
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative study of singlet oxygen production by photosensitiser dyes encapsulated in silicone: towards rational design of anti-microbial surfaces†
Sacha
Noimark‡
 a,
Enrico
Salvadori‡
bc,
Rafael
Gómez-Bombarelli‡
d,
Alexander J.
MacRobert
e,
Ivan P.
Parkin
a and
Christopher W. M.
Kay
*b
a,
Enrico
Salvadori‡
bc,
Rafael
Gómez-Bombarelli‡
d,
Alexander J.
MacRobert
e,
Ivan P.
Parkin
a and
Christopher W. M.
Kay
*b
aMaterials Chemistry Research Centre, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
bInstitute of Structural and Molecular Biology and London Centre for Nanotechnology University College London, Gower Street, London WC1E 6BT, UK. E-mail: c.kay@ucl.ac.uk; Tel: +44 207 679 7312
cSchool of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK
dDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
eUCL Division of Surgery and Interventional Science, University College London, 74 Huntley St, London WCIE 6AU, UK
First published on 23rd September 2016
Abstract
Surfaces with built-in antimicrobial activity have the potential to reduce hospital-acquired infections. One promising strategy is to create functionalised surfaces which, following illumination with visible light, are able to generate singlet oxygen under aerobic conditions. In contrast to antibiotics, the mechanism of bacterial kill by species derived from reactions with singlet oxygen is completely unselective, therefore offering little room for evolutionary adaptation. Here we consider five commercially available organic photosensitiser dyes encapsulated in silicone polymer that show varied antimicrobial activity. We correlate density functional theory calculations with UV-Vis spectroscopy, electron paramagnetic resonance spectroscopy and singlet oxygen production measurements in order to define and test the elements required for efficacious antimicrobial activity. Our approach forms the basis for the rational in silico design and spectroscopic screening of simple and efficient self-sterilising surfaces made from cheap, low toxicity photosensitiser dyes encapsulated in silicone.
1 Introduction
The high incidence of hospital-acquired infections poses a significant problem for healthcare services. Not only do they prolong patient discomfort and jeopardise their health, but they also present a taxing burden on resources. In the UK it has been estimated that the treatment of these infections cost NHS hospitals over £1 billion each year.1 In England, an estimated 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients contract a hospital-acquired infection annually within the NHS, with 9000 deaths associated with methicillin-resistant Staphylococcus aureus bloodstream infections and Clostridium difficile infections in 2007.2,3
000 patients contract a hospital-acquired infection annually within the NHS, with 9000 deaths associated with methicillin-resistant Staphylococcus aureus bloodstream infections and Clostridium difficile infections in 2007.2,3
Hospital surfaces are significant reservoirs of bacteria and contact by healthcare personnel and patients aids the transfer of bacteria and facilitates the spread of infection.4 Hence, the use of self-sterilising surface technology is one strategy that may help disrupt the transmission of bacteria between surfaces, patients and healthcare professionals.
Current research involves the use of photosensitiser dyes immobilised in polymers as part of an infection-control strategy in healthcare environments.5–19 Cationic dyes such as methylene blue or crystal violet are proven antimicrobial agents against both Gram-positive and Gram-negative bacteria.8–19 Porphyrins and phthalocyanines are also used for photodynamic therapy (PDT) applications, however, although they have been successfully incorporated into polymers,20–22 the procedure is non-trivial as compared to swell-encapsulation. Moreover, some porphyrins can be significantly more expensive than widely available dyes considered in the present study.
Photobactericidal polymers can be synthesised using a simple dipping approach to incorporate photosensitiser dye molecules including methylene blue, toluidine blue O and crystal violet into polymers.5,11–19 Such dye-incorporated polymers have induced the lethal photosensitisation of both Gram-positive and Gram-negative bacteria, with samples demonstrating strong antimicrobial efficacy under laser irradiation and standard hospital lighting conditions.5–19Table 1 summarises representative examples of the light-activated antimicrobial activity of these dyes against both Gram-positive and Gram-negative bacteria. Some of these polymers also induce significant bacterial kill under dark conditions.14–18 The non-site-specific mode of bacterial attack utilised by this photochemical kill approach reduces the risk of bacterial resistance. The photosensitiser dyes used in this strategy are cheap and commercially available and the method of incorporation is easy to scale up for industrial production. Consequently, dye-containing polymers are attractive candidates for use as antimicrobial surfaces.
Antimicrobial activity is predominantly induced by a light-activated mechanism, see Fig. 1. The dye molecules absorb light in the visible region of the electromagnetic spectrum, and subsequently undergo intersystem crossing (ISC) to the triplet state.23 The triplet state can also undergo one of two photochemical processes, historically termed ‘Type I’ and ‘Type II’ mechanisms. The Type I mechanism involves one-electron redox processes that occur between the excited triplet state dye molecule and any molecule with the appropriate redox potential; in this context, proteins, unsaturated lipids, DNA etc. on the target cell.24 The Type II mechanism involves an energy transfer process between the excited triplet state dye molecule and molecular oxygen in the vicinity, resulting in the generation of singlet oxygen (1O2) species.24 Highly electrophilic, 1O2 rapidly reacts with electron-rich substrates such as unsaturated hydrocarbons, amines, sulphides and phenols which are abundant in cell membrane proteins, lipids and in DNA.25–29 The Type II process is the desirable mechanism, as the chromophore returns to the ground state following 1O2 production and can subsequently generate more 1O2 in a catalytic fashion.
