
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexibility unleashed in acyclic monoterpenes: conformational space of citronellal revealed by broadband rotational spectroscopy†
Sérgio R.
Domingos
 abc,
Cristóbal
Pérez
abc,
Chris
Medcraft‡
a,
Pablo
Pinacho
ad and
Melanie
Schnell
*abc
abc,
Cristóbal
Pérez
abc,
Chris
Medcraft‡
a,
Pablo
Pinacho
ad and
Melanie
Schnell
*abc
aMax Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: melanie.schnell@mpsd.mpg.de
bCenter for Free-Electron Laser Science, Luruper Chaussee 149, 22761 Hamburg, Germany
cThe Hamburg Centre for Ultrafast Imaging, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
dDepartment of Physical Chemistry and Inorganic Chemistry, Universidad de Valladolid, Valladolid, Spain
First published on 7th June 2016
Abstract
Conformational flexibility is intrinsically related to the functionality of biomolecules. Elucidation of the potential energy surface is thus a necessary step towards understanding the mechanisms for molecular recognition such as docking of small organic molecules to larger macromolecular systems. In this work, we use broadband rotational spectroscopy in a molecular jet experiment to unravel the complex conformational space of citronellal. We observe fifteen conformations in the experimental conditions of the molecular jet, the highest number of conformers reported to date for a chiral molecule of this size using microwave spectroscopy. Studies of relative stability using different carrier gases in the supersonic expansion reveal conformational relaxation pathways that strongly favour ground-state structures with globular conformations. This study provides a blueprint of the complex conformational space of an important biosynthetic precursor and gives insights on the relation between its structure and biological functionality.
1 Introduction
Understanding the interplay between the three-dimensional structure of a biomolecule and its functionality is critical for rationalising ligand–receptor mechanisms in biochemistry.1,2 Aspects such as conformational flexibility and chirality come into play if one is to understand these ligand–receptor interactions. Exploration and mapping of the potential energy surface of such key biomolecular systems is thus of utmost importance in revealing the underpinning mechanisms. Citronellal is a prominent acyclic monoterpene, predominantly formed by the secondary metabolism of plants, and a versatile precursor in many key biochemical reactions.3–5 A known example in acid catalysis is the Prins reaction of citronellal where the protonation of the carbonyl group results in the synthesis of several isopulegol isomers as a consequence of the many conformations that citronellal can adopt.6 The conformational flexibility of citronellal, arising from five C–C single bonds along the backbone (Fig. 1), results in many stable energy minima and thus plays a pivotal role in biosynthesis of monoterpenes. | ||
| Fig. 1 Schematic drawing of the chemical structure of citronellal, also known as rhodinal or 3,7-dimethyloct-6-en-1-al (C10H18O). | ||
Citronellal is a known insect repellent7,8 and the main component found in citronella oil giving it its distinctive lemon scent, a property that is regularly exploited in the cosmetics industry. Citronellal is chiral and as such exists as one of two stereoisomers. As an odourant molecule (−)-citronellal has been subject of multiple studies that aim at the elucidation of its binding interactions with olfactory receptors.9 The biological selectivity of the olfactory system is well known,10 for example the olfactory discrimination between the stereoisomers of carvone: while the R enantiomer smells like spearmint, the S enantiomer smells like caraway seeds. Studies suggest that selective docking to olfactory receptors is governed by the handedness and conformation of the ligand.10,11 In particular, recent investigations addressing binding efficiencies of citronellal to olfactory receptors indicate that the orientation of the reactive carbonyl group is crucial and governs the docking efficiency to the receptors.9,11
Rotational spectroscopy is a powerful spectroscopic method to obtain information on the structure of molecules in the gas phase with high precision and resolution.12,13 Recent advances such as the implementation of short and intense microwave chirps in broadband excitation spectroscopic schemes revolutionised the field of rotational spectroscopy.14 Using chirped-pulse Fourier-transform microwave (CP-FTMW) spectroscopy, it is possible to record rotational spectra of complex, flexible molecules spanning several GHz with a single acquisition. Complemented by substantial improvements in quantum–chemical calculations and the aid of automated spectral assignment programs,15,16 an unprecedented amount of structural information and internal dynamics of isolated molecules and clusters can now be extracted from a broadband rotational spectrum. We refer to some recent work on terpenes,15,17–20 pharmaceutical compounds,21,22 amino acids,23,24 sugars,25,26 water clusters27,28 and micro-solvated organic compounds.29–32 In addition, broadband rotational spectroscopy can now also be exploited to investigate chiral molecules and determine the absolute configuration of molecular species within chiral mixtures with high precision.33,34
In this work, we use CP-FTMW spectroscopy to investigate the conformational flexibility of citronellal under the conditions of a cold molecular beam. Citronellal, with its acyclic and extremely flexible structure, is a good candidate to benchmark both the quality of quantum–chemical calculations for the prediction of gas-phase conformations and the sensitivity of the spectroscopic technique to unambiguously identify molecular species with complex conformational spaces. In the following, we discuss the observed conformations, their structural similarities, stabilisation mechanisms and how they are energetically ordered. Additionally, we investigate how the relative population of each conformer is influenced by the carrier gas used for the expansion and tentatively devise conformational relaxation trajectories to interpret our observations. We find that citronellal has a preference for certain types of conformations and we discuss its implications regarding flexibility and functionality.
