
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocatalytic activity of a self-stabilized synthetic flavin anchored on a TiO2 surface†
Manjula
Pandiri
a,
Mohammad S.
Hossain
b,
Frank W.
Foss
Jr.
b,
Krishnan
Rajeshwar
b and
Yaron
Paz
*a
aDepartment of Chemical Engineering, Technion, Haifa 32000, Israel. E-mail: paz@tx.technion.ac.il
bDepartment of Chemistry and Biochemistry, The University of Texas at Arlington, Arlington, 76019-0065, TX, USA
First published on 13th June 2016
Abstract
Synthetic flavin molecules were anchored on Degussa P25 titanium dioxide (TiO2). The effect of their presence on the photocatalytic (PC) activity of TiO2 was studied. Under UV light, an increase in the degradation rate of ethanol was observed. This increase was accompanied by stabilization of the anchored flavin against self-degradation. The unprecedented stabilization effect was found also in the absence of a reducing agent such as ethanol. In contrast, under the less energetic visible light, fast degradation of the anchored flavin was observed. These rather surprising observations were attributed to the propensity for charge transport from excited flavin molecules to the semiconductor and to the role that such charge transfer may play in stabilizing the overall assembly. Anchored flavins excited by UV light to their S2, S3 electronic states were able to transfer the excited electrons to the TiO2 phase whereas anchored flavin molecules that were excited by visible light to the S1 state were less likely to transfer the photo-excited electrons and therefore were destabilized. These findings may be relevant not only to anchored flavins in general but to other functionalized photocatalysts, and may open up new vistas in the implementation of sensitizers in PC systems.
1. Introduction
The photocatalytic decontamination of air, water and surfaces has been the subject of thousands of academic and technological works.1,2 Based on its high activity, non-toxicity, cost-effectivity and earth abundancy, titanium dioxide (TiO2) is considered to be the photocatalyst of choice. Nevertheless, this material has a large bandgap (3.0–3.2 eV), which limits its photocatalytic (PC) activity to the UV part of the spectrum, making the use of solar light quite impractical. During the last two decades, therefore, an intense effort has been dedicated to extend the PC activity of TiO2 to the visible part of the spectrum. This was achieved mostly by doping with metallic3,4 and non-metallic5,6 dopants, but also by promoting oxygen vacancies7 and by surface modification.8A complementary approach to the use of dopants is to anchor sensitizing molecules on the photocatalyst surface. In this instance, the concept is based on absorption of visible light by the sensitizer, and the transport of charge from the sensitizing molecule to the inorganic semiconductor (simultaneously serving to reduce the rate of carrier recombination in the semiconductor phase). The surface chemical properties of the semiconductor in terms of substrate (e.g., pollutant) adsorption and the formation kinetics of reactive oxygen species are also exploited. This concept should not be misconstrued with the use of dyes as self-degrading sensitizers to evaluate the visible-light activity of new photocatalyst material candidates. Such evaluation is inadequate as pointed out recently.9,10
Synthetic flavins, attached to bismuth oxychloride semiconductor surfaces, were used by us as sensitizers for the photodegradation of salicylic acid.11 In what follows, we examine the PC performance of a synthetic flavin attached to TiO2 from the perspective of its photostability, as stability issues are of utmost importance in the use of sensitizers for the PC degradation of environmental pollutants.
Flavins are molecules containing a three-ring isoalloxazine motif. This motif is very common in a variety of biologically active molecules such as riboflavin (RF, vitamin B2), flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD).12 Generically, natural flavins are characterized by side-groups attached to the isoalloxazine motif at positions N10 and lack of side-groups attached at position N3 (Fig. 1). As a consequence, the N3 imide is acidic, and as such might contribute to intra-molecular proton transfer events leading to irreversible reduction.13 In contrast, synthetic flavins, such as the one used in this study, are immune to such deleterious pathways.
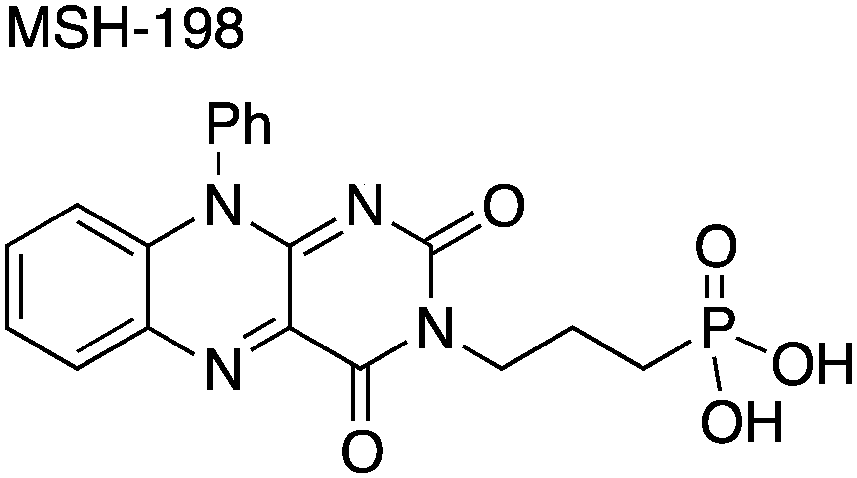 | ||
| Fig. 1 The chemical structure of the synthetic flavin, MSH-198. | ||
Ethanol was chosen as our model “pollutant” in this study based on the extensive literature that exists on its PC degradation mechanism, both in the liquid14 and in the gas phase containing both pristine TiO2 and TiO2 loaded with metallic catalyst islands serving as electron sinks.14,15
Specifically, we present findings related to the remarkable self-stabilization of the surface-confined flavin molecules under irradiation with visible light. This self-stabilization occurs irrespective of whether a reducing agent (such as ethanol) is present in the adjoining phase contacting the oxide semiconductor surface.
2. Experimental section
2.1. Flavin synthesis
The synthetic MSH-198 flavin used in this study is shown in Fig. 1. It is an isoalloxazine derivative, which lacks the parent methyl substituents at C7 and C8 ring positions in natural flavins, has an aromatic phenyl substituent at position N10, and bears a three-carbon aliphatic linker chain and a phosphonic acid anchor group attached at N3 position. The compound was prepared by modifying earlier work.16 Synthetic details of the preparation of this compound appears are contained in the ESI.†2.2. Preparation of TiO2/flavin composite
In a typical synthesis, P25 TiO2/MSH-198 composite particles (hereafter designated simply as: P25/MSH-198) were prepared by adding 20 ml of 1 × 10−4 M flavin solution (made up from methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water 0.01:1 v/v) to 300 mg of the commercially available (Degussa P25) TiO2 particles and stirring the mixture for 2 h in the dark. The resultant composite was filtered, washed with ethanol and deionized water to remove unattached molecules. The material was then dried in air at 60 °C.
:water 0.01:1 v/v) to 300 mg of the commercially available (Degussa P25) TiO2 particles and stirring the mixture for 2 h in the dark. The resultant composite was filtered, washed with ethanol and deionized water to remove unattached molecules. The material was then dried in air at 60 °C.
