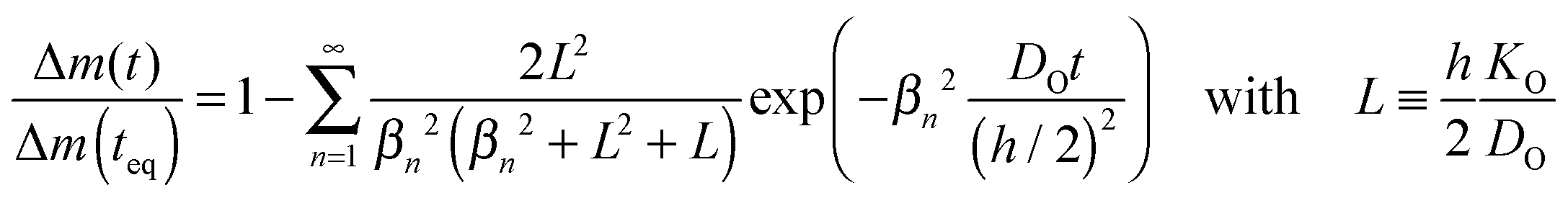DOI:
10.1039/C6CP02131J
(Communication)
Phys. Chem. Chem. Phys., 2016,
18, 22031-22038
Oxygen exchange at gas/oxide interfaces: how the apparent activation energy of the surface exchange coefficient depends on the kinetic regime
Received
31st March 2016
, Accepted 20th July 2016
First published on 21st July 2016
Abstract
In the dedicated literature the oxygen surface exchange coefficient KO and the equilibrium oxygen exchange rate ![[Fraktur R]](https://www.rsc.org/images/entities/char_e18f.gif) 0O are considered to be directly proportional to each other regardless of the experimental circumstances. Recent experimental observations, however, contradict the consequences of this assumption. Most surprising is the finding that the apparent activation energy of KO depends dramatically on the kinetic regime in which it has been determined, i.e. surface exchange controlled vs. mixed or diffusion controlled. This work demonstrates how the diffusion boundary condition at the gas/solid interface inevitably entails a correlation between the oxygen surface exchange coefficient KO and the oxygen self-diffusion coefficient DO in the bulk (“on top” of the correlation between KO and 0O for the pure surface exchange regime). The model can thus quantitatively explain the range of apparent activation energies measured in the different regimes: in the surface exchange regime the apparent activation energy only contains the contribution of the equilibrium exchange rate, whereas in the mixed or in the diffusion controlled regime the contribution of the oxygen self-diffusivity has also to be taken into account, which may yield significantly higher apparent activation energies and simultaneously quantifies the correlation KO ∝ DO1/2 observed for a large number of oxides in the mixed or diffusion controlled regime, respectively.
0O are considered to be directly proportional to each other regardless of the experimental circumstances. Recent experimental observations, however, contradict the consequences of this assumption. Most surprising is the finding that the apparent activation energy of KO depends dramatically on the kinetic regime in which it has been determined, i.e. surface exchange controlled vs. mixed or diffusion controlled. This work demonstrates how the diffusion boundary condition at the gas/solid interface inevitably entails a correlation between the oxygen surface exchange coefficient KO and the oxygen self-diffusion coefficient DO in the bulk (“on top” of the correlation between KO and 0O for the pure surface exchange regime). The model can thus quantitatively explain the range of apparent activation energies measured in the different regimes: in the surface exchange regime the apparent activation energy only contains the contribution of the equilibrium exchange rate, whereas in the mixed or in the diffusion controlled regime the contribution of the oxygen self-diffusivity has also to be taken into account, which may yield significantly higher apparent activation energies and simultaneously quantifies the correlation KO ∝ DO1/2 observed for a large number of oxides in the mixed or diffusion controlled regime, respectively.
As early as 1996 Kilner et al.1 showed that there is, for a large number of different materials, a quite obvious correlation KO ∝ DO1/2 between the oxygen surface exchange coefficient KO and the oxygen diffusion coefficient DO (over almost 10 (!) orders of magnitude for DO) for a number of perovskite compositions (with predominant electronic conductivity). Despite this empirical evidence it is assumed until to date,2–4 however, that KO and DO are independent parameters, each of which reflects an independent microscopic mechanism. A major reason for this picture can be identified in the methodology of standard oxygen exchange experiments:5 Here the oxygen flux jKO through a surface element subject to a concentration difference ΔcO is expressed as follows
The phenomenological eqn (1) defines KO without recurring to any microscopic mechanism. As eqn (1) is only valid for surface reactions at x = 0 it seemed plausible that KO depends exclusively on the defect chemistry of the surface. In 1998 Maier6 proposed a relation between KO and the equilibrium oxygen exchange rate 0O at the gas/solid interface (s. Maier,6 p. 222, eqn (149))
| | 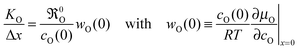 | (2) |
where
cO(0) is the oxygen equilibrium concentration near the surface,
wO(0) is the thermodynamic factor near the surface,
R and
T have the usual meaning, and
μO is the chemical potential of oxygen.
0O is the oxygen exchange rate of the rate determining step in a consecutive reaction sequence.
7,8 Without violating Maier's universally valid original approach, in the literature
4,9–14 also an effective oxygen exchange rate (
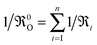
,
n = number of consecutive reactions) is used to describe so-called consecutive “heterogeneous” reactions. The quantitative measure of the parameter Δ
x (with the dimension of a length) having been left undefined, Maier's suggestion essentially means that
KO is proportional to
0O.
15
For the oxygen isotope exchange the thermodynamic factor is unity and eqn (2) yields ![[K with combining tilde]](https://www.rsc.org/images/entities/i_char_004b_0303.gif) O = w(0)KO*. (Throughout the text an asterisk characterizes a parameter determined in an isotope exchange experiment, whereas a tilde refers to a parameter determined in a chemical exchange experiment.) From
O = w(0)KO*. (Throughout the text an asterisk characterizes a parameter determined in an isotope exchange experiment, whereas a tilde refers to a parameter determined in a chemical exchange experiment.) From ![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) O = w(0)DO = w(0)DO*/fO, where DO is the self-diffusion coefficient and DO* is the tracer diffusion coefficient of oxygen, respectively, follows with the correlation factor fO ≃ 1 a simple theoretical correlation.15
O = w(0)DO = w(0)DO*/fO, where DO is the self-diffusion coefficient and DO* is the tracer diffusion coefficient of oxygen, respectively, follows with the correlation factor fO ≃ 1 a simple theoretical correlation.15
| | 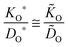 | (3) |
In 2006 De Souza
7 explicitly pointed out that there is a generally observed discrepancy between experimentally determined
KO/
DO ratios and the theoretically expected ratios according to
eqn (3). Instead of fulfilling
eqn (3) the experimental data yield
KO*/
DO* >
O/
O. This was a first significant experimental indication that
eqn (2) might not fully take into account all aspects of the problem.
Armstrong et al.12,13 used in 2011 and 2013 a novel experimental approach, called isothermal isotope exchange (IIE), to extract accurate KO* values in a surface exchange controlled regime. This technique relies on the exchange of isotopically labelled oxygen for lattice oxygen in a similar way to isotope exchange depth profiling (IEDP) with secondary ion mass spectrometry (SIMS), but is conducted in situ on powder samples (particle size 200–300 nm). In this way the exchange kinetics can be expected to be clearly in the surface exchange regime. Very unexpectedly Armstrong et al.12,13 found apparent activation energies of KO* with negative (!) or very small positive values for different lanthanum based MIEC perovskite oxides like LC, LF, LSM, LSF, LSC or LSCF, but also for the standard oxygen ion conductors YSZ and GDC, which have fluorite structure. The reported range of apparent activation energies of the oxygen tracer surface exchange coefficient is −97 kJ mol−1 to +12 kJ mol−1 and is in strong contrast to the reported range from +60 kJ mol−1 to +333 kJ mol−1 measured by conventional methods generally using macroscopic sample geometries, i.e. IEDP/SIMS and electrical conductivity relaxation (ECR) (s. Armstrong et al.,13 Table 5), but also measured by other powder-based methods like pulsed isotope exchange (PIE)9–11 and long-term gas phase exchange/analysis GPE/GPA.4,14,16 These experimental results suggest that the measured oxygen surface exchange coefficient KO, if measured in the mixed regime and in the diffusion controlled regime, may depend not only on the equilibrium oxygen exchange rate 0O at the gas/solid interface but also on the oxygen diffusion coefficient DO in the bulk. Such a behaviour would clearly contradict eqn (2) which postulates that KO depends on 0O only in all kinetic regimes.