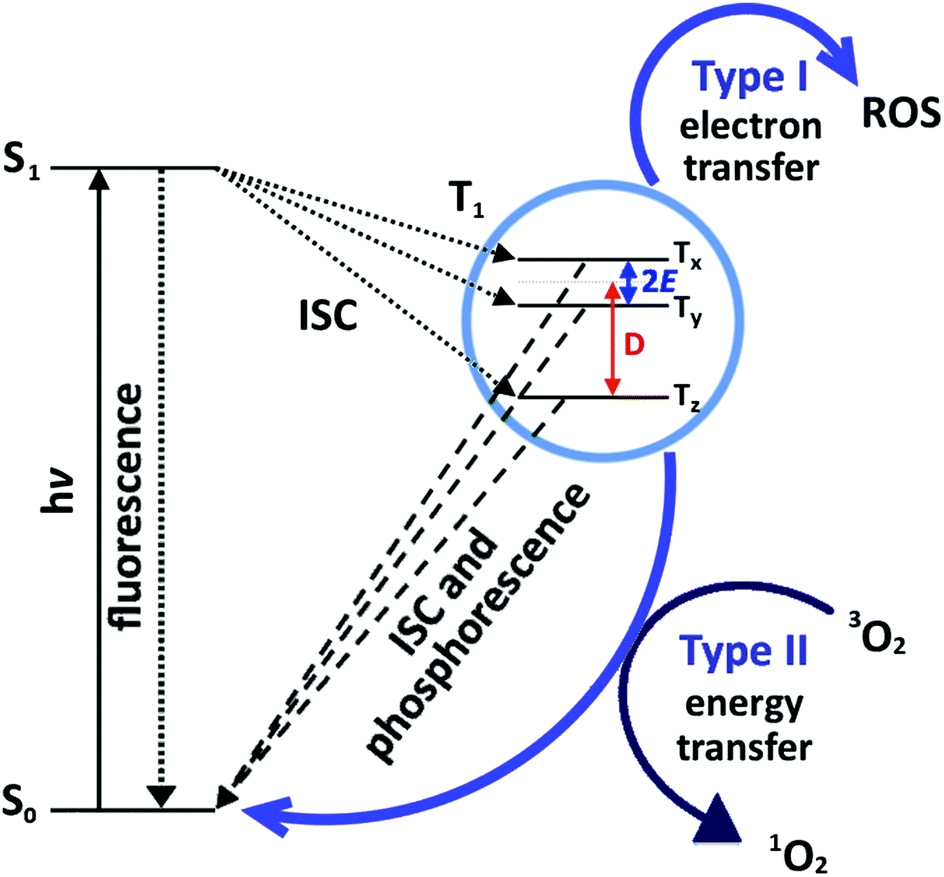 | ||
| Fig. 1 Jablonski diagram and accompanying photochemical processes subsequent to photoexcitation of a photosensitizer. The ground state molecule (S0) absorbs a photon of energy and is promoted to an excited singlet state (S1). The excited singlet state molecule can release energy radiatively (fluorescence), via internal conversion or can undergo ISC to the excited triplet state (T1) which has three sublevels: Tx, Ty, Tz. D and E are the zero field splitting parameters, which describe the magnetic dipolar interaction between the two unpaired electrons. The triplet state molecule can release energy as radiation (phosphorescence) or transfer its energy to surrounding molecules by either Type I or Type II photoprocesses and return to the ground state. A Type I photoprocess involves electron transfer between the triplet state and a substrate, whereas a Type II photoprocess involves the quenching of the triplet state by molecular oxygen. | ||
Suitable dyes for the development of photobactericidal polymers should exhibit properties including: (i) strong absorption in the visible region of the electromagnetic spectrum, (ii) good photostability under prolonged irradiation, (iii) high ISC yield for efficient production of the triplet state, (iv) long triplet lifetime to allow for the reaction with surrounding molecules such as molecular oxygen, (v) selectively towards a Type II photoprocess, and (vi) when incorporated into a polymer, the dyes should not leach into the environment.
Here we explore the structure–activity relationships of several (i) phenothiazine dyes: acridine orange (AO), methylene blue (MB) and toluidine blue O (TBO) and (ii) triarylmethane dyes: crystal violet (CV) and malachite green (MG) encapsulated in silicone. We investigated both their electronic structures, obtained by DFT calculations, and their photochemical properties, using several experimental techniques, in order to gain insight into the reasons for the differing photobactericidal activity. Time-resolved electron paramagnetic resonance (TR-EPR) spectroscopy was used to investigate the properties of the photoexcited triplet states, while, time-resolved near infrared (TR-NIR) spectroscopy was employed to monitor 1O2 phosphorescence. Our approach provides a straightforward methodology for a rapid and effective materials design strategy for light-activated antimicrobial surfaces.
2 Experimental
2.1 DFT calculations
Calculations were performed with the Gaussian 09 suite of programmes at the DFT level of theory using several hybrid correlation-exchange functionals: B3LYP, long-range corrected CAM-B3LYP,30 meta-GGA M062X,31 the extended hybrid functional X3LYP,32 ω-B97XD. Unrestricted open-shell calculations were performed for the triplet states. Harmonic analysis was performed to check the nature of equilibrium geometries. The effect of aqueous solution on equilibrium geometries, energies and optical properties was included via the IEF-PCM approach using the default parametrisation. Magnetic properties were calculated using the ORCA 2.9 suite of programmes, with solvation effects included via the COSMO method. The dipole–dipole zero field splitting (ZFS) was predicted using the methods described by Neese et al33 over several basis sets (TZVP, IGLO-II and also EPR-II for the molecules lacking sulphur atoms) and DFT functionals (PBE, B3LYP, B2PLYP). The URO option (unrestricted natural orbitals) was used.2.2 Materials synthesis and characterisation
:1 ratio and an excess (4–5 g) was spread into a channel on a clean glass surface. The system was centrifuged for 5 minutes (1100 rpm, 60 mbar vacuum) and any excess was removed. The polymer was cured at 150 °C for 6 minutes after which it was allowed to cool and subsequently, was cut into 1 cm × 3 cm strips.