2 Methods
2.1 Experimental methods
The sample was purchased from Sigma-Aldrich (99% purity) and used without further purification. All the broadband rotational spectra used in this study were obtained with the Hamburg COMPACT spectrometer,35 which has been described elsewhere and thus only a brief description is given here. A supersonic expansion brings the sample into the vacuum chamber where a 4 μs chirp spanning 2 → 8 GHz polarises the ensemble of molecules. The seeding of molecules into a supersonic expansion is done using a pulsed nozzle (Parker General Valve Series 9) operating at 3 Hz with a constant flow of carrier gas (neon or argon) at stagnation pressures of 3 bar to generate a cold molecular jet. To create sufficient vapour pressure the sample was heated to 80 °C directly at the nozzle. The chirp is generated with an arbitrary waveform generator (AWG) and amplified in a 300 W traveling wave tube (TWT) amplifier before being broadcast into the vacuum chamber using a horn antenna. The horn antennas used here have higher gain and improved directionality compared with those used previously. The reader is referred to ref. 28 for further details on the horn antennas. Upon chirped microwave excitation we record the free induction decay (FID) of the macroscopic dipole moment of the ensemble of molecules. For this experiment, we utilised a new data acquisition scheme using the ‘fast frame’ option of the digital oscilloscope.28 In short, eight back-to-back excitation chirps are performed on each pulse of sample and the subsequent eight FID acquisitions are co-added and averaged. This scheme decreases the measurement time and sample consumption, resulting in an effective repetition rate of 24 Hz for the experiment. We record 40 μs of the FIDs which, after a Fourier transformation (FT), gives us a resolution of 25 kHz in our microwave spectrum. 1.5 million FIDs (equivalent to 14 hours of measurement time) were co-added to obtain the final spectrum.2.2 Computational methods
The conformational space of citronellal was explored in two stages. Initially, a semi-empirical search was performed using the Austin Model 1 (AM1) that is implemented in Spartan.36 More than one hundred conformations were obtained and further re-optimised at higher levels of theory using Gaussian09.37 Fifteen conformations were found within an energy cut-off of 6 kJ mol−1 using the B3LYP-D3 (Becke, three-parameter, Lee–Yang–Parr) exchange–correlation functional, including Grimme's empirical dispersion correction, and the aug-cc-pVTZ basis set. Additionally, those fifteen structures were also optimised using second-order Moller–Plesset perturbation theory (MP2) with the 6-311++G** basis set. Frequency calculations were performed to ascertain the valid energy minima and obtain the zero-point corrected energies (see ESI†).Secondly, a more localised and systematic search of the conformational space was performed at the B3LYP/3-21G level of theory by scanning combinations of two dihedral angles in steps of 10 degrees and allowing the remaining coordinates to relax during the optimisation at each step. The potential energy surface obtained from one of these scans is shown in Fig. 2 (top panel). Cross sections of the maps were performed at the B3LYP-D3/aug-cc-pVTZ level of theory (see Fig. 2, lower panels). This methodology revealed two additional conformations, XI and XII, that were not identified semi-empirically and have energies within the 6 kJ mol−1 energy cut off.
