
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs the chemistry of lawrencium peculiar?†
Wen-Hua
Xu
ab and
Pekka
Pyykkö
*b
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry and Molecular Science, Northwest University, 710127 Xi’an, China. E-mail: xuwenhua.qf@gmail.com
bDepartment of Chemistry, University of Helsinki, POB 55 (A. I. Virtasen aukio 1), 00014 Helsinki, Finland. E-mail: Pekka.Pyykko@helsinki.fi
First published on 8th June 2016
Abstract
It is explicitly verified that the atomic 7p1 ground-state configuration of Lr originates from relativistic effects. Without relativity one has 6d1. All three ionization potentials IP1–3 of Lr resemble those of Lu. Simple model studies on mono- and trihydrides, monocarbonyls or trichlorides suggest no major chemical differences between Lr and the lanthanides.
1 Introduction
The periodic table is about chemistry. The group is related to the number of valence electrons and the period is related to the number of nodes in the radial functions of these electrons. In lawrencium, 103Lr, counting the filled 5f shell as the ‘core’, there are three valence electrons. It had been debated for some time, whether they are 7s26d1 or 7s27p1/21, until both an experiment1 and also the latest calculations supported the latter alternative. That 7p1 atomic ground state was first surmised by Brewer2 and first calculated by Desclaux and Fricke.3 Large MCDF calculations by Zou and Froese Fischer4 support the 5d1 and 7p1 ground states for Lu and Lr, respectively, and yield very different oscillator strengths. For Lr, however, they are not yet experimentally confirmed.This does not yet settle the question on the chemical behaviour. If all three valence electrons are formally ionized away, in an Lr(III) compound, lawrencium clearly belongs to Group 3 in Period 7, and nothing unexpected has happened in its chemistry.
