
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraordinary stability of hemocyanins from L. polyphemus and E. californicum studied using infrared spectroscopy from 294 to 20 K†
Mireille
Khalil
a,
Zahia
Boubegtiten-Fezoua
a,
Nadja
Hellmann
*b and
Petra
Hellwig
*a
aLaboratoire de Bioélectrochimie et Spectroscopie, UMR 7140 Université de Strasbourg CNRS, 1 Rue Blaise Pascal 67070, France. E-mail: hellwig@unistra.fr; Tel: +33 368 85 12 73
bInstitute for Molecular Biophysics, University of Mainz, Jakob-Welder-Weg 26, 55128 Mainz, Germany. E-mail: nhellmann@uni-mainz.de; Tel: +49 6131 392 3567
First published on 22nd September 2016
Abstract
Hemocyanins are large oligomeric respiratory proteins found in many arthropods and molluscs. Here we give infrared spectroscopic evidence of a high stability towards exposure to sub-zero temperatures for hemocyanins from the arthropods Limulus polyphemus and Eurypelma californicum at different pH values. Small but distinct temperature induced changes of the secondary structure were observed, but a stable core of at least 40% α-helical structure is preserved as identified in the infrared spectra obtained between 294 and 20 K. The structural changes differ in detail somewhat for the two hemocyanins, with overall fewer changes observed in the case of E. californicum. Notably, in both cases the overall changes in the α-helical content are found to be fully reversible. The small changes in the secondary structure and reversibility upon cold treatment seem to be a particular property of the two hemocyanins, since it was not observed for myoglobin studied in the same way.
Introduction
Respiratory proteins facilitate oxygen transport and storage and are thus key elements of oxygen supply in vertebrates and higher invertebrates. Three types can be distinguished based on the structure of the active site: hemoglobins (Hb), where oxygen is bound via an iron-ion to a porphyrin ring; hemerythrins, which contain merely two iron-ions and hemocyanins (Hcs) where oxygen is bound between two copper ions, which are held in the protein by six histidines.1 Here we focus on hemocyanins (Hcs). These extracellular multi-subunit proteins are found in the hemolymph of several species of molluscs and arthropods.2,3 Subunits of arthropod Hcs are arranged in hexamers or multiples of hexamers such as 2 × 6mers, 4 × 6mers, 6 × 6mers and 8 × 6mers.3,4 Each subunit of a molecular mass around 72–75 KDa carries one active site.5–7 To date it has not been clear why some species regulate their oxygen supply based on hexameric Hcs while others developed larger oligomers. Even in a particular species two different variants might exist, which in a few cases have been shown to play a slightly different physiological role.8 A clear correlation of the size of hemocyanins with the living conditions of the host organism seems to be lacking. However, based on the data available so far there seems to be a tendency that larger oligomers display a higher stability with respect to temperature-induced denaturation. A remarkable stability of the oxy-forms of Limulus polyphemus and Eurypelma californicum Hcs (91 and 92.5 °C respectively) was revealed using differential scanning spectroscopy.9,10Eurypelma californicum is a spider found in the deserts of southwest North America, a particularly hostile environment which is characterized by large temperature fluctuations between day and night. E. californicum Hc is a 24-mer complex with a molecular mass of 1.7 MDa including seven types of subunits.11,12 Each subunit shows similar oxygen binding behavior despite their unique immunological and physicochemical properties.11,13 The horseshoe crab, Limulus polyphemus Hc, lives in the eastern coast of North and Central America in varying habitats depending on the life cycle.14 The Hc from L. polyphemus, with a molecular mass of 3.5 MDa, is composed of 48 subunits. The crystal structure of the homohexamer of subunit II of L. polyphemus Hc was resolved with a resolution of 2.4 Å.15–17 A relatively high sequence homology of 60% was reported between the Lpol-II subunit from L. polyphemus and the analogous subunit “a” in E. californicum.18
E. californicum Hc was shown to be more stable with regard to chemical denaturation and temperature induced unfolding in the native oligomer than as an isolated subunit.9,19 Pavlina et al. investigated the stability of the quaternary structure of L. polyphemus and P. vulgaris Hcs using fluorescence spectroscopy in the presence of denaturing agents.20 They concluded that the quaternary structure of the Hcs proteins is stabilized by hydrophilic and polar forces and that the denaturation process consists of two steps: the dissociation of the native molecule into its subunits and the denaturation of the subunits. Stabilization due to oligomer formation was also reported for other arthropod Hcs.21,22
In the study presented here, we investigated the structural changes induced in these Hcs by very low temperatures. Infrared spectroscopy is an established tool for the characterization of the secondary structure of proteins and peptides. Here we monitored the amide I signal at different pH values as a function of temperature from room temperature down to 20 K for Hcs from E. californicum and L. polyphemus. For comparison, we also studied myoglobin as an example of small monomeric proteins. Using IR-spectroscopy, samples can be studied at high concentrations, which are close to the in vivo concentrations (up to 100 mg mL−1). To the best of our knowledge no data for the cold stability of L. polyphemus and E. californicum Hcs or any other large oligomeric proteins have been reported yet.
Experimental
Sample preparation
The Hc from E. californicum was obtained from the hemolymph by dorsal punctuation of the heart. The sample was diluted immediately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v) with 0.1 M Tris/HCl pH 7.8, 5 mM CaCl2, 5 mM MgCl2 in order to stabilize the protein. The sample was then centrifuged for 30 min to remove blood cells. The hemocyanins were purified from the supernatant by gel filtration (S300 26/60, GE Healthcare) in 0.1 M Tris/HCl pH 7.8, 5 mM CaCl2, 5 mM MgCl2 at 4 °C.19 Hc from L. polyphemus was obtained from the hemolymph (obtained as described in Martin et al.23) by centrifugation at 4 °C: 15 min at 10000g, the resulting supernatant was spun again for 15 min at 10000g and the supernatant from this step spun for 2 h at 300000g. The pellet was resuspended in 10 mM Tris/HCl, 10 mM CaCl2, 30 mM MgCl2 (pH 7.8 at 20 °C) and kept at 4 °C. Purity of the samples was verified by SDS-gel-electrophoresis (Fig. S1, ESI†). The Hcs were studied in 20 mM Tris, 5 mM CaCl2, 5 mM MgCl2 (E. californicum, pH 7.0, 7.8, 8.5) and 20 mM Tris, 10 mM CaCl2, 10 mM MgCl2, 100 mM NaCl (L. polyphemus, pH 7.0, 7.5, 8.5) with the pH adjusted at 20 °C. Samples were transferred into the respective buffer by ultrafiltration using centrifugal filters (100 kDa cut-off) shortly before the experiment. Sample concentration was 2 mM referring to subunits, corresponding to about 150 mg mL−1. Myoglobin from equine heart was purchased from sigma Aldrich (France). The sample was dissolved in 20 mM Tris, pH 7.5 with a final concentration of 2 mM.