2.3. Characterization
Chemisorption of flavins on the TiO2 surface was confirmed by Fourier transform infrared (FTIR) transmission spectroscopy (Model Vertex 70v, Bruker) of P25 TiO2 thin films on which MSH-198 was chemisorbed. To this end, thin films of transparent TiO2 were coated on a ZnSe substrate. Coating was performed by spin-coating of a precursor Ti(OPr)4 acetylacetonate complex prepared according to published procedure.17 The flavin, MSH-198, was directly tethered on the TiO2 layer by 2 h immersion of the oxide film in a solution (made of methanol:water 0.01:1 v/v) containing 1 × 10−4 M solution of the flavin. The chemically-modified samples were washed with ethanol and water and dried in an air oven.
Diffuse reflectance spectra of the P25/MSH-198 particles were taken by a Shimadzu UV2600 spectrophotometer, equipped with an integrating sphere. Solid state 121.8 MHz 31P magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy was carried out on powdered samples on a 300 MHz AVANCE III (Bruker) solid-state NMR spectrometer equipped with triple-resonance probes using 4 mm (outer diameter) zirconia rotors. 31P cross polarization (CP) MAS echo experiments (indirect excitation) were carried with a 10.0 μs π pulse width, an echo interval τ (200 μs) identical to the rotational period TR (rotor spin rate: 5000 Hz), with a 1H decoupling level of 100 kHz and 5.0 μs π/2 pulse. Hartmann–Hahn radio-frequency levels were matched at 50 kHz, with 2 ms contact time. Relaxation delay was 3 s and up to 16k transients were collected. Direct excitation (DE) MAS echo experiments were carried out employing the same pulse parameters, however, now with a repetition delay of 60 s.
2.4. Photocatalysis measurements
The photocatalytic (PC) oxidation kinetics measurements of ethanol using the various types of particles were performed in a stainless steel reactor of 83 ml capacity, designed in a manner that enabled to locate the samples within the compartment of a Bruker IFS55 FTIR instrument (Fig. 2). In a typical experiment, 20 mg of the oxide catalyst were spread uniformly over a 0.79 cm2 area of a custom-built photocatalyst holder, placed at the bottom of the reactor (Fig. 2), thus yielding optically thick conditions. A 2 μL aliquot of ethanol was injected into the reactor by a microsyringe through a septum. Prior to illumination, the reaction system was kept in the dark for 2 h to ensure adsorption–desorption equilibrium on the photocatalyst surface. Various types of experiments were carried out, all of which utilized well-defined light-emitting diode (LED) sources (LED-R Ltd): a 365 nm (full width at half maximum, FWHM, of emission band: 10 nm) LED and a 435 nm (FWHM 15 nm) LED. The output of the LEDs, in terms of the photon flux, could be tuned at will. Accordingly, the photon flux impinging on the photocatalyst surfaces ranged from 1.1 × 1015 photons per s per cm2 to 7.85 × 1015 photons per s per cm2. Whenever a comparison between the behaviors at different wavelengths was sought, extreme care was taken to maintain the same photon flux rather than the same energy flux.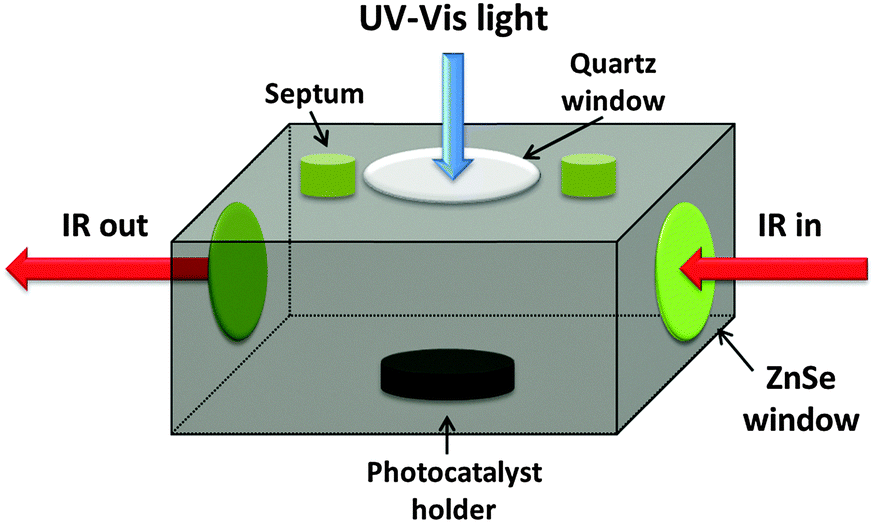 | ||
| Fig. 2 The in situ photocatalytic reactor custom designed for this study. | ||
3. Results
3.1. Chemisorption of MSH-198 on the TiO2 surface
Fig. 3B presents the FTIR spectrum of MSH-198 (as measured in a KBr pellet) as well the spectrum of MSH-198 attached to TiO2 thin film supported on ZnSe (Fig. 3C). Generally speaking, the spectrum of the MSH-198 can be divided into isoalloxazine-related peaks, linker-related peaks and anchoring (phosphonic acid) group-related peaks. The vibrational spectrum of N10-derivatized isoalloxazine has been studied thoroughly in the literature. Although tethering at the N3 position slightly shifts some of the peaks, it is still possible to rather easily assign most of the peaks in the powdered MSH-198 spectrum, according to published assignments.18–20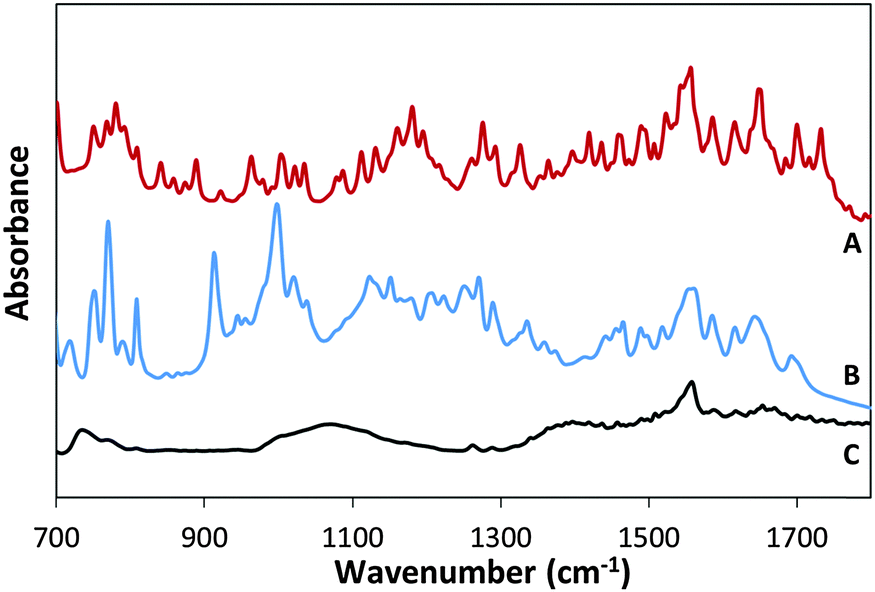 | ||
| Fig. 3 FT-IR spectra of synthetic flavin derivative (A) MSH-197, (B) MSH-198 and (C) P-25/MSH-198. | ||
This statement is bolstered by comparing the spectrum with that of a similar synthetic flavin, (denoted here as MSH-197, see Fig. 3A), that differs from the MSH-198 only by its anchor group, i.e., it has a carboxylic acid as an anchor group instead of phosphonic acid. The spectra of the two flavins were very similar. However, peaks at 914 cm−1, 999 cm−1, 1207 cm−1 and 1225 cm−1 appeared in the MSH-198 spectrum but not in that of MSH-197. The 914 cm−1 and 999 cm−1 features can be assigned to P–OH whereas the 1207 cm−1 and the 1225 cm−1 peaks represent P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties.21
O moieties.21
Upon chemisorption (Trace C, Fig. 3), both the P–OH-related peaks and the PO-related peaks disappeared, and instead, a broad (974–1232 cm−1) peak appeared, centered at 1076 cm−1. A similar peak was observed in crystallized α-zirconium phosphate as well as in phenyl phosphonic acid adsorbed on zirconia, and was interpreted as indicating tridentate adsorption symmetry.22 This scenario applies in the adsorption of MSH-198 as well. It is worth noting that bidentate chemisorption is unlikely since the doublet below 970 cm−1, known to be a characteristic of phosphonic acid adsorption on TiO2 through a bridging bidentate mode,23 was not observed here in the P25/MSH-198 spectrum. Another hallmark of chemisorption, was a reduction in the intensity of almost all isoalloxazine – related peaks relative to the intensity of the 1564 cm−1 peak (assigned as C–N stretch coupled with the C–C stretch mode). Other relatively pronounced peaks were observed at 1668 cm−1 and 1672 cm−1 (assigned to C(4)O and C(2)O, respectively24) as well as a strong peak at 740 cm−1.
