state optimizations,31 and vertical excitation energies and properties.32 Spin-component scaling assumes different scaling of energy contributions of the same and opposite-spin components. This procedure enhances the accuracy of adiabatic energies for both ππ* and nπ* states.33 In a recent benchmark study, it was shown that the SCS-CC2 method provided accurate 0–0 transition energies for a set of organic molecules with a standard deviation of 0.06 eV.34 Besides, the SCS-CC2 method was the only method that reproduced all the properties of the S1 state of cytosine.35 Only valence electrons were correlated in calculations.The optimization of excited-state equilibrium geometries is a difficult and computationally demanding task. Therefore, we have to compromise with respect to the choice of the one-particle basis sets. A rather small Dunning's36,37 correlation-consistent basis set cc-pVDZ (C, O: 9s4p1d/3s2p1d; H: 4s1p/2s1p) was employed for all geometry optimizations. In addition, a larger basis set with diffuse functions, aug-cc-pVDZ (C, O: 10s5p2d/4s3p2d; H: 5s2p/3s2p), was used for single point calculations. Auxiliary basis sets for the resolution-of-identity approximation of the two-electron integrals were taken from the TURBOMOLE library.38
The conductor-like screening model (COSMO) implemented in TURBOMOLE39,40 was utilized in the present work to study AB in acetonitrile. This continuum model has been proven to be very reliable in modeling spectral shifts that are consequences of electrostatic interactions in polar solvents.41–45
The ground state geometries were optimized in gas phase and acetonitrile. Vertical electronic excitation energies at the optimized ground state geometries were computed employing both environments. Excited state geometries were solely optimized in gas phase. In order to estimate their energies in acetonitrile we applied the solvatochromic shifts computed from the ground state vertical excitation spectra.
3 Results and discussion
3.1 Ground state geometries and vertical electronic excitation energies
The optimized ground state geometries in acetonitrile with the most significant bond lengths are presented in Fig. 2. The bond lengths of the gas phase structures are very similar to the ones in acetonitrile (cf. Fig. S1 in the ESI†) and therefore will not be discussed. Chelated enol structure is found to be nonplanar with a butterfly shape. The intramolecular hydrogen bond length is 1.59 Å. In our previous work,24 we characterized this hydrogen bonded species and its spectral properties. The complete failure of the harmonic approximation for the hydrogen motion was demonstrated. Besides the asymmetric O–H⋯O conformer, we found that the symmetric O⋯H⋯O conformer is also present in the gas phase at room temperature. The computed lengths of the O1–C1, C1–C2, C2–C3 and C3–O2 bonds are found to be 1.27, 1.45, 1.39 and 1.34 Å, respectively. Since the double bonds are longer and the single bonds are shorter than their typical values, these values indicate that there is π electron delocalization of the O1–C1–C2–C3–O2–H pseudocycle. In addition, the benzene groups allow electron delocalization over the entire molecule. The average C–C bond length in the benzene rings is 1.41 Å. The methoxy group does not influence the geometry of the benzene ring to which it is attached. The length of the C7–O bond is 1.36 Å. It is well known that a small basis set overestimates the CC2 bond lengths46 and therefore it is expected that a larger basis set would contract SCS-CC2 bond lengths. If we compare the optimized bond lengths of chelated enol in the present work with the ones obtained with the cc-pVTZ basis set from our previous work,24 we find that there is a small difference of ≈0.01 Å between them. | ||
| Fig. 2 Geometries of the optimized ground state in acetonitrile: (a) chelated enol, (b) non-chelated enol, (c) rotamer E, (d) rotamer Z and (e) keto. All bond lengths are in Å. | ||
We note that the optimized ground state geometry of the non-chelated enol is more distorted than that of the chelated form. Comparison of these two geometries reveals that the O2–C3–C4–C5 dihedral angle increases its value from 19 to 35 degrees whereas the O1–C1–C10–C11 angle remains unchanged. The π electron delocalization of the O1–C1–C2–C3–O2–H pseudocycle present in the chelated enol decreases due to the absence of the intramolecular hydrogen bond. In line with this observation, we find that double O1–C1 and C2–C3 bonds shrink by ≈0.02 Å whereas the single C1–C2 and C3–O2 bonds elongate by the same value. In addition, the C1–C10 and C3–C4 bonds are stretched by 0.02 and 0.01 Å compared to those of the chelated enol structure.
Computed nuclear arrangement of rotamers E and Z suggests a certain difference in their electronic structure. The electron delocalization present in both enol tautomers is still noticeable in rotamer E. Thus, the rotation around the double C2–C3 bond partially conserves electron delocalization although the pseudocycle is decomposed. By comparing the bond lengths of rotamer E with those of the non-chelated enol structure we find that only O2–C3 and C3–C4 bonds change their lengths by 0.01 and −0.01 Å. On the other hand, the bond lengths of rotamer Z differ more significantly from those of the non-chelated enol implying that the π electron delocalization is absent. The lengths of the C1–O1 and C2–C3 double bonds are 1.22 and 1.34 Å, respectively. Note that the C–C bonds in the benzene rings of rotamer Z are shorter by ≈0.02–0.03 Å than those of rotamer E and both enol forms.
In the conformational search of the keto tautomer we found two structures that differ by 5 kJ mol−1. The more stable geometry is characterized by its symmetric structure with respect to the benzene rings and is shown in Fig. 2. The C–O bond length in the optimized geometry of the keto tautomer is 1.24 Å. The O2–C3–C2–C1 dihedral angle is found to be 102 degrees.