:1 acetone (VWR):water swelling solution made up to a concentration of 0.002 mol dm−3 dye. The samples were allowed to swell in the dark under room temperature conditions for 24 h, after which they were removed and air-dried in the dark (24 h). Residual dye was removed by gently washing the samples in water and the towel-dried samples were stored under dark conditions.
The triarylmethane dyes, crystal violet (Sigma-Aldrich) and malachite green oxalate salt (Sigma-Aldrich), were incorporated into the silicone polymer substrate using a simple dipping process.14 The silicone samples were immersed in dye solutions (0.002 mol dm−3) made up in water for 24 h, after which they were air-dried in the dark for 24 hours. Residual dye was washed off using water and the samples were towel-dried and stored under dark conditions. The uptake of dye into the polymers can be controlled by varying the dipping time, until polymer saturation is achieved.14
000 laser pulses and was analysed using the curve fitting App in MATLAB™.
We note that a rigorous approach has been developed to take into account the spectral overlap between the photosensitiser characteristic absorption spectrum and the emission profile of the light source employed.37 This is particularly relevant for: steady state studies; comparisons between different experimental set ups; and the determination of absolute triplet quantum yields when light sources with complex emission profiles are used (e.g. fluorescent with light). However, here we focus solely on relative quantum yields as we aim to pinpoint the effect of the encapsulation into a polymeric matrix on singlet oxygen production.
3 Results and discussion
Numerous studies have examined 1O2 generation by photosensitisers.43–46 However, to date there has been scant investigation of the triplet states responsible for 1O2 generation in PDT. In the following, the likelihood of whether the triplets of the five dyes considered will undergo a Type I or Type II mechanism is assessed using DFT, and their triplet states, which are directly responsible for the generation of reactive oxygen species, are detected by TR-EPR. This combination of experimental (optical and magnetic spectroscopies) and theoretical studies is then examined to establish trends in the resultant 1O2 yields (TR-NIR) of dye embedded polymers. The impact of these findings on the design of new antimicrobial materials is addressed and discussed in relation to the spatial distribution of the dyes within the polymer.3.1 DFT studies
A theoretical analysis of the five dyes was conducted to identify structure–property trends and analyse their initial suitability as photosensitisers. For that, we assessed their electronic structure, lowest singlet and triplet energy levels, electron-donating and accepting ability and ZFS in the lowest triplet state.The most significant differentiating feature in terms of molecular structure across the dyes is the rigid, planar geometry of TBO, AO and MB, as opposed to the rotatable phenyl groups of CV and MG. Two opposing effects determine the arrangement of the aryl rings in a propeller-like structure: minimising steric repulsion and maximising π conjugation. CV is predicted to have a ground state belonging to the D3 point group, with the three subunits at an angle δ = 32.5° (compared to δ = 28.0° in the crystal structure).47 In the case of MG, the ring lacking the nitrogen group is positioned at δ = 43.5° to the central plane, whereas the other two rings with a nitrogen group substituent are at δ = 29.1° from the plane, and thus more strongly conjugated.
