
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticles in aqueous solutions: influence of size and pH on hydrogen dissociative adsorption and Au(III) ion reduction†
B. G.
Ershov
*,
E. V.
Abkhalimov
 *,
R. D.
Solovov
and
V. I.
Roldughin
*,
R. D.
Solovov
and
V. I.
Roldughin
A. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Science, Leninsky pr. 31-4, Moscow, 119071, Russia. E-mail: ershov@ipc.rssi.ru; abkhalimov@ipc.rssi.ru
First published on 20th April 2016
Abstract
The shift of the localized surface plasmon resonance (LSPR) band of gold nanoparticles to shorter wavelengths upon saturation of the hydrosol with hydrogen is used as a tool to study the electrochemical processes on the particle surface. It is shown that dissociative adsorption of hydrogen takes place on the surface of a particle and results in the migration of a proton into the dispersion medium, while the electron remains on the nanoparticle, i.e., a hydrogen-like nanoelectrode is formed. It is shown that Au(III) ions can be reduced on the gold nanoelectrodes. A thermodynamic scheme explaining the shift of the LSPR band is used to explain the peculiarities of the Au(III) ion reduction. The reduction rate does not depend on the ion concentration and varies linearly with pH. The observed correlations are explained in terms of a simple model of electrochemical processes taking place on the nanoparticle as an electrode. It is shown that with an increase in the particle size, its capacity for dissociative adsorption of hydrogen decreases and the Au(III) reduction slows down.
Introduction
Bulk gold is a chemically inert metal exhibiting no catalytic activity, while nanosized gold (up to 5 nm) deposited on various substrates catalyzes a variety of reactions of organic synthesis.1–4The gold hydrosol shows intensive absorption in the visible region caused by collective excitation of the conduction electrons by light. The optical properties of the hydrosol depend substantially on the size of nanoparticles, density of electrons in them and properties of the dispersion medium and interface.5 It is clear that the change in the electron density (concentration) in the particles results in a shift of the localized surface plasmon resonance (LSPR) band.6–10 The electron density can be changed in many ways, for example, by adsorption of electron acceptors or donors on the nanoparticle surface. Thus, the shift of the plasmon resonance could serve as a tool for the study of the processes taking place at the surface of the nanoparticles. In particular, it was used to study the adsorption of gases on gold nanoparticles in aqueous medium.11–15 It has been shown that saturation of the hydrosol with ozone induces a reversible shift of the LSPR band to longer wavelength. This shift of the band manifests a size effect: the smaller the nanoparticle size, the greater the shift. The shift of the band was explained by variations of the electron density.13 That is, the smaller the gold particles, the greater the decrease in the electron density of nanoparticles as a result of shifting of conduction electrons to adsorbed ozone molecules. Conversely, the adsorption of oxygen or nitrous oxide does not induce noticeable change in the LSPR band that indicates their weak bonding with the surface of nanoparticles.13
Hydrogen adsorption and dissociation are elementary steps in many reactions involving hydrogen and catalyzed by noble metal (in particular, gold) nanoparticles. We considered it promising to study these phenomena by electron spectroscopy in the gold hydrosol. The mechanism of hydrogen adsorption on gold nanoparticles can be identified by the shift of the plasmon resonance. The influence of various factors (first of all, particle size and pH) on the process can also be revealed by the pattern of plasmon absorption variation. Previously,16 it was shown that the saturation of a gold hydrosol (3 nm particles were used) with hydrogen induces a shift of the LSPR band to the shorter wavelengths. This indicates that the density (or concentration) of electrons in the nanoparticles increases. In other words, hydrogen molecules, unlike ozone, are electron donors. This suggests the dissociative adsorption of the hydrogen on the nanoparticle surface. The use of gold particles of different size and solutions with different pH (from 3.5 to 11.5) confirmed this assumption. A critical size (20–30 nm) above which gold particles are unable to adsorb hydrogen by the dissociative mechanism was established. It was also shown that overpotential on nanoparticles predetermines the possibility of Au(III) reduction. The particle overpotential and the rate of the catalytic reduction of Au(III) ions with hydrogen were shown to depend linearly on the pH.
Experimental
Preparation of Au nanoparticles
The gold hydrosol was synthesized using tetrachloroauric(III) acid trihydrate, sodium borohydride, and sodium citrate, all ACS Reagent Grade chemicals (Aldrich). Solutions were prepared with distilled water, which was additionally deionized on an Arium 611 unit (Sartorius, Germany) and had a specific electrical conductivity of not more than 0.056 μS cm−1.Gold nanoparticles with average sizes of 4.6, 7.8, and 12.2 nm were prepared by pulsed photochemical reduction of Au(III) in aqueous solutions.17 Particles of different sizes were obtained by varying the initial Au(III) concentration and pulsed radiation time. A previous study18 has indicated that the particles with an average size of 4.6 nm prepared by this technique were not twinned but single domain crystals.
Gold particles with sizes of 19 and 28.2 nm were synthesized by the Turkevich procedure.19,20 Gold hydrosols with a mean particle size of 4.6 and 28.2 nm were used as primary subjects of this study.
Saturation with hydrogen
The experiments on hydrogen adsorption on Au nanoparticles were performed as follows. An aliquot of a desired gold colloidal solution (4 mL) was placed into a special cell equipped with a quartz cuvette with a light path of 10.0 mm. The cell design allowed evacuation of it and isolation of its internal volume from the environment. Then hydrogen was injected into the cell. After the optical spectrum was measured, hydrogen was removed from the colloidal solution by vacuum pumping for 20–30 s. After this, the residual hydrogen concentration in the solution did not exceed 10−6 mol L−1.Catalysis
The catalysis was carried out as follows. For the catalytic reduction of Au(III), 1 mL gold hydrosol was mixed with a 2 mL of Au(III) aqueous solution, and a 1 mL trisodium citrate solution. The concentrations of the gold catalyst and trisodium citrate in all solutions were 0.05 mmol L−1 and 1 mmol L−1 respectively. The resulting solution was stirred using a magnetic stirrer for 1 h; then, it was deaerated and saturated with hydrogen.For keeping a constant pH value in solutions, the Britton–Robinson buffer was used.21
Characterization
The absorption spectra of gold colloidal solutions were measured at specified time intervals (up to several days) using a Cary 100 Scan UV-Visible spectrophotometer (Varian Inc.) equipped with a thermostated cuvette compartment at 20 °C (wavelength range 190–900 nm, bandwidth 2 nm, integration time 0.1 s, data interval 1 nm, scan speed 600 nm min−1).The sizes and shapes of the gold nanoparticles were also analyzed using a Leo-912 AB Omega (Carl Zeiss) and JEM-2100 (JEOL) transmission electron microscope. For this purpose, a droplet of a colloidal solution was kept on a formvar-coated copper grid for 30 s and then removed with filter paper. The images were processed using the Gwyddion software.22 The histograms were computed on a minimum of 100 randomly selected nanoparticles.
