
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic study on the active site of a SiO2 supported niobia catalyst used for the gas-phase Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam
M. M.
Maronna†
*ab,
E. C.
Kruissink
a,
R. F.
Parton
a,
F.
Soulimani
c,
B. M.
Weckhuysen
c and
W. F.
Hoelderich†
d
aDSM Chemical Technology R&D B.V., Urmonderbaan 22, 6167 RD Geleen, The Netherlands
bTU Dortmund, Lehrstuhl für Technische Chemie B, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: marius.maronna@tu-dortmund.de; Tel: +49-2403-8094146
cUtrecht University, Inorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
dTCHK Consultancy, 67227 Frankenthal, Germany
First published on 27th July 2016
Abstract
NbOx/SiO2 with a very high catalytic activity for the gas-phase Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam, was investigated by different spectroscopic methods in order to obtain new insights in the formation and nature of the active sites. FT-IR spectroscopy in combination with pyridine adsorption measurements revealed that the catalyst material contains Lewis-acidic sites, most probably related to the Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of isolated tetrahedral NbO4 surface species, whereas no Brønsted-acidic sites were observed. Results from in situ Raman and complementary FT-IR measurements strongly suggest that Brønsted-acidic Nb–OH sites can be generated from NbO groups by reaction with ethanol. This is in agreement with the observation that ethanol is essential for obtaining a very good catalyst performance. However, the Brønsted-acidic sites can be detected in significant amounts in particular in the presence of a Lewis-base, e.g. pyridine, most probably because the formation and/or the stability of these Brønsted-acidic sites are enhanced by a basic molecule. Assuming that cyclohexanone oxime, being a base, can play a similar role as pyridine, we propose on the basis of the spectroscopic findings obtained in this work and our kinetic results published recently, a reaction scheme for the formation of the active site at the NbO group as well as for the recovery of the NbO site during the final stage of the gas-phase Beckmann rearrangement.
O groups of isolated tetrahedral NbO4 surface species, whereas no Brønsted-acidic sites were observed. Results from in situ Raman and complementary FT-IR measurements strongly suggest that Brønsted-acidic Nb–OH sites can be generated from NbO groups by reaction with ethanol. This is in agreement with the observation that ethanol is essential for obtaining a very good catalyst performance. However, the Brønsted-acidic sites can be detected in significant amounts in particular in the presence of a Lewis-base, e.g. pyridine, most probably because the formation and/or the stability of these Brønsted-acidic sites are enhanced by a basic molecule. Assuming that cyclohexanone oxime, being a base, can play a similar role as pyridine, we propose on the basis of the spectroscopic findings obtained in this work and our kinetic results published recently, a reaction scheme for the formation of the active site at the NbO group as well as for the recovery of the NbO site during the final stage of the gas-phase Beckmann rearrangement.
1. Introduction
The Beckmann rearrangement of cyclohexanone oxime is the final step of the industrial synthesis of ε-caprolactam, the monomer for polyamide 6. In the classical process this reaction is carried out in the liquid phase, homogeneously catalysed by oleum.1 Since the 1940's gas-phase processes have been investigated as an alternative, utilizing pure and doped amorphous oxides, zeolites or MMS materials as heterogeneous catalysts. The main objective of all these research efforts is to avoid the formation of a substantial amount of ammonium sulphate as co-product and the use of hazardous and corrosive liquids. After important developments concerning the zeolite catalysed gas-phase Beckmann rearrangement (GPBR) reported by the Hoelderich group,1–5 Sumitomo Chemicals Co. Ltd (Japan) started-up the first industrial GPBR plant in 2003. Silicalite is used as heterogeneous catalyst in a fluidized bed reactor system with continuous regeneration in a second fluidized bed with an ε-caprolactam capacity of about 60 kta.6–8Ushikubo et al. were the first to publish the use of bulk tantalum and niobium oxides as catalysts for the GPBR.9,10 Later on, catalyst materials containing niobia (e.g. Nb-MCM-41, Nb-SBA-15, Nb-beta and NbOx/SiO2) were investigated by Hoelderich et al.11,12 In particular the niobia on silica catalyst material, i.e. NbOx/SiO2, displayed a very promising catalytic performance in the GPBR.13–15
In a previous publication we have reported on the modification and optimization of the NbOx/SiO2 catalyst material.16 We found that the optimized catalytic material provides a very high catalytic performance in the GPBR, and that the performance, observed in an easily manageable and cheap fixed bed reactor, is in terms of productivity and ε-caprolactam selectivity in a range comparable with that of the silicalite catalyst currently used by Sumitomo Chemicals Co. Ltd. Additionally, it was demonstrated that the presence of niobia on the silica support is essential for a high catalytic performance. Furthermore, we concluded from Raman spectroscopy results that, in active and selective catalysts, a substantial fraction of the niobia is present as isolated tetrahedral NbO4 surface species containing a NbO group. These findings lead to the suggestion that isolated NbO4 sites play an important role in the catalysis of the GPBR. In previous research on the kinetics of the GPBR over the NbOx/SiO2 material we showed that the presence of ethanol (or higher linear alcohols) in the reactant gas-phase is essential for a high catalytic performance.17 The obtained kinetic data did not fit to a Langmuir–Hinshelwood mechanism (equilibrium adsorption of cyclohexanone oxime on the active site) but in contrast it could be described very well by assuming that an adsorption reaction occurs (where no equilibrium, but a steady state is attained) involving cyclohexanone oxime, the alcohol component and the isolated tetrahedral mono-oxo NbO4 surface species. This adsorption reaction as first step is then followed by the Beckmann rearrangement reaction.
The objective of the present work is to obtain more information on the formation and nature of the active site. In literature, it is generally accepted that Brønsted-acidity is required for the Beckmann rearrangement. We mentioned above the role of isolated NbO4 surface sites containing a NbO group as well as the possible adsorption reaction between this site, ethanol and cyclohexanone oxime. Interestingly, work published by Iwasawa et al. suggests that (potentially) Brønsted-acidic sites can be formed by the reaction of a NbO group with ethanol. These authors proposed, in a publication on ethanol dehydrogenation over a silica supported niobia catalyst that a NbO group reacts dissociatively with an ethanol molecule under formation of an ethoxy- and a hydroxyl group both attached to the Nb central-atom.18,19 In the work cited, this reaction was proposed for a NbO group as part of a dioxo Nb(O)2 surface species, but possibly the same reaction could occur for a NbO group as part of a mono-oxo surface species. Thus, on the basis of the kinetic observations and literature data presented above, we hypothesized that the required Brønsted-acidity could be provided in our case by a reaction of a NbO group with ethanol, as shown in Fig. 1. In the following we will discuss spectroscopic results to gain information on the validity of this hypothesis and more general, on the formation and nature of the active site(s). The techniques applied are Raman spectroscopy and FT-IR measurements in combination with pyridine adsorption, ethanol adsorption and combined pyridine–ethanol adsorption. Finally, on the basis of this information a reaction scheme for the formation of the active site at the NbO group will be proposed as well as for the recovery of the NbO group during the final stage of the Beckmann rearrangement.