The structures, the frontier molecular orbitals (HOMO and LUMO), and the spin density distribution of the lowest triplet state of the five dye molecules are depicted in Fig. 2. The three planar molecules have similar electronic density distributions. Despite the offset in dihedral angle between the aryl substituents, the π systems of the triarylmethane dyes are highly conjugated and show similar distribution to the planar dyes. Because of the D3 symmetry, CV has two degenerate HOMOs belonging to the E irreducible representation, whereas the LUMO belongs to the A1 irreducible representation with delocalisation over all three aryl units.
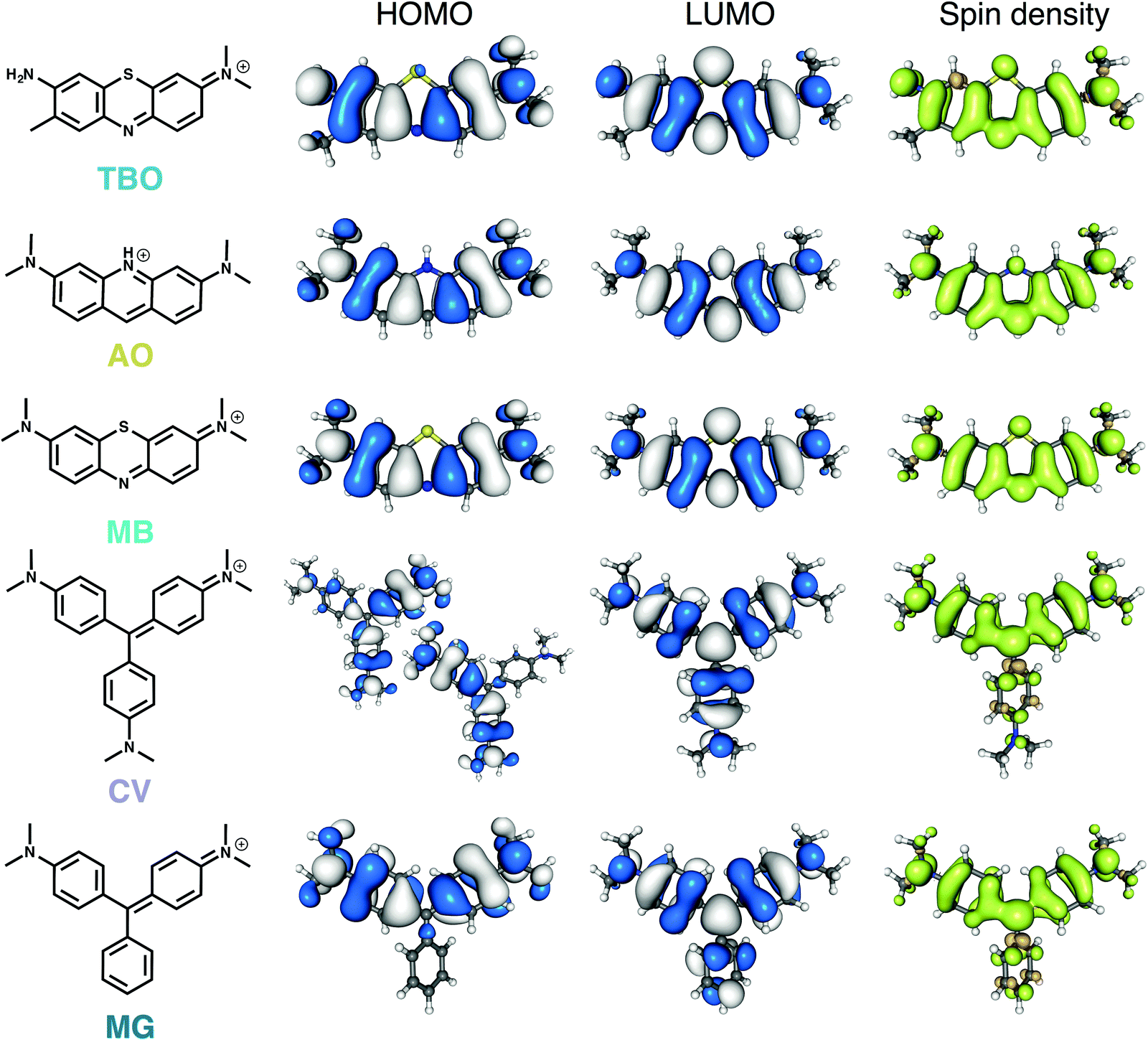 | ||
| Fig. 2 Orbital electronic densities (contour at 0.02 au) for the ground state and spin densities (contour at 0.002 au) for the first triplet state of MB, TBO, AO, CV and MG at the CAM-B3LYP/cc-pVDZ level of theory. | ||
We assessed the accuracy of TD-DFT calculations for these cationic dyes (see ESI†). The experimental trends are recovered, with an absolute deviation of around 0.25 eV with respect to excitation energies measured in solution. This deviation of TD-DFT calculations for cationic dyes in solution has been described in the literature.48
The distribution of spin densities is similar across all dyes (Fig. 2). The three planar dyes have almost identical frontier orbital distributions, and in the case of MG only the rings bearing dimethylamino groups participate in the π overlap. Whereas two degenerate E triplet states exist in the case of CV, relaxing their geometries breaks the symmetry as one of the dimethylaminophenyl groups moves away from planarity (δ = 47°) thus lowering its contribution to the π system. The co-planarity is increased for the other two rings (δ = 25°), which affords a geometry similar to that of the relaxed MG triplet, and thus a spatial spin distribution similar to the other dyes considered here.
Triplet energies were estimated using TD-DFT and UDFT calculations (Table 2) in implicit solvent and span from around 1.30 (TBO and MB) to 2.17 (AO) eV. Comparison with available phosphorescence spectra in the literature suggests that UDFT performs better, with average deviations around 0.1 eV. Since the lowest singlet state of molecular oxygen lies at approximately 0.98 eV, the Type II mechanism is energetically available for all five dyes. Note that, the lowest triplet excited state of AO (ca. 2.2 eV) lies above both the first and second singlet states of molecular oxygen (0.98 and 1.63 eV respectively), and hence AO has an additional energy channel to transfer the excitation. Overall, the energies of both the lowest singlet and triplet states of the triarylmethane dyes are amongst those of the planar compounds.
| Theoretical | Experimental | ||||||
|---|---|---|---|---|---|---|---|
| B3LYP cc-pVDZ | B3LYP cc-pVTZ | CAM-B3LYP cc-pVDZ | X3LYP cc-pVDZ | M062X cc-pVDZ | ωB97XD cc-pVDZ | ||
| TBO | 1.34 | 1.35 | 1.32 | 1.34 | 1.44 | 1.32 | N/A |
| AO | 1.93 | 1.96 | 2.13 | 1.94 | 2.21 | 2.17 | 2.0838,39–2.1340 |
| MB | 1.28 | 1.28 | 1.28 | 1.28 | 1.39 | 1.28 | 1.3941–1.5040 |
| CV | 1.67 | 1.70 | 1.70 | 1.68 | 1.86 | 1.84 | 1.6942 |
| MG | 1.45 | 1.48 | 1.58 | 1.46 | 1.66 | 1.62 | N/A |
In order to analyse the potential competing role of the Type I versus the Type II mechanism, we have computed the electron affinity and ionisation potential of the dyes in their triplet state (Table 3, see ESI† for values referenced to the ground state). The results are in keeping with experimental results, MB has a standard reduction potential of +0.011 V, compared with −0.357 V for CV and their B3LYP/cc-pVDZ aqueous ground state electron affinities are 3.79 eV and 3.29 eV respectively.49 As occurs with the singlet and triplet excitation energies, both triarylmethane dyes lie within the range spanned by the planar dyes both as excited-state electron donors and acceptors.