:2 (v/v) with 0.1 M Tris/HCl pH 7.8, 5 mM CaCl2, 5 mM MgCl2 in order to stabilize the protein. The sample was then centrifuged for 30 min to remove blood cells. The hemocyanins were purified from the supernatant by gel filtration (S300 26/60, GE Healthcare) in 0.1 M Tris/HCl pH 7.8, 5 mM CaCl2, 5 mM MgCl2 at 4 °C.19 Hc from L. polyphemus was obtained from the hemolymph (obtained as described in Martin et al.23) by centrifugation at 4 °C: 15 min at 10000g, the resulting supernatant was spun again for 15 min at 10000g and the supernatant from this step spun for 2 h at 300000g. The pellet was resuspended in 10 mM Tris/HCl, 10 mM CaCl2, 30 mM MgCl2 (pH 7.8 at 20 °C) and kept at 4 °C. Purity of the samples was verified by SDS-gel-electrophoresis (Fig. S1, ESI†). The Hcs were studied in 20 mM Tris, 5 mM CaCl2, 5 mM MgCl2 (E. californicum, pH 7.0, 7.8, 8.5) and 20 mM Tris, 10 mM CaCl2, 10 mM MgCl2, 100 mM NaCl (L. polyphemus, pH 7.0, 7.5, 8.5) with the pH adjusted at 20 °C. Samples were transferred into the respective buffer by ultrafiltration using centrifugal filters (100 kDa cut-off) shortly before the experiment. Sample concentration was 2 mM referring to subunits, corresponding to about 150 mg mL−1. Myoglobin from equine heart was purchased from sigma Aldrich (France). The sample was dissolved in 20 mM Tris, pH 7.5 with a final concentration of 2 mM.
Fourier transform infrared spectroscopy
The mid-infrared (MIR) spectra were obtained using a Vertex 70 FTIR spectrometer (Bruker, Germany) equipped with a liquid nitrogen MCT detector. The spectrometer was purged with dry air to avoid the spectral contribution from humidity. Typically five spectra of 256 scans were recorded at a resolution of 4 cm−1 in the spectral range from 1800 to 1000 cm−1.The FTIR-ATR measurements of Hcs were performed at different pH values (7, 7.5/7.8 and 8.5) in the attenuated total reflection mode (ATR Harrick crystal, Diamond Prism). 2 μL of Hcs protein solution were deposited in order to obtain a film on the diamond crystal. It is noted that after drying the samples on the window the concentration was probably significantly higher than 150 mg mL−1 mentioned above and the final pH was not defined.
Temperature-dependent measurements
For temperature-dependent measurements, 2 μL of the protein sample were air-dried on a 2 mm thick CaF2 window and placed in a copper sample holder mounted on the cold finger of a closed cycle Helium-cryostat (Model DE-202 AE, Advanced Research Scientific, Allentown, PA, USA). The cryostat system and the sample holder operate at a pressure of 10−4 mTorr.The temperature close to the sample was controlled using a silicon diode (Scientific Instruments Calibration, precision of ± (0.5 K)). The temperature was regulated from 294 to 20 K with a heating resistor monitored using a digital temperature controller (Model 9700-1-1, Scientific Instruments, West Palm Beach, FL, USA).
Determination of oxygen saturation after freezing
Samples at pH 7.5 and 7.8 were exposed to −80 and −194 °C overnight at concentrations similar to the ones for FTIR measurements. The samples were thawed and then diluted with the corresponding buffer, to final concentrations between 0.25 and 0.8 mg mL−1. The absorption spectrum at ambient oxygen pressure (about 150 Torr) was measured using a Cary 1E (Varian, path length 1 cm) at 20 °C. The oxygenation level is expressed as the ratio of the absorbance of the copper–oxygen band at 340 nm to the aromatic band at 280 nm.Data analysis
In order to obtain the relative contribution of each secondary structure element, the deconvolution of the amide I signal is necessary. Several different procedures have been probed and the following found to be best suited for our data. First the wavenumber of each component was determined from the second derivative of the individual spectrum. Seven Gaussian curves were fitted to each spectrum (Origin 8.5 software) while the identified position was kept constant during the first fitting. Then, a consecutive optimization of amplitudes, band positions and the half-width of the individual bands was performed.24,25The individual amide I band components were obtained at 1610, 1618, 1633, 1644, 1660, 1679 and 1694 cm−1 and have been assigned to the secondary structure elements as follows: α-helices (1660 cm−1), unordered structures (1644 cm−1), parallel β-sheets (1633 cm−1), anti-parallel β-sheets (two bands in 1618 and 1694 cm−1) and β-turns (1679 cm−1), on the basis of a large body of experimental data.26–29 A complete fit of the amide I bands was only possible when adding a band at 1610 cm−1. The position of this signal is not typical for any known amide group implicated in common secondary structure elements. Finally, the area of each individual band was used to calculate its relative contribution to the overall area of the measured spectrum between 1729 and 1591 cm−1. The absolute error due to the deconvolution variability and the baseline correction was estimated to be about ±3.5%. All spectra shown were normalized on the basis of the area of the amide I band. No smoothing procedures were applied.
Standard deviation
The standard deviation was calculated for each element of the secondary structure following this formula.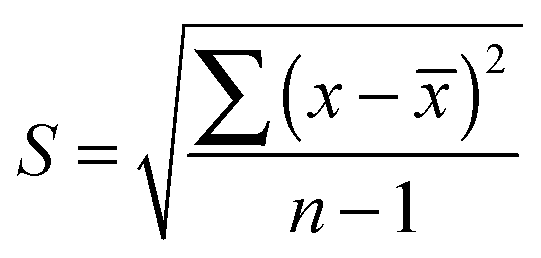
![[x with combining overline]](https://www.rsc.org/images/entities/i_char_0078_0305.gif) , represent the average of the difference between the two measurements made by the cryostat and the FTIR-ATR averaged over all conditions (pH) and over both species, x, the difference between both values: (ATR − cryostat) for any particular condition and species and n, the total number of differences.
, represent the average of the difference between the two measurements made by the cryostat and the FTIR-ATR averaged over all conditions (pH) and over both species, x, the difference between both values: (ATR − cryostat) for any particular condition and species and n, the total number of differences.
