
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A heterojunction photocatalyst composed of zinc rhodium oxide, single crystal-derived bismuth vanadium oxide, and silver for overall pure-water splitting under visible light up to 740 nm†
Ryoya
Kobayashi
a,
Toshihiro
Takashima
b,
Satoshi
Tanigawa
a,
Shugo
Takeuchi
c,
Bunsho
Ohtani
d and
Hiroshi
Irie
*bd
aSpecial Doctoral Program for Green Energy Conversion Science and Technology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 4-3-11 Takeda, Kofu, Yamanashi 400-8511, Japan
bClean Energy Research Center, University of Yamanashi, 4-3-11 Takeda, Kofu, Yamanashi 400-8511, Japan. E-mail: hirie@yamanashi.ac.jp
cGraduate School of Environmental Science, Hokkaido University, Nishi 10, Nishi 5, Sapporo, Hokkaido 060-0810, Japan
dInstitute for Catalysis, Hokkaido University, Nishi 10, Kita 21, Sapporo, Hokkaido 001-0021, Japan
First published on 30th June 2016
Abstract
We recently reported the synthesis of a solid-state heterojunction photocatalyst consisting of zinc rhodium oxide (ZnRh2O4) and bismuth vanadium oxide (Bi4V2O11), which functioned as hydrogen (H2) and oxygen (O2) evolution photocatalysts, respectively, connected with silver (Ag). Polycrystalline Bi4V2O11 (p-Bi4V2O11) powders were utilized to form ZnRh2O4/Ag/p-Bi4V2O11, which was able to photocatalyze overall pure-water splitting under red-light irradiation with a wavelength of 700 nm (R. Kobayashi et al., J. Mater. Chem. A, 2016, 4, 3061). In the present study, we replaced p-Bi4V2O11 with a powder obtained by pulverizing single crystals of Bi4V2O11 (s-Bi4V2O11) to form ZnRh2O4/Ag/s-Bi4V2O11, and demonstrated that this heterojunction photocatalyst had enhanced water-splitting activity. In addition, ZnRh2O4/Ag/s-Bi4V2O11 was able to utilize nearly the entire range of visible light up to a wavelength of 740 nm. These properties were attributable to the higher O2 evolution activity of s-Bi4V2O11.
Introduction
Since the first report of photo-induced water splitting by titanium dioxide (TiO2) and platinum (Pt) electrodes,1 the potential to convert photon energy into hydrogen (H2) energy by photocatalytic water-splitting using photoelectrodes and powdered photocatalysts has been extensively investigated due to the simplicity of the process and the possibility for large-scale H2 production. Numerous studies have attempted to identify powdered photocatalysts that are able to split water into H2 and oxygen (O2) at a molar ratio of ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (overall water splitting),2–26 particularly under visible-light irradiation for the efficient utilization of sunlight energy.8–26 One of the major approaches for overall water splitting is the construction of “Z-scheme” systems, which are composed of two visible-light sensitive photocatalysts. These systems were first reported by Sayama and coworkers, who used Pt-deposited strontium titanate co-doped with chromium and tantalum (Pt/SrTiO3:Cr,Ta) as an H2 evolution photocatalyst (H2 photocatalyst) and Pt-deposited tungsten trioxide (Pt/WO3) as an O2 evolution photocatalyst (O2 photocatalyst).20 To date, numerous Z-scheme systems consisting of various H2 and O2 photocatalysts have been reported.21–23
:1 (overall water splitting),2–26 particularly under visible-light irradiation for the efficient utilization of sunlight energy.8–26 One of the major approaches for overall water splitting is the construction of “Z-scheme” systems, which are composed of two visible-light sensitive photocatalysts. These systems were first reported by Sayama and coworkers, who used Pt-deposited strontium titanate co-doped with chromium and tantalum (Pt/SrTiO3:Cr,Ta) as an H2 evolution photocatalyst (H2 photocatalyst) and Pt-deposited tungsten trioxide (Pt/WO3) as an O2 evolution photocatalyst (O2 photocatalyst).20 To date, numerous Z-scheme systems consisting of various H2 and O2 photocatalysts have been reported.21–23
Despite the potential of Z-scheme systems for photocatalytic water splitting, most systems reported to date are only able to utilize visible light up to a wavelength of 520 nm. In addition, because conventional Z-scheme systems require a suitable redox mediator, such as iodate (IO3−)/iodide (I−) or ferric (Fe3+)/ferrous (Fe2+) ions, these systems are not capable of splitting “pure” water (i.e., distilled water without chemicals). Recently, solid-state Z-scheme systems that function in the absence of a redox mediator have been reported.24,25 However, because the pH of the system required adjustment to 3.5 using sulfuric acid (H2SO4), pure-water splitting was not accomplished.
Recently, we developed novel solid-state photocatalysts by inserting silver (Ag) as an electron mediator between zinc rhodium oxide (ZnRh2O4, Eg of 1.2 eV) as an H2 photocatalyst and defective silver antimonate (Ag1−xSbO3−y, Eg of 2.7 eV) or bismuth vanadium oxide (Bi4V2O11) as an O2 photocatalyst (ZnRh2O4/Ag/Ag1−xSbO3−y26 or ZnRh2O4/Ag/Bi4V2O1127). Using these systems, overall pure-water splitting proceeded via the inserted Ag, which mediated the transfer of photoexcited electrons from the conduction band (CB) of the O2 photocatalyst to the valence band (VB) of the H2 photocatalyst. ZnRh2O4/Ag/Ag1−xSbO3−y and ZnRh2O4/Ag/Bi4V2O11 utilized visible light up to wavelengths of 545 and 700 nm, respectively, a property that was determined by the photoactivity of Ag1−xSbO3−y and Bi4V2O11. In the previous study, we utilized polycrystalline Bi4V2O11 (p-Bi4V2O11) powder, which was prepared by a normal solid-state reaction, for the synthesis of ZnRh2O4/Ag/Bi4V2O11.