The pH of colloidal solution was measured using an Econix 120.
Results and discussion
Gold nanoparticles had a spherical shape (Fig. S1, ESI†) and unimodal size distribution. The average sizes of particles mainly studied were 4.6 ± 0.8 nm and 28.2 ± 3.3 nm.It was found that in the pH range from 3.5 to 11.5, the optical spectrum of the gold hydrosol exhibits LSPR absorption bands identical in the position and shape with a maximum at 514 ± 1 nm. At pH < 3.5 and pH > 12, the hydrosol loses the aggregate stability, which finally results in precipitation of the metal nanoparticle aggregates (see Fig. S2, ESI†). After injection of hydrogen into the gold hydrosol at pH from 3.6 to 11.5, the LSPR band shifts to shorter wavelengths. Over a period of 15 min, a limiting and stable position of the band is achieved (Fig. 1). The band shape virtually does not change, while the shift of the maximum (Δλmax, nm) increases with an increase in the pH. It was found that Δλmax increases almost linearly with pH (from 2 ± 0.5 nm at pH 3.6 to 9 ± 0.5 nm at pH 11.1) (Fig. 2).
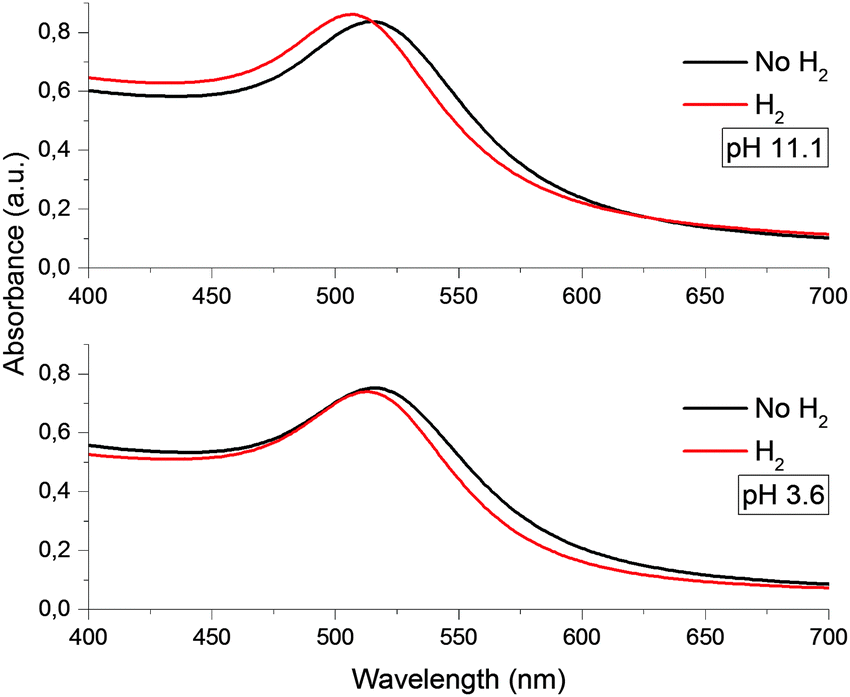 | ||
| Fig. 1 Optical absorption spectra of gold nanoparticles with an average size of 4.6 nm at various pH. | ||
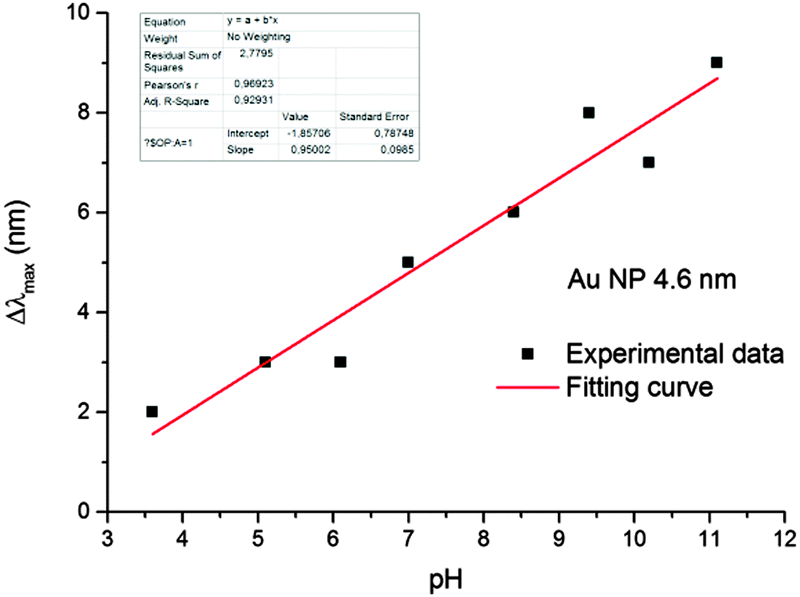 | ||
| Fig. 2 The pH dependence of the shift of LSPR bands of the gold hydrosol in the solutions saturated by the hydrogen at [Aucoll] = 2 × 10−4 mol L−1. | ||
After evacuation of the hydrosol, the LSPR band returns to the initial position. TEM measurements demonstrated that the injection and subsequent removal of hydrogen have almost no effect on the average size of the gold particles in solution (≈4.6 nm) or on the size distribution. Hence, we can conclude that the reversible shift of the LSPR band is not caused by the aggregation or peptization of nanoparticles.
As will be shown below, the detected shift of the LSPR band of the gold hydrosol is due to dissociative adsorption of hydrogen, resulting in an increase in the density (concentration) of electrons in the nanoparticles.
In order to confirm the dissociative mechanism of hydrogen interaction with gold nanoparticles in aqueous solution, experiments on Au(III) reduction with gold nanoparticles were performed. In the absence of nanoparticles, Au(III) is not reduced with hydrogen. This is due to the fact that the standard electrode potential of hydrogen E0(2H+/H2) is much higher than the gold potential E0(AuCl4−/Au0) = −0.05 V.23 Thus, molecular hydrogen cannot reduce Au(III) ions to atoms in bulk solution. Meanwhile, in the presence of nanoparticles, Au(III) ions are efficiently reduced on their surface, as E0(AuCl4−/Aubulk) = 1.08 V. That is, the gold nanoparticles catalyze the reduction. Fig. 3a shows the time variation of the absorption spectrum of the hydrosol measured during the reduction of Au(III) ions with hydrogen in a solution with an initial pH of 10.2.