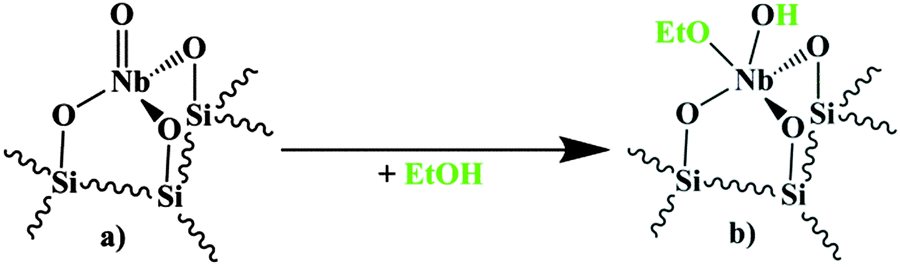 | ||
| Fig. 1 Reaction of the tetrahedral mono-oxo NbO4 surface species (a) with ethanol under formation of a potentially Brønsted-acidic surface complex (b), based on the mechanism proposed by Iwasawa et al.18,19 | ||
2. Experimental
2.1. Catalyst material preparation
As support material, commercially available silica (DAVICAT SI 1151, Grace Davison) was used. After crushing and sieving to obtain the desired fraction, the silica was dried at 150 °C for 3 h and subsequently loaded via the incipient wetness impregnation method with a solution of Nb(OEt)5 (Sigma Aldrich, 98%) in ethanol (VWR, 99%, dried). After short intensive mixing in a rotary evaporator, resulting in a homogeneous wetting of the particles, vacuum was applied followed by ventilation (repeated three times) to push the precursor solution deep into the pore system of the carrier material. Then, the loaded silica was calcined in static air at a predetermined temperature (range: 400 °C ≤ T ≤ 800 °C) for 15 h (heating rate from room temperature to target temperature 2 °C min−1) and finally cooled down slowly to room temperature again. The textural properties of the catalyst materials calcined at different temperatures and results of elemental analysis of the silica support are given in Tables 1 and 2, respectively.| Element | Content/wt% |
|---|---|
| Na | 0.064 |
| Al | 0.009 |
| Fe | 0.003 |
| K | 0.003 |
| Ca | 0.038 |
| Mg | 0.010 |
| Mo | <7 × 10−5 |
| Ni | <7 × 10−5 |
2.2. Catalyst material characterization
Raman spectra were recorded with a Kaiser RXN1 spectrometer using a green laser (λ = 532 nm, P = 100 mW, 5 s exposure time, 20 accumulations) and a noncontact 5.5′′ objective for laser focusing and collecting the scattered radiation (CCD detector). The catalyst was located in a quartz glass reactor with connections to direct the flow of gases through the reactor. The spectra were recorded at 400 °C reactor temperature under flowing nitrogen (pure or saturated with ethanol or water). The flow of pure nitrogen could be switched to ethanol or water saturated nitrogen (saturation at atmospheric pressure and 27 °C room temperature) by redirecting the nitrogen flow to a bubbler, filled with dried ethanol (VWR, 99%) or deionized water, respectively.FT-IR spectra were recorded with a Perkin Elmer Spectrum One spectrometer (resolution: 1.0 cm−1, 5 accumulations, FR-DTGS detector) including a stainless steel measuring cell with KBr windows and connections to direct the flow of gases through the cell. The materials were pressed into cyclic self-supporting wafers (∼10 mg material pressed at 8 t and room temperature, Ø = 8 mm) and fixed into the holder of the measuring cell. The catalyst wafers were heated at 200 °C for 2 h under flowing nitrogen atmosphere to exclude humidity. Blank spectra were recorded under flowing nitrogen atmosphere (40 ml min−1) for 200, 250, 300 and 400 °C cell temperature (equilibration time 5 min). These blank spectra were later used to produce the difference spectra of the pyridine- and combined pyridine–ethanol adsorption experiments. Then, the system was cooled down to 200 °C again. In order to carry out the adsorption measurements, the constant nitrogen flow was directed to a bubbler, filled with pyridine (spectroscopically grade) or dried ethanol (VWR, 99%) or an ethanol–pyridine mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio), to saturate the nitrogen flow at 21 °C room temperature at atmospheric pressure. After 5 min to establish the adsorption–desorption equilibrium on the material, a spectrum was recorded and the cell was heated up to the next higher temperature while constantly flushing the cell with the saturated nitrogen atmosphere.
:1 molar ratio), to saturate the nitrogen flow at 21 °C room temperature at atmospheric pressure. After 5 min to establish the adsorption–desorption equilibrium on the material, a spectrum was recorded and the cell was heated up to the next higher temperature while constantly flushing the cell with the saturated nitrogen atmosphere.
Nitrogen adsorption isotherms were measured with a Micromeritics Gemini VII 2390 Surface Analyser. The material samples were evacuated at 300 °C for 2 h and cooled down to −196 °C under vacuum. To obtain the adsorption–desorption isotherms, the nitrogen pressure was varied in the relative pressure range (P/P0) between 0.01 and 0.96. Brunauer–Emmett–Teller (BET) surface areas were calculated from the part of the adsorption isotherm between 0.05 and 0.25P/P0. Mesopore diameters were calculated from the desorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) method.
The elemental analyses to determine the niobia loading of the catalysts were executed using inductively coupled plasma optical emission spectroscopy (ICP-OES) utilizing a Thermo Scientific iCAP6500 system. In order to perform the measurements the catalysts (∼0.2 wt%) were dissolved in an aqueous solution of hydrofluoric acid and nitric acid.
3. Results and discussion
3.1. Raman spectroscopy: NbOx/SiO2 interacting with ethanol- or water-saturated nitrogen atmosphere
Fig. 2 presents the Raman spectra of catalyst material B (see Table 1) under flowing nitrogen atmosphere (Fig. 2a) and under flowing ethanol saturated nitrogen atmosphere (Fig. 2b), both at 400 °C. In the case of a pure nitrogen atmosphere a Raman band is clearly observed at 980 cm−1. According to the literature this band can be assigned to the NbO stretching vibration of isolated tetrahedral mono-oxo NbO4 surface species.16,20,21 Additionally, Raman bands of network vibrations of the silica support at 1045 and 1160 cm−1 (transverse and longitudinal optical stretch respectively) are present.16,22
 | ||
| Fig. 2 In situ Raman spectra of catalyst material B at 400 °C cell temperature under (a) nitrogen flow (after 3 h) and (b) ethanol saturated nitrogen flow (after 10 h). | ||
Upon switching from pure nitrogen to ethanol saturated nitrogen atmosphere, first strong fluorescence was observed, but fortunately the intensity decreased with time. Spectrum (b) in Fig. 2 was recorded after 10 h. It can be seen that the NbO vibrational band at 980 cm−1 disappeared nearly completely, only a weak shoulder being left, whereas the silica network bands remained. This observation suggests that ethanol could have reacted with the NbO group or, at least that the NbO vibration was suppressed due to the adsorption of ethanol molecules on the NbO group. Adsorption could disturb the NbO vibration and thus affect the Raman intensity.