Let us briefly compare ground state electronic energies of the optimized tautomers in acetonitrile and the gas phase. We find that the chelated enol tautomer in acetonitrile is 41 kJ mol−1 more stable than the non-chelated form due to the intramolecular hydrogen bond. This energy difference amounts to 63 kJ mol−1 in gas phase. Thus polar solvents considerably stabilize breaking of the hydrogen bond. The same trend is observed for ground state energies of rotamers Z and E. The energies of non-chelated enol, rotamer E and rotamer Z differ by up to 6 kJ mol−1 regardless of the environment. Surprisingly, the keto tautomer is 7–9 kJ mol−1 more stable than the chelated enol. The thermal and entropic contributions assuming harmonic approximation are almost equal for all tautomers and amount to ≈560 kJ mol−1. Hence, our results reveal that the free energy of the keto tautomer is lower than that of the chelated enol tautomer by several kJ mol−1. This small value is within the error of the CC2 method and thus we cannot decisively estimate the equilibrium constant. Besides, in order to quantitatively determine the relative free energies of keto and chelated enol tautomers, it is necessary to compute anharmonic frequencies,24,47 which is beyond the scope of the present work.
Vertical electronic singlet and triplet excitation spectra at the ground state geometries are given in Table 1. The most important frontier Hartree–Fock orbitals are presented in Fig. S2–S6 in the ESI.† Rydberg states are not observed among the first three excited singlet and triplet states. In the graphical representation of the electron densities of the orbitals, we therefore omitted the Rydberg-type orbitals and denominate the lowest unoccupied valence molecular orbital by LUMO. Inspection of the orbitals reveals a difference in the electron density of rotamers Z, E and both enol tautomers. In the latter case, all tautomers have a similar electron density distribution in all orbitals. In the case of rotamer Z, the πH−1 and πH−2 orbitals exchange their order. Furthermore, πH, πH−4 and nH−5 have a somewhat different electron density distribution in comparison with that of rotamer E.
| State | Gas phase | Acetonitrile | ||||||
|---|---|---|---|---|---|---|---|---|
| Character | Excitation | f | Energy | Character | Excitation | f | Energy | |
| Chelated enol | ||||||||
| S0 | GS | 0.00 | GS | 0.00 | ||||
| S1 | ππ* | H → L | 0.901 | 3.95 | ππ* | H → L | 0.979 | 3.78 |
| S2 | nπ* | H−5 → L | 0.017 | 4.06 | nπ* | H−5 → L | 0.002 | 4.17 |
| S3 | ππ* | H → L+1, H−3 → L | 0.006 | 4.55 | ππ* | H → L+1, H−3 → L | 0.005 | 4.55 |
| T1 | ππ* | H → L | 3.18 | ππ* | H → L | 3.16 | ||
| T2 | nπ* | H−5 → L | 3.82 | ππ* | H−2 → L | 3.84 | ||
| T3 | ππ* | H−2 → L | 3.94 | ππ* | H−4 → L, H−5 → L | 3.99 | ||
| Non-chelated enol | ||||||||
| S0 | GS | 0.65 | GS | 0.42 | ||||
| S1 | nπ* | H−5 → L | 0.000 | 4.25 | nπ* | H−5 → L | 0.000 | 4.21 |
| S2 | ππ* | H → L, H → L+1 | 0.308 | 5.20 | ππ* | H → L | 0.939 | 4.59 |
| S3 | ππ* | H → L | 0.559 | 5.24 | ππ* | H → L+1 | 0.004 | 5.00 |
| T1 | ππ* | H → L | 3.92 | ππ* | H → L | 3.69 | ||
| T2 | nπ* | H−5 → L | 4.05 | nπ* | H−5 → L | 4.00 | ||
| T3 | ππ* | H−1 → L | 4.55 | ππ* | H−2 → L | 4.33 | ||
| Rotamer Z | ||||||||
| S0 | GS | 0.61 | GS | 0.43 | ||||
| S1 | nπ* | H−5 → L | 0.018 | 4.42 | nπ* | H−5 → L, H−5 → L+1 | 0.069 | 4.42 |
| S2 | ππ* | H → L+1 | 0.042 | 5.16 | ππ* | H → L, H → L+1 | 0.674 | 4.78 |
| S3 | ππ* | H−2 → L, H−1 → L | 0.008 | 5.26 | ππ* | H → L+1 | 0.017 | 5.01 |
| T1 | ππ* | H → L | 3.90 | ππ* | H → L+1, H → L | 3.72 | ||
| T2 | nπ* | H−5 → L | 4.26 | nπ* | H−5 → L+1 | 4.21 | ||
| T3 | ππ* | H−1 → L, H−2 → L | 4.50 | nπ* | H−5 → L, H−2 → L | 4.42 | ||
| Rotamer E | ||||||||
| S0 | GS | 0.59 | GS | 0.47 | ||||
| S1 | nπ* | H−5 → L | 0.008 | 4.25 | nπ* | H−5 → L | 0.024 | 4.19 |
| S2 | ππ* | H → L | 0.633 | 5.10 | ππ* | H → L | 0.636 | 4.71 |
| S3 | ππ* | H → L+1 | 0.005 | 5.23 | ππ* | H−3 → L | 0.013 | 5.09 |
| T1 | ππ* | H → L | 3.89 | ππ* | H → L | 3.76 | ||
| T2 | nπ* | H−5 → L | 4.14 | nπ* | H−5 → L | 4.07 | ||
| T3 | ππ* | H−1 → L | 4.47 | ππ* | H−4 → L | 4.37 | ||
| Keto | ||||||||
| S0 | GS | −0.07 | GS | −0.09 | ||||
| S1 | nπ* | H−4 → L | 0.006 | 3.80 | nπ* | H−4 → L | 0.009 | 3.85 |
| S2 | nπ* | H−4 → L+1 | 0.001 | 4.03 | nπ* | H−5 → L | 0.001 | 4.07 |
| S3 | ππ* | H−1 → L | 0.020 | 4.50 | ππ* | H → L | 0.314 | 4.37 |
| T1 | ππ* | H → L+2 | 3.51 | ππ* | H → L+2 | 3.47 | ||
| T2 | nπ* | H−4 → L | 3.54 | nπ* | H−4 → L | 3.60 | ||
| T3 | ππ* | H−2 → L | 3.72 | ππ* | H−2 → L | 3.69 | ||
The first excited singlet state of the chelated enol tautomer originates from πH → πL* transition. It is a bright (i.e. with a large oscillator strength, f ≈ 0.9) state with an excitation energy of 3.95 eV in the gas phase. In acetonitrile, the S1 energy is red-shifted to 3.78 eV. This value is close to the observed maximum of the absorption band in acetonitrile solution (≈3.50 eV10,15,19). It is worthwhile to note that the difference between experimental and computed energies partially comes from neglecting the tert-butyl group. In simpler β-diketones, such as malonaldehyde and acetylacetone, the bright state is the second excited state.18,48–51 Thus, the extended electronic conjugation present in AB shifts the bright state below the dark state (i.e. the nπ* state with a small oscillator strength). The excitation energy of the nπ* state is 4.06 and 4.17 eV in the gas phase and acetonitrile, respectively. This state originates from nH−5 → πL* excitation. Approximately 0.4–0.5 eV above the dark state, there is another ππ* state. The lowest triplet state of the chelated enol tautomer has ππ* character that comes from the same excitation as the S1 state. The T1 vertical energy is 3.18 eV and it exhibits a negligible solvatochromic shift in acetonitrile. The triplet nπ* state is the second state in the gas phase and is located 0.13 eV below the S1 state. In acetonitrile, it shifts to the fifth triplet state.
