
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-field thermal mixing in [1-13C] pyruvic acid for brute-force hyperpolarization
David T.
Peat
a,
Matthew L.
Hirsch
b,
David G.
Gadian
a,
Anthony J.
Horsewill
a,
John R.
Owers-Bradley
*a and
James G.
Kempf
 *b
*b
aSchool of Physics & Astronomy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: John.Owers-Bradley@nottingham.ac.uk
bBruker Biospin Corp., 15 Fortune Drive, Billerica, MA 01821, USA. E-mail: James.Kempf@bruker.com
First published on 24th June 2016
Abstract
We detail the process of low-field thermal mixing (LFTM) between 1H and 13C nuclei in neat [1-13C] pyruvic acid at cryogenic temperatures (4–15 K). Using fast-field-cycling NMR, 1H nuclei in the molecule were polarized at modest high field (2 T) and then equilibrated with 13C nuclei by fast cycling (∼300–400 ms) to a low field (0–300 G) that activates thermal mixing. The 13C NMR spectrum was recorded after fast cycling back to 2 T. The 13C signal derives from 1H polarization via LFTM, in which the polarized (‘cold’) proton bath contacts the unpolarised (‘hot’) 13C bath at a field so low that Zeeman and dipolar interactions are similar-sized and fluctuations in the latter drive 1H–13C equilibration. By varying mixing time (tmix) and field (Bmix), we determined field-dependent rates of polarization transfer (1/τ) and decay (1/T1m) during mixing. This defines conditions for effective mixing, as utilized in ‘brute-force’ hyperpolarization of low-γ nuclei like 13C using Boltzmann polarization from nearby protons. For neat pyruvic acid, near-optimum mixing occurs for tmix ∼ 100–300 ms and Bmix ∼ 30–60 G. Three forms of frozen neat pyruvic acid were tested: two glassy samples, (one well-deoxygenated, the other O2-exposed) and one sample pre-treated by annealing (also well-deoxygenated). Both annealing and the presence of O2 are known to dramatically alter high-field longitudinal relaxation (T1) of 1H and 13C (up to 102–103-fold effects). Here, we found smaller, but still critical factors of ∼(2–5)× on both τ and T1m. Annealed, well-deoxygenated samples exhibit the longest time constants, e.g., τ ∼ 30–70 ms and T1m ∼ 1–20 s, each growing vs. Bmix. Mixing ‘turns off’ for Bmix > ∼100 G. That T1m ≫ τ is consistent with earlier success with polarization transfer from 1H to 13C by LFTM.
Introduction
Low-field thermal mixing (LFTM) is the process by which dissimilar spins in a solid sample are brought to mutual equilibrium by exposure to a magnetic field small enough that their magnetic resonance lineshapes come into overlap.1–3 In this way, mutual spin flips can occur with conservation of energy, allowing the noted equilibration, e.g., among heteronuclei in NMR (nuclear magnetic resonance). This phenomenon is of special recent interest as a way to hyperpolarize low-γ nuclear spins (γ = gyromagnetic ratio) such as 13C, 15N or 31P, using spin order originally established in high-γ nuclei like 1H.4,5 From a thermodynamic viewpoint, LFTM is a process of rapid cooling, in which a highly polarized (cold) bath of spins is made to strongly couple with poorly polarized (hot) spins. For mixing 1H with low-γ nuclei, the protons have much larger specific heat (proportional to γ2), and thus dominate in establishing the final ‘spin temperature’.1–3 The result is a cold (highly polarized) set of low-γ spins.Hyperpolarization is a potentially transformative approach to dramatically enhance sensitivity in solution NMR and magnetic resonance imaging (MRI). Orders-of-magnitude gains are available from solids-into-liquids methods like the ‘brute-force’ approach4–7 (yielding 102 to 104-fold enhancements) and dissolution dynamic nuclear polarization8 (d-DNP, for >104-fold). Methods to hyperpolarize directly in the liquid state are also promising, especially using parahydrogen (p-H2).9,10 This can yield >104-fold polarization gains in molecules reacting with p-H2,11 but has limits due to chemical specificity in transferring p-H2 spin order into a molecule of interest. Direct DNP in liquids is also well known, but typically limited to non-polar solvents and lower fields,12,13 while optically pumped methods also warrant interest,14 as are approaches in which molecular carriers of hyperpolarized xenon can enable in vivo imaging applications.15–19
Among generally applicable methods of hyperpolarization, the brute-force approach is a natural fit with LFTM. In brute force, a large Boltzmann polarization is built up on protons at high field and low temperature (e.g., B = 14 T, T = 100 mK to 2 K). Protons are the preferred starting point, offering polarization build-up that can be >10-fold faster than for low-γ nuclei. And yet the ultimate goal is usually hyperpolarization of a low-γ species like 13C for use as an ultrasensitive, background-free agent for MRI.20–22 LFTM is a way to get the best of both worlds without resorting to an NMR pulse sequence, which could likewise effect a polarization transfer but at the expense of limiting the amount of sample by confinement to a radiofrequency (RF) coil.
In recent experiments,5,7 brute-force was married with LFTM in application to 13C-labeled pyruvic acid. Protons equilibrated to noted high-B, low-T conditions were used to hyperpolarize 13C when the sample was ejected from the polarizer and through a low field region (<100 G). After either immediate aqueous dissolution,5 or off-site transport followed by dissolution,7 hyperpolarized 13C was observed by solution NMR. Enhancements of 100–1000× were obtained in pyruvic acid and other molecules. That corresponded to up to 0.2% 13C polarization, with potential to reach >10%, which is more than 104 times more than thermal equilibrium levels (∼0.0001% to 0.001%) for 13C in MRI and solution NMR.
Pyruvic acid is the hottest current target for medical imaging with hyperpolarization.23 It and other small-molecule metabolites have rates of cellular uptake and chemical conversion that vary with tissue health.20–22 Tracking such processes by MRI already enables detection and grading of various cancers23–28 or cardiac function.29–31 These approaches depend absolutely on hyperpolarization. Though labelling with a low-γ nucleus provides the huge advantages of chemically specific, background-free detection, as well as much longer polarization lifetimes, it also requires hyperpolarization to overcome the γ2-to-γ3 dependence of sensitivity, on top of the disadvantage of low metabolite concentration.
Here, we define conditions necessary for effective LFTM in [1-13C] pyruvic acid and compare to those used in recent brute-force experiments. We start by detailing various physical conditions of frozen, neat pyruvic acid relevant to brute force. That includes samples that are or are not well-deoxygenated, and those rapidly frozen or subsequently annealed. Temperature-dependent 1H longitudinal relaxation times (T1) are presented to highlight differences among these samples. We then extend that knowledge by exploring effects of O2-exposure and annealing on LFTM. That includes quantifying build-up and decay with LFTM vs. the size of the mixing field and the duration of exposure. Time constants for 1H–13C equilibration and eventual decay are quantified via the build-up and loss of 13C signal intensity vs. duration of the mixing period. Finally, in addition to investigating optimum conditions for LFTM, we also determine the threshold field above which mixing becomes inactive due to removal of 1H–13C spectral overlap.