In the present study, Bi4V2O11 powder was obtained by pulverizing single crystals of Bi4V2O11 (s-Bi4V2O11) to form the powdered photocatalyst ZnRh2O4/Ag/s-Bi4V2O11, which displayed enhanced water-splitting activity compared to ZnRh2O4/Ag/p-Bi4V2O11. Notably, ZnRh2O4/Ag/s-Bi4V2O11 was able to utilize visible light up to wavelengths of 740 nm, which represents nearly the entire visible light spectrum and to our knowledge, is the longest wavelength reported to date (ESI 1†).
Experimental
Sample preparation
A melting–slow cooling method was applied to grow single crystals of Bi4V2O11 (s-Bi4V2O11) by melting (940 °C) and slow cooling (4 °C h−1 to 740 °C) of stoichiometric mixtures of commercial bismuth oxide (Bi2O3, purity 99.9%; Kanto Kagaku) and vanadium oxide (V2O5, purity 99.0%; Kanto Kagaku) powders.28 The obtained single crystals adopted a thin plate-like sheet structure and had a thickness of ∼0.2 mm and transverse dimensions of ∼1 mm × 2 mm. The thin sheets of Bi4V2O11 were used directly for Laue photography and X-ray diffractometer analyses, and were further pulverized for use in all other experiments.ZnRh2O4 powders were synthesized using a solid-state reaction method. Briefly, stoichiometric amounts of commercial zinc oxide (ZnO, purity 99.0%; Kanto Kagaku) and rhodium oxide (Rh2O3, purity 99.9%; Kanto Kagaku) powders were mixed and calcined at 1000 °C for 24 h and the obtained powders were thoroughly ground. As a reference, the Bi2O3 and V2O5 powders were mixed and calcined at 850 °C for 8 h to prepare poly-crystal powders of Bi4V2O11 (p-Bi4V2O11), which were thoroughly ground prior to experimental use.
A powdered heterojunction photocatalyst composed of ZnRh2O4, s-Bi4V2O11, and Ag (ZnRh2O4/Ag/s-Bi4V2O11) was prepared using the following simple method.27 ZnRh2O4, Ag2O, and s-Bi4V2O11 powders were mixed at a molar ratio of 1:1:1.2 in an identical manner to that described above. The mixed powders were pressed into pellets by applying a force of 60 kN, and the pellets were then heated at 750 °C for 2 h. The pellets were ground into a fine powder, which was soaked in an aqueous solution (50 mL) of 3 M nitric acid (HNO3, Kanto Kagaku) for 15 min. The powder was filtered and thoroughly washed with distilled water, and was then dried at 65 °C for 12 h. As a reference, ZnRh2O4/Ag/p-Bi4V2O11 was also prepared using the same procedure.27
Characterization
A Laue camera (SA-HFM3, Rigaku) was used for the characterization of Bi4V2O11 single crystals. The crystal structures of the single crystals and powder obtained by pulverizing single crystals were determined by X-ray diffraction (XRD) using a PW-1700 X-ray diffractometer (PANalytical). The structure of the ZnRh2O4/Ag/Bi4V2O11 powder was also determined by XRD. The Brunauer–Emmett–Teller (BET) surface area was determined using a nitrogen adsorption apparatus (Micromeritics, TriStar 3000, Shimadzu). UV-visible absorption (UV-vis) spectra were obtained by the diffuse reflection method using a V-650 spectrometer (Jasco) with barium sulfate (BaSO4) as the reflectance standard. Photoacoustic (PA) spectroscopy measurements were conducted in a nitrogen atmosphere under monochromic light generated from a 300 W xenon (Xe) lamp (LX300, Eagle) equipped with a monochromator (CT-101T, Jasco) and modulated by a light chopper at 80 Hz. The PA signal was amplified and monitored using a digital lock-in amplifier (LI5640, NF).29,30 A scanning electron microscope (SEM, JSM-6500F, JEOL Ltd) was used to observe the morphology of the prepared photocatalysts. A scanning transmission electron microscope (STEM, Tecnai Osiris, FEI) was also utilized with element maps obtained by energy-dispersive X-ray spectrometry (EDS).O2 evolution activity by the half reaction of water
Action spectra for O2 evolution were measured in 3 mL water containing either s-Bi4V2O11 or p-Bi4V2O11 powder (30 mg) and Ce4+ (cerium sulfate (Ce(SO4)2), 0.1 mol L−1; Kanto Kagaku) as a sacrificial agent. The pH of the solution was not adjusted prior to the spectral measurements, which were performed with constant stirring using a magnetic stirrer and under illumination with monochromatic light (570 ± 5 to 750 ± 5 nm) generated from a diffraction grating-type illuminator (CRM-FD, Jasco) equipped with a 300 W Xe lamp (C2578-02, Hamamatsu Photonics). Higher-order diffracted light was cut off using an appropriate glass filter. The amount of evolved O2 was monitored using a gas chromatograph (GC-8A, Shimadzu). The wavelength dependence of the AQE was then evaluated. AQE values for O2 evolution were calculated using the equation: AQE (%) = 100 × 4 × O2 evolution rate/incident photon rate, because O2 evolution is represented by the formula: 2H2O + 4h+ → O2 + 4H+.Water-splitting reactions
To evaluate the photocatalytic activity, ZnRh2O4/Ag/s-Bi4V2O11 or ZnRh2O4/Ag/p-Bi4V2O11 composite powder (60 mg) was suspended in 12 mL pure water (pH unadjusted) under an argon atmosphere (50 kPa) and constant stirring using a magnetic stirrer. A Xe lamp (LA-251Xe, Hayashi Tokei) equipped with an optical filter (irradiated wavelength >420 nm; Y-44, Hoya) and light-emitting diode (LED) lamps with wavelengths of 545 (LEDH60-545, Hamamatsu Photonics), 610 (LEDH60-600, Hamamatsu Photonics), 700 (LEDH60-700, Hamamatsu Photonics), and 740 nm (LEDH60-740, Hamamatsu Photonics) were used for light irradiation. The amount of evolved H2 and O2 was monitored using an online GC-8A gas chromatograph. AQE values were calculated using the amount of evolved O2 in the same way as described above.Photocatalytic overall water-splitting by ZnRh2O4/Ag/s-Bi4V2O11 was also evaluated using pure water containing 33% isotopic water (H218O, purity 97 atm% 18O; Sigma-Aldrich) under light irradiation emitted from the 740 nm LED. The evolved gas was detected using a gas chromatograph/mass spectrometer (GC/MS; GCMS-QP 2010 Ultra, Shimadzu), which was operated in the selective-ion mode and monitored ions with masses of 28 (14N14N), 32 (16O16O), 34 (18O16O), and 36 (18O18O). The measurement conditions, other than those used for pure water containing isotropic water, were the same as those used in the photocatalytic overall pure-water splitting tests.