The breaking of the intramolecular hydrogen bond and rotation of the hydroxyl group strongly destabilizes the ππ* states. Particularly, the S1 and T1 states of the chelated enol blueshift by 1.29 and 0.74 eV in the gas phase non-chelated form. These shifts are somewhat smaller in acetonitrile. The excitation energy of the first bright state of non-chelated enol is 4.55 and 4.17 eV in the gas phase and acetonitrile, respectively. Since nπ* states arise from the nonbonding orbitals of oxygen atoms, their energy is almost unchanged by the torsion of the hydroxyl group. The consequence of this behavior is that the singlet and triplet nπ* states of the non-chelated enol are the first and second excited states in both environments. From the similarity of the ground state electronic structure of non-chelated enol and rotamer E, we predict that their electronic excitation spectra will be very similar. The computed energies are found to differ at most by 0.14 eV. In the case of rotamer Z, the singlet and triplet nπ* states are destabilized by ≈0.2 eV relative to those of the chelated enol and rotamer E.
The geometry of the keto tautomer is particularly favorable for nπ* states. The lowest two excited singlet states originate from the nonbonding orbitals of oxygen atoms. Their excitation energies are 3.87 and 4.10 eV in the gas phase and 3.94 and 4.16 eV in acetonitrile. The first ππ* state is the third excited singlet state with energy that amounts to 4.57 in the gas phase. In acetonitrile the third excited singlet state comes from πH → πL* transition. Its excitation energy is 4.46 eV. The maximum of the keto absorption band in acetonitrile is found at a somewhat higher energy of 4.68 eV.10,15,19 In the triplet manifold, the first excited state has ππ* character whereas the second state has nπ* character coming from nH−4 → πL* excitation. The energy difference between these two states is found to be only 0.03 eV in the gas phase, indicating a strong vibronic coupling between them. In acetonitrile, these two states move away so that the energy gap between them increases to 0.13 eV.
The most noticeable feature of the electronic absorption spectrum is that the tautomerization of chelated enol shifts the bright ππ* state above the dark nπ* state. Since the ππ* state mainly stems from the πH → πL* transition the cause of this spectral shift should be attributed to the changes of the πH and πL* orbitals upon tautomerization. The electron density of the πH orbital is partially localized on the benzene ring to which the methoxy group is attached and thus its energy is not significantly modified upon torsions. On the other hand, the situation of the πL* orbital is the opposite. Its electron density is extended on the O1–C1–C2–C3–O2–H pseudocycle, particularly on the O1, C1 and C3 atoms. We have already addressed that tautomerization reduces the electronic delocalization of this pseudocycle. Specially, the O1–C1 bond gains more double bond character and becomes stronger. As a consequence, the πL* orbital becomes less stable than in the chelated enol tautomer and the excitation into the πL* orbital has larger energies than in the chelated enol tautomer. Therefore, the loss of UVA absorption upon tautomerization is directly related to the change of the electron density distribution of the pseudocycle.