Results
The pyruvic acid samples studied here were all neat, frozen solids (no additives or co-solvents) as utilized in recent brute-force experiments5 to hyperpolarize 13C and in further work to transport7 the hyperpolarization from the polarizer to a remote imaging centre. Here, in addition to detailing LFTM of 1H and 13C in such samples, we also explore the importance of certain sample-handling protocols. In particular, both annealing32 and deoxygenation of samples are known to induce large changes in longitudinal relaxation times (T1) of 1H and 13C. In this study, we explore LFTM in [1-13C] pyruvic acid, and also changes in the mixing that occur in cases of partial sample annealing and exposure to oxygen.The relevant temperature range of the current study is >4 K. Although brute force relies on polarization in conditions of low (<2 K) or ultralow (<500 mK) temperature, higher cryogenic temperatures are critical in post-polarization steps of sample extraction, thermal mixing, transport and dissolution. For example, during the lead-up to extraction from a brute-force polarizer, sample warming occurs (5–15 K for 30–60 s), and further changes may occur in the step of ejection in concert with LFTM. Finally, 4–80 K is relevant for transport.
As background to the thermal-mixing story, Fig. 1 displays temperature-dependent impacts of both oxygenation and annealing on the high-field T1 of protons in pyruvic acid. Fig. 1(a) focuses on oxygenation. Because O2 is paramagnetic, it can induce nuclear spin relaxation via motions relative to the surrounding bath of frozen pyruvic acid. T1vs. temperature is shown for two well-deoxygenated, well-sealed samples and one that had been exposed to air. For each, a characteristic ‘valley’ profile is apparent with minimum near 75 K, the temperature at which methyl rotations have the greatest spectral density near relevant NMR frequencies. Below ∼45 K, sharply rising T1 values result in >100-fold slower relaxation near 5–10 K in well-deoxygenated cases. If instead, O2 is present, the steep rise is significantly attenuated, as seen in data from a sample that, although originally deoxygenated, was exposed to air before its introduction to the helium-atmosphere cryostat of the fast field-cycling (FFC) apparatus. That resulted in ∼30-fold faster T1 relaxation near 4.2 K compared to the well-sealed sample. [See Fig. 1 caption and Materials and Methods for details on deoxygenation, sample sealing and the mild exposure to O2/air that yielded such changes.]
 | ||
Fig. 1 Profiles of T1(1H) vs. temperature for [1-13C] pyruvic acid. (a) From non-annealed samples. The full profile from a deoxygenated sample ( ) is as previously described,7 and was collected in a flame-sealed quartz tube and distinct NMR apparatus. Here, in the fast-field-cycling (FFC) apparatus, we collected T1 profiles from two non-annealed samples: one O2-exposed ( ) is as previously described,7 and was collected in a flame-sealed quartz tube and distinct NMR apparatus. Here, in the fast-field-cycling (FFC) apparatus, we collected T1 profiles from two non-annealed samples: one O2-exposed ( ), the other (□) deoxygenated and well-sealed. (See Materials and Methods for alternative sealing approach needed for samples tested in the FFC apparatus.) O2 exposure gave up to >10× faster relaxation at low temperatures (4–30 K). The present deoxygenated sample (□) exhibits a T1 profile matching earlier results ( ), the other (□) deoxygenated and well-sealed. (See Materials and Methods for alternative sealing approach needed for samples tested in the FFC apparatus.) O2 exposure gave up to >10× faster relaxation at low temperatures (4–30 K). The present deoxygenated sample (□) exhibits a T1 profile matching earlier results ( ), thus demonstrating on-par exclusion of O2 for a sample to be studied by FFC. (b) T1 profile following partial/intermediate annealing ( ), thus demonstrating on-par exclusion of O2 for a sample to be studied by FFC. (b) T1 profile following partial/intermediate annealing ( ) of the well-deoxygenated sample [i.e., of same sample as □ in (a)]. This annealing yielded tremendous change in the T1 profile, matching earlier results (▲) obtained in noted separate apparatus.7 The intermediate annealing protocol is described in Materials and Methods. It yielded nearly 102-fold slower relaxation at the centre of the ‘T1 valley’ vs. a non-annealed sample [ ) of the well-deoxygenated sample [i.e., of same sample as □ in (a)]. This annealing yielded tremendous change in the T1 profile, matching earlier results (▲) obtained in noted separate apparatus.7 The intermediate annealing protocol is described in Materials and Methods. It yielded nearly 102-fold slower relaxation at the centre of the ‘T1 valley’ vs. a non-annealed sample [ , same data as in (a)]. All data in (a and b) were collected at 2.0 or 2.1 T, excepting one set (▲) in (b) from 4.2 T. The latter is intended as overview of the overall pattern of intermediate-annealed behaviour. The distinct field has insignificant impact (∼2×) on T1 compared to ∼100× changes induced by this degree of annealing. , same data as in (a)]. All data in (a and b) were collected at 2.0 or 2.1 T, excepting one set (▲) in (b) from 4.2 T. The latter is intended as overview of the overall pattern of intermediate-annealed behaviour. The distinct field has insignificant impact (∼2×) on T1 compared to ∼100× changes induced by this degree of annealing. | ||
Next, we explored annealing directly in the FFC apparatus. Prolonged exposure to temperature above a glass transition (Tg) enables structural organization at the atomic and molecular scale. Resulting morphology can be fixed by subsequent cooling and maintenance of the sample below Tg. In brute-force hyperpolarization, such annealing can be critical in order to obtain spin-relaxation properties that are favourable during periods used to prepare for and execute sample extraction and/or transport of a hyperpolarized sample.7 For neat pyruvic acid, Tg of ∼215–230 K was previously discovered.32
Fig. 1(b) shows dramatic changes in the T1 profile for frozen pyruvic acid after thermal conversion to a form intermediate between non- and fully annealed states.7,32 For example, intermediate annealing here increases T1 by >100× at 75 K, which is the valley centre for the non-annealed form. The conditioning that led to this change (see Materials and Methods) is akin to that used in recent brute-force experiments,† where corresponding changes in T1 were critical to success.5,7 In particular, intermediate annealing removes the T1 valley that otherwise would range across 4 to 150 K in non-annealed pyruvic acid. The changed shape of the 1H profile [Fig. 1(b)] occurs very similarly at the 1-13C site.32
This apparent removal of the valley mitigates post-polarization losses during preparation for ejection as well as thermal mixing, storage, transport and ultimate dissolution. The physical origins of the changes in T1 relate to structural rearrangement during annealing (or partial annealing). A consistent picture is that a new morphology results that restricts methyl rotation. As a consequence, higher temperature is required to match the spectral density of rotations with NMR transitions. Correspondingly, Fig. 1(b) shows the T1 valley minimum shifted up to ∼200 K vs. ∼75 K for non-annealed pyruvic acid. More-complete annealing7,32 (not shown) similarly positions the T1 minimum near 200 K, but also yields a steeper rise when the temperature drops below ∼125 K. With that, a fully annealed sample maintains the 102-fold T1 gap relative to non-annealed all the way down to 4–10 K. Thus far, only intermediate-annealed samples of pyruvic acid have been tested with brute-force hyperpolarization, and the current study of LFTM is similarly focused on this degree of annealing.
