
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adsorption in zeolites using mechanically embedded ONIOM clusters†
Ryan E.
Patet
ab,
Stavros
Caratzoulas
*ab and
Dionisios G.
Vlachos
ab
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: cstavros@udel.edu; Tel: +1-302-831-0362
bCatalysis Center for Energy Innovation (CCEI), Web: http://www.efrc.udel.edu
First published on 1st September 2016
Abstract
We have explored mechanically embedded three-layer QM/QM/MM ONIOM models for computational studies of binding in Al-substituted zeolites. In all the models considered, the high-level-theory layer consists of the adsorbate molecule and of the framework atoms within the first two coordination spheres of the Al atom and is treated at the M06-2X/6-311G(2df,p) level. For simplicity, flexibility and routine applicability, the outer, low-level-theory layer is treated with the UFF. We have modelled the intermediate-level layer quantum mechanically and investigated the performance of HF theory and of three DFT functionals, B3LYP, M06-2X and ωB97x-D, for different layer sizes and various basis sets, with and without BSSE corrections. We have studied the binding of sixteen probe molecules in H-MFI and compared the computed adsorption enthalpies with published experimental data. We have demonstrated that HF and B3LYP are inadequate for the description of the interactions between the probe molecules and the framework surrounding the metal site of the zeolite on account of their inability to capture dispersion forces. Both M06-2X and ωB97x-D on average converge within ca. 10% of the experimental values. We have further demonstrated transferability of the approach by computing the binding enthalpies of n-alkanes (C1–C8) in H-MFI, H-BEA and H-FAU, with very satisfactory agreement with experiment. The computed entropies of adsorption of n-alkanes in H-MFI are also found to be in good agreement with experimental data. Finally, we compare with published adsorption energies calculated by periodic-DFT for n-C3 to n-C6 alkanes, water and methanol in H-ZSM-5 and find very good agreement.
I. Introduction
Zeolites are among the most widely studied inorganic materials. Their wide-ranging industrial applications (e.g., separations, catalysis)1,2 have stimulated work which aims at optimizing the properties of existing zeolites via framework atom modifications, or at designing new zeolite-based materials for specific applications.1,3–5 Electronic structure calculations, molecular dynamics, and Monte Carlo simulations have provided tremendous insights into the properties of these materials, host–guest interactions, and catalytic activity.6–9 Accurate description of site-specific binding is critical to the calculation of catalytic pathways and the development of new catalysts but remains a challenge for theoretical models because the host–guest interactions are determined by long-range electrostatic interactions and the long-range electron correlation effects that give rise to dispersion forces.Periodic density-functional theory calculations can adequately address the electrostatic problem, but LDA and GGA functionals fail to capture the rather weak dispersion forces, and advanced, meta-hybrid functionals (with demonstrated ability to capture dispersion, e.g., M06-2X or ωB97X-D) are not easy to utilize because of algorithmic difficulties in computing the exact exchange. Although empirical dispersion corrections (DFT-D)10 have been parameterized for various GGA functionals, and more accurate semilocal exchange functionals (vdW-DF2) are being developed designed to capture dispersion interactions in a non-empirical fashion,11 periodic-DFT calculations remain computationally expensive for zeolites (cubic scaling with the number of atoms), especially if one wishes to map out catalytic pathways.
While it is widely recognized that the extended aluminosilicate framework around the active site of a zeolite-based catalyst is as important for the description of site-specific binding as the active site itself, it has also been argued that quantum mechanical description of atoms far from the active site and the substrate might not be critical. Hybrid QM/MM approaches capitalize on this premise in order to reduce the computational cost by layering the extended system (active site and a large part of the framework) into regions that are treated with varying degrees of accuracy.12–14 These layering strategies are in essence subtractive computational schemes, whereby the low-level-theory energy of an individual layer is subtracted from the energy of the real system and is substituted by a high-level-theory estimate. Typically, the reactive region is treated quantum mechanically (QM) and the surrounding environment is treated with a molecular mechanics force field (MM). However, it is not unusual to treat the environment quantum mechanically as well (QM/QM), either with low-level theories or with periodic-DFT, with concomitant increase in the computational cost. Notable in this respect is the QM/QM strategy of Tuma and Sauer (MP2/CBS:DFT + dispersion), whereby a small-size cluster is used to model the reactive region at the MP2 level, while the rest of the system is treated with periodic-DFT.15,16 This strategy has the advantage of handling long-ranged electrostatic interactions more accurately and of ameliorating the problem of GGA functionals not capturing dispersion interactions. One can surely envisage the development of variants with MP2 replaced by meta-GGA or hybrid-meta-GGA functionals specifically developed and parameterized to take into account dispersion and hydrogen bonding.
Hybrid QM/MM methodologies are inherently approximate as they primarily aim at routine applicability in large systems (e.g., enzymes, zeolites). The size of the QM domain, the theory level at which the QM domain is treated, the size of the overall system, the complexity of the MM force field or of the low-level quantum theory (e.g., HF or semi-empirical) in the environmental domain, and the interaction between the layers (e.g., mechanical versus electrostatic embedding) are all the result of a compromise between accuracy and computational efficiency. Choices that work universally remain a challenge. For example, what works for non-polar probe or substrate molecules might not work as well for polar ones, or when the adsorbate is basic enough to deprotonate proton-exchanged zeolites, leading to formation of an ion-pair structure.
Two-layer QM/MM studies with small, high-theory layers and medium-size, low-theory layers (containing on the order of 50 tetrahedral atoms or 50T) have been valuable at demonstrating capabilities and exposing limitations.17–31 ONIOM(B3LYP/6-31++G(d,p):HF/3-21G) single point calculations on ONIOM(B3LYP/6-31++G(d,p):MNDO) optimized structures of a (12T:48T) cluster model of acidic CHA for the adsorption of H2O and NH3 correctly predict proton donation to NH3 and ion-pair formation.25 A comparison of computed binding enthalpies with experimental data was not made, but one expects that the deviation from experiment should be significant given that B3LYP does not capture dispersive and hydrogen-bonding interactions. The binding energy was, nonetheless, in satisfactory agreement with periodic-B3LYP calculations – albeit somewhat underestimated – which is remarkable considering the rather small size of the embedded cluster and the level of theory at which the outer-layer was modelled (HF/3-21G). Nevertheless, the reasonable agreement between periodic-B3LYP calculations and ONIOM(B3LYP/6-31++G(d,p):HF/3-21G//B3LYP/6-31++G(d,p):MNDO) suggests that electrostatic interactions are rather well captured, although, convergence with the outer-layer basis set size was not reported and thus we are unable to assess the role of error cancellation in the ONIOM estimates. Interestingly, when the same methodology was applied to a host of probe molecules within a wide range of proton affinities (i.e., gas-phase basicities), and upon comparison with experimental data, the binding was severely underestimated, especially that of ion-pairs.24
Boekfa et al. have used a 5T:34T embedded model of H-MFI and computed binding energies for ethylene, benzene, ethylbenzene, and pyridine considering a combination of MP2 for the high-layer and various methods for the outer layer, including UFF (Universal Force Field), HF, B3LYP and M06-2X, with various combinations of triple-ζ-plus-diffuse and double-ζ quality basis sets, respectively.28 They have asserted that ONIOM(MP2/6-311+G(2df,2p):M06-2X/6-31G(d,p)//MP2/6-31G(d,p):M06-2X/6-31G(d,p)) gives good binding energies. This assertion was based on a comparison of the computed energies with experimental energies in H-FAU and not in H-MFI (the zeolite that was actually modelled) and thus it is difficult to ascertain the accuracy of the method. In fact, the rather acceptable error of ca. +5 kcal mol−1 (under-binding) should be expected to be higher in H-MFI, given that H-MFI has stronger acidity and smaller pores than H-FAU.32–35
Recently, Head-Gordon and co-workers partially re-parameterized the CHARMM force field for QM/MM calculations of electrostatically embedded zeolite cluster models.29–31 By investigating convergence with respect to the size of the framework QM domain, they have suggested that the QM region does not have to extend beyond the first coordination sphere and proposed the use of a 5T cluster which they model at the ωB97X-D/6-311++G(3df,3pd)//ωB97X-D/6-31G(d,p) theory level (namely, optimization at the ωB97X-D/6-31G(d,p) level followed by a single-point energy calculation at the ωB97X-D/6-311++G(3df,3pd) level). Thus the burden of capturing the interaction between the substrate molecule and the framework of the zeolite is on the force field that models the MM region, typically consisting of up to 44 tetrahedral atoms. They have computed binding enthalpies within 10% of the reported experimental values for species that are both physisorbed and chemisorbed in MFI, H-MFI, and H-BEA zeolites.31 This is a promising methodology which has demonstrated considerable improvements in accuracy, although it remains to be tested for a wider class of polar probe molecules in order to ascertain its accuracy and whether further re-parameterization of the CHARMM force field will be required.
Having in mind wide and routine applicability, in this article we explore a mechanically-embedded, three-layer QM/QM/MM ONIOM approach which retains the simplicity of the Universal Force Field (UFF) for the MM region. Philosophically, our decision to explore a QM/QM/MM approach stems from the understanding that, for polar and strongly binding substrate molecules, the binding energy greatly depends on QM–MM interactions, dispersive, and electrostatic. Electrostatic interactions, in particular, polarize the active site (always part of the QM region) and a reliable approach toward capturing such interactions more adequately would be to replace part of the MM region with a quantum region that would be treated at a medium theory level which, nevertheless, is capable of capturing some of the dispersion interactions as well. By considering a broad class of probe molecules, our intent is to understand the range of these interactions in the sense of how extended the intermediate layer needs to be and of the “minimal” theory level one needs to employ to describe it, while maintaining a good balance between reliability and computational cost.