 | ||
| Fig. 3 Geometries of the optimized S1 state: (a) keto, (b) non-chelated enol, (c) rotamer E, and (d) rotamer Z. All bond lengths are in Å. | ||
 | ||
| Fig. 4 Geometries of the optimized T1 state: (a) chelated enol, (b) non-chelated enol, (c) rotamer E, (d) rotamer Z, (e) keto nπ*, and (f) keto ππ*. All bond lengths are in Å. | ||
| State | Gas phase | Acetonitrile | ||
|---|---|---|---|---|
| Character | Energy | Character | Energy | |
| Chelated enol | ||||
| T1 | ππ* | 2.82 | ππ* | 2.80 |
| Non-chelated enol | ||||
| S1 | nπ* | 2.96 | nπ* | 3.15 |
| T1 | ππ* | 2.46 | ππ* | 2.46 |
| Rotamer Z | ||||
| S1 | nπ* | 3.16 | nπ* | 3.35 |
| T1 | ππ* | 2.35 | ππ* | 2.36 |
| Rotamer E | ||||
| S1 | nπ* | 2.79 | nπ* | 2.85 |
| T1 | ππ* | 2.47 | ππ* | 2.46 |
| Keto | ||||
| S1 | nπ* | 3.38 | nπ* | 3.46 |
| T1 | nπ* | 3.18 | nπ* | 3.27 |
| T1 | ππ* | 3.25 | ππ* | 3.24 |
The optimization of the local minimum of the S1 state of chelated enol at the SCS-CC2 level failed since all attempts ended in structures whose C1–C2–C3–C4 dihedral angles were close to 90 degrees. At these geometries, there is a conical intersection between the S1 (ππ*) state and the ground state. On the other hand, our preliminary optimization of the S1 state at the CAM-B3LYP/TZVP level revealed the almost planar ππ* minimum of the chelated enol tautomer (cf. Fig. S6 in the ESI†) whose adiabatic energy is 3.93 eV, which is only 0.16 eV more stable than the ground state vertical energy at this level of theory. Computation of the SCS-CC2 S1 energy at CAM-B3LYP optimized geometry coincidentally gave the same value of 3.93 eV. The existence of the S1 minimum on the computational level indicates that the potential energy surface in its vicinity is very shallow. Thus, it does not have major effects on the photochemistry of AB since the excited state population will not be trapped in such minimum. In addition, phototautomerization may effectively deplete the population in the Franck–Condon point before it arrives at the S1 minimum. We also note that optical experiments in ethanol at 77 K13,17 revealed the fluorescence spectrum of chelated enol with well-resolved vibronic peaks. The origin of the spectrum was estimated to be 3.17 eV, approximately 0.3 eV lower than the vertical excitation energy. In that case, steric effects in the solid matrix may hinder phototautomerization and allow the S1 minimum to be populated.
The S1 state of non-chelated, rotamers Z and E, and keto tautomers has nπ* character coming from nH−5 → πL* transition. In the case of the keto tautomer, there is also contribution from nH−4 → πL* excitation. The considerable C1–O1 bond elongation found in the geometries of these molecules is a consequence of excitation from the nonbonding lone-pair oxygen orbital to the antibonding orbital with respect to this bond. It is known that the CC2 methods somewhat overemphasize C–O bond elongation42,52 and therefore the computed bond lengths are probably too long. In non-chelated enol the C1–O1 bond is elongated by 0.16 Å relative to the ground state value. The other C–O bond exhibits only negligible elongation. Besides the C–O bond stretching, the excitation to the S1 state of non-chelated enol leads to shrinking of the C1–C2 and C3–C4 bonds by 0.08 and 0.03 Å. The adiabatic energy of this tautomer in the lowest excited singlet state amounts to 2.96 eV in the gas phase. This energy is blueshifted by 0.19 eV in acetonitrile. The S1 adiabatic energy of rotamer Z is 3.16 and 3.35 eV in the gas phase and acetonitrile, respectively. In a similar fashion to non-chelated enol, the most noticeable changes occur at the O1–C1 (0.18 Å), C1–C2 (−0.09 Å) and C2–C3 (0.04 Å) bonds. The adiabatic energy of the rotamer E tautomer is 2.79 eV in the gas phase whereas it increases to 2.85 eV in acetonitrile. The largest geometry changes of the S1 state in comparison with the ground state are observed for the C1–O1 and C2–C3 bonds which increase by 0.14 and 0.06 Å, respectively. On the other hand, the C1–C2 and C3–C4 bonds shrink by 0.09 and 0.06 Å. Concerning the S1 geometry of the keto tautomer, substantial elongation of the O1–C1 bond by 0.16 Å is found, accompanied by contraction of the neighboring C1–C10 and C1–C2 bonds by 0.08 and 0.03 Å. The S1 state of the keto tautomer stabilizes by 0.49 eV upon geometry relaxation in the gas phase. Its adiabatic energy is 3.38 eV. In acetonitrile, this energy exhibits a relatively small solvatochromic shift of 0.08 eV.
All optimized local minima on the T1 potential surface mainly originate from πH → πL* excitation except that of the keto tautomer which has nπ* character and comes from the same excitation as the S1 state. In the case of chelated enol, besides πH → πL* excitation there is a contribution from πH → πL+1* as well. The nuclear arrangement of this tautomer in the lowest triplet state is relatively similar to the ground state geometry. The intramolecular hydrogen bond becomes weaker in the T1 state since its length increases by 0.14 Å. The C3–O2 and C4–C9 bonds are longer by 0.03 Å than their ground state values. On the other hand, the C3–C4 bond shrinks by 0.06 Å. The T1 adiabatic energy of chelated enol amounts to ≈2.8 eV, irrespective of the environment. This minimum represents a global minimum on the T1 potential energy surface of AB. The experimentally determined T1 adiabatic energy from phosphorescence spectra in ethanol at 77 K has been reported to be somewhat lower (2.52 eV).13,17 We also note that the energy difference between the computed and experimental energies of the T1 state is approximately the same as that found for the S1 vertical excitation energy at the ground state geometry.
The lowest triplet state geometry of non-chelated enol is considerably twisted relative to the ground state. The most outstanding characteristic of the T1 geometry is that the C1–C2–C3–C4 and C1–C2–C3–O2 dihedral angles are increased by 100 and 112 degrees relative to the ground state values. This geometry distortion is followed by the C2–C3 and C3–O2 bond stretching by 0.11 and 0.03 Å, whereas the C3–C4 bond shrinks by 0.06 Å. The adiabatic energy in gas phase is 2.46 eV. Taking into account the ground state energy difference this corresponds to an increase of 0.29 eV relative to the T1 energy of chelated enol. The T1 adiabatic energy is also 2.46 eV in acetonitrile.
Both rotamers E and Z in the T1 state exhibit similar bond length changes relative to their ground state geometries. The most notable are the C2–C3 bond stretching and the C3–C4 bond shrinking. The adiabatic energies of the T1 state of rotamers E and Z are 2.47 and 2.35 eV, respectively, in the gas phase. Both energies are almost unchanged in acetonitrile.