The three first ionization potentials of Lr are compared with those of La–Lu in Fig. 1. They are quite similar, especially with Lu. Therefore the ionic chemistry of Lr could be expected to be similar to that of the lanthanides.
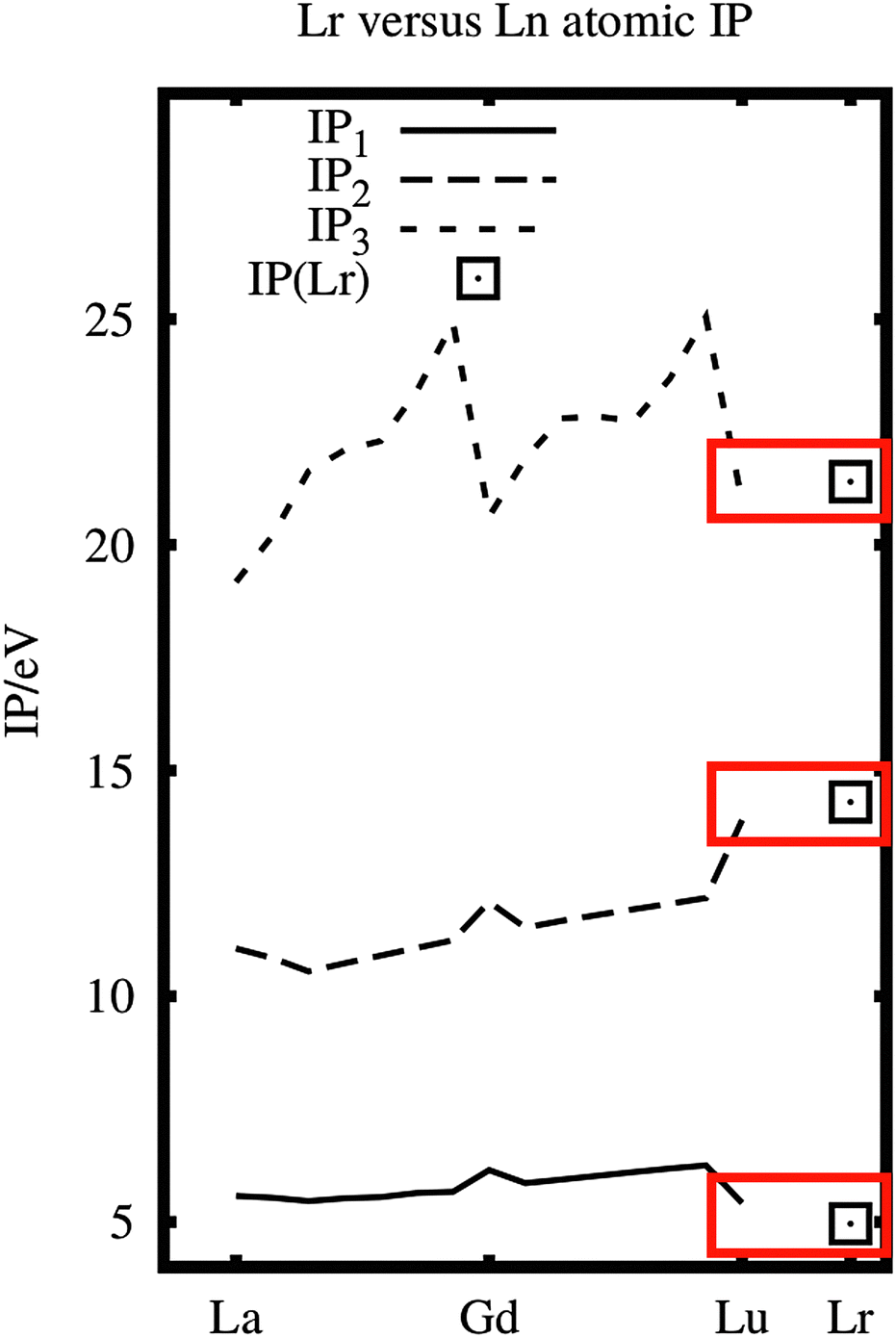 | ||
| Fig. 1 The ionization potentials of La–Lu (experimental data5) and Lr (experimental and calculated IP1;1 calculated IP2,36). Note the similarity of Lu and Lr. | ||
Experimentally this is what happened, see Brüchle et al.,7 Hoffman et al.8 and Scherer et al.,9 all in 1988. Brüchle and Hoffman found that Lr(III) had a similar elution behaviour to the latter Ln(III) and Scherer found no evidence for a possible reduction to lower oxidation states than Lr(III) in aqueous solution. Recently, in reductive surroundings, all the divalent lanthanide oxidation states Ln(II) have also been obtained.10 These divalent lanthanide, Ln(II), compounds are mostly 5d1. No such experiments exist on Lr.
Calculations suggest that the free-atom Lr(I) and Lr(II) are 7s2 and 7s1, respectively,5,6,11 in contrast to the quoted 5d1 for Ln(II) in compounds.10 The stabilisation of the 7s shell in Group 3 can be compared with that of the 6s shell in Group 13, which contributes to the chemical difference between indium and thallium, having the main oxidation states In(III) and Tl(I), respectively. Similarly, for lead, the relativistic stabilization of the 6s shell favours the divalent Pb(II) state in PbO or PbSO4 and destabilizes the Pb(IV) state in PbO2, thereby explaining most of the voltage of the lead battery.12 In these main-group cases the relativistic stabilization of an ns shell leads to different main oxidation states in Periods 5 and 6. One possibility considered here is whether one could have a similar change between lanthanides and actinides. Recall that the relativistic stabilization of valence s shells down the same column increases as Z2, where Z is the nuclear charge.