To define the low-field range appropriate for thermal mixing, as well as associated time constants for equilibration and loss of polarization in the 1H–13C system, we used the NMR sequence of Fig. 2. The time sequence of FFC events is shown, as synchronized with an RF pulse sequence. Although pulses are only applied to 13C, both 1H and 13C have clearly defined roles in the experiment. The FFC sequence moves the spin system through events
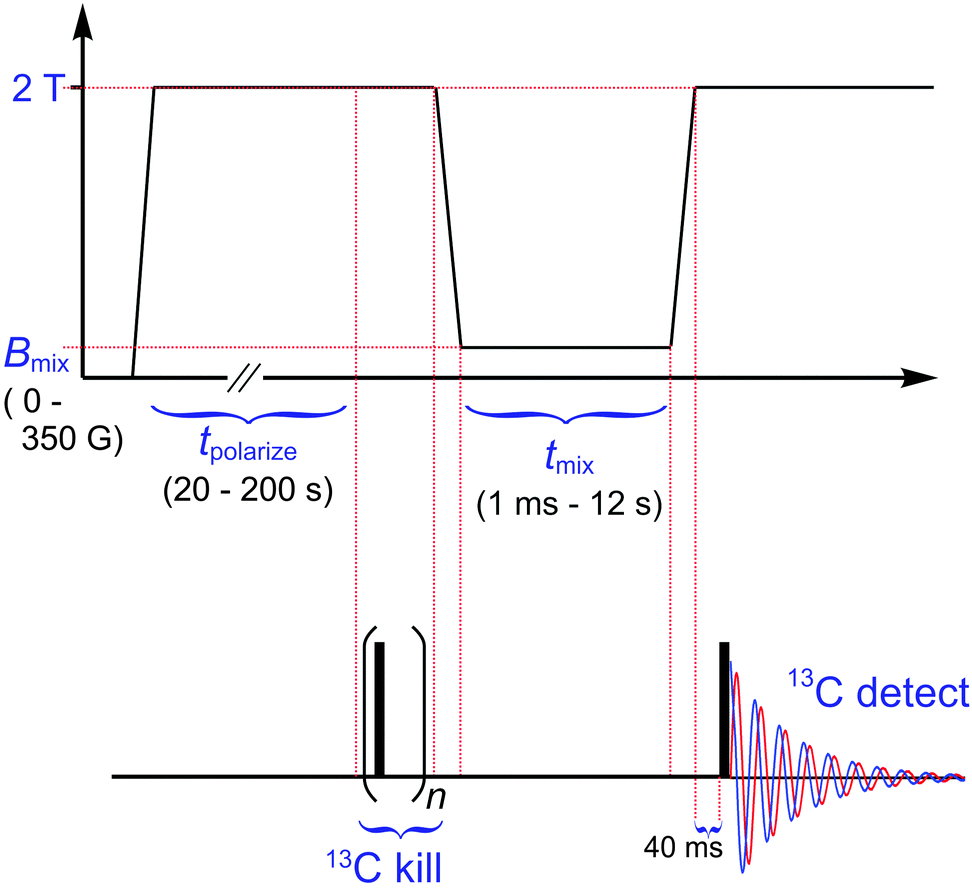 | ||
| Fig. 2 Pulse sequence for characterization of low-field thermal mixing. The opening period (∼1 s) at zero field erases the spin magnetization of all nuclei. The subsequent period (20–200 s) at 2.0 T polarizes 1H sufficiently for good signal-to-noise in 13C after equilibration of the two nuclei. To ensure zero input polarization from 13C for the mixing period, a preceding 13C kill sequence (n = 40 π/2 pulses at 350 μs intervals) was incorporated.‡ Subsequent fast field cycling (∼330 ms at 6 T s−1) brings the sample to the field of choice (e.g., Bmix = 0–350 G) for thermal mixing at duration tmix = 0.001–12 s. At the shortest of these time points, the exact field value may be skewed somewhat due to a settling time expected to be up to 40 ms. The return to 2.0 T (∼400 ms at 5 T s−1) after mixing incorporates a 40 ms settling period to ensure stable, resonant 13C detection after the single π/2 pulse indicated. | ||
(i) to ensure all 1H and 13C spin order is destroyed via an initial period at zero field,
(ii) to polarize 1H nuclei at high field (e.g., 2 T),
(iii) to mix 1H polarization with 13C via exposure to low-field (Bmix = 0–350 G) for time tmix = 0.001–12 s,
and, finally,
(iv) to detect resulting 13C magnetization via NMR after returning to high field.
For present experiments at or below 20 K, the field cycling is fast (<400 ms) compared to the timescale of high-field (e.g., >500 G = 0.05 T) longitudinal relaxation. Faster processes turn on at some point below 500 G (e.g., thermal mixing). However, the cycling rate requires only ∼10 ms to cover 500 G plus similar time for settling. (See caption to Fig. 2 and Materials and Methods for details.) Observations ahead indicate that this is fast enough for at least semi-quantitative determination of all time constants operative during the LFTM experiment.§
Results of mixing studies are shown in Fig. 3. 2D plots of 13C signal intensity vs. Bmix and tmix are shown in (a–c) for three of the same frozen [1-13C] pyruvic acid samples used in the above T1 study. Namely, Fig. 3(a) is from the O2-exposed, non-annealed sample [ from Fig. 1(a)], Fig. 3(b) is from well-deoxygenated, non-annealed [□ from Fig. 1(a)], and Fig. 3(c) is also well-deoxygenated, but intermediate-annealed [
from Fig. 1(a)], Fig. 3(b) is from well-deoxygenated, non-annealed [□ from Fig. 1(a)], and Fig. 3(c) is also well-deoxygenated, but intermediate-annealed [ from Fig. 1(b)]. All three samples reveal a thermal-mixing ‘hot spot’ that results in greatest 13C intensity centered near (Bmix, tmix) ∼ (50 G, 100 ms).
from Fig. 1(b)]. All three samples reveal a thermal-mixing ‘hot spot’ that results in greatest 13C intensity centered near (Bmix, tmix) ∼ (50 G, 100 ms).