 | ||
| Fig. 2 Top panel: Potential energy landscape of citronellal obtained at the B3LYP/3-21G level of theory by scanning the dihedral coordinates ϕ1 and ϕ2 (indicated in blue and red in the molecular structure, respectively) in steps of 10°. Lower panels: Linear transit scans of ϕ1 (in blue) and ϕ2 (in red) obtained at the B3LYP-D3/6-311++G** level of theory. | ||
3 Results and discussion
3.1 Conformational space and assignment
In Fig. 3 we show a portion of the measured rotational spectrum (Ne-seeded, upper trace, in black) plotted against the fitted rotational lines of fifteen conformations of citronellal (lower traces, in colours). The assignment of observed experimental lines to rotational transitions of a certain conformer was performed through a recurrent fit based on Watson's A-reduced rigid rotor Hamiltonian as implemented in PGOPHER38 and JB95.39 As a result, rotational constants (A, B, C) and quartic centrifugal distortion constants (DJ, DK, DJK, δJ, δK) were well determined for most of the conformers. The experimentally determined parameters are shown in Table 1. We note that line splittings due to methyl group internal rotation are present for a number of conformers. However, further analysis concerning internal dynamics is out of the scope of this paper. | ||
| Fig. 3 Portion of the 2–8 GHz spectrum of citronellal (1.5 M acquisitions, 14 h of measurement time). The upper trace (in black) shows the experimental spectrum obtained using neon as a carrier gas. The lower traces (in colour) represent simulations employing the fitted parameters of citronellal as given in Table 1. Intensities are based on calculated dipole moment components and estimated relative populations. | ||
| I | II | III | IV | VI | |
|---|---|---|---|---|---|
| A (MHz) | 1317.4649(13) | 1580.7079(14) | 1546.5213(18) | 1362.2074(34) | 1660.9330(646) |
| B (MHz) | 692.52126(73) | 522.02024(65) | 567.18188(53) | 584.81168(50) | 496.59102(68) |
| C (MHz) | 558.53524(84) | 472.84994(84) | 495.88040(54) | 481.86671(44) | 451.12094(65) |
| D J (kHz) | 0.209(16) | 0.2442(65) | 0.3316(42) | 0.2551(36) | 0.5479(97) |
| D JK (kHz) | −0.188(71) | −1.185(23) | −1.706(33) | −0.595(27) | −6.84(11) |
| D K (kHz) | 0.454(69) | 3.635(70) | 5.19(17) | 2.33(71) | −0.3979(650) |
| δ J (kHz) | — | 0.0487(29) | 0.0831(30) | 0.0822(23) | −0.1214(36) |
| δ K (kHz) | — | — | — | 0.371(71) | 0.0211(50) |
| a/b/c | n/y/y | n/y/n | y/y/n | y/y/n | y/y/y |
| μ a/μb/μc† | −0.13/1.37/−1.81 | 0.12/−2.20/−0.64 | −0.79/−2.30/−0.55 | 2.91/1.37/−0.03 | −2.61/1.78/−1.25 |
| N | 53 | 50 | 51 | 49 | 31 |
| σ (kHz) | 12.3 | 14.5 | 9.1 | 6.3 | 8.9 |
| κ | −0.647 | −0.911 | −0.864 | −0.766 | −0.925 |
| VII | IX | X | XI | XII | |
|---|---|---|---|---|---|
| A (MHz) | 1605.4178(12) | 1893.5114(11) | 1820.05(40) | 3118.2631(31) | 2574(10) |
| B (MHz) | 509.32052(60) | 432.31046(42) | 447.53527(86) | 344.60535(30) | 356.34111(43) |
| C (MHz) | 418.49286(46) | 375.05049(40) | 424.92546(89) | 330.63279(31) | 352.28168(41) |
| D J (kHz) | 0.2299(45) | 0.0765(32) | 0.0957(49) | 0.0453(17) | 0.0315(16) |
| D JK (kHz) | −0.366(27) | −0.549(12) | — | −0.578(51) | — |
| D K (kHz) | — | 2.94(11) | — | — | — |
| δ J (kHz) | 0.0754(29) | — | — | — | — |
| δ K (kHz) | — | — | — | — | — |
| a/b/c | y/y/y | n/y/y | y/n/n | y/n/n | y/n/n |
| μ a/μb/μc† | −1.35/−1.33/1.22 | −0.22/2.10/−1.66 | −3.10/−0.85/−0.73 | −1.67/−0.76/1.99 | −1.70/0.01/1.85 |
| N | 77 | 64 | 28 | 43 | 26 |
| σ (kHz) | 13.0 | 9.7 | 14.9 | 9.3 | 8.8 |
| κ | −0.847 | −0.925 | −0.968 | −0.990 | −0.996 |
| XIII | XIV | XV | XVI | XVII | |
|---|---|---|---|---|---|
| A (MHz) | 2423.0569(23) | 2623.4(49) | 1679.6748(77) | 1513.7868(22) | 1434.95789(53) |
| B (MHz) | 380.05537(50) | 350.1327(11) | 576.72284(69) | 589.84641(60) | 634.32380(91) |
| C (MHz) | 352.68703(46) | 339.9565(10) | 523.05132(69) | 452.20259(66) | 533.68888(54) |
| D J (kHz) | 0.0597(30) | 0.0698(34) | 0.1399(73) | 0.0358(66) | 0.183(14) |
| D JK (kHz) | — | −1.22(15) | — | 0.589(64) | — |
| D K (kHz) | — | — | — | — | — |
| δ J (kHz) | — | 0.0125(36) | — | — | 0.0280(81) |
| δ K (kHz) | — | — | — | — | — |
| a/b/c | y/y/y | y/n/n | y/n/y | y/y/y | n/y/y |
| μ a/μb/μc† | −2.24/−0.87/1.55 | −1.39/−0.43/−2.05 | 2.03/0.06/−1.81 | 1.74/1.82/−0.73 | 0.04/−2.26/−1.41 |
| N | 54 | 20 | 21 | 41 | 21 |
| σ (kHz) | 16.1 | 8.4 | 8.7 | 12.8 | 5.8 |
| κ | −0.974 | −0.991 | −0.907 | −0.741 | −0.777 |
A complete assignment of the fifteen reported conformers was facilitated using the automated spectral fitting program AUTOFIT16 that uses estimated rotational constants and dipole moments as a starting point and delivers refined candidates based on a least-squares fitting algorithm. We note that within the set of fifteen assigned conformers, in addition to their distinct rotational constants, unique signatures differentiate them solely based on the type of spectrum observed (see Table 1). As an example, conformers I, II and III have distinct spectral types: whilst conformer I shows both b- and c-type rotational transitions, conformer II reveals only b-type and conformer III shows a- and b-type transitions. This is a consequence of the extreme flexibility of the molecule itself expressed by the changing orientation of the dipole moment. The large number of flexible coordinates, that are dependent on each other, results in a rich and complex conformational landscape. Therefore, the distinct types of rotational transitions observed for different conformers of citronellal facilitate the reliable assignment of multiple conformations in very dense and congested spectra as the one investigated here.