II. Computational methods
A. Embedded cluster design
Zeolite clusters were cut out from crystals taken from the database of structures maintained by the IZA.36 Dangling silicon bonds from this structure were saturated with hydrogen atoms at a bond length of 1.47 Å along the corresponding Si–O bonds of the crystal. One silicon atom was replaced by an aluminum atom and an H+ cation was introduced to the adjacent oxygen atom in the lowest-energy configuration.The QM/MM zeolite models were developed using the ONIOM method, as implemented in the Gaussian 09 suite of programs.37–41 We have investigated both two- and three-layer ONIOM models, whereby the total enthalpy of the system is given by
| H(ONIOM2) = H(H,SL) + H(L,RL) − H(L,SL) | (1) |
| H(ONIOM3) = H(H,SL) + H(M,IL) + H(L,RL) − H(M,SL) − H(L,IL) | (2) |
The small layer was chosen to include the first two tetrahedral coordination spheres around the Al atom, shown for H-MFI in Fig. 1a and b. The small layer was mechanically embedded into a real layer which includes all the atoms up to the sixth tetrahedral coordination sphere of the Al atom, again shown for H-MFI in Fig. 1c–e. For the three-layer ONIOM models, an intermediate layer was investigated to include the third, fourth, and in the case of H-FAU, fifth tetrahedral coordination spheres around the Al atom. The real layer of the model was investigated to include the fourth, fifth, sixth, and in the case of H-FAU, seventh tetrahedral coordination spheres around the Al atom. Representations of H-MFI, H-BEA, and H-FAU can be seen in Fig. 2a–c, respectively. In all models, the small layer of the model was allowed to relax while the intermediate and real layers were frozen to maintain the integrity of the zeolite framework structure. We should note that, in preliminary studies, relaxation of the intermediate layer occasionally converged to structures with multiple imaginary frequencies.
 | ||
| Fig. 1 Representations of the (a) 1st (5T), (b) 2nd (17T), (c) 3rd (40T), (d) 4th (78T), (e) 5th (137T), and (f) 6th (226T) coordination spheres surrounding an Al-atom substituted in the T12 position of H-MFI. | ||
 | ||
| Fig. 2 ONIOM representations of (a) H-MFI, (b) H-BEA, and (c) H-FAU zeolites showing the layers of the standard models. The small layer is shown as a ball-and-stick representation, the intermediate layer is shown as a tubeframe representation, and the real layer is shown as a wireframe representation. | ||
B. Theory levels
In both the two-layer and three-layer models we treated the QM region with the hybrid-meta-GGA functional M06-2X, a global functional with 54% HF exchange, with demonstrated ability to capture dispersion interactions.42 We have considered two basis sets: 6-31G(d,p) and 6-311G(2df,p). We have not considered augmented basis sets, not only because diffuse functions increase the computational cost, but also because they can increase the basis set superposition error (BSSE). Extensive studies by Truhlar and co-workers have shown that better accuracy could be achieved if the extra cost were instead invested in a larger valence space (e.g., triple-zeta basis), or more polarization.43,44In the two-layer model, the outer (low-level) layer has been simulated with the UFF. The UFF has enjoyed significant popularity in mechanically embedded QM/MM electronic structure calculations of large systems because of its simplicity. Additionally, because of the absence of partial charges on the UFF atoms, it affords us the flexibility to partition the system into QM and MM regions in a number of ways without worrying about the net charge of the MM region not being zero. An issue that is often overlooked is that a non-neutral MM region can be a serious source of error in the binding energies, especially for polar molecules.20–23,26,28,45 We have investigated the effect of the cluster size by keeping the QM region size constant and varying the MM region. So, in the case of H-MFI, we have considered the ONIOM2 models M06-2X/6-31G(d,p):UFF and M06-2X/6-311G(2df,p):UFF and the clusters 17T:120T and 17T:209T.
In the three-layer QM/QM/MM ONIOM model, we have investigated layer size effects and theory/basis set effects for the intermediate layer while the outer layer has been simulated with the UFF. For the intermediate layer, we have considered HF theory and the three functionals B3LYP, ωB97X-D and M06-2X, the basis sets 3-21G, 6-31G, 6-311G, 6-31G(d,p), and 6-31G(2df,p), and the clusters 17T:23T:97T, 17T:61T:59T, and 17T:23T186T for H-MFI, 16T:18T:77T for H-BEA and 14T:16T:188T for H-FAU.
Calculated binding strengths have been benchmarked against available experimental values provided in Table 1, measured using a combination of TPD and microcalorimetry between 323 and 480 K.46–50 Vibrational frequencies were used to calculate thermal corrections to the binding enthalpies. For the spurious soft vibrational modes of the framework and the wrong asymptotic behavior of the enthalpy at low frequencies, we have employed the quasi-rigid rotor harmonic oscillator (qRRHO) approximation proposed by Grimme51 and Head-Gordon31 (see the ESI† for more details). Binding enthalpies were calculated by eqn (3) and are reported only at atmospheric pressure and 25 °C, as changing the temperature within the 50–100 °C range results in enthalpy changes of less than 0.5 kcal mol−1 in all cases.
| ΔHads = HZ+ads(Elec,VibqRRHO) − HZ(Elec,VibrqRRHO) − Hads(Elec,Trans,VibHO,Rot) | (3) |
| Zeolite | H-MFI | H-BEA | H-FAU | |
|---|---|---|---|---|
| Molecule | Proton affinity (kcal mol−1) | ΔHads,Exp (kcal mol−1) | ΔHads,Exp (kcal mol−1) | ΔHads,Exp (kcal mol−1) |
| a Microcalorimetry at 323–480 K.46–50 b Average of IR and calorimetry experiments at 300–650 K.32–35 | ||||
| Watera | 165.0 | −21.5 ± 2.4 | — | — |
| Benzenea | 179.3 | −15.5 ± 1.2 | — | — |
| Methanola | 180.3 | −27.5 ± 1.2 | — | — |
| Ammoniaa | 204.0 | −34.7 ± 1.2 | — | — |
| 2-Fluoropyridinea | 211.8 | −32.3 ± 3.1 | — | — |
| Methylaminea | 214.1 | −44.2 ± 1.2 | — | — |
| 3-Fluoropyridinea | 214.8 | −45.4 ± 1.7 | — | — |
| 3-Chloropyridinea | 215.7 | −45.4 ± 2.2 | — | — |
| Ethylaminea | 218.0 | −46.6 ± 1.2 | — | — |
| n-Butylaminea | 219.0 | −52.6 ± 1.2 | — | — |
| Dimethylaminea | 220.5 | −49.0 ± 1.2 | — | — |
| Isopropylaminea | 220.8 | −49.0 ± 1.2 | — | — |
| Pyridinea | 222.0 | −47.8 ± 1.2 | — | — |
| 3-Methylpyridinea | 222.8 | −53.8± 2.4 | — | — |
| 2-Methylpyridinea | 223.7 | −58.6 ± 2.2 | — | — |
| Trimethylaminea | 226.8 | −49.0 ± 1.2 | — | — |
| Methaneb | 129.9 | — | — | — |
| Ethaneb | 142.5 | — | — | — |
| Propaneb | 149.5 | −10.8 | — | −7.4 |
| n-Butaneb | — | −14.2 | — | −9.4 |
| n-Pentaneb | — | −17.1 | — | −11.0 |
| n-Hexaneb | — | −20.4 | −15.3 | −12.7 |
| n-Heptaneb | — | −22.5 | — | −16.4 |
| n-Octaneb | — | −25.4 | — | — |
A full study of cluster and layer size, and of theory level effects on the calculated ΔHads has been conducted using an H-MFI model because the greatest amount of experimental data was available for all sixteen probe molecules used here (top 16 molecules in Table 1). H-MFI a QM/QM/MM model that was deemed accurate for H-MFI was then applied to H-BEA and H-FAU zeolite frameworks in order to study how changing the pore size and shape affected binding enthalpies.32–35 In order to investigate transferability across different frameworks, and because full sets of experimental ΔHads for the sixteen probe molecules were not available, we also investigated the binding of a number of alkanes (see Table 1) in H-MFI, H-BEA and H-FAU and compared with experimental data, when available.
The calculations involving quantum layers are subject to the basis set superposition error (BSSE). There is no exact way to correct for BSSE in ONIOM calculations. The most obvious way for both two- and three-layer ONIOM models is to isolate the QM layer (capped with H atoms to saturate the dangling bonds) and perform a counterpoise correction on it and the substrate molecule; we follow this approach here as well.21,23,24,26 The adsorption schemes are summarized in eqn (4) and (5).
| ΔHads(2 layer) = [H(H,SL) + H(L,RL) − H(L,SL)]Z+ads + BSSE(H,SL)Z+ads − [H(H,SL) + H(L,RL) − H(L,SL)]Z − H(H)ads | (4) |
| ΔHads(3 layer) = [H(H,SL) + H(M,IL) + H(L,RL) − H(M,SL) − H(L,IL)]Z+ads + BSSE(H,SL)Z+ads − [H(H,SL) + H(M,IL) + H(L,RL) − H(M,SL) − H(L,IL)]Z − H(H)ads | (5) |
III. Results
A. Quantum cluster and two-layer ONIOM
In Table 2, we present binding energies in H-MFI, modeled with a 17T quantum cluster and two two-layer ONIOM clusters, 17T:120T and 17T:209T. For the 17T cluster, the calculations are performed at the M06-2X/6-31G(d,p) and M06-2X/6-311G(2df,p) theory levels; the ONIOM calculations are performed at the M06-2X/6-31G(d,p):UFF and M06-2X/6-311G(2df,p):UFF levels.| Small layer theory | Exp. | M06-2X/6-31G(d,p) | M06-2X/6-311G(2df,p) | M06-2X/6-31G(d,p) | M06-2X/6-311G(2df,p) | ||
|---|---|---|---|---|---|---|---|
| Real layer tetrahedral atoms | None | None | 120T | 209T | 120T | 209T | |
| Water | −21.5 | −20.9 (−27.4) | −19.0 (−24.9) | −20.9 (−27.2) | −21.0 (−27.3) | −19.8 (−25.2) | −19.9 (−25.3) |
| Benzene | −15.5 | −6.1 (−10.9) | −7.5 (−10.0) | −12.1 (−16.2) | −13.0 (−17.0) | −13.5 (−15.8) | −14.4 (−16.6) |
| Methanol | −27.5 | −23.7 (−30.0) | −21.8 (−26.7) | −26.8 (−33.0) | −26.9 (−33.2) | −25.8 (−30.6) | −26.0 (−30.8) |
| Ammonia | −34.7 | −35.5 (−37.7) | −32.7 (−34.0) | −31.0 (−33.4) | −31.1 (−33.5) | −29.3 (−30.7) | −29.2 (−30.6) |
| 2-Fluoropyridine | −32.3 | −20.5 (−26.1) | −21.4 (−24.7) | −30.4 (−35.1) | −31.1 (−35.8) | −31.3 (−34.5) | −32.1 (−35.2) |
| Methylamine | −44.2 | −39.6 (−42.3) | −38.1 (−39.6) | −38.6 (−41.5) | −39.4 (−42.3) | −38.0 (−39.9) | −38.3 (−40.2) |
| 3-Fluoropyridine | −45.4 | −26.3 (−30.7) | −26.9 (−29.2) | −33.2 (−38.0) | −34.1 (−39.0) | −34.0 (−37.2) | −34.8 (−37.9) |
| 3-Chloropyridine | −45.4 | −25.3 (−29.1) | −26.1 (−28.5) | −36.6 (−40.1) | −37.7 (−41.1) | −38.1 (−40.6) | −38.4 (−40.8) |
| Ethylamine | −46.6 | −41.7 (−45.1) | −40.0 (−41.8) | −44.3 (−47.9) | −45.0 (−48.6) | −43.8 (−45.9) | −44.1 (−46.2) |
| n-Butylamine | −52.6 | −41.9 (−47.6) | −40.8 (−43.7) | −50.6 (−55.3) | −51.1 (−55.7) | −49.9 (−52.5) | −50.7 (−53.3) |
| Dimethylamine | −49.0 | −41.0 (−44.5) | −40.4 (−42.3) | −43.9 (−47.5) | −44.3 (−47.8) | −44.1 (−46.2) | −44.4 (−46.6) |
| Isopropylamine | −49.0 | −41.9 (−46.0) | −40.3 (−42.5) | −47.0 (−50.5) | −47.4 (−51.0) | −46.1 (−48.2) | −46.7 (−48.8) |
| Pyridine | −47.8 | −31.5 (−35.3) | −31.9 (−34.1) | −35.9 (−40.0) | −36.7 (−40.7) | −37.2 (−39.7) | −37.9 (−40.4) |
| 3-Methylpyridine | −53.8 | −31.0 (−35.0) | −31.9 (−34.1) | −42.9 (−46.4) | −43.7 (−47.2) | −43.9 (−46.2) | −45.0 (−47.3) |
| 2-Methylpyridine | −58.6 | −32.0 (−36.2) | −32.8 (−35.4) | −45.2 (−49.0) | −46.1 (−50.0) | −45.6 (−48.2) | −46.5 (−49.1) |
| Trimethylamine | −49.0 | −37.2 (−41.5) | −37.1 (−39.4) | −35.4 (−39.8) | −36.1 (−40.5) | −36.2 (−38.8) | −36.4 (−39.0) |
| Mean signed error | 11.0 (6.7) | 11.5 (8.9) | 6.1 (2.0) | 5.5 (1.4) | 6.0 (3.3) | 5.5 (2.8) | |
| Mean unsigned error | 11.1 (8.1) | 11.5 (9.3) | 6.1 (4.5) | 5.5 (4.3) | 6.0 (4.5) | 5.5 (4.3) | |
By the mean signed error (MSE) of 6.7 kcal mol−1 and the mean unsigned error (MUE) of 8.1 kcal mol−1, we can see that the 17T quantum cluster under-binds almost systematically when we do not correct for the BSSE. Both the MSE and MUE for BSSE-corrected energies increase to 11 kcal mol−1, indicating that, for the 17T quantum cluster, M06-2X/6-31G(d,p) under-binds systematically. This poor performance does not seem to be basis set related, as increasing the basis set size from 6-31G(d,p) to 6-311G(2df,p) increases the average BSSE-corrected error by 0.5 kcal mol−1. The shortcomings of small or medium-size cluster calculations of adsorption energies are of course well known and here are presented mainly for comparison purposes. Small and medium-sized quantum clusters have been quite reliable for the calculation of activation energies of reactions in zeolites primarily because the long-range interactions essentially cancel out, leaving an energy controlled by local electronic interactions, provided that significant changes in geometry do not take place.53 However, long-range interactions and confinement phenomena remain an issue when we calculate adsorption energies, because similar error cancellation does not occur.