 | ||
Fig. 3
13C-detected low-field thermal mixing signal intensity derived from 1H polarization in [1-13C] pyruvic acid. Upper (2D) and lower (1D) plots are pairs corresponding to samples that were (a and d) O2-exposed, non-annealed, (b and e) well-deoxygenated, non-annealed, and (c and f) well-deoxygenated, intermediate-annealed. Each sample corresponds to a T1 profile in Fig. 1, as noted in the main text. In 2D plots of (a–c), a shading scale maps the tmix and Bmix dependencies of 13C signal intensity gained by low-field contact with the proton bath. [The shading scale shown in (a) also applies to (b and c), although each data set was normalized individually and no quantitative intensity comparison among sample types is implied, as recycle delays shorter than the sample-dependent values of T1(1H) were used. The normalized scale is, however, identical for each upper/lower pair of plots.]. Corresponding fixed-field slices in (d–f) are plotted for Bmix = 0 G ( ), 40 G ( ), 40 G ( ) and 80 G ( ) and 80 G ( ), where each curve is a fit to eqn (1). All data sets were obtained using the NMR pulse sequence of Fig. 2. Data in (a and d) and (b and e) were collected at 4.2 K, whereas the rightmost set [(c and f), intermediate annealed] was collected at 10 K. That these sets are comparable, in spite of the temperature difference, was demonstrated by repetition of (b) at 10 K. In spite of poorer signal-to-noise, this did show the same intensity pattern as here, as well as nearly indistinguishable results from fits to eqn (1) (see ahead, Fig. 5). Furthermore, one slice (at 50 G) was repeated for the partially annealed sample (c) at 4.2 K and yielded rise and fall times indistinguishable from those at 10 K (see Materials and Methods). ), where each curve is a fit to eqn (1). All data sets were obtained using the NMR pulse sequence of Fig. 2. Data in (a and d) and (b and e) were collected at 4.2 K, whereas the rightmost set [(c and f), intermediate annealed] was collected at 10 K. That these sets are comparable, in spite of the temperature difference, was demonstrated by repetition of (b) at 10 K. In spite of poorer signal-to-noise, this did show the same intensity pattern as here, as well as nearly indistinguishable results from fits to eqn (1) (see ahead, Fig. 5). Furthermore, one slice (at 50 G) was repeated for the partially annealed sample (c) at 4.2 K and yielded rise and fall times indistinguishable from those at 10 K (see Materials and Methods). | ||
Qualitative comparison of Fig. 3(a) to the 2D plots in (b and c) reveals that O2-exposure limits the degree of 13C intensity that LFTM is able to establish for mixing, particularly below Bmix = 50 G. For more quantitative analysis, we fit individual time slices at constant Bmix to
 | (1) |
Individual fits to eqn (1) of slices from Fig. 3(a–c) are correspondingly shown in Fig. 3(d–f). Each data set and curve shows an initial rise in 13C intensity (typically τ < 50 ms) followed by the much slower decay process (T1m typically ranging 1–20 s). Initial rises in Fig. 3(d–f) often deviate from the fitted curve. This may be due to moderately skewed early response caused by ramping to and settling at Bmix. Thus, fitted τ values are likely less accurate than the T1m values. Nonetheless, detailed accuracy is not essential to assess the general timescale needed for effective mixing, nor are we prevented from making reliable relative comparisons of trends between samples with varied oxygenation and/or annealing.
For closer analysis, Fig. 4 plots all fitted parameters for the three varieties of sample. Fig. 4(a) gives overall intensity vs. Bmix. (Each sample normalized for I0,max = 1.) This again emphasizes that O2 exposure reduces the 13C polarization achievable for Bmix < 50 G. In fact, LFTM efficacy is sharply peaked in that case, and so oxygenation, even by apparently mild exposure to air, can require especially precise conditions to achieve productive 1H–13C equilibration.
 | ||
Fig. 4 Parameters of thermal mixing at 4.2 K. Showing (a) I0, the 13C intensity, (b) τ, the 1H–13C mixing time constant, and (c) T1m, the common relaxation time constant for decay of order in the 1H–13C spin system. Each plot shows results for a non-annealed and O2-exposed sample (closed circle,  ) along with those for non-annealed, de-oxygenated (open square, ) along with those for non-annealed, de-oxygenated (open square,  ) and intermediate-annealed de-oxygenated (closed triangle, ) and intermediate-annealed de-oxygenated (closed triangle,  ) samples. Fit parameters correspond to eqn (1). Uncertainties are the asymptotic standard error reported by Mathematica's nonlinear regression routine. Large uncertainties for the highest T1 values are due primarily to collection of data up to only tmix = 12 s. ) samples. Fit parameters correspond to eqn (1). Uncertainties are the asymptotic standard error reported by Mathematica's nonlinear regression routine. Large uncertainties for the highest T1 values are due primarily to collection of data up to only tmix = 12 s. | ||
The situation is more forgiving for a well-deoxygenated sample. Fig. 4(a) shows that the intensity profiles for the two O2-free cases tested here are relatively flat over Bmix = 0–50 G. Partial annealing yields particularly consistent intensity over this range. Meanwhile both non- and intermediate-annealed samples show similar falloff as Bmix increases from 50 to 100 G. Slight 13C intensity lingers up to 200 G, especially in the non-annealed case, a fact that is especially apparent in the corresponding 2D profile of Fig. 3(b). However, signal-to-noise begins to limit fit quality for traces vs. tmix at the largest Bmix values. Finally, we observed almost no 13C intensity for traces collected at Bmix = 250, 300 and 350 G for each of the three samples (data not shown). That indicates that LFTM is no longer effective above a threshold of approximately 200 G for this sample.
The intensity variations are explained by sample-to-sample variation of τ and T1m. First, in Fig. 4(b), the O2-exposed sample shows significantly reduced equilibration times (τ ∼ 5–15 ms), all less than the smallest value from each of the well-deoxygenated samples (τ ∼ 20 ms at Bmix = 0). Furthermore, those two profiles continue to rise from τ ∼ 30 to >60 ms as Bmix increases over the LFTM-active range (i.e., up to ∼150–200 G). The faster equilibration observed when paramagnetic O2 is present is likely due to electron-assisted LFTM. For example, few-spin processes may occur involving mutual flips of 1H, 13C and an electron(s), as resonances of the latter overlap with nuclear lines at these low fields. Also, electron-induced broadening may extend the range of Bmix over which 1H–13C overlap is sufficient for LFTM.
Similar impact is apparent in the plots of decay time constants, T1mvs. Bmix in Fig. 4(c). At all values of Bmix, the O2-exposed sample exhibits fastest relaxation and partially annealed exhibits the slowest. Deoxygenation appears to be especially critical. For example, in a typical mixing event employed in recent brute-force studies (500 ms, near 50 G),5,7T1m losses would be roughly 40% for O2-exposed, 10% for deoxygenated and <5% for partially annealed (and deoxygenated) sample variants. Even worse can be expected at mixing fields <50 G, where the same transit time can yield >90% loss for an O2-exposed sample. These facts echo our point above: although thermal mixing can work in the presence of O2, great care would be needed to select and reproduce successful conditions. It is simply much better and more straightforward to carefully exclude O2.
The results also quantify the value of annealing a sample to maintain 13C polarization gained by LFTM. Fig. 4(b) already shows that the partial annealing has no drawback on the rate of 1H–13C equilibration. Only modest impact was observed, i.e., an average of ∼20% longer τ relative to non-annealed. Because the added build-up time is on the tens-of-milliseconds scale and occurs within a hundreds-of-milliseconds event (i.e., sample extraction in a brute-force experiment), there is no sacrifice. More important is the benefit from partial annealing, which approximately doubles T1m over a range of Bmix values. For the most important mixing fields (0–100 G), T1m runs from ∼2.6 s to 30 s for the partially annealed form, vs. only 0.8 s to 15 s for the non-annealed and deoxygenated sample. Annealing is thus a very effective protection against polarization loss in brute-force experiments. For example, extracting a sample from a high-field polarizing environment (∼14 T) through a mixing field of <100 G is practical on the 1 s time scale, whereas dropping to the 100 ms timescale in order to achieve similar loss for a non-annealed sample would push the limits of practical sample ejection, or require modifications to keep B above the mixing threshold for a greater portion of the eject path.
A final point of interest on parameter variation vs. Bmix is that all three sample variations exhibit a rising T1m for Bmix up to about 100 G. Subsequent drop-offs occur near 100–150 G, depending on the sample. For now, this is unexplained, but reproducible. Four other 2D profiles collected from O2-exposed samples had the same roughly parabolic increase of T1m up to Bmix ∼ 100 G, followed by a dip between 100–200 G. Similar behavior is apparent in the other two sample variations tested, but with a T1m ‘peak’ at slightly higher Bmix. The abrupt apparent change might be caused by entry into a new regime near or above the LFTM threshold, where eqn (1) no longer applies. That transition may reflect a switching off of LFTM or new importance of 3-spin mixing events involving 1H and two 13C nuclei. The latter process has been used to explain mixing of 7Li and 19F in LiF crystals at 75 G.1–3 It might also be that 3-spin mixing explains the long tail of 13C intensity vs. Bmix, e.g., where I0 does not quite go to zero at 200 G in Fig. 4(a).