| Energy (eV) | ||||
|---|---|---|---|---|
| Ionisation potential | Electron affinity | |||
| GAS | PCM | GAS | PCM | |
| TBO | 8.70 | 4.45 | 6.47 | 5.13 |
| AO | 7.83 | 3.61 | 6.17 | 4.89 |
| MB | 8.58 | 4.45 | 6.36 | 5.07 |
| CV | 7.37 | 3.76 | 5.98 | 4.95 |
| MG | 7.93 | 4.07 | 6.22 | 5.00 |
ZFS were computed for the five dyes, and are presented in Table 4. For each molecule, the sign of D was independent of the functional and the basis set. For the phenothiazine dyes, the theoretical D values were approximately 30% smaller than the experimental values. This deviation is similar to that found for excited triplets with low D values, such as chlorophyll a.33 This underestimation has been attributed to the lack of medium-range correlation effects in the DFT calculations. Structurally similar molecules such as anthracene, are known to have a positive D values, whereas rod-like molecules, such as carotenoids, have negative D values.50 The negative D value predicted for the phenothiazine dyes implies that their principal zero-field axis is rotated from being perpendicular to the molecular plane, to parallel. This is probably due to the presence of the hetero-atoms and associated lone pairs. MG is also predicted to have a negative D, which we can understand in so far as the non-substituted phenyl ring plays little role in the π system, as it is rotated by 43° away from the central methenium plane. On the other hand, CV is predicted to have a far smaller and positive D value. Presumably, this difference is due to the equivalence of the three rings and the larger delocalisation over them.
| Method | D (MHz) | |||||
|---|---|---|---|---|---|---|
| B3LYP TZVP | B3LYP IGLO-II | COSMO B3LYP IGLO-II | PBE TZVP | PBE IGLO-II | Experimentala | |
| a This work. Note that the sign of D cannot be determined directly from the TR-EPR experiments. | ||||||
| TBO | −1325 | −1296 | −1186 | −1353 | −1319 | 1790 |
| AO | −1301 | −1267 | −1205 | −1364 | −1325 | 1800 |
| MB | −1295 | −1269 | −1151 | −1322 | −1291 | 1740 |
| CV | 808 | 796 | 991 | 767 | 756 | 1650 |
| MG | −1138 | −1117 | −1014 | −1117 | −1097 | — |
3.2 Spectroscopic characterisation
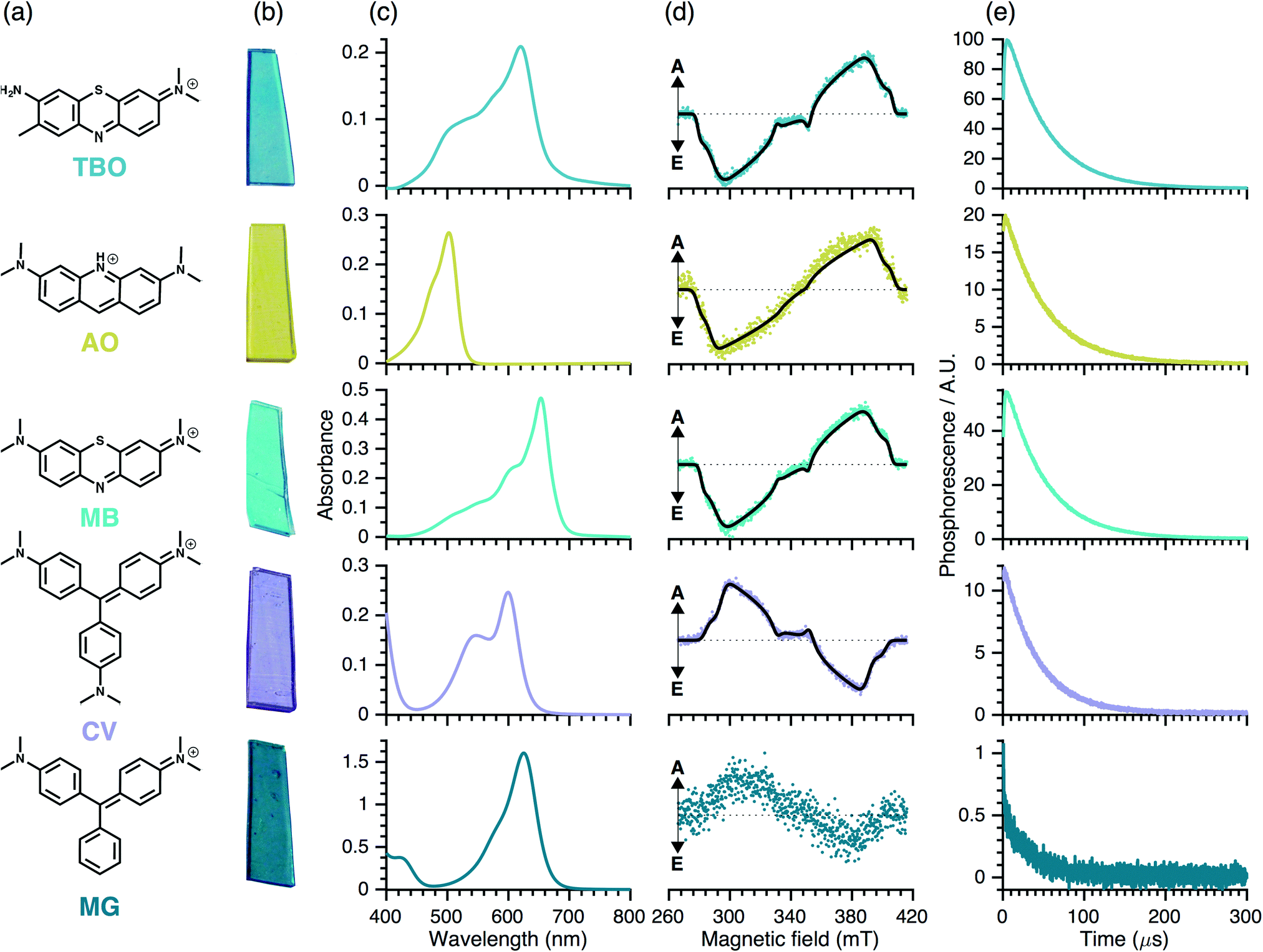 | ||
| Fig. 3 (a) Molecular structure and (b) photographs of photosensitiser dye molecules encapsulated within medical grade silicone. Characterisation of their photochemical properties using: (c) UV-Vis absorbance spectroscopy, (d) TR-EPR spectroscopy and (e) TR-NIR spectroscopy. In (d), the simulated TR-EPR spectra are superimposed onto the experimental data (black line) and A and E stand for absorption and emission, respectively. | ||
The TR-EPR spectra (Fig. 3(d)) are typical of spin-polarised (i.e. non-Boltzmann populated) triplet states of aromatic molecules. Although they differ in detail and intensity, the spectra are similar within each dye group, implying that chemical variability has only minor influence on the electronic structure of the triplet states. The spectra were simulated to obtain the ZFS parameters, |D| and |E|, and the relative populations of the three triplet sublevels, px:py:pz (see Table 5). Note that we have excluded MG from the simulations due the weak intensity of the TR-EPR spectrum.