The two-layer models M06-2X/6-31G(d,p):UFF and M06-2X/6-311G(2df,p):UFF show a marked improvement (Table 2). The incorporation of the third, fourth, and fifth tetrahedral UFF coordination spheres around the Al atom improves the BSSE-corrected MUE to 6.1 and 6.0 kcal mol−1 for the 6-31G(d,p) and 6-311G(2df,p) small layers basis sets, respectively, but we still see systematic under-binding, despite the fact that the UFF is known to overestimate the van der Waals interactions. It is notable that, when we do not include BSSE corrections, the MUE is lower, 4.5 kcal mol−1 for both the 6-31G(d,p) and 6-311G(2df,p) small layers basis sets (Table 2). That notwithstanding, there is clear improvement over the 17T cluster which is most evident in the adsorbates with the larger proton affinities, which bind more strongly in the two-layer ONIOM model – even though the binding energies are still underestimated. Adsorbates with larger proton affinities are able to accept the proton from the zeolite and the resulting ion pair seems to be stabilized through interactions with the pore.
Increasing the size of the real layer to also include the sixth tetrahedral coordination sphere reduces the BSSE-corrected MSE and MUE by ca. 0.5 kcal mol−1 both in the case of M06-2X/6-31G(d,p):UFF and in the case of M06-2X/6-311G(2df,p):UFF (Table 2). Overall, we see that by extending the size of the real layer (i.e., by including more of the framework) we have additional stabilization of the adsorbed stated on account of attractive van der Waals interactions between the substrate molecule and the walls of the zeolite. However, both ONIOM2 models investigated here tend to under-bind on average, a behavior that becomes more pronounced and more systematic when we correct for the BSSE.
B. Three-layer ONIOM – method, basis set and layer size effects
In the following we present results for a number of three-layer QM/QM/MM ONIOM models using a combination of theories and basis sets with a mechanically embedded 17T:23T:97T cluster model of H-MFI. The 17T:23T:97T cluster is of the same size as the 17T:120T cluster used in the ONIOM2 calculations but with 23T atoms of the real layer reassigned to an intermediate quantum layer. In all the models considered, the small layer (17T) is treated at the M06-2X/6-311G(2df,p) level. For the intermediate layer (23T), we assess HF theory and the three functionals B3LYP, M06-2X and ωB97x-D, and the basis sets 3-21G, 6-31G, 6-311G, 6-31G(d,p) and 6-31G(2df,p). The outer layer is modelled with the UFF.At the ILT/3-21G, ILT/6-31G and ILT/6-311G (ILT = HF, B3LYP, M06-2X, ωB97x-D) levels, we see a dramatic change compared to the ONIOM2 calculations as all four models now over-bind (negative MSE) with MUE in the range 5–10.5 kcal mol−1, without BSSE corrections (Tables 3 and 4). M06-2X and ωB97x-D over-bind systematically and, in fact, we see an increase in the MUE and in the degree of over-binding as we move from the minimal basis set 3-21G (MUE ca. 8.0 kcal mol−1) to 6-31G (MUE ca. 10 kcal mol−1). For the 6-311G basis set, however, the MUE drops to the 3-21G level – which is still significantly high and almost twice as large as in the ONIOM2 calculations. For the same basis sets, HF and B3LYP appear better balanced, with MUE in the range 5–6 kcal mol−1. Correcting for the BSSE is more beneficial to M06-2X and ωB97x-D than to HF and B3LYP. For example, the MUE at the M06-2X/6-311G and ωB97x-D/6-311G intermediate levels drops by 2 kcal mol−1 (to 5.5 and 6.2 kcal mol−1, respectively) when we correct for the BSSE. At the HF/6-311G and B3LYP/6-311G intermediate levels, the MUE drops by 0.4 kcal mol−1 (to 4.9 and 5.1 kcal mol−1, respectively) when we correct for the BSSE. Interestingly, when one considers the BSSE-corrected MUE, all four models appear quite equivalent.
| Small layer theory | Exp. | M06-2X/6-311G(2df,p) | |||
|---|---|---|---|---|---|
| Intermediate layer theory | HF/3-21G | B3LYP/3-21G | M06-2X/3-21G | ωB97x-D/3-21G | |
| Water | −21.5 | −24.6 (−30.4) | −25.3 (−31.2) | −26.4 (−32.3) | −26.0 (−31.9) |
| Benzene | −15.5 | −7.7 (−9.8) | −8.8 (−10.9) | −15.5 (−17.8) | −16.0 (−18.1) |
| Methanol | −27.5 | −28.2 (−33.1) | −29.4 (−34.3) | −31.3 (−36.2) | −31.0 (−35.9) |
| Ammonia | −34.7 | −39.4 (−40.6) | −40.3 (−41.5) | −42.0 (−43.2) | −41.6 (−42.8) |
| 2-Fluoropyridine | −32.3 | −35.4 (−38.0) | −36.5 (−39.1) | −40.9 (−43.6) | −42.5 (−45.2) |
| Methylamine | −44.2 | −48.9 (−50.3) | −50.2 (−51.6) | −52.4 (−53.8) | −52.7 (−54.1) |
| 3-Fluoropyridine | −45.4 | −40.9 (−43.4) | −42.4 (−44.9) | −46.8 (−49.3) | −48.5 (−50.9) |
| 3-Chloropyridine | −45.4 | −42.7 (−44.9) | −43.3 (−45.6) | −47.0 (−49.2) | −49.3 (−51.5) |
| Ethylamine | −46.6 | −53.9 (−55.4) | −55.4 (−56.9) | −58.1 (−59.6) | −59.1 (−60.6) |
| n-Butylamine | −52.6 | −60.0 (−62.3) | −62.6 (−64.7) | −67.1 (−69.1) | −68.4 (−70.4) |
| Dimethylamine | −49.0 | −53.6 (−55.4) | −55.2 (−57.0) | −56.9 (−58.7) | −58.3 (−60.0) |
| Isopropylamine | −49.0 | −57.9 (−59.7) | −60.2 (−61.9) | −63.9 (−65.6) | −65.3 (−67.1) |
| Pyridine | −47.8 | −43.4 (−45.5) | −44.4 (−46.4) | −48.9 (−51.0) | −50.0 (−52.0) |
| 3-Methylpyridine | −53.8 | −49.7 (−51.8) | −50.7 (−52.8) | −54.5 (−56.6) | −56.8 (−58.9) |
| 2-Methylpyridine | −58.6 | −49.9 (−52.1) | −50.7 (−52.9) | −53.5 (−55.8) | −55.5 (−57.8) |
| Trimethylamine | −49.0 | −45.1 (−47.4) | −47.1 (−49.2) | −53.5 (−55.7) | −53.0 (−55.0) |
| Mean signed error | −0.5 (−2.9) | −1.8 (−4.2) | −5.4 (−7.8) | −6.3 (−8.7) | |
| Mean unsigned error | 5.0 (5.5) | 5.4 (5.9) | 6.0 (8.1) | 6.7 (8.8) | |
| Small layer theory | Exp. | M06-2X/6-311G(2df,p) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Intermediate layer theory | HF/6-31G | B3LYP/6-31G | M06-2X/6-31G | ωB97x-D/6-31G | HF/6-311G | B3LYP/6-311G | M06-2X/6-311G | ωB97x-D/6-311G | |
| Water | −21.5 | −25.2 (−31.0) | −25.5 (−31.3) | −27.1 (−32.9) | −26.9 (−32.7) | −23.9 (−29.6) | −24.2 (−30.0) | −25.9 (−31.7) | −25.5 (−31.3) |
| Benzene | −15.5 | −6.4 (−8.5) | −7.0 (−9.1) | −13.1 (−15.3) | −14.6 (−16.6) | −4.6 (−6.8) | −5.0 (−7.2) | −11.6 (−13.8) | −12.9 (−15.0) |
| Methanol | −27.5 | −28.6 (−33.5) | −28.9 (−33.8) | −31.5 (−36.4) | −32.0 (−36.9) | −26.7 (−31.5) | −27.1 (−32.0) | −30.0 (−34.9) | −30.1 (−35.0) |
| Ammonia | −34.7 | −41.6 (−42.8) | −42.0 (−43.2) | −44.0 (−45.1) | −44.3 (−45.5) | −39.0 (−40.2) | −40.0 (−41.2) | −42.0 (−43.2) | −42.0 (−43.2) |