Fig. 3a presents the Raman spectrum of catalyst material B (see Table 1) under flowing pure nitrogen atmosphere. The NbO vibrational band at 980 cm−1 is clearly observable, besides the two silica network bands. Upon switching the atmosphere to water saturated nitrogen no fluorescence appeared, and after 3 h equilibration time the spectrum was recorded shown as Fig. 3b. Obviously, no significant difference is observed between spectrum (a) and spectrum (b) in Fig. 3. Apparently, the NbO groups in the catalyst under study do not react with water and thus cannot form Brønsted-acidic sites by reaction with water under the given conditions. This finding is in agreement with the stability against water of Lewis-acid sites in niobic acid (also containing NbO groups), reported by Nakajima et al.23
 | ||
| Fig. 3 In situ Raman spectra of catalyst material B at 400 °C cell temperature under (a) nitrogen flow (after 3 h) and (b) water saturated nitrogen flow (after 3 h). | ||
3.2. FT-IR spectroscopy: NbOx/SiO2 and SiO2 interacting with pyridine, ethanol and a combination of pyridine and ethanol
In order to explore the acidic properties of NbOx/SiO2 and to verify the hypothesis mentioned above about the formation of Brønsted-acidic sites, FT-IR spectra of adsorbed pyridine were measured before and after treatment with ethanol, and in addition (co-) adsorption of ethanol and pyridine was investigated. In the following, the results of these FT-IR experiments will be presented. Measurements of the unloaded silica support were carried out for comparison. | ||
| Fig. 4 Pyridine adsorption FT-IR difference spectra under constant pyridine saturated nitrogen flow at subsequently increasing cell temperatures (equilibration time 5 min) for (a) NbOx/SiO2 material B and (b) pure silica carrier (calcined at 600 °C). | ||
| Pyridine species//νCC(N) vibration type//band intensity24 | Absorbance maximum/cm−1 |
|---|---|
| a Pyridine coordinated to Lewis-acid site. b Pyridine protonated by Brønsted-acidic sites. c H-bonded pyridine to H-bonded and isolated silanol groups. | |
| LPya | |
| 8a/strong | 1600–1633,24,27 1610,29 1620,25,28 1606,30 160231 |
| 8b/variable | 1580,24 1577,25,28–30 157231 |
| 19a/variable | 1488–1503,24 1490,25,28,29 148431 |
| 19b/very strong | 1445–1465,24,27 1446,29,30 1450,25 144231 |
| BPyb | |
| 8a/strong | 1630–1640,27 1638,25 1635,28,31 164024,29 |
| 8b/strong | 1620,24,25 1578,31 157629 |
| 19a/very strong | 1485–1500,24 1490,25,28,29 148231 |
| 19b/strong | 1500–1540,27 1545,25,30 1538,29,31 1540,24 153130 |
| HPyc | |
| 8a | 161425 |
| 8b/strong | 1580–1600,24,27 1593,25 1595,26 159830 |
| 19a/weak | 1485–1490,24 149025 |
| 19b/very strong | 1440–1447,24,27 1438,25 144626,30 |
The pyridine IR adsorption spectrum of the niobia loaded silica (Fig. 4a) displays two bands with high intensity, at 1444 cm−1 and 1596 cm−1, in agreement with the IR spectrum published by Datka et al.32 for pyridine adsorbed on 2 wt% Nb2O5/SiO2 (bands at around 1450 and 1600 cm−1, respectively). These bands can be assigned to pyridine bonded to Lewis-acidic sites, as is also the case for the less intense bands at 1483 cm−1 and 1576 cm−1 (shoulder, see Table 3).
Considering the pyridine adsorption spectrum of the silica support (Fig. 4b) the highest absorption intensity is observed for a doublet at 1444 and 1448 cm−1.
For increasing cell temperature, the intensity of the 1444 cm−1 band decreases much faster than the intensity of the band at 1448 cm−1, resulting in a decrease of the ratio of these intensities from larger than 1 to smaller than 1. Apparently, the two bands correspond to different species of adsorbed pyridine, the adsorption being much weaker for the 1444 cm−1 band than for the other. Looking into literature (see Table 3), it seems obvious to identify the weakest adsorbed pyridine (1444 cm−1) as pyridine H-bonded to surface silanol groups, while the band at 1448 cm−1 then can be ascribed to pyridine relatively more strongly bonded to a Lewis-acidic site.
According to the same reasoning, the intense band at 1596 cm−1, displaying a strongly decreasing intensity for increasing cell temperature, can be assigned to H-bonded pyridine and the band at 1607 cm−1 to Lewis-acidic sites.
Apparently, the unloaded silica support contains some Lewis-acidic sites (main bands at 1448 and 1607 cm−1). Since this is not expected for pure silica, the detected Lewis-acidity most probably originates from incorporated Al and/or Fe in the silica, in agreement with the detected metal impurities listed in Table 2. The elemental analysis also detected Na-and Ca-impurities (see Table 2) which is not unusual for commercially produced silicas. Most probably the main fraction of the alkali- and earth alkali-ions will not be present at the surface but in the bulk of the silica since a high oxygen coordination is energetically most favourable. The fraction present close to the surface might give rise to slightly Lewis-basic sites. To the best of our knowledge no effects have been reported in literature of such weakly basic sites neither on pyridine FT-IR spectra nor on the selectivity of the Beckmann rearrangement. The exposure of incompletely coordinated Na- and/or Ca-ions directly at the surface (as is observed e.g. for zeolite Y) is very improbable for the silica and the catalyst materials investigated in this work. If present, such ions would promote the formation of nitriles.33 However, as published previously, nitrile selectivity of our catalyst amounts only to about 0.8%, and was ascribed to the presence of minor concentrations of isolated silanol groups.16,17 Summarizing, the contribution of Na- and Ca-ions to the catalytic performance is most probably absent or very minor.
The bands related to Lewis-acidic impurities in the silica at 1448 and 1607 cm−1 are also observed in the spectrum of the catalyst material (Fig. 4a), but always with much lower intensities than the niobia related bands. Note also that in the spectrum of the catalyst material (Fig. 4a) the intensity ratios of the bands at 1444 relative to 1448 cm−1 and 1596 relative to 1607 cm−1 remain larger than 1 with increasing cell temperature, in contrast to the observations for these bands in the silica spectrum (Fig. 4b). This supports the assignment of the 1444 cm−1 and 1596 cm−1 band to niobia related Lewis-acidity in case of the catalyst material, while these were ascribed to H-bonded pyridine for the unloaded support. It should be noted that in case of the catalyst material the bands at 1444 and 1596 cm−1 must be a superposition of the bands ascribed to H-bonded pyridine and to pyridine coordinated to Lewis-acidic sites related to niobia. From the intensity ratios follows that the contribution of the latter is dominating in particular at the highest temperatures. It should also be taken into account that all measurements were carried out with a pyridine saturated flow for all temperatures. This explains why H-bonded pyridine can still be observed at rather high cell temperatures.
Summarizing, the NbOx surface species provides Lewis-acidity, most probably related to the NbO structure unit of tetrahedral mono-oxo NbO4 surface species and possibly also to oligomeric niobia surface species,16 expressed in pyridine bands at 1444 (ν19b), 1483 (ν19a), 1576 (ν8b, shoulder) and 1596 cm−1 (ν8a).