2 Atomic results
We first verify the relativistic origin of the ground-state change from 6d1 to 7p1, see Table S1 in the ESI.† Compared with the non-relativistic results, Dirac–Fock (DF) shifts down the relative energy of (n + 1) 2P to n2D by nearly 3 eV, and changes the ground state configuration. The relativistic effect is so large that already DF-level evidence makes sense. MCDF results were reported by Fritzsche et al.13The calculated orbital energies for Tl and Lr atoms are shown in Fig. 2. It is seen that the relativistic stabilization of the Tl 6s shell is substantial, making its energy comparable to the ligand orbital energy (here H). In contrast, the Lr 7s orbital energy is small, despite a larger Z.
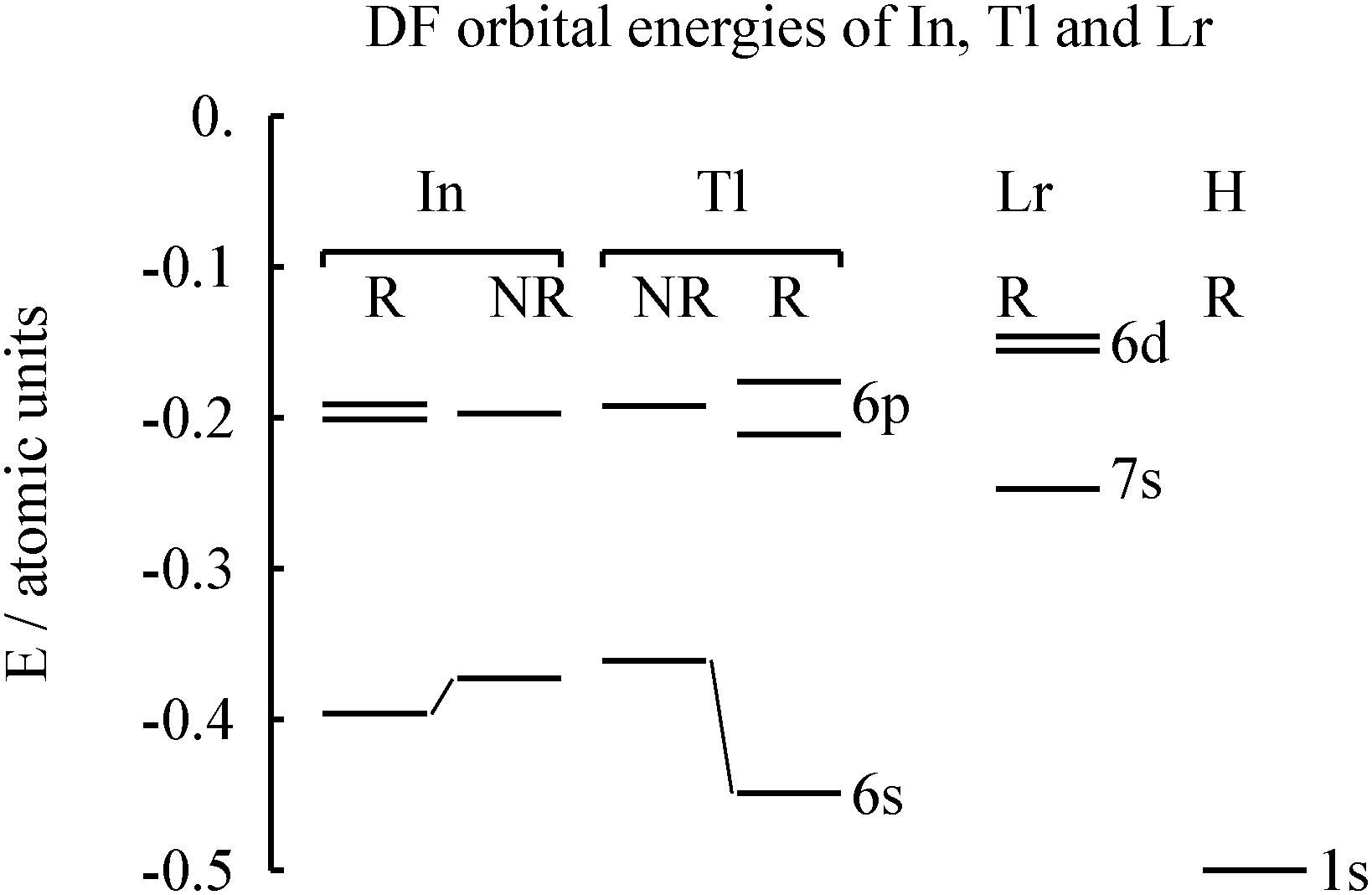 | ||
| Fig. 2 The calculated relativistic (R) and non-relativistic (NR) Dirac–Fock orbital energies of neutral In, Tl and Lr atoms. For lawrencium, the electron configuration 7s26d1 is assumed. Values from Desclaux.15 | ||
Does the atomic ground state matter in chemistry? As seen in Fig. 2, the valence orbital energies of the electropositive element Lr are small and hence in compounds these electrons, whether 7s, 6d or 7p, will largely go away, anyway. Group 13 is more electronegative14 than Group 3.