| 2-Fluoropyridine | −32.3 | −37.4 (−39.9) | −37.6 (−40.2) | −42.7 (−45.4) | −44.8 (−47.4) | −34.4 (−37.0) | −35.2 (−37.8) | −40.7 (−43.3) | −42.3 (−44.9) |
| Methylamine | −44.2 | −51.0 (−52.3) | −51.5 (−52.8) | −54.3 (−55.7) | −55.1 (−56.4) | −48.0 (−49.3) | −48.9 (−50.2) | −51.9 (−53.3) | −52.5 (−53.8) |
| 3-Fluoropyridine | −45.4 | −41.9 (−44.4) | −42.5 (−44.9) | −47.7 (−50.2) | −49.8 (−52.2) | −39.0 (−41.4) | −40.0 (−42.3) | −45.6 (−47.9) | −47.0 (−49.4) |
| 3-Chloropyridine | −45.4 | −44.8 (−47.0) | −45.0 (−47.1) | −49.3 (−51.5) | −51.5 (−53.7) | −42.2 (−44.4) | −42.7 (−44.9) | −47.3 (−49.5) | −49.4 (−51.5) |
| Ethylamine | −46.6 | −56.3 (−57.9) | −56.5 (−58.0) | −60.1 (−61.8) | −61.7 (−63.2) | −53.2 (−54.7) | −53.8 (−55.3) | −57.4 (−59.1) | −58.6 (−60.1) |
| n-Butylamine | −52.6 | −60.7 (−62.9) | −61.2 (−63.3) | −67.2 (−69.2) | −69.1 (−71.0) | −57.7 (−59.9) | −58.7 (−60.8) | −64.5 (−66.5) | −66.1 (−68.1) |
| Dimethylamine | −49.0 | −55.9 (−57.7) | −56.2 (−58.0) | −59.3 (−61.2) | −60.9 (−62.7) | −52.6 (−54.4) | −53.5 (−55.3) | −56.7 (−58.5) | −58.1 (−59.9) |
| Isopropylamine | −49.0 | −58.6 (−60.4) | −59.2 (−61.0) | −64.4 (−66.2) | −66.3 (−68.0) | −55.2 (−57.0) | −56.3 (−58.1) | −61.9 (−63.6) | −63.4 (−65.2) |
| Pyridine | −47.8 | −45.7 (−47.7) | −45.8 (−47.8) | −51.0 (−53.1) | −52.4 (−54.5) | −42.6 (−44.6) | −43.2 (−45.3) | −49.2 (−51.2) | −50.1 (−52.1) |
| 3-Methylpyridine | −53.8 | −52.3 (−54.4) | −52.4 (−54.5) | −57.1 (−59.1) | −59.4 (−61.5) | −48.9 (−50.9) | −49.7 (−51.7) | −54.7 (−56.7) | −56.8 (−58.8) |
| 2-Methylpyridine | −58.6 | −53.1 (−55.2) | −53.5 (−55.6) | −56.9 (−59.2) | −59.0 (−61.3) | −50.3 (−52.4) | −51.2 (−53.4) | −54.9 (−57.2) | −56.9 (−59.1) |
| Trimethylamine | −49.0 | −47.2 (−49.5) | −47.2 (−49.3) | −55.3 (−57.3) | −55.8 (−57.8) | −43.6 (−45.9) | −43.4 (−45.6) | −52.0 (−54.1) | −52.1 (−54.2) |
| Mean signed error | −2.1 (−4.5) | −2.4 (−4.8) | −6.8 (−9.2) | −8.2 (−10.5) | 0.7 (−1.7) | 0.0 (−2.4) | −4.6 (−7.0) | −5.7 (−8.0) | |
| Mean unsigned error | 5.1 (5.9) | 5.2 (6.0) | 7.3 (9.2) | 8.3 (10.5) | 4.9 (5.3) | 5.1 (5.5) | 5.5 (7.4) | 6.2 (8.1) | |
The rather large fluctuations in the MUE with the intermediate layer basis sets 3-21G, 6-31G and 6-311G have led us to investigate the effect of adding polarization (Tables 5 and 6). Specifically, we have performed single-point energy calculations with the intermediate layer basis sets 6-31G(d,p) and 6-31G(2df,p) on geometries optimized at the M06-2X/6-311G(2df,p):ILT/3-21G:UFF and M06-2X/6-311G(2df,p):ILT/6-31G:UFF (ILT = HF, B3LYP, M06-2X, ωB97x-D). For M06-2X and ωB97x-D, the MUE reaches a plateau value of ca. 4 kcal mol−1, irrespective of the optimization geometry on which the single-point calculations were performed (Tables 5 and 6; see also Fig. 3a and b). Further, correcting for the BSSE leaves the MUE practically unaffected. In contrast, the MUE for HF and B3LYP in the intermediate layer are largely unconverged with respect to the basis set (Fig. 3). Adding polarization seems less beneficial, as the MUE rises to ca. 9 kcal mol−1 accompanied by significant under-binding, with MSE of ca. 6 kcal mol−1 without BSSE correction and ca. 9 kcal mol−1 with BSSE correction. Clearly, due their intrinsic shortcomings and despite the attendant computational savings, neither HF nor B3LYP are suitable for three-layer QM/QM/MM ONIOM calculations of binding energies; the dispersive interactions between the substrate molecule and the framework surrounding the active site play a significant role.
| Small layer theory | Exp. | M06-2X/6-311G(2df,p) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Intermediate layer theory | HF/6-31G(d,p)//HF/3-21G | B3LYP/6-31G(d,p)//B3LYP/3-21G | M06-2X/6-31G(d,p)//M06-2X/3-21G | ωB97x-D/6-31G(d,p)//ωB97x-D/3-21G | HF/6-31G(2df,p)//HF/3-21G | B3LYP/6-31G(2df,p)//B3LYP/3-21G | M06-2X/6-31G(2df,p)//M06-2X/3-21G | ωB97x-D/6-31G(2df,p)//ωB97x-D/3-21G | |
| Water | −21.5 | −20.4 (−26.3) | −20.9 (−26.8) | −21.9 (−27.8) | −22.1 (−28.0) | −19.4 (−25.3) | −19.8 (−25.7) | −20.7 (−26.6) | −21.0 (−26.9) |
| Benzene | −15.5 | −4.0 (−6.1) | −4.4 (−6.5) | −9.0 (−11.3) | −12.6 (−14.7) | −3.5 (−5.6) | −3.8 (−5.9) | −8.2 (−10.5) | −12.2 (−14.2) |
| Methanol | −27.5 | −21.5 (−26.4) | −22.3 (−27.2) | −24.5 (−29.4) | −24.7 (−29.6) | −20.0 (−24.9) | −20.8 (−25.7) | −22.9 (−27.8) | −23.2 (−28.1) |
| Ammonia | −34.7 | −33.1 (−34.4) | −33.8 (−35.0) | −35.2 (−36.4) | −35.4 (−36.7) | −31.0 (−32.2) | −31.7 (−32.9) | −32.9 (−34.1) | −33.3 (−34.5) |
| 2-Fluoropyridine | −32.3 | −28.4 (−31.0) | −29.4 (−32.0) | −33.8 (−36.5) | −36.1 (−38.8) | −26.2 (−28.9) | −27.4 (−30.0) | −31.7 (−34.4) | −34.1 (−36.8) |
| Methylamine | −44.2 | −41.3 (−42.7) | −42.3 (−43.7) | −44.6 (−46.0) | −45.6 (−47.0) | −38.9 (−40.3) | −39.9 (−41.3) | −42.2 (−43.6) | −43.2 (−44.6) |
| 3-Fluoropyridine | −45.4 | −32.4 (−34.9) | −33.3 (−35.8) | −37.8 (−40.3) | −40.1 (−42.6) | −30.3 (−32.8) | −31.5 (−33.9) | −35.8 (−38.3) | −38.2 (−40.7) |
| 3-Chloropyridine | −45.4 | −37.0 (−39.2) | −37.7 (−39.9) | −41.8 (−44.1) | −44.4 (−46.6) | −35.0 (−37.3) | −36.0 (−38.2) | −40.1 (−42.3) | −42.7 (−44.9) |
| Ethylamine | −46.6 | −46.1 (−47.6) | −47.1 (−48.6) | −49.8 (−51.3) | −51.5 (−53.0) | −43.5 (−45.0) | −44.6 (−46.1) | −47.3 (−48.8) | −49.0 (−50.5) |
| n-Butylamine | −52.6 | −49.6 (−51.8) | −51.3 (−53.5) | −56.3 (−58.3) | −58.4 (−60.3) | −46.9 (−49.1) | −48.7 (−50.9) | −53.7 (−55.6) | −55.8 (−57.8) |
| Dimethylamine | −49.0 | −46.1 (−47.8) | −47.1 (−48.8) | −49.7 (−51.4) | −51.6 (−53.3) | −43.6 (−45.4) | −44.8 (−46.5) | −47.3 (−49.0) | −49.3 (−51.0) |
| Isopropylamine | −49.0 | −47.8 (−49.6) | −49.7 (−51.4) | −54.4 (−56.1) | −56.4 (−58.2) | −45.3 (−47.1) | −47.3 (−49.0) | −52.0 (−53.7) | −54.0 (−55.8) |
| Pyridine | −47.8 | −36.7 (−38.8) | −37.5 (−39.5) | −42.1 (−44.1) | −43.7 (−45.7) | −34.5 (−36.6) | −35.4 (−37.4) | −39.9 (−41.9) | −41.6 (−43.7) |
| 3-Methylpyridine | −53.8 | −42.9 (−45.0) | −43.5 (−45.6) | −48.0 (−50.1) | −50.7 (−52.8) | −40.6 (−42.7) | −41.4 (−43.5) | −45.8 (−47.9) | −48.7 (−50.7) |
| 2-Methylpyridine | −58.6 | −45.2 (−47.4) | −45.9 (−48.1) | −49.2 (−51.5) | −51.6 (−53.9) | −43.3 (−45.4) | −44.2 (−46.4) | −47.4 (−49.7) | −50.0 (−52.3) |
| Trimethylamine | −49.0 | −37.2 (−39.5) | −37.7 (−39.9) | −43.5 (−45.6) | −45.3 (−47.4) | −35.0 (−37.3) | −35.4 (−37.6) | −40.5 (−42.7) | −43.0 (−45.1) |