| Relative populations [px:py:pz] |
ZFS parameters D, |E| (MHz) | |
|---|---|---|
| TBO | 0.81:0.19:0 |
−1790, 400 |
| AO | 0.68:0.32:0 |
−1800, 430 |
| MB | 0.76:0.24:0 |
−1738, 390 |
| CV | 0:0.46:0.54 |
+1650, 350 |
The triplet states of the planar phenothiazine dyes and propeller-like CV have similar ZFS parameters (see Table 5) despite differences in the molecular shapes; this suggests that in the case of CV, the spin density is mostly localised over two of the three rings (see DFT section). However, the relative sublevel populations give inverse polarisation patterns of the type EEAEAA for the phenothiazine dyes compared to AAEAEE for CV (see Table 5). This inversion can be rationalised in two alternative ways: either the change in the molecular geometry – planar as opposed to propeller-like – influences the sign of the ZFS parameters, or it changes the population rates without affecting the sign of ZFS parameters. In light of the DFT calculations, we conclude that the sign of D is indeed reversed between the two groups.
Inspection of Fig. 3(d) indicates that the quality (signal-to-noise ratio, S/N) of the TR-EPR spectra is somewhat worse for AO and substantially worse for MG. This may be due to either lower triplet yield (i.e. ISC efficiency) or a more isotropic ISC, leading to a less polarised spin system. However, the similar relative triplet sub-level populations for the two classes of dyes implies that the relative rates of ISC are similar. Hence, the worse S/N observed for AO in comparison with TBO and MB is likely to stem from reduced ISC efficiency, which could be due to reduced spin–orbit coupling due to the lack of the sulphur atom. Similar considerations could hold for MG, which within the triarylmethane dye group, lacks an amine substituent and displays worse S/N and hence has lower triplet yield than CV and can hardly be detected experimentally.
Quantum yield. Using the integrated signal of the TR-NIR-1O2 phosphorescence, in combination with the UV-Vis absorbance data, the relative 1O2 quantum yields were calculated using eqn (1):
| I = κ × P × ΦΔ × (1 − 10−A) | (1) |
| ΦΔTBO > ΦΔAO > ΦΔMB > ΦΔCV > ΦΔMG |
| TRNIR-1O2 phosphorescence signal analysis | |||
|---|---|---|---|
| τ 1 (μs) | τ 2 (μs) | Φ Δ normalised | |
| TBO | 48.23 ± 0.03 | 1.46 ± 0.01 | 1.00 |
| AO | 49.59 ± 0.08 | 0.62 ± 0.01 | 0.85 |
| MB | 49.62 ± 0.08 | 1.40 ± 0.01 | 0.56 |
| CV | 40.00 ± 0.12 | 0.78 ± 0.01 | 0.10 |
| MG | — | — | 0.01 |
To determine the effects of immobilisation of the dyes on 1O2 generation, TR-NIR measurements were performed on dye solutions. These showed a differing trend in 1O2 quantum yields compared to polymer-encapsulated dyes. The 1O2 generation by AO encapsulated in a polymer was markedly better than in solution. This behaviour may be due either to more efficient deactivation pathways (e.g. vibrational relaxation and collisional quenching) or to intermolecular interactions (π-stacking and/or dimerisation) that hinder ISC efficiency and subsequently lead to reduced 1O2 quantum yields in solution.
The poor performance of MB with respect to TBO encapsulated in polymers may be due to comparatively higher concentrations and thus, a dimerisation effect. Indeed, Baptista et al. have summarised the mechanisms for ROS generation following MB photoexcitation:511O2 arises from the Type 2 energy transfer mechanism between the excited triplet state of the MB monomer and molecular oxygen. MB dimers may also be photoexcited with visible light since they exhibit peak absorption near 590 nm, however the excited MB dimers are too short-lived to enable significant interaction with oxygen to generate any 1O2. In the present study, a 532 nm excitation source was used: both dimers and monomers absorb at this wavelength. If dimers are present in the polymer the relative singlet oxygen generation efficiency will be reduced accordingly. However, the results presented in this work are relevant for potential antimicrobial applications as they suggest further materials optimisation should be undertaken to reduce the presence of dimers within the polymer to enhance 1O2 generation.
The higher AO 1O2 quantum yield in the polymer is also in contrast to the poor S/N observed in TR-EPR. It may be that this is due to a better energy coupling with molecular oxygen, resulting in efficient 1O2 production despite triplet state yields that are poorer than MB. As mentioned in the DFT section, the energy of the AO triplet state is high enough also to couple with the second singlet state of molecular oxygen, which may account for the increased yield.
A similar effect is observed for the triarylmethane dye, CV, which shows negligible 1O2 production in low viscosity solvents where the group can rotate freely, but is markedly improved when encapsulated in the polymer. This can be understood in that rotor molecules such as MG and CV can form twisted intramolecular charge transfer (TICT) states in solution, rather than undergoing ISC to the triplet state,52 resulting in negligible observed 1O2 yields. Thus, it appears that when encapsulated in the polymer, formation of TICT states are suppressed in CV (but not in MG), resulting in higher triplet yield and comparatively enhanced 1O2 production.