Results and discussion
Characterization
A back-reflection Laue pattern taken from normal to the plate surface of a Bi4V2O11 single crystal is shown in Fig. S1 (ESI 2†). The distribution of diffraction spots revealed that Bi4V2O11 was a single crystal. XRD patterns for a single crystal of Bi4V2O11 and the powders obtained by pulverizing the single crystals of Bi4V2O11 were also determined (Fig. 1). For the single Bi4V2O11 crystal in the direction normal to the plate surface, only (00l) peaks were detected, indicating that the plate face of the single crystal corresponded to the (001) plane. This assumption is reasonable because Bi4V2O11 is an Aurivillius-type bismuth layer-structured oxide (BLSO) and consists of alternating (Bi2O2)2+ layers and oxygen-deficient pseudo-perovskite blocks of (VO3.5□0.5)2− along the c-axis.31 Thus, because the growth rate of BLSO crystals in the c-axis direction is much lower than that along the a(b)-axis, single crystals of Bi4V2O11 in the form of thin plate-like sheets with plate faces in the (001) plane were obtained. For the pulverized powder, all of the observed XRD peaks could be assigned to those for Bi4V2O11. The BET surface areas of pulverized s-Bi4V2O11 and as-prepared ZnRh2O4 powders were 0.179 and 3.60 m2 g−1, respectively.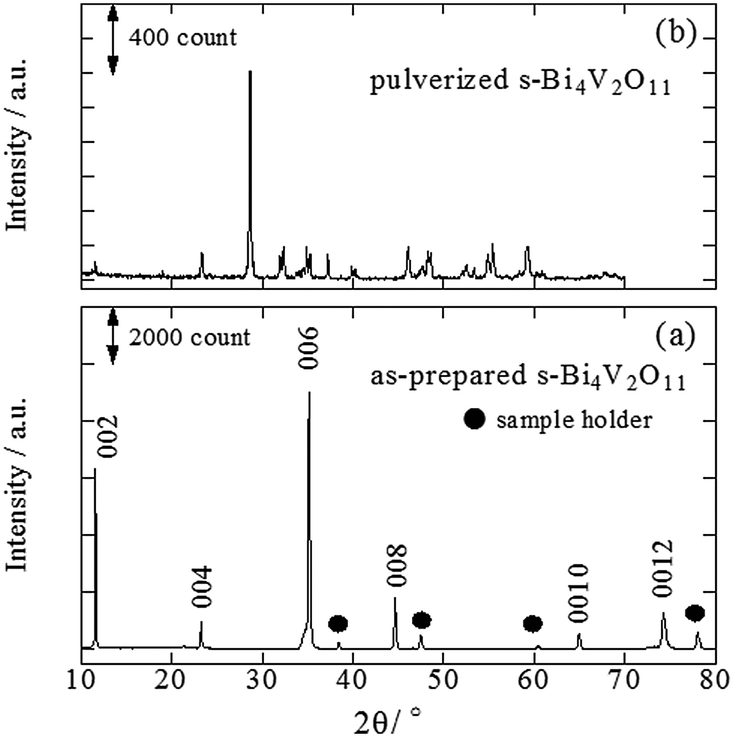 | ||
| Fig. 1 XRD patterns of the as-prepared (a) and pulverized s-Bi4V2O11 (b). | ||
UV-vis absorption and PA spectra of s-Bi4V2O11 are shown in Fig. 2. Although the UV-vis absorption intensity decreased with increasing wavelength, relatively strong intensity was observed in longer wavelength regions. In contrast, the PA absorption of s-Bi4V2O11 decreased with increasing wavelength, a property that reflects true photoabsorption,30 which will be discussed later.
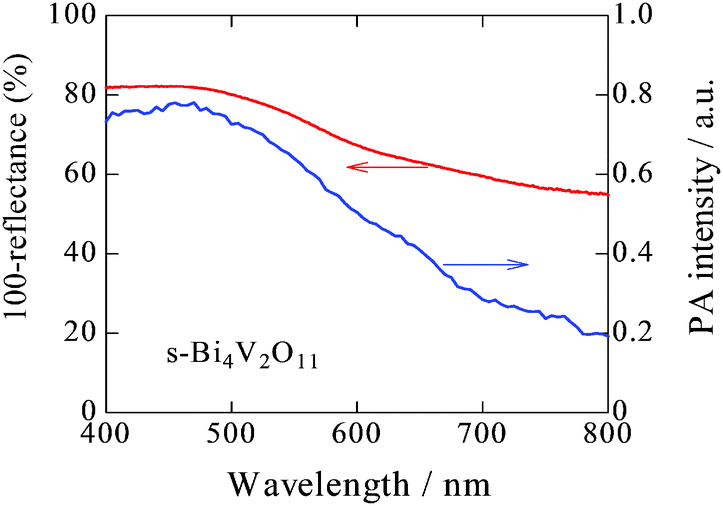 | ||
| Fig. 2 UV-visible absorption and PA spectra of s-Bi4V2O11. | ||
The XRD pattern of ZnRh2O4/Ag/s-Bi4V2O11 following HNO3 treatment is shown in Fig. 3. The XRD peaks mainly corresponded to two phases originating from ZnRh2O4 and Bi4V2O11, although trace peaks attributable to a type of Ag oxide, such as AgVO3, were also detected. However, Ag peaks were not observed. This result is plausible because most of the Ag was likely removed by the HNO3 treatment and the amount of Ag remaining in the composite was below the limit of detection.