Results
The cold stability of L. polyphemus and E. californicum Hcs was monitored using FTIR spectroscopy at pH 7, pH 7.5/7.8 and pH 8.5 at temperatures between 294 K and 20 K (Fig. 1A–D). In the spectral range between 1450 and 1750 cm−1 two main signals dominate the spectra of proteins, amide I and amide II modes.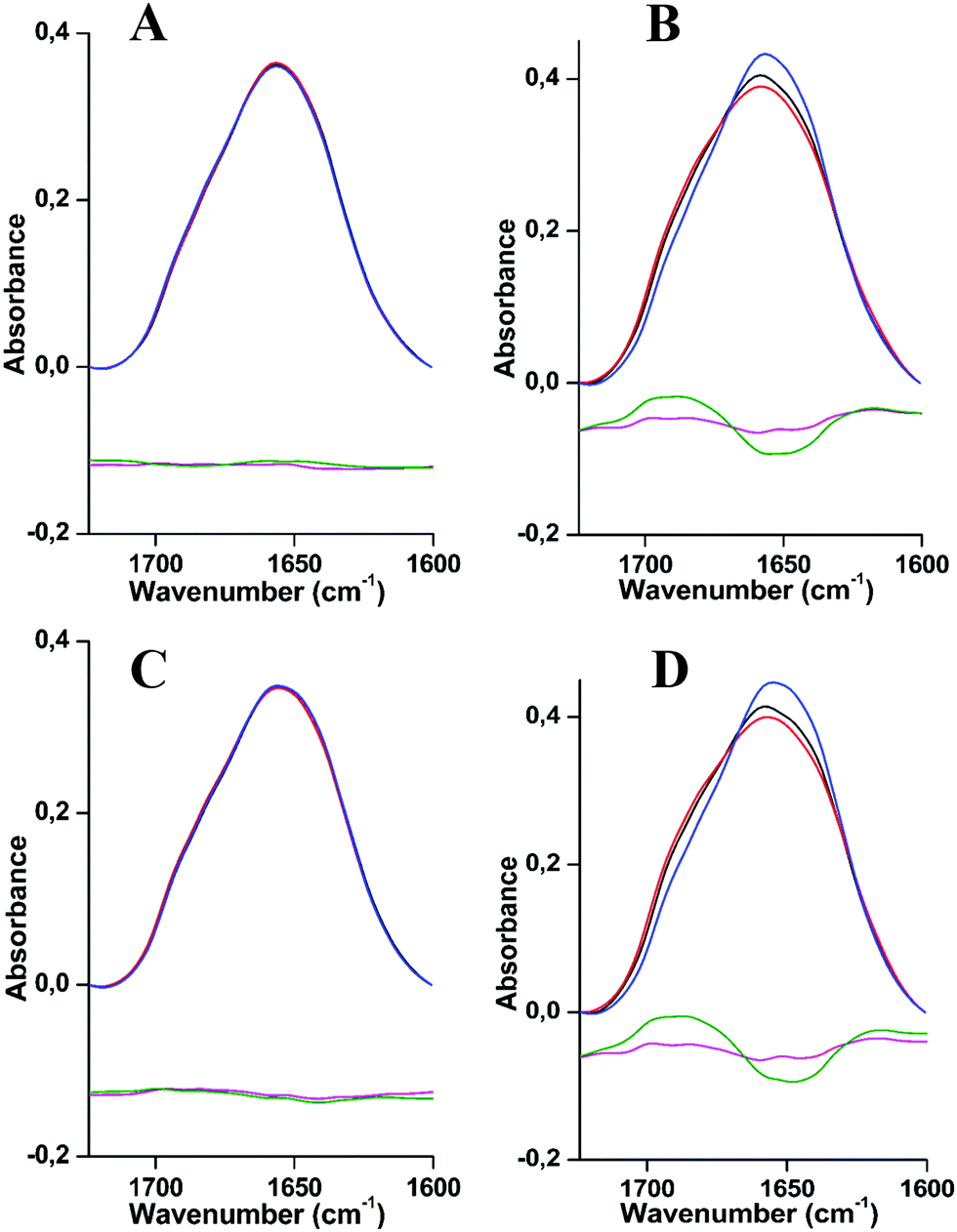 | ||
| Fig. 1 Absorbance FTIR spectra of L. polyphemus and E. californicum Hcs at 294 K (A and B) and 20 K (C and D), respectively, at pH 7 in black, pH 7.5/7.8 in red and 8.5 in blue. The difference spectra are represented in violet and in green by subtracting the spectra at pH values (7 and 8.5) from the spectrum at the intermediate pH (7.5/7.8) respectively. FTIR spectra are normalized by the surface of amide I bands. | ||
The amide II band is seen at around 1537 cm−1 (Fig. S2, ESI†). It includes the coupled stretching ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and bending δ(N–H) vibrational modes and it is influenced by the tertiary structure. This is in line with previous reports on the low temperature behavior of proteins, a shift of about 9 cm−1 towards higher wavenumbers, reflecting the changes in the hydrogen bonding structure.30 Here, we will focus on the amide I vibrational mode, which involves the contribution of the ν(CO) stretching vibration of the protein polypeptide chains, specific to the secondary structure of a protein.31,32 This band can be used to follow conformational changes induced by an external parameter, such as temperature.33,34
N) and bending δ(N–H) vibrational modes and it is influenced by the tertiary structure. This is in line with previous reports on the low temperature behavior of proteins, a shift of about 9 cm−1 towards higher wavenumbers, reflecting the changes in the hydrogen bonding structure.30 Here, we will focus on the amide I vibrational mode, which involves the contribution of the ν(CO) stretching vibration of the protein polypeptide chains, specific to the secondary structure of a protein.31,32 This band can be used to follow conformational changes induced by an external parameter, such as temperature.33,34
Fig. 1 shows the comparison of the amide I region for both Hcs at 294 K and 20 K and for different pH values (pH 7, 7.5/7.8 and 8.5). The data reveal slight changes in the overall structure. In the case of Hc from L. polyphemus, the amide I band centered at 1660 cm−1 adopts essentially the same shape for the three pH values measured (Fig. 1A and C), while a slight change occurs in the case of E. californicum Hc (Fig. 1B and D). In order to highlight these changes, difference spectra were calculated by subtracting the absorbance spectra at extreme pH values (7 and 8.5) from the spectrum at the intermediate pH (7.5 or 7.8). The details of the deconvoluted amide I bands are shown in Fig. S3 and S4, ESI.† Each component was assigned to secondary structure elements as follows: α-helices (1659–1661 cm−1), unordered structures (1642–1646 cm−1), parallel β-sheets (1629–1635 cm−1), anti-parallel β-sheets (two bands in 1616–1625 cm−1 and 1693–1695 cm−1 regions) and β-turns (1678–1682 cm−1), on the basis of a large body of experimental data.26–29
Effects of vacuum on the secondary structure
The data presented in this study were obtained on the basis of experiments of protein samples under vacuum. In order to exclude an effect on the integrity of the protein structure by the vacuum, control experiments with an ATR cell were performed (see the Materials and methods part). Table 1 summarizes the comparison of the spectra obtained at 294 K with those of a film of the same protein on an ATR crystal (Table 1). The content deviation was calculated for each secondary structure element under these conditions and then averaged over both Hcs at three pH values, yielding fATR−fcryo ± stddev. This procedure delivered the following values: −4.7 ± 3.1 (α-helix), 3.5 ± 2.4 (unordered), 1.3 ± 