O group with ethanol (see Introduction and Fig. 1). Iwasawa and co-workers applied the same technique to investigate the reaction between ethanol and a di-oxo Nb(O)2 surface species and claimed the formation of Nb–OH and Nb–OEt groups during reaction at 250 °C.18,19 A band observed at 3440 cm−1 was ascribed to a Nb–OH group (νOH) and a number of bands between 2979 and 2885 cm−1 (νCH) to an Nb–OEt group. Use of deuterated ethanol (EtOD) resulted in formation of Nb–OD, characterized by a νOD vibration with an absorbance maximum at 2577 cm−1.
Fig. 5 shows our FT-IR spectra of the catalyst material B and the silica support (see Table 1) during exposure to a flow of ethanol-saturated nitrogen and nitrogen without ethanol at 200 °C.
 | ||
| Fig. 5 FT-IR spectra of catalyst material B (a, b, e and f) and silica carrier (c and d) under nitrogen flow (a and c), EtOD saturated nitrogen flow (b, d and e) and EtOH saturated nitrogen flow (f) at 200 °C cell temperature after 3 h equilibration time. | ||
Deuterated ethanol (EtOD) was used in nearly all experiments. The reason for this was that a Nb–OD band probably can be detected more sensitively than a Nb–OH band besides the intense silanol bands, as a consequence of the much larger distance between the band positions of Nb–OD and silanols (band positions Nb–OD: 2577 cm−1, Nb–OH: 3440 cm−1, Si–OH: about 3680–3740 cm−1).
From Fig. 5b follows that, unfortunately, there is no indication for the presence of an Nb–OD group, since no band can be observed at about 2577 cm−1.
The following other effects of ethanol (EtOD) treatment can be derived from Fig. 5. The band for isolated silanol groups (3738 cm−1) was almost completely removed, both for the catalyst material and the silica support (compare Fig. 5 spectra a with b, and c with d, respectively).
Furthermore, two new band groups appear, the same for catalyst material and silica support (Fig. 5b and d, respectively), consisting of a two-membered band group with absorbance maxima at 2723 and 2753 cm−1 and a four-membered band group with absorbance maxima at 2881 (shoulder), 2907, 2938 and 2980 cm−1. The peak positions of the four-membered band group are very similar to those ascribed by the Iwasawa group to the Nb–OEt νCH-vibrations (published peak positions: 2885, 2900, 2935 and 2979 cm−1).18,19 However, we remarked already that these peaks are not exclusively observed in case of the NbOx/SiO2 catalyst material but also for the unloaded silica support. Thus, it seems obvious that the four-membered band group does not originate from Nb–OEt νCH-vibrations but from Si–OEt νCH-vibrations. In addition, the very substantial intensity loss of the isolated silanol band at 3738 cm−1 is in agreement with the occurrence of a reaction between isolated silanol groups and ethanol. Such an esterification reaction has been known since a long time for silanol groups of silica and alcohols.34
The two-membered band group with absorbance maxima at 2723 and 2753 cm−1, appearing when treating the catalyst and silica material with deuterated ethanol, can be assigned to D-bridged and isolated Si–OD groups. These deuterated silanol groups are formed by a H–D-exchange reaction between Si–OH and HOD. The partly deuterated water (HOD) is formed in the esterification reaction between an isolated silanol group (Si–OH) and the deuterated ethanol (EtOD) in the cell atmosphere, mentioned above. When applying an ethanol (non-deuterated) atmosphere this two-membered band group was not observed, as can be seen when comparing the spectra e (EtOD) and f (EtOH) in Fig. 5. Finally, it should be noted that no band for Nb–OH can be observed after catalyst treatment with ethanol (Fig. 5f, no band at 3440 cm−1).
In summary, we could demonstrate that ethanol is reacting with the silica support. However, there were no indications for the formation of Nb–OEt and Nb–OH (Nb–OD) groups according to the reaction presented in Fig. 1. Possibly, the mono-oxo NbO group is less reactive towards ethanol than the dioxo Nb(O)2 group, presented by Iwasawa et al.18,19 In Section 3.1 it was reported that the NbO Raman band at 980 cm−1 nearly completely disappears upon contact with an ethanol containing atmosphere. Since no reaction seems to occur, most probably this observation can be explained by adsorption of ethanol on the NbO group.
Fig. 6 displays pyridine adsorption FT-IR difference spectra measured after the pre-treatment of catalyst material B (see Table 1) with a flow of ethanol saturated nitrogen at 200 °C cell temperature. Comparing Fig. 6 with Fig. 4a (pyridine adsorption on untreated catalyst material B) all bands of pyridine adsorbed on Lewis-acidic sites are observed again at the same wavenumber positions. However, a remarkable difference is the increase of the band at 1488 cm−1, which before was a shoulder. According to Table 3, this band corresponds to the most intense band of pyridine bonded to a Brønsted-acidic site. Bands with lower intensity would be expected then at positions around 1540 cm−1 and 1630–1640 cm−1. Indeed, the presence of some very weak bands at these positions cannot be excluded, but cannot be established with certainty in view of the general noise observed in this part of the spectrum.
 | ||
| Fig. 6 FT-IR spectra of catalyst material B under pyridine saturated nitrogen flow at 200 °C cell temperature after (a) 5 min and (b) after 30 min. The catalyst material was pre-treated with ethanol saturated nitrogen flow at 200 °C in situ cell temperature for 30 min before switching to pyridine atmosphere. | ||
Summarizing, it cannot be excluded that Brønsted-acidic sites are formed during treatment in an ethanol-containing atmosphere, albeit at rather low concentrations since mainly the most intense pyridine adsorption band corresponding to such sites is observed. In Section 3.2.2 it was reported that no Nb–OH groups could be detected by FT-IR at the same reaction conditions in measurements without subsequent addition of pyridine. This suggests that Brønsted-acidic Nb–OH groups can be detected more sensitively by FT-IR after adsorption of pyridine. Another explanation could be that some residual adsorbed ethanol, still present after changing the gas atmosphere from ethanol to pyridine, reacts with the NbO groups catalysed by the Lewis-basic pyridine (see further below).
To explore the possibility of the formation of Brønsted-acidic sites further, in situ FT-IR adsorption experiments were carried out where the catalyst material was exposed to a gas containing both ethanol and pyridine.