The SEM and TEM images of the P25 particles were taken prior to and following the chemisorption of the flavins, in order to assure that the flavins do not form chunks on the surface (see ESI†). The images of the pre- and post-adsorption particles were found to be identical, i.e. there was no evidence for three dimensional structures of flavins.
The 31P NMR spectra of MSH-198 prior to and after adsorption on P25 TiO2 are presented in Fig. 4 (A and B, respectively). The reported signal is relative to 85% H3PO4. At room temperature, the P peak of the chemisorbed MSH-198 was found to be shifted relative to that of free MSH-198 from 30.4 ppm to 29.08 ppm, indicating chemisorption. The P lines of the chemisorbed molecule were also broader. This may indicate that at room temperature, free rotation of the phosphonic groups was hindered on chemiadsorption on the TiO2 surface. Similar trends were observed by previous authors in the chemisorption of phenyl phosphonic acid.25
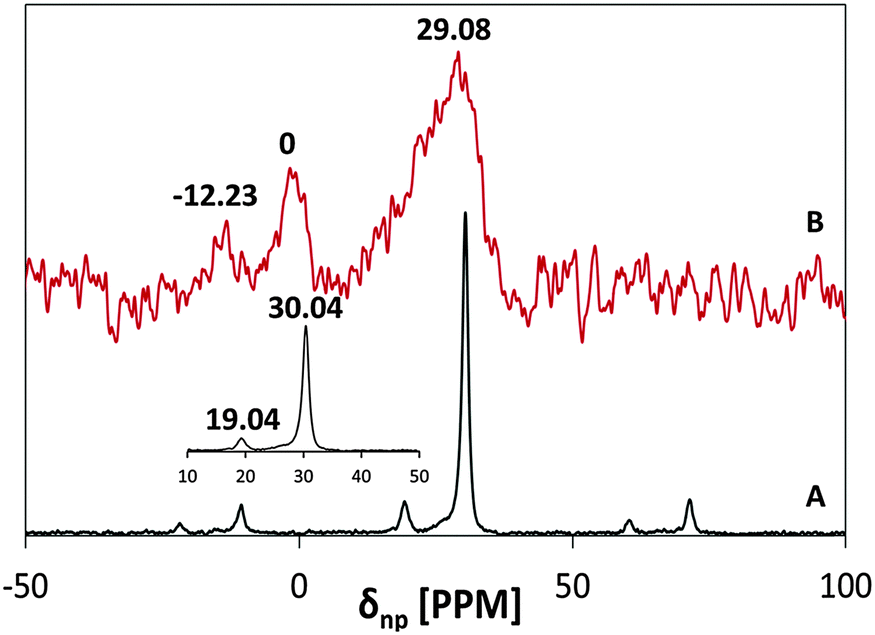 | ||
| Fig. 4 31P MAS NMR spectra of: (A) free MSH-198 and (B) P25/MSH-198. | ||
The spectral changes upon adsorption were followed by performing UV-vis diffuse reflectance spectroscopy measurements on P-25, MSH-198 and P-25/MSH-198; Fig. 5 shows the corresponding spectra. The spectrum of the free (pristine) MSH-198 was characterized by high molar extinction coefficients, typical of π–π* type transitions. It comprised of a shoulder at 235 nm and well-resolved peaks at 260 nm, 350 nm and 437 nm. This set of UV-vis peaks was very similar to the peaks in riboflavin26 corresponding to the vibronic transitions: S0 → S4, S0 → S3, S0 → S2, S0 → S1, respectively. A hump between 420 nm and nm was clearly observed for the P25/MSH-198 sample, reflecting chemisorption of MSH-198 on the oxide surface. It is noteworthy that a shoulder that was located at 490 nm in the spectrum of the free flavin disappeared upon chemisorption. Such a feature was not observed in the spectrum of free natural flavins such as riboflavin.26
 | ||
| Fig. 5 UV-diffuse reflectance spectra of (A) P-25 TiO2 (B) P-25/198 (C) MSH-198. | ||
3.2. Photocatalysis measurements
The photocatalytic (PC) activity of P25/MSH-198 was investigated by following the degradation kinetics of ethanol in air using in situ FTIR (Fig. 6). The photooxidation of ethanol was characterized by the gradual disappearance of C–OH characteristic peaks at 1055 cm−1 and 1230 cm−1 as well as C–H (s, a) stretching modes at 2899 cm−1 and 2967 cm−1 respectively. In parallel, absorption at ∼2750 cm−1 and 2700 cm−1, usually assigned to the C–H stretching mode in aldehydes,27 was observed. The increase was also correlated with the appearance of a strong peak at 1740 cm−1 assigned to CO stretch in aldehydes. A constant growth in the sharp rotational-vibrational peaks assigned to water vapor (1400–1700 cm−1), and in the CO2-related peaks at 2357 cm−1 (stretching), 2335 cm−1 (stretching) and 670 cm−1 (bending) completes the description of the in situ FTIR features accompanying photocatalysis. Based on these observations, it can be concluded that PC degradation of ethanol involves the formation of acetaldehyde (CH3–CHO) as an intermediate, leading to complete mineralization.28 While mineralization continues through deeper photooxidation of acetaldehyde to acetic acid, no acetic acid was observed in this study, except for minute amounts under high light intensity. This can be explained by strong adsorption of acetic acid and acetate ions on the TiO2 surface.29
 | ||
| Fig. 6 Changes in the FTIR spectrum of ethanol during its PC degradation with P25/MSH-198 (A) prior to light exposure, (B) following 60 min exposure (C) following 120 min exposure. | ||
Fig. 7 presents ethanol photodegradation kinetics data for P25 TiO2 and for P25/MSH-198 culled from the FTIR results. The ordinate data were scaled using the amplitude of the 1055 cm−1 peak (C–O stretching) of ethanol (see Fig. 6, for example). Also shown in Fig. 7 are the kinetics data under nitrogen environment, i.e., in the absence of dioxygen. Blank experiments, i.e., in the absence of a photocatalyst, did not reveal a significant decrease in ethanol concentration. A considerable increase in the ethanol photodegradation rate of ethanol upon using P25/198 instead of P25 TiO2 is clearly seen in these data. In the absence of dioxygen, the degradation rate was hindered and the rate with flavin particles was equal to that with pristine P25 TiO2 (Traces A and B, Fig. 7). In all the cases, an apparent first order kinetics model could be fit to the data. Based on comparison of the first-order rate constants, the PC enhancement factor on using flavinated particles ranged between 1.4 and 2.0.