2.6 (parallel β-sheets), 5.3 ± 1.3 (anti-parallel β-sheets) and −5.5 ± 2.3 (β-turns) (Table 1). A small increase of contributions from α-helices, β-turns and a decrease of unordered and anti-parallel β-sheet conformations appear to be induced by the vacuum. It seems that the native α-helical conformation is more stable in a vacuum and that the proteins studied here tend to adopt a more organized structure. Indeed, it was previously demonstrated with α-helical polyalanine that a native α-helical conformation is stabilized under vacuum conditions.35 On the other hand, different orientations of the proteins on the ATR crystal compared to the samples measured in transmission in the cryostat could also contribute to these changes. Overall, the changes are small enough to be ignored.| Secondary structure (%) | Hemocyanins | pH 8.5 | pH 7.5/7.8 | pH 7 | Average deviation ATR − cryostat ± stddev. | |||
|---|---|---|---|---|---|---|---|---|
| ATR | Cryostat | ATR | Cryostat | ATR | Cryostat | |||
| α-Helix | L. polyphemus | 36 | 43 | 36 | 43 | 35 | 43 | −4.7 ± 3.1 |
| E. californicum | 39 | 42 | 37 | 37 | 36 | 39 | ||
| Unordered | L. polyphemus | 29 | 23 | 25 | 23 | 29 | 22 | 3.5 ± 2.4 |
| E. californicum | 25 | 23 | 24 | 21 | 24 | 23 | ||
| Parallel β-sheet | L. polyphemus | 11 | 10 | 13 | 9 | 13 | 9 | 1.3 ± 2.6 |
| E. californicum | 11 | 14 | 12 | 12 | 13 | 11 | ||
| Anti//β-sheet | L. polyphemus | 9 | 4 | 11 | 4 | 8 | 4 | 5.3 ± 1.3 |
| E. californicum | 10 | 5 | 11 | 7 | 11 | 4 | ||
| β-Turns | L. polyphemus | 15 | 20 | 15 | 21 | 15 | 22 | −5.5 ± 2.3 |
| E. californicum | 15 | 16 | 16 | 23 | 16 | 23 | ||
In order to compare the stability of hemocyanins with another respiratory protein, an experiment on myoglobin was performed under the same experimental conditions as Hcs (see Fig. 2A). Table 2 shows the contribution of each secondary structure elements obtained on the basis of the deconvolution of the amide I band. The major contribution corresponds to α-helical structures of 64% plus 7% of the unordered structure. This result is in very good agreement with the FTIR analysis reported by Meersman et al. which show that the secondary structure consists of 73.5% of the α-helical plus unordered structure.34 The contribution of the extended chain, β-turns and the anti-parallel β-sheet remains stable upon lowering the temperature. Upon cooling the native α-helical structure decreases by (−28%) in favor of the unordered structure (+26%) at 10 K (see Table 2 and Fig. 2B).
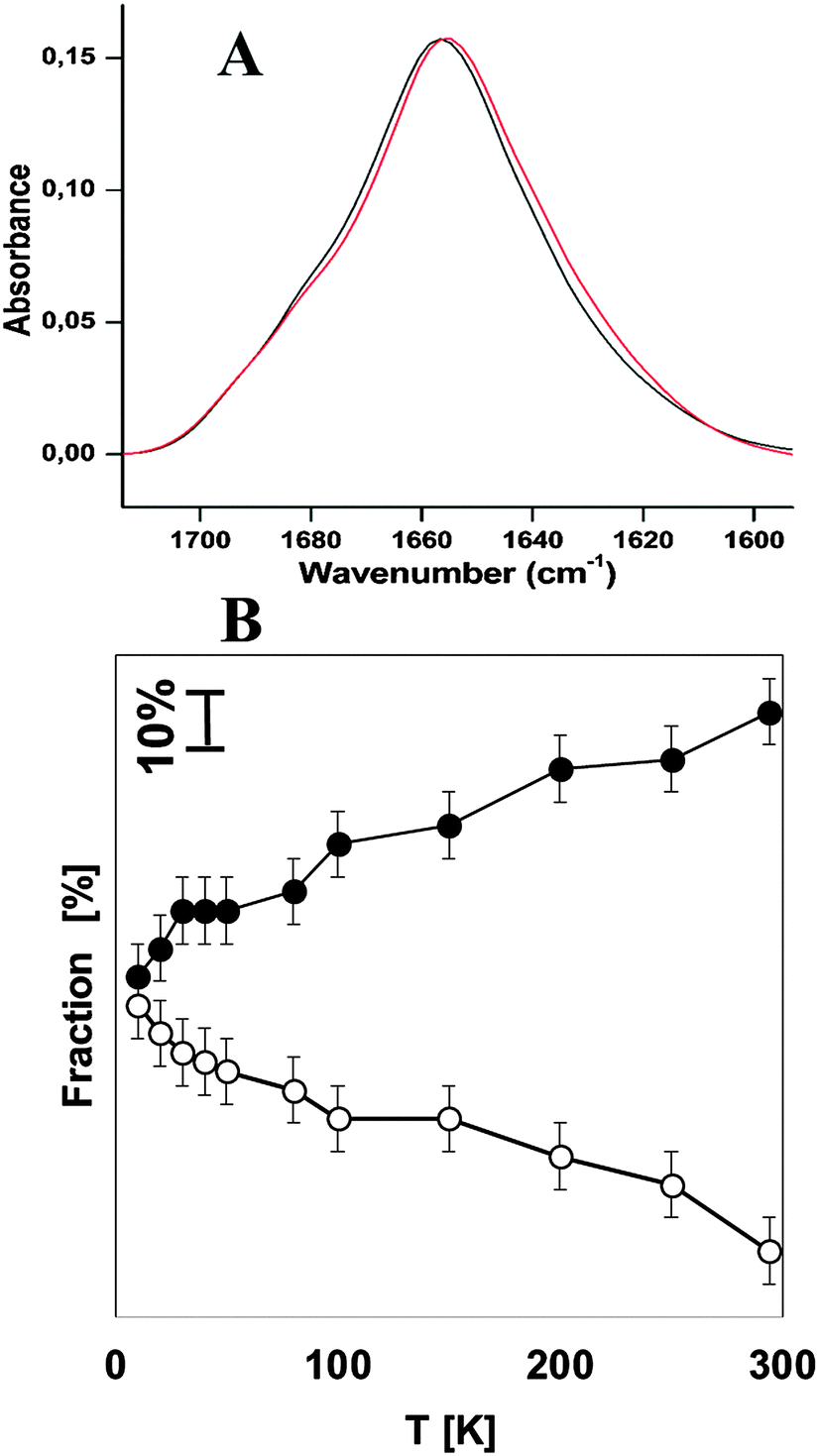 | ||
| Fig. 2 (A) Absorbance FTIR spectra of myoglobin at 294 K in black and 20 K in red. (B) The temperature dependence of the α-helix and unordered structure of myoglobin at pH 7.5. Filled circles: α-Helices; open circles: unordered structure. | ||
| Temperature (K) | α-Helices | Unordered | Extended chain | β-turns | Anti-//β-sheet |
|---|---|---|---|---|---|
| 294 | 64 (49) | 7 (20) | 10 (10) | 13 (15) | 6 (6) |
| 250 | 59 (49) | 14 (22) | 10 (11) | 13 (14) | 4 (4) |
| 200 | 58 (47) | 17 (26) | 9 (10) | 12 (13) | 4 (4) |
| 150 | 52 (44) | 21 (26) | 10 (11) | 13 (14) | 4 (5) |
| 100 | 50 (45) | 21 (24) | 11 (11) | 13 (14) | 5 (6) |
| 80 | 45 (45) | 24 (24) | 10 (9) | 15 (15) | 6 (7) |
| 50 | 43 (43) | 26 (26) | 12 (12) | 14 (14) | 5 (5) |
| 40 | 43 (42) | 27 (27) | 11 (11) | 14 (14) | 5 (6) |
| 30 | 43 (42) | 28 (28) | 11 (11) | 14 (14) | 4 (5) |
| 20 | 39 (41) | 30 (28) | 11 (11) | 14 (14) | 6 (6) |
| 10 | 36 (36) | 33 (33) | 11 (11) | 13 (13) | 7 (7) |
Analysis of the effect of pH on the native structures of L. polyphemus and E. californicum Hcs
In the first series of experiments, the secondary structure of Hcs was probed at different pH values since their oxygen binding affinities are pH dependent.36,37 At all pH values probed (pH 7, 7.5/7.8 and 8.5), the proteins are functional at standard temperature.Fig. 3 shows the relative contribution of each secondary structure element obtained on the basis of the deconvolution. The raw data used to prepare the graphs are summarized in Fig. S3 and S4 and Table S1 (ESI†). In all cases, the major contribution results from the band centered at around 1659–1661 cm−1, reflecting an α-helical structure of 35 to 43%.