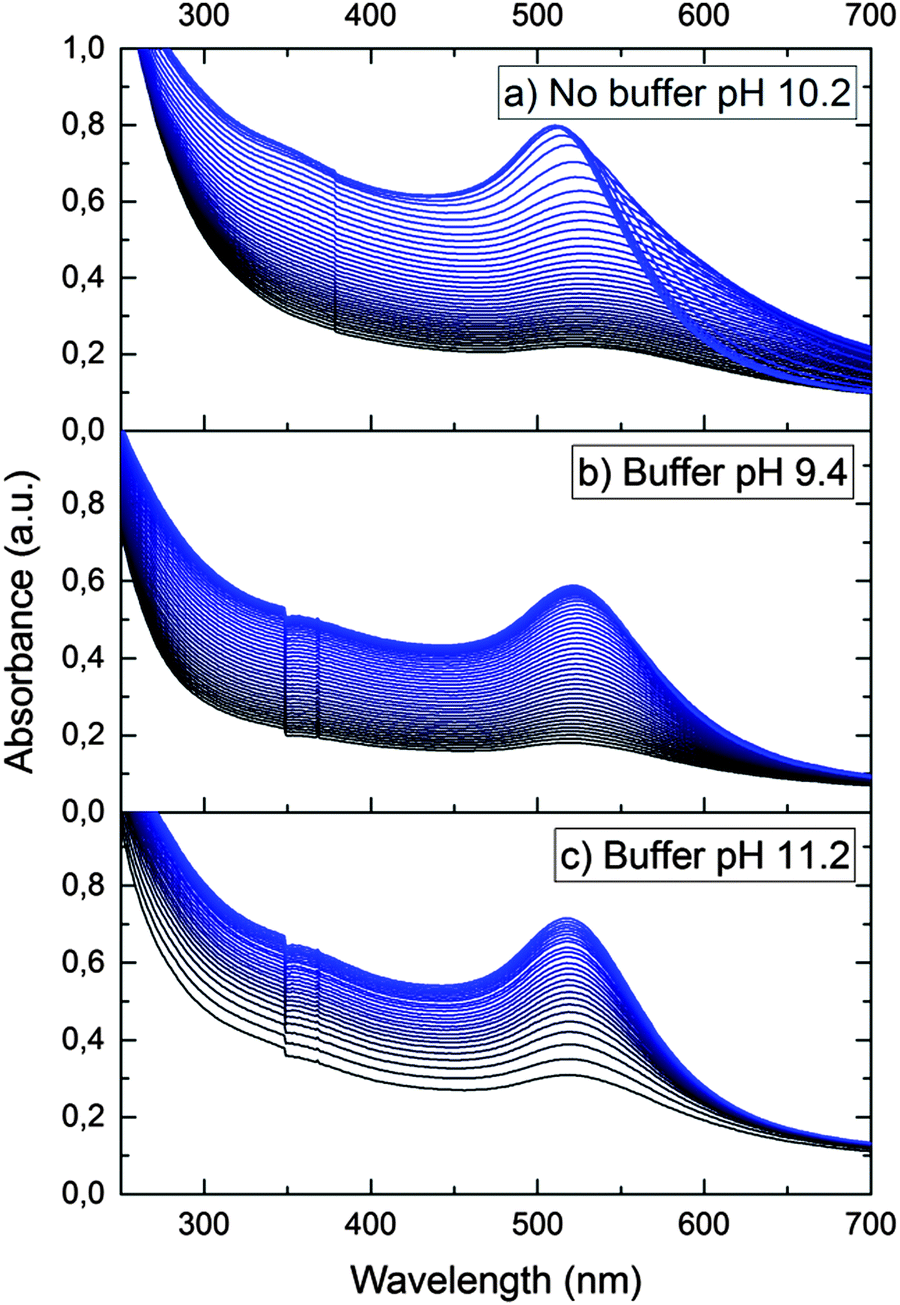 | ||
| Fig. 3 The change in the absorption spectrum of the gold hydrosol during reduction of Au(III) by hydrogen. The solution [Au(III)] = 2 × 10−4 mol L−1, [Aucoll] = 5 × 10−5 mol L−1, [CitNa3] = 1.0 × 10−3 mol L−1. Time interval between curves is 2 min. The average size of gold nanoparticles is 4.6 nm. | ||
It can be seen that the absorption intensity increases with time. The LSPR band first shifts to longer wavelengths and then gradually narrows down and shifts to shorter wavelengths. After 150 min, the reduction is completed. The observed alteration of the LSPR band is due to the change in the composition of the solution caused by the reaction:
| 2AuCl4− + 3H2 → 2Au0 + 8Cl− + 6H+ | (1) |
Fig. 4–7 show the kinetics of plasmon absorption increase caused by reduction of Au(III) ions with hydrogen in the presence of gold nanoparticles. Since, as noted above, new nanoparticles cannot arise in the system, the increase in the absorption can be fully attributed to the growth of the existing particles. This is confirmed by TEM data (Fig. S4, ESI†). As expected, the particle sphericity is simultaneously improved.
Analysis of the kinetics of Au(III) reduction shows a linear proportionality between the reduction rate (determined by the rate of absorption increase in the maximum) and the initial concentration of colloidal gold (Fig. 4). That is, the reaction occurs on the catalyst surface and, as expected, the reaction rate is proportional to the particle surface area. This fact also supports the assumption that the increase in the solution absorbance is due to the increase in the particle size rather than to an increase in the particle concentration. The emergence of new particles would lead to significant deviations from the linear dependence shown in Fig. 4.