3.2 Theoretical predictions and structural similarities
Our theoretical predictions using quantum–chemical calculations anticipate a total of seventeen conformations within an energetic window of 6 kJ mol−1. In Fig. 4 we show the calculated structures, depicting also the relative energies and strongest intramolecular interactions (dashed lines) present for each conformer. We find that the predicted conformations can be grouped based on their backbone structural similarities, i.e., dihedral angles with similar values. We thus define families of conformers based on their similarity for the first two dihedral angles (ϕ0, ϕ1). These dihedrals define the general folding of the molecule into a more prolate or globular shape while the precise conformation is determined by structural changes localised on the aldehyde terminus. With this condition we find a total of seven families grouped as follows: (I, IV, V, XVII), (II, III, VI, X), (VII), (VIII), (IX, XI, XII, XVI), (XIII, XIV) and (XV). Table S1 in the ESI† contains all the dihedral angles considered here. It is interesting to note that within their respective families, the following pairs of conformers (I, IV), (II, X) and (III, VI) can be interconverted between one another by a single rotation along ϕ4 (C6–C7–C8–O9), and allowing a relaxation of all other dihedral coordinates to take place (see Table S1 and Fig. S3 of the ESI†). This rotation effectively controls the relative orientation of the carbonyl functional group with respect to the backbone. Conformers I, II and III, where the oxygen lone pairs are pointing inwards in a folded-like conformation, result in an overall globular structure. The rotated equivalents IV, X and VI, respectively, have the carbonyl group pointing outwards assuming an exposed prolate-like structure, as indicated by the asymmetry parameters (κ) of these conformers (see Table 1). | ||
| Fig. 4 Molecular structures of seventeen conformations of citronellal obtained from B3LYP-D3/aug-cc-pVTZ calculations. The structures are ordered based on their predicted relative zero-point corrected energies (in kJ mol−1 and cm−1, respectively). Results of MP2 calculations are summarised in Table S2 of the ESI.† The dashed coloured lines depict the predominant intramolecular interactions. | ||
Both quantum–chemical methods (B3LYP-D3 and MP2, see Table S2 in the ESI†) predict the global minimum to be conformer I, which is the conformation with the lowest absolute value for κ, reflecting its pronounced globular shape (see Fig. 4). The strong b- and c-type transitions observed (see Table 1 and ESI† for the full broadband spectrum) also support the preliminary assignment of conformer I as the global minimum.