 | ||
| Fig. 3 Secondary structure elements (α-helix, unordered, parallel β-sheet, anti-parallel β-sheet and β-turns) found on the basis of the deconvolution of the amide I band of L. polyphemus and E. californicum Hcs at 294 K (A and B) and 20 K (C and D), respectively, and at different pH values: in black at pH 7, grey at pH 7.5/7.8 and white at pH 8.5. | ||
The direct comparison of the data of the two species at different pH values reveals that at 294 K the α-helical content is a little higher for the Hc from L. polyphemus (differences of 4, 6 and 1% for pH 7, 7.5/7.8 and 8.5, respectively, seen in Fig. 3A and B and Table S1, ESI†); the shift of the later being lower than the calculated error. The parallel β-sheet, anti-parallel β-sheet and β-turn structures are approximately the same in the structures of L. polyphemus and E. californicum Hcs at 294 K and at pH 7, 7.5/7.8. At pH 8.5, the contribution of the β-turn structure decreases for the sample from E. californicum at 294 K, explaining the change in the shape of the amide I band seen in Fig. 1. In contrast, the relative contribution of β-turns in the L. polyphemus Hc structure remains stable under the same conditions.
The same pH dependence was then studied at 20 K. A similar behavior was observed for both proteins with respect to changing pH when compared to the data obtained at 294 K. One exception was the unordered structure that decreases for the protein from L. polyphemus at 20 K (see Fig. 3C and D and Table S1, ESI†).
Secondary structure composition changes reversibly between 20 K and 294 K
In order to investigate the reversibility of the small, but clear structural changes observed under the effect of temperature, difference spectra were calculated for the data obtained for Hc from L. polyphemus at pH 8.5 from 294 to 20 K. This revealed consistent temperature dependent changes in the absorption spectrum (Fig. 4). The difference spectra include the sum of all changes induced by cooling/heating.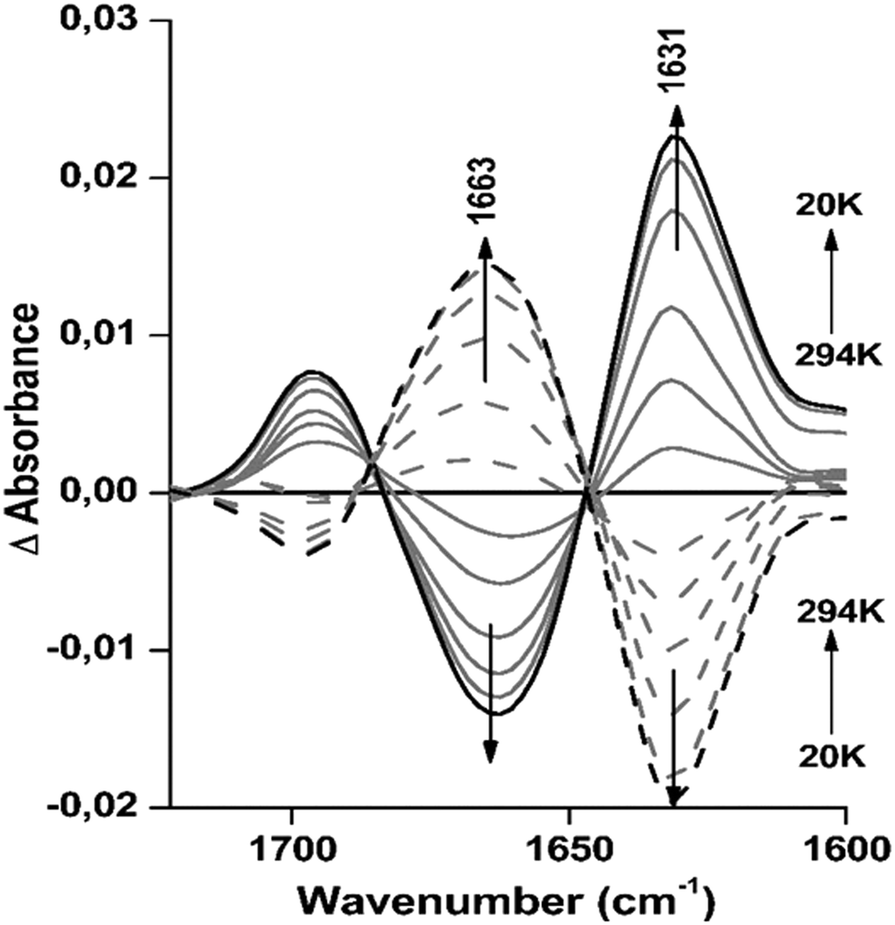 | ||
| Fig. 4 Difference FTIR spectra of L. polyphemus Hc at pH 8.5. The cooling experiments from 294 to 20 K are represented as full lines and the heating up from 20 to 294 K as dashed lines. For the cooling cycle, FTIR spectra were obtained by subtracting the spectrum at 294 K from the spectra at lower temperatures. For the heating cycle, the spectra at lower temperatures were subtracted from the spectrum at 294 K. FTIR spectra obtained for the cooling and the heating experiments were normalized to the area of the amide I band. | ||
More precisely, the strong peak at 1663 cm−1 decreases, leading to a negative peak in the difference spectra, while the absorbance at 1631 cm−1 increases, producing a positive peak (see full lines in Fig. 4). These structural changes are completely reversible, as seen from the spectra of L. polyphemus Hcs obtained after reheating the sample (dashed lines in Fig. 4 and Table S4, ESI†). The signals at 1696 and 1631 cm−1, which include the parallel and antiparallel β-sheets, partially overlap with the decrease in the unordered structure leading to a signal at 1663 cm−1. These structural changes are in agreement with the percentages of parallel β-sheets and unordered structures given by deconvolution (Table S4, ESI†). It is important to point out that the shifts observed in the secondary structure for both proteins during cooling correspond to small global changes in the magnitude of 3 to 7% of the corresponding absorbance spectra. For E. californicum Hc we also observed nearly fully reversible difference spectra as a function of temperature at pH 8.5, as seen in Fig. S5 (ESI†).