Why is Lr, like other lanthanides and actinides, so electropositive? A broad-brush explanation could be that they all belong to Group 3 and the electronegativities in the Periodic table increase from left to right (from Group 1 to Group 18), probably due to increasing partial screening by the fellow valence electrons.
3 Molecular results
3.1 Hydrides
We first consider the simple hydride models and calculate the reaction energy, ΔE for the model reaction| MH3 → MH + H2 | (1) |
The structural parameters are given in Table 2.
| Molecule | Symmetry | Bond length | Bond angle |
|---|---|---|---|
| a Ref. 17. b Ref. 10. c Shortest M–C. | |||
| LuH | Linear | 1.895 | |
| LrH | Linear | 1.960 | |
| LuH2 | C 2v | 1.915 | 113.5 |
| LrH2 | C 2v | 1.954 | 110.6 |
| C 2v | 2.015 | 117.5 | |
| LuH3 | C 3v | 1.921 | 112 |
| LrH3 | C 3v | 1.940 | 107 |
| LuCO | Linear | 2.297 (Lu–C), 1.167 | 180 |
| LrCO | Linear | 2.384 (Lr–C), 1.169 | 180 |
| LuCl3 | D 3h | 2.394 | 120 |
| LrCl3 | C 3v | 2.424 | 113 |
| [(Cp′)3Lu]− | C 1 | 2.635b,c | |
| [(Cp′)3Lr]− | C 1 | 2.697c | |
3.2 Monocarbonyls
We then compare LrCO with the series LnCO, Ln = La–Lu, studied both experimentally and theoretically by Xu et al.19 There the three last members Ln = Tm, Yb, and Lu which could not be made, and they had theoretically weak bonds, for Lu with a σ2π1 valence configuration. We now find that Lr behaves just like Lu, which further supports putting it under Lu in the Periodic table. At the CCSD(T) level, LrCO is 1.0 eV below Lr + CO, while LuCO is 0.77 eV below Lu + CO. The attempts to produce LuCO19 nevertheless failed.Population analyses are shown in Tables 3–5. Projection analysis is stable regarding different types of functionals. In this study, Mulliken populations agree well with the projection analysis. There is a high correlation between Lu and Lr electronic configurations in all the hydrides and carbonyls.
