
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Far-infrared spectra of the tryptamine A conformer by IR-UV ion gain spectroscopy†
Michael
Schmitt
*a,
Frans
Spiering
b,
Vitali
Zhaunerchyk‡
b,
Rienk T.
Jongma
b,
Sander
Jaeqx
b,
Anouk M.
Rijs
*b and
Wim J.
van der Zande
*b
aHeinrich-Heine-Universität Institut für Physikalische Chemie, D-40225 Düsseldorf, Germany. E-mail: mschmitt@uni-duesseldorf.de
bRadboud University, Institute for Molecules and Materials, Felix Laboratory, Toernooiveld 7c, 6525 ED Nijmegen, The Netherlands. E-mail: a.rijs@science.ru.nl; wim.van.der.zande@asml.com
First published on 10th November 2016
Abstract
We present far infrared spectra of the conformer A of tryptamine in the 200 to 500 cm−1 wavenumber range along with resonant photoionization spectra of the far-infrared excited conformer A of tryptamine. We show that single-far-infrared photon excited tryptamine has highly structured resonance enhanced multi-photon ionization spectra, revealing the mode composition of the S1-state. Upon multiple-far-infrared photon absorption, the resonance enhanced multi-photon ionization spectrum broadens allowing ion gain spectroscopy to be performed. In the ion gain spectrum we detect the fundamental far-infrared modes but also combination and overtone bands with high efficiency. The implications to dip spectroscopy using a free electron laser compared to more conventional light sources are discussed.
1 Introduction
Ion-dip and fluorescence-dip spectroscopy are powerful tools for recording molecular spectra from the microwave (MW)1 over the far infrared (far-IR),2 the infrared (IR),3,4 to the ultraviolet (UV)5 region. In all these techniques, the first laser (or microwave) pulse resonantly depopulates a given (ro)vibronic state of a molecule. The modulation of the population by the pump pulse is then probed by a second laser pulse via resonant two-photon ionization (R2PI), laser induced fluorescence (LIF), or IR predissociation/photodissociation.6,7 In most cases, the modulated state is the ground state, but also the excited state (S1) and the ionic state (D0) can be investigated by this technique depending on the timing of the lasers.8 Depopulation of the ground state results in a decreased ion or fluorescence signal (the so-called dip), if the pump and probe laser share a common ground state level. These techniques are referred to either as MW-UV double resonance (DR) spectroscopy, IR ion-dip (IR-ID)/IR-UV DR spectroscopy, or UV-UV DR spectroscopy in the literature. The latter is also known as spectral hole-burning.9,10Surprisingly, sharp features in the R2PI spectrum are not required for IR-ID spectroscopy.11–14 In fact, even with a completely featureless and unstructured R2PI spectrum, as was, for example, demonstrated for the Watson–Crick base pair of guanine–cytosine, IR-ID spectroscopy yields narrow IR resonances.15 This observation suggests large changes in the R2PI spectrum either in the form of a large shift or a large strength increase.
The conformer space of tryptamine, which is the chromophore of the aromatic amino acid tryptophan, has been studied in great detail.16–32 The formation efficiency of individual conformers strongly depends on the expansion and evaporation (or laser desorption) conditions as well as on the cooling rates in a molecular beam in which the tryptamine molecules are captured.25,26 Park et al.16 were the first to use the rotational contours in the fluorescence spectrum to deduce that the excitation spectrum of tryptamine contains seven different conformers out of the possible 27 conformers, which they named A, B, C(1), C(2), D, E, and F.
The low energy vibrations of tryptamine have been studied using single vibronic level fluorescence (SVLF) spectroscopy24 and conformation-specific ionization-detected stimulated Raman spectroscopy.31,32 In the present paper we report the low energy excitations between 200 and 500 cm−1 in tryptamine A using an intense Free Electron Laser (FEL) light source. We concentrate on the most abundant conformer A studied under conditions where the contribution of other conformers is small. We first present far infrared spectra with ion dip spectroscopy using R2PI as probe in the UV. We continue with evidence on the number of IR photons being absorbed and with the evolution of the IR-UV DR spectra upon increasing the IR intensity. We conclude that in 200–500 cm−1 excitation range intramolecular vibrational redistribution (IVR) is a slow process. We find that tuning the IR intensity provides an opportunity for a novel form of sensitive ion gain spectroscopy, in which, surprisingly, many combination bands are detectable. These insights may well lead to new methodology for investigating low energy vibrations and the IVR dynamics.
2 Methods
2.1 Experimental methods
In order to obtain the R2PI spectra, tryptamine was evaporated by direct heating to about 410 K prior to the expansion. Using helium or argon as a carrier gas, tryptamine was entrained in a molecular beam, cooled and transported through a skimmer to the excitation region, where the counterpropagating UV and IR laser beams are crossed perpendicularly with the pulsed molecular beam. The pressure in the interaction region is of the order of 7 × 10−7 mbar during the experiment, ensuring collision free conditions. Ions are extracted in vertical direction into a reflectron time-of-flight mass spectrometer with unit mass resolution up to m/z = 1500. The output of a Nd-YAG (Spitlight 120, Innolas) laser pumped dye laser (Narrow Scan, Radiant Dye) was doubled to generate nanosecond UV light. The UV output was attenuated to 100 μJ to avoid saturation and power broadening of the (1 + 1) R2PI spectra of the different conformers. The R2PI laser was scanned between 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 and 34960 cm−1 which covers the vibrationless S1 state origins of the tryptamine conformers.
820 and 34960 cm−1 which covers the vibrationless S1 state origins of the tryptamine conformers.
IR-ID spectra were taken by fixing the UV frequency of the dye laser to the S1 origin transition of a specific conformer and scanning the IR frequency of FELIX in the range of 200 to 500 cm−1. The ion signal from the UV laser is monitored and shows dips if IR resonances are excited that share a common ground state level with the UV transition.