In 2015 Knoblauch et al.17 performed thermogravimetric relaxation experiments on sintered CeO2 pellets (1 mm thickness, 93% of theoretical density) with a grain size of about 20 μm. Because of the strongly enhanced chemical diffusivity18 during the oxygen reduction step of nominally undoped CeO2 Knoblauch et al.17 concluded that their reduction experiments had been performed in the surface exchange regime. They also reported an apparent negative activation energy of −64 kJ mol−1 for O. That is, the work of Knoblauch et al.17 showed that the apparent activation energy of the chemical oxygen surface exchange coefficient O can also have negative values if it is measured in the surface exchange controlled regime.
The experimental contradictions to the general validity (i.e. in all kinetic regimes) of eqn (2) necessitate a rediscussion of the meaning of the parameter KO. In order to facilitate a fundamental theoretical discussion the experimental situation of the oxygen exchange experiments is idealized as far as possible. In analogy to Maier6 jump relaxation effects, space charge effects, structural changes and trapping effects are considered secondary. Likewise the mathematical treatment is simplified by considering a homogeneous semi-infinite solid oxide (s. Fig. 1). In this case one has to consider only one gas/oxide interface at x = 0 where defect chemical reactions at the interface are quantified by the equilibrium oxygen exchange rate 0O. The diffusion process of oxygen ions in the solid oxide is quantified by the oxygen self-diffusion coefficient DO. The oxygen partial pressure in the gas phase is given by pO2. That is, the considered oxide is phenomenologically described by two independent material parameters
| | | 0O = 0O(T,pO2), DO = DO(T,pO2) | (4) |
which are functions of the temperature
T and the oxygen partial pressure
pO2, which control (together with unintentionally introduced impurities and/or deliberately added dopants) the concentration of oxygen vacancies
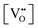
or interstitials
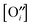
(and of the other ionic and electronic defects). The oxygen surface exchange coefficient
KO, however, is a phenomenological parameter which, a priori, is not yet related to a specific microscopic process
viaeqn (1). In view of the experimental findings outlined above it can be tentatively suggested that, at least for IEDP/SIMS and ECR experiments, but most probably also for PIE
9–11 and GPE/GPA
4,14,16 experiments,
KO should depend on the two independent parameters
DO and
0O as follows
| | | KO = KO(DO, 0O) | (5) |
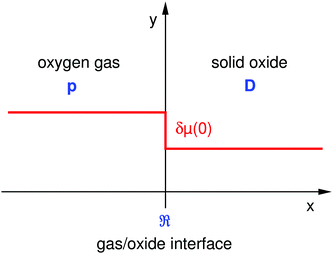 |
| | Fig. 1 Schematic representation of a gas/oxide interface of a homogeneous semi-infinite solid oxide. D is the self-diffusion coefficient of oxygen ions (DO) in the solid oxide, p is the oxygen partial gas pressure (pO2) in the oxygen gas phase and is the equilibrium oxygen exchange rate (0O) at the interface. At t = 0 a small step-like change δμ(0) of the chemical potential of oxygen (δμO(x = 0)) is experimentally induced at the gas/oxide interface at x = 0. | |
In order to derive the hypothetical function (5) from fundamental principal relations it is appropriate to adopt Maier's6 notation and his very general procedure: the oxygen flux jKO(0) through the surface (at x = 0) which is induced by a small change of the chemical potential of oxygen δμO(0) becomes6
| | 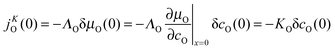 | (6) |
where
ΛO is the conductance and ∂
μO/∂
cO the reciprocal “chemical” capacity. It is important to keep in mind that
eqn (6) is an approximation near chemical equilibrium. It is very instructive to recall Maier's comment on
eqn (6): “It is worth noting that these relationships [
eqn (6)] are quite general in that they do not preselect a certain mechanism.”
6 This statement implicitly comprises the fact that the parameter
KO principally can be the result of a combination of several mechanisms.
The diffusive oxygen flux19jDO(0) in the near-surface region of thickness δx(0), which is driven by the gradient of the chemical potential of oxygen ∂μO/∂x|x=0, becomes
| | 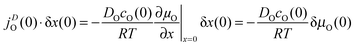 | (7) |
where
DO is the oxygen self-diffusion coefficient and
cO(0) is the oxygen concentration (at the surface).
The first step in the derivation of the hypothetical function (5) is to study the correlation between KO and DO which principally results from the boundary condition at the gas/solid interface
which connects surface exchange and bulk transport and which therefore has to be respected when solving the respective diffusion equation.
20 With
eqn (6) and (7) the boundary condition
eqn (8) yields
| | 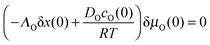 | (9) |
This means that for a moderate value of δ
μO(0),
i.e. a near-equilibrium situation at the surface, the (standard) boundary condition
eqn (8) yields the relation
DOcO(0)/
RT =
ΛOδ
x(0). With the thermodynamic factor
wO(0) (s.
eqn (2)) and
KO =
ΛO∂
μO/∂
cO|
x=0 (s.
eqn (6)) one finally obtains the following general correlation between
KO and
DO which is valid in the
near-equilibrium situation
In the second step the meaning of the parameter δx(0) will be clarified. For an isotope exchange experiment wO(0) = 1 is valid and eqn (10) simply becomes δx*(0) = DO*/KO*, which is equivalent to the so-called characteristic length l0c used in the literature to roughly estimate in which kinetic regime a given oxygen exchange experiment is performed. Adopting this notation the subsequent discussion will be facilitated for the reader as eqn (10) now becomes
| | 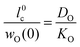 | (11) |
Here the upper case index 0 means that
l0c refers to a near-surface layer (at
x = 0). Until now
eqn (11) was only a formal mathematical rule to calculate
l0c and results as such directly from the boundary condition
eqn (8). The
physical meaning of
l0c will become evident if one defines with
eqn (11) a characteristic time
t0c (valid likewise for the near-surface region at
x = 0) as follows
| | 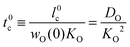 | (12) |
In Appendix A it is demonstrated that the characteristic time
t0c physically corresponds to the relaxation time of a (sufficiently small) change in the oxygen potential δ
μ(0) at the surface.
Eqn (12) leads to the following two relations
| |  | (13) |
The characteristic length
l0c is thus proportional to the diffusion length the oxygen ions cover in the near-surface region during the characteristic time
t0c. The term
KO/Δ
x (as formulated by Maier
6) in
eqn (2) can be considered equivalent to the inverse characteristic time
t0c. This relation suggests to
postulate| | 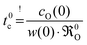 | (14) |
Combining
eqn (13) and (14) yields the missing correlation anticipated as “the hypothetical function” in
eqn (5)| | 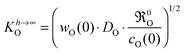 | (15) |
It should be recalled that
eqn (15) is a consequence of a boundary condition, and hence, is valid in all experimental situations where the considered boundary condition (
eqn (8)) is valid. The upper case index
h → ∞ refers to the fact that this boundary condition is often applicable to plate-like samples of large thickness
h, that is, if
h ≫
l0c.