Finally, thermal-mixing behavior at somewhat higher temperatures is relevant to the conditions of sample extraction for brute-force hyperpolarization. As in recent work,5,7 preparation for sample ejection requires bringing the sample-handling system to positive pressure with concurrent warming to about 10–12 K. In spite of a pre-cooled sample path, some additional warming likely also occurs during ejection itself.
Thus, to extend the relevance of the current work, we compared LFTM at 4.2, 10 and 15 K by collecting full 2D sets vs. tmix and Bmix from the non-annealed, well-deoxygenated sample. Fig. 5(a–c) plots the I0, τ and T1m, respectively, as obtained from fitting each constant-Bmix slice at each temperature to eqn (1). Comparing 4.2 and 10 K, little-to-no differences were observed in the profiles of these parameters vs. Bmix. This is consistent with our separate observation of essentially identical mixing at 50 G in the intermediate-annealed sample at 4.2 and 10 K (see Materials and Methods). Characterizations at higher temperatures were challenging due to poor signal-to-noise. Without resorting to long signal averaging, the 15 K set was near the threshold of detection and attempts at 20 K were not quantifiable. Nonetheless, 15 K results in Fig. 5 show the same general patterns as at the lower temperatures. In particular, no significant changes in τ are apparent and the few T1m points available (limited by data quality) do not suggest increased rates of polarization loss.
 | ||
Fig. 5 Impact of temperature on mixing parameters vs. Bmix, including data at 4.2 K ( ), 10 K ( ), 10 K ( ) and 15 K (▲). Showing (a) I0, the 13C intensity, (b) τ, the 1H–13C mixing time constant, and (c) T1m, the common relaxation time constant for decay of order in the 1H–13C spin system. The 4–20 K range is thought to be most relevant for mixing as utilized in recent brute-force hyperpolarization experiments.5,7 In (c), T1m values at 15 K include only fitted results whose uncertainty was <100%. Large uncertainties there are due to a combination of poor-signal-to-noise and a maximum value of tmix that was insufficient for full decay. ) and 15 K (▲). Showing (a) I0, the 13C intensity, (b) τ, the 1H–13C mixing time constant, and (c) T1m, the common relaxation time constant for decay of order in the 1H–13C spin system. The 4–20 K range is thought to be most relevant for mixing as utilized in recent brute-force hyperpolarization experiments.5,7 In (c), T1m values at 15 K include only fitted results whose uncertainty was <100%. Large uncertainties there are due to a combination of poor-signal-to-noise and a maximum value of tmix that was insufficient for full decay. | ||
Discussion
A key overall finding here is that beneficial 13C polarization build-up from 1H–13C equilibration (at 1/τ) is much faster than the detrimental approach (at 1/T1m) to the near-zero thermal-equilibrium polarizations of low mixing fields (e.g., <500 G). We observed (1/τ) ∼ 103 × (1/T1m) all tested conditions (T = 4–15 K and Bmix = 0–200 G). This is consistent with earlier analysis5 of the very low losses observed in seminal brute-force experiments, where it was argued that no more than 5% loss could be due to thermal mixing. Low losses (∼10–30%) were also observed by Gadian et al.4 using LFTM to equilibrate 1H polarization with either 13C or 31P in field-swept brute-force studies without sample ejection. In spite of those successes, high-efficiency LFTM should not be taken for granted, as evidenced by the poor equilibration of hyperpolarized 129Xe with co-solidified 13C-labeled molecules (variously reported at 0.1–5% efficient33–35). Those difficulties might be partly attributed to distinct spin physics with 129Xe, but were most likely due to poor dipolar contact in inhomogeneous mixtures. That problem was uniquely imposed by the method of condensing dissimilar components (xenon and target molecule) and is of no concern in our exploration of neat molecules, nor is it likely to be important on extension to frozen solutions.It is, however, valuable to consider what guidance the present results provide for LFTM in other samples. For example, brute force hyperpolarization has been demonstrated in other molecules, although enhancements were smaller and the role of LFTM was not clear.5 First, one lesson here can readily be taken as general: avoid opportunities for O2 or other paramagnetic impurities to infiltrate the sample. The resulting electron-induced relaxation should be similarly detrimental across a variety of samples. Barring that, consider how the quality of unadulterated mixing may vary vs. sample type. The first key factors are the dipolar linewidths of interacting nuclei. The LFTM equilibration time τ is a function of the spectral overlap of the interacting spins, which is set by the linewidths and the degree of separation between lines.1–3 The widths are independent of field, whereas separation is given by Bmix(γh–γl), where γh and γl are for high- and low-γ nuclei. Of course, linewidths vary somewhat among molecules. Indeed, [1-13C] pyruvic acid exhibits different values vs. its morphology, e.g., 1H ranging from 25 to 35 kHz from non- to fully annealed. For molecular targets with a distinct low-γ nucleus, the magnitude of dipolar interactions and degree of spectral separation vs. Bmix will also differ. That can alter τ and shift optimum position for LFTM vs. (Bmix, tmix).
In spite of differing linewidths, effects on τ are slight, as seen for non- vs. intermediate-annealed forms in the present study [Fig. 4(b)]. For distinct molecules, linewidth variations will typically be of similar scale as with pyruvic acid morphologies, thus similarly small changes in τ (∼20%) are anticipated. Beyond neat samples, the same general timescale is reasonably expected for solvated molecules, assuming a solvent with similar proton density. That is consistent with the findings of Gadian et al.,4 which demonstrated effective LFTM for sodium [1-13C] acetate, both in water–glycerol solution and in powder form, on timescales (∼100 ms) similar to τ values observed here. In solution, the low-γ spin on the target molecule will equilibrate according to dominant solvent 1H polarization and the dipolar interactions of those protons with the target nucleus. Even in challenging cases of particularly narrow lineshapes, resulting slower equilibration would be tolerable. For example, a rough requirement is that build-up be about 10× faster than decay, and the factor of ∼103 observed in the work presented here leaves significant flexibility.
Variations in T1m from molecule to molecule and for solvated vs. neat molecules may be more important for future considerations. Unlike τ, which depends on coherent dipolar evolution (similar to spin diffusion), T1m reflects relaxation governed by incoherent fluctuations of the dipolar interaction. The cause is molecular motion. Of course, that can vary significantly among sample types. Importantly, we have also shown here that in [1-13C] pyruvic acid T1m sits just above a comfort threshold. That is, for at least the non-annealed form, T1m is only a bit longer than the ∼1 s required to eject a sample from a brute-force polarizer (400–500 ms of that in an LFTM active field). Thus it will be valuable in future work to characterize and understand variations in T1m among a variety of targets for brute-force hyperpolarization. One may also consider simply applying a field to part of the ejection path in order to reduce transit time through LFTM-active conditions.