The source of far infrared radiation is the Free Electron Laser for Infrared eXperiments (FELIX) in Nijmegen, providing tunable light from 2000 cm−1 to 60 cm−1 (5–167 μm). The infrared light is transported from FELIX to the interaction region through evacuated tubing to avoid absorption due to moist air. The relative bandwidth of the far-IR radiation is 0.5% full width at half maximum of the wavelength. The wavelength is calibrated using a spectrometer with an accuracy of 5 cm−1; however, the relative frequency is known to better than 1 cm−1. In the following, IR is used to imply the full IR + far IR part of the spectrum. The output of FELIX consists of at ten macropulses per second. Each macropulse has a duration of about 7 microseconds and consists of about seven thousand micro-pulses each with a duration of a few picoseconds separated by one nanosecond. The energy per macropulse is of the order of 100 mJ. The molecular beam velocity with argon as carrier gas is such that most of the molecules interact with the full IR macropulse. Both IR and UV wavelengths can be tuned independently. The delay between the IR pulse and the UV pulse can be controlled as well. In our experiments, FELIX was operated at 5 Hz and the ionization laser at 10 Hz to enhance the signal quality of the ion-dip and other double resonance signals. FELIX was blocked regularly to control that both channels do not have a different off-set.
In all experiments, the molecules were subjected to the full FEL pulse of 7 microseconds duration; near the end of this pulse the UV laser was fired. This relative timing of IR and UV pulses was the same or all experiments.
2.2 Theoretical methods
For assignment of the R2PI spectra of vibrationally excited ground state molecules, the structures of the S0 and S1 states were required. Structure optimizations were performed employing Dunning's correlation consistent polarized valence triple zeta (cc-pVTZ) basis set from the Turbomole library.33,34 The equilibrium geometries of the electronic ground and the lowest excited singlet states are optimized using the approximate coupled cluster singles and doubles model (CC2) employing the resolution-of-the-identity approximation.35–37 The Hessians and harmonic vibrational frequencies for both electronic states, which are utilized in the Franck–Condon (FC) simulations, have been obtained from numerical second derivatives using the NumForce script.38 implemented in the Turbomole program suite.39 The vibrational state density is calculated using a direct count-algorithm from an anharmonic analysis of the vibrational spectrum. This allows for the determination of vibrational averaging effects due to the individual vibrational motion.40 Such an anharmonic analysis is implemented in the Gaussian program package.41 The procedure for the calculation of cubic and of some of the quartic force constants utilizes third derivatives of the potential energy with respect to the normal coordinates. They were calculated for the electronic ground state of the tryptamine A conformer using numerical derivatives of analytical second derivatives of the MP2 energies using the 6-31G(d,p) basis set. According to the FC principle the relative intensity of a vibronic band depends on the square of the overlap integral of the vibrational wave functions in both electronic states. The simulation of the R2PI spectrum has been performed using the program FCFit, which has been described in detail before.42,43 Briefly, the program computes the FC integrals of multidimensional, harmonic oscillators mainly based on the recursion formula given in the papers of Doktorov, Malkin, and Man'ko.44,453 Results
3.1 Infrared-ion dip spectra of tryptamine
IR-ID spectra were recorded by scanning the far-IR laser while probing with a fixed UV laser. The frequency of the UV laser was tuned to transition of a certain conformer, in order to conformer selectively detect the IR resonances. Fig. 1 shows the IR-ID spectrum of the A conformer of tryptamine with the UV laser set to the vibronic origin of tryptamine A at 34915.6 cm−1. The far-IR pulse energy used influences the dynamics of IR absorption and intramolecular vibrational redistribution (IVR), as will be shown below. For this spectrum a macropulse energy of 37 mJ was chosen such that the R2PI peak shows a significant decrease that is approximately linear in IR power.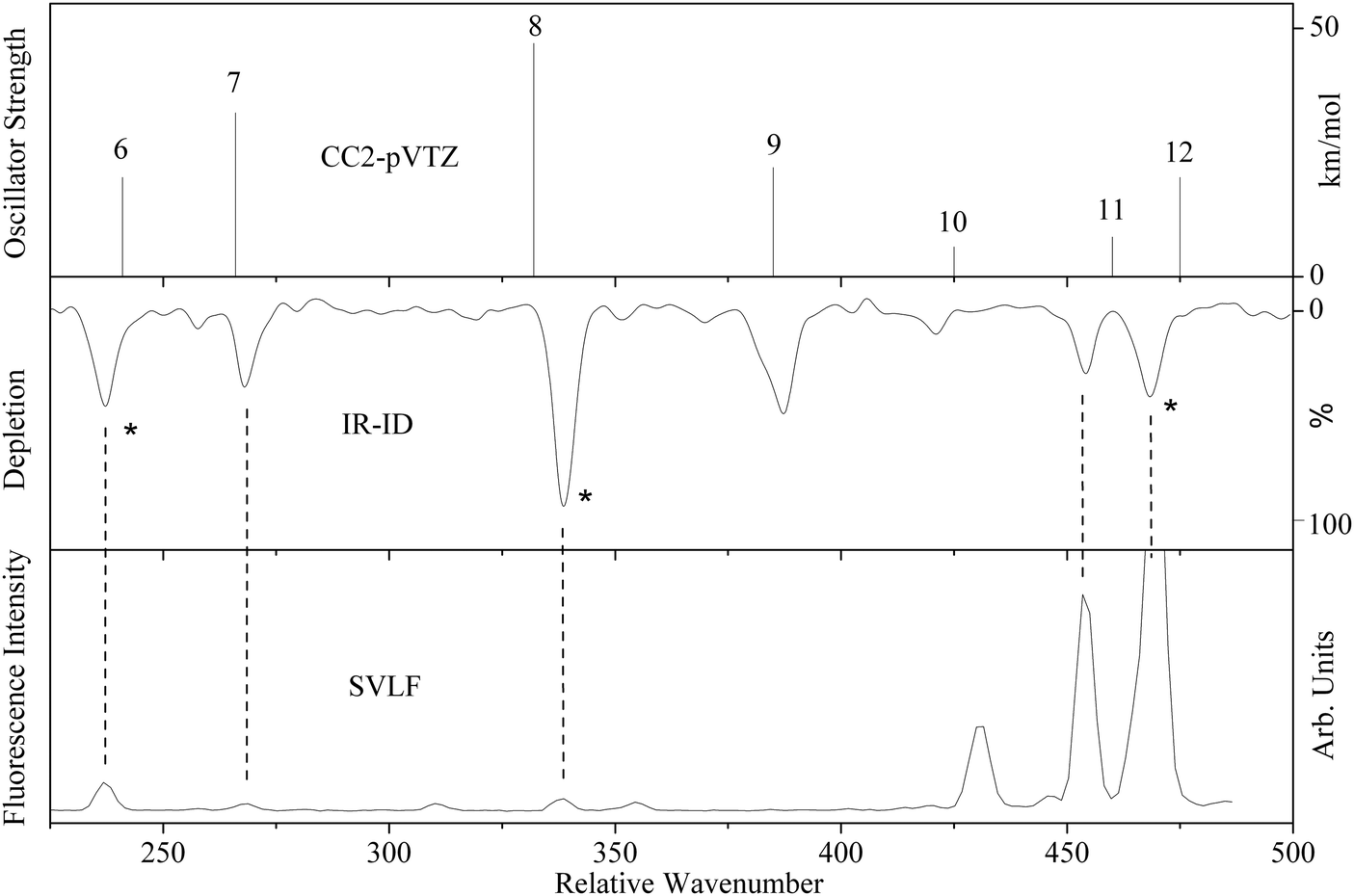 | ||
| Fig. 1 Comparison of the CC2/cc-pVTZ calculation (upper trace) with the IR-ID spectrum (middle trace) and the SVLF spectrum from ref. 24 (lower trace) of tryptamine A. The asterisks in the IR-ID spectrum mark the spectral positions which are used for the IR-UV double resonance spectra described in Section 3.2. The mode numbering refers to that given in Fig. S1 and Table S1 of the ESI.† | ||
The IR-ID spectrum is compared on the same wavenumber scale with single vibronic level emission spectra from ref. 24, and the CC2/cc-pVTZ ab initio calculated vibrational spectrum. The mode numbering used in Fig. 1 refers to the normal modes, given in Table S1 (ESI†) and discussed in detail later (Section 4).