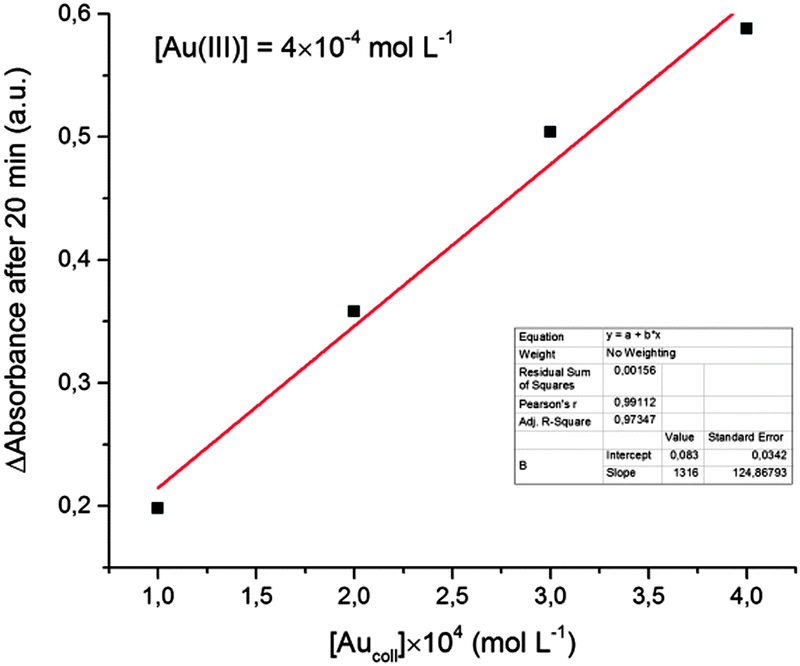 | ||
| Fig. 4 Absorbance change vs. [Aucoll] concentration. | ||
It was also shown (Fig. 5) that the rate of reduction does not depend on the concentration of Au(III) ions in solution, i.e. the reaction follows a zero-order kinetics. The rate constant for reduction increases with solution pH (Fig. 6). Moreover, there is a strict linear relationship between these parameters (Fig. 7).
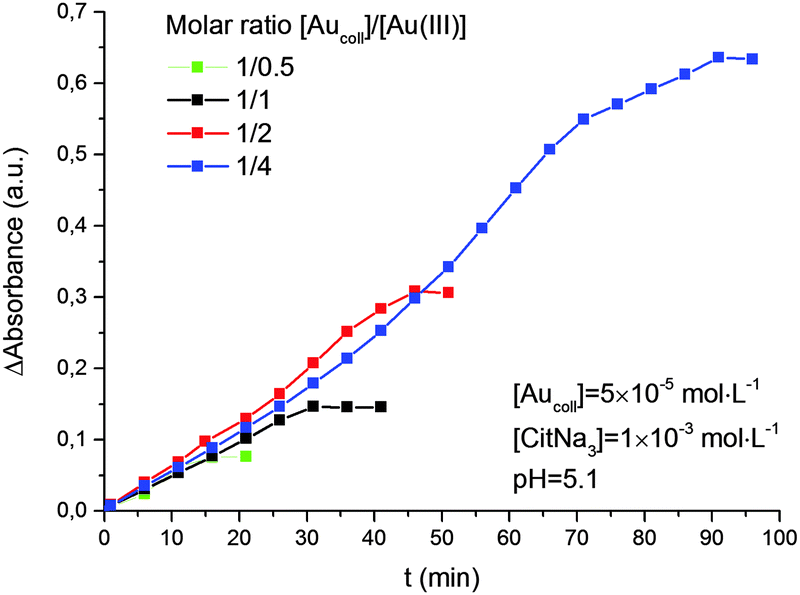 | ||
| Fig. 5 Kinetics of Au(III) reduction by hydrogen with different molar ratios of [Aucoll]/[Au(III)]. The average size of gold nanoparticles is 4.6 nm. | ||
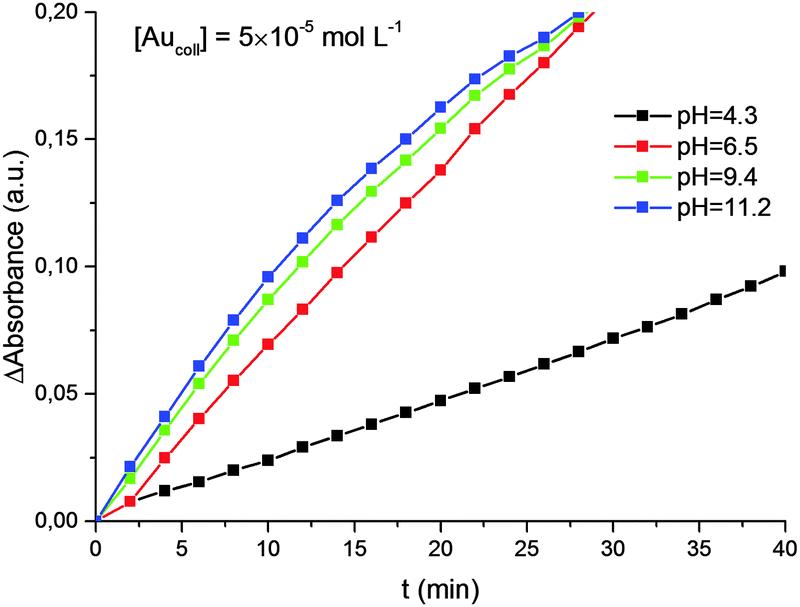 | ||
| Fig. 6 Kinetics of Au(III) reduction by hydrogen at different pH. The average size of gold nanoparticles is 4.6 nm. | ||
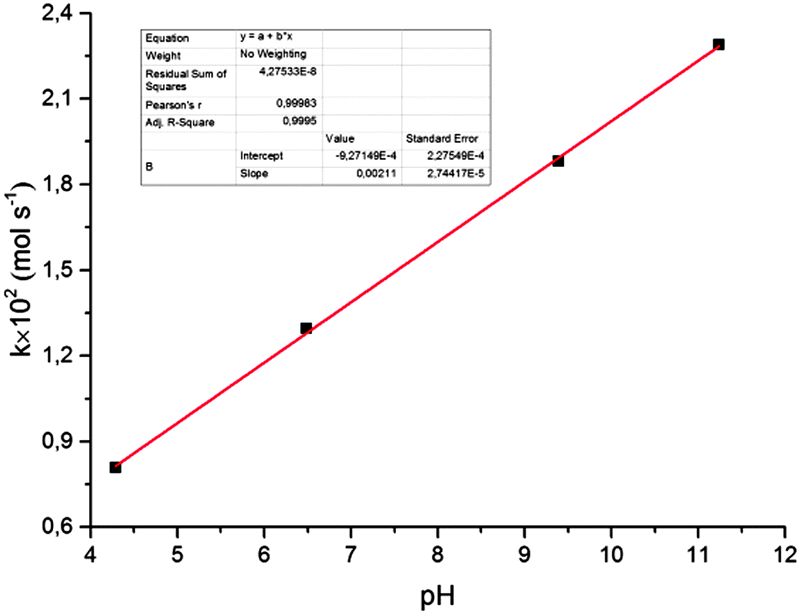 | ||
| Fig. 7 Rate constant for the reduction of Au(III) ions vs. pH. | ||
The properties of the gold hydrosol described above were studied in detail using 4.6 nm particles. The experiments with other particles demonstrated that an increase in the particle size (from 7.8 to 12.2 nm) is accompanied by a substantial decrease in the shift of the LSPR band caused by saturation of solution with hydrogen. Too small value of Δλmax shift did not allow us to establish its quantitative correlation with the particle size. Nevertheless, it was reliably established that in the case of particles larger than 20 nm, the saturation of the gold hydrosol with hydrogen does not cause a shift of the LSPR band (see Fig. 8). Particles of such size were also shown to be much less efficient catalysts for the reduction of Au(III) ions with hydrogen. This is demonstrated by Fig. 8b, which presents the initial kinetics of Au(III) reduction in the gold hydrosol with 4.6 nm (black curve) and 28.2 nm (red curve) particles. For 28.2 nm gold particles, the rate of reduction of Au(III) ions is 10.5 ± 1.4 times lower than for 4.6 nm particles. A similar trend was found in the catalytic properties of the gold nanoparticle supported on γ-Al2O3. It has been shown that the gold nanoparticles of 3.8, 4.6, and 19.4 nm sizes, unlike the bulk metal, can adsorb hydrogen and catalyze the deuterium–protium exchange in the 77–523 K range. The specific catalytic activity of fine particles (4.6 nm) is 20 times as great as that for coarse particles (19.4 nm).24 Thus, the catalytic activity substantially decreases with an increase in the gold particle size.