For an oxygen isotope exchange experiment (with wO(0) = 1 and DO* = fO·DO) eqn (15) becomes (dropping the index h → ∞ in order to simplify the notation in eqn (16)–(19))
| | 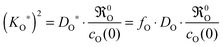 | (16) |
Here (
KO*,
DO*) are data couples determined
via an isotope exchange experiment. In a chemical oxygen exchange experiment (with
w(0) ≫ 1
21,22 and
O =
wO(0)·
DO) the data couples (
O,
O) are determined. In this case
eqn (15) yields
| | 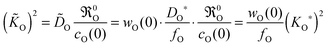 | (17) |
that is, one has
O = (
wO(0)/
fO)
1/2·
KO* in contrast to
O = (
wO(0)/
fO)·
DO*. Comparing
KO/
DO ratios for the isotope exchange and for the chemical exchange experiment, respectively, one obtains from
eqn (16) and (17) the following theoretical relations
| | 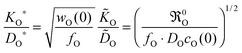 | (18) |
Because of
fO ≈ 1 and
wO(0) ≫ 1
21,22 one immediately obtains
KO*/
DO* >
O/
O, which is in agreement with a large number of experimental results.
7 Thus
eqn (18) supports the postulate expressed in
eqn (14).
For more complicated sample geometries and correspondingly more complicated boundary conditions a more complicated correlation than eqn (15) is to be expected. The formal calculation of a characteristic length l0c by eqn (11) is mathematically always possible, but the result would mask the physical meaning of l0c which is explicitly expressed in eqn (13) only. Likewise the characteristic time t0c has been (almost completely) neglected in the materials science literature, which focuses practically exclusively on KO. It is, however, principally preferable for the discussion of experimental studies to consider the two characteristic parameters t0c and l0c compiled together with KO in eqn (19)
| | 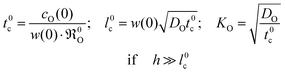 | (19) |
where
h is the thickness of a plate-like sample. The set of
eqn (19) shows that the condition
h ≫
l0c is realised if the oxygen self-diffusion
DO is low and/or
0O is high and/or
w(0) is small. This means that oxygen exchange occurs practically only in a near-surface region of characteristic thickness
l0c during the characteristic time
t0c. Consequently the oxygen exchange of the
whole sample is hindered due to the slow oxygen self-diffusion in the bulk. This situation corresponds to the so-called diffusion controlled regime.
The very specific condition h ≈ l0c means that within the characteristic time t0c practically all the oxygen of the sample is exchanged. This scenario is called mixed regime as the oxygen exchange rate at the surface (0O) and the self-diffusion coefficient in the bulk (DO) hinder the overall oxygen exchange simultaneously. Note that the condition h ≈ l0c is more easily reached during chemical exchange (with w(0) ≫ 1) than during isotope exchange (with w(0) = 1) because during chemical exchange oxygen self-diffusion is enhanced by the thermodynamic factor, O = wO(0)DO.
The condition h ≪ l0c means for a given value of h that the oxygen exchange at the sample surface is sufficiently slow and/or oxygen bulk transport is sufficiently rapid. In this case the oxygen exchange of the whole sample is entirely dominated by the (apparent equilibrium) oxygen exchange rate 0O at the surface, and this corresponds to the so-called surface exchange regime. For a thin plate-like sample with thickness h and a sphere with radius r the solution of the diffusion equation for the oxygen exchange kinetics in the surface exchange regime can be approximated by
| | 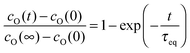 | (20) |
where
τeq is the time constant to reach chemical equilibrium.
17 For a plane sheet one gets
τeq = (
h/2)/
KO and for a sphere
τeq = (
r/3)/
KO.
17 Recalling that
KO is a consequence of the boundary condition one concludes
| | 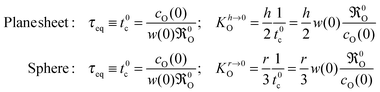 | (21) |
Comparing
Kh→∞O for a thick plane sheet in
eqn (15) with
Kh,r→0O for an extremely thin plane sheet or for powder samples, respectively, shows that, in the latter case, the oxygen exchange coefficient becomes independent of
DO. This means that the oxygen exchange rate
0O can be directly determined from the time constant
τeq in
eqn (20),
e.g. in an isothermal isotope exchange (IIE) experiment
13 or in a thermogravimetric relaxation experiment
17 carried out in the surface exchange regime. Further,
eqn (21) suggests a direct experimental check of the postulate in
eqn (14): if the postulate is valid the time constant
τeq should not depend on the surface-to-volume ratio of the sample in the surface exchange regime.
The arguments displayed above offer a clue to explain the differences in the reported range of apparent activation energies (s. Armstrong et al.,13 Table 5). Armstrong et al.13 determined KO values via IIE clearly in the surface exchange regime (spheres: 200–300 nm, wO(0) = 1) according to eqn (21)
| | 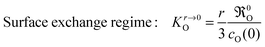 | (22) |
Other studies (s. references compiled in Table 5 of Armstrong
et al.13) which determined
KO values
via IEDP/SIMS or ECR, respectively, as well as PIE-based
9–11 and GPE/GPA-based
4,14,16 investigations often remained in the diffusion controlled regime (
h ≫
l0c) and therefore unintentionally tended to determine a parameter corresponding to the following expression for the surface exchange coefficient
| | 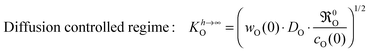 | (23) |
The totally different temperature dependencies which are expected (activation energy for
0O only in
eqn (22) and the mean value of the activation energy for
0Oand for
DO in
eqn (23)) comprise the reported range of apparent activation energies for the two entirely different experimental scenarios and further supports the postulate in
eqn (14).
Eqn (23) obviously allows to rationalise the
KO ∝
DO1/2 correlation reported in the literature.
1
In the following section it is demonstrated how the oxygen exchange rate 0O can also be measured via an 18O exchange experiment on a thick plate-like sample with SIMS depth profiling, that is, in an experimental situation where the oxygen exchange is clearly in the diffusion controlled regime, h ≫ l0c. In 2001 Fielitz and Borchardt,23 without, however, discussing the physical meaning of τeq*, pointed out that the corresponding solution of the diffusion equation as given by Crank20 should be used in the following form
| | 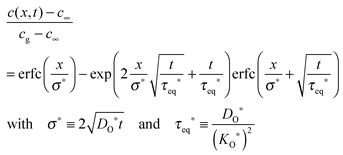 | (24) |
to fit SIMS depth profiles (s. elsewhere
23 for further details) in order to extract the tracer isotope diffusion length
σ* and the time constant
τeq* which is
the time necessary to obtain isotope equilibrium at the surface (
x = 0). As the boundary condition
eqn (8) was used to solve the diffusion equation
20 one can use the set of
eqn (19) (with
wO(0) = 1 for an isotope exchange experiment)
| | 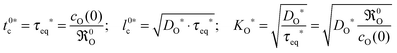 | (25) |
This means that the oxygen exchange rate
0O can be directly determined by the time constant
τeq* in
eqn (24). It should be underlined that the specific form of the solution Fielitz and Borchardt
23 proposed explicitly contains the function
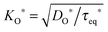
(compare to
τeq* in
eqn (24)), whereas this fact is not evident in the original form given by Crank.
20
The presented revision of the principal nature of the surface exchange coefficient has been essentially triggered by the publications of Armstrong et al.12,13 since these investigations demonstrated clearly that the experimentally determined apparent activation energies for KO* strongly depend on the kinetic regime of the respective experiments. Armstrong et al.12,13 studied a relatively broad spectrum of materials (various perovskite MIECs like LC, LF, LSM, LSF, LSC or LSCF as well as two widely used oxygen electrolyte materials, namely YSZ and GDC). The similarities of the regime dependency of KO* therefore suggests that their observation does not reflect a specific property of a class of materials, but rather an underlying universal phenomenon. It should, however, be recalled that there is another method to determine KO* on powders which does not find any dependence of the apparent activation energies on the kinetic regime. Bouwmeester et al.9 and Yoo et al.10,11 used a novel pulse 18O–16O isotopic exchange (PIE) technique for the rapid determination of the oxygen surface exchange rate of oxide ion conductors. For this method powders with an average particle size in the range 0.4–5.0 μm are used. The reported apparent activation energies did not significantly differ from the values determined on bulk samples via IEDP/SIMS (or ECR, respectively). Recent GPE/GPA-based work4,14,16 comprises similar activation energies for the surface exchange coefficient and for the oxygen diffusivity.