| Mol. | Type | Functional | Valence population |
|---|---|---|---|
| LuH | P, G | PBE | Lu 6s(1.78)5d(0.71)6p(0.22) H 1s(1.26) |
| M, S | PBE | Lu 6s(1.77)5d(0.68)6p(0.20) H 1s(1.34) | |
| LrH | P, G | PBE | Lr 7s(1.82)6d(0.63)7p(0.28) H 1s(1.25) |
| P, G | PBE0 | Lr 7s(1.80)6d(0.57)7p(0.28) H 1s(1.30) | |
| P, G | CAMB3LYP | Lr 7s(1.80)6d(0.53)7p(0.27) H 1s(1.34) | |
| M, S | PBE | Lr 7s(1.83)6d(0.61)7p(0.21) H 1s(1.34) | |
| LuH2 | M, S | PBE | Lu 6s(1.03)5d(0.96)6p(0.27) H 1s(1.35) |
| LrH2 | M, S | PBE | Lr 7s(1.19)6d(0.87)7p(0.25) H 1s(1.33) |
| LuH3 | P, G | PBE | Lu 6s(0.74)5d(1.06)6p(0.27) H 1s(1.29) |
| M, S | PBE | Lu 6s(0.66)5d(0.97)6p(0.25) H 1s(1.36) | |
| LrH3 | P, G | PBE | Lr 7s(0.89)6d(1.05)7p(0.25) H 1s(1.26) |
| M, S | PBE | Lr 7s(0.84)6d(0.94)7p(0.17) H 1s(1.34) | |
| LuCO | M, S | PBE | Lu 6s(1.77)5d(0.75)6p(0.18) C 2sp(3.90) O 2sp(6.25) |
| LrCO | M, S | PBE | Lr 7s(1.82)6d(0.66)7p(0.20) C 2sp(3.91) O 2sp(6.27) |
| Mol. | Natural electron configuration |
|---|---|
| LrH | [core]7s(1.92) 6d(0.34) 7p(0.04) |
| LrH3 | [core]7s(0.84) 6d(0.67) 7p(0.01) |
| LrCO | [core]7s(1.93) 6d(0.51) 7p(0.05) |
| LuCO | [core]6s(1.90) 5d(0.56) 6p(0.06) |
| Mol. | Orb. | Energy | Populations |
|---|---|---|---|
| LrH | h | −6.31 | Lr s(64), Lr p(14), Lr d(13), H s(7) |
| h-1 | −10.61 | Lr s (28), Lr p(5), Lr d(10), H s(56) | |
| LrH3 | h | −9.65 | Lr p(8), Lr d(26), H s(67) |
| h-1 | −10.16 | Lr p(14), Lr d(18), H s(65) | |
| h-2 | −11.65 | Lr s(39), H s(55) | |
| LrCO | h | −6.31 | Lr p(10), Lr d(26), C p(47), O p(16) |
| h-1 | −6.92 | Lr s(86), Lr p(7), Lr d(6) | |
| LuCO | h | −6.72 | Lu p(11), Lu d(23), C p(44), O p(15) |
| h-1 | −6.38 | Lu s(84), Lu p(8), Lu d(8) |
The C–O stretching frequencies are 1897 and 1921 cm−1 for LrCO and LuCO, respectively. The valence orbitals of LuCO and LrCO are compared in Fig. 3 and found to be very similar. We conclude that although the p populations strongly depend on the method of calculation, Mulliken, NBO (Natural Bond Orbital) or projection, the results for Lu 6p and Lr 7p are closely similar.