Kinetics. The TR-NIR response can be fitted with two components with lifetimes τ1 and τ2. τ1 corresponds to collisional quenching of 1O2 with other species such as the polymer matrix (τΔ). τ2, corresponds to the formation of 1O2 by energy transfer from the triplet state of the dye (τT). In silicone, the calculated lifetimes, τ1 and τ2 are similar for the three phenothiazine dyes, whereas τ1 is 20% shorter for CV. This possibly reflects a different spatial distribution: CV (and MB) are predominantly surface bound and hence more exposed to quenching by molecular oxygen, whereas the other dyes are encapsulated within the polymer bulk.
Overall, the TR-EPR and TR-NIR measurements indicate that polymer-encapsulated phenothiazine dyes AO, TBO and MB are all potential light-activated antimicrobial agents, as they yield efficient triplet-state production and 1O2 generation. However, the 1O2 diffusion distance of the ROS is very short (<1 micron).11,14 Since the incorporation strategy for phenothiazine dyes results in a uniform dispersion through the polymer bulk, the relative dye concentration and ROS generation at the surface is low. In contrast, CV has a high surface dye concentration14 and – despite its lower triplet yield – is suprisingly effective as a ROS generator. In healthcare environments, bacteria colonise surfaces and therefore, a material that generates ROS at the surface–bacteria interface is key for enhancing kills. Location of the photosensitiser in these photo-active polymers is important, however, it is necessary to consider scalability for their widespread use in healthcare applications. Although when TBO and MB are surface immobilised9 (Table 1) their antimicrobial efficacies greatly surpass that of CV, their surface immobilisation is both costly and non-trivial, requiring lengthy and challenging functionalisation steps.
4 Conclusion
Using a simple encapsulation strategy, we incorporated five commercially available organic photosensitiser dyes into medical grade silicone. DFT predictions, that the triplet states of these dyes molecules should be 1O2 photosensitisers, were confirmed spectroscopically. The DFT calculations shed light onto the underlying photophysical mechanism by showing that Type II reactions are more favourable than Type I. Moreover, there was no indication of a radical signal appearing in the EPR spectra following prolonged irradiation. This not only demonstrates the photostability of the dyes, but also suggests that Type II processes are predominant over Type I. Furthermore, the experiments correlated with the DFT predictions and with the exception of MG, all dyes showed a Type II mechanism. The experiments also indicated that the photoactivity of rotor dyes such as CV can be enhanced by immobilisation in polymer. However, and more crucially, a high concentration of dye should be localised at the surface of the polymer so that the 1O2 produced may be utilized. These critical aspects should be considered when designing new photobactericidal polymers. One strategy to achieve this would be to optimise the swell-encapsulation procedure so that good 1O2 generators, such as MB and TBO, are preferentially located at the surface. Alternatively, covalently linking the dye onto the polymer surface would also provide a high local concentration, but this approach is both challenging and not easily scalable for industrial production.Optimisation by the current empirical approach is not effective as a long-term strategy and achieving a mechanistic understanding is critical to underpin future advances in the development of photobactericidal surfaces. We suggest that the combination of theory and experiment presented here will facilitate the rapid design of more effective light-activated antimicrobial materials.
Acknowledgements
IPP and SN would like to thank the EPSRC and Ondine Biopharma for their financial support.References
- R. Plowman, Eurosurveillance, 2000, 5, 49–50 Search PubMed.
- National Audit Office: Reducing Healthcare Associated Infections in Hospitals in England, 12 June 2009.
- NICE QS61; Infection prevention and control, http://publications.nice.org.uk/infection-prevention-and-control-qs61, accessed: 11-03-2016.
- K. Page, M. Wilson and I. P. Parkin, J. Mater. Chem., 2009, 19, 3819–3831 RSC.
- S. Perni, J. Pratten, M. Wilson, C. Piccirillo, I. P. Parkin and P. Prokopovich, J. Biomater. Appl., 2011, 25, 387–400 CrossRef CAS PubMed.
- S. Ismail, S. Perni, J. Pratten, I. Parkin and M. Wilson, Infect. Control Hosp. Epidemiol., 2011, 32, 1130–1132 CrossRef PubMed.
- A. J. T. Naik, S. Ismail, C. Kay, M. Wilson and I. P. Parkin, Mater. Chem. Phys., 2011, 129, 446–450 CrossRef CAS.
- S. Perni, C. Piccirillo, A. Kafizas, M. Uppal, J. Pratten, M. Wilson and I. P. Parkin, J. Cluster Sci., 2010, 21, 427–438 CrossRef CAS.
- C. Piccirillo, S. Perni, J. Gil-Thomas, P. Prokopovich, M. Wilson, J. Pratten and I. P. Parkin, J. Mater. Chem., 2009, 19, 6167–6171 RSC.
- S. Perni, P. Prokopovich, I. P. Parkin, M. Wilson and J. Pratten, J. Mater. Chem., 2010, 20, 8668–8673 RSC.
- S. Perni, C. Piccirillo, J. Pratten, P. Prokopovich, W. Chrzanowski, I. P. Parkin and M. Wilson, Biomaterials, 2009, 30, 89–93 CrossRef CAS PubMed.
- S. Perni, P. Prokopovich, C. Piccirillo, J. Pratten, I. P. Parkin and M. Wilson, J. Mater. Chem., 2009, 19, 2715–2723 RSC.
- S. Noimark, C. W. Dunnill, C. W. M. Kay, S. Perni, P. Prokopovich, S. Ismail, M. Wilson and I. P. Parkin, J. Mater. Chem., 2012, 22, 15388–15396 RSC.
- S. Noimark, M. Bovis, A. J. MacRobert, A. Correia, E. Allan, M. Wilson and I. P. Parkin, RSC Adv., 2013, 3, 18383–18394 RSC.