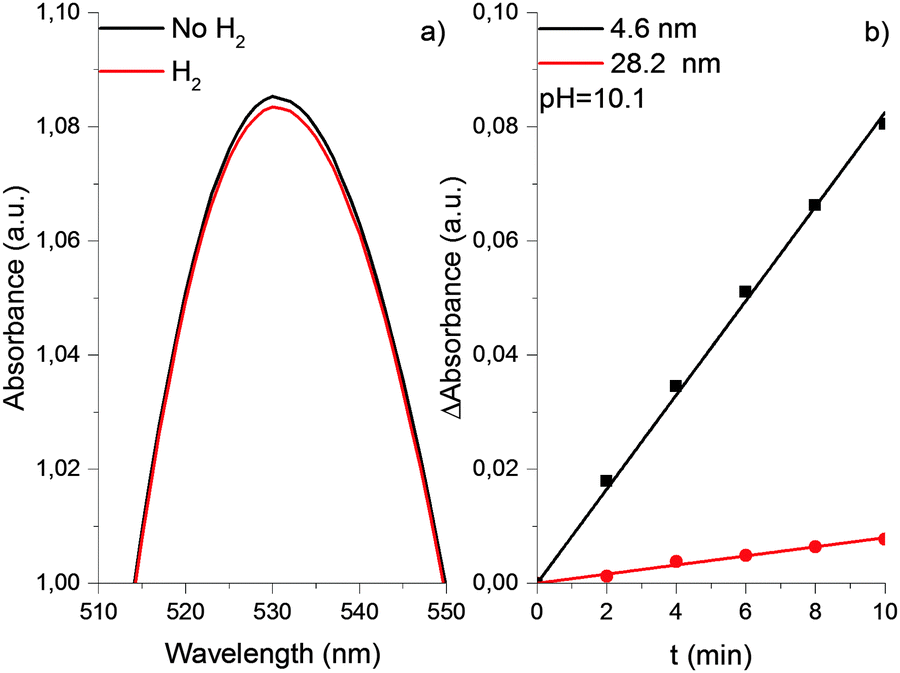 | ||
| Fig. 8 Optical absorption spectra of gold nanoparticles with an average size of 28.2 nm before and after saturation with hydrogen (a). Kinetics of catalytic reduction of Au(III) on gold nanoparticles with different size (b). | ||
Our investigations indicate that dissociative hydrogen adsorption efficiently takes place on gold nanoparticles with small size. The dissociative adsorption results in accumulation of electrons in nanoparticles. Particles acquire a charge and the higher the pH, the higher the charge. When Au(III) ions are present in the solution, electrons “accrued” by nanoparticles are consumed for reduction of these ions. In other words, nanoparticles act as a sort of “hydrogen-like nanoelectrode” performing electrochemical discharge/ionization reactions. As the nanoparticle size increases, the dissociative adsorption of hydrogen on gold decreases, and for large particles (≥20 nm), it becomes hardly detectable. Reduced dissociative adsorption is largely responsible for the significant decrease in the Au(III) reduction rate following the increase in the nanoparticle size. Besides, the decrease in the reduction rate is caused by the decrease in the specific surface area of nanoparticles (at fixed gold mass). Calculations show that the ratio of surface atoms in 4.6 and 28.2 nm spherical particles at the same concentration of gold atoms in the hydrosol is equal to approximately 5.5. Experiments show that the Au(III) reduction rate is actually 10.5 ± 1.4 times higher for 4.6 nm particles. This significant increase in the reduction rate with a decrease in the particle size is, apparently, due to the change in their surface structure. As the particle size decreases, the concentration of structural elements able to atomize the adsorbed hydrogen molecule increases. Indeed, according to published data,25,26 decreasing gold particle size leads to a decrease in the coordination of gold atoms and an increase in the number of the low-coordination edge and corner sites. A larger number of low-coordination atoms in smaller particles results in enhanced activation and atomization of H2 molecules on the gold surface. It is generally considered that hydrogen reacts and dissociates on low-coordination gold atoms located at the corners and edges on the surface of gold nanoparticles.27,28 Ionization of hydrogen atoms is accompanied by accumulation of electrons in the particle, which is manifested as the shift of the LSPR band. This conclusion is supported by the results of ref. 29. It was demonstrated that the active sites for H2 dissociation are located at the corners and edges on the surface of the gold nanoparticles supported on rutile TiO2(110). A computational investigation confirmed that the efficiency of dissociative adsorption of H2 on Aun clusters increases with decreasing cluster size.
We used the approach developed previously25,26 to calculate the percentages of edge and corner atoms versus the total number of atoms on the nanoparticle surface (see Fig. S3, ESI†). It was found that the proportion of these low-coordination sites, which are likely to catalyze atomization of hydrogen molecules, is approximately 18% for 4.6 nm particles. For 28.2 nm particles, it is only 3%. The decrease in the concentration of the active sites on the gold nanoparticle surface with increasing particle size is apparently responsible for the observed noticeable decrease in the rate of Au(III) reduction with hydrogen.