 | ||
| Fig. 3 The HF π (HOMO), σ (HOMO−1) and σ donation molecular orbitals (from left to right) of the monocarbonyls LuCO and LrCO with ECP. They are fairly similar to each other.19 Isodensity value = 0.05 a.u. | ||
3.3 Lawrencium trichloride and a divalent complex
One feature of the bonding in lanthanide chlorides is the pπ–dπ bond. It is also observed in LrCl3. Note that unlike in D3h LuCl3, the geometry of LrCl3 is C3v, with an out-of-plane vibrational frequency of only 48 cm−1. For the bonding molecular orbitals, see Fig. 4.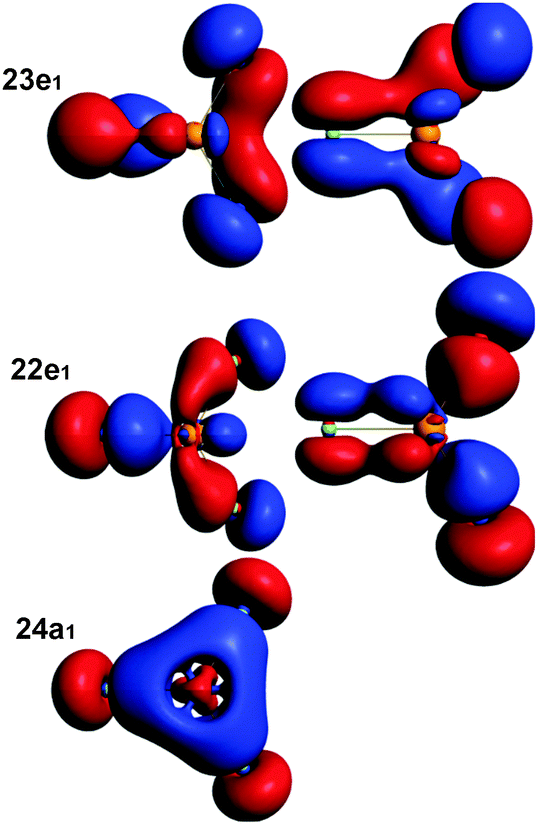 | ||
| Fig. 4 The molecular orbitals of the C3v LrCl3 with 3p(Cl)–6d(Lr) bonds. They are fairly similar to those of LuCl3. The ‘e’ orbitals are doubly degenerate and both components are shown. Isodensity value = 0.05. | ||
In recent years, one breakthrough in lanthanide chemistry is that divalent complexes were synthesized and characterized for all lanthanides. We now studied an Lr complex with the same ligand as that for Ln, i.e. C5H4SiMe3 (Cp′). Experimentally, a potassium atom in a crown ether10 functioned as the counterion of [Ln(Cp′)3]−.
A stable geometry was found for this complex anion. The electronic structure is similar to that of Lu. The metal configuration is 6d1. Spin–orbit effects were included in the calculation. As seen from Fig. 5, this HOMO is a d1 orbital on Lr.
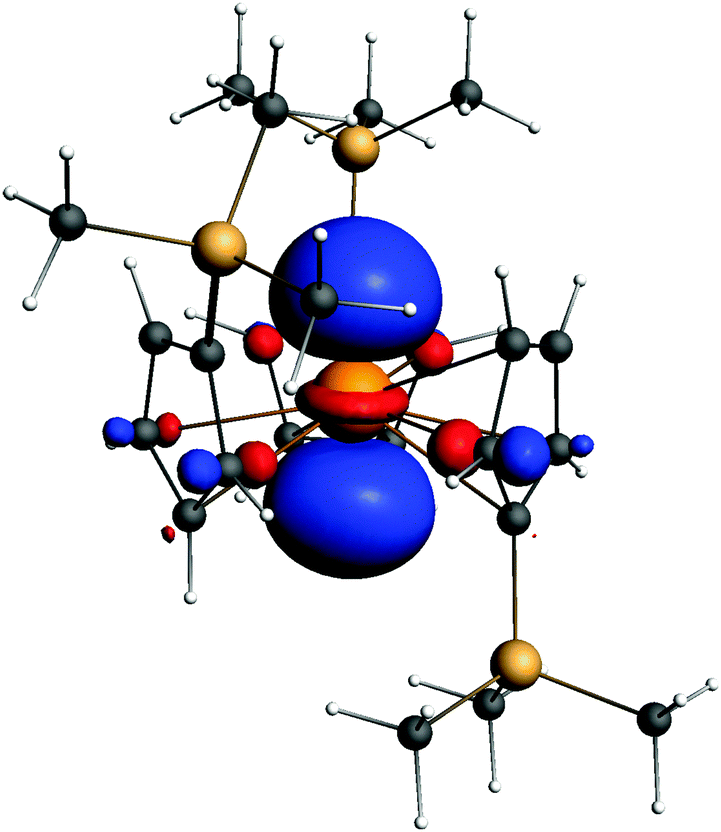 | ||
| Fig. 5 The HOMO orbital of [(Cp′)3Lr]−. Isodensity value = 0.05 a.u. | ||
The structures of all systems are given in the ESI.†
4 Relation to the periodic table
Three different choices can be outlined for the f-element rows:1. Fourteen-element rows, La–Yb and Ac–No. Put Lu and Lr in Group 3. Chosen by Jensen20 and currently Wikipedia.