- S. Noimark, E. Allan and I. P. Parkin, Chem. Sci., 2014, 5, 2216–2223 RSC.
- S. Noimark, J. Weiner, N. Noor, E. Allan, C. K. Williams, M. S. P. Shaffer and I. P. Parkin, Adv. Funct. Mater., 2015, 25, 1367–1373 CrossRef CAS.
- S. K. Sehmi, S. Noimark, J. C. Bear, W. J. Peveler, M. Bovis, E. Allan, A. J. MacRobert and I. P. Parkin, J. Mater. Chem. B, 2015, 3, 6490–6500 RSC.
- S. J. Beech, S. Noimark, K. Page, N. Noor, E. Allan and I. P. Parkin, RSC Adv., 2015, 5, 26364–26375 RSC.
- M. J. Bovis, S. Noimark, J. H. Woodhams, C. W. M. Kay, J. Weiner, W. J. Peveler, A. Correia, M. Wilson, E. Allan, I. P. Parkin and A. J. MacRobert, RSC Adv., 2015, 5, 54830–54842 RSC.
- A. Felgenträger, T. Maisch, A. Späth, J. A. Schröder and W. Bäumler, Phys. Chem. Chem. Phys., 2014, 16, 20598–20607 RSC.
- F. van Laar, F. Holsteyns, I. Vankelecom, S. Smeets, W. Dehaen and P. A. Jacobs, J. Photochem. Photobiol., A, 2001, 144, 141–151 CrossRef CAS.
- S. Artarsky, S. Dimitrova, R. Bonnett and M. Krysteva, Sci. World J., 2006, 6, 374–382 CrossRef CAS PubMed.
- M. Wainwright and K. B. Crossley, Int. Biodeterior. Biodegrad., 2004, 53, 119–126 CrossRef CAS.
- C. S. Oliveira, R. Turchiello, A. J. Kowaltowski, G. L. Indig and M. S. Baptista, Free Radicals Biol. Med., 2011, 51, 824–833 CrossRef CAS PubMed.
- A. Jimenez-Banzo, X. Ragas, P. Kapusta and S. Nonell, Photochem. Photobiol. Sci., 2008, 7, 1003–1010 CAS.
- A. Michaeli and J. Feitelson, Photochem. Photobiol., 1994, 59, 284–289 CrossRef CAS PubMed.
- A. Michaeli and J. Feitelson, Photochem. Photobiol., 1995, 61, 255–260 CrossRef CAS PubMed.
- J. L. Ravanat, P. Di Mascio, G. R. Martinez, M. H. G. Medeiros and J. Cadet, J. Biol. Chem., 2000, 275, 40601–40604 CrossRef CAS PubMed.
- X. S. Zhang, B. S. Rosenstein, Y. Wang, M. Lebwohl and H. C. Wei, Free Radicals Biol. Med., 1997, 23, 980–985 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- X. Xu and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2673–2677 CrossRef CAS PubMed.
- S. Sinnecker and F. Neese, J. Phys. Chem. A, 2006, 110, 12267–12275 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- L. M. Rossi, P. R. Silva, L. L. R. Vono, A. U. Fernandes, D. B. Tada and M. S. Baptista, Langmuir, 2008, 24, 12534–12538 CrossRef CAS PubMed.
- O. Planas, N. Macia, M. Agut, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2016, 138, 2762–2768 CrossRef CAS PubMed.
- F. Cieplik, A. Pummer, J. Regensburger, K.-A. Hiller, A. Spath, L. Tabenski, W. Buchalla and T. Maisch, Front. Microbiol., 2015, 6, 706 Search PubMed.
- I. Goryacheva, G. Mel'nikov and S. Shtykov, J. Anal. Chem., 2000, 55, 874–878 CrossRef CAS.
- I. Goryacheva, S. Shtykov, G. Melnikov and E. Fedorenko, Environ. Chem. Lett., 2003, 1, 82–85 CrossRef CAS.
- H.-J. Timpe and S. Neuenfeld, J. Chem. Soc., Faraday Trans., 1992, 88, 2329–2336 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- G. N. Lewis, T. T. Magel and D. Lipkin, J. Am. Chem. Soc., 1942, 64, 1774–1782 CrossRef CAS.
- X. Ragàs, X. He, M. Agut, M. Roxo-Rosa, A. R. Gonsalves, A. C. Serra and S. Nonell, Molecules, 2013, 18, 2712–2725 CrossRef PubMed.
- C. S. Oliveira, R. Turchiello, A. J. Kowaltowski, G. L. Indig and M. S. Baptista, Free Radicals Biol. Med., 2011, 51, 824–833 CrossRef CAS PubMed.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS PubMed.
- R. Pottier, R. Bonneau and J. Joussor-Dubien, Photochem. Photobiol., 1975, 22, 59–61 CrossRef CAS PubMed.
- S. Lovell, B. J. Marquardt and B. Kahr, J. Chem. Soc., Perkin Trans. 2, 1999, 2241–2247 RSC.
- P. Homem-de Mello, B. Mennucci, J. Tomasi and A. B. F. da Silva, Theor. Chem. Acc., 2005, 113, 274–280 CrossRef CAS.
- P. Rao and E. Hayon, J. Am. Chem. Soc., 1974, 3794, 1287–1294 CrossRef.
- E. Salvadori, M. Di Valentin, C. W. M. Kay, A. Pedone, V. Barone and D. Carbonera, Phys. Chem. Chem. Phys., 2012, 14, 12238–12251 RSC.
- H. C. Junqueira, D. Severino, L. G. Dias, M. S. Gugliotti and M. S. Baptista, Phys. Chem. Chem. Phys., 2002, 4, 2320–2328 RSC.
- M. A. Haidekker and E. A. Theodorakis, J. Biol. Eng., 2010, 4, 11 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02529c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2016 |