The nature of particular active sites on the nanoparticle surface and the extent to which they influence the hydrogen atomization and the rate of Au(III) reduction remain obscure. It needs to be kept in mind that the specific surface area and the percentages of edge and corner atoms versus the total atoms were calculated with the assumption that the nanoparticles are spherical and have the same size, and the surface corresponds to a perfect crystal structure. In reality, this is not the case. The particle size distribution is fairly broad and the particle shape and crystal structure are not perfect especially for coarse particles. We do not exclude the formation of twinned crystals that can also have an impact on common quantity of edge and corner atoms. It has been also shown that the shape of the gold particles affects the catalytic activity.30 However, we consider that the small particles observed on TEM images of large particles can exert a stronger impact on kinetics than twinned particles (see Fig. S1, ESI†). Perhaps, it is the reason for catalytic reduction of the Au(III) ions by hydrogen observed for coarse particles.
It is noteworthy that, according to ref. 31, nanogold catalysts exhibit a unique preference toward forming cis-2-butene compared to the trans isomer, and the ratio of cis/trans isomers is structure-sensitive in terms of the size of gold nanoparticles. A proportional relationship was revealed: an increase in the fraction of edge + corner atoms following a decrease in the particle size from 9 to 2.7 nm entails a proportional increase in the cis-/trans-2-butene ratio in the products.
The plasmon absorption of light by gold nanoparticles is a sensitive tool for analysis of the electron state in the metal and redox reactions catalyzed by the nanoparticles. According to the Mie–Drude theory,32,33 the position of the LSPR peak λmax is inversely proportional to (ne)1/2, where ne is the concentration of conduction electrons in the metal. The theory predicts a blue shift of the plasmon absorption band with increasing ne and, conversely, a red shift with decreasing ne. Correspondingly, upon the adsorption of hydrogen, the LSPR band actually shows a hypsochromic shift, whereas the adsorption of ozone induces, conversely, a bathochromic shift.14,15 Unfortunately, due to the uncertainty of many parameters used in this theory, it is impossible to calculate the absolute change in the concentration of electrons in gold nanoparticles from the Δλmax value. However, the positions of bands and the relative concentrations of electrons in the metal core are related by the simple expression
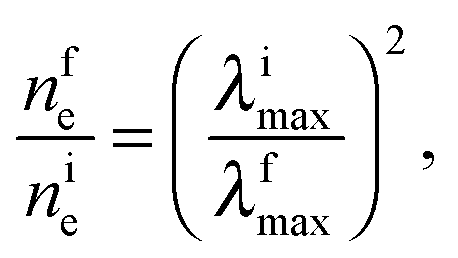 | (2) |
Eqn (2) can be rearranged as follows. Considering that nfe = nie + Δne and λimax = λfmax + Δλ and neglecting the higher powers of small value Δλ/λfmax (≤6%), one gets
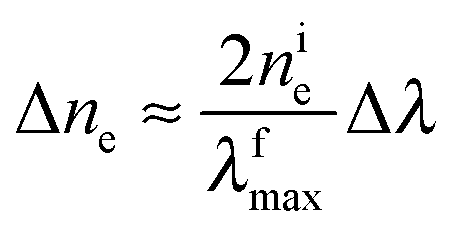 | (3) |
It is evident that the change in the potential is proportional to the change in the concentration of excess electrons in the particle,
| Δφp = −keΔne | (4) |
Formula (4) can be substantiated as follows. We will use the known formula describing the potential variation near a charged spherical particle of radius R placed into a bipolar environment (electrolyte, plasma)34