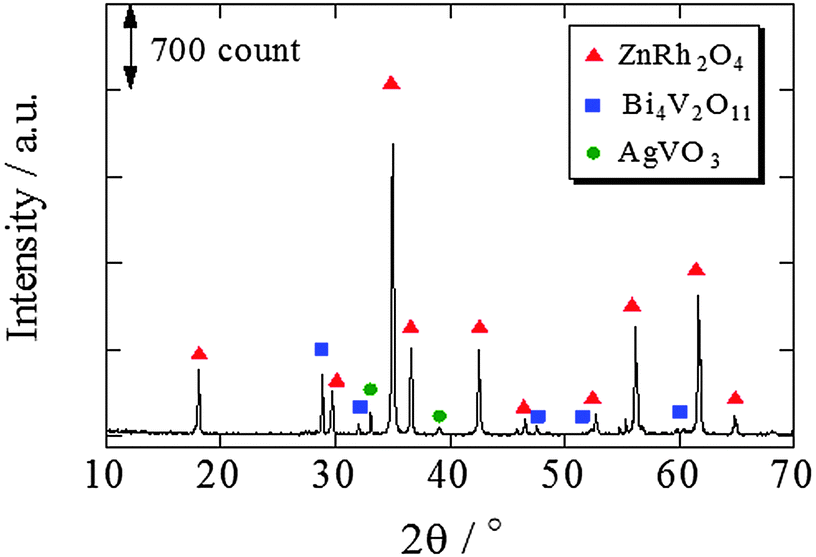 | ||
| Fig. 3 XRD patterns of ZnRh2O4/Ag/s-Bi4V2O11 after HNO3 treatment. | ||
UV-vis absorption spectra of s-Bi4V2O11, ZnRh2O4, and ZnRh2O4/Ag/s-Bi4V2O11 with and without HNO3 treatment are shown in Fig. 4. As can be seen in the spectra, untreated ZnRh2O4/Ag/s-Bi4V2O11 showed greater absorption at wavelengths longer than ∼600 nm compared to HNO3-treated ZnRh2O4/Ag/s-Bi4V2O11. After the HNO3 treatment, the absorption by ZnRh2O4/Ag/s-Bi4V2O11 at wavelengths longer than ∼600 nm decreased and the spectral profile was in between those of ZnRh2O4 and s-Bi4V2O11. Photos of pulverized s-Bi4V2O11, as-prepared ZnRh2O4, and ZnRh2O4/Ag/p-Bi4V2O11 after HNO3 treatment are shown in Fig. S2 (ESI 3†).
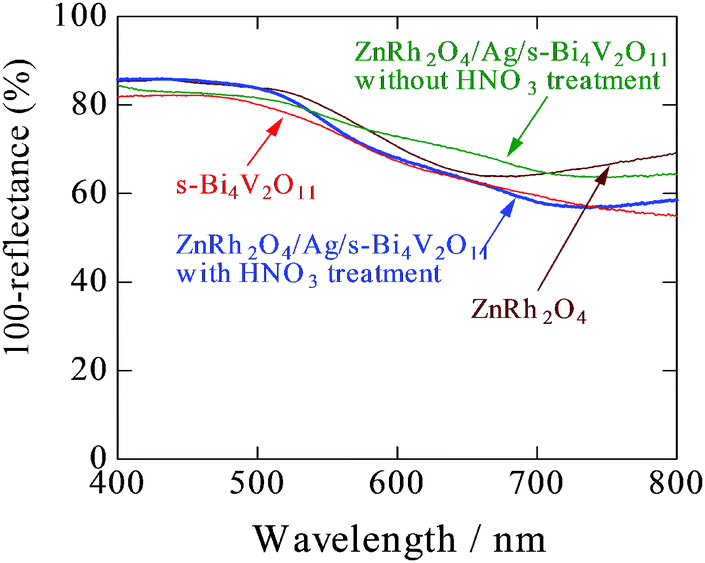 | ||
| Fig. 4 UV-visible absorption spectra of s-Bi4V2O11, ZnRh2O4, and ZnRh2O4/Ag/s-Bi4V2O11 (with and without HNO3 treatment). | ||
In an SEM image of HNO3-treated ZnRh2O4/Ag/s-Bi4V2O11 powder, small ZnRh2O4 (∼50–100 nm) and large Bi4V2O11 particles (∼10 μm) were clearly visible (Fig. 5a). Notably, the particle size of each photocatalytic material was identical to that before preparation of the heterojunction photocatalyst (Fig. 5b and c). STEM imaging and EDS-based elemental mapping of HNO3-treated ZnRh2O4/Ag/Bi4V2O11 were also performed (Fig. 6a–e). As was observed in the SEM image, the s-Bi4V2O11 and ZnRh2O4 particles were clearly distinguishable (Fig. 6a and b) based on size differences. In addition, Ag was distributed (Fig. 6e) between the areas of Bi (Fig. 6c) and Rh (Fig. 6d), indicating that the Ag was inserted between the particles of ZnRh2O4 and Bi4V2O11.