IR-ID spectra of the other conformers (B to F) of tryptamine were also recorded. They are shown in the ESI.† In the present work however, we will concentrate on the A conformer of tryptamine.
3.2 IR-UV double resonance spectra of tryptamine
Subsequently, we reversed the experiment and scanned the ionization laser, while fixing FELIX to specific far-IR transitions of tryptamine, obtained from IR-ID spectroscopy. This experiment yields R2PI spectra, which are comprised of transitions from the (cold) vibrational ground state and from vibrationally hot molecules. We call this in the following IR-UV DR spectroscopy. Fig. 2 shows the results of three such experiments. The spectra show the change in the R2PI spectrum upon exciting the A conformer at far-IR resonances at 237.0 cm−1, 341.5 cm−1, and 468.0 cm−1, respectively. The red trace shows the double resonance R2PI spectrum after IR excitation, the black trace shows the R2PI spectrum without IR present. The right hand side displays the difference spectra. These spectra contain the R2PI features of the seven different conformers (labelled A, B, C, D, E, and F). Conformer A is the most abundant.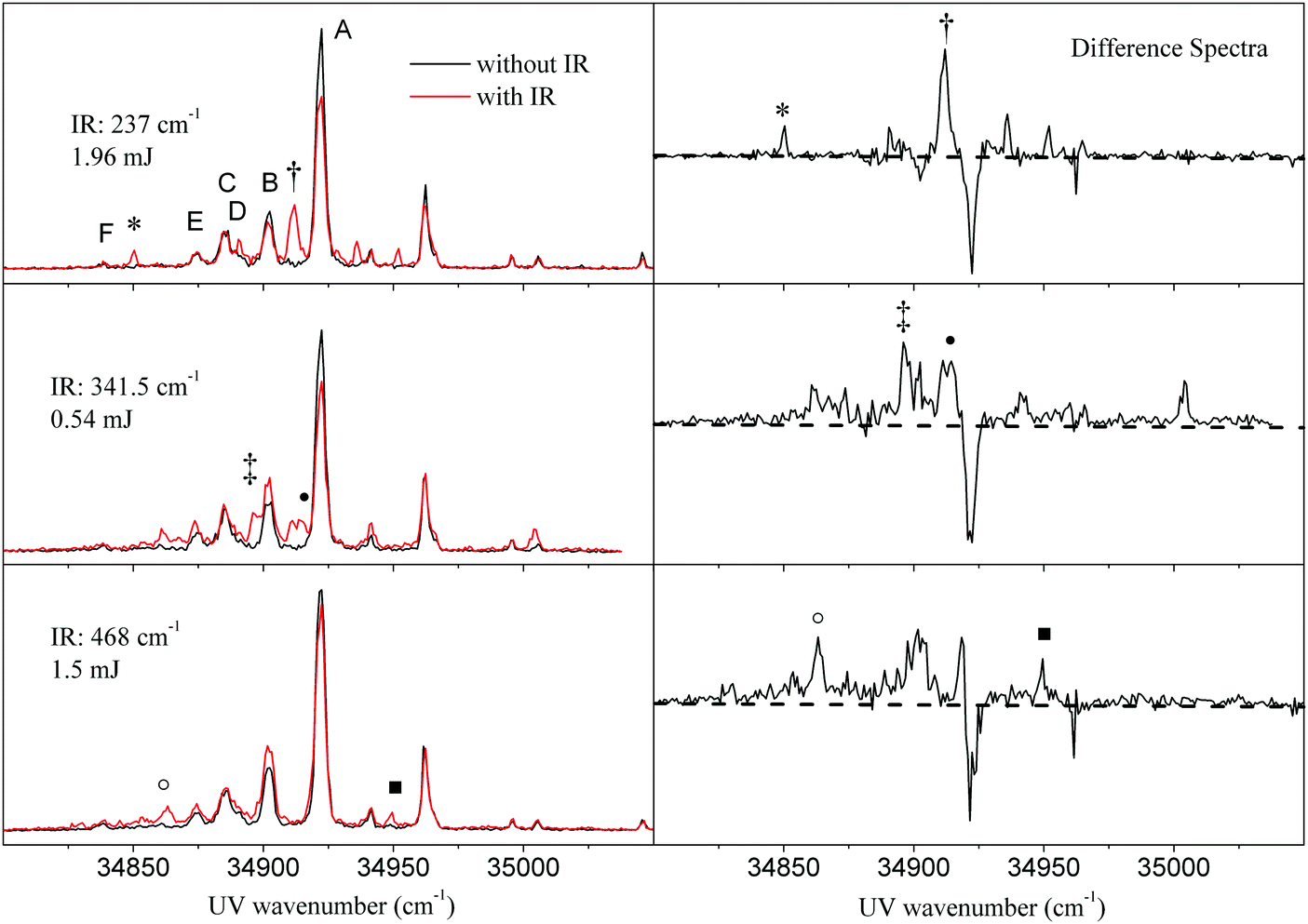 | ||
| Fig. 2 Left side: R2PI spectra of tryptamine after pumping IR resonances of the A conformer at low power at 237.0 cm−1, 341.5 cm−1, and 468.0 cm−1. Right side: Difference spectra of R2PI with IR and without IR. Negative signals are due to signal decrease upon IR action, while positive signals are due to ionization from the vibrational excited states. | ||
All three IR-UV DR spectra of Fig. 2 show a decrease of the signal at the origin band of the A conformer at 34915.6 cm−1 caused by the reduction of the ground state population upon IR excitation. We note here that the far-IR radiation from FELIX is strongly attenuated to the values given in Fig. 2. Therefore, the decrease of the ion signal of conformer A is smaller than what has been obtained for the IR-ID spectra in Fig. 1. The three experiments excite vibration 6 at 237.0 cm−1, vibration 8 at 341.5 cm−1 and vibration 12 at 468.0 cm−1, respectively. The slight reduction of the B conformer origin at 34895.9 cm−1 can be traced back to a partial overlap of the 237.0 cm−1 band with a vibrational band of the B conformer at 238.5 cm−1 (cf. Fig. S2 of the ESI†). In all three spectra, a number of sharp new bands appeared in the R2PI spectrum associated with the excitation of vibrational excited tryptamine A molecules.