 | ||
| Fig. 7 Kinetics of ethanol PC degradation under 365 nm light with (A) P-25, nitrogen environment, (B) P-25/198, nitrogen environment, (C) P-25 in air (D) P-25/198 in air. | ||
The rate constant for ethanol degradation was also measured as a function of light intensity in the range: 100–600 μW cm−2. Here again, the rate constant was calculated based on changes in the 1055 cm−1 peak. As expected, increasing light intensity had a positive effect on the degradation rate. Plotting the rate constants on a log–log scale revealed that the dependence of the degradation rate constant on the intensity had 0.46 order (Fig. 8). This value may be compared with previous publications claiming first order kinetics behavior at low intensities going down to 0.5 order at high intensities,30 and with a measured31 value for the kinetics order of 0.64 for ethanol degradation on Hombifine TiO2 under 0.05 to 1.4 × 10−7 E s−1 cm−2 light flux.
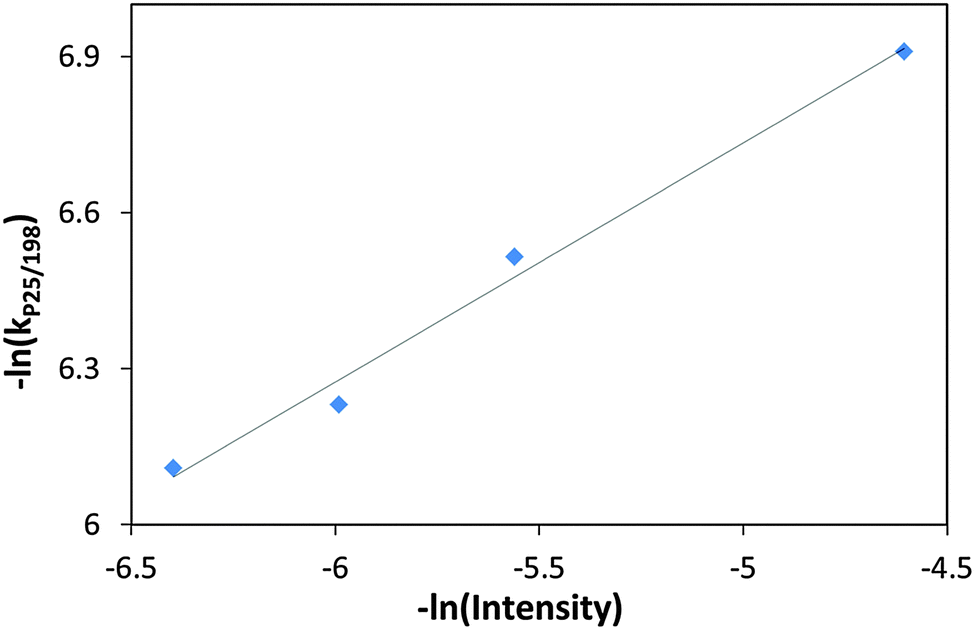 | ||
| Fig. 8 (−ln) of the rate constant with P25/MSH-198 versus (−ln) of the UV (365 nm) intensity during the PC degradation of ethanol. | ||
3.3. Photostability of oxide-anchored flavin
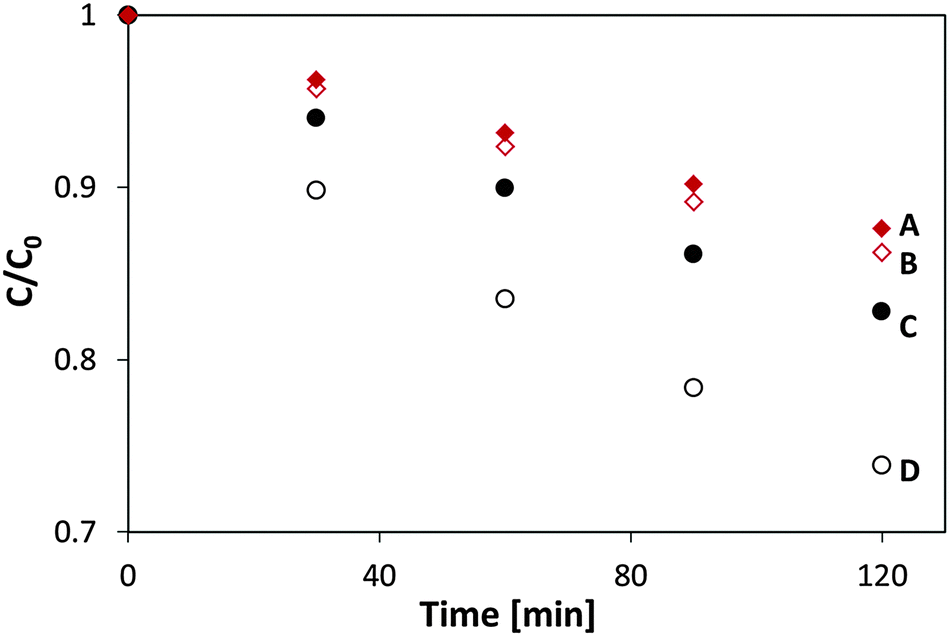
The stability of the MSH-198 molecules overcoating the P25 TiO2 particles was tested by repeating the ethanol PC degradation measurements and comparing the observed kinetics behavior with corresponding measurements on pristine P25 TiO2. The decrease in the rate of ethanol degradation upon repeating the UV-induced photocatalytic tests was found to be minute. This is clearly demonstrated by presenting the (normalized) ratio of the PC rate constant with P25/MSH-198 to that with P25 TiO2, as a function of reaction cycle (Fig. 9). From the figure, the positive kinetics effect achieved by flavinating the photocatalyst particles was clearly seen to be maintained under successive repetitions. This indicates that the synthetic flavin molecules attached to the P25 TiO2 particles were not photodegraded while serving to increase the PC activity of the native TiO2 particles via chemical modification of their surfaces. | ||
| Fig. 9 The normalized rate constant ratio for PC degradation for P25/MSH-198 compared with P25 TiO2 (see text) as a function of the number of repeat reaction cycles with the same set of samples. All measurements were performed under exposure to UV (365 nm) LED light. | ||
That the attached flavins were hardly damaged during the long (750 min) exposure to the UV light can be also deduced by measuring the UV-vis diffuse reflection spectrum of the particles prior to and after five repeat cycles of exposure. As presented in Fig. 10, the signature “hump” at 400 nm–500 nm in the spectrum of the flavinated particles measured prior to exposure, remained also after exposure to UV light for 750 min. For comparison, the spectrum of pristine P25 particles is given as well in Fig. 10.