Fig. 5A–C give an overview of the relative contribution of the unordered structure and parallel β-sheets of L. polyphemus Hc at all pH values as a function of temperature. It seems that from 294 to 100 K a rather stable secondary structure composition exists, whereas between 50 and 100 K a transition of the secondary structure takes place. This transition involves a gradual decrease of the unordered structure and a simultaneous increase of parallel β-sheets, a tendency that is most pronounced for pH 8.5. For E. californicum Hc, however, no transition temperature was observed for any pH values studied (Fig. 5D–F); the structural ensemble was stable during freezing.
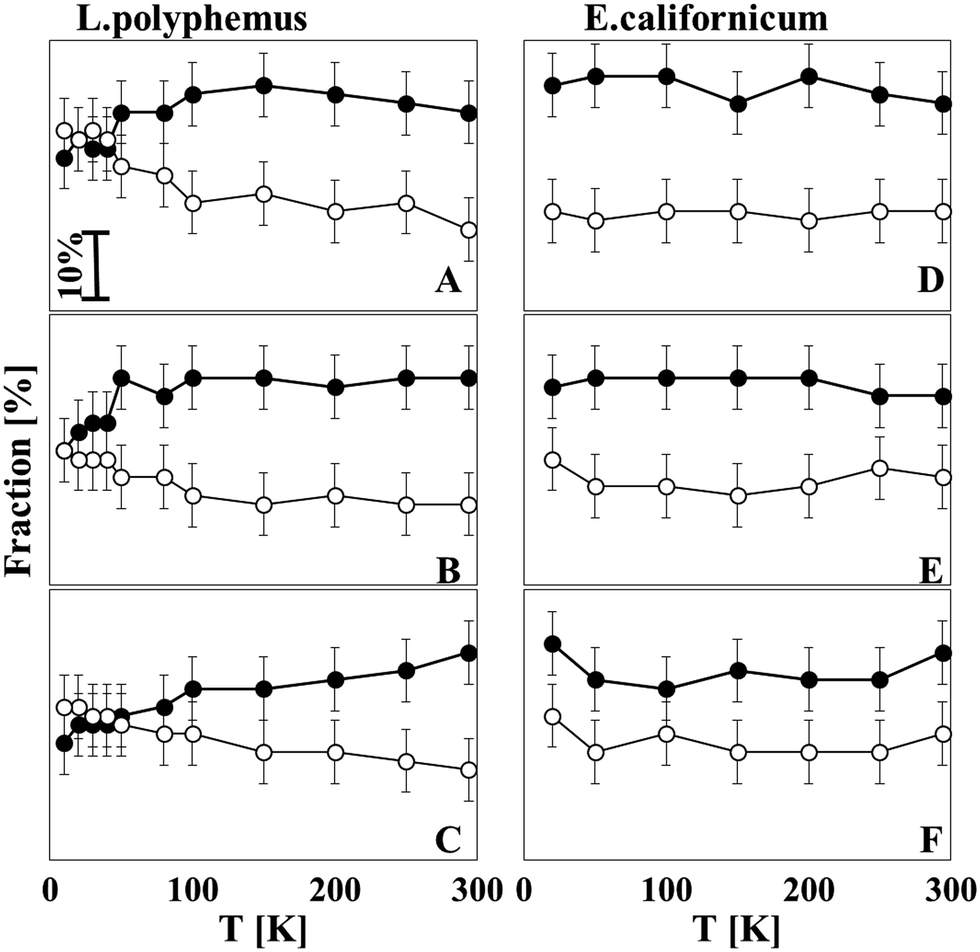 | ||
| Fig. 5 Temperature dependence of the unordered and parallel β-sheet structures of L. polyphemus Hc (A) at pH 7, (B) pH 7.5 and (C) at pH 8.5 and E. californicum Hc (D) at pH 7, (E) pH 7.8 and (F) at pH 8.5. Filled circles: Unordered structures; open circles: parallel β-sheet structure. | ||
Effects of low temperatures on the oxygen binding level
Fig. 6 shows that, in line with the structural integrity of the protein at low temperatures, only very weak changes can be reported when the oxygen saturation level is analyzed after thawing. Analytical ultracentrifugation shows that the oligomeric structure is affected to some extent, with up to 30% of the proteins being dissociated (Table S8, ESI†). This is no contradiction, since at ambient oxygen pressure individual subunits are also fully oxygenated for these two Hcs.13,38 We can conclude that the oxygen binding capacity is maintained, indicating that the second domain which contains the active site and is mainly α-helical remains intact for both Hcs.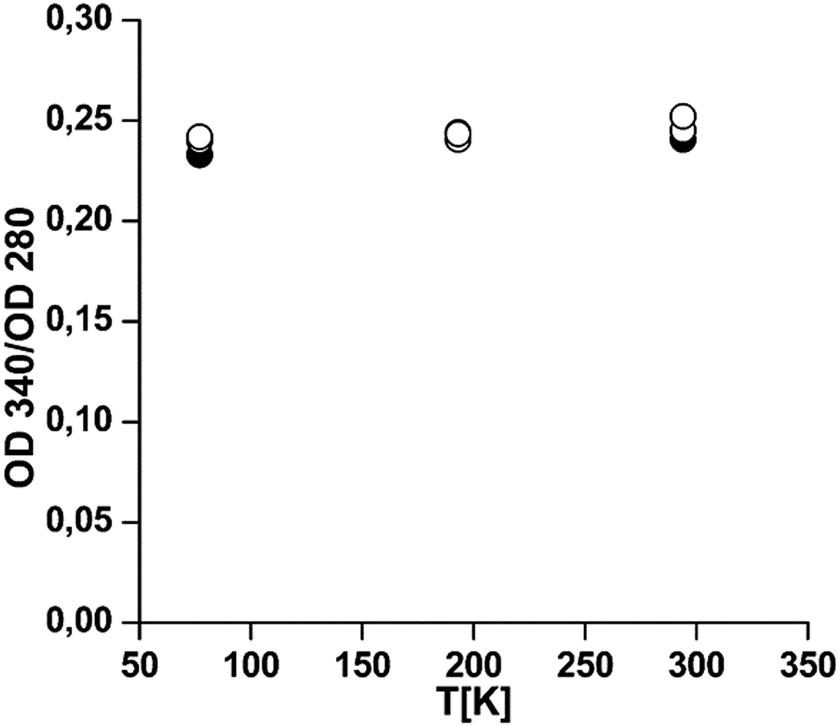 | ||
| Fig. 6 Oxygen binding level is not altered by cold treatment. Samples were exposed to −80 and −194 °C overnight. After thawing an appropriate dilution the UV-visible absorption spectra were measured. The oxygenation level is expressed as the ratio of the absorbance of the copper–oxygen band at 340 nm to the aromatic band at 280 nm. Filled circles: E. californicum Hc at pH 7.8; open circles: L. polyphemus Hc at pH 7.5. The value at 290 K represents the control. | ||
Discussion
In the current study, the structural stability of the Hcs from L. polyphemus and E. californicum in sub-zero temperatures was investigated. Both proteins were previously shown to be very stable with respect to heat-induced unfolding, with transition temperatures at 92 and 93 °C respectively.9,10 In control experiments, we obtained (91 and 93 °C, respectively, see Fig. S6, ESI†).FTIR spectroscopy showed that upon cooling down to 20 K only small, but largely reversible changes in the secondary structure elements occur. The oxygen binding level is not altered by cold treatment down to −197 °C. Thus, overall both proteins are remarkably stable upon exposure to freezing temperatures. In contrast, freezing of myoglobin and monoclonal antibody 1 (mAb1) in solution was reported to result in protein unfolding with transition temperatures of −12.7 and −23 °C, respectively.34,39 In general, cold denaturation unfolding experiments are performed under conditions which circumvent the crystallization of ice in the relevant temperature region (e.g. by choosing destabilizing solvent conditions or high pressure). A possible explanation for the absence of unfolding in our study could then be the employment of thin films with a high protein concentration (about 150 mg mL−1 or even higher, due to drying of the sample to a film), which could lead to a stabilization of the protein structure. We note, however, that the oxygen binding experiments have been performed in solution; thus once back under physiological conditions this putative stabilization is not required any more. Thus it seems that the high stability and activity under extreme conditions is an intrinsic property of the protein. This is supported by the observation that myoglobin exposed to the same treatment recovers some of the α-helical structure having lost at low temperatures, but not all: the level at 294 K is only 49% instead of 64% as observed before cold treatment, thus having irreversibly lost about 15% α-helical structure. The corresponding values for the two Hcs are 1 to 2%.