During the PIE experiment, a powder sample is loaded in a packed-bed micro-reactor. Maintaining conditions of chemical equilibrium, the response to an 18O-enriched pulse fed through the reactor under continuous flow conditions is measured by mass spectrometric analysis of the gas phase fractions of oxygen isotopomers (18O2, 16O18O and 16O2) at the exit of the reactor. The essential difference between PIE9–11 and IIE12,13 is the residence time between the 18O2 atmosphere and the powder sample: for PIE the mean residence time is 5 to 40 ms,9–11 whereas in an IIE experiment the residence time is 15 to 40 min.12,13 In this context, it is worth recalling that the surface exchange regime is not reached before the 18O concentration gradient (in the bulk of a powder particle) practically disappears, which means that the value of DO* has become no longer relevant for the 18O exchange process. Since principally a certain (finite) diffusion time t has to elapse before an 18O concentration gradient in a particle with nominal radius r disappears, IIE seems to be more suitable than PIE to operate in a true surface exchange regime. This can be easily demonstrated by a straightforward estimation: the condition 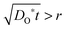 relates the necessary diffusion length, which is required to reach the surface exchange regime, to the particle radius. Estimating an average particle radius rPIE ≈ 3 μm9–11 for the PIE experiment and an average particle radius rIIE ≈ 0.3 μm12,13 for the IIE experiment one has tPIE/tIIE = (rPIE/rIIE)2 ≈ 100 as a condition to reach the surface exchange regime in the published PIE studies compared to the published IIE studies. Since, however, the experimentally realized residence times tPIE during the PIE experiment are only in the millisecond range,9–11 so that one can estimate the ratio tPIE/tIIE ≈ 40 ms/40 min = 1.7 × 10−5, it cannot be excluded that PIE operates in a transitory, i.e. mixed controlled regime, similarly to the great majority of the published IEDP/SIMS studies.
relates the necessary diffusion length, which is required to reach the surface exchange regime, to the particle radius. Estimating an average particle radius rPIE ≈ 3 μm9–11 for the PIE experiment and an average particle radius rIIE ≈ 0.3 μm12,13 for the IIE experiment one has tPIE/tIIE = (rPIE/rIIE)2 ≈ 100 as a condition to reach the surface exchange regime in the published PIE studies compared to the published IIE studies. Since, however, the experimentally realized residence times tPIE during the PIE experiment are only in the millisecond range,9–11 so that one can estimate the ratio tPIE/tIIE ≈ 40 ms/40 min = 1.7 × 10−5, it cannot be excluded that PIE operates in a transitory, i.e. mixed controlled regime, similarly to the great majority of the published IEDP/SIMS studies.
Likewise the GPE/GPA-based investigations4,14,16 use macroscopic samples and fairly long annealing times (for details see Appendix B). This inevitably implies a shift to L values typical for a mixed controlled or diffusion controlled regime (see Table 1 and Appendix C).
Table 1 Characteristic dimensionless parameters of the kinetic regimes
| Kinetic regime |
L = (h/2)(KO/DO) |
ϕ(β1,L) |
β
1
2
|
| Surface control |
<0.3 |
1 |
L
|
| Mixed regime |
0.3 to 30 |
— |
— |
| Diffusion regime |
>30 |
8/π2 |
π2/4 |
Summary
In 1998 Maier6 proposed eqn (2) according to which the exchange rate 0O (for oxygen) is proportional to the surface exchange coefficient KO. Since then two well documented significant experimental findings became known which contradicted the general validity of eqn (2):
(i) Instead of the theoretically expected relation15KO*/DO* ≅ O/O the experiments yield7KO*/DO* > O/O.
(ii) If measured in the surface exchange regime (h ≪ l0c) the apparent activation energies for KO* and O are very small13,17 whereas the apparent activation energies measured in the mixed controlled regime (h ≈ l0c) or in the diffusion controlled regime (h ≫ l0c) are considerably larger (s. Armstrong et al.,13 Table 5).
These two surprising experimental observations, but especially the latter one, raise doubtful questions about the general validity of eqn (2), which necessitate a thorough fundamental rediscussion of the surface exchange coefficient for oxygen (on oxide surfaces).
Following principally Maier's6 procedure the simplest possible experimental situation was chosen, i.e. an oxide surface close to chemical equilibrium (s. eqn (6)). For a complete picture, one has, however, to include the diffusive transport of oxygen (s. eqn (7)) and the fact that at the gas/solid interface there is a boundary condition to be respected. For the gas/solid interface of a homogeneous semi-infinite oxide this is eqn (8). This boundary condition inevitably leads to a mathematical correlation between KO and DO (s. eqn (11)), which, at first glance, seems to be only a formal recipe to calculate the so-called characteristic length l0c. Through the definition of a characteristic time t0c, this parameter can be attributed a physical meaning in eqn (13) as t0c is the time necessary for the relaxation of a small change of the chemical potential of oxygen at the surface, δμO(0). Further, it becomes obvious that in eqn (2) the term KO/Δx (as formulated by Maier6) is equivalent to the inverse characteristic time t0c. It is therefore consistent to correlate the oxygen exchange rate 0O and the characteristic time t0c directly, as postulated through eqn (14). This postulate and the boundary condition (eqn (8)) entail an explicit dependency between the (surface) exchange rate and the (bulk) diffusivity of oxygen KO = KO(DO,0O) (s. eqn (15)). At the same time such a dependency implies that for more complicated diffusion boundary conditions more complicated functions for KO have to be expected.
The relations for KO in the different kinetic regimes derived in the framework of the proposed novel interpretation of the standard theoretical concept6 for KO finally enable to unambiguously explain the two experimental observations (i) and (ii). In the presented model eqn (2) is only valid for the special case of the true surface exchange regime (s. eqn (21)). The parameter Δx in eqn (2), however, then is no longer a constant, but corresponds to the surface-to-volume ratio of the sample. As a consequence, the time constant τeq to reach equilibrium (s. eqn (20) and (21)) should not depend on the surface-to-volume ratio of the sample in the surface exchange regime.
Appendix A
For a thin surface layer δx′(x = 0) the following expression can be formulated| | 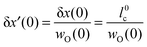 | (A1) |
If δx′(0) is sufficiently small and/or if the oxygen flux jKO(0) is approximately constant within the layer of thickness δx′(0) one obtains with eqn (6)| | 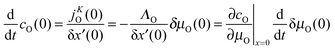 | (A2) |
That is, the time constant t0c for the exponential relaxation kinetics of a small oxygen potential change δμO(0) at the surface is given by| | 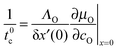 | (A3) |
Together with KO = ΛO∂μO/∂cO|x=0 and eqn (A1), (A3) yields eqn (12).