 | ||
| Fig. 10 UV-diffuse reflectance spectra of (A) P25 TiO2, (B) P25/MSH-198 and (C) P25/MSH-198 after 5 cycles of PC degradation of ethanol under 365 nm UV LED irradiation. | ||
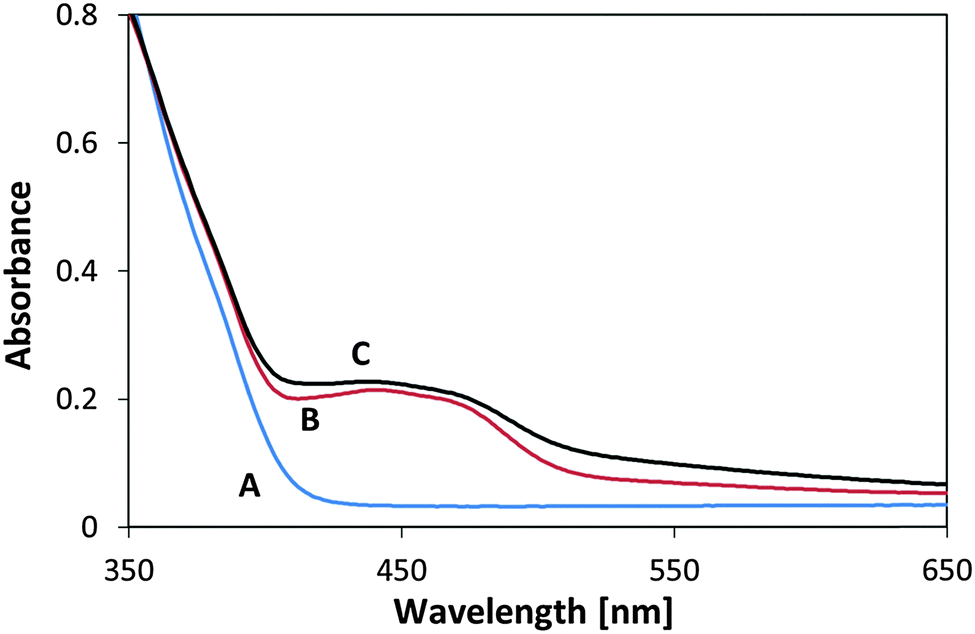
To further test the photostability of the flavinated P25 TiO2/MSH-198 particles, they were exposed to UV light under ambient conditions and their diffuse reflection UV-vis spectrum was measured following exposure. In this case no reducing agent (such as ethanol) were introduced into the system. Two light sources were used: a UV 365 nm LED and 435 nm LED. In both cases the same photon flux (1.1 × 1015 photons per sec per cm2) and the same time of exposure (2 h) was used. As presented in Fig. 11, a clear change in the spectrum upon exposure to visible light was observed, manifested by the disappearance of the 400–500 “hump” in the spectrum. In contrast, the spectrum of flavinated particles exposed to UV light was hardly changed. This (enhanced) stability under 365 nm light is far from a trivial observation if one takes into account the facts that the flavin molecules were attached to a UV-active photocatalyst, that UV photons are more energetic than the visible-light photons and that powdered MSH-198 has similar extinction coefficient at 435 nm and 365 nm as shown in Fig. 5C above. It should be noted, however, that the observation that the attached flavins were relatively stable under UV light should not be taken as proof for complete immunity towards long term exposure to UV light.
 | ||
| Fig. 11 UV-vis diffuse reflectance spectra of P25/MSH-198 (A) as synthesized (B) after exposure to UV light (C) following exposure to visible light. | ||
Another set of flavin self-degradation tests was performed on un-attached MSH-198 particles, which were exposed to UV light and to visible light under the same conditions prevailing in the measurements of Fig. 11 (i.e., flux of 1.1 × 1015 photons per s per cm2), exposure time = 2 h. As presented in Fig. 12, upon exposure, a decrease in the whole absorption envelope was observed. Further, several changes in the relative intensities and in the peak locations were observed.
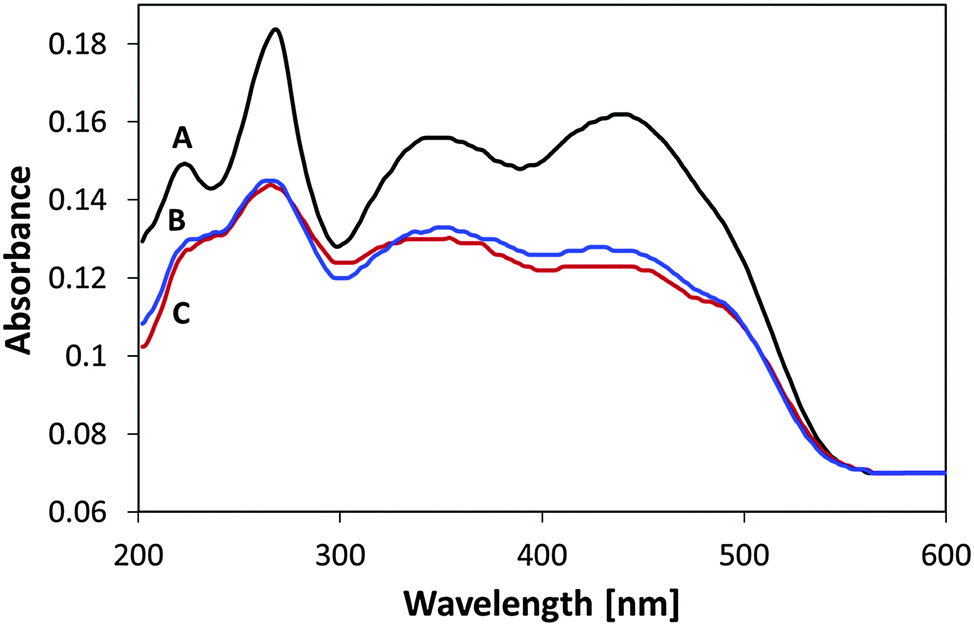 | ||
| Fig. 12 UV-vis diffuse reflectance spectra of MSH-198 (A) as-synthesized, (B) following exposure to UV light, (C) following exposure to visible light. | ||
The decrease in the 438 nm peak was more pronounced than the decrease in the 349 nm peak. The decrease was accompanied by a red shift of the peak to 427 nm. In parallel, the 268 nm peak was slightly blue-shifted to 265 nm, and the well-resolved peak at 222 nm became upon exposure, no more than a shoulder, manifesting a broadening of the 265 nm peak. Overall, MSH-198 that was exposed to light behaved quite similarly under UV light and under visible light (except for a very small change in the location of the 349 nm peak, which was shifted to 345 nm under visible light only). The observation that MSH-198 had the same changes in its spectrum regardless of the exposure wavelength is quite in contrast to its behavior once chemisorbed on the TiO2 particles (Fig. 11). This very important difference is discussed in the Discussion section below.