3.3 Conformational relaxation
All fifteen fitted spectra can be assigned to calculated structures as the numbering in Table 1 and Fig. 4 indicate. Two predicted low-energy conformations (conformers V and VIII) could not be identified in the experimental spectrum. A tentative explanation for this has been devised by evaluating potential cooling pathways that could allow the interconversion between conformers V and VIII to other lower-lying conformations. To plot such cooling pathways we performed systematic scans of candidate dihedral coordinates that could facilitate interconversion between conformers. In Fig. 5 we show the results of a suggested cooling trajectory that allows conformational relaxation of conformer V into conformer I, and conformer VIII into conformer II. Interestingly, we find that for both cases, a two-step cooling pathway through sequential rotation of dihedral coordinates is possible. Panels A and C of Fig. 5 show that (1) concerted relaxation by simultaneous rotation of two dihedrals is not energetically favourable compared with that of sequential relaxation steps, and (2) multiple trajectories require attention. We find that for each conformation, the depicted relaxation trajectories are energetically favoured. In Fig. 5 (panel B) we show that conformer V may transit from its initial conformation to an intermediate state through rotation of ϕ3. Consequently, this intermediate state V′ relaxes to the final state as conformation I by rotation of ϕ4. In a similar way, conformation VIII initially relaxes to an intermediate state VIII′ through rotation of ϕ3, following rotation of ϕ2 leading to a new lower local minimum as conformation II (see panel D). The magnitude of the predicted interconversion energy barriers reported here are in line with previously reported studies.40–42 Moreover, we note that from a purely energetic point of view, our tentative cooling trajectories (V → I and VIII → II) are in agreement with the energy ordering for conformers obtained from the B3LYP-D3 calculation.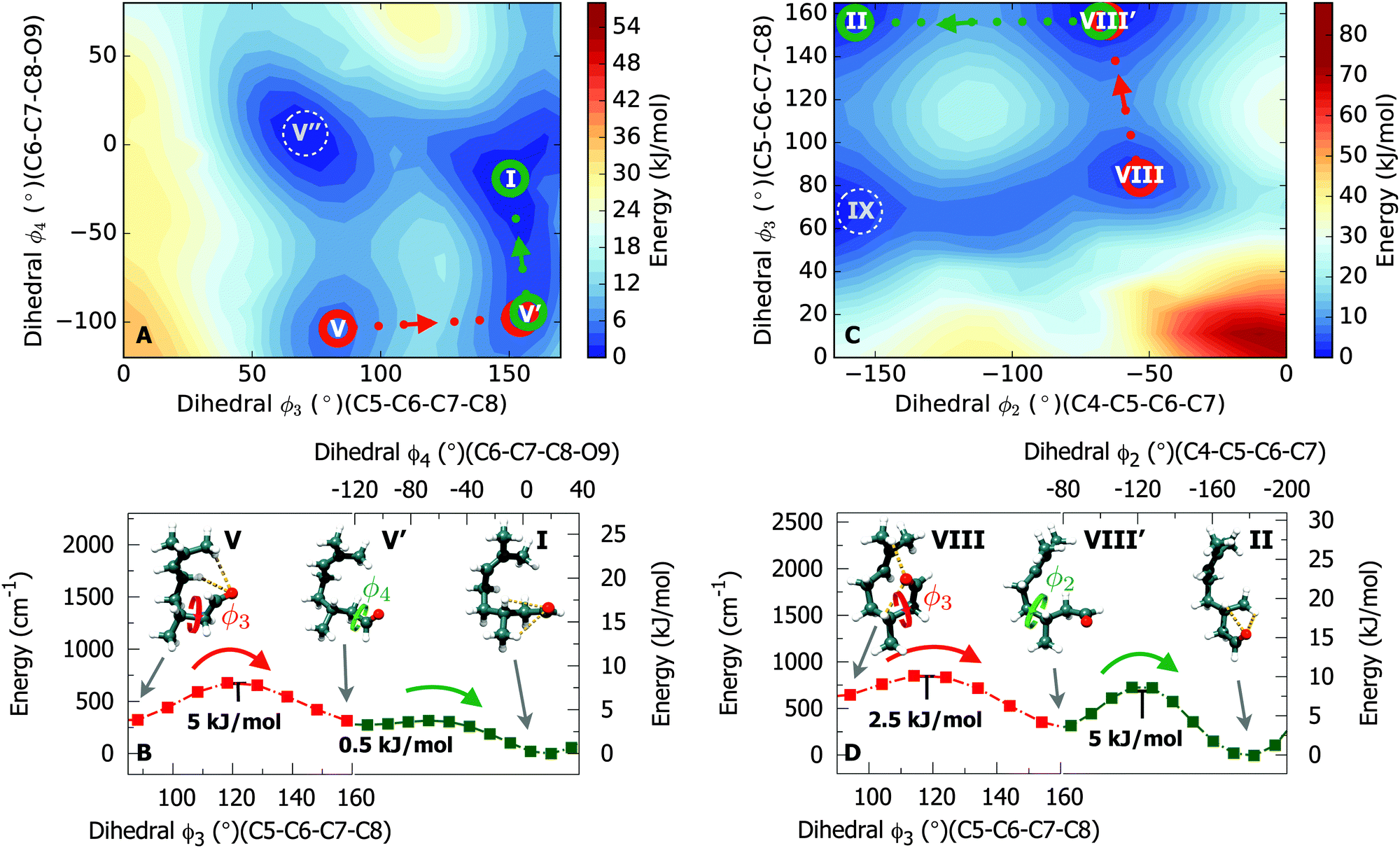 | ||
| Fig. 5 Conformational relaxation pathways for conformer V (panels A and B) and conformer VIII (panels C and D). The potential energy surfaces (panels A and C) were predicted at the B3LYP/3-21G level of theory. Panels B and D show the relevant cross sections of the surfaces obtained at the B3LYP-D3/6-311++G** level of theory. Relaxation pathways involving two sequential steps were plotted for conformers V → I and VIII → II, respectively, using rotations of dihedral coordinates in a sequential manner. | ||
3.4 Relative stability of the energy minima
As Fig. 2 illustrates, the potential-energy landscape of citronellal is shaped by deep pockets, where conformers are confined by energy barriers that in most cases are higher than 10 kJ mol−1. Furthermore, nine low-energy minima are predicted by theory within an energy window of less than 3 kJ mol−1, an indication that a correlation between the predicted energy ordering and a confident assignment of a global minimum is not trivial. Therefore, to gain insight into the relative stabilities of the energy minima, we performed the same experiment but using argon as the seeding gas for the supersonic expansion.Previous studies have shown that heavier inert gases have an enhanced cooling efficiency in