The triplet state of the keto tautomer is responsible for photodegradation of AB.14 This process proceeds mainly via α bond cleavages of carbonyl groups. Schwank and Rudolph found that AB is photolabile in non-polar solvents whereas in polar solvents photodegradation was low.4 Kikuchi et al. measured the phosphorescence spectra of AB in ethanol at 77 K. The authors assigned the lowest triplet state of the keto form to the ππ* state with an energy of 3.03 eV. The optimization of the keto tautomer revealed that there are two local T1 minima corresponding to ππ* and nπ* states. This is not surprising since both states are found to be very close in the ground state vertical excitation spectrum. The more stable minimum in gas phase belongs to the nπ* state. The geometry is similar to that of the S1 state. The C1–O1 bond elongates by 0.13 Å whereas the C1–C10 and C1–C2 bonds shrink by 0.06 and 0.02 Å relative to the ground state geometry. On the other hand, the ππ* local minimum is characterized by bond distortions that mainly include the benzene ring to which the methoxy group is attached as well as the C3–C4 and C3–O2 bonds. The largest bond stretching is observed for the C4–C5 bond and amounts to 0.08 Å. In addition to πH → πL* excitation, the triplet ππ* state also originates from πH → πL+1* and πH → πL+2* transitions. The adiabatic energy of the nπ* local minimum is 3.18 eV while the ππ* minimum is located only 0.07 eV above it in the gas phase. However, due to different solvatochromic shifts, these two minima exchange their order of stability in acetonitrile. The ππ* state then becomes more stable by 0.03 eV. Its adiabatic energy is estimated to be 3.24 eV, close to the value of 3.03 eV measured in solid ethanol.13,17 The solvent dependent stability of the lowest triplet state of the keto tautomer sheds new light on the degradation mechanism in non-polar and polar solvents. Thus, photodegradation in non-polar solvents proceeds from the triplet nπ* state, while the less reactive ππ* state is responsible for the modest degradation in polar solvents.
In order to examine the proposed relaxation mechanisms, we computed potential energy profiles of the ground, singlet and triplet excited states along selected reaction coordinates. Due to computational constraints, in all cases we kept the values of all coordinates of the initial geometry fixed except the reaction coordinate. The computed pathways represent an extreme case in which the photorelaxation is infinitely fast so that other coordinates have no time to adjust. Thus, the computed barriers' heights represent an upper limit of the actual values. A similar approach has previously been employed by Plötner and Dreuw in their study of photoisomerization of hemithioindigo hemistilbene.53
Fig. 5 displays potential energy profiles along the C1–C2–C3–C4 dihedral angle in gas phase and acetonitrile. The initial geometry is the Franck–Condon (FC) point, i.e. the ground state chelated enol structure. We find an avoided crossing between the ground and S1 state at a dihedral angle of around 90 degrees. The avoided crossing is likely associated with a conical intersection54 by changes of other coordinates such as pyramidalization55,56 at one C atom on the C2–C3 double bond. This intersection is a funnel for an efficient internal conversion to the ground state. We assigned this process to a time constant of 1.18 ps observed in the experiment performed by Brixner and coworkers on DBM.18 Upon internal conversion to the ground state, the molecule can relax to the chelated enol or rotamer E local minima. Our calculations revealed that there is a small barrier on the S1 state (≈0.2 eV) between the FC point and the avoided crossing. This barrier will most probably disappear upon relaxation of other coordinates. Note that in the gas phase, there is also a crossing between the S1 and triplet nπ* states. Since these two states have different orbital types, the intersystem crossing is supposed to be fast.57 This crossing does not exist in acetonitrile since the triplet nπ* state is blue-shifted above the S1 state.
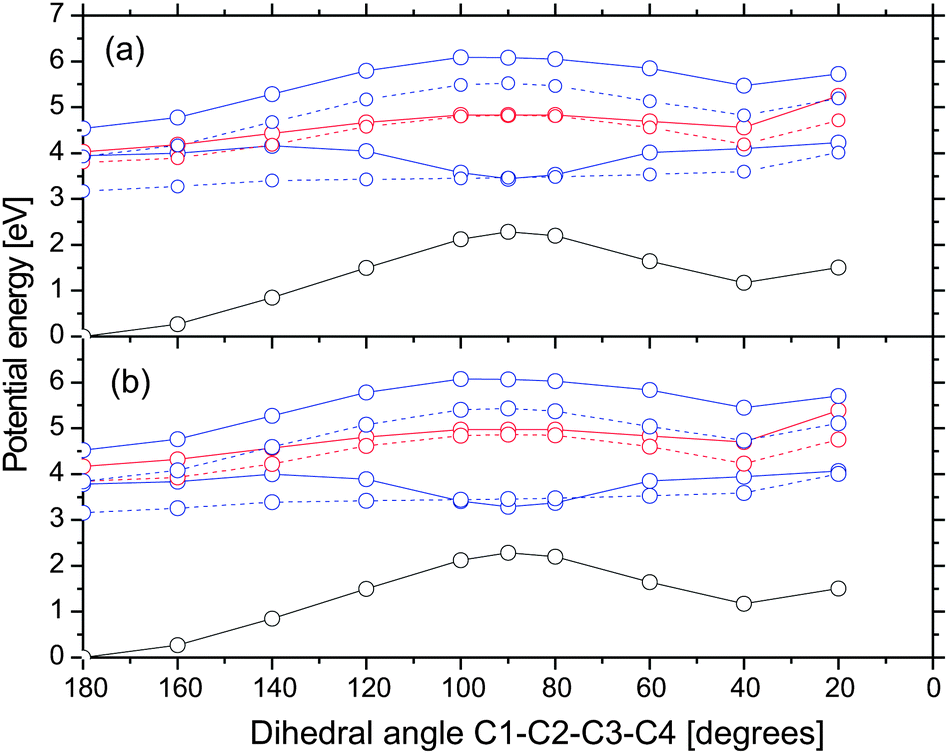 | ||
| Fig. 5 Potential energy profiles along the dihedral angle C1–C2–C3–C4 in the gas phase (a) and acetonitrile (b). The initial geometry (angle = 180 degrees) is the Franck–Condon point. Color scheme: nπ*, red; ππ*, blue; ground state, black; singlet state, full line; triplet states, dashed line. | ||
The dark nπ* state is responsible for the creation of non-chelated enol. In order to qualitatively estimate the position of the crossing between the bright and dark states, we calculated potential energy profiles along the dihedral angle H–O2–C3–C4 (cf.Fig. 6). The initial geometry corresponds to the FC point. Indeed, we find that the crossing between the bright and dark states is very close to the FC point. In gas phase, the crossing occurs at ≈165 degrees. Its energy is only 0.14 eV (=1130 cm−1) higher than the FC point. Since this value is lower than the zero-point energy of in-plane OH bending motion, we believe that internal conversion to the nπ* state readily occurs. In acetonitrile, the crossing is blue-shifted by 0.36 eV, making it slightly less accessible. We attribute this internal conversion to a process with a time constant of 147 fs seen in the femtosecond transient absorption experiment of DBM.18 The formation of non-chelated enol on the nπ* state proceeds almost barrierless. Note that the singlet nπ* state is very close to the triplet ππ* state which opens the possibility of intersystem crossing to the triplet state.