2. Fourteen-element rows, Ce–Lu and Th–Lr. Put La and Ac in Group 3. Chosen by Lavelle,21 the Royal Society of Chemistry and the American Chemical Society.
3. Fifteen-element rows, La–Lu and Ac–Lr. This includes f0 among the f1 to f14 series. All elements are mostly trivalent. Their ionic and covalent22 radii form a continuous series. Now chosen by IUPAC23 and by us. To us the atomic ground state is less important than the chemical bonding, in the systems so far considered.
5 Computational details
The geometries were optimized at the ZORA2c24 level, DFT (PBE functional25) with TZ2P26 Slater basis sets. The vibrational frequencies were obtained to confirm the minima. However, [(Cp′)3Lr]− was optimized with the TPSSh functional27,28 to compare with the published [(Cp′)3Lu]− results. Solvent effects were considered by the COSMO model29 with tetrahydrofurane (THF) parameters. For more details, see the computational part of ref. 10. ADF 201630,31 and Turbomole 7.0232 packages were used. To calculate more accurate energetics, the two-component (2c)-MP233,34 and (2c)-CCSD(T)35 as implemented in Dirac 15.0,36 and CCSD(T) implemented in Molpro 2015.137,38 were employed. The basis sets are Dyall all-electron double zeta39 and ECP from the Stuttgart/Cologne group,40,41 respectively.6 Conclusion
All three ionization potentials of the lawrencium atom resemble those of the lanthanides, especially lutetium. Despite the different atomic ground states of d1 and (p*)1 for Lu and Lr, respectively, their chemical behaviour in the present systems is found to be similar. Nothing prevents one from keeping a fifteen-element trivalent actinide row Ac–Lr, under the trivalent lanthanide row La–Lu. This entirely avoids the issues arising from fourteen-element rows.20,21Acknowledgements
This project is supported by grant 275845 from The Academy of Finland to Professor Dage Sundholm. WHX thanks the China Scholarship Council and the Northwest University, Xi'an for a leave of absence and support (No. 13NW24). CSC, Espoo, Finland, provided both software and computer time. PP is grateful to the Magnus Ehrnrooth Foundation for a travel grant.References
- T. K. Sato, M. Asai, A. Borschevsky, T. Stora, N. Sato, Y. Kaneya, K. Tsukada, Ch. E. Düllmann, K. Eberhardt, E. Eliav, S. Ichikawa, U. Kaldor, J. V. Kratz, S. Miyashita, Y. Nagame, K. Ooe, A. Osa, D. Renisch, J. Runke, M. Schädel, O. Thörle-Pospiech, A. Toyoshima and N. Trautmann, Nature, 2015, 520, 209–211 CrossRef CAS PubMed.
- L. Brewer, J. Opt. Soc. Am., 1971, 61, 1101–1111 CrossRef CAS.
- J. P. Desclaux and B. Fricke, J. Phys., 1980, 41, 943–946 CrossRef CAS.
- Y. Zou and C. Froese Fischer, Phys. Rev. Lett., 2002, 88, 183001 CrossRef PubMed.
- A. Kramida, Yu. Ralchenko, J. Reader and the NIST ASD Team, Atomic Spectra Database (ver. 5.2), 2016, v 5.2.
- X.-Y. Cao and M. Dolg, Mol. Phys., 2003, 101, 961–969 CrossRef CAS.