Appendix B
A widely applicable reaction model comprises the following consecutive steps:7
(1) adsorption dissociation (eq.)
| | 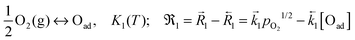 | (B1) |
(2) charge transfer (rds)
| | 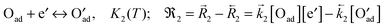 | (B2) |
(3) incorporation (eq.):
| | 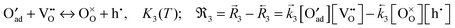 | (B3) |
The reactions are formulated according to the Kröger–Vink notation, using square brackets for the respective concentrations,
Kn for the mass action constants (
n = 1, 2, 3),
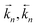
for the forward/backward rate constants,
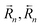
for the forward/backward reaction rates and
n for the net reaction rates of these reactions. The so called apparent or effective equilibrium exchange rate
0O of the rate determining step (charge transfer
(B2) is often assumed as the rate determining step
7) is defined by
| | 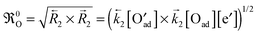 | (B4) |
After inserting the mass action constants
| | 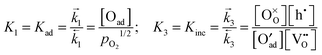 | (B5) |
into
eqn (B4) and defining notational abbreviations
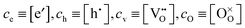
one finally gets if,
e.g., charge transfer is rate determining
| | 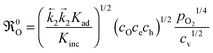 | (B6) |
Eqn (B6) demonstrates in a very convincing manner that the surface exchange coefficient will have a non-trivial temperature dependence in the surface controlled regime (
KO ∝
0O, see
eqn (21)). The resulting apparent activation energy should generally be less than the average energy of
DO and
0O. In the diffusion controlled regime (
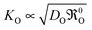
, see
eqn (15)) the resulting apparent activation energy is the average activation energy of
DO and
0O.
We will now demonstrate that there is an equivalence of the definitions of the phenomenological parameters used in Maier's approach6 and the approach of Klier and Kučera.24 Comparing both approaches it becomes evident that recent work,4,14 based on the work of Klier and Kučera,24 uses the same phenomenological description for their consecutive model as Maier6 and the present authors (neglecting, however, the charge transfer step, which seems to be a weakness of this approach). With rH for the “heterogeneous” reaction rate, ra for the (dissociative) “adsorption” rate, and ri for the “incorporation” rate they obtain (see eqn (8) and (12) in Farlenkov et al.14) rH = ra·ri/(ra + ri), i.e. they set 1/rH = 1/ra + 1/ri as expected for a consecutive model. In Maier's notation this would become 1/0 = 1/1 + 1/3 if the charge transfer step (eqn (B2)) is neglected (in order to facilitate the comparison). In eqn (13) of Farlenkov et al.14 the surface exchange coefficient, k, without specifying the kinetic regime, is defined as k = const. × rH (where the constant contains the specific data of the material, which have only a negligible temperature dependence). This corresponds formally to the relation originally proposed by Maier6 as well as to our result for the mere surface controlled regime, KO = const. × 0O, as expressed by eqn (21). It is, however, debatable (see Appendix C) whether the pure surface controlled regime can anyhow be studied if the dense (polycrystalline and single crystalline) samples have a thickness of 5 mm and undergo annealing times of more than 5 × 104 s.14
Appendix C
The most direct way to follow the kinetics of oxygen uptake or release in situ at high temperatures is a gravimetric experiment.17 For plate-like samples with thickness h the mass change, Δm(t), if normalized to the mass change at equilibrium, Δm(teq), is given by (Crank20 p. 60)| | 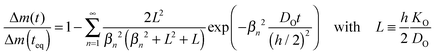 | (C1) |
The dimensionless quantities βn are positive roots of βn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) tanβn = L. As the infinite sum in eqn (C1) converges rapidly one has approximately17
tanβn = L. As the infinite sum in eqn (C1) converges rapidly one has approximately17| | 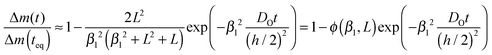 | (C2) |
where DO is the effective oxygen diffusion coefficient in the solid and KO is the oxygen surface exchange coefficient defined by eqn (1). Positive roots of β1tanβ1 = L are plotted in Fig. 2 which allows a graphical visualisation of the kinetic regimes. The values of the resulting characteristic dimensionless parameters of the kinetic regimes are compiled in Table 1.
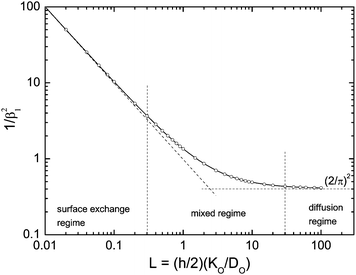 |
| | Fig. 2 Graphical visualisation of the kinetic regimes17 (open points are positive roots of β1tanβ1 = L tabulated by Carslaw and Jaeger25). The diffusion regime is characterised by β12 ≃ π2/4 and the surface exchange regime by β12 ≃ L. | |
Acknowledgements
We are indebted to Martin Schmücker, Aldo Steinfeld, Robert Pitz-Paal, Clemens Feldmann and their co-workers for valuable discussions. Financial support by the Initiative and Networking Fund of the Helmholtz Association of German Research Centers within the Virtual Institute SolarSynGas as well as by Deutsche Forschungsgemeinschaft (DFG) for one of the authors (P. F.) is gratefully acknowledged.
References
- J. A. Kilner, R. A. De Souza and I. C. Fullarton, Solid State Ionics, 1996, 86–88, 703 CrossRef CAS.
- J. A. Kilner, S. J. Skinner and H. H. Brongersma, J. Solid State Electrochem., 2011, 15, 861 CrossRef CAS.
- J. Blair and D. S. Mebane, Solid State Ionics, 2015, 270, 47 CrossRef CAS.
- M. V. Ananyev, E. S. Tropin, V. A. Eremin, A. S. Farlenkov, A. S. Smirnov, A. A. Kolchugin, N. M. Porotnikova, A. V. Khodimchuk, A. V. Berenov and E. Kh. Kurumchin, Phys. Chem. Chem. Phys., 2016, 18, 9102 RSC.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53–56, 859 CrossRef CAS.
- J. Maier, Solid State Ionics, 1998, 112, 197 CrossRef CAS.
- R. A. De Souza, Phys. Chem. Chem. Phys., 2006, 8, 890 RSC.
- R. Merkle and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 3874 CrossRef CAS PubMed.
- H. J. M. Bouwmeester, C. Song, J. Zhu, J. Yi, M. van Sint Annaland and B. A. Boukamp, Phys. Chem. Chem. Phys., 2009, 11, 9640 RSC.
- C.-Y. Yoo, B. A. Boukamp and H. J. M. Bouwmeester, J. Solid State Electrochem., 2011, 15, 231 CrossRef CAS.
- C.-Y. Yoo and H. J. M. Bouwmeester, Phys. Chem. Chem. Phys., 2012, 14, 11759 RSC.
- E. N. Armstrong, K. L. Duncan, D. J. Oh, J. F. Weaver and E. D. Wachsman, J. Electrochem. Soc., 2011, 158, B283 CrossRef CAS.
- E. N. Armstrong, K. L. Duncan and E. D. Wachsman, Phys. Chem. Chem. Phys., 2013, 15, 2298 RSC.
- A. S. Farlenkov, M. V. Ananyev, V. A. Eremin, N. M. Porotnikova, E. K. Kurumchin and B.-T. Melekh, Solid State Ionics, 2016, 290, 108 CrossRef CAS.
- J. Maier, Solid State Ionics, 2000, 135, 575 CrossRef CAS.
- A. Berenov, A. Atkinson, J. Kilner, M. Ananyev, V. Eremin, N. Porotnikova, A. Farlenkov, E. Kurumchin, H. J. M. Bouwmeester, E. Bucher and W. Sitte, Solid State Ionics, 2014, 268, 102 CrossRef CAS.
- N. Knoblauch, L. Dörrer, P. Fielitz, M. Schmücker and G. Borchardt, Phys. Chem. Chem. Phys., 2015, 17, 5849 RSC.
- S. Ackermann, J. R. Scheffe and A. Steinfeld, J. Phys. Chem. C, 2014, 118, 5216 CAS.
-
H. Schmalzried, Solid State Reactions, Verlag Chemie, Weinheim, 1974, p. 54 Search PubMed.
-
J. Crank, The Mathematics of Diffusion, Oxford University Press, 2nd edn, 1975, p. 36 Search PubMed.
- J. Claus, M. Leonhardt and J. Maier, J. Phys. Chem. Solids, 2000, 61, 1199 CrossRef CAS.