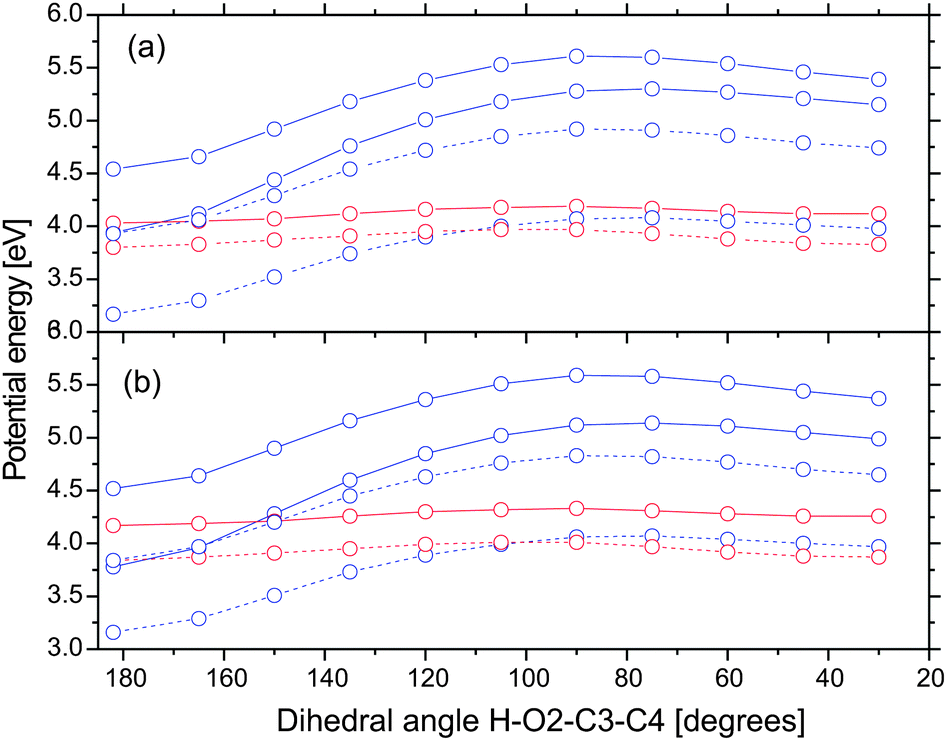 | ||
| Fig. 6 Potential energy profiles along the dihedral angle H–O2–C3–C4 in the gas phase (a) and acetonitrile (b). The initial geometry (angle = 182 degrees) is the Franck–Condon point. Color scheme: nπ*, red; ππ*, blue; singlet state, full line; triplet states, dashed line. | ||
In order to examine whether the formation of rotamer Z proceeds on the S1 nπ* state, we computed potential energy profiles along the C10–C1–C2–C3 dihedral angle starting from the optimized structure of non-chelated enol in the S1 state. They are shown in Fig. 7. Note that energies are computed relative to the ground state chelated enol. The energy increment at an angle of 40 degrees is due to the proximity of the hydroxyl group and the nearby hydrogen atom. The energy minimization would certainly separate these atoms and decrease the energies. The computed energy profiles indicate that there is a barrier of ≈0.7 eV for the rotation about the single C1–C2 bond in the S1 nπ* state. Since the barrier is 0.33 and 0.76 eV higher than the vertical excitation energy of the bright state of chelated enol at the ground state geometry in gas phase and acetonitrile, respectively, we find that this pathway is less probable. Note that the barrier for the ground state rotation about the single C1–C2 bond is only 0.2 eV. Thus, rotamer Z is likely to be formed in the ground state. Chen et al. argued that rotational isomerization of acetylacetone proceeds easily on the T1 ππ* state. We do not exclude this possibility but remark that the single C1–C2 bond of rotamer Z is shorter in the T1 state than in the ground state by 0.01 Å, indicating that it is slightly stiffer as well.