Fig. 7 presents the FT-IR difference spectra of catalyst material B (see Table 1) during exposure to a gas flow containing both ethanol and pyridine at 200 °C for different contact times. When comparing Fig. 7 with Fig. 4a it is clear that new bands appear at 1488, 1540 (weak and broad) and 1633 cm−1. These bands can most probably be assigned to pyridine protonated by Brønsted-acidic sites (ν19a, ν19b and ν8a vibrations respectively, see Table 3). The intensity of the bands at 1488 and 1633 cm−1 increase significantly with contact time (see Fig. 7) which is not the case at all for the other bands observed. Furthermore the bands at 1444 and 1596 cm−1, assigned to NbO related Lewis-acidity, clearly decreased strongly in intensity. The remaining bands at 1448 and 1607 cm−1 belong to the Lewis-acidic impurities of the silica support. Those bands do not seem to be affected by the addition of ethanol in combination with pyridine. It has to be remarked that the intensity (after base-line subtraction) of the band at 1607 cm−1 in Fig. 7 is in reasonable agreement with its intensity in Fig. 4 (for 200 °C cell temperature).
 | ||
| Fig. 7 Combined ethanol–pyridine adsorption FT-IR difference spectra under constant ethanol–pyridine saturated nitrogen flow after different contact times at 200 °C cell temperature for catalyst material B (see Table 1). | ||
Thus, these observations suggest that as a consequence of the interaction of ethanol and pyridine with the Lewis-acidic NbO groups, new Brønsted-acidic Nb–OH groups are formed, leading to a pyridinium ion. The increasing intensities of the bands at 1488 and 1633 cm−1 with contact time could be interpreted as progress of the formation reaction of the Brønsted-acidic Nb–OH surface sites. Remarkably, the bands corresponding to NbO related Lewis-acidity decreased strongly, even at the shortest reaction time (1 min, Fig. 7). This suggests that preceding the reaction, ethanol is adsorbed on the NbO groups thereby affecting the vibrational states and destroying the IR bands due to Lewis-acidity related to the NbO group. The concentration of pyridine bonded to Brønsted-acidic sites appears to be higher after 30 min treatment with a gas containing both ethanol and pyridine (Fig. 7) than after first 30 min ethanol and then 30 min pyridine contact (Fig. 6), as follows from the relatively higher intensity of the bands at 1488 and 1633 cm−1 in the former case (Fig. 7). This suggests that the presence of pyridine has a catalysing effect on the formation of the Brønsted-acidic sites.
When repeating the above discussed measurement with catalyst materials A and C, differing in calcination temperature (see Table 1), band positions observed as well as the increase of band intensities with time are very similar to those displayed in Fig. 7 for catalyst material B (results not shown). Fig. 8 presents the pyridine–ethanol adsorption difference spectra for the catalyst materials A, B and C after 30 min contact time. All bands, except the weak and broad band at 1540 cm−1, observed in Fig. 6 and 7 are also visible for the catalyst materials A and C in Fig. 8. Main differences between the spectra presented in Fig. 8 are the much lower band intensities of the bands at 1448 and 1607 cm−1 observed for the catalyst material with the lowest calcination temperature (material A).
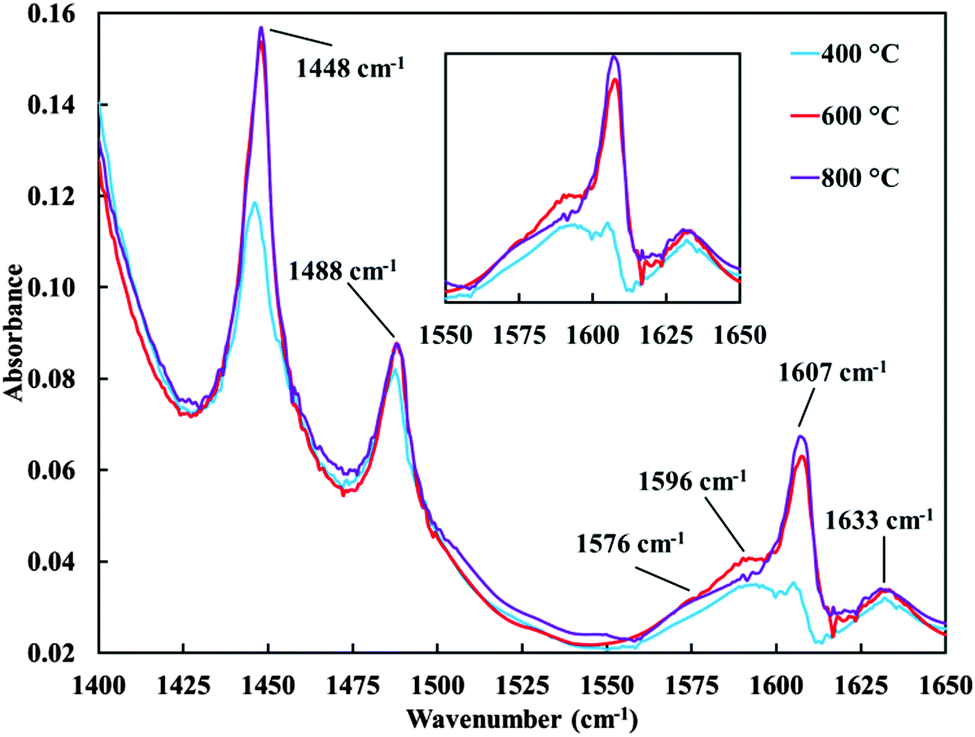 | ||
| Fig. 8 Combined ethanol–pyridine adsorption FT-IR difference spectra under constant ethanol–pyridine saturated nitrogen flow at 200 °C in situ cell temperature after 30 min contact time for catalyst materials with different calcination temperatures (materials A (400 °C), B (600 °C) and C (800 °C), see Table 1). | ||
These bands are assigned to pyridine coordinated to Lewis-acidic impurities of the silica. A lower intensity might be explained by the higher degree of hydration of the silica surface. Due to hydration, Lewis-acidic sites related to Al and/or Fe impurities can be transformed into very weak Brønsted-acidic sites.
Finally, also FT-IR spectra were recorded during interaction of the catalyst material with an ethanol and pyridine containing gas for subsequently increasing cell temperatures.
Fig. 9 illustrates these spectra for the catalyst material B (Fig. 9a) and for the silica support (Fig. 9b). In case of the silica support (Fig. 9b) the main difference with the spectra recorded without ethanol (Fig. 4b) is the lower intensity ratio of the bands at 1444 relative to 1448 cm−1 and 1596 relative to 1607 cm−1, these ratios being smaller than 1 already at 200 °C cell temperature. This can be explained by the reaction of the isolated silanol groups with ethanol to ethylsilicate surface groups (see Section 3.2.2) and in consequence a reduced surface fraction of isolated silanol groups leading to less H-bonded pyridine.
 | ||
| Fig. 9 Combined ethanol–pyridine adsorption FT-IR difference spectra under constant ethanol–pyridine saturated nitrogen flow at subsequently increasing in situ cell temperatures (5 min equilibration time) for (a) catalyst material B and (b) silica support (calcined at 600 °C). | ||
Comparing the spectra of the catalyst material (Fig. 9a) with the reference spectra without ethanol (see Fig. 4a) it can be seen that the bands around 1444 and 1596 cm−1 are much broader in particular for the lowest temperatures investigated. Nevertheless, the bands related to the Brønsted-acidic sites at 1488 and 1633 cm−1, are again very well observable. Regarding the temperature dependence of the band intensities it follows that the normally expected decrease of intensity with increasing cell temperature is relatively small for the bands at 1488 and 1633 cm−1 in comparison to the bands around 1444 and 1596 cm−1. Thus, the intensities of the bands related to the Brønsted-acidic Nb-site increase relative to the other bands corresponding to Lewis-acidic sites. This is in particular the case for the band at 1633 cm−1, which did not change at all in intensity with increasing cell temperature.