supersonic expansions due to the larger collision energies they provide.43,44 Consequently, an argon-seeded expansion is expected to favour conversion of higher energy conformers to lower ones, facilitating an assignment of the ground-state structure(s) and assisting in quantitatively evaluate our quantum–chemical predictions. In Fig. 6 we show four sections (panels A → D) of the broadband spectrum of citronellal using neon (in red) and argon (in blue) as carrier gases and relevant rotational transitions of conformers I, II, III, IV, VI and X. These six conformations can undergo interconversion by means of rotation of a single dihedral coordinate (ϕ4). Interconversion barriers for IV → I, VI → III and X → II can be found in Fig. S3 of the ESI.† Panel A displays two rotational transitions of conformer III (JKaKc ← JKaKc′: 321 ← 212 and 625 ← 524) and one transition of conformer IV (625 ← 524). Whilst the rotational transitions from conformer III can be observed in both neon- and argon-seeded spectra, the transition from conformer IV is only observed in the neon spectrum thus indicating that IV has relaxed to a lower energy conformation. Similar conclusions can be drawn for the remaining conformers by evaluating the panels B → D. Based on these observations, we conclude that improved cooling is achieved through collisions with argon during the supersonic expansion and thus conformational relaxation of conformers IV, VI and X (in addition to V and VIII) is facilitated, strongly suggesting that conformers I, II and III are the lower-lying energy conformations.
 | ||
| Fig. 6 Coloured traces show portions of the CP-FTMW spectra of 1.5 million co-added FIDs using neon (red trace) and argon (blue trace) as backing gas in the supersonic expansion. The black traces show simulations based on the experimental molecular parameters (Table 1) for the numbered conformers. | ||
Comparison of the rotational spectra with Ne and Ar provides additional information: whilst in the Ne spectrum we observe fifteen conformations, only eight of those fifteen conformers (I, II, III, VII, XI, XIII, XV and XVI) are observed in the Ar spectrum. Interestingly, we find that in general only one conformer is observed in the Ar spectrum for each of the families. Particular cases are those of conformers II and III, and of conformers XI and XVI, belonging to the same families, which are observed in both Ne and Ar spectra (see Table S1 of the ESI†). Regardless of their similarities, conformers II and III are separated by an energy barrier of 10 kJ mol−1 (see Fig. S2, panel D in the ESI†) and thus conformational relaxation is not achieved through higher energy collisions with Ar compared with that of Ne. The same stands for conformations XI and XVI, where dihedrals ϕ2, ϕ3 and ϕ4 assume very distinct values, resulting in higher energy barriers not favouring conformational relaxation. These observations are nevertheless consistent with energetically-favoured relaxation trajectories that allow interconversion between conformers of the same family due to their intrinsic structural similarities. Moreover, it becomes more evident that the relative orientation of the terminus containing the aldehyde functional group – reflected mostly in the dihedral coordinates ϕ3 and ϕ4 – is responsible for most of the conformational richness of citronellal.