 | ||
| Fig. 5 SEM images of ZnRh2O4/Ag/s-Bi4V2O11 powder after HNO3 treatment (a), as-prepared ZnRh2O4 (b) and pulverized s-Bi4V2O11 (c) powders. | ||
 | ||
| Fig. 6 STEM images of ZnRh2O4/Ag/s-Bi4V2O11 after HNO3 treatment. STEM image (a), its enlargement (b), and EDS element maps (c–e), in which blue (c), red (d), and yellow (e) colors correspond to Bi, Rh, and Ag, respectively. | ||
Action spectra for O2 evolution of Bi4V2O11
To examine and compare the activity of s-Bi4V2O11 with that of p-Bi4V2O11 as an O2 evolution photocatalyst, the half reaction of water over s-Bi4V2O11 and p-Bi4V2O11 was evaluated using an aqueous Ce(SO4)2 solution containing Ce4+ as the sacrificial agent (Fig. 7). Aqueous Ce(SO4)2 is yellowish and transparent, and absorbs visible light up to a wavelength of ∼550 nm. Thus, we irradiated s-Bi4V2O11 and p-Bi4V2O11 with light at wavelengths of longer than 570 nm and found that both materials were able to produce O2 under visible light up to 750 nm. This property indicates that the maximum Eg value of Bi4V2O11 was 1.65 eV. The AQE values of s-Bi4V2O11 were ∼2–3-fold larger than those of p-Bi4V2O11. As s-Bi4V2O11 is considered to have higher crystallinity and anisotropy than p-Bi4V2O11, photogenerated electrons and holes would therefore have enhanced mobility and separation (Fig. S3 in the ESI 4†).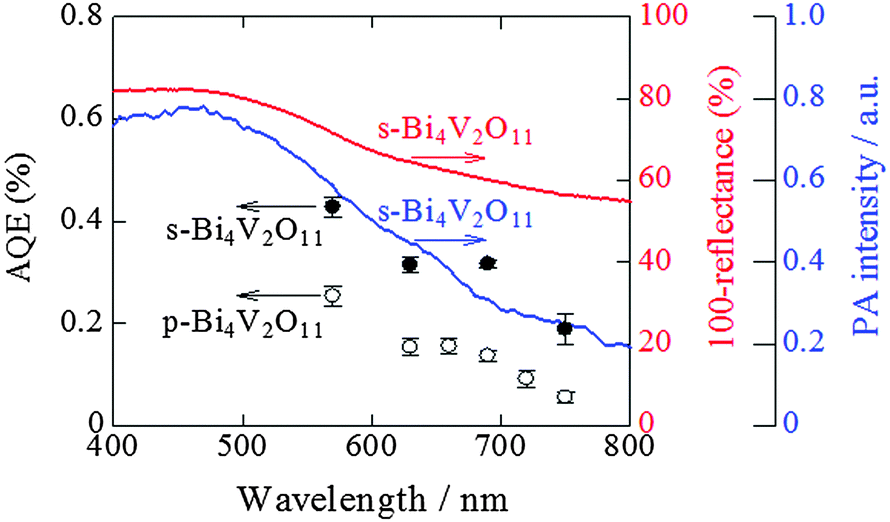 | ||
| Fig. 7 Action spectra for O2 evolution by the half reaction of water over s-Bi4V2O11 (solid circles) and p-Bi4V2O11 (open circles) in the presence of Ce4+. The UV-visible absorption (red line) and PA (blue line) spectra of s-Bi4V2O11 are also shown. | ||
It should be noted that the AQE values for O2 evolution by s-Bi4V2O11 were not consistent with the corresponding UV-vis absorption spectrum. A discrepancy between AQE values and absorption is frequently encountered, particularly for absorption in longer wavelength regions.32,33 In contrast, the AQE values for O2 evolution coincided well with the PA absorption spectrum, which is generally accepted as corresponding to true absorption.30 Thus, it can be concluded that the O2 evolution reaction proceeded via photoinduced band excitation (Fig. S3 in the ESI 4†). This excitation can be assigned to that originating from the VB, which contains O 2p and Bi 6s orbitals from the (Bi2O2)2+ layers, to the CB, which is composed of a mixture of Bi 6p orbitals and anti-bonding O 2p and V 3d orbitals of (VO3.5□0.5)2− blocks.34 The obtained Eg value (1.65 eV) is smaller than the previously reported values of ∼1.96–2.2 eV.34–37 Although the reason for this difference is unclear, it may be attributable to the greater amount of oxygen defects in our samples (larger than 0.5 in VO3.5□0.5)2−. The oxygen defect states form an isolated band below the CB bottom, whose width increases with the concentration of the defects, reaches the CB bottom, and finally starts to overlap with the CB, similar to the case of TiO2.38 In addition, it was reported that the band-gap of Bi4V2O11 changes depending on the morphologies and preparation procedures.37
We previously reported that ZnRh2O4 can utilize visible light at wavelengths up to 770 nm and is also likely capable of utilizing infrared light for H2 evolution.32 Thus, ZnRh2O4/Ag/s-Bi4V2O11 is expected to be sensitive to visible light up to at least 750 nm.
Overall water splitting
The time courses of H2 and O2 evolution from pure water by ZnRh2O4/Ag/s-Bi4V2O11 powder under visible-light irradiation (>420 nm) are shown in Fig. S4 (ESI 5†). Under these conditions, the linear generation of H2 and O2 at a molar ratio of 2 to 1 was observed. We performed this experiment with newly prepared powders and observed similar H2 and O2 evolution rates, confirming the reproducibility of the data. In addition, we characterized the prepared ZnRh2O4/Ag/s-Bi4V2O11 powder after the water-splitting reaction under the same conditions as used in Fig. S4 (ESI†) by XRD and STEM/EDS and found that the sample did not appear to have changed (Fig. S5 and S6 in the ESI 5†). We also confirmed that the water-splitting reaction did not occur in the dark (data not shown).The time courses of H2 and O2 evolution resulting from water splitting by ZnRh2O4/Ag/s-Bi4V2O11 and ZnRh2O4/Ag/p-Bi4V2O11 under irradiation with monochromic light with wavelengths of 545, 610, 700, and 740 nm were next measured (Fig. 8a–d). Under all conditions, ZnRh2O4/Ag/s-Bi4V2O11 evolved H2 and O2 from water at a molar ratio of 2 to 1. The H2 and O2 evolution rates of ZnRh2O4/Ag/s-Bi4V2O11 irradiated with 610 nm and 700 nm light were ∼two- and three-fold larger, respectively, than those of ZnRh2O4/Ag/p-Bi4V2O11. In addition, the induction period for linear H2 and O2 evolution was shortened. Upon irradiation with 740 nm light, ZnRh2O4/Ag/s-Bi4V2O11 evolved H2 and O2 at a ratio of 2 to 1 after the induction period, whereas ZnRh2O4/Ag/p-Bi4V2O11 only produced a trace amount of H2. Thus, overall water splitting induced by 740 nm light was only achieved with ZnRh2O4/Ag/s-Bi4V2O11 and was attributable to the enhanced O2-evolution activity of s-Bi4V2O11. Although the reason for the delay in H2 and O2 evolution is unclear, this phenomenon has been observed with other materials.27 The delay might be due to existing defects,39 redox reactions other than water-splitting reactions,39–42 reconstruction of surface states,43 or adsorption of H2 and O2 molecules on the photocatalyst surface.43,44 However, the delay can be shortened by enhancing the activity.