| Mean signed error | 6.4 (4.0) | 5.6 (3.2) | 2.0 (−0.4) | 0.2 (−2.2) | 8.5 (6.1) | 7.5 (5.1) | 4.0 (1.6) | 2.1 (−0.3) | |
| Mean unsigned error | 6.4 (4.8) | 5.7 (4.5) | 3.9 (4.0) | 3.6 (3.8) | 8.5 (6.5) | 7.5 (5.6) | 4.6 (3.8) | 3.7 (3.3) | |
| Small layer theory | Exp. | M06-2X/6-311G(2df,p) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Intermediate layer theory | HF/6-31G(d,p)//HF/6-31G | B3LYP/6-31G(d,p)//B3LYP/6-31G | M06-2X/6-31G(d,p)//M06-2X/6-31G | ωB97x-D/6-31G(d,p)//ωB97x-D/6-31G | HF/6-31G(2df,p)//HF/6-31G | B3LYP/6-31G(2df,p)//B3LYP/6-31G | M06-2X/6-31G(2df,p)//M06-2X/6-31G | ωB97x-D/6-31G(2df,p)//ωB97x-D/6-31G | |
| Water | −21.5 | −20.7 (−26.5) | −21.4 (−27.2) | −22.5 (−28.3) | −22.5 (−28.3) | −19.6 (−25.4) | −20.2 (−26.1) | −21.3 (−27.1) | −21.3 (−27.2) |
| Benzene | −15.5 | −4.1 (−6.2) | −5.1 (−7.2) | −9.9 (−12.2) | −12.7 (−14.8) | −3.7 (−5.8) | −4.6 (−6.7) | −9.2 (−11.5) | −12.3 (−14.4) |
| Methanol | −27.5 | −22.1 (−27.0) | −23.0 (−27.9) | −25.1 (−30.0) | −25.7 (−30.6) | −20.5 (−25.4) | −21.4 (−26.3) | −23.6 (−28.4) | −24.1 (−29.0) |
| Ammonia | −34.7 | −33.4 (−34.6) | −34.2 (−35.4) | −35.6 (−36.8) | −35.9 (−37.1) | −31.3 (−32.4) | −32.1 (−33.3) | −33.3 (−34.5) | −33.7 (−34.9) |
| 2-Fluoropyridine | −32.3 | −29.0 (−31.5) | −30.1 (−32.7) | −34.7 (−37.3) | −37.0 (−39.6) | −26.9 (−29.5) | −28.3 (−30.8) | −32.7 (−35.4) | −35.1 (−37.7) |
| Methylamine | −44.2 | −41.9 (−43.2) | −43.2 (−44.5) | −45.4 (−46.7) | −46.2 (−47.5) | −39.4 (−40.7) | −40.9 (−42.2) | −42.9 (−44.3) | −43.8 (−45.1) |
| 3-Fluoropyridine | −45.4 | −33.5 (−35.9) | −35.1 (−37.5) | −39.8 (−42.3) | −41.8 (−44.2) | −31.6 (−34.0) | −33.4 (−35.8) | −38.1 (−40.5) | −40.0 (−42.4) |
| 3-Chloropyridine | −45.4 | −37.1 (−39.2) | −37.8 (−40.0) | −41.9 (−44.1) | −44.0 (−46.2) | −35.0 (−37.2) | −36.1 (−38.2) | −40.1 (−42.3) | −42.2 (−44.4) |
| Ethylamine | −46.6 | −46.9 (−48.5) | −47.9 (−49.5) | −50.7 (−52.4) | −52.5 (−54.0) | −44.3 (−45.8) | −45.5 (−47.0) | −48.2 (−49.9) | −50.0 (−51.5) |
| n-Butylamine | −52.6 | −50.7 (−52.9) | −52.1 (−54.3) | −57.5 (−59.5) | −59.4 (−61.4) | −48.1 (−50.3) | −49.5 (−51.7) | −54.9 (−56.9) | −56.9 (−58.8) |
| Dimethylamine | −49.0 | −46.6 (−48.4) | −47.5 (−49.3) | −50.1 (−51.9) | −51.8 (−53.6) | −44.0 (−45.8) | −45.1 (−46.9) | −47.6 (−49.4) | −49.4 (−51.2) |
| Isopropylamine | −49.0 | −48.5 (−50.3) | −50.3 (−52.0) | −55.1 (−56.8) | −57.0 (−58.7) | −46.0 (−47.7) | −47.9 (−49.6) | −52.6 (−54.3) | −54.5 (−56.2) |
| Pyridine | −47.8 | −37.1 (−39.1) | −38.1 (−40.1) | −42.7 (−44.8) | −44.3 (−46.3) | −34.9 (−36.9) | −36.0 (−38.1) | −40.7 (−42.7) | −42.3 (−44.3) |
| 3-Methylpyridine | −53.8 | −43.6 (−45.6) | −44.4 (−46.5) | −48.5 (−50.6) | −51.0 (−53.0) | −41.3 (−43.3) | −42.4 (−44.5) | −46.4 (−48.5) | −49.0 (−51.0) |
| 2-Methylpyridine | −58.6 | −45.6 (−47.7) | −46.5 (−48.6) | −49.4 (−51.7) | −51.8 (−54.0) | −43.6 (−45.7) | −44.8 (−46.9) | −47.7 (−50.0) | −50.1 (−52.4) |
| Trimethylamine | −49.0 | −37.6 (−39.9) | −37.7 (−39.8) | −44.4 (−46.5) | −45.6 (−47.7) | −35.3 (−37.6) | −35.4 (−37.5) | −41.7 (−43.8) | −43.2 (−45.3) |
| Mean signed error | 5.9 (3.5) | 4.9 (2.5) | 1.2 (−1.2) | −0.4 (−2.8) | 8.0 (5.6) | 6.8 (4.5) | 3.2 (0.8) | 1.6 (−0.8) | |
| Mean unsigned error | 5.9 (4.6) | 5.2 (4.5) | 3.9 (4.1) | 3.7 (4.0) | 8.0 (6.1) | 6.8 (5.1) | 4.2 (3.7) | 3.6 (3.5) | |
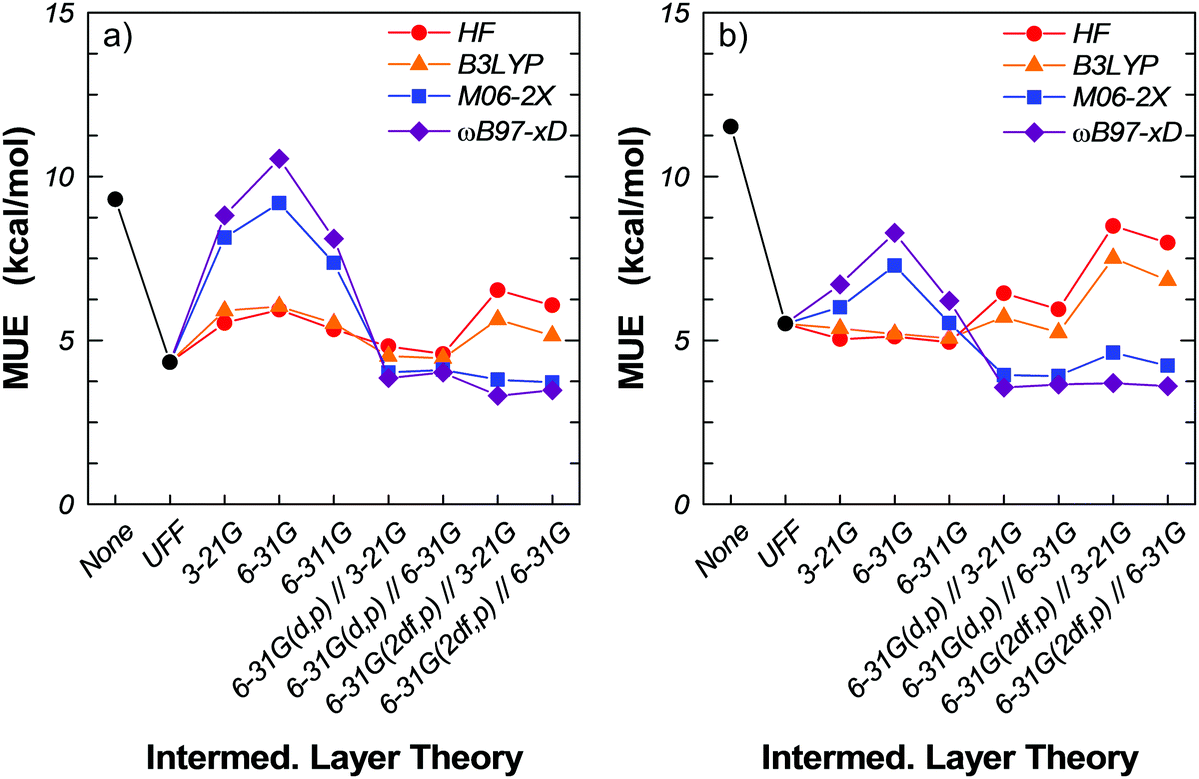 | ||
| Fig. 3 Mean unsigned error at various intermediate layer theories. (a) BSSE-uncorrected and (b) BSSE-corrected values. | ||
Of all the probe molecules investigated for this study, pyridine and its chloro- fluoro- and methyl-derivatives and the amines posed a great challenge to the two-layer ONIOM methodology. The MSE calculated over the subset of the pyridines and amines is 8.1 and 5.0 kcal mol−1, respectively. In contrast, the three-layer models perform significantly better. For example, at the M06-2X/6-311G(2df,p):M06-2X/6-31(d,p):UFF//M06-2X/6-311G(2df,p):M06-2X/6-31G:UFF level, the MSE dropped to 4.3 and −2.1 kcal mol−1, showing the importance of the intermediate quantum mechanical layer for the binding of basic molecules. The decrease in the error relative to the ONIOM2 models – of the same size – and the almost similar behavior of the intermediate layer theories in the ONIOM3 models suggest the importance of electrostatic interactions and of charge polarization in particular. QM/MM methods with mechanically embedded QM regions do not consider the polarization of the quantum region (i.e. of the active site) by the framework and vice versa, force fields with fixed partial charges on the atoms do not react to changes in the electron density of the quantum region. QM/MM formulations with electrostatic embedding address the former problem because the Hamiltonian includes an extra term with the electrostatic potential, but they still suffer from the latter, which is of some significance when we calculate transition states, as there is no reason to assume that the polarization of the MM region will not be affected along the reaction pathway. However, by introducing a quantum mechanical intermediate layer, which replaces part of the MM layer, we partly ameliorate the lack of polarization; at the same time, we are also treating the dispersion interactions more accurately than with the UFF.54
We have also investigated the effects of the size of the intermediate and real layers on the binding energy (Table 7). We have considered three H-MFI clusters, 17T:23T:97T, 17T:61T:59T and 17T:23T:186, modelled at the M06-2X/6-311G(2df,p):M06-2X/6-31(d,p):UFF//M06-2X/6-311G(2df,p):M06-2X/3-21G:UFF level. In the 17T:61T:59T cluster, we have reassigned 38 atoms of the real layer of 17T:23T:97T to the intermediate layer, while keeping the total number of atoms fixed. In the 17T:23T:186T cluster, we have added atoms within the sixth coordination sphere of 17T:23T:97T. The results are presented in Table 7 and in Fig. 4. The MUE, with or without BSSE corrections, remains practically flat and independent of the size of the cluster, indicating that, overall, it is not affected in any significant way when we extend the MM layer beyond the sixth coordination sphere around the Al atoms of the active site, fluctuating around the value of ca. 3.8 kcal mol−1. We have seen similar behavior in the two-layer ONIOM models presented earlier. Extending the intermediate layer can lead to minor improvement.