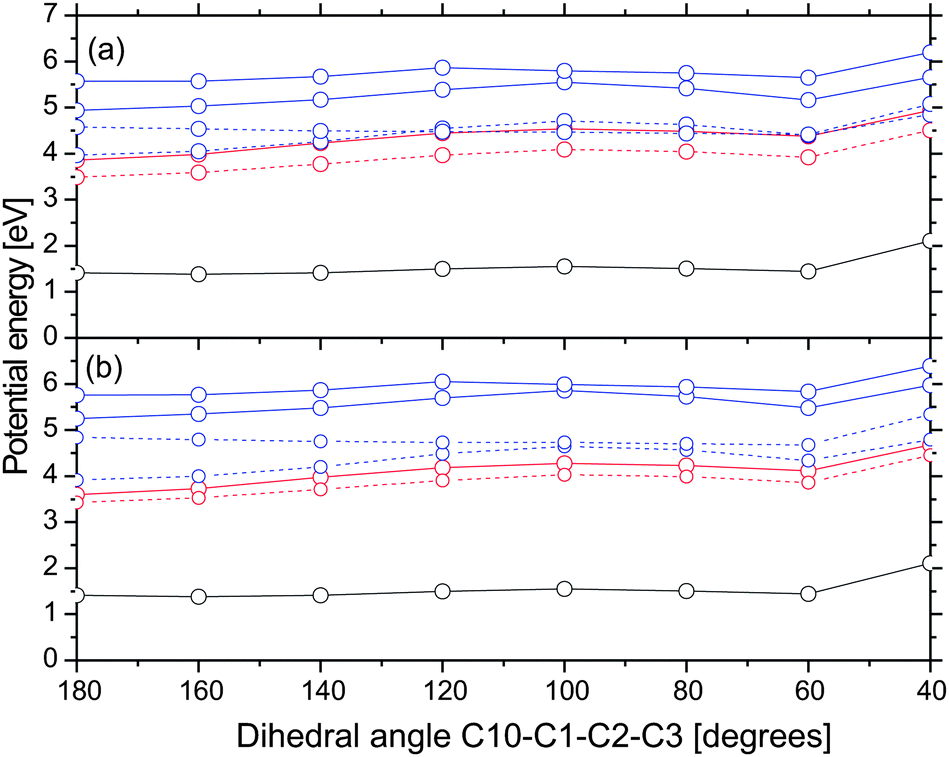 | ||
| Fig. 7 Potential energy profiles along the dihedral angle C10–C1–C2–C3 in acetonitrile (a) and the gas phase (b). The initial geometry (angle = 180 degrees) is the optimized structure of non-chelated enol in the S1 state. Color scheme: nπ*, red; ππ*, blue; ground state, black; singlet state, full line; triplet states, dashed line. | ||
The proposed decay pathways of the photoexcited chelated enol in gas phase and acetonitrile are summarized in Fig. 8. The decay pathways do not depend on the environment although the quantum yield of tautomerization will be different. After the initial photoexcitation to the S1 (ππ*) state of chelated enol, AB can relax back to the ground state of chelated enol and rotamer E tautomers by internal conversion. The other decay pathway of photoexcited chelated enol involves internal conversion to the dark nπ* state by torsion of the hydroxyl group. Our finding suggests that tautomerization of non-chelated enol into rotamer Z is unlikely to proceed in the first excited singlet state.
 | ||
| Fig. 8 The proposed decay pathways of photoexcited chelated enol. | ||
4 Conclusions
Avobenzone is used in sunscreens as a UVA filter due to its ability to absorb radiation and efficiently dissipate the absorbed energy. Nevertheless, photochemical reactions can reduce its efficiency as a UVA filter. Its photochemistry is complex due to the appearance of several tautomers. In this contribution, we have studied the chelated enol, non-chelated enol, rotamers Z and E, and keto tautomers in the ground, S1 and T1 states by means of the coupled cluster theory. We have been able to show that the torsion about the double C2–C3 bond of photoexcited chelated enol leads to internal conversion to the ground state. The molecule can subsequently relax to the chelated enol or rotamer E local minima. The other channel for the relaxation of photoexcited chelated enol is found to be an opening of the hydrogen chelated ring by torsion of the hydroxyl group. This motion leads to internal conversion to the dark state and creation of non-chelated enol. Our findings show that it is very unlikely that rotamer Z is formed by rotation of the singlet C1–C2 bond in the first excited state. The more probable mechanism includes rotation in the ground state although we do not exclude the role of the triplet state. Our results suggest that solvent dependent photolability of AB is related to the relative order of the lowest triplet ππ* and nπ* states of the keto tautomer.Acknowledgements
The authors acknowledge the Ministry of Education and Science of Serbia for the financial support (Contract No. 172040).References
- G. P. Pfeifer and A. Besaratinia, Photochem. Photobiol. Sci., 2012, 11, 90–97 CAS.
- M. M. Ristić, M. Petković and M. Etinski, J. Serb. Chem. Soc., 2012, 77, 1037–1045 CrossRef.
- N. A. Shaath, Photochem. Photobiol. Sci., 2010, 9, 464–469 CAS.
- W. Schwack and T. Rudolph, J. Photochem. Photobiol., B, 1995, 28, 229–234 CrossRef CAS.
- P. Gacoin, J. Chem. Phys., 1972, 57, 1418–1425 CrossRef CAS.
- D. Veierov, T. Bercovici, E. Fischer, Y. Mazur and A. Yogev, J. Am. Chem. Soc., 1977, 99, 2723–2729 CrossRef CAS.
- H. Gonzenbach, T. J. Hill and T. G. Truscott, J. Photochem. Photobiol., B, 1992, 16, 377–379 CrossRef CAS.
- S. Tobita, J. Ohba, K. Nakagawa and H. Shizuka, J. Photochem. Photobiol., A, 1995, 92, 61–67 CrossRef CAS.
- I. Andrea, A. Bringhen, F. Böhm, H. Gonzenbach, T. Hill, L. Mulroy and T. G. Truscott, J. Photochem. Photobiol., B, 1997, 37, 147–150 CrossRef.
- A. Cantrell and D. J. McGarvey, J. Photochem. Photobiol., B, 2001, 64, 117–122 CrossRef CAS.
- A. Aspée, C. Aliaga and J. C. Scaiano, Photochem. Photobiol., 2007, 83, 481–485 CrossRef PubMed.