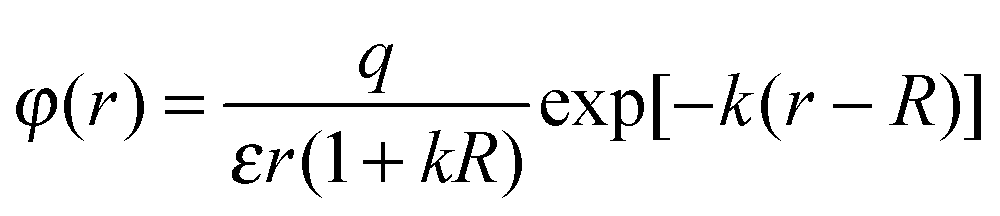 | (5) |
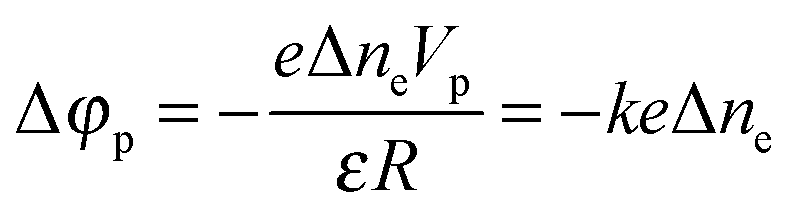 | (6) |
Thus, we assume that hydrogen molecules are dissociatively adsorbed on gold nanoparticles that lead to accumulation of electrons in the metal. Hydrogen is ionized on colloid gold particles as on nanoelectrodes, according to the equation
| H2 + Aucoll(ne) ⇄ 2H+ + 2e | (7) |
The equilibrium constant for reaction (7) is given by
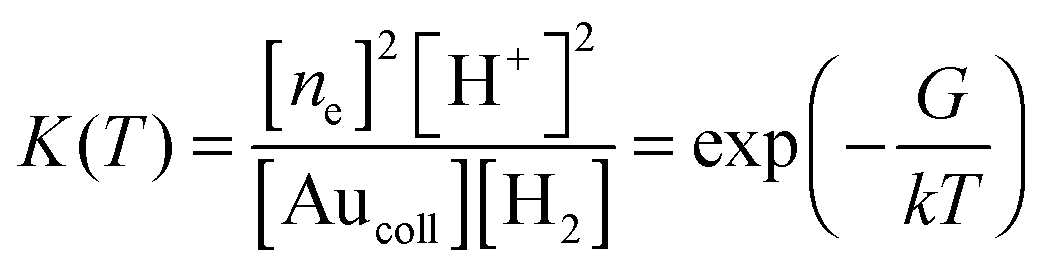 | (8) |
Furthermore, electron transfer towards a nanoparticle (or back) can occur repeatedly. Therefore, an increase and/or decrease in the concentration of electrons in the particle (unlike the bulk electrode) leads to a change in ΔG and the particle potential. Therefore, we will write ΔG in the form
| ΔG = ΔG* − eΔφp, | (9) |
Having employed the above equations, one has
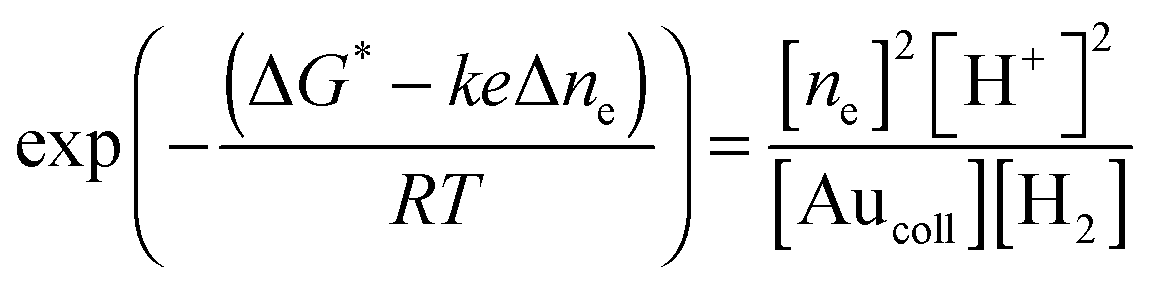 | (10) |
Δne = A* − B*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[H+] = A + B pH, ln[H+] = A + B pH, | (11) |
In view of the relationship between Δne and Δλ (eqn (3)), one gets
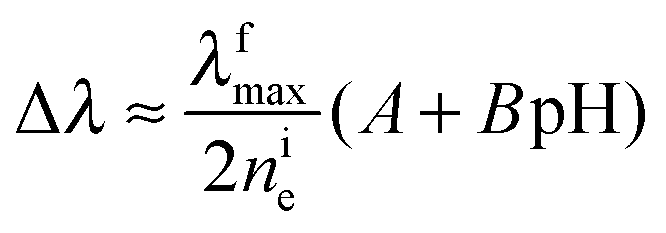 | (12) |
| Δφp = −ke(A + B pH) | (13) |
Now we will consider the reduction of Au(III) ions with hydrogen on gold nanoparticles. This process can be treated, with a sufficient reason, as an electrode reaction, because it is based on the step of transfer of charged species (electrons) through an interface typical of electrochemical processes. Moreover, discharge/ionization is the rate-limiting step in the complex electrode process. It can be described by the following reaction scheme. As a result of dissociative adsorption of hydrogen, electrons are accumulated on the gold particle (particle charging). When a sufficient number of electrons has been accumulated on the nanoparticle, the potential of the particle reaches a value at which reduction of Au(III) ions takes place. The electrochemical process on the gold nanoelectrode is vividly depicted in Fig. 9.
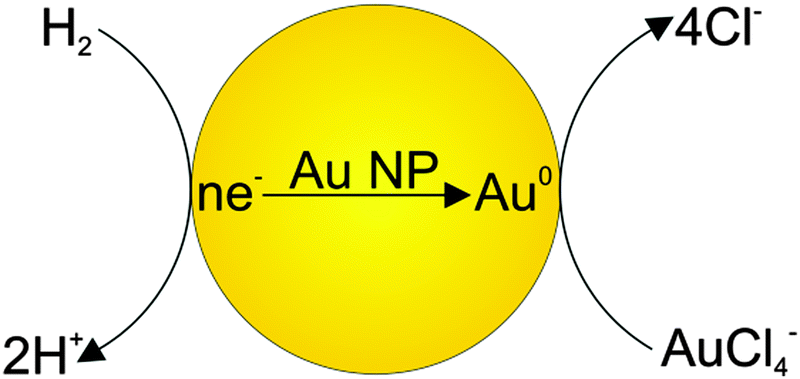 | ||
| Fig. 9 Scheme of the electrochemical process on the gold nanoelectrode. | ||
During this reaction, the excess of electrons decreases (particle discharge). This results in a decrease in the particle potential. Hence, the Au(III) reduction slows down. Finally, a dynamic equilibrium is established in which the particle charging and discharge rates are equal. This gives rise to a quasi-steady state in which the particle potential is somewhat smaller (in magnitude) than that specified by formula (13) and which corresponds to thermodynamic equilibrium in the absence of Au(III) reduction. The potential of the quasi-steady state is determined by the ratio of rate constants for particle charging and discharge.
As noted above, the rate of reaction does not depend on the Au(III) concentration and linearly increases with an increase in the pH. The former fact indicates that particle charging, i.e., dissociative adsorption and the related electron transfer, is the rate-limiting step in the Au(III) reduction. The linear dependence of the rate on the pH can be established from consideration of electrochemical processes with the assumption that the properties of gold particles (electrodes) do not change significantly during the process.