4 Conclusions and outlook
We have successfully used broadband rotational spectroscopy and quantum–chemical calculations to probe the complex conformational space of citronellal and assigned fifteen stable conformations in the cold conditions of a molecular jet. To date, structural studies on acyclic terpenes were limited due to the many degrees of freedom allowed by their backbone configuration. This study demonstrates the outstanding performance of the technique to resolve the structure of flexible molecules. We have evaluated the relative stability of energy minima by comparing rotational spectra of citronellal using different seeding gases. Evidence is found for enhanced cooling efficiency in an Ar-seeded expansion compared with that of Ne, which facilitates the assignment of the global minimum structure of citronellal, and aids in the prediction of conformational relaxation trajectories. Globular structures, instead of linear ones, are found to be more stable due to intramolecular stabilisation of the aldehyde functional group.Putative bindings of citronellal to olfactory receptors have been previously investigated and assume a docking with different hydrogen bond connections to the aldehyde group.9 The relative orientation of the aldehyde moiety with respect to the rest of the molecule is critical in establishing a relation between conformation and functionality.11 Our findings reveal that citronellal has strong preference for conformations that have the aldehyde moiety pointing inwards, assuming a stable chair-like folded conformation, in prejudice to linear prolate-like forms with the aldehyde group pointing outwards. We also note that regardless if the preferred conformations are of the folded or unfolded type, the great majority of observed species utilises the aldehyde functional group to stabilise the overall structure, resulting in less electronic density available for intermolecular interactions to take place. However, we show that rotation of the aldehyde terminus can be achieved with significant ease for many conformers, resulting in a rich and rather complex conformational space. This appears to be in contrast with a recent microwave study on another acyclic monoterpene, linalool, that has revealed a single conformer under similar experimental conditions.45 Our observations are also consistent with previous reports that show that the origin of isopulegol isomer formation is strongly dependent on the energetically most-favoured chair-like conformations of (S)-citronellal.6
The role of conformational flexibility and molecular handedness in the selectivity of olfactory receptors upon docking of ligands such as citronellal is yet not fully understood. This case study should not be considered exceptionally unusual due to its large conformational flexibility, but an example of how maneuverable and complex potential-energy landscapes can be even for molecules of this size. Moreover, this study provides particular structural insights that may aid in assessing the roles of conformational flexibility and docking efficiency of systems with bio-selectivity.
Acknowledgements
We acknowledge financial support by the Deutsche Forschungsgemeinschaft in the context of the priority program SPP1807 (SCHN1280/4-1). Furthermore, this work has been supported by the excellence cluster “The Hamburg Centre for Ultrafast Imaging – Structure, Dynamics and Control of Matter at the Atomic Scale” of the Deutsche Forschungsgemeinschaft. S. R. D. and C. P. acknowledge a Postdoctoral Research Fellowship from the Alexander von Humboldt Foundation. We acknowledge the use of the GWDG computer cluster.References
- B. Malnic, J. Hirono, T. Sato and L. B. Buck, Cell, 1999, 96, 713–723 CrossRef CAS PubMed.
- W. B. Floriano, N. Vaidehi, W. A. Goddard, M. S. Singer and G. M. Shepherd, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10712–10716 CrossRef CAS.
- G. Siedenburg, M. Breuer and D. Jendrossek, Appl. Microbiol. Biotechnol., 2012, 97, 1571–1580 CrossRef PubMed.
- G. Siedenburg, D. Jendrossek, M. Breuer, B. Juhl, J. Pleiss, M. Seitz, J. Klebensberger and B. Hauer, Appl. Environ. Microbiol., 2012, 78, 1055–1062 CrossRef CAS PubMed.
- P. Mäki-Arvela, N. Kumar, V. Nieminen, R. Sjöholm, T. Salmi and D. Y. Murzin, J. Catal., 2004, 225, 155–169 CrossRef.
- S. C. Hammer, A. Marjanovic, J. M. Dominicus, B. M. Nestl and B. Hauer, Nat. Chem. Biol., 2015, 11, 121–126 CrossRef CAS PubMed.
- U. Sakulku, O. Nuchuchua, N. Uawongyart, S. Puttipipatkhachorn, A. Soottitantawat and U. Ruktanonchai, Int. J. Pharm., 2009, 372, 105–111 CrossRef CAS PubMed.
- W. S. Leal, Y.-M. Choo, P. Xu, C. S. B. da Silva and C. Ueira-Vieira, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18704–18709 CrossRef CAS PubMed.
- A. Stary, C. Suwattanasophon, P. Wolschann and G. Buchbauer, Biochem. Biophys. Res. Commun., 2007, 361, 941–945 CrossRef CAS PubMed.
- J. C. Brookes, A. Horsfield and A. Stoneham, J. R. Soc., Interface, 2009, 6, 75–86 CrossRef CAS PubMed.
- R. C. Araneda, A. D. Kini and S. Firestein, Nat. Neurosci., 2000, 3, 1248–1255 CrossRef CAS PubMed.
- C. H. Townes and A. L. Schawlow, Microwave Spectroscopy, Dover, New York, 1975 Search PubMed.
- W. Gordy and R. L. Cook, Microwave Molecular Spectra, Wiley, New York, 1984 Search PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79, 053103 CrossRef PubMed.
- D. Schmitz, V. A. Shubert, T. Betz and M. Schnell, Front. Chem., 2015, 3, 15 Search PubMed.
- N. A. Seifert, I. A. Finneran, C. Pérez, D. P. Zaleski, J. L. Neill, A. L. Steber, R. D. Suenram, A. Lesarri, S. T. Shipman and B. H. Pate, J. Mol. Spectrosc., 2015, 312, 13–21 CrossRef CAS.
- J. R. A. Moreno, T. R. Huet and J. J. L. Gonzalez, Struct. Chem., 2012, 24, 1163–1170 CrossRef.