| SL:IL:RL tetrahedral atoms | Exp. | 17T:23T:97T | 17T:61T:59T | 17T:23T:186T | 17T:23T:97T | 17T:61T:59T | 17T:23T:186T |
|---|---|---|---|---|---|---|---|
| Theory | M06-2X/6-311G(2df,p):M062X/3-21G:UFF | M06-2X/6-311G(2df,p):M06-2X/6-31G(d,p):UFF//M06-2X/6-311G(2df,p):M062X/3-21G:UFF | |||||
| Water | −21.5 | −26.4 (−32.3) | −25.9 (−31.8) | −26.5 (−32.4) | −21.9 (−27.8) | −21.3 (−27.2) | −22.0 (−27.9) |
| Benzene | −15.5 | −15.5 (−17.8) | −18.5 (−20.4) | −16.2 (−18.5) | −9.0 (−11.3) | −11.2 (−13.2) | −9.6 (−11.9) |
| Methanol | −27.5 | −31.3 (−36.2) | −31.2 (−36.2) | −31.5 (−36.4) | −24.5 (−29.4) | −23.6 (−28.6) | −24.7 (−29.6) |
| Ammonia | −34.7 | −42.0 (−43.2) | −43.4 (−44.6) | −42.1 (−43.3) | −35.2 (−36.4) | −36.0 (−37.2) | −35.2 (−36.4) |
| 2-Fluoropyridine | −32.3 | −40.9 (−43.6) | −44.6 (−47.4) | −41.7 (−44.3) | −33.8 (−36.5) | −32.3 (−35.1) | −34.5 (−37.2) |
| Methylamine | −44.2 | −52.4 (−53.8) | −53.7 (−55.0) | −52.6 (−54.0) | −44.6 (−46.0) | −44.8 (−46.1) | −44.8 (−46.2) |
| 3-Fluoropyridine | −45.4 | −46.8 (−49.3) | −50.8 (−53.2) | −47.5 (−50.0) | −37.8 (−40.3) | −37.4 (−39.9) | −38.5 (−41.0) |
| 3-Chloropyridine | −45.4 | −47.0 (−49.2) | −50.9 (−53.1) | −48.0 (−50.2) | −41.8 (−44.1) | −40.6 (−42.8) | −42.7 (−44.9) |
| Ethylamine | −46.6 | −58.1 (−59.6) | −60.2 (−61.6) | −58.5 (−60.0) | −49.8 (−51.3) | −49.0 (−50.4) | −50.1 (−51.6) |
| n-Butylamine | −52.6 | −67.1 (−69.1) | −67.6 (−69.4) | −67.9 (−69.9) | −56.3 (−58.3) | −52.0 (−53.8) | −57.0 (−59.0) |
| Dimethylamine | −49.0 | −56.9 (−58.7) | −59.1 (−60.8) | −57.3 (−59.0) | −49.7 (−51.4) | −49.7 (−51.4) | −49.9 (−51.7) |
| Isopropylamine | −49.0 | −63.9 (−65.6) | −64.8 (−66.4) | −64.5 (−66.2) | −54.4 (−56.1) | −52.8 (−54.4) | −55.0 (−56.7) |
| Pyridine | −47.8 | −48.9 (−51.0) | −52.7 (−54.6) | −49.6 (−51.7) | −42.1 (−44.1) | −40.8 (−42.7) | −42.7 (−44.7) |
| 3-Methylpyridine | −53.8 | −54.5 (−56.6) | −58.7 (−60.9) | −55.5 (−57.5) | −48.0 (−50.1) | −47.2 (−49.3) | −48.8 (−50.9) |
| 2-Methylpyridine | −58.6 | −53.5 (−55.8) | −58.3 (−60.7) | −54.5 (−56.8) | −49.2 (−51.5) | −48.3 (−50.7) | −50.1 (−52.4) |
| Trimethylamine | −49.0 | −53.5 (−55.7) | −55.8 (−57.9) | −54.0 (−56.2) | −43.5 (−45.6) | −42.6 (−44.7) | −43.9 (−46.1) |
| Mean signed error | −5.4 (−7.8) | −7.7 (−10.1) | −5.9 (−8.3) | 2.0 (−0.4) | 2.7 (0.3) | 1.5 (−0.9) | |
| Mean unsigned error | 6.0 (8.1) | 7.7 (10.1) | 6.4 (8.6) | 3.9 (4.0) | 3.8 (3.7) | 3.8 (3.9) | |
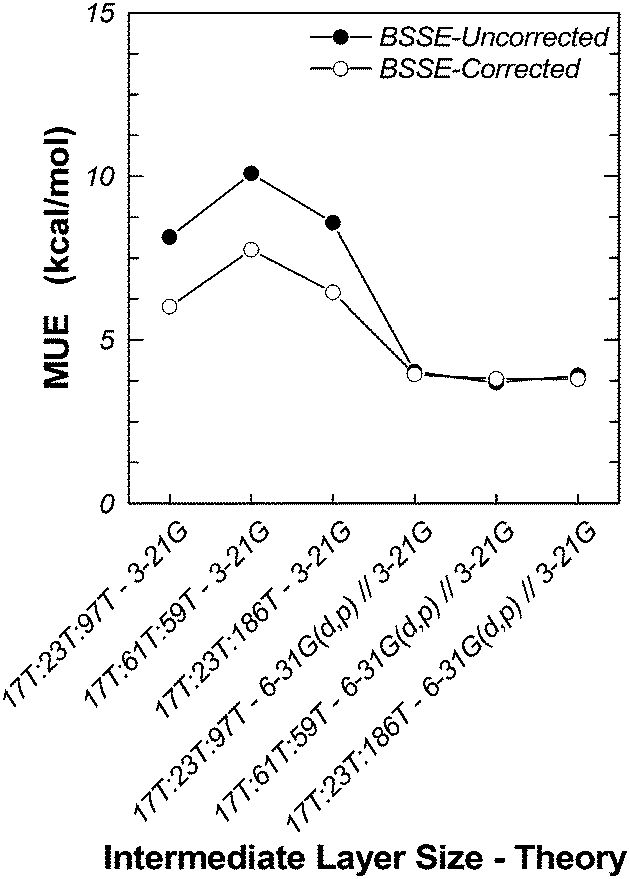 | ||
| Fig. 4 Behavior of mean unsigned error with intermediate and real layer sizes and intermediate layer theory. | ||
C. Adsorption of alkanes in H-MFI, H-BEA and H-FAU
To test transferability, we have extended our calculations to include frameworks of different pore sizes, specifically H-BEA and H-FAU. Experimentally determined adsorption strengths for the series of 16 adsorbates investigated above were not available for H-BEA and H-FAU. Numerous studies have, however, investigated the adsorption of linear alkanes in H-MFI, H-BEA and H-FAU. Measurement of their adsorption enthalpies is not without issues, as the acidic zeolite environments cause alkanes to rapidly undergo isomerization, alkylation, and other reactions, and thereby the average of these experimental adsorption enthalpies has been taken as a best estimate for the real system.32–35 In Table 8, we present calculations with and without polarization in the intermediate layer using a 17T:23T:97T cluster for H-MFI, a 16T:18T:77T cluster of H-BEA and a 14T:16T:188T cluster for H-FAU.| Zeolite | H-MFI | H-BEA | H-FAU | H-MFI | H-BEA | H-FAU | H-MFI | H-BEA | H-FAU | H-MFI | H-BEA | H-FAU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SL:IL:RL tetrahedral atoms | Exp.a | 17T:23T:97T | 16T:18T:77T | 14:16T:188T | 17T:23T:97T | 16T:18T:77T | 14:16T:188T | 17T:23T:97T | 16T:18T:77T | 14:16T:188T | ||
| Theory | M06-2X/6-311G(2df,p):M062X/3-21G:UFF | M06-2X/6-311G(2df,p):M06-2X/6-31G(d,p):UFF//M06-2X/6-311G(2df,p):M062X/3-21G:UFF | M06-2X/6-311G(2df,p):M06-2X/6-311G(2df,p):UFF//M06-2X/6-311G(2df,p):M062X/3-21G:UFF | |||||||||
| a Values in parantheses are projected based on a linear fit of the remaining alkane values. | ||||||||||||
| Methane | (−5.3) | (−2.7) | (−2.9) | −8.8 (−9.6) | −6.5 (−7.4) | −5.5 (−6.1) | −7.3 (−8.2) | −5.9 (−6.8) | −4.0 (−4.6) | −7.0 (−7.8) | −5.8 (−6.7) | −3.7 (−4.3) |
| Ethane | (−8.2) | (−5.2) | (−5.0) | −13.4 (−14.6) | −9.1 (−10.3) | −8.3 (−9.8) | −11.9 (−13.2) | −8.3 (−9.5) | −8.1 (−9.5) | −11.6 (−12.9) | −8.1 (−9.4) | −7.9 (−9.4) |
| Propane | −10.8 | (−7.8) | −7.4 | −20.0 (−21.5) | −13.9 (−15.2) | −10.5 (−12.0) | −17.3 (−18.8) | −13.2 (−14.4) | −8.9 (−10.4) | −16.8 (−18.3) | −13.0 (−14.3) | −8.6 (−10.1) |
| n-Butane | −14.2 | (−10.3) | −9.4 | −23.8 (−25.5) | −15.7 (−17.5) | −12.5 (−14.3) | −20.4 (−22.1) | −14.9 (−16.7) | −10.9 (−12.6) | −19.9 (−21.6) | −14.8 (−16.7) | −10.4 (−12.1) |
| n-Pentane | −17.1 | (−12.8) | −11.0 | −28.1 (−29.6) | −18.6 (−19.8) | −15.6 (−17.6) | −23.7 (−25.3) | −16.9 (−18.1) | −13.8 (−15.8) | −23.1 (−24.7) | −16.7 (−17.9) | −13.2 (−15.2) |
| n-Hexane | −20.4 | −15.3 | −12.7 | −32.7 (−34.4) | −18.0 (−19.8) | −18.8 (−20.8) | −27.0 (−28.7) | −15.8 (−17.6) | −16.1 (−18.1) | −26.4 (−28.1) | −15.5 (−17.3) | −15.4 (−17.5) |
| n-Heptane | −22.5 | (−17.8) | −16.4 | −35.4 (−37.2) | −23.5 (−25.1) | −21.7 (−23.7) | −30.0 (−31.8) | −21.4 (−23.0) | −18.5 (−20.6) | −29.2 (−31.0) | −21.0 (−22.6) | −17.8 (−19.9) |
| n-Octane | −25.4 | (−20.3) | (−17.8) | −38.9 (−40.6) | −25.9 (−27.5) | −23.2 (−25.3) | −32.7 (−34.5) | −22.4 (−24.0) | −20.0 (−22.1) | −32.1 (−33.8) | −22.0 (−23.6) | −19.2 (−21.3) |
| Mean signed error | −9.6 (−11.1) | −4.9 (−6.3) | −4.2 (−5.9) | −5.8 (−7.3) | −3.3 (−4.8) | −2.2 (−3.9) | −5.3 (−6.8) | −3.1 (−4.5) | −1.7 (−3.4) | |||
| Mean unsigned error | 9.6 (11.1) | 4.9 (6.3) | 4.2 (5.9) | 5.8 (7.3) | 3.3 (4.8) | 2.2 (3.9) | 5.3 (6.8) | 3.1 (4.5) | 1.7 (3.4) | |||
Two trends are observed, both in the experimental and computational results. First, as the alkane chain length increases, the adsorption strength increases. When comparing the adsorption strengths with increasing alkane chain length for a given zeolite system, the slope of the computational and experimental results are similar, suggesting that the model does capture this effect. Adding polarization to the intermediate layers in this model was very important, as can be seen in the case of H-MFI. Without polarization H-MFI has a MUE of 9.6 kcal mol−1, which decreases to 5.8 and 5.3 kcal mol−1 for the 6-31G(d,p) and 6-311G(2df,p) basis sets. In the case of H-BEA, the MUE is equal to 3.1 kcal mol−1, while in H-FAU the MUE is 1.7 kcal mol−1. In the case of H-MFI, the error is somewhat larger than in the other two zeolites because of the smallest pore and the higher proximity of the adsorbate to the wall of the pore, which is treated with the UFF. As the pore size increases, the adsorbate does not interact as much with the UFF layer, and the error decreases. Nonetheless, the accuracy is quite satisfactory in all three cases, especially if one considers the uncertainty in the experimental enthalpies of adsorption.