The three difference spectra show ion dips (negative signals) as well as ion gains (positive peaks). As will be shown in the discussion, the ion gain peaks in the ionization spectrum can be understood from ionization of vibrationally excited states in the electronic ground state to vibrational levels in the S1 state. While exciting at 341.5 cm−1 and at 468.0 cm−1, the beginning of a continuum can be observed at the red of the origin band next to more sharp features. Some of the ion gain peaks are marked to indicate that at these UV wave numbers also IR-UV ion gain spectra (see Section 3.3) have been taken.
In a subsequent experiment, we varied the fluence of the IR photon beam in order to check whether we can increase the number of IR photons being absorbed within a macropulse. Fig. 3 shows the R2PI spectra with and without interaction with 341.5 cm−1 IR radiation at different IR macropulse energies (0.54, 5.4, and 37 mJ, respectively). Increasing the IR pulse energy leads to nearly full depletion of the origin band signal and hence a 100% dip signal of the pumped band. Simultaneously, the shape of the R2PI spectrum of vibrational excited molecules changes into a broad featureless background. The narrow peaks observed at low power do not increase, but seem to disappear in a broad background.
 | ||
| Fig. 3 Macropulse energy dependence of the R2PI spectra after pumping the IR resonance of the A conformer at 341.5 cm−1. The right column shows the difference spectra of R2PI with IR and without IR. | ||
The observation that an increased IR power changes the spectra qualitatively indicates that the number of absorbed IR photons is different. The possible processes are ladder climbing and IVR with subsequent second absorption in the fundamental band. The process of IVR results in a heterogenous collection of molecules in different modes giving rise to a multitude of transitions between different excited low energy vibrational levels in both electronic states. We note that near 34884 cm−1 there is a dip in the broad background of the R2PI signal (cf.Fig. 3). The zero signal in the difference spectra reflects probably a spurious compensation of a reduction in signal due to depletion of the C conformers, which also has a resonance around 341 cm−1 (cf. Fig. S2 of the ESI†) and an increase in the R2PI response of the vibrational excited A conformer. This disappearance of the additional sharp R2PI lines and the transformation of the R2PI spectrum into a nearly continuous background around the origin band (cf. right column of Fig. 3) inspired us to obtain ion gain spectra using ionization wavelengths different from the origin band.
3.3 IR-UV ion gain spectra of tryptamine
Instead of probing one of the vibronic bands in the R2PI spectrum of cold ground state molecules as in IR-ID spectroscopy, we fixed the UV laser at the frequency of one of the bands, which newly appear in the far-IR pumped R2PI spectra shown in Fig. 2.Fig. 4 compares the IR-ID spectrum obtained via excitation of the origin band of the tryptamine A conformer and scanning the IR laser (a) with the IR spectra taken upon UV excitation at the positions marked with † (b), * (c), ‡ (d), ● (e), ○ (f), and □ (g) in Fig. 2. The intensity of the UV laser was chosen such that the UV laser alone gave no detectable ionization of ground state molecules. The IR laser macropulse energy was chosen at the intermediate value of 5.4 mJ with respect to the results in Fig. 3 such that the R2PI spectrum shows broadening.