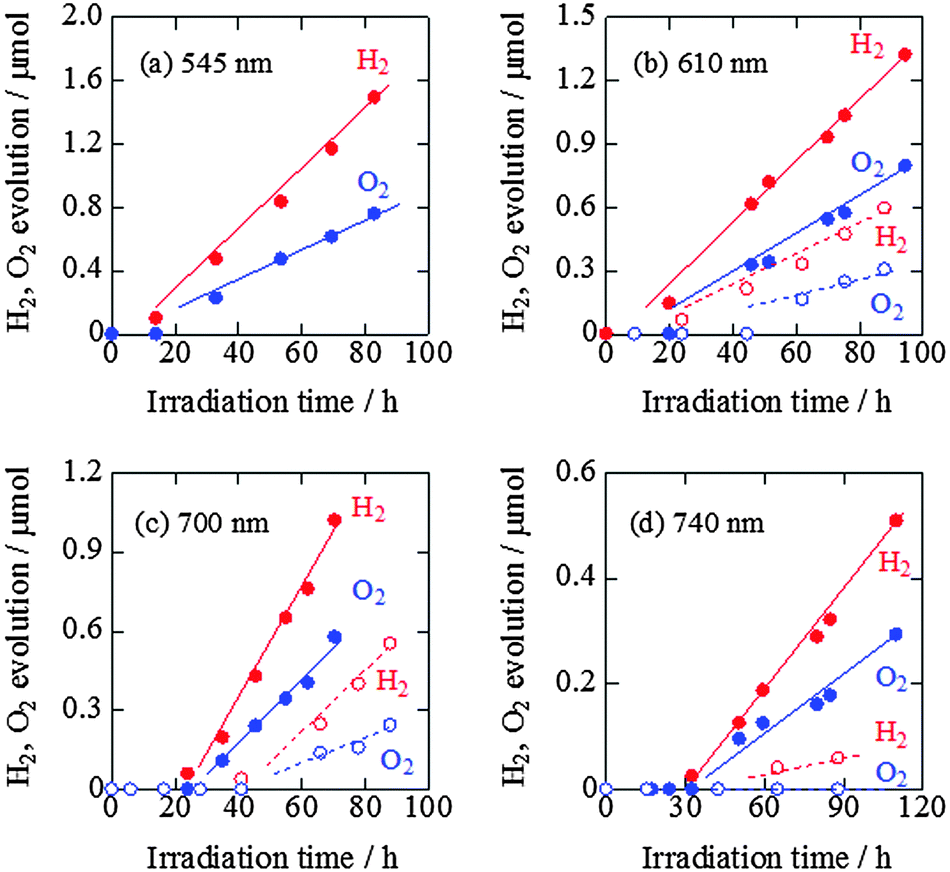 | ||
| Fig. 8 Time courses of H2 and O2 evolution resulting from water splitting by ZnRh2O4/Ag/s-Bi4V2O11 (closed circles with solid lines) and ZnRh2O4/Ag/p-Bi4V2O11 (open circles with broken lines) under irradiation with 545 nm LED (a), 610 nm LED (b), 700 nm LED (c), and 740 nm LED light (d). The H2 and O2 evolution data of ZnRh2O4/Ag/p-Bi4V2O11 with 610 nm LED and 700 nm LED light are cited from ref. 27. | ||
As shown in Fig. 7, the AQE value of s-Bi4V2O11 for O2 was ∼0.2% even at 750 nm and s-Bi4V2O11 was expected to evolve O2 under the entire range of visible light (∼770 nm). Thus, further enhancement of O2 evolution activity by s-Bi4V2O11 would allow ZnRh2O4/Ag/s-Bi4V2O11 to utilize the entire range of visible light and even near-infrared light, as the Eg value of ZnRh2O4 is 1.2 eV,33 for the overall water-splitting.
The mechanisms of O2 evolution on BiVO4(010) and (011) facets were previously proposed.45 On both surfaces, water molecules favorably adsorb on Bi sites. On the (010) surface, photogenerated holes induce the removal of two protons from the adsorbed water molecule in a stepwise manner, and the resulting O adatom obtains an electron by bonding with adjacent O anions to form a peroxo intermediate (Bi–O–O–V). Following the adsorption of a second water molecule at the same Bi site, a second O adatom is formed by the identical process and then obtains an electron from the nearby peroxo group, leading to the formation of a superoxo intermediate. A rapid rearrangement results in the formation of O2, which is then desorbed from the surface. On the (010) surface, the V–O bond does not participate in the reaction, and a hydroperoxo intermediate (Bi–O–O–H) is formed after the adsorption of the second water molecule. Two protons are removed from the adsorbed water molecule in a stepwise manner, and O2 is released from the surface. For both the (010) and (011) surfaces, it is likely that O2 forms via peroxo species on BiVO4, similar to the process that occurs on TiO2.46,47 In the case of Bi4V2O11, because the VB is composed of hybrid O 2p and Bi 6s orbitals of the (Bi2O2)2+ layers, the intermediate species would be the hydroperoxo group.