Second, as the average pore size of the zeolite decreases, the adsorption strength of a given alkane increases, in qualitative agreement with the expectation for these zeolite systems.32–35 It is well established that binding enthalpy increases (in absolute value) as the pore size decreases and becomes more confining. For these three frameworks, the pore size varies in the order H-FAU > H-BEA > H-MFI,36 and thereby, the relative binding would be expected to follow the order H-FAU < H-BEA < H-MFI.
D. Relationship between proton affinity and enthalpy of adsorption
By making use of thermodynamic cycles, differences in the binding strengths of probe molecules in Brønsted acidic zeolites have been correlated with differences in the proton affinities (PA) of the probe molecules themselves. The thermodynamic cycle involves a state in which the acidic proton has been donated to the substrate molecule (see Fig. 5 inset). The result of this assumption is a linear correlation of unit slope between ΔHads and the PA. For any given zeolite, this is not a strong correlation, because not all substrate molecules are strong enough bases to accept the acidic proton. Thus, while the correlation might be useful in assessing relative binding strengths of strong bases or of a strong base and of a weak one, the PA of the adsorbate is not a reliable descriptor of the relative binding strength of molecules with weak basicity.28,48,49,55–57 Adsorbates with gas phase PA greater than ∼200 kcal mol−1 demonstrate the ability to abstract the proton from the zeolite and form an ion pair structure. In this range, the thermodynamic cycle holds and we see that both the experimental and computational data sets correlate well with the proposed model in which the PA of an adsorbate correlates linearly to its adsorption strength in a zeolite with unit slope.48,49,55–57 In this range of PA, the quantum intermediate layer has contributed to the electrostatic stabilization of the ion pair. At low PA of less than ∼180 kcal mol−1, the adsorbate is unable to abstract the proton of the active site and binding is primarily affected through dispersion and hydrogen bonding interactions in the pore. The thermodynamic cycle no longer holds, and we see that both experimental and computational models deviate from the predicted linear trend of unit slope, as expected.48,49,55–57 In these cases, the use of a theory in the intermediate layer able to account for dispersion forces between the adsorbate and zeolite walls (M06-2X and ωB97x-D), as well as the benefit of adding polarization to the basis set to allow directional interaction between the adsorbate and pore wall can be seen in the accuracy of these models.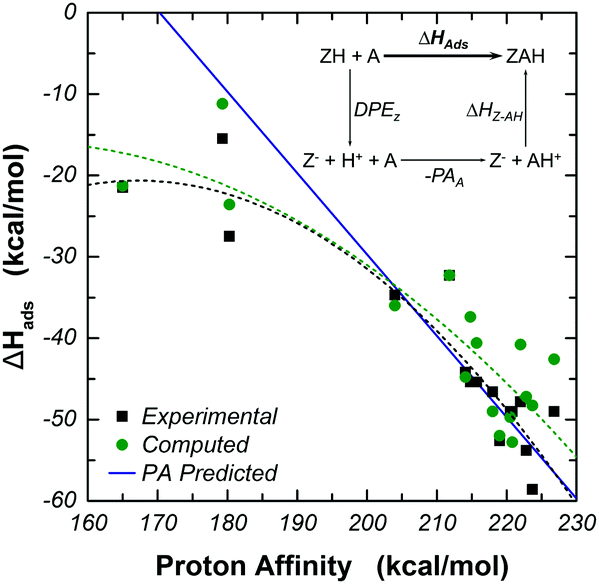 | ||
| Fig. 5 Correlation between gas phase proton affinity and ΔHads in H-MFI zeolites. Calculations performed using the 17T:61T:59T model of H-MFI at the M06-2X/6-311G(2df,p):M06-2X/6-31G(d,p):UFF//M06-2X/6-311G(2df,p):M06-2X/3-21G:UFF theory level with BSSE correction. In the catalytic cycle, DPEZ is the deprotonation energy of the zeolite, PAA is the gas phase proton affinity of the adsorbate, and ΔHZ–AH is the interaction energy between the deprotonated zeolite and the protonated adsorbate. Dotted lines show second order fits of the experimental and computational data to guide the eye when compared to the blue line, showing the predicted binding strength using the gas phase proton affinity (PA) correlation with a slope of 1.48,49,55–57 | ||
IV. Discussion
Although detailed quantitative comparisons with experiment can at times prove quite challenging, as the experimental values are a statistical average over a distribution of acid sites, adsorbate-site geometries and adsorbate configurations whereas the calculated values refer to optimized binding geometries at a particular acid site and adsorbate configuration, comparisons between different computational strategies can, on the other hand, prove quite instructive. The handling of long-ranged electrostatic interactions by embedded cluster QM/MM strategies has been a source of concern to critics of this methodology. Below, we compare our results with published periodic-DFT calculations and find very good agreement, which should allay some of the reservations about hybrid QM/MM calculations.For the adsorption of n-C3 to n-C6 alkanes at the T12 site of the zigzag channel of H-ZSM-5, Tranca et al.58 have reported binding energies of −11.5, −15.1, −19.1 and −22.0 kcal mol−1, respectively, using periodic-DFT calculations at the PBE-D theory level. At the same theory level, Chiu et al.59 have more recently reported binding energies of −16.3, −19.9, −21.8 and −24.9 kcal mol−1, respectively, which are very close to those calculated by us (−16.8, −19.9, −23.1 and −26.4 kcal mol−1, respectively) with a 17T:23T:97T three-layer ONIOM model at the M06-2X/6-311G(2df,p):M06-2X/6-311G(2df,p):UFF//M06-2X/6-311G(2df,p):M062X/3-21G:UFF (Table 8). Chiu et al. have also calculated binding energies with the semilocal exchange–correlation functional vdW-DF2, which is designed to take into account dispersion interactions in a non-empirical way.11 The reported values of −16.0, −20.8, −23.2 and −28.2 kcal mol−1, respectively, are very close to PBE-D, and to those calculated by us.
Chiu et al. have also reported binding energies of water and methanol in H-ZSM-5 using periodic-DFT. For water, the PBE-D value of −22.2 kcal mol−1 fares very well upon comparison with the experimental value of −21.5 kcal mol−1 and is close to our value of −21.3 kcal mol−1. On the other hand, the vdW-DF2 value of −17.2 kcal mol−1 somewhat underestimates water adsorption. For methanol, the PBE-D value of −24.4 is close to our value of −23.6 kcal mol−1, whereas the vdW-DF2 functional somewhat underestimates binding, predicting an energy of −21.3 kcal mol−1.
Even though more systematic benchmarking is required to fully access strengths and weaknesses in the performance of QM/QM/MM embedded cluster ONIOM models for predicting adsorption in zeolites, they do not seem to under-perform relative to periodic-DFT calculations.
In Table S1 of the ESI,† we show the calculated adsorption entropies of n-C1 to n-C8 alkanes in H-MFI, assuming a molecular surface area of 200 × 600 (pm × pm)60 for 2-dimensional free translations of the guest molecule. In the temperature range of 25 to 200 °C, the computed values vary little with temperature. Upon comparison with experimental values reported by De Moor et al. for n-C3 to n-C6 alkanes,60 the computed values systematically overestimate the entropic losses, but they are, nevertheless, in good agreement with experiment, with an error of no more than 10 J mol−1 K−1. Despite the good agreement with experiment, we are of the opinion that the entropic error that is associated with the notoriously inaccurate low-frequencies in the harmonic approximation cannot be fully addressed by the qRRHO approximation. Sauer and co-workers,61–63 in recent work that was inspired by earlier work by Njegic and Gordon,64 have convincingly argued and demonstrated that correcting for the error in the adsorption entropy due to the soft modes requires not only consideration of the strong anharmonic character of these modes, but also that the diagonalization of the Hessian be performed in the curvilinear space of the intrinsic coordinates of the system. This approach is not entirely without problems, as it is not always easy to find appropriate intrinsic coordinates, nor is it entirely computationally inexpensive for large systems (factor of 10 computational overhead)63 as the anaharmonicity calculation requires a number of single-point energy calculations to sample the potential energy surface along a normal mode in curvilinear space.