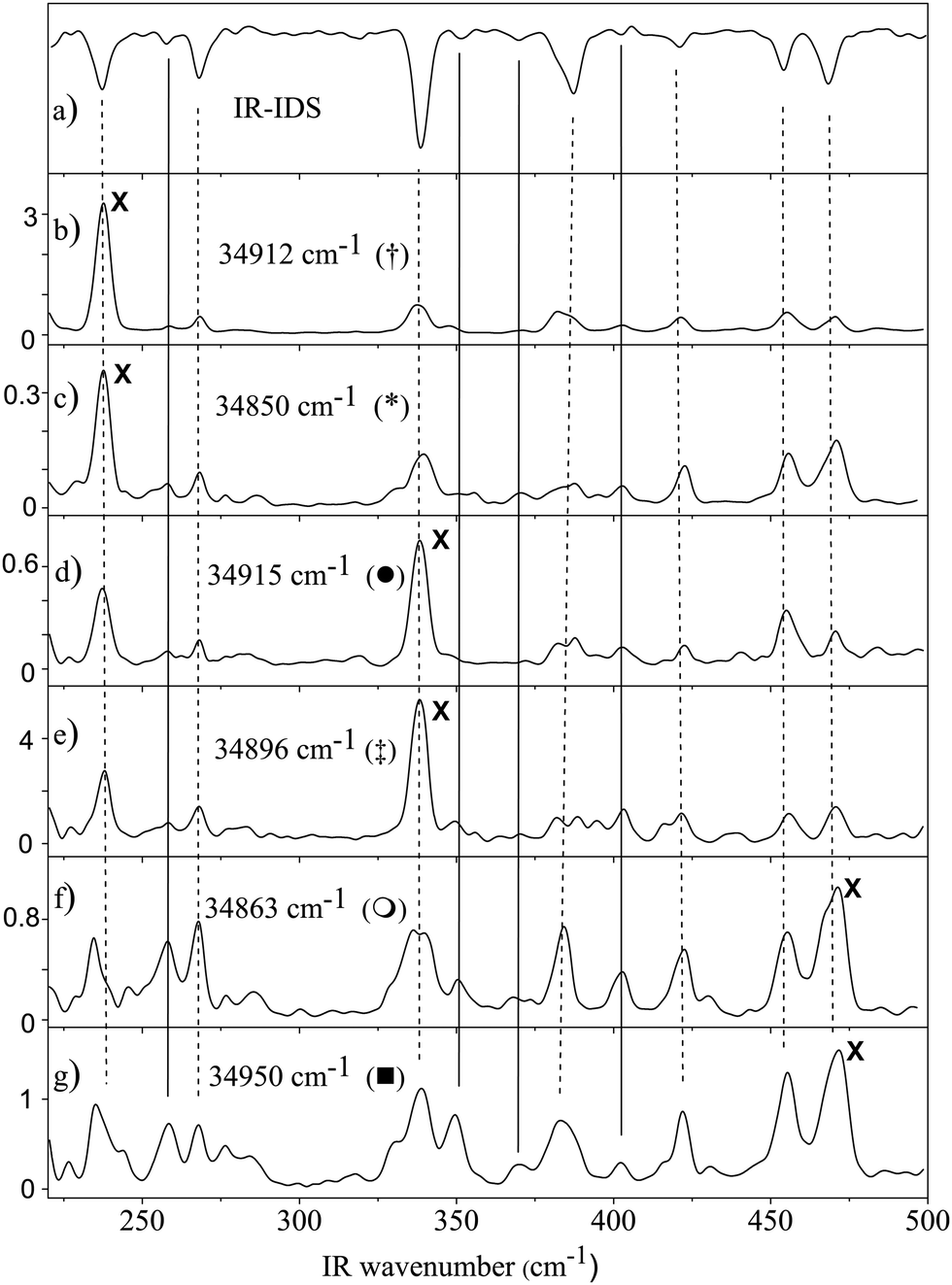 | ||
| Fig. 4 Trace (a) shows the IR-ID spectrum of tryptamine A. Traces (b) and (c) show the IR-UV gain spectra obtained by pumping the newly detected bands † (34912 cm−1) and * (34850 cm−1), from Fig. 2, while scanning the IR laser. Traces (d) and (e) IR-UV gain spectra obtained by pumping the newly detected bands ‡ (34897 cm−1) and ● (34915 cm−1). (f and g) IR-UV gain spectra obtained by pumping the newly detected bands ○ (34863 cm−1) and □ (34950 cm−1). The symbols refer to the bands in Fig. 2. The crosses mark the IR wavenumbers, at which the new UV bands were detected. Dashed lines connect fundamental vibrations, straight lines combination bands. | ||
The peaks, which are present in the IR-ID spectrum and associated with the fundamental bands of tryptamine A (Fig. 1 and trace a of Fig. 4), are again observed in the ion gain spectra with even a better signal to noise ratio. The dashed lines in Fig. 4 connect these fundamental excitations. The spectra in Fig. 4b–g were obtained by exciting the new bands at 34912 cm−1 and 34850 cm−1 († and * in Fig. 2) connected to the new bands upon IR pumping at 237 cm−1. The UV wavenumbers of 34915 and 34896 cm−1 refer to new bands upon IR pumping at 341.5 cm−1 (‡ and ● in Fig. 2). UV excitation at 34863 cm−1 and 34950 cm−1 (○ and □ in Fig. 2) refers to IR pumping at 468 cm−1. The crosses (X) in the ion gain spectra indicate the far-IR wavenumbers, which are linked to the respective additional UV bands in the R2PI spectra of Fig. 2. For example, when pumping the band marked with † in Fig. 2, which appears only upon FIR pumping at 237 cm−1, there is a strong propensity to this band in the IR-UV gain spectra, shown in Fig. 4. This propensity between the UV wavenumber at which a new band in the IR spectrum appears and the intensity of the respective band in the IR-UV gain spectra is symbolized by the crosses in Fig. 4.
4 Discussion
The overall aim of taking IR spectra of large molecules is to obtain insight in low frequency backbone type motion, which is expected to be very sensitive to the molecular structure. IR-ID spectroscopy is one way to obtain such spectra.46–49 Alternatively, dispersed fluorescence emission spectra can also be used to determine the positions of vibrational resonances.29 Recently, results on tryptamine have been reported where stimulated Raman has been used to study low energy vibrational modes31,32 in an ion dip spectroscopy fashion as pioneered in the group of Felker.50 Also here, the spectra show apparent interference between stimulated Raman spectra of the different conformers and ion gain and ion loss signals. In this case, the presence of multiple lasers with high energy photons creates also other sources of ionization signal. However, only direct IR excitation makes it possible to also interpret the strength of the IR resonances.4.1 Infrared-ion dip spectra
Assignment of the vibrational bands observed in the IR-ID spectra of conformer A of tryptamine (Fig. 1), based on the calculated frequencies from the CC2/cc-pVTZ calculations, is straightforward. The mode numbering, along with the computed ground state vibrational wavenumbers from the CC2/cc-pVTZ calculations are shown in the second column of Table S1 of the ESI.† The displacement vectors of the modes discussed here are shown in Fig. S1 of the ESI.† The band observed at 237 cm−1 is assigned to vibration 6 (calculated at 240.9 cm−1). The energetically following band at 260 cm−1 can be assigned as an overtone of vibration 3 calculated at 135.7 cm−1. The band observed at 268 cm−1 is again assigned to a fundamental mode, namely vibration 7. At 341.5 cm−1, vibration 8 is observed (calculated at 332.2 cm−1). The relative wavenumbers in the IR-ID and SVLF spectra are identical, but the intensities are very different. Some of the vibrational bands with considerable intensity in the IR-ID spectra are practically absent in the SVLF spectrum due to their unfavorable FC factors (e.g. vibrations 9 and 10 in Fig. 1). Others show up strongly in the fluorescence emission spectrum but are absent in the IR-ID spectrum (e.g. the vibration at 428 cm−1 in Fig. 1).In our experiment, we observed 100% depletion of the ion signal. However, the maximum depletion possible with single-photon absorption in a closed two-level system is only 50%. This suggests that the excited state undergoes a reaction either in the form of fragmentation, IVR, or subsequent excitation by a second IR photon in a ladder climbing process. After careful examination of the mass spectra, we exclude fragmentation for the pulse and photon energies used here. A macropulse of the FEL consists of more than five thousand micro-pulses of picosecond duration, separated by one nanosecond intervals. Using reasonable excitation strengths of 5–50 km mol−1 for a far-IR vibrational mode, one can estimate that a single micropulse has an excitation probability of 0.15–1.50%, implying that absorption of a few photons at full FELIX power during the microsecond macropulse is possible and the ion signal depletion can indeed exceed 50%. Hence, multiple photon absorption is a possible process. Regarding the possibility of IVR, a certain density of states is required. IVR becomes in general exponentially faster with increasing excitation energies. From a normal mode analysis, we predict that the vibrational state density calculated by direct count is below one state per wavenumber for the region of the present study, which is considered insufficient for IVR to take place. The nature of the vibrational modes does play a significant role and may affect the state count. The modes of lowest frequencies in tryptamine are torsional modes of the ethylamino side chain. Their frequencies are overestimated, even considering anharmonic corrections. Nonetheless, the state density at far-IR one-photon conditions is not large enough to enable IVR. Indeed, one can observe well-structured DR-R2PI spectra after pumping far IR transitions at low fluences, which requires the survival of the energy in the excited modes during a macropulse (7 μs).