Fig. 9 shows the action spectrum for overall pure-water splitting by ZnRh2O4/Ag/s-Bi4V2O11. We calculated the total number of incident photons from each LED light source and the O2-evolution rates from the slopes of the plots in Fig. 8a–d and then estimated the AQE values (Table S1 in the ESI 6†). Because the H2 and O2 evolution rates were 2:1 for all wavelengths, the obtained AQE values for O2 evolution were nearly identical to those for H2 evolution. The shape of the action spectrum for overall water splitting by ZnRh2O4/Ag/s-Bi4V2O11 was more similar to that of the PA spectrum of s-Bi4V2O11 than that of the UV-vis absorption spectrum of s-Bi4V2O11. An identical phenomenon was observed for the action spectrum of s-Bi4V2O11 for O2 evolution in aqueous Ce4+ solution (Fig. 7), indicating that the overall pure-water splitting reaction proceeded via the band-gap excitation of s-Bi4V2O11, as well as ZnRh2O4, which has an Eg value of 1.2 eV, and can therefore absorb longer wavelength light than s-Bi4V2O11. For this reason, the AQE values reflected the active photoabsorption capacity of s-Bi4V2O11.
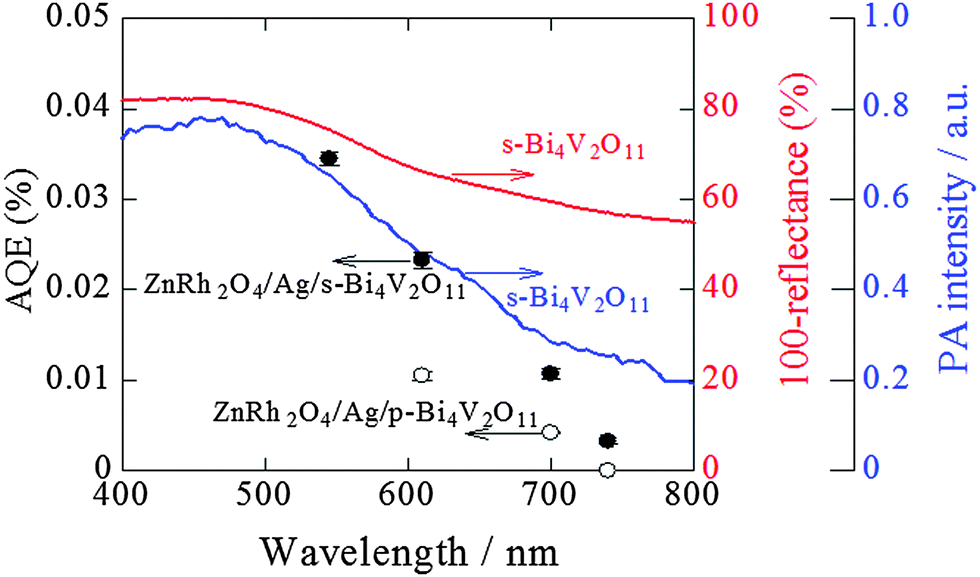 | ||
| Fig. 9 Action spectra for O2 evolution from pure water by ZnRh2O4/Ag/s-Bi4V2O11 (solid circles) and ZnRh2O4/Ag/p-Bi4V2O11 (open circles) under irradiation with LED light sources. Note that the AQE values for H2 evolution were almost identical to those for O2 evolution, as H2 and O2 evolution at a ratio of 2:1 was observed. The UV-visible absorption (red line) and PA (blue line) spectra of s-Bi4V2O11 are also shown. The AQE values of ZnRh2O4/Ag/p-Bi4V2O11 irradiated with 610 nm and 700 nm LED are cited from ref. 27. | ||
To confirm the liberation of O2 from water by ZnRh2O4/Ag/s-Bi4V2O11, water splitting with water containing 33% H218O under irradiation with 740 nm light (740 nm LED, the same intensity in Fig. 8d) was next examined (Fig. 10). The amount of 16O16O (calc. 16O16O) was obtained using the equation calc. 16O16O = obs. 16O16O − (obs. 14N14N/0.78) × 0.21, because the detected 14N14N (N2) originated from external air that entered the GC/MS system rather than from air dissolved in water, as was demonstrated in our previous report.26,27 Under irradiation with 740 nm light for 120 h, 16O16O, 16O18O, and 18O18O were detected at a ratio of 4.00:3.97:1.03, which was close to the theoretical ratio of 4:4:1 for overall water-splitting. In addition, the time course of total O2 evolution (16O16O + 16O18O + 18O18O) using water containing 33% H218O was consistent with that using normal H216O (solid circles with a solid line in Fig. 10, the same data as in Fig. 8d with the same symbol). Therefore, it was confirmed that O2 was evolved from water by ZnRh2O4/Ag/s-Bi4V2O11 under irradiation with 740 nm light. Based on these results, we confidently concluded that ZnRh2O4/Ag/s-Bi4V2O11 was capable of overall pure-water splitting under visible-light irradiation with a wavelength of 740 nm (Scheme S1 in the ESI 7†).
 | ||
| Fig. 10 GC/MS spectrometry results for the splitting of water containing 33% H218O by ZnRh2O4/Ag/s-Bi4V2O11 irradiated with 740 nm LED light. | ||
In a previous study,27 we attempted to scale up the overall pure-water splitting reaction by increasing the amounts of ZnRh2O4/Ag/p-Bi4V2O11 and pure water by a factor of 10 (600 mg of photocatalyst was suspended in 120 mL pure water). Using this approach, the amounts of evolved H2 and O2 increased ∼40 fold, although the amounts of photocatalyst and water were increased only tenfold. Further improvements in the yields of H2 and O2 evolution may be possible by optimization of the ratio of the photocatalyst and water, modifying the shape and/or volume of the reaction vessel, how the light is irradiated, and other chemical engineering-based modifications.