It is worth noting that the effect of light exposure on the flavin spectrum was studied before with an aqueous solution of riboflavin (RF), where the main photoproduct was claimed to be lumichrome (LC).32 A comparison between the MSH-198 spectrum following exposure to that of post-illuminated RF reveals some similarities (decrease in the 438 nm peak, blue-shift in the 349 nm peak etc.). Nonetheless, the 222 nm peak in LC is quite strong relative to the other peaks, which is in contrast to our case here where the 222 nm peak lost its intensity (Fig. 12).
4. Discussion
4.1. Photostability of flavins: general
The photochemical reactions of flavins have been studied quite thoroughly, in particular, in natural flavins such as FAD, FMN and RF.26 Generically the degradation pathways can be divided into three categories: photoreduction, photodealkylation and photoaddition. Intermolecular photoreduction occurs under anaerobic conditions in the presence of amino acids, α-hydroxycarboxylic acids, thiols, aldehydes and unsaturated hydrocarbons, leading to the formation of 1,5-dihydroflavins or its alkyl derivatives. Flavin reduction is usually not observed spectrally under ambient conditions due to re-oxidation upon exposure to air.33 In contrast, intramolecular photoreduction of natural flavins involves dehydrogenation of the side-chain at N10 position to yield a variety of ketonic or aldehydic functions in the side chain with or without loss of side-chains carbon atoms.34 The rate of intramolecular photoreduction was found to be very sensitive to the polarity of the solvent, as this altered conformation of the side chain.Photodealkylation is also an intramolecular process, involving, in natural flavins, synchronous breakage of the N10–C1′ and C2′–H bonds. The excited singlet state is considered to be the major intermediate, yet there are cases, in particular under low pH, where the dealkylation process goes through a mediated triplet state. Photoaddition of ROH molecules was reported to occur under conditions where the ROH is the solvent. In that case the solvent residue adds to the flavin molecule at positions C6 or C9.35
In the presence of dioxygen, illuminated flavins may catalyze the oxidation of many substrates such as amino acids, proteins, nucleotides and lipids. One mechanism is direct oxidation by electron abstraction from the substrate. In that case, the reduced flavins are often recycled by re-oxidation. A second mechanism, which operates at high concentration of dioxygen, is based on energy transfer from excited flavin in its triplet state to a ground (triplet) molecular oxygen yielding an excited (singlet) oxygen molecule. The latter is capable of attacking a large variety of organic molecules.
4.2. Mechanism of photocatalytic activity enhancement and self-stabilization mechanism
In terms of the data presented above on oxide/synthetic flavin/electrolyte interfaces, any mechanistic discussion has to explain three facts: (a) the remarkable stability of the flavinated particles during exposure to UV light, regardless of the presence of ethanol or not. (b) The instability of the flavinated particles under visible light, as opposed to their stability under UV light. (c) The faster kinetics for ethanol degradation with the flavinated particles under UV light in comparison with pristine P25 TiO2. Recall also from a variety of evidences presented earlier, the adsorbed MSH-198 forms strong interaction with the TiO2 surface.While the ensuing discussion is based on thermodynamics, i.e., on the location of the relevant energy bands and levels, one should bear in mind that the actual measurements reflect not only thermodynamics but also kinetics. To explain the thermodynamics, one has to refer to the energy levels scheme, presented in Fig. 13. The location of the valence band (Ev) of TiO2 is known to be 2.75 versus SHE at pH 036 and is shifted negatively (upward) by 59 mV per pH unit. The redox level of oxygen (O2 + e → O2−) can be taken as −0.16 V versus SHE (for a standard state of 1 M O2)37 and is pH independent. The location of the relevant energy levels of the MSH-198 is more obscure. Nevertheless, values can still be deduced as follows.
 | ||
| Fig. 13 The thermodynamics scheme for ethanol photocatalytic degradation on P25 TiO2/-MSH-198 particles at pH 7: (A) under UV light (B) under visible light. | ||
The linear correlation between the electrochemical E1/2 of an organic molecule (which is a good approximation for the redox potential) and its ionization potential [E1/2 = 0.89 (IP)–6.04]38 facilitates location of its HOMO level. For our case, at pH 7, the E1/2 values of the flavin lies around −0.1 V to −0.25 V SHE.39,40 The E1/2 of synthetic flavins, having functional groups at their N3 position is slightly more negative; this gives an estimated IP of 6.5–6.7 eV. Taking the SHE level at 4.45 V relative to vacuum41 yields a value of 2.05 V SHE for the HOMO level of the flavin at pH 7.
Once the HOMO level (S0) is estimated, locations of the singlet excited states S1, S2, S3 can be deduced based on the absorption spectra of the flavin (Fig. 5). Here, the S0 → S1, S0 → S2, S0 → S3, S0 → S4 transitions correspond to 2.83 eV, 3.54 eV, 4.77 eV and 5.25 eV, respectively. Accordingly, the excited states S1, S2, S3, S4 at a pH of 7 can be pinpointed at −0.78 V, −1.49 V, −2.72 V, and −3.2 V, respectively vs. SHE. At the same pH, the valence band of TiO2 is at 2.35 V vs. SHE and the conduction band is at −0.85 V SHE. Therefore, from a thermodynamics point of view, flavin molecules excited to the S2 state can inject electrons to the TiO2 phase, whereas flavins excited to the S1 state are incapable of injecting electrons to the semiconductor.
As described in the preceding paragraphs, the self-photodegradation mechanism of flavins hinges on the flavins being in an electronically-excited state. For intramolecular photoreduction to occur, there is no need for S2 excitation and in fact photodegradation processes may occur (upon intersystem crossing) at the triplet state, which is considerably lower in energy than the S1 state. Hence, similar extinction coefficients under 360 nm and 450 nm tend to be manifested as similar reaction cross sections in un-attached flavins, as indeed found in this study.
This indifference to the exposure wavelength is altered upon anchoring the flavin molecules to the TiO2 particles. Under UV light, electrons excited to the S2 level may be easily transferred to the semiconductor phase, thus hindering intramolecular rearrangement. In contrast, under visible light, the photogenerated electrons are excited to the S1 level. Since this level is lower in energy than the conduction band of TiO2, it is unlikely that these electrons are transferred to the semiconductor phase. As a consequence, under visible light, anchoring of the flavin molecules is expected not to have any benevolent effect on their photostability. The same explanation holds also in the presence of ethanol. As is well known, ethanol can be degraded both by direct injection of electrons (i.e., hole injection to the alcohol) or indirectly, by OH radicals formed on the surface of the photocatalyst.15 The rate constant of the photocatalytic degradation of ethanol on pristine TiO2 under UV light is quite high, reflecting the redox potential of ethanol [1.145 V]42 and its high proclivity to adsorption on the polar oxide. It is reasonable to expect that anchoring of the flavin molecules on the oxide surface would reduce the number of adsorbed ethanol molecules, leading to a decrease in the oxidation rate of ethanol by this mechanism.