Conclusions
We detailed 1H–13C thermal mixing in neat [1-13C] pyruvic acid at the cryogenic temperatures (≤15 K) operative during sample extraction from a brute-force hyperpolarization apparatus.5,7 There, LFTM is used to take advantage of ∼10× faster 1H build-up times compared to the target of hyperpolarization, 13C. Such low-γ nuclei are excellent candidates as imaging agents due to long solution-state polarization lifetimes, the opportunities for background-free detection and tracing metabolic conversion or other signatures of tissue health.Results here map out parameters for effective LFTM with unprecedented detail. This reveals an excellent match with fields and exposure times employed for sample ejection in noted brute-force studies. Importantly, we also quantified the beneficial impacts of annealing and deoxygenation for LFTM. Excluding O2 was clearly valuable in order to limit decay during mixing, doubling of the lifetime (T1m) of spin order during active LFTM. At the same time, the intermediate-annealed state of frozen pyruvic acid exhibited a further doubling of T1m. Rapid growth of 13C polarization during mixing, combined with much slower decay of 1H–13C order (i.e., T1m ≫ τ), suggests that LFTM can be exploited with almost no loss.
It was also essential to determine the low-field threshold for active mixing. That is because, some circumstances require turning off mixing in order to prevent destruction of polarization on the low-γ nucleus via contact with unpolarised protons. For example, long 13C lifetimes can enable transport of 13C hyperpolarization from the polarizer to an imaging centre, although faster relaxing 1H polarization typically disappears during transport. In the recent demonstration of such transport with hyperpolarized [1-13C] pyruvic acid,7 the sample was transported in modest high field and then passed in to a dissolution apparatus before imaging. In that final passage a 300 G ‘magnetic tunnel’ was used to prevent destructive LFTM. Here we have shown that this was more than sufficient and also set guidelines for future work.
Finally, it is worthwhile to comment on roles for LFTM beyond brute-force. LFTM is operative in solids only, yet it cannot be generally utilized with other solids-to-liquids hyperpolarization methods, such as d-DNP. That is because the vast majority of current d-DNP experiments require intimate contact of electron spins with the nuclear bath.¶ For LFTM conditions, the electrons would cause rapid relaxation to zero of all nuclear polarization,37,38 much as we observed with O2. Thus, d-DNP utilizes dissolution at high field within the polarizing cryostat, extracting the sample via the dissolution process itself. Brute force has been unique for its ability to extract a hyperpolarized solid, and thereby utilize LFTM and also enable remote transport. We also note one last advantage of LFTM vs. more-familiar NMR techniques to transfer polarization from 1H to low-γ spins (e.g., spin-locked cross polarization39). LFTM avoids both RF heating and confinement of the sample to an RF coil, and thus maintains the scalability of brute force to larger samples and/or to multi-sample production. This is a key advantage of brute force, especially important in comparison to higher throughput hyperpolarization methods like d-DNP.
Experimental
Samples
Neat 1-13C (99%) pyruvic acid was purchased from Cambridge Isotope Labs (Tewksbury, MA, USA). Purchased quantities were deoxygenated by 5–10 freeze–pump–thaw cycles and kept in the −30 °C freezer of our N2-atmosphere drybox. All samples originated from this deoxygenated stock, including the O2-exposed case.Sample sealing and O2 exposure
Space available in the FFC apparatus required a very short (<20 mm) sample tube, and alternative sealing approach vs. the flame-sealing approach noted in separate publications.7 Here, short sample tubes were 5 mm OD (4 mm ID) Suprasil (synthetic quartz) purchased from Wilmad (Vineland, NJ, USA). These included a custom 5 mm-long constriction to about half of the ID and centered 18 mm from the bottom of the tube. Tubes were broken off by scoring and snapping at the constriction, and then the surface was roughed by gentle external filing at the neck and top surface, as shown in Fig. 6(a). Tubes were then soaked several minutes in 0.25 M ethylenediaminetetraacetic acid (EDTA), thoroughly rinsed with ultralow-metal-content Chromosolv LC-MS water and vacuum dried. The EDTA and water were from Sigma Aldrich (St. Louis, MO, USA). | ||
| Fig. 6 Sample tubes for cryo-tolerant sealing and exclusion of O2. (a) Empty tube and cap as prepared for sample loading. Opaque quality of the cap is due to addition of finely ground quartz. (b) Two tubes, as filled with neat [1-13C] pyruvic acid, capped and cured in the dry box. | ||
A cap for airtight, cryo-tolerant sealing was formed as follows. An approximately 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of Stycast 1266 epoxy (Ellsworth Adhesives, Germantown, WI, USA) and finely ground quartz was cured in a cylindrical Teflon form with 5 mm diameter. A 0.5 mm wide circular groove was machined out of the hardened cylinder so the cap [Fig. 6(a)] mates inside and out with the constricted sample tube. A similar clean-and-rinse protocol was applied to the cap as for sample tubes. In the drybox, ∼160 μL of deoxygenated [1-13C] pyruvic acid was carefully pipetted to each sample tube. Stycast 1266 resin and hardener were freshly mixed in the drybox, and a thin layer was spread outside the tube constriction, on the top surface, and on the cap. These were press fit, held upright while curing in the drybox for >24 hours at room temperature. The final product is shown in Fig. 6(b).
:1 mix of Stycast 1266 epoxy (Ellsworth Adhesives, Germantown, WI, USA) and finely ground quartz was cured in a cylindrical Teflon form with 5 mm diameter. A 0.5 mm wide circular groove was machined out of the hardened cylinder so the cap [Fig. 6(a)] mates inside and out with the constricted sample tube. A similar clean-and-rinse protocol was applied to the cap as for sample tubes. In the drybox, ∼160 μL of deoxygenated [1-13C] pyruvic acid was carefully pipetted to each sample tube. Stycast 1266 resin and hardener were freshly mixed in the drybox, and a thin layer was spread outside the tube constriction, on the top surface, and on the cap. These were press fit, held upright while curing in the drybox for >24 hours at room temperature. The final product is shown in Fig. 6(b).
The ‘O2-exposed’ sample was not sealed in this manner. It was likewise prepared in the drybox using deoxygenated [1-13C] pyruvic acid. However, it was then sealed using only tightly packed, degassed Teflon tape at the end of a cut (∼20 mm long) and cleaned Suprasil NMR tube filled with 200–300 μL of sample. Finally, tight external taping with Teflon (or parafilm to similar result) was used. Evidently, and in spite of only brief (≤5–10 min) exposure to air at room-temperature, such ‘seals’ were too permeable to maintain deoxygenation. Several samples prepared in this way yielded T1 profiles as shown for the ‘O2-exposed’ sample in Fig. 1(a). Nonetheless, these poorly sealed cases proved serendipitous in that corresponding data enabled us to explore the effects of O2 contamination on both T1vs. temperature and LFTM.
Finally, we note that present, relatively large samples must be less sensitive to O2 exposure than the high surface-to-volume ratio samples used in prior brute-force work.5 Thus, the demonstrated detrimental effects of O2-exposure urge particular caution for such preparations of a sample.