V. Conclusions
We have explored mechanically embedded three-layer QM/QM/MM ONIOM models for computational studies of binding in metal-substituted zeolites. In all the models considered, the high-level-theory layer consists of the adsorbate molecule and of the framework atoms within the first two coordination spheres of the metal atom and is treated at the M06-2X/6-311G(2df,p) level. For simplicity and flexibility in partitioning the system into QM and MM regions in a number of ways without worrying about the net charge of the MM region not being zero, the outer, low-level-theory layer is treated with the UFF. We have modelled the intermediate-level layer quantum mechanically and investigated the performance of HF theory and of three DFT functionals, B3LYP, M06-2X and ωB97x-D, for different layer sizes and various basis sets, with and without BSSE corrections. We have studied the binding of sixteen probe molecules with a broad range of basicities in H-MFI and compared the computed adsorption enthalpies with published experimental data. We have demonstrated that HF and B3LYP are inadequate for the description of the interactions between the probe molecules and the framework surrounding the metal site of the zeolite, on account of being unable to capture dispersion forces. Both M06-2X and ωB97x-D on average converge within ca. 10% of the experimental values, at the M06-2X/6-311G(2df,p):M06-2X/6-31G(d,p):UFF//M06-2X/6-311G(2df,p):M06-2X/3-21:UFF and ωB97x-D/6-311G(2df,p):ωB97x-D/6-31G(d,p):UFF//ωB97x-D/6-311G(2df,p):ωB97x-D/3-21:UFF theory levels, respectively. In particular, the three-layer ONIOM models perform significantly better than the two-layer model M06-2X/6-311G(2df,p):UFF in stabilizing ion-pair structures of adsorbate molecules with high proton affinities. The mean errors over the subsets of the pyridines and of the amines drop significantly showing the importance of the intermediate quantum mechanical layer for the stabilization of ion-pairs. We have further computed the binding enthalpies of n-alkanes (C1–C8) in H-MFI, H-BEA and H-FAU at the M06-2X/6-311G(2df,p):M06-2X/6-31G(d,p):UFF//M06-2X/6-311G(2df,p):M06-2X/3-21:UFF theory level and found very good agreement with experiment, with mean unsigned errors of 5.3, 3.1 and 1.7 kcal mol−1, respectively, demonstrating transferability of the model across zeolite frameworks and classes of molecules. The transferability is further demonstrated by the good agreement between computed entropies of adsorption and experimental values reported by De Moor et al. for the adsorption of n-C3 to n-C6 alkanes in H-ZSM-5.Acknowledgements
We acknowledge support from the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award number DE-SC0001004. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399–434 CrossRef CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- M. Guisnet and P. Magnoux, Appl. Catal., 1989, 54, 1–27 CrossRef CAS.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- A. Simperler, R. G. Bell, M. D. Foster, A. E. Gray, D. W. Lewis and M. W. Anderson, J. Phys. Chem. B, 2004, 108, 7152–7161 CrossRef CAS.
- A. Miyamoto, Y. Kobayashi, M. Elanany, H. Tsuboi, M. Koyama, A. Endou, H. Takaba, M. Kubo, C. A. D. Carpio and P. Selvam, Microporous Mesoporous Mater., 2007, 101, 324–333 CrossRef CAS.
- L. Liu, L. Zhao and H. Sun, J. Phys. Chem. C, 2009, 113, 16051–16057 CAS.
- A. Janda, B. Vlaisavljevich, L.-C. Lin, S. M. Sharada, B. Smit, M. Head-Gordon and A. T. Bell, J. Phys. Chem. C, 2015, 119, 10427–10438 CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- É. D. M. Kyuho Lee, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B, 2010, 82, 081101(R) CrossRef.
- F. Maseras and K. Morokuma, J. Comput. Chem., 1995, 16, 1170–1179 CrossRef CAS.
- H. Lin and D. G. Truhlar, Theor. Chem. Acc., 2007, 117, 185–199 CrossRef CAS.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- C. Tuma and J. Sauer, Phys. Chem. Chem. Phys., 2006, 8, 3955–3965 RSC.
- C. Tuma and J. Sauer, Chem. Phys. Lett., 2004, 387, 388–394 CrossRef CAS.
- E. M. Evleth, E. Kassab, H. Jessri, M. Allavena, L. Montero and L. R. Sierra, J. Phys. Chem., 1996, 100, 11368–11374 CrossRef CAS.
- T. Fujino, M. Kashitani, J. N. Kondo, K. Domen, C. Hirose, M. Ishida, F. Goto and F. Wakabayashi, J. Phys. Chem., 1996, 100, 11649–11653 CrossRef CAS.
- M. Krossner and J. Sauer, J. Phys. Chem., 1996, 100, 6199–6211 CrossRef CAS.
- W. Panjan and J. Limtrakul, J. Mol. Struct., 2003, 654, 35–45 CrossRef CAS.
- S. Kasuriya, S. Namuangruk, P. Treesukol, M. Tirtowidjojo and J. Limtrakul, J. Catal., 2003, 219, 320–328 CrossRef CAS.
- C. Raksakoon and J. Limtrakul, THEOCHEM, 2003, 631, 147–156 CrossRef CAS.
- S. Namuangruk, P. Pantu and J. Limtrakul, J. Catal., 2004, 225, 523–530 CrossRef CAS.
- X. Solans-Monfort, M. Sodupe, O. Mó, M. Yáñez and J. Elguero, J. Phys. Chem. B, 2005, 109, 19301–19308 CrossRef CAS PubMed.
- X. Solans-Monfort, M. Sodupe, V. Branchadell, J. Sauer, R. Orlando and P. Ugliengo, J. Phys. Chem. B, 2005, 109, 3539–3545 CrossRef CAS PubMed.
- R. Rungsirisakun, B. Jansang, P. Pantu and J. Limtrakul, J. Mol. Struct., 2005, 733, 239–246 CrossRef CAS.
- S. Namuangruk, D. Tantanak and J. Limtrakul, J. Mol. Catal. A: Chem., 2006, 256, 113–121 CrossRef CAS.
- B. Boekfa, S. Choomwattana, P. Khongpracha and J. Limtrakul, Langmuir, 2009, 25, 12990–12999 CrossRef CAS PubMed.
- P. M. Zimmerman, M. Head-Gordon and A. T. Bell, J. Chem. Theory Comput., 2011, 7, 1695–1703 CrossRef CAS PubMed.
- S. M. Sharada, P. M. Zimmerman, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2013, 117, 12600–12611 Search PubMed.
- Y.-P. Li, J. Gomes, S. M. Sharada, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2015, 119, 1840–1850 CAS.
- T. F. Narbeshuber, H. Vinek and J. A. Lercher, J. Catal., 1995, 157, 388–395 CrossRef CAS.
- F. Eder and J. A. Lercher, Zeolites, 1997, 18, 75–81 CrossRef CAS.
- F. Eder, M. Stockenhuber and J. A. Lercher, J. Phys. Chem. B, 1997, 101, 5414–5419 CrossRef CAS.
- S. Kotrel, M. P. Rosynek and J. H. Lunsford, J. Phys. Chem. B, 1999, 103, 818–824 CrossRef CAS.
- Ch. Baerlocher and L. B. McCusker, Database of Zeolite Structures, International Zeolite Association, 1996, http://www.iza-structure.org/databases/, accessed 9 February 2015 Search PubMed.
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS.
- S. Dapprich, I. Komaromi, K. S. Byun, K. Morokuma and M. J. Frisch, THEOCHEM, 1999, 461, 1–21 CrossRef.
- T. Vreven, K. Morokuma, O. Farkas, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 2003, 24, 760–769 CrossRef CAS PubMed.
- T. Vreven, K. S. Byun, I. Komaromi, S. Dapprich, J. A. Montgomery, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580–1591 CrossRef CAS PubMed.
- S. Namuangruka, D. Tantanak and J. Limtrakul, J. Mol. Catal. A: Chem., 2006, 256, 113–121 CrossRef.
- A. Ison and R. J. Gorte, J. Catal., 1984, 89, 150–158 CrossRef CAS.
- D. J. Parrillo and R. J. Gorte, J. Phys. Chem., 1993, 97, 8786–8792 CrossRef CAS.
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615–635 CrossRef CAS.
- C. C. Lee, R. J. Gorte and W. E. Farneth, J. Phys. Chem. B, 1997, 101, 3811–3817 CrossRef CAS.
- S. Savitz, A. L. Myers and R. J. Gorte, J. Phys. Chem. B, 1999, 103, 3687–3690 CrossRef CAS.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. C, 2008, 112, 6860–6868 CAS.
- J. T. Fermann, T. Moniz, O. Kiowski, T. J. McIntire, S. M. Auerbach, T. Vreven and M. J. Frisch, J. Chem. Theory Comput., 2005, 1, 1232–1239 CrossRef CAS PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- D. J. Parrillo, R. J. Gorte and W. E. Farneth, J. Am. Chem. Soc., 1993, 115, 12441–12445 CrossRef CAS.
- C. Lee, D. J. Parrillo, R. J. Gorte and W. E. Farneth, J. Am. Chem. Soc., 1995, 118, 3262–3268 CrossRef.
- A. Jentys, R. R. Mukti, J. A. Tanaka and J. A. Lercher, Microporous Mesoporous Mater., 2006, 90, 284–292 CrossRef CAS.
- D. C. Tranca, N. Hansen, J. A. Swisher, B. Smit and F. J. Keil, J. Phys. Chem. C, 2012, 116, 23408–23417 CAS.
- C.-c. Chiu, G. N. Vayssilov, A. Genest, A. Borgna and N. Rosch, J. Comput. Chem., 2014, 35, 809–819 CrossRef CAS PubMed.
- B. A. D. Moor, M.-F. Reyniers, O. C. Gobin, J. A. Lercher and G. B. Marin, J. Phys. Chem. C, 2011, 115, 1204–1219 Search PubMed.
- G. Piccini and J. Sauer, J. Chem. Theory Comput., 2013, 9, 5038–5045 CrossRef CAS PubMed.
- G. Piccini and J. Sauer, J. Chem. Theory Comput., 2014, 10, 2479–2487 CrossRef CAS PubMed.
- G. Piccini, M. Alessio, J. Sauer, Y. Zhi, Y. Liu, R. Kolvenbach, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2015, 119, 6128–6137 CAS.
- B. Njegic and M. S. Gordon, J. Chem. Phys., 2006, 125, 224102 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp03266d |
| This journal is © the Owner Societies 2016 |