On conventional electrodes, electrochemical reduction of metal ions proceeds if some overpotential (in magnitude) is present on the electrode. According to the Faraday law, the rate of electrolytic deposition of the metal V does not depend on the concentration of cations in solution and is determined by the current intensity:
| V(M) = μI, | (14) |
The same pattern is quite applicable to nanoparticles. Note only that the overpotential on gold nanoparticles appears in our case as a result of reaction (7) and is caused by excess concentration of electrons determined by relation (11). As has already been noted, the rate-limiting step is the accumulation of excess electrons upon hydrogen adsorption and dissociation, whereas Au(III) reduction is comparatively fast. This means that the steady-state potential established on the particles is close to E0 (for nanoparticles, this value can certainly differ from that found for the bulk electrode but we will designate it by the same symbol). Since the overpotential and the concentration of electrons are interrelated, some stationary concentration of excess electrons on nanoparticles is obviously also established, so that the rate of removal of the excess electrons viareaction (1) is equal to the rate of their arrival by reaction (7):
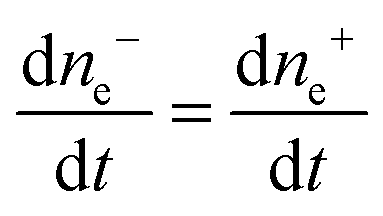 | (15) |
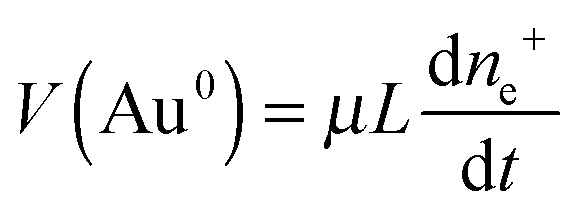 | (16) |
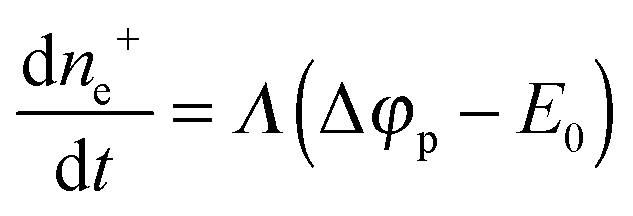 | (17) |
Then, taking account of relations (14)–(17) gives
| V(Au0) = μLΛ(Δφp − E0) = Aef + Bef pH | (18) |
Conclusions
The presented results indicate that dissociative adsorption of hydrogen molecules accompanied by accumulation of electrons in the metal takes place on gold nanoparticles in colloid solution. That is, the situation is similar to electrochemical reactions on a hydrogen electrode. The processes are reflected and recorded in the variation of the LSPR. This fact provides very important information on the mechanisms of reactions catalyzed by gold nanoparticles. In particular, accumulation of electrons in the particles induces an increase (in magnitude) of the overpotential of particles and a shift of the plasmon resonance band to shorter wavelengths. The dependence of the overpotential on the pH typical of a hydrogen electrode is manifested. If Au(III) ions are present in the solution, they are deposited on gold particles. The reduction rate does not depend on the concentration of gold ions. The rate-limiting step of the reaction is dissociative adsorption of hydrogen molecules. The reduction rate is determined by the overpotential and depends linearly on the pH. With increasing size of gold nanoparticles, their ability to accumulate excess electrons by the dissociative mechanism decreases and the Au(III) reduction rate becomes lower. High sensitivity of optical absorption of gold nanoparticles to electron donors and acceptors and to the composition of the environment allows effective use of LSPR spectroscopy to study the mechanism of catalytic reactions.Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. 15-03-02068-a.Notes and references
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev.: Sci. Eng., 1999, 41, 319–388 CAS.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- Y.-S. Bao, M. Baiyin, B. Agula, M. Jia and B. Zhaorigetu, J. Org. Chem., 2014, 79, 6715–6719 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- A. Henglein, P. Molvaney and T. Linnert, Faraday Discuss., 1991, 92, 31–44 RSC.
- A. Henglein and D. Meisel, Langmuir, 1998, 14, 7392–7396 CrossRef CAS.
- B. G. Ershov and A. V. Gordeev, Mendeleev Commun., 2001, 4, 147–148 CrossRef.
- T. Ung, M. Giersig, D. Dunstan and P. Mulvaney, Langmuir, 1997, 13, 1773–1782 CrossRef CAS.
- P. Mulvaney, J. Pérez-Juste, M. Giersig, L. M. Liz-Marzán and C. Pecharromán, Plasmonics, 2006, 1, 61–66 CrossRef CAS.
- P. A. Morozov, B. G. Ershov, E. V. Abkhalimov, O. V. Dement'eva, T. B. Rumyantseva, V. M. Rudoy and V. I. Roldughin, Colloid J., 2011, 73, 668–675 CrossRef CAS.
- P. A. Morozov, B. G. Ershov, E. V. Abkhalimov, O. V. Dement'eva, M. A. Filippenko, V. M. Rudoy and V. I. Roldughin, Colloid J., 2012, 74, 502–509 CrossRef CAS.
- B. G. Ershov, E. V. Abkhalimov, V. I. Roldughin, V. M. Rudoy, O. V. Dement'eva and R. D. Solovov, Phys. Chem. Chem. Phys., 2015, 17, 18431–18436 RSC.
- S. D. Puckett, J. A. Heuser, J. D. Keith, W. U. Spendel and G. E. Pacey, Talanta, 2005, 66, 1242–1246 CrossRef CAS PubMed.
- A. N. Pisarenko, W. U. Spendel, R. T. Taylor, J. D. Brown, J. A. Cox and G. E. Pacey, Talanta, 2009, 80, 777–780 CrossRef CAS PubMed.
- B. G. Ershov, V. I. Roldughin, E. V. Abkhalimov, R. D. Solovov, O. V. Dement'eva and V. M. Rudoy, Colloid J., 2014, 76, 308–313 CrossRef CAS.
- R. D. Solovov and B. G. Ershov, Colloid J., 2014, 76, 595–599 CrossRef CAS.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- B. V. Enutsun and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef.
- H. T. S. Britton and R. A. Robinson, J. Chem. Soc., 1931, 1456–1462 RSC.
- D. Necas and P. Klapetek, Cent. Eur. J. Phys., 2012, 10, 181–188 Search PubMed.
- B. G. Ershov, Russ. Chem. Rev., 1981, 50, 1119–1133 CrossRef.
- O. A. Boeva, B. G. Ershov, K. N. Zhavoronkova, A. A. Odintsov, R. D. Solovov, E. V. Abkhalimov and N. D. Evdokimenko, Dokl. Phys. Chem., 2015, 463, 165–167 CrossRef CAS.
- S. Schimpf, M. Lucas, C. Mohr, U. Rodemerck, A. Bruckner, J. Radnik, H. Hofmeister and P. Claus, Catal. Today, 2002, 72, 63–78 CrossRef CAS.
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef CAS.
- C. Mohr, H. Hofmeister, J. Radnik and P. Claus, J. Am. Chem. Soc., 2003, 125, 1905–1911 CrossRef CAS PubMed.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS PubMed.
- A. Lyalin and T. Taketsugu, Faraday Discuss., 2011, 152, 185–201 RSC.
- C. Mohr, H. Hofmeister and P. Claus, J. Catal., 2003, 213, 86–94 CrossRef CAS.
- X.-F. Yang, A.-Q. Wang, Y.-L. Wang, T. Zhang and J. Li, J. Phys. Chem. C, 2010, 114, 3131–3139 CAS.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, Willey, New York, 1983 Search PubMed.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, 1995 Search PubMed.
- R. Kubo, Statistical Mechanics, Amsterdam, North-Holland, 1965 Search PubMed.
- D. H. Everett, Basic Principles of Colloid Science, The R. Soc. Chem., London, 1988 Search PubMed.
- S. R. de Groot and P. Mazur, Non-Equilibrium Thermodynamics, Amsterdam, North-Holland, 1962 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp01996j |
| This journal is © the Owner Societies 2016 |