- M. Leonhardt, R. A. De Souza, J. Claus and J. Maier, J. Electrochem. Soc., 2002, 149, J19 CrossRef CAS.
- P. Fielitz and G. Borchardt, Solid State Ionics, 2001, 144, 71 CrossRef CAS.
- K. Klier and E. Kučera, J. Phys. Chem. Solids, 1966, 27, 1087 CrossRef CAS.
-
H. S. Carslaw and J. C. Jaeger, Conduction of Heat in Solids, Clarendon Press, 2nd edn, 1959 Search PubMed.
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 
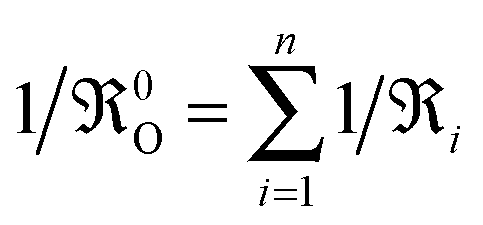 , n = number of consecutive reactions) is used to describe so-called consecutive “heterogeneous” reactions. The quantitative measure of the parameter Δx (with the dimension of a length) having been left undefined, Maier's suggestion essentially means that KO is proportional to
, n = number of consecutive reactions) is used to describe so-called consecutive “heterogeneous” reactions. The quantitative measure of the parameter Δx (with the dimension of a length) having been left undefined, Maier's suggestion essentially means that KO is proportional to 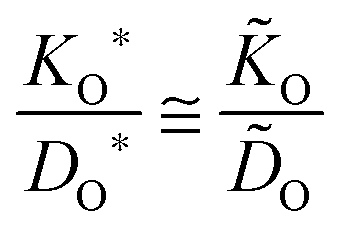
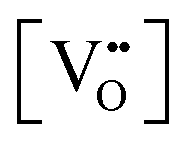 or interstitials
or interstitials 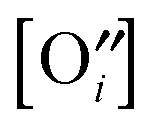 (and of the other ionic and electronic defects). The oxygen surface exchange coefficient KO, however, is a phenomenological parameter which, a priori, is not yet related to a specific microscopic process viaeqn (1). In view of the experimental findings outlined above it can be tentatively suggested that, at least for IEDP/SIMS and ECR experiments, but most probably also for PIE9–11 and GPE/GPA4,14,16 experiments, KO should depend on the two independent parameters DO and