Sample annealing
The procedure was similar to that presented elsewhere,7,32 here implemented directly in the FFC apparatus. It was applied to the sample already characterized as non-annealed (and well-deoxygenated) without melting or removing it from the helium-flow cryostat and NMR probe. The initial non-annealed state was generated by freezing the liquid sample (m.p. = 285 K) from room temperature to <200 K in ∼20–30 min. Non-annealed, intermediate- and fully annealed forms are stable below ∼215 K for at least as long as yet monitored (>4 days). After ∼2 days characterization of the non-annealed form, measuring T1(1H) between 4–120 K, and LFTM over 4–20 K, we then proceeded to generate the intermediate-annealed form. For this, the sample was left overnight (∼8 h) at ∼243 K and then dropped quickly (∼20 m) to 200 K. After this, T1 values were obtained at 200, 80 and 10 K before proceeding to characterization of LFTM. The 243 K annealing temperature was chosen to correspond with the freezer temperature where samples were held in preparation for earlier brute-force hyperpolarization experiments.5Fast field cycling (FFC) and cryogenic systems
Experiments were performed on the FFC apparatus at the University of Nottingham, as described previously by Horsewill and Xue.40 Low-inductance (20 mH) superconducting magnet at 4.2 K and fast-ramping power supply from Cryogenic, Ltd (London, UK) provide fields between 0 to 2.5 T with ramping rate up to 10 T s−1. The system includes an integrated helium flow cryostat that sips helium from the magnet reservoir. Temperature is maintained via a Lakeshore 331 controller coupled to resistive heater in the helium flow path. Sample temperature is reported by a calibrated Cernox sensor in good thermal contact with a brass block surrounding the NMR sample and coil. Temperature stability was ≤(±0.05 K) for runs approaching 24 h. Min, max and mean values were recorded with each fixed-Bmix profile of intensity vs. tmix, and with each recovery trace to measure T1.Field stability and reproducibility in this apparatus have been estimated to be about ±1.5 G based on the standard deviation of repeated 1H NMR measurements, each collected following a field-cycling event.40 Rechecking here, we similarly estimated reproducibility of ±5 G via the RMS deviations of fitted peak positions from in 13C spectra represented in the 2D arrays of Fig. 3(a–c). Only spectra above signal-to-noise of 60 were used, or about ∼75 spectra from each array. (Note, the observed range was ±20 G.) Each spectrum involved cycling events and settling time (40 ms) described in Fig. 2. Finally, we believe any offset field (e.g., due to flux trapping by the superconducting magnet of the ∼0.25–0.65 G earth field), was likely smaller than the noted variation in field stability. This is based in the indistinguishability of the 2D data set (vs. Bmix and tmix) in Fig. 3(a) from another collected immediately after on the same sample, but with reversed polarity of the magnet.
NMR apparatus
The NMR probe contains a single solenoid coil tuned to about 21.6 MHz. Different nuclei were addressed by adjusting the field to B0 = γn−1 × (21.6 MHz), where γn is the gyromagnetic ratio. The NMR spectrometer was operated by home-written code in visual basic. Automated data-collection steps through values of Bmix, tmix and temperature were similarly controlled by home-written software.Data processing and analysis
Processing and analysis was performed using Mathematica. Time-domain signals were multiplied by a matched exponential window (20 and 5 kHz for 1H and 13C), Fourier transformed and autophased. 13C intensities were obtained by integrating the frequency domain over ±50 kHz about the center frequency, and similarly for 1H spectra in T1 experiments.Parameters of thermal mixing were determined by fitting to eqn (1) using Mathematica's standard routine for nonlinear regression. Error bars in Fig. 4 and 5 are the asymptotic standard errors reported by the fitting routine These may underestimate uncertainty in cases where data trends do not strictly follow eqn (1), as discussed in the main text.
NMR experiment parameters
Thermal-mixing experiments using the pulse sequence of Fig. 2 had tpolarize = 40 s at 4.2 K in the case of non-annealed, O2-exposed samples. Due to the longer T1 of the non-annealed, well-deoxygenated sample at 4.2 K (see Fig. 1), we used correspondingly longer polarization time of 150 s. Meanwhile, experiments on that sample at 10, 15 and 20 K used tpolarize = 100, 40 and 20 s, respectively. For the intermediate-annealed (and well-deoxygenated) sample, we used tpolarize = 200 s at 4.2 K and 150 s at 10 K.The NMR excitation and detection frequency was 21.6 MHz, corresponding to B0 = 2.018 T for 13C or 0.507 T for 1H. T1 experiments in the FFC apparatus were by saturation recovery, the same as performed in earlier experiments7 with a static-field system at 2 and 4 T. Here, 1H saturation recovery occurred at 2.0 T, followed by detection at the resonant field (0.507 T). That was achieved using a field-cycling pulse sequence similar to that in Fig. 2. Pulse times for 1H and 13C were typically t90 = 2 and 8 μs. Single-scan 13C spectra were collected in 512 complex time-domain points at 0.3 s dwell time (3.3 MHz spectral width). Similar parameters were used for 1H-detected experiments to measure T1(1H).
The time required to collect a full 2D series of thermal-mixing results depended on the polarization time and the size of the set of tmix and Bmix values used. The longest running 2D series [Fig. 3(c), tpolarize = 150 s] required ∼17 h for 18 values of tmix from 1 ms to 12 s in regular increments of 0.24 log units and 19 values of Bmix ranging 0 to 100 G in increments of 10 G, then 120 to 200 G in increments of 20 G, and finally 250, 300 and 350 G. Plots in Fig. 3(a–c) also incorporate slices at Bmix = 110, 130, 150, 170, 190 G that were linearly interpolated from nearest neighbors in order to plot regular 10 G intervals over 0–200 G. Data sets above 200 G revealed very little 13C intensity, and are not shown in either Fig. 3(a–c) or in the plots of fitted parameters vs. Bmix in Fig. 4 and 5.
Finally, due to longer polarization times for the intermediate-annealed sample, collection of a full 2D set at 4.2 K was less practical than at 10 K. Nonetheless, as noted in the caption to Fig. 3, this sample exhibited nearly indistinguishable LFTM behavior at these two temperatures for the 50 G ‘mixing optimum’. Specifically, τ = (40.0 ± 8.4) ms and T1m = (6.1 ± 1.2) s at 4.2 K, vs. (38.5 ± 8.5) ms and (5.0 ± 1.1) s at 10 K.
Acknowledgements
We thank Andy Stewart from the Univ. of Nottingham Physics and Astronomy electronics shop for occasional repairs of the NMR probe. We thank Werner Maas of Bruker Biospin for encouragement and support of the project.Notes and references
- A. Abragam and W. G. Proctor, Phys. Rev., 1957, 106, 160–161 CrossRef CAS.
- A. Abragam and W. G. Proctor, Phys. Rev., 1958, 109, 1441–1458 CrossRef.
- A. Abragam, Principles of Nuclear Magnetism, Clarendon Press, Oxford, 1961 Search PubMed.
- D. G. Gadian, K. S. Panesar, A. J. P. Linde, A. J. Horsewill, W. Kockenberger and J. R. Owers-Bradley, Phys. Chem. Chem. Phys., 2012, 14, 5397–5402 RSC.
- M. A. Hirsch, N. Kalechofsky, A. Belzer, M. M. Rosay and J. G. Kempf, J. Am. Chem. Soc., 2015, 137, 8428–8434 CrossRef CAS PubMed.
- J. R. Owers-Bradley, A. J. Horsewill, D. T. Peat, K. S. K. Goh and D. G. Gadian, Phys. Chem. Chem. Phys., 2013, 15, 10413–10417 RSC.
- M. L. Hirsch, B. A. Smith, M. Mattingly, A. G. Goloshevsky, M. Rosay and J. G. Kempf, J. Magn. Reson., 2015, 261, 87–94 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS PubMed.