- G. Mturi and B. S. Martincigh, J. Photochem. Photobiol., A, 2008, 200, 410–420 CrossRef CAS.
- A. Kukuchi, N. Oguchi and M. Yagi, J. Phys. Chem. A, 2009, 113, 13492–13497 CrossRef PubMed.
- C. Paris, V. Lhiaubet-Vallet, O. Jiménez, C. Trullas and M. A. Miranda, Photochem. Photobiol., 2009, 85, 178–184 CrossRef CAS PubMed.
- M. Yamaji and M. Kida, J. Phys. Chem. A, 2013, 117, 1946–1951 CrossRef CAS PubMed.
- L. P. da Silva, P. Ferreira, D. Duarte, M. Miranda and J. E. da Silva, J. Phys. Chem. A, 2014, 118, 1511–1518 CrossRef PubMed.
- A. Kukuchi, N. Oguchi-Fujiyama, K. Miyazawa and M. Yagi, Photochem. Photobiol., 2014, 90, 511–516 CrossRef PubMed.
- P. K. Verma, F. Koch, A. Steinbacher, P. Nuernberger and T. Brixner, J. Am. Chem. Soc., 2014, 136, 14981–14989 CrossRef CAS PubMed.
- A. D. Dunkelberger, R. D. Kieda, B. M. Marsh and F. F. Crim, J. Phys. Chem. A, 2015, 119, 6155–6161 CrossRef CAS PubMed.
- P. K. Verma, A. Steinbacher, F. Koch, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2015, 17, 8459–8466 RSC.
- G. H. G. Trossini, V. G. Maltarollo, R. D. Garcia, C. A. S. O. Pinto, M. V. R. Velasco, K. M. Honorio and A. R. Baby, J. Mol. Model., 2015, 21, 319 CrossRef PubMed.
- S. Mandal, R. Bera, S. Das, S. K. Nayak, A. Pramanik and A. Patra, ChemPhysChem, 2015, 16, 3618–3624 CrossRef CAS PubMed.
- J. Zawadiak and M. Mrzyczek, Spectrochim. Acta, Part A, 2010, 75, 925–929 CrossRef PubMed.
- M. Petković and M. Etinski, RSC Adv., 2014, 4, 38517–38526 RSC.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154 CrossRef.
- C. Hättig, J. Chem. Phys., 2003, 118, 7751 CrossRef.
- A. Köhn and C. Hättig, J. Chem. Phys., 2003, 119, 5021 CrossRef.
- C. Hättig and A. Köhn, J. Chem. Phys., 2002, 117, 6939 CrossRef.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119–4127 RSC.
- N. O. C. Winter, N. K. Graf, S. Leutwyler and C. Hättig, Phys. Chem. Chem. Phys., 2013, 15, 6623–6630 RSC.
- S. Lobsiger, M. Etinski, S. Blaser, H.-M. Frey, C. Marian and S. Leutwyler, J. Chem. Phys., 2015, 143, 234301 CrossRef PubMed.
- T. H. J. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- R. A. Kendall, T. H. J. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 9, 6796 CrossRef.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Schäfer, A. Klamt, D. Sattel, J. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- M. Etinski and C. M. Marian, Phys. Chem. Chem. Phys., 2010, 12, 4915–4923 RSC.
- M. Etinski and C. M. Marian, Phys. Chem. Chem. Phys., 2010, 12, 15665–15671 RSC.
- V. Rai-Constapel, M. Etinski and C. M. Marian, J. Phys. Chem. A, 2013, 117, 3935–3944 CrossRef CAS PubMed.
- C. M. Marian, M. Etinski and V. Rai-Constapel, J. Phys. Chem. A, 2014, 118, 6985–6990 CrossRef CAS PubMed.
- M. Etinski, J. Tatchen and C. M. Marian, Phys. Chem. Chem. Phys., 2014, 16, 4740–4751 RSC.
- T. Fleig, S. Knecht and C. Hättig, J. Phys. Chem. A, 2007, 111, 5482–5491 CrossRef CAS PubMed.
- M. Petković, M. Ristić and M. Etinski, J. Phys. Chem. A, 2016, 120, 1536–1544 CrossRef PubMed.
- A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 1999, 103, 4494–4504 CrossRef CAS.
- X.-B. Chen, W.-H. Fang and D. L. Phillips, J. Phys. Chem. A, 2006, 110, 4434–4441 CrossRef CAS PubMed.
- J. D. Coe and T. J. Martínez, J. Phys. Chem. A, 2006, 110, 618–630 CrossRef CAS PubMed.
- A. Trivella, T. N. Wassermann, J. M. Mestdagh, C. M. Tanner, F. Marinelli, P. Roubin and S. Coussan, Phys. Chem. Chem. Phys., 2010, 12, 8300–8310 RSC.
- M. Etinski, T. Fleig and C. M. Marian, J. Phys. Chem. A, 2009, 113, 11809–11816 CrossRef CAS PubMed.
- J. Plötner and A. Dreuw, J. Phys. Chem. A, 2009, 113, 11882–11887 CrossRef PubMed.
- D. G. Truhlar and C. A. Mead, Phys. Rev. A: At., Mol., Opt. Phys., 2003, 68, 032501 CrossRef.
- J. Q. M. Ben-Nun and T. J. Martínez, J. Phys. Chem. A, 2000, 104, 5161–5175 CrossRef.
- M. Barbatti, J. Paier and H. Lischka, J. Chem. Phys., 2004, 121, 11614–11624 CrossRef CAS PubMed.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The bond lengths of the ground state structures optimized in the gas phase, the most important frontier Hartree–Fock orbitals, the geometry of the optimized S1 state of chelated enol at the CAM-B3LYP/TZVP level and the Cartesian coordinates of all structures. See DOI: 10.1039/c6cp03533g |
| This journal is © the Owner Societies 2016 |