(and of the other ionic and electronic defects). The oxygen surface exchange coefficient KO, however, is a phenomenological parameter which, a priori, is not yet related to a specific microscopic process viaeqn (1). In view of the experimental findings outlined above it can be tentatively suggested that, at least for IEDP/SIMS and ECR experiments, but most probably also for PIE9–11 and GPE/GPA4,14,16 experiments, KO should depend on the two independent parameters DO and 

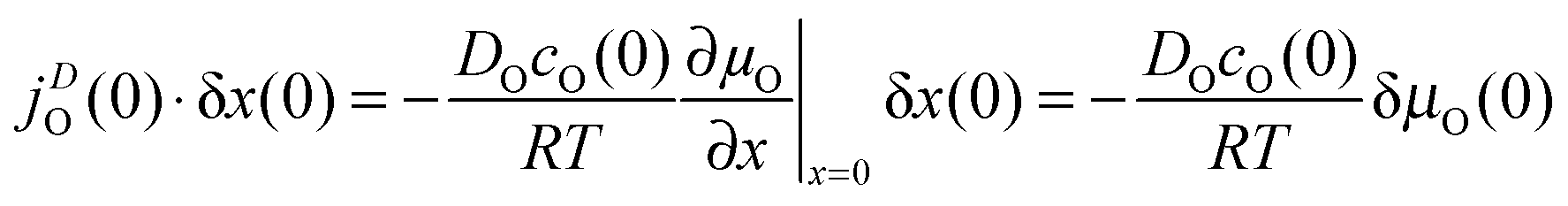
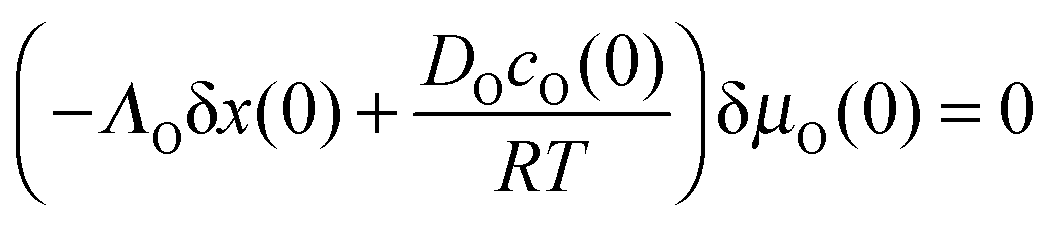
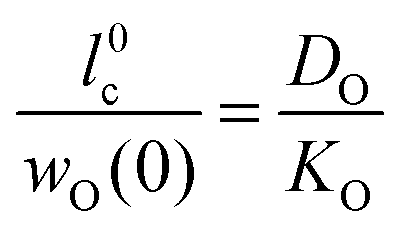



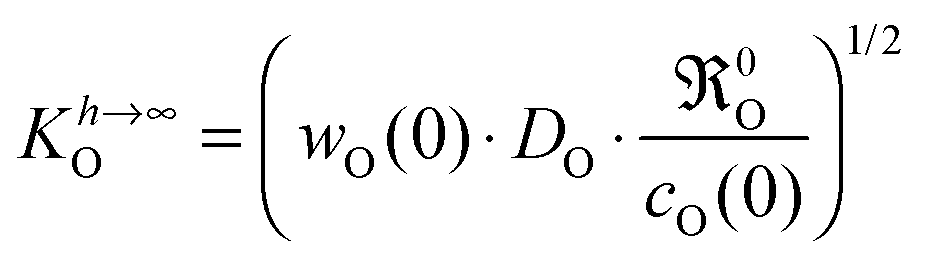

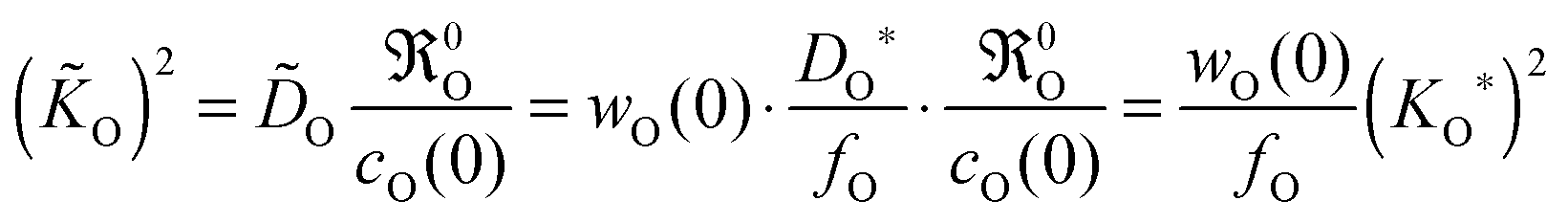
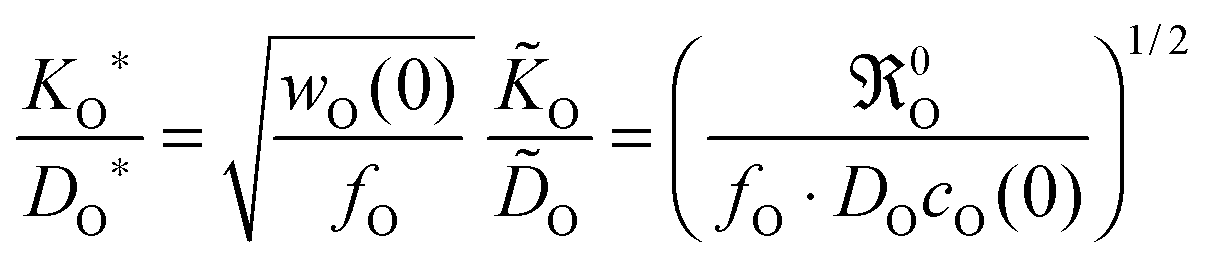



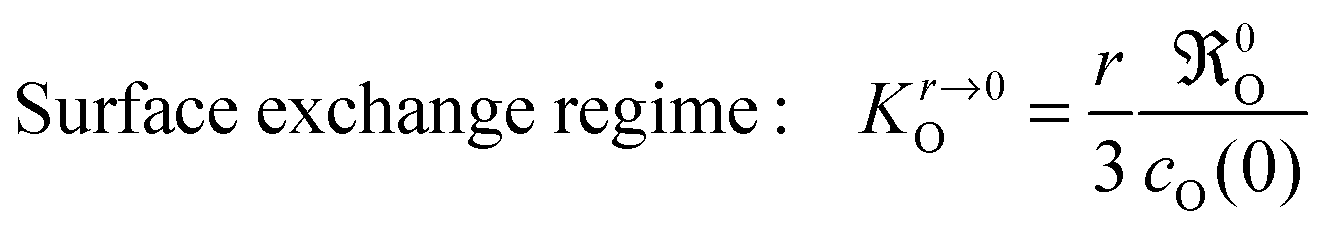
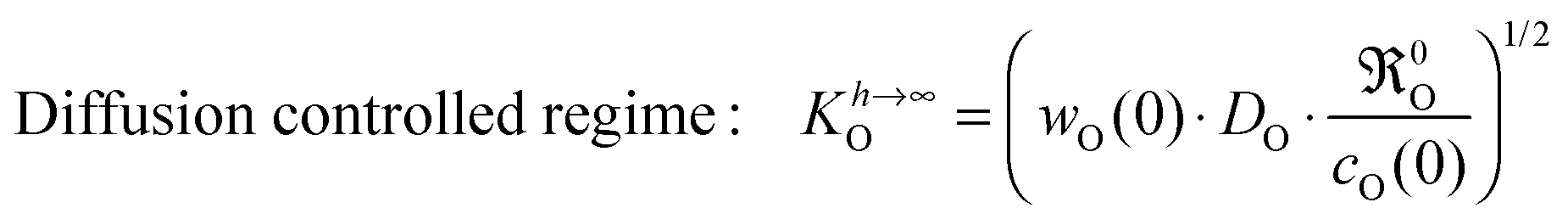
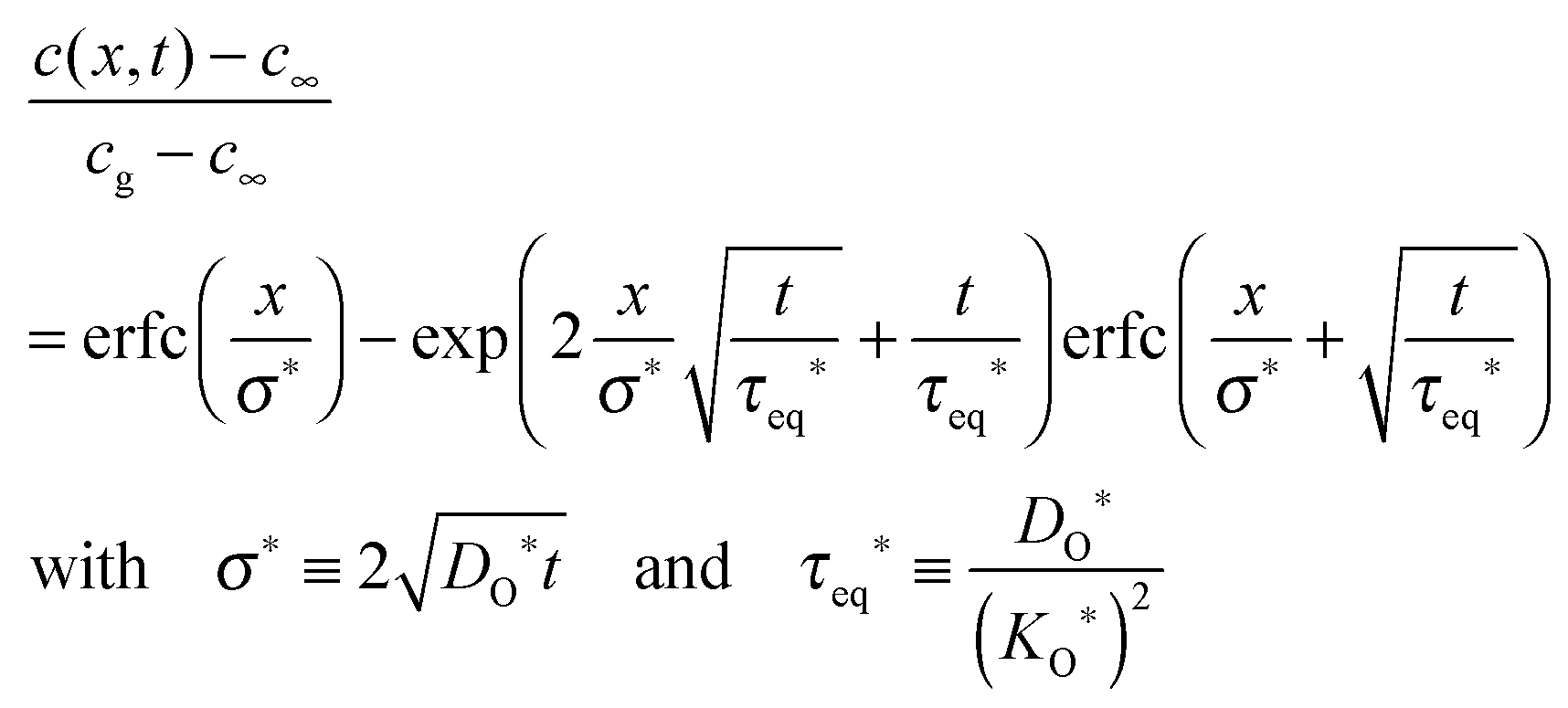
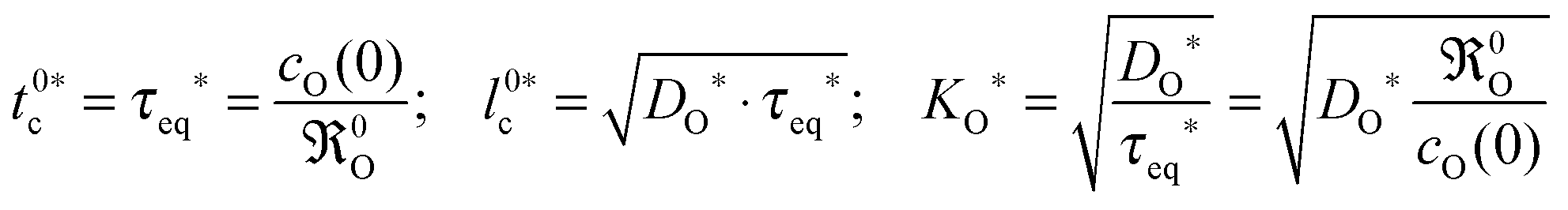
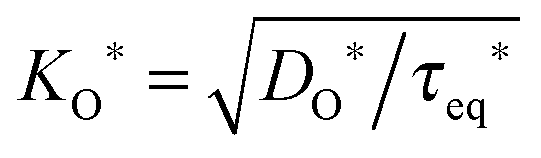 (compare to τeq* in eqn (24)), whereas this fact is not evident in the original form given by Crank.20
(compare to τeq* in eqn (24)), whereas this fact is not evident in the original form given by Crank.20
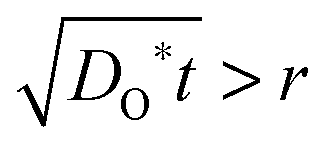 relates the necessary diffusion length, which is required to reach the surface exchange regime, to the particle radius. Estimating an average particle radius rPIE ≈ 3 μm9–11 for the PIE experiment and an average particle radius rIIE ≈ 0.3 μm12,13 for the IIE experiment one has tPIE/tIIE = (rPIE/rIIE)2 ≈ 100 as a condition to reach the surface exchange regime in the published PIE studies compared to the published IIE studies. Since, however, the experimentally realized residence times tPIE during the PIE experiment are only in the millisecond range,9–11 so that one can estimate the ratio tPIE/tIIE ≈ 40 ms/40 min = 1.7 × 10−5, it cannot be excluded that PIE operates in a transitory, i.e. mixed controlled regime, similarly to the great majority of the published IEDP/SIMS studies.