- P. Höfer, G. Parigi, C. Luchinat, P. Carl, G. Guthausen, M. Reese, T. Carlomagno, C. Griesinger and M. Bennati, J. Am. Chem. Soc., 2008, 130, 3254–3255 CrossRef PubMed.
- K. G. Valentine, G. Mathies, S. Bédard, N. V. Nucci, I. Dodevski, M. A. Stetz, T. V. Can, R. G. Griffin and A. J. Wand, J. Am. Chem. Soc., 2014, 136, 2800–2807 CrossRef CAS PubMed.
- L. T. Kuhn, Top. Curr. Chem., 2013, 338, 229–300 CrossRef CAS PubMed.
- B. M. Goodson, Y. Song, R. E. Taylor, V. D. Schepkin, K. M. Brennan, G. C. Chingas, T. F. Budinger, G. Navon and A. Pines, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14725–14729 CrossRef CAS.
- M. M. Spence, S. M. Rubin, I. E. Dimitrov, E. J. Ruiz, D. E. Wemmer, A. Pines, S. Q. Yao, F. Tian and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10654–10657 CrossRef CAS PubMed.
- C. Boutin, H. Desvaux, M. Carrière, F. Leteurtre, N. Jamin, Y. Boulard and P. Berthault, NMR Biomed., 2011, 24, 1264–1269 CrossRef CAS PubMed.
- Y. Bai, P. A. Hill and I. J. Dmochowski, Anal. Chem., 2012, 84, 9935–9941 CrossRef CAS PubMed.
- N. S. Khan, B. A. Riggle, G. K. Seward, Y. Bai and I. J. Dmochowski, Bioconjugate Chem., 2015, 26, 101–109 CrossRef CAS PubMed.
- R. Sriram, J. Kurhanewicz and D. B. Vigneron, eMagRes, 2014, 3, 311–324 Search PubMed.
- S. Meier, P. R. Jensen, M. Karlsson and M. H. Lerche, Sensors, 2014, 14, 1576–1597 CrossRef CAS PubMed.
- K. M. Brindle, J. Am. Chem. Soc., 2015, 137, 6418–6427 CrossRef CAS PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Larson, A. L. Harzstark, M. Ferrone, M. van Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjaer-Larsen, A. P. Chen, R. E. Hurd, L. I. Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 5, 198ra108 Search PubMed.
- S. E. Day, M. I. Kettunen, F. A. Gallagher, D.-E. Hu, M. Lerche, J. Wolber, K. Golman, J. H. Ardenkjaer-Larsen and K. M. Brindle, Nat. Med., 2007, 13, 1382–1387 CrossRef CAS PubMed.
- T. Harris, G. Eliyahu, L. Frydman and H. Degani, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18131–18136 CrossRef CAS PubMed.
- K. M. Brindle, S. E. Bohndiek, F. A. Gallagher and M. I. Kettunen, Magn. Reson. Med., 2011, 66, 505–519 CrossRef PubMed.
- D. M. Wilson and J. Kurhanewicz, J. Nucl. Med., 2014, 55, 1567–1572 CrossRef CAS PubMed.
- Y. Li, I. Park and S. J. Nelson, Cancer J., 2015, 21, 123–128 CrossRef CAS PubMed.
- O. J. Rider and D. J. Tyler, J. Cardiovasc. Magn. Reson., 2013, 15, 1–9 CrossRef PubMed.
- C. Purmal, B. Kucejova, A. D. Sherry, S. C. Burgess, C. R. Malloy and M. E. Merritt, Am. J. Physiol., 2014, 307, H1134–H1141 CAS.
- A. J. Bakermans, D. Abdurrachim, R. P. M. Moonen, A. G. Motaal, J. J. Prompers, G. J. Strijkers, K. Vandoorne and K. Nicolay, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 88–89, 1–47 CrossRef CAS PubMed.
- J. G. Kempf, N. Kalechofsky and M. Rosay, US Pat., US2015061666 (A1), 2014 Search PubMed.
- C. R. Bowers, H. W. Long, T. Pietrass, H. C. Gaede and A. Pines, Chem. Phys. Lett., 1993, 205, 168–170 CrossRef CAS.
- A. Cherubini, G. S. Payne, M. O. Leach and A. Bifone, Chem. Phys. Lett., 2003, 371, 640–644 CrossRef CAS.
- N. Lisitza, I. Muradian, E. Frederick, S. Patz, H. Hatabu and E. Y. Chekmenev, J. Chem. Phys., 2009, 131, 044508 CrossRef PubMed.
- A. Bornet, X. Ji, B. Vuichoud, J. Milani, D. Gajan, A. J. Rossini, L. Emsley, G. Bodenhausen and S. Jannin, Joint 5th International DNP Symposium and COST Action EuroHyperpol, Egmond aan Zee, The Netherlands, 2015.
- D. T. Peat, A. J. Horsewill, W. Kockenberger, A. J. P. Linde, D. G. Gadian and J. R. Owers-Bradley, Phys. Chem. Chem. Phys., 2013, 15, 7586–7591 RSC.
- S. Macholl, H. Johannesson and J. H. Ardenkjaer-Larsen, Phys. Chem. Chem. Phys., 2010, 12, 5804–5817 RSC.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1972, 56, 1776–1777 CrossRef CAS.
- A. J. Horsewill and Q. Xue, Phys. Chem. Chem. Phys., 2002, 4, 5475–5480 RSC.
Footnotes |
| † Distinct dimensions and geometry of the sample may impact annealing. Samples here were ∼4 mm OD by 10 mm long solid cylinders. Earlier brute-force experiments used samples of similar diameter and length, but frozen as a hollow, thin (∼1 mm) cylinder. It has not been discounted that such geometry might change response to annealing. T1vs. temperature has not been measured for the hollow cylinder case. |
| ‡ The 13C kill sequence might be thought unnecessary because an effective mixing period will peg 13C polarization only according to the pre-mixing polarization level on 1H, almost regardless of starting 13C polarization.5 The reason is that the ‘spin specific heat’ [ref. 1, 2 and 5] of protons is 100-fold larger than for 13C in pyruvic acid (and most all protonated molecules, even at 99% 13C). Nonetheless, a signal of dominant origins in 1H polarization is only guaranteed when mixing is active, i.e., for just a portion of the full (tmix, Bmix) space we tested. Thus, the 13C kill is needed when tmix is not on the timescale of equilibration and/or when Bmix is above the mixing threshold. |
| § About 3–4 ms was required to drop from 200 G to zero field, where 200 G is conservatively high for the threshold above which mixing ceases for [1-13C] pyruvic acid. This few ms ramping through LFTM-active fields might skew interpretation of the mixing rise time (τ), especially for data collected at the lowest values of Bmix. |
| ¶ As promising alternative, Jannin and coworkers demonstrated ‘remote’ DNP,36 which starts with DNP to solvent 1H spins in contact with solute free radicals. That traditional step is followed by spin diffusion to protons in molecules forming dispersed crystallites that are isolated from the free radicals. Finally, cross polarization yields hyperpolarized 13C in the isolated targets. The physical separation can allow controlled two-spin LFTM (e.g., of 1H and 13C), as well as production of transportable hyperpolarization. |
| This journal is © the Owner Societies 2016 |