- D. Schmitz, V. A. Shubert, B. M. Giuliano and M. Schnell, J. Chem. Phys., 2014, 141, 034304 CrossRef CAS PubMed.
- C. Medcraft and M. Schnell, Z. Phys. Chem., 2015, 230, 1 CrossRef.
- V. A. Shubert, D. Schmitz, C. Medcraft, A. Krin, D. Patterson, J. M. Doyle and M. Schnell, J. Chem. Phys., 2015, 142, 214201 CrossRef PubMed.
- M. Varela, C. Cabezas, J. C. Lopez and J. L. Alonso, J. Phys. Chem. A, 2013, 117, 13275–13278 CrossRef CAS PubMed.
- T. Betz, S. Zinn and M. Schnell, Phys. Chem. Chem. Phys., 2015, 17, 4538–4541 RSC.
- C. Pérez, S. Mata, C. Cabezas, J. C. Lopez and J. L. Alonso, J. Phys. Chem. A, 2015, 119, 3731–3735 CrossRef PubMed.
- C. Pérez, S. Mata, S. Blanco, J. C. Lopez and J. L. Alonso, J. Phys. Chem. A, 2011, 115, 9653–9657 CrossRef PubMed.
- E. J. Cocinero, A. Lesarri, P. Écija, F. J. Basterretxea, J.-U. Grabow, J. A. Fernandez and F. Castaño, Angew. Chem., Int. Ed., 2012, 51, 3119–3124 CrossRef CAS PubMed.
- E. J. Cocinero, A. Lesarri, P. Écija, A. Cimas, B. G. Davis, F. J. Basterretxea, J. A. Fernandez and F. Castaño, J. Am. Chem. Soc., 2013, 135, 2845–2852 CrossRef CAS PubMed.
- C. Pérez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Science, 2012, 336, 897–901 CrossRef PubMed.
- C. Pérez, S. Lobsiger, N. A. Seifert, D. P. Zaleski, B. Temelso, G. C. Shields, Z. Kisiel and B. H. Pate, Chem. Phys. Lett., 2013, 571, 1–15 CrossRef.
- J. Thomas, O. Sukhorukov, W. Jäger and Y. Xu, Angew. Chem., Int. Ed., 2014, 53, 1156–1159 CrossRef CAS PubMed.
- S. Ghosh, J. Thomas, W. Huang, Y. Xu and W. Jäger, J. Phys. Chem. Lett., 2015, 6, 3126–3131 CrossRef CAS.
- C. Pérez, J. L. Neill, M. T. Muckle, D. P. Zaleski, I. Peña, J. C. Lopez, J. L. Alonso and B. H. Pate, Angew. Chem., Int. Ed., 2015, 54, 979–982 CrossRef PubMed.
- C. Pérez, A. Krin, A. L. Steber, J. C. Lopez, Z. Kisiel and M. Schnell, J. Phys. Chem. Lett., 2016, 7, 154–160 CrossRef PubMed.
- D. Patterson, M. Schnell and J. M. Doyle, Nature, 2013, 497, 475–477 CrossRef CAS PubMed.
- V. A. Shubert, D. Schmitz, C. Pérez, C. Medcraft, A. Krin, S. R. Domingos, D. Patterson and M. Schnell, J. Phys. Chem. Lett., 2016, 7, 341–350 CrossRef CAS PubMed.
- D. Schmitz, V. Alvin Shubert, T. Betz and M. Schnell, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS.
- Spartan 10, http://www.wavefun.com.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, O. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian 09, 2009 Search PubMed.
- C. M. Western, PGOPHER, a Program for Simulating Rotational, Vibrational and Electronic Structure, http://pgopher.chm.bris.ac.uk.
- D. Plusquellic, JB95 Spectral fitting program, 2001.
- P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1998, 120, 10724–10732 CrossRef CAS.
- G. M. Florio, R. A. Christie, K. D. Jordan and T. S. Zwier, J. Am. Chem. Soc., 2002, 124, 10236–10247 CrossRef CAS PubMed.
- J. L. Alonso, C. Pérez, M. Eugenia Sanz, J. C. Lopez and S. Blanco, Phys. Chem. Chem. Phys., 2009, 11, 617–627 RSC.
- R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, J. Chem. Phys., 1990, 93, 3142–3150 CrossRef CAS.
- U. Erlekam, M. Frankowski, G. von Helden and G. Meijer, Phys. Chem. Chem. Phys., 2007, 9, 3786–3789 RSC.
- H. V. L. Nguyen, H. Mouhib, S. Klahm, W. Stahl and I. Kleiner, Phys. Chem. Chem. Phys., 2013, 15, 10012–10018 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fi. See DOI: 10.1039/c6cp02876d |
| ‡ Current address: School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| This journal is © the Owner Societies 2016 |