Yet, the opposite trend was observed (Fig. 7) and the rate of degradation of ethanol with P25 TiO2/MSH-198 was found to be higher than for pristine P25 TiO2. It is proposed that the observed enhancement in the degradation rate stems from the injection of photogenerated electrons from the excited flavin molecule to the semiconductor phase, which in turn facilitates oxidation of ethanol by electron transfer to the flavin radical cation (see Fig. 13). Such electron transfer replenishes the flavin cation with a ground state electron, contributing to its stability.
In this context, a report that the presence of methanol facilitated the reduction of halogenated ethylenes43 can be viewed as if the presence of chlorinated ethylenes assisted in the oxidation of the alcohol molecules. Along this line, the TiO2 support phase in the P25 TiO2/MSH-198 assembly plays the same role that the chlorinated compounds played in the reported methanol experiments, namely as a sink for excited electrons. That charge transport between flavins and TiO2 is very efficient can be deduced also from the ease by which FAD was reduced electrochemically in a layered TiO2–FAD structure.44
While the faster rate of ethanol degradation was explained above by thermodynamics, i.e., by the locations of the energy levels, one should take into consideration also the contribution of kinetics. In this context, our FTIR measurements indicate strong interaction between the MSH-198 and the P25 phases via the formation of tridentate P–O–Ti interactions. This strong interaction may contribute to facile charge transport between the MSH-198 phase and the oxide. Ultrafast spectroscopy would address this possibility and such experiments are planned as a follow-up to this study.
From the energy diagram in Fig. 13, it is clear that reduction of O2 by excited flavin molecules is also thermodynamically possible. However, this by itself does not imply that the process indeed occurs, due to the presence of a kinetic barrier. This kinetic barrier is eliminated if reduction takes place on the oxide surface, where dioxygen is easily adsorbed. It should be noted that fast “trapping” of electrons during dioxygen reduction is crucial not only because it reduces the rate of recombination at the interface but also since superoxides (or other ROS), formed by this process, may play a dominant role in the more advanced steps of ethanol degradation (i.e., from acetaldehyde to carbon dioxide and water).45
Photoexcited flavin cations are known to react with alcohols in a photoaddition manner that yields hydroxyl- or alkoxy-dihydroflavins.35 Our data indicate that such addition reaction is irrelevant for the anchored MSH-198. First, this mechanism is relevant only to excited flavin cations. In our case, to obtain an excited flavin cation might require the absorption of more than one photon, which is quite unlikely under the experimental conditions. Second, if this was the situation, one could have expected the enhancement effect of the anchored flavin to eventually disappear as more and more ethanol molecules are added to the solution. In contrast, the degradation rate with flavinated photocatalyst remained constant upon performing the experiments five times sequentially. Moreover, the distribution of intermediates and the ratio between the rate of CO2 release versus the rate of ethanol consumption do not support the notion of a photoaddition pathway.
Previous works reported that upon illumination, FMN dissolved in methanol, undergoes two-electron, two-proton reduction to FMNH2, while the methanol is oxidized to formaldehyde,43 thus describing a direct route for alcohol degradation in the absence of a semiconductor. Such direct mechanism of alcohol oxidation upon light absorption by the flavin compound was not observed by us with unattached MSH-198. The lack of evidence for direct oxidation of ethanol by unattached flavins may be attributed to the lower concentration of the alcohol in our gas phase studies in comparison with the liquid phase studies of the former report.
A different scenario is based on TiO2 excitation and transport of electrons from the conduction band of the photocatalyst to the ground state of the anchored flavin. Here, the large number of ways by which flavins can accommodate charge may assist in stabilizing the flavin molecules. This property may have to do with the large extent of delocalization within the tricyclic structure together with the large number of amine groups that may accept H+ reversibly. Therefore, one may envisage a situation where, over time, electrons are accumulated on the attached flavin molecules, thus minimizing recombination of holes generated at the photocatalyst.
Continuing with this line of thought, one could envision a two-photon scenario for the P25 TiO2/MSH-198 assembly that is very similar to the Z-scheme in the coupled photosystem I/photosystem II scheme. Generally speaking, the number of artificial Z-scheme photocatalytic systems that were reported so far is not large. Most of the reports have focused on photocatalytic water splitting for hydrogen production.46,47 Only a handful of reports are on the PC degradation of pollutants.48 Within the context of our observations, the Z-scheme mechanism means the absorption of one photon by the TiO2 photocatalyst and a second photon by the flavin molecule. The photon that is absorbed by the photocatalyst is responsible for initiating the oxidation of the ethanol (both directly and indirectly). The second photon, exciting an electron to an excited state in the flavin molecule, facilitates fast charge transport from the conduction band of the TiO2 particle to the empty ground level of the flavin and at the same time reduces dioxygen to ROS. Fast injection of electrons to the ground state of the flavin molecule is critical to its stabilization in the absence of ethanol. Clearly, such “self-stabilization” is impossible under visible light as the energy of visible light photons is insufficient for photoexciting the oxide component.
At first glance, the Z-scheme mechanism seems to go hand in hand with the kinetics data on the degradation of ethanol. Yet, a second look on the data negates this scenario. The number of photons absorbed by the photocatalyst is approximately 1 × 1015 per cm2 per s. Taking the diameter of one particle as 0.1 micrometer and the absorption by a thickness of 0.1 micrometer as 10% gives that the number of photons absorbed in one particle within a second is approximately 104. Hence, the time between two successive absorption events is, on the average, 10−4 s. This time is significantly larger than the radiative life time in the lowest excited singlet state, which was estimated as 12 ns.49 Hence, the probability for synchronous absorption of two photons, one by TiO2 and a second by a flavin molecule attached to this particle is practically zero. Moreover, if the Z-scheme scenario was dominant, one could have expected a second order dependence of the rate constant on the light intensity due to the requirement of synchronous absorption of two photons. In contrast, a square root dependence was observed (Fig. 8).
5. Conclusions
Anchoring of a synthetic flavin to the TiO2 surface increased the photocatalytic degradation kinetics of ethanol under UV light, and at the same time stabilized the anchored flavin molecules against photodegradation. Under UV light, this stability effect was found also in the absence of a sacrificial agent (e.g., ethanol), whereas visible light was found to be quite harmful to the anchored flavin molecules. These rather surprising observations were attributed by us to the propensity for charge transport from excited flavin molecules to the semiconductor and to the role that such transport may play in stabilizing the system. Per this reasoning, anchored flavins that were excited by UV light to their S2, S3 states were able to transfer the excited electrons to the oxide phase whereas anchored flavins that were excited by visible light to their S1 state were less likely to transfer the excited electron and hence were destabilized. These findings may be relevant not only to anchored flavins but to other functionalized photocatalysts, and may assist in the implementation of sensitizers in photocatalytic systems.Acknowledgements
This project was supported by the BSF-NSF program (CHE-1303803). The support of the P. and E. Nathan Research Fund and the Russel Berrie Nanotechnology Institute at the Technion is gratefully acknowledged. We thank the Shimadzu Center for Advanced Analytical Chemistry (University of Texas at Arlington) for mass spectrometry analysis.Notes and references
- Advances in Chemical Engineering, ed. H. D. Lasa and B. S. Rosales, 2009, vol. 36 Search PubMed.