This increase of relative intensity suggests again, just as in the case discussed above, that Lewis-acidic sites can be transformed into Brønsted-acidic sites, in this case to a greater extent at higher temperature.
Summarizing, the ethanol–pyridine adsorption measurements demonstrated that a Brønsted-acidic site, stabilized by adsorbed pyridine, can be formed by reaction of a tetrahedral mono-oxo NbO4 surface species with ethanol in the presence of pyridine. This reaction is “catalysed” by pyridine, but in addition pyridine is essential as part of the adsorption complex. Most probably, the Lewis-basic properties of pyridine are responsible for this effect, and therefore other Lewis-basic molecules such as cyclohexanone oxime might in a similar way result in the formation of an adsorption complex.
3.3. Proposed reaction cycle for the formation of the active site at the Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O group and the recovery of the NbO group during the final stage of the gas-phase Beckmann rearrangement
O group and the recovery of the NbO group during the final stage of the gas-phase Beckmann rearrangement
In the following, a reaction scheme will be proposed for the formation of an adsorption complex, at the NbO group, consisting of a Brønsted-acidic site and adsorbed cyclohexanone oxime. Furthermore, the proposal includes, after a general discussion of the GPBR, the recovery of the NbO group during the final stage of the reaction. The discussion will account for all our observations. These include not only the spectroscopic data presented in this research work, but also the recently published kinetic results.17 The kinetic data could be described very well by a kinetic scheme with an adsorption reaction as first step (154 kJ mol−1 activation energy), involving the NbO containing surface groups, cyclohexanone oxime and an alcohol, leading to a Brønsted-acidic surface adsorption complex. The second step of this reaction path is the formation of ε-caprolactam by the H+-catalysed Beckmann rearrangement (68 kJ mol−1 activation energy). Furthermore, the kinetic results can be described best by assuming that a steady state is reached, where the rate of formation of the Brønsted-acidic surface complex and of Beckmann rearrangement are equal. The observation that no adsorption equilibrium is attained, but an adsorption complex is formed by a reaction, is in good agreement with the spectroscopic results described in the previous sections, where the extent of transformation of the NbO sites together with pyridine and ethanol into a Brønsted-acidic site increased with increasing temperature. It should be remarked that the spectroscopic results were obtained with pyridine, while the kinetic data refer to a reaction involving cyclohexanone oxime.
However, we assume that cyclohexanone oxime and pyridine, being both Lewis-bases, can play a similar role in the formation of the Brønsted-acidic surface complex.
The formation of the surface complex is depicted in Fig. 10. It can be assumed that ethanol molecules coordinate via the O-atom (see Fig. 10b) to the Lewis-acidic Nb-atom of the NbO4 surface species (Fig. 10a). Since ethanol is only weakly acidic (slightly less acidic than water) it cannot hydrolyse the NbO bond of the NbO4 surface species.
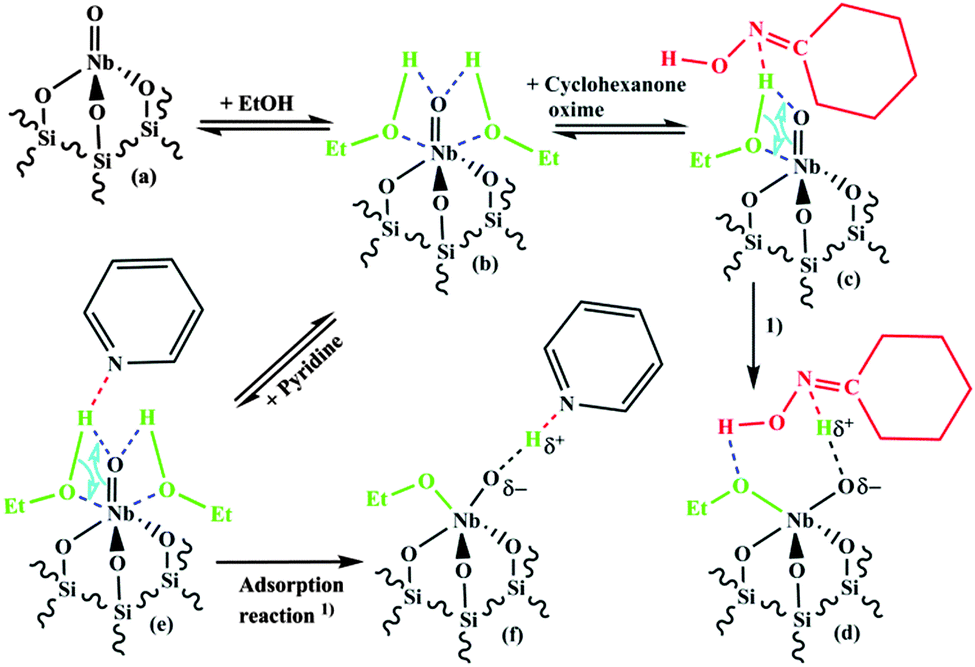 | ||
| Fig. 10 Proposed scheme for the adsorption reaction of cyclohexanone oxime or pyridine with ethanol and the tetrahedral mono-oxo NbO4 surface species. | ||
When a Lewis-basic molecule e.g. cyclohexanone oxime or pyridine is present in the gas-phase besides the alcohol it could coordinate to the H-atom of ethanol (Fig. 10c or e). Abstraction of a proton from the adsorbed ethanol makes the O-atom of the adsorbed ethanol negative and increases its reactivity towards the Nb-central atom. Then, a Nb–OEt bond can be formed together with a “Nb–Oδ−⋯Hδ+-base” Brønsted-acidic group containing the adsorbed base (Fig. 10d or f). It should be noted that not much data are available on the base strength of oximes. According to Politzer and Murray35 the basicity of acetoxime, (H3C)2CN–OH, should be close to or slightly lower than that of pyridine, and the basicities of cyclohexanone oxime and acetoxime are probably not much different. Thus, the base strength of cyclohexanone oxime would enable a similar reaction as observed for pyridine, resulting in an adsorption complex consisting of a Brønsted-acid site and adsorbed cyclohexanone oxime.
Cyclohexanone oxime is most probably N-protonated in the adsorption complex (Fig. 10d and 11B), since this leads to an energetically more favourable state than O-protonation. According to calculations for different oxime molecules the energy difference amounts to about 70–80 kJ mol−1.36–39
Regarding the mechanism of the Beckmann rearrangement, following after the adsorption reaction, the reaction scheme, most commonly accepted for solid acid catalysts, comprises the following steps. Starting from the N-protonated adsorbed state, a 1,2-H shift leads to the O-protonated oxime molecule. Then follows dehydration and N-insertion (rearrangement step), rehydration (formation of an enol–amid complex) and finally tautomerization (formation of a keto–amide complex).36 Landis et al. proposed for the zeolite catalysed GPBR a direct formation of O-protonated oxime.40 However, Shinohara et al. concluded in a theoretical study that with oxides (e.g. SiO2/Al2O3) as catalyst the reaction starts with N-protonation, while the O-protonation is more favourable for the liquid phase reaction.41 In addition, other computational studies42–45 and experimental studies46–49 (including FT-IR and NMR measurements) of oxime molecules adsorbed on Brønsted-acidic sites of different zeolites confirm that N-protonation takes place as first step. Actually, there is no agreement in literature about the rate-determining step and published (calculated) activation energies differ considerably, probably due to the calculation method and model applied. In several publications the 1,2-H shift is identified as rate determining step, but the activation energy for this reaction step seems to increase with increasing acid strength of the Brønsted-acidic adsorption site. Shinohara et al.41 reports 20.1 kJ mol−1 for silica-alumina, whereas Sirijaraensre et al.44 presents 101.8 kJ mol−1 for a H-[B]-ZSM-5 and 131.6 kJ mol−1 for a H-[Al]-ZSM-5 zeolite as activation energy for the 1,2-H shift reaction.