The duration of our macropulses makes the present experiment different from experiments using nanosecond pulse IR sources, where the note can be added that for now no such lasers exist in the 200–500 cm−1 wavenumber region. In general, nanosecond laser sources require a much higher IVR rate to allow multiple photon absorption. The possibility of second (IR) photon excitation during the macropulse is discussed below.
4.2 IR-UV double resonance spectra
We observe that the IR excitation process prior to taking a R2PI spectrum leads to the appearance of additional sharp bands in the IR-UV DR spectra (Fig. 2 and 3). These new bands may arise from diagonal (vn′′ = vn′) transitions starting from the IR excited vibrational level in the electronic ground state (vn′′). In order to check this assumption, we calculated the absorption spectrum after IR excitation at 237 cm−1 in the Franck–Condon approximation (Fig. 5). The required Hessians have been taken from a normal mode analysis performed at the CC2/cc-pVTZ optimized structures of ground and excited state. No frequency scaling has been used. | ||
| Fig. 5 R2PI spectrum of tryptamine after pumping IR resonances of the A conformer at 237.0 cm−1 (black trace) along with a Franck–Condon simulation using the vibrationless origin and the vibration 6 as ground state levels. For details of the simulation see text. The bands marked with * are those, that appear only upon concerted action of IR and UV radiation. IR pulse energy used here is 1.96 mJ. The quantum numbers refer to the simulated FC spectrum. | ||
The simulated spectrum (in red) in Fig. 5 consists of the (cold) transitions from the vibrationless ground state (mainly the electronic origin 000 and the fundamental transition to vibration 110), and of transitions originating from the excited level of vibration 6 in the ground state, the most prominent one being the diagonal 611 transition and also some off-diagonal transitions like 601510. Saturation of IR excitation is assumed, leading to the same population in the ground and vibrational state. The agreement between the positions and intensities of the additional lines in the R2PI spectrum is surprising considering the simple parameter free model used. Only one of the additional lines in the R2PI spectrum is missing in the simulation. An explanation of the missing peak might be slight contributions from the B conformer, since the 237 cm−1 band has overlap with the 239 cm−1 band of the B conformer, cf. Fig. S2 of the ESI.† Also the absorption spectrum after IR excitation at 341.5 cm−1 was calculated with the same assumption in the Franck–Condon approximation. It is shown in the ESI† (Fig. S3).
As mentioned above, the fact that the spectrum at low macropulse energy can be interpreted by Franck–Condon arguments implies that the IVR rate after a one photon absorption is sufficiently slow that IVR does not occur during the macropulse. Furthermore, the structured and well-resolved R2PI spectra show that one far-IR photon excitation is not followed by IVR (with a possible exception of the 468 cm−1 resonance, where the R2PI spectrum in Fig. 2 already shows a weak continuous background). The IR laser full width at half maximum bandwidth of 0.5% implies that with an anharmonicity up to a few percent a second photon excitation is possible. As the state density increases sharply with excitation energy, the IVR rate after two-photon absorption may allow IVR to occur more easily. After absorption of a second ladder photon, the strongly enhanced IVR may facilitate absorption of multiple photons in the original fundamental mode again.
The fact that the IR-UV double resonance spectra show a strong dependence on the FELIX power indicates multiple IR photon excitation processes to operate. To obtain the IR-ID spectrum of Fig. 1, FELIX was operated at rather large macropulse energies. In the experimental practice, the IR energy is chosen such that depletion of the origin band in the R2PI spectrum is significant and simultaneously that the decrease is approximately linear with IR power. This means that a rather high power, in the order of 50 mJ per macropulse, is chosen.