- Y. Paz, Appl. Catal., B, 2010, 99, 448–460 CrossRef CAS.
- S. Kim, S. J. Hwang and W. Choi, J. Phys. Chem. B, 2005, 109, 24260–24267 CrossRef CAS PubMed.
- H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, K. Ikeue and M. Anpo, J. Photochem. Photobiol., A, 2002, 148, 257–261 CrossRef CAS.
- X. Wu, S. Yin, Q. Dong, C. Guo, H. Li, T. Kimura and T. Sato, Appl. Catal., B, 2013, 142–143, 450–457 CrossRef CAS.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- E. M. Samsudin, S. B. Abd-Hamid, J. C. Juan, W. J. Basirun and A. E. Kandjani, Appl. Surf. Sci., 2015, 359, 883–896 CrossRef CAS.
- J. Wen, X. Li, W. Liu, Y. Fang, J. Xie and Y. Xu, Chin. J. Catal., 2015, 36, 2049–2070 CrossRef CAS.
- M. Rochkind, S. Pasternak and Y. Paz, Molecules, 2015, 20, 88–110 CrossRef CAS PubMed.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21–28 CrossRef CAS.
- M. Rochkind, M. Pandiri, M. S. Hossain, F. W. Foss Jr., K. Rajeshwar and Y. Paz, J. Phys. Chem. C DOI:10.1021/acs.jpcc.5b12768.
- C. A. Abbas and A. A. Sibirny, Microbiol. Mol. Biol. Rev., 2011, 75, 321–360 CrossRef CAS PubMed.
- A. Niemz, J. Imbriglio and V. M. Rotello, J. Am. Chem. Soc., 1997, 119, 887–892 CrossRef CAS.
- P. Pichat, Catal. Today, 1994, 19, 313–334 CrossRef CAS.
- Z. Yu and S. S. C. Chuang, J. Catal., 2007, 246, 118–126 CrossRef CAS.
- S. Shinkai, H. Nakao, I. Kuwahara, M. Miyamoto, T. Yamaguchi and O. Manabe, J. Chem. Soc., Perkin Trans. 1, 1988, 313–319 RSC.
- Y. Paz, Z. Luo, L. Rabenberg and A. Heller, J. Mater. Res., 1995, 10, 2842–2848 CrossRef CAS.
- Y. Zheng, J. Dong, B. A. Palfey and P. R. Carey, Biochemistry, 1999, 38, 16727–16732 CrossRef CAS PubMed.
- M. Unno, R. Sano, S. Masuda, T. Ono and S. Yamauchi, J. Phys. Chem. B, 2005, 109, 12620–12626 CrossRef CAS PubMed.
- A. S. Eisenberg and J. P. M. Schelvis, J. Phys. Chem. A, 2008, 112, 6179–6189 CrossRef CAS PubMed.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS.
- J. Randon, P. Blanc and R. Paterson, J. Membr. Sci., 1995, 98, 119–129 CrossRef CAS.
- A. Michelmore, W. Gong, P. Jenkins and J. Ralston, Phys. Chem. Chem. Phys., 2000, 2, 2985–2992 RSC.
- C. Thoing, A. Pfeifer, S. Kakorin and T. Kottke, Phys. Chem. Chem. Phys., 2013, 15, 5916–5926 RSC.
- P. Falaras, I. M. Arabatzis, T. Stergiopoulos, G. Papavassiliou and M. Karagianni, J. Mater. Process. Technol., 2005, 161, 276–281 CrossRef CAS.
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15–39 RSC.
- F. R. Dubois, V. D. Voort, I. Sedman, A. A. Ismail and H. R. Ramaswamy, J. Am. Oil Chem. Soc., 1996, 73, 787 CrossRef.
- J. M. Coronado, S. Kataoka, I. Tejedor-Tejedor and M. A. Anderson, J. Catal., 2003, 219, 219–230 CrossRef CAS.
- D. V. Kozlov, E. A. Paukshtis and E. N. Savinov, Appl. Catal., B, 2000, 24, 7–12 CrossRef.
- R. Terzian and N. Serpone, J. Photochem. Photobiol., A, 1995, 89, 163–175 CrossRef CAS.
- A. V. Vorontsov and V. P. Dubovitskaya, J. Catal., 2004, 221, 102–109 CrossRef CAS.
- V. I. Birss, S. Guha-Thakurta, C. E. McGarvey, S. Quach and P. Vanysek, J. Electroanal. Chem., 1997, 423, 13–21 CrossRef CAS.
- V. Massey, G. Palmer and D. Ballow, in ‘Oxidases and related redox systems’, ed. T. E. King, H. S. Mason and M. Morrison, University Park Press, Baltimore, 1973, p. 25 Search PubMed.
- W. L. Cairns and D. E. Metzler, J. Am. Chem. Soc., 1971, 93, 2772–2777 CrossRef CAS PubMed.
- G. Schollhammer and P. Hemmerich, Eur. J. Biochem., 1974, 44, 561–577 CrossRef.
- S. Pasternak and Y. Paz, ChemPhysChem, 2013, 14, 2059–2070 CrossRef CAS PubMed.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- L. L. Miller, G. D. Nordblom and E. A. Maybda, J. Org. Chem., 1972, 37, 916–918 CrossRef CAS.
- H. Shinohara, M. Gratzel, N. Vlachopoulos and M. Aizawa, Bioelectrochem. Bioenerg., 1991, 26, 307–320 CrossRef CAS.
- L. Gorton and G. Johansson, J. Electroanal. Chem., 1980, 113, 151–158 CrossRef CAS.
- W. N. Hansen and G. J. Hansen, Phys. Rev. A: At., Mol., Opt. Phys., 1987, 36, 1396–1402 CrossRef CAS.
- T. Iwasita, The Electrocatalysis of Ethanol Oxidation, 3rd LAMNET Workshop – Brazil, 2002, pp. 2–4.
- C. E. Ciptadjaya, W. Guo, J. Angeli and S. Obare, Environ. Sci. Technol., 2009, 43, 1591–1597 CrossRef CAS PubMed.
- E. V. Milsom, H. R. Perrott, L. M. Peter and F. Marken, Langmuir, 2005, 21, 9482–9487 CrossRef CAS PubMed.
- I. Sopyan, M. Watanabe, S. Murasawa, K. Hashimoto and A. Fujishima, J. Photochem. Photobiol., A, 1996, 98, 79–86 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- J. K. Eweg, F. Müller, A. J. W. G. Visser, C. Veeger, D. Bebelaar and J. D. W. Voorst, Photochem. Photobiol., 1979, 30, 463–471 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02060g |
| This journal is © the Owner Societies 2016 |