Our measured activation energy17 for the GPBR, 68 kJ mol−1, is rather low as compared to the calculated literature data. Note, that our experimental value is just the overall effective value combining the several steps mentioned above. For an interpretation of this activation energy a detailed knowledge of the structure of the adsorption complex including surrounding silanol groups would be required. The low activation energy suggests a small difference between N-protonation and O-protonation (see above) and participation of silanol groups in the dehydration and rehydration steps (formation of H-bonds). A very low activation energy (45.2 kJ mol−1) was also calculated (albeit for a liquid phase situation) by Yamabe et al.50 The mechanism turned out to be concerted (N-insertion, rehydration and tautomerization occurring in one single step) in particular in the case of cyclohexanone oxime.
Since we don't have sufficient data to establish in detail which reaction steps occur and are rate determining in our case, we present only a very general reaction scheme in Fig. 11. After the adsorption reaction between cyclohexanone oxime, ethanol and the tetrahedral mono-oxo NbO4 surface site (Fig. 11A), cyclohexanone oxime is most probably present in N-protonated state (Fig. 11B). Then, the Beckmann rearrangement takes place according to (a combination of) the steps described above.
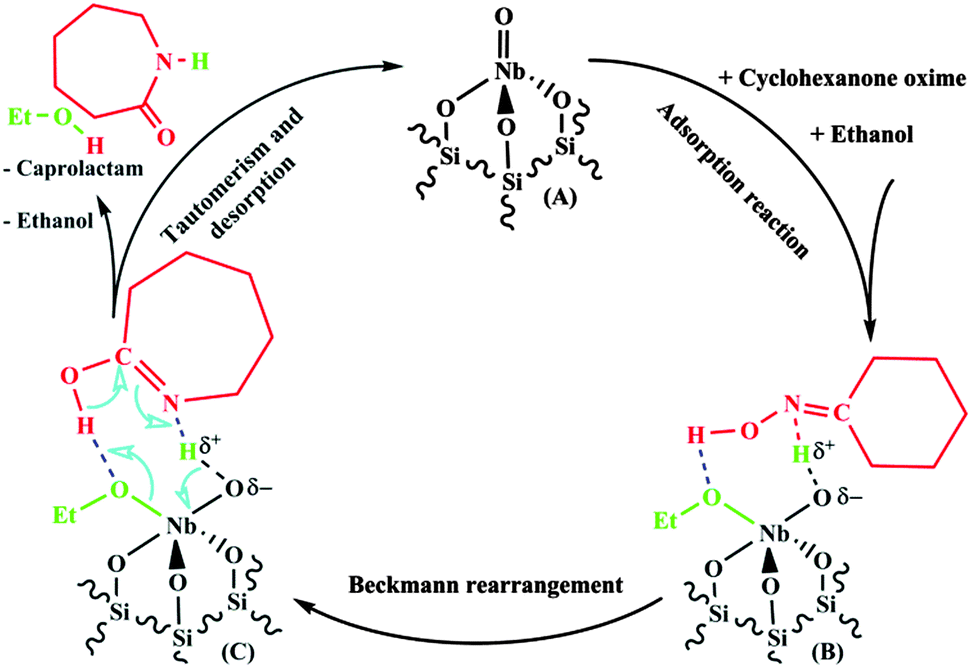 | ||
| Fig. 11 Proposed reaction cycle of the GPBR reaction over a tetrahedral mono-oxo NbO4 surface species. | ||
According to the structure proposed in Fig. 11C the Beckmann rearrangement results in the formation of the caprolactam molecule in the enol-form. Similar to the situation with pyridine (Fig. 10f) and cyclohexanone oxime (Fig. 10d and 11B), the enol-caprolactam is bonded to the Nb–OH Brønsted-acid site via the N-atom. However, the protonation of the N-atom of the enol-caprolactam initializes an acid-catalysed enol–keto tautomerization. The resulting ε-caprolactam is only very weakly basic due to the strongly electron withdrawing nature of the carbonyl group.
Due to the very weakly basic character of ε-caprolactam the adsorption complex is destabilized and decomposes, in one concerted step, to ε-caprolactam, ethanol and the tetrahedral mono-oxo NbO4 surface species.
The weak interaction of ε-caprolactam with the acidic sites of the NbOx/SiO2 catalyst explains why we did not observe any effect of the presence of ε-caprolactam in the feed (no product inhibition).17
To gain a better insight into the reaction path of the GPBR over a NbOx/SiO2 catalyst material further spectroscopic measurements (e.g. in situ FT-IR) should be performed, including the adsorption of cyclohexanone oxime as well as the subsequent and co-adsorption of cyclohexanone oxime and ethanol.
4. Conclusions
NbOx/SiO2 catalyst materials were investigated spectroscopically in order to obtain information about the formation and nature of the surface sites responsible for the gas-phase Beckmann rearrangement of cyclohexanone oxime to ε-caprolactam. In situ pyridine adsorption FT-IR measurements revealed that the catalyst contains Lewis-acidic sites, most probably related to isolated tetrahedral NbO4 surface species containing a NbO group, while no Brønsted-acidic sites were observed. In situ Raman and in situ adsorption FT-IR results came to the conclusion that Brønsted-acidic sites, essential for the Beckmann rearrangement, can be generated from the Lewis-acidic NbO surface groups by reaction with ethanol. However, these Brønsted-acidic sites can only be detected in significant amounts if the NbOx/SiO2 catalyst material is exposed to a reactant gas containing not only ethanol, but also pyridine which initiates the reaction with ethanol. From this observation it can be concluded that the formation and/or the stability of the Brønsted-acidic site are enhanced by the presence of a Lewis-basic molecule. Assuming that cyclohexanone oxime can play a similar role as pyridine, and based on both the spectroscopic findings reported in this work and our kinetic results published recently, a general reaction cycle for the gas-phase Beckmann rearrangement over an isolated tetrahedral mono-oxo NbO4 surface species was proposed. This cycle starts with an adsorption reaction of ethanol and cyclohexanone oxime on the NbO4 site under formation of the Brønsted-acidic site. Then, the Beckmann rearrangement of the adsorbed cyclohexanone oxime takes place followed by the desorption of ε-caprolactam as final step.