relates the necessary diffusion length, which is required to reach the surface exchange regime, to the particle radius. Estimating an average particle radius rPIE ≈ 3 μm9–11 for the PIE experiment and an average particle radius rIIE ≈ 0.3 μm12,13 for the IIE experiment one has tPIE/tIIE = (rPIE/rIIE)2 ≈ 100 as a condition to reach the surface exchange regime in the published PIE studies compared to the published IIE studies. Since, however, the experimentally realized residence times tPIE during the PIE experiment are only in the millisecond range,9–11 so that one can estimate the ratio tPIE/tIIE ≈ 40 ms/40 min = 1.7 × 10−5, it cannot be excluded that PIE operates in a transitory, i.e. mixed controlled regime, similarly to the great majority of the published IEDP/SIMS studies.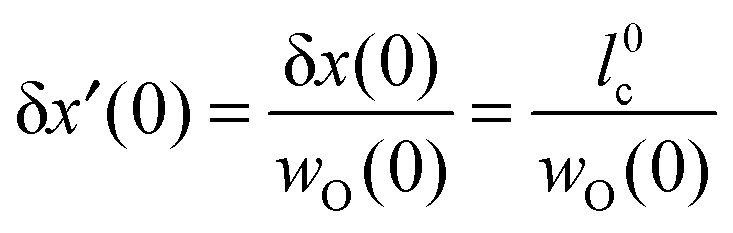
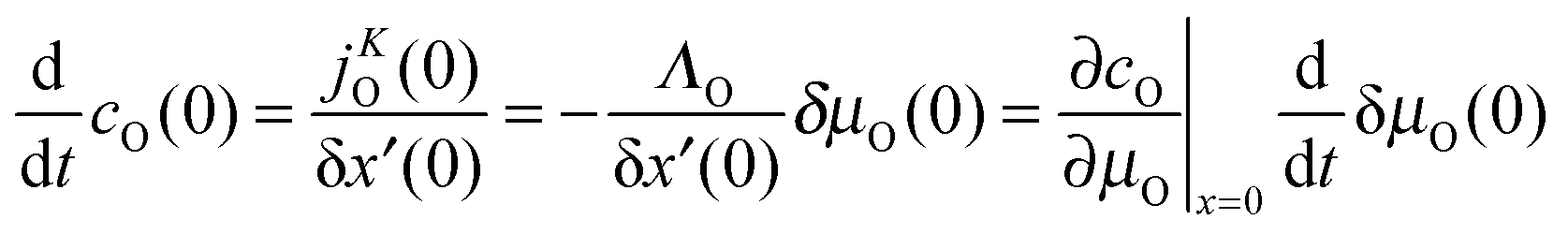
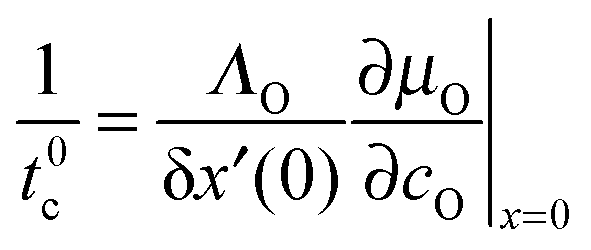
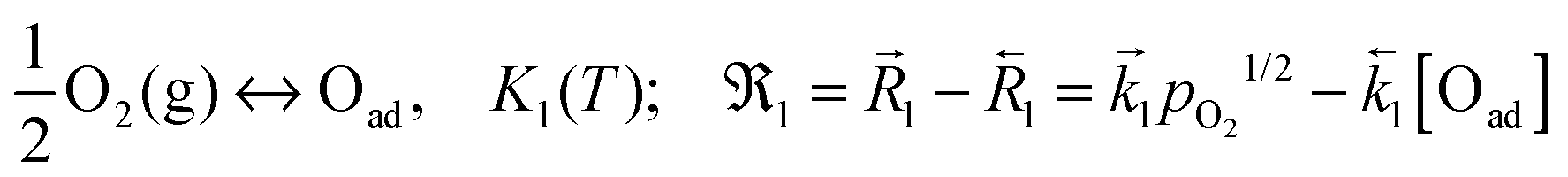
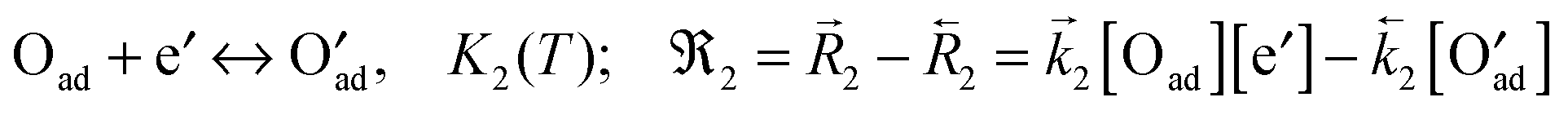

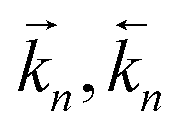 for the forward/backward rate constants,
for the forward/backward rate constants,  for the forward/backward reaction rates and
for the forward/backward reaction rates and 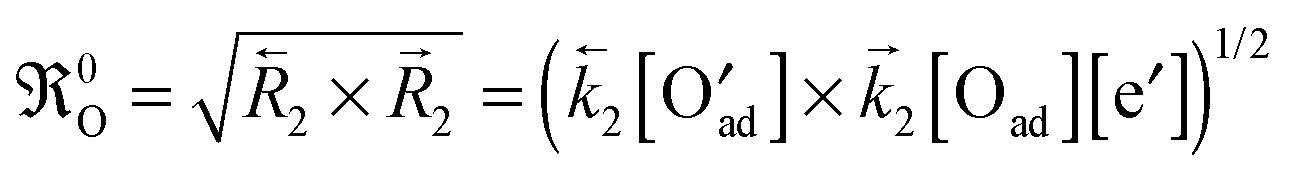

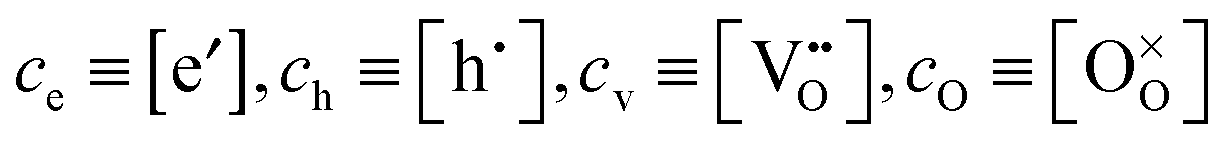 one finally gets if, e.g., charge transfer is rate determining
one finally gets if, e.g., charge transfer is rate determining
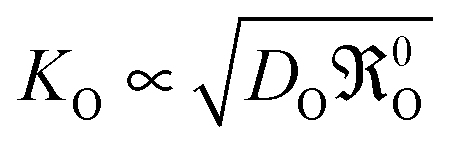 , see eqn (15)) the resulting apparent activation energy is the average activation energy of DO and
, see eqn (15)) the resulting apparent activation energy is the average activation energy of DO and