4.3 IR-UV ion gain spectra
The large information content of the IR-UV ion gain spectra forms the most intriguing result of the present work; we can not only employ ion gain spectra to determine fundamental bands with good signal-to-noise, but also observe more spectral features in the far-IR spectra, which we assign to combination bands and overtones. Along with the known fundamental frequencies in the 0–200 cm−1 range observed using single vibronic laser induced fluorescence24 and the resonances presented from IR-ID spectroscopy between 200 and 500 cm−1, we have been able to assign nearly all bands in the IR-UV gain spectra of Fig. 4 to fundamental bands (connected by dashed lines) and to overtones and combination bands of the known fundamentals (connected by straight lines), based on the comparison to the results of a CC2/cc-pVTZ ab initio normal mode analysis as shown in Fig. 6. The given assignment is based on a harmonic normal mode analysis, and there might be equally plausible assignments, regarding the number and the density of lines. | ||
| Fig. 6 IR-UV ion gain spectrum after excitation at 34950 cm−1 (expanded view of trace g of Fig. 4) along with the assignment of fundamentals, overtones, and combination bands. | ||
The intensities of the overtones and combination bands do not reflect their oscillator strengths, otherwise these would have been observed in the IR-ID spectrum (trace a of Fig. 4). The intensities of the fundamental modes can be compared among themselves and reveal their oscillator strength only by comparing the ion gain spectra with the IR-ID spectrum. Why would overtones and combination modes be more easily detected in the gain spectra? First, molecules excited in combination bands will have more complex R2PI spectra because of the many more combinations. Hence, single IR photon absorption into a combination band may already suffice to create ionization at many UV wavelengths different from diagonal transitions. The occurrence of fundamental and combination bands in one spectrum may reflect that under our conditions fundamental bands require two IR photon excitation to be observable, whereas for excitation of combination bands possibly a single photon may suffice. We note that at different ionization wavelengths, the relative intensities of the combination bands in the gain spectrum of Fig. 4 vary significantly. The detection efficiency of the combination band excited molecules depend on the UV laser wavelength, hence the R2PI spectrum is structured.
In obtaining the data in the ion gain spectra, we have selected ionization wavelengths coinciding with new features obtained in R2PI spectra at very low fluence. Indeed, propensity survives in the spectra of the chosen UV wavelength. This propensity rule is quite similar to the one in fluorescence emission. While in the latter, the propensity rule states, that the excited vibronic band has the largest FC factor with the respective vibration in the ground state, in IR-UV ion gain spectra the far-IR excitation of a certain vibrational mode leads to a higher probability for ionization through this vibronic band in the excited state. The present results certainly give rise to many possible experiments. Our next attempts will be to broaden the application of IR-UV gain spectroscopy to other systems than tryptamine. Also a more quantitative determination of IVR rates is of high interest in this energy range.
If we compare the present results with the more abundant body of IR-ID spectroscopy performed at wavelengths below 5 micron using standard laser systems, the amount of energy of a single photon is 5 to 10 times as large as used in our studies. At these high energies IVR distributes internal energy over a much larger number of low energy modes. Probably, features in the IR-excited R2PI spectra are so much broadened that at any individual UV wavelength the ionization yield is simply negligible. This interpretation explains also why for high frequency IR modes ion-dip spectroscopy still operates even when a R2PI spectrum of the cold ground state molecules is structureless. Upon excitation around 5 micron, the R2PI spectrum of the hot vibrational species effectively disappears.
5 Conclusions
In the present article, we determined the changes of the R2PI spectrum of conformer A of tryptamine upon far-IR excitation of low frequency modes at different far-IR power levels. These R2PI spectra allowed for a mode decomposition of the excited S1 state, as confirmed by Franck–Condon calculations. Our findings made it possible to record far-IR spectra between 200 cm−1 and 500 cm−1 using ion gain spectroscopy. We showed that this yields both the fundamental modes but also many combination bands, the latter with surprising sensitivity. We used a Free Electron Laser (FEL) as a light source, which produces very short pulses in pulse trains of typically 7 microseconds. Thus, these pulses are considerably longer than experiments with nanosecond lasers normally use. The long duration of the FEL pulse train increases the chance for IVR after absorption of a second IR photon in the IR-ID experiments. An FEL enables direct excitation of low energy vibrational modes, which makes it possible to verify not only the positions but also relative intensities in far infrared spectra.46–49 Our results allow for a rationalization of the success of IR-ID spectroscopy at shorter IR wavelengths.One may wonder whether these observations are unique to tryptamine and why these effects have not been observed in the large body of work on IR spectroscopy using ID spectroscopy. Firstly, one should realize that FELIX is a special source in contrast to table top IR sources. Not only does FELIX allow routinely taking spectra up to wavelengths of 50 micron, but also the pulse structure with a pulse length of many microseconds makes IVR processes also at low IR photon energies important. The present study of tryptamine shows a clear distinction between two IR energy regimes: one-photon (far-IR) excitation well below 500 cm−1, where no IVR is observed; and the two-photon excitation, at energies that are high enough to facilitate IVR.
The above results indicate that the low frequency range in large molecules can be studied in a more sensitive way by making use of changes in structured R2PI ionization spectra. We believe that these results are general for medium sized molecules such as tryptamine. In spite of the easy access to light sources that allow IR-ID at these low IR photon energies, our results show that high intensity sources, such as FELIX, are critical for gaining insight in low frequency backbone motions that are highly relevant for structural questions in complex molecules.
Acknowledgements
All authors acknowledge the Stichting voor Fundamenteel Onderzoek der Materie (FOM) for Support of the FELIX laboratory. We highly appreciate the skillful assistance by the FELIX laboratory staff. The work was financially supported by the Deutsche Forschungsgemeinschaft SCHM1043/12-2. V. Z. acknowledges the Swedish Research Council for providing him with a postdoctoral fellowship grant (Grant No. 623-2008-7180) and the funding received from the European Communities Seventh Framework Program (FP7/2007-2013, no 312284). Granted computing time at the Universitätsrechenzentrum Köln is gratefully acknowledged.References
- G. Berden, W. L. Meerts and W. Kreiner, Chem. Phys., 1993, 174, 247 CrossRef CAS
.