The active site is mainly α-helical17 and does not seem to suffer distortions as indicated by the preservation of oxygen binding capacity at least down to –197 °C. This is in line with the observation that even at 20 K the α-helical structure that is observed for all three pH values is stable with values close to those reported on crystal structures (43%, 1NOL.pdb), namely 43–40% at pH 8.5, 43–35% at pH 7.5 /7.8 and 41–37% for pH 7.
A small difference can be noted between the Hcs from the two organisms in terms of stability of the unordered and β-sheets structures. In the case of E. californicum Hc, practically no changes in the secondary structure content were found in the whole temperature range, whereas in L. polyphemus Hc the amount of unordered and parallel β-sheet structures changes below 50 K, especially at pH 8.5. Studies employing treatment with SDS revealed that Hc from L. polyphemus in this case is less stable than the Hc from E. californicum.40 Together with the result of our study this might indicate that this lowered stability is a general feature.
Whether the changes in the structure upon exposure to low temperatures and pH observed for the two proteins relate to differences in the biological function cannot be answered based on the present study. It is interesting to note, however, that L. polyphemus Hc is exceptional in the sense that it is one of the few Hcs with an inversed Bohr-effect and regulated by chloride, while Hc from E. californicum has a normal Bohr-effect.38,41
Another point that needs to be discussed is the effect of temperature on the pH value in the solution, since the pH value of buffer solutions may depend on temperature. For Tris–HCl buffer, in which both Hcs are solubilized, an increase of ∼1.2 units in the temperature range from +25 to −30 °C was reported by Kolhe et al.42 Estimations of the pH change of Tris-buffer when changing temperature from 20 to −194 °C were identified to correspond to +2.3 units in buffer solution alone, but much less pronounced in the presence of 40 mg mL−1 albumin (+0.1).43 Due to the high protein concentration employed in our study, a similar stabilization of the pH might be expected, although this effect is most likely dependent on the protein type. The structural changes studied here are thus essentially based on the effect of external parameters like temperature or pH, but not on the temperature dependent pH change of the buffer.
Conclusion
In this study we showed that Hcs from two species under conditions of high protein concentration and low water content retained their secondary structure down to 50 K, and their oxygen binding capacity down to 77 K. It seems that Hcs function as their own cryoprotectant under these conditions. Below 50 K the two hemocyanins respond differently to the decreased temperature: L. polyphemus Hc showed a re-arrangement of its secondary structure upon decreasing temperature at all pH values, but no structural changes were observed upon increasing the pH; E. californicum Hc is characterized by a decrease of the β-turn structure at pH 8.5, a structural change rarely reported in the literature so far.Acknowledgements
We are indebted to the Institute Universitaire de France (IUF), the icFRC, Fondation Recherche Medicale, the University of Strasbourg and the CNRS, for financial support.References
- S. N. Vinogradov, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1985, 82, 1–15 CrossRef CAS.
- K. E. Van Holde and K. I. Miller, in Adv. Protein Chem, ed. F. M. R. J. T. E. C. B. Anfinsen and S. E. David, Academic Press, 1995, vol. 47, pp. 1–81 Search PubMed.
- J. Markl and H. Decker, Blood and Tissue Oxygen Carriers, Springer, 1992 Search PubMed.