Acknowledgements
M. Maronna would like to thank DSM for the financial foundation of the GPBR project and for providing the laboratory facilities for executing experimental work. The authors are grateful to Dr Stephan Küppers and Dr Hannelore Lippert (Forschungszentrum Jülich ZEA-3 Analytik, Germany) for ICP-MS measurements, Dr Jürgen Dornseiffer (Forschungszentrum Jülich, IEK-1, Germany) for applying N2-adsorption analysis and Mr Frank Schmidberger from Grace Davison (Germany) for providing the silica material.References
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev., 2001, 4, 381–441 Search PubMed.
- G. P. Heitmann, G. Dahlhoff and W. F. Hoelderich, Appl. Catal., A, 1999, 185, 99–108 CrossRef CAS.
- W. F. Hoelderich, J. Röseler and G. Heitmann, Catal. Today, 1997, 37, 353–366 CrossRef.
- J. Röseler, G. Heitmann and W. F. Hoelderich, Appl. Catal., A, 1996, 144, 319–333 CrossRef.
- W. F. Hoelderich, J. Roeseler and D. Arntz, US Pat., 5741904, 1998, first assigned to Degussa, then taken over by Sumitomo Search PubMed.
- Y. Izumi, H. Ichihashi and Y. Shimazu, Bull. Chem. Soc. Jpn., 2007, 7, 1280–1287 CrossRef.
- H. Ichihashi and M. Kitamura, Catal. Today, 2002, 73, 23–28 CrossRef CAS.
- H. Ichihashi and H. Sato, Appl. Catal., A, 2001, 221, 359–366 CrossRef CAS.
- T. Ushikubo and K. Wada, J. Catal., 1994, 148, 138–148 CrossRef CAS.
- K. Sugiyama, G. Anan and T. Ushikubo, Surf. Coat. Technol., 1999, 112, 76–79 CrossRef CAS.
- M. Anilkumar and W. F. Hoelderich, J. Catal., 2008, 260, 17–29 CrossRef CAS.
- M. Anilkumar and W. F. Hoelderich, Catal. Today, 2012, 198, 289–299 CrossRef CAS.
- M. Anilkumar and W. F. Hoelderich, J. Catal., 2012, 293, 76–84 CrossRef CAS.
- M. Anilkumar, PhD thesis, RWTH Aachen University, 2009.
- A. Mettu and W. F. Hoelderich, WO2010063276A1, 2010.
- M. M. Maronna, E. C. Kruissink, R. F. Parton, J. T. Tinge, F. Soulimani, B. M. Weckhuysen and W. F. Hoelderich, Appl. Catal., B, 2016, 185, 272–280 CrossRef CAS.
- M. M. Maronna, E. C. Kruissink, J. T. Tinge, D. W. Agar and W. F. Hoelderich, Ind. Eng. Chem. Res., 2016, 55, 1202–1214 CrossRef CAS.
- N. Ichikuni, M. Shirai and Y. Iwasawa, Catal. Today, 1996, 28, 49–58 CrossRef CAS.
- N. Ichikuni and Y. Iwasawa, J. Phys. Chem., 1994, 98, 11576–11581 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 14410–14425 CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 6487–6498 CAS.
- J. C. Mikkelsen, F. L. Galeener and W. J. Mosby, J. Electron. Mater., 1981, 10, 631–651 CrossRef CAS.
- K. Nakajima, Y. Baba and R. Noma, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
- M. R. Basila, T. R. Kantner and K. H. Rhee, J. Phys. Chem., 1964, 68, 3197–3207 CrossRef CAS.
- A. Ramirez, B. L. Lopez and L. Sierra, J. Phys. Chem. B, 2003, 107, 9275–9280 CrossRef CAS.
- M. I. Zaki, M. A. Hasan and F. A. Al-Sagheer, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS.
- T. Barzetti, E. Selli and D. Moscotti, J. Chem. Soc., Faraday Trans., 1996, 92, 1401–1407 RSC.
- F. Hatayama, T. Ohno and T. Maruoka, J. Chem. Soc., Faraday Trans., 1991, 87, 2629–2633 RSC.
- T. Kataoka and A. Dumesic, J. Catal., 1988, 112, 66–79 CrossRef CAS.
- H. Miyata, K. Fujii and T. Ono, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 3121–3128 RSC.
- J. Datka, A. M. Turek and J. M. Jehng, J. Catal., 1992, 135, 186–199 CrossRef CAS.
- A. Aucejo, M. C. Burguet, A. Corma and V. Fornes, Appl. Catal., 1986, 22, 187–200 CrossRef CAS.
- R. K. Iler, The Chemistry of Silica, John Wiley & Sons, Hoboken, 1979 Search PubMed.
- P. Politzer and J. S. Murray, Structural Analysis of Hydroxylamines, Oximes and Hydroxamic Acids: Trends and Patterns. Patai's Chemistry of Functional Groups, John Wiley & Sons, 2010 Search PubMed.
- M. T. Nguyen, G. Raspoet and L. G. Vanquickenborne, J. Am. Chem. Soc., 1997, 119, 2552–2562 CrossRef CAS.
- M. T. Nguyen, G. Raspoet and L. G. Vanquickenborne, J. Chem. Soc., Perkin Trans. 2, 1997, 821–825 RSC.
- M. T. Nguyen, G. Raspoet and L. G. Vanquickenborne, J. Chem. Soc., Perkin Trans. 2, 1995, 1791–1795 RSC.
- M. T. Nguyen and L. G. Vanquickenborne, J. Chem. Soc., Perkin Trans. 2, 1993, 1969–1972 RSC.
- P. S. Landis and P. B. Venuto, J. Catal., 1966, 6, 245–252 CrossRef CAS.
- Y. Shinohara, S. Mae, D. Shouro and T. Nakajima, J. Mol. Struct., 2000, 497, 1–9 CrossRef CAS.
- J. Sirijaraensre and J. Limtrakul, ChemPhysChem, 2006, 7, 2424–2432 CrossRef CAS PubMed.
- J. Sirijaraensre, T. N. Truong and J. Limtrakul, J. Phys. Chem. B, 2005, 109, 12099–12106 CrossRef CAS PubMed.
- J. Sirijaraensre and J. Limtrakul, Phys. Chem. Chem. Phys., 2009, 11, 578–585 RSC.
- T. Bucko, J. Hafner and L. Benco, J. Phys. Chem. A, 2004, 108, 11388–11397 CrossRef CAS.
- Y. M. Chung and H. K. Rhee, J. Mol. Catal. A: Chem., 2001, 175, 249–257 CrossRef CAS.
- A. B. Fernandez, M. Boronat, T. Blasco and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2370–2373 CrossRef CAS PubMed.
- V. R. Marthala, Y. Jiang, J. Huang, W. Wang and R. Gläser, J. Am. Chem. Soc., 2006, 128, 14812–14813 CrossRef CAS PubMed.
- C. Flego and L. Dalloro, Microporous Mesoporous Mater., 2003, 60, 263–271 CrossRef CAS.
- S. Yamabe, N. Tsuchida and S. Yamazaki, J. Org. Chem., 2005, 70, 10638–10644 CrossRef CAS PubMed.
Footnote |
| † Formerly: RWTH Aachen, Lehrstuhl für Technische Chemie und Heterogene Katalyse, Worringerweg 1, 52074 Aachen, Germany. |
| This journal is © the Owner Societies 2016 |