- P. Çarçabal, R. T. Kroemer, L. C. Snoek, J. P. Simons, J. M. Bakker, I. Compagnon, G. Meijer and G. von Helden, Phys. Chem. Chem. Phys., 2004, 6, 4546–4552 RSC
- R. H. Page, Y. R. Shen and Y. T. Lee, J. Chem. Phys., 1988, 88, 5362 CrossRef CAS
- S. Tanabe, T. Ebata, M. Fujii and N. Mikami, Chem. Phys. Lett., 1993, 215, 347–352 CrossRef CAS
- H. Saigusa and E. C. Lim, J. Phys. Chem., 1991, 95, 2364 CrossRef CAS
- C. M. Leavitt, A. B. Wolk, J. A. Fournier, M. Z. Kamrath, E. Garand, M. J. V. Stipdonk and M. A. Johnson, J. Phys. Chem. Lett., 2012, 3, 1099–1105 CrossRef CAS PubMed
- N. Heine, M. R. Fagiani and K. R. Asmis, J. Phys. Chem. Lett., 2015, 6, 2298–2304 CrossRef CAS PubMed
- M. Gerhards, M. Schiwek, C. Unterberg and K. Kleinermanns, Chem. Phys. Lett., 1998, 297, 515 CrossRef CAS
- R. J. Lipert and S. D. Colson, J. Phys. Chem., 1989, 93, 3894 CrossRef CAS
- M. Schmitt, H. Müller and K. Kleinermanns, Chem. Phys. Lett., 1994, 218, 246 CrossRef CAS
- H. Fricke, A. Funk, T. Schrader and M. Gerhards, J. Am. Chem. Soc., 2008, 130, 4692 CrossRef CAS PubMed
- A. M. Rijs, M. Kabelac, A. Abo-Riziq, P. Hobza and M. S. de Vries, ChemPhysChem, 2011, 12(10), 1816–1821 CrossRef CAS PubMed
- A. M. Rijs, E. R. Kay, D. A. Leigh and W. J. Buma, J. Phys. Chem. A, 2011, 115, 9669–9675 CrossRef CAS PubMed
- A. M. Rijs, G. Ohanessian, J. Oomens, G. Meijer, G. von Helden and I. Compagnon, Angew. Chem., Int. Ed., 2010, 49, 2332–2335 CrossRef CAS PubMed
- A. Abo-Riziq, L. Grace, M. K. E. Nir, P. Hobza and M. S. de Vries, PNAS, 2005, 102, 20 CrossRef CAS PubMed
- Y. D. Park, T. R. Rizzo, L. A. Peteanu and D. H. Levy, J. Chem. Phys., 1986, 84, 6539–6549 CrossRef CAS
- J. R. Carney and T. S. Zwier, J. Phys. Chem. A, 2000, 104, 8677 CrossRef CAS
- T. Nguyen, T. Korter and D. Pratt, Mol. Phys., 2005, 103, 1603–1613 CrossRef CAS
- T. Nguyen and D. Pratt, J. Chem. Phys., 2006, 124, 054317 CrossRef PubMed
- L. A. Philips and D. H. Levy, J. Chem. Phys., 1988, 89, 85–90 CrossRef CAS
- L. A. Philips, S. P. Webb, S. J. Martinez, G. R. Fleming and D. H. Levy, J. Am. Chem. Soc., 1988, 110, 1352 CrossRef CAS
- M. Schmitt, M. Böhm, C. Ratzer, C. Vu, I. Kalkman and W. L. Meerts, J. Am. Chem. Soc., 2005, 127, 10356–10364 CrossRef CAS PubMed
- M. Schmitt, R. Brause, C. Marian, S. Salzmann and W. L. Meerts, J. Chem. Phys., 2006, 125, 124309 CrossRef PubMed
- M. Schmitt, K. Feng, M. Böhm and K. Kleinermanns, J. Chem. Phys., 2006, 125, 144303 CrossRef PubMed
- Y. K. Sturdy and D. C. Clary, Phys. Chem. Chem. Phys., 2007, 9, 2065–2074 RSC
- M. Böhm, R. Brause, C. Jacoby and M. Schmitt, J. Phys. Chem. A, 2009, 113, 448–455 CrossRef PubMed
- B. C. Dian, J. Clarkson and T. S. Zwier, Science, 2004, 303, 1169–1173 CrossRef CAS PubMed
- J. Alonso, V. Cortijo, C. P. S. Mata, C. Cabezas, J. Lpez and W. Caminati, J. Mol. Struct., 2011, 269, 41–48 CAS
- M. Schmitt, U. Henrichs, H. Müller and K. Kleinermanns, J. Chem. Phys., 1995, 103, 9918–9928 CrossRef CAS
- W. Roth, C. Jacoby, A. Westphal and M. Schmitt, J. Phys. Chem. A, 1998, 102, 3048–3059 CrossRef CAS
- N. Mayorkas, S. Izbitski, A. Bernat and I. Bar, J. Phys. Chem. Lett., 2012, 3, 603–607 CrossRef CAS PubMed
- N. Mayorkas, A. Bernat, S. Izbitski and I. Bar, J. Chem. Phys., 2013, 138, 124312 CrossRef PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef
- C. Hättig and F. Weigend, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef
- C. Hättig and A. Köhn, J. Chem. Phys., 2002, 117, 6939–6951 CrossRef
- C. Hättig, J. Chem. Phys., 2002, 118, 7751–7761 CrossRef
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS
-
TURBOMOLE V6.3 2012, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian 03, revision a.1, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed
- D. Spangenberg, P. Imhof and K. Kleinermanns, Phys. Chem. Chem. Phys., 2003, 5, 2505–2514 RSC
- R. Brause, M. Schmitt, D. Spangenberg and K. Kleinermanns, Mol. Phys., 2004, 102, 1615–1623 CrossRef CAS
- E. V. Doktorov, I. A. Malkin and V. I. Man'ko, J. Mol. Spectrosc., 1975, 56, 1–20 CrossRef CAS
- E. V. Doktorov, I. A. Malkin and V. I. Man'ko, J. Mol. Spectrosc., 1977, 64, 302–326 CrossRef
- X. Li, P. Claes, M. Haertelt, P. Lievens, E. Janssens and A. Fielicke, Phys. Chem. Chem. Phys., 2016, 18, 6291 RSC
- S. Jaeqx, J. Oomens, A. Cimas, M. Gaigeot and A. Rijs, Angew. Chem., Int. Ed., 2014, 53, 3663–3666 CrossRef CAS PubMed
- J. A. Fournier, C. T. Wolke, C. J. Johnson, M. A. Johnson, N. Heine, S. Gewinner, W. Schöllkopf, T. K. Esser, M. R. Fagiani, H. Knorke and K. R. Asmis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18132–18137 CrossRef CAS PubMed
- M. Cirtog, A. Rijs, Y. Loquais, V. Brenner, B. Tardivel, E. Gloaguen and M. Mons, J. Phys. Chem. Lett., 2012, 3, 3307–3311 CrossRef CAS
- G. V. Hartland, B. F. Henson, V. A. Venturo and P. M. Felker, J. Phys. Chem., 1992, 96, 1164–1173 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02358d |
| ‡ Current address: Department of Physics, University of Gothenburg, 412 96 Gothenburg, Sweden. |
| This journal is © the Owner Societies 2016 |