Conclusions
We prepared the solid-state heterojunction photocatalyst ZnRh2O4/Ag/s-Bi4V2O11 and demonstrated that this material simultaneously evolves H2 and O2 from pure water at a molar ratio of ∼2 to 1 under irradiation with nearly the entire range of visible light at wavelengths of up to 740 nm. Through the use of s-Bi4V2O11 in place of p-Bi4V2O11, the AQE for overall water splitting was increased and the sensitivity of the system was increased to wavelengths up to 740 nm. ZnRh2O4 was used as the H2 photocatalyst in the present system because it has an Eg value of 1.2 eV and is therefore capable of effectively utilizing both visible and infrared light. Despite this desirable property, ZnRh2O4 is not suitable for large-scale systems because it contains Rh, which is a rare chemical element. Moreover, in this system, HNO3 treatment is required for removing the excess Ag to achieve overall pure-water splitting, because Ag acts as a sacrificial agent for O2 evolution. Such treatment might damage ZnRh2O4 and Bi4V2O11, leading to decreased photocatalytic activity. To avoid such damage and potentially further improve the activity, we replaced Ag with gold (Au), because Au is stable and does not act as a sacrificial agent, and confirmed that an Au-inserted ZnRh2O4 and Bi4V2O11 (ZnRh2O4/Au/s-Bi4V2O11) system is capable of overall pure-water splitting. Detailed studies of ZnRh2O4/Au/s-Bi4V2O11 are currently underway in our laboratory.Acknowledgements
This study was partially supported by the Cooperative Research Program of Institute for Catalysis, Hokkaido University (Grant #15A1002). We express gratitude to Prof. Dr Tanaka and Ms Maruyama for their support with the back-reflection Laue pattern measurements. We also thank Mr G. Newton for the careful reading of the manuscript.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Sato, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2001, 105, 6061–6063 CrossRef CAS.
- K. Domen, A. Kudo, T. Onishi, N. Kosugi and H. Kuroda, J. Phys. Chem., 1986, 90, 292–295 CrossRef CAS.
- H. Kato, K. Asakusa and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- K. Domen, J. Kondo, M. Hara and T. Takata, Bull. Chem. Soc. Jpn., 2000, 73, 1307–1331 CrossRef CAS.
- A. Kudo, Int. J. Hydrogen Energy, 2007, 32, 2673–2678 CrossRef CAS.
- K. Maeda, R. Abe and K. Domen, J. Phys. Chem. Lett., 2011, 115, 3057–3064 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, T. Takata, M. Hara, N. Saito, K. Toda, Y. Inoue, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 20504–20510 CrossRef CAS PubMed.
- K. Teramura, K. Maeda, T. Saito, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2005, 109, 21915–21921 CrossRef CAS PubMed.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042–1048 CAS.
- H. Liu, J. Yuan, W. Shangguan and Y. Teraoka, J. Phys. Chem. C, 2008, 112, 8521–8523 CAS.
- N. Lei, M. Tanabe and H. Irie, Chem. Commun., 2013, 49, 10094–10096 RSC.
- P. Dhanasekaran and N. M. Gupta, Int. J. Hydrogen Energy, 2012, 37, 4897–4907 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng and Q. Yu, et al. , Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 1–6 CrossRef.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, J. Photochem. Photobiol., A, 2002, 148, 71–77 CrossRef CAS.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
- A. Kudo, Pure Appl. Chem., 2007, 79, 1917–1927 CrossRef CAS.
- S. Tanigawa and H. Irie, Appl. Catal., B, 2016, 180, 1–5 CrossRef CAS.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CAS.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- R. Kobayashi, S. Tanigawa, T. Takashima, B. Ohtani and H. Irie, J. Phys. Chem. C, 2014, 118, 22450–22456 CAS.
- R. Kobayashi, K. Kurihara, T. Takashima, B. Ohtani and H. Irie, J. Mater. Chem. A, 2016, 4, 3061–3067 CAS.
- N. Yasuda, M. Miyayama and T. Kudo, Mater. Res. Bull., 2001, 36, 323–333 CrossRef CAS.
- N. Murakami, O. O. P. Mahaney, T. Torimoto and B. Ohtani, Chem. Phys. Lett., 2006, 426, 204–208 CrossRef CAS.
- N. Murakami, O. O. P. Mahaney, R. Abe, T. Torimoto and B. Ohtani, J. Phys. Chem. C, 2007, 111, 11927–11935 CAS.
- G. Mairesse, P. Roussel, R. N. Vannier, M. Anne, C. Pirovano and G. Nowogrocki, Solid State Sci., 2003, 5, 851–859 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- Y. Takimoto, T. Kitta and H. Irie, Int. J. Hydrogen Energy, 2012, 37, 134–138 CrossRef CAS.
- Y. Lu, Y. Pu, J. Wang, C. Qin, C. Chen and H. J. Seo, Appl. Surf. Sci., 2015, 347, 719–726 CrossRef CAS.
- V. Thakral and S. Uma, Mater. Res. Bull., 2010, 45, 1250–1254 CrossRef CAS.
- S. Kumar and P. D. Sahare, NANO: Brief Rep. Rev., 2013, 8, 1350007 Search PubMed.
- X. Chen, J. Liu, H. Wang, Y. Ding, Y. Sun and H. Yan, J. Mater. Chem. A, 2013, 1, 877–883 CAS.
- I. Justicia, P. Ordejon, G. Canto, J. L. Mozos, J. Fraxedas, G. A. Battiston, R. Gerbasi and A. Figueras, Adv. Mater., 2002, 14, 1399–1402 CrossRef CAS.
- T. Ishii, H. Kato and A. Kudo, J. Photochem. Photobiol., A, 2004, 163, 181–186 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- H. W. Kang, S. N. Lim and S. B. Park, Int. J. Hydrogen Energy, 2012, 37, 4026–4035 CrossRef CAS.
- H. W. Kang and S. B. Park, Mater. Sci. Eng., B, 2016, 211, 67–74 CrossRef CAS.
- T. Uchihara, M. Matsumura, A. Yamamoto and H. Tsubimura, J. Phys. Chem., 1989, 93, 5870–5874 CrossRef CAS.
- J. F. Gomes, C. A. Martins, M. J. Giz, G. Tremiliosi-Filho and G. A. Camera, J. Catal., 2013, 301, 154–161 CrossRef CAS.
- J. Yang, D. Wang, X. Zhou and C. Li, Chem. – Eur. J., 2013, 19, 1320–1326 CrossRef CAS PubMed.
- T. Kisumi, A. Tsujiko, K. Murakoshi and Y. Nakato, J. Electroanal. Chem., 2003, 545, 99–107 CrossRef CAS.
- R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2014, 126, 1290–1298 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02903e |
| This journal is © the Owner Societies 2016 |