- W. Brüchle, M. Schädel, U. W. Scherer, J. V. Kratz, K. E. Gregorich, D. Lee, M. Nurmia, R. M. Chasteler, H. L. Hall, R. A. Henderson and D. C. Hoffman, Inorg. Chim. Acta, 1988, 146, 267–276 CrossRef.
- D. C. Hoffman, R. A. Henderson, K. E. Gregorich, D. A. Bennett, R. M. Chasteler, C. M. Chasteler, C. M. Gannett, H. L. Hall, D. M. Lee, M. Nurmia, S. Cai, R. Agarwal, A. W. Charlop, Y. Y. Chu, G. T. Seaborg and R. J. Silva, J. Radioanal. Nucl. Chem., 1988, 124, 135–144 CrossRef CAS.
- U. W. Scherer, J. V. Kratz, M. Schädel, W. Brüchle, K. E. Grigorich, R. A. Henderson, D. Lee, M. Nurmia and D. C. Hoffman, Inorg. Chim. Acta, 1988, 146, 249–254 CrossRef CAS.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
- E. Eliav, U. Kaldor and Y. Ishikawa, Phys. Rev. A: At., Mol., Opt. Phys., 1995, 52, 291–296 CrossRef CAS.
- R. Ahuja, A. Blomqvist, P. Larsson, P. Pyykkö and P. Zaleski-Ejgierd, Phys. Rev. Lett., 2011, 106, 018301 CrossRef PubMed.
- S. Fritzsche, C. Z. Dong, F. Koike and A. Uvarov, Eur. Phys. J. D, 2007, 45, 107–113 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell Univ. Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- J. P. Desclaux, At. Data Nucl. Data Tables, 1973, 12, 311–406 CrossRef CAS.
- B. Vest, K. Klinkhammer, C. Thierfelder, M. Lein and P. Schwerdtfeger, Inorg. Chem., 2009, 48, 7953–7961 CrossRef CAS PubMed.
- K. Balasubramanian, J. Chem. Phys., 2002, 116, 3568–3575 CrossRef CAS.
- S. Dubillard, J.-B. Rota, T. Saue and K. Faegri, J. Chem. Phys., 2006, 124, 154307 CrossRef CAS PubMed.
- W.-H. Xu, X. Jin, M.-H. Chen, P. Pyykkö, M.-F. Zhou and J. Li, Chem. Sci., 2012, 3, 1548–1554 RSC.
- W. B. Jensen, Found. Chem., 2015, 17, 23–31 CrossRef CAS.
- L. Lavelle, J. Chem. Educ., 2008, 85, 1482–1483 CrossRef CAS.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- IUPAC, iupac.org/what-we-do/periodic-table-of-elements/, Downloaded 4 May 2016.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. Staroverov, G. Scuseria, J. Tao and J. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- ADF2016, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- TURBOMOLE V7.02 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- S. Knecht and T. Saue, X2Cmod: A modular code for Exact-Two-Component Hamiltonian Transformations. 2010–2013 with contributions from M. Ilias, H. J. Aa. Jensen and M. Repisky, 2010.
- J. K. Laerdahl, T. Saue and K. Faegri Jr, Theor. Chem. Acc., 1997, 97, 177–184 Search PubMed.
- L. Visscher, T. J. Lee and K. G. Dyall, J. Chem. Phys., 1996, 105, 8769–8776 CrossRef CAS.
- R. Bast, T. Saue, L. Visscher, H. J. A. Jensen et al., DIRAC, a relativistic ab initio electronic structure program, Release DIRAC15 (2015), see http://www.diracprogram.org.
- H.-J. Werner, P. Knowles, G. Knizia, F. Manby and M. Schütz, et al. MOLPRO, version 2015.1, a package of ab initio programs, 2015, see http://www.molpro.net Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- K. G. Dyall, Theor. Chem. Acc., 2012, 131, 1–11 Search PubMed.
- X.-Y. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- X.-Y. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02706g |
| This journal is © the Owner Societies 2016 |