- K. E. v. Holde and K. I. Miller, Q. Rev. Biophys., 1982, 15, 1–129 CrossRef PubMed.
- H. Decker, N. Hellmann, E. Jaenicke, B. Lieb, U. Meissner and J. Markl, Integr. Comp. Biol., 2007, 47, 631–644 CrossRef CAS PubMed.
- J. Markl, Biol. Bull., 1986, 171, 90–115 CrossRef CAS.
- A. Volbeda and W. G. J. Hol, J. Mol. Biol., 1989, 209, 249–279 CrossRef CAS PubMed.
- A. Kolsch, J. Hornemann, C. Wengenroth and N. Hellmann, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 1853–1859 CrossRef PubMed.
- R. Sterner, T. Vogl, H. J. Hinz, F. Penz, R. Hoff, R. Foll and H. Decker, FEBS Lett., 1995, 364, 9–12 CrossRef CAS PubMed.
- K. Idakieva, Y. Raynova, F. Meersman and C. Gielens, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2013, 164, 201–209 CrossRef CAS PubMed.
- J. Markl, A. Savel, H. Decker and B. Linzen, Hoppe-Seyler's Z. Physiol. Chem., 1980, 361, 649–660 CrossRef CAS PubMed.
- R. Voit, G. Feldmaier-Fuchs, T. Schweikardt, H. Decker and T. Burmester, J. Biol. Chem., 2000, 275, 39339–39344 CrossRef CAS PubMed.
- H. Decker, J. Markl, R. Loewe and B. Linzen, Hoppe-Seyler's Z. Physiol. Chem., 1979, 360, 1505–1507 CAS.
- G. S. Ehlinger and R. A. Tankersley, Biol. Bull., 2004, 206, 87–94 CrossRef PubMed.
- R. Topham, S. Tesh, G. Cole, D. Mercatante, A. Westcott and C. Bonaventura, Arch. Biochem. Biophys., 1998, 352, 103–113 CrossRef CAS PubMed.
- K. A. Magnus, B. Hazes, H. Ton-That, C. Bonaventura, J. Bonaventura and W. G. J. Hol, Proteins: Struct., Funct., Bioinf., 1994, 19, 302–309 CrossRef CAS PubMed.
- B. Hazes, K. H. Kalk, W. G. J. Hol, K. A. Magnus, C. Bonaventura, J. Bonaventura and Z. Dauter, Protein Sci., 1993, 2, 597–619 CrossRef CAS PubMed.
- A. Buzy, J. Gagnon, J. Lamy, P. Thibault, E. Forest and G. Hudryclergeon, Eur. J. Biochem., 1995, 233, 93–101 CAS.
- R. Hubler, B. Fertl, N. Hellmann and H. Decker, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1998, 1383, 327–339 CrossRef CAS.
- P. Dolashka-Angelova, A. Dolashki, S. Stevanovic, R. Hristova, B. Atanasov, P. Nikolov and W. Voelter, Spectrochim. Acta, Part A, 2005, 61, 1207–1217 CrossRef PubMed.
- A. Dolashki, M. Radkova, E. Todorovska, M. Ivanov, S. Stevanovic, L. Molin, P. Traldi, W. Voelter and P. Dolashka, Mar. Biotechnol., 2015, 17, 743–752 CrossRef CAS PubMed.
- P. Dolashka-Angelova, R. Hristova, S. Stoeva and W. Voelter, Spectrochim. Acta, Part A, 1999, 55, 2927–2934 CrossRef.
- A. G. Martin, F. Depoix, M. Stohr, U. Meissner, S. Hagner-Holler, K. Hammouti, T. Burmester, J. Heyd, W. Wriggers and J. Markl, J. Mol. Biol., 2007, 366, 1332–1350 CrossRef CAS PubMed.
- W. Yassine, N. Taib, S. Federman, A. Milochau, S. Castano, W. Sbi, C. Manigand, M. Laguerre, B. Desbat, R. Oda and J. Lang, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1722–1730 CrossRef CAS PubMed.
- S. Castano and B. Desbat, Biochim. Biophys. Acta, Biomembr., 2005, 1715, 81–95 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS PubMed.
- J. L. R. Arrondo, A. Muga, J. Castresana and F. M. Goni, Prog. Biophys. Mol. Biol., 1993, 59, 23–56 CrossRef CAS PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369–430 CrossRef CAS PubMed.
- S. A. Tatulian, in Lipid–Protein Interactions: Methods and Protocols, ed. J. H. Kleinschmidt, Springer, New York, 2013, ch. 9, pp. 177–218, DOI:10.1007/978-1-62703-275-9_9.
- E. S. Manas, Z. Getahun, W. W. Wright, W. F. DeGrado and J. M. Vanderkooi, J. Am. Chem. Soc., 2000, 122, 9883–9890 CrossRef CAS.
- E. Kauffmann, N. C. Darnton, R. H. Austin, C. Batt and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6646–6649 CrossRef CAS PubMed.
- D. Reinstadler, H. Fabian and D. Naumann, Proteins: Struct., Funct., Genet., 1999, 34, 303–316 CrossRef CAS.
- B. T. Yesilyurt, C. Gielens and F. Meersman, FEBS J., 2008, 275, 3625–3632 CrossRef CAS PubMed.
- F. Meersman, L. Smeller and K. Heremans, Biophys. J., 2002, 82, 2635–2644 CrossRef CAS PubMed.
- Y. Levy, J. Jortner and O. M. Becker, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2188–2193 CrossRef CAS PubMed.
- H. Decker and R. Sterner, J. Mol. Biol., 1990, 211, 281–293 CrossRef CAS PubMed.
- B. Hazes, K. A. Magnus, C. Bonaventura, J. Bonaventura, Z. Dauter, K. H. Kalk and W. G. J. Hol, Protein Sci., 1993, 2, 597–619 CrossRef CAS PubMed.
- M. Brenowitz, C. Bonaventura and J. Bonaventura, Arch. Biochem. Biophys., 1984, 230, 238–249 CrossRef CAS PubMed.
- K. L. Lazar, T. W. Patapoff and V. K. Sharma, mAbs, 2010, 2, 42–52 CrossRef PubMed.
- S. Baird, S. M. Kelly, N. C. Price, E. Jaenicke, C. Meesters, D. Nillius, H. Decker and J. Nairn, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1380–1394 CrossRef CAS PubMed.
- B. Sullivan, J. Bonaventura and C. Bonaventura, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 2558–2562 CrossRef CAS.
- P. Kolhe, E. Amend and S. K. Singh, Biotechnol. Prog., 2010, 26, 727–733 CrossRef CAS PubMed.
- D. L. Williamssmith, R. C. Bray, M. J. Barber, A. D. Tsopanakis and S. P. Vincent, Biochem. J., 1977, 167, 593–600 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp03510h |
| This journal